 Jenny Sassone
Jenny Sassone Flavia Valtorta
Flavia Valtorta Andrea Ciammola
Andrea Ciammola- 1Division of Neuroscience, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 2Vita-Salute San Raffaele University, Milan, Italy
- 3Department of Neurology, IRCCS Istituto Auxologico Italiano, Milan, Italy
Mutations in the PARKIN gene cause early-onset Parkinson’s disease (PD). Despite the high proportion of still missing phenotyping data in the literature devoted to early-onset PD, studies suggest that, as compared with late-onset PD, PARKIN patients show dystonia at onset and extremely dose-sensitive levodopa-induced dyskinesia (LID). What pathophysiological mechanisms underpin such early and atypical dyskinesia in patients with PARKIN mutations? Though the precise mechanisms underlying dystonia and LID are still unclear, evidence suggests that hyperkinetic disorders in PD are a behavioral expression of maladaptive functional and morphological changes at corticostriatal synapses induced by long-term dopamine (DA) depletion. However, since the dyskinesia in PARKIN patients can also be present at onset, other mechanisms beside the well-established DA depletion may play a role in the development of dyskinesia in these patients. Because cortical and striatal neurons express parkin protein, and parkin modulates the function of ionotropic glutamatergic receptors (iGluRs), an intriguing explanation may rest on the potential role of parkin in directly controlling the glutamatergic corticostriatal synapse transmission. We discuss the novel theory that loss of parkin function can dysregulate transmission at the corticostriatal synapses where they cause early maladaptive changes that co-occur with the changes stemming from DA loss. This hypothesis suggests an early striatal synaptopathy; it could lay the groundwork for pharmacological treatment of dyskinesias and LID in patients with PARKIN mutations.
Introduction
Loss of function mutations in the PARKIN gene cause autosomal recessive juvenile parkinsonism (ARJP) (Kitada et al., 1998). Autopsy studies have shown that neuropathological changes in patients with PARKIN mutations differ from those observed in idiopathic Parkinson’s disease (iPD). Unlike iPD, neuronal loss is greater in the substantia nigra pars compacta (SNpc) than in the locus coeruleus in most parkin cases. Also, Lewy bodies are not detected in the majority of parkin cases, suggesting a difference in the pathogenic processes between PARKIN-related disease and iPD (Schneider and Alcalay, 2017). Also, PARKIN-associated, early-onset PD is phenotypically distinct from iPD. Recent reviews in the clinical literature on genetic PD have pointed to the lack of a detailed description of individual phenotypic features (Kasten et al., 2017, 2018). Evidence suggests, however, that unlike late-onset PD patients, parkin patients display dystonia at early disease stages and dyskinesia at exceedingly low dosages of levodopa (Wickremaratchi et al., 2011). Early dystonia and dyskinesia are also reported in other early-onset genetic forms of PD such as PINK1 and DJ1 (Lohmann et al., 2009). Since PARKIN mutations account for about three-quarters of PD cases with an age at onset of 30 years or younger, understanding the pathophysiological mechanisms that underlie early and atypical dystonia and dyskinesia in PARKIN patients is a key step toward achieving a more personalized treatment for this genetic condition and toward understanding the biological basis of this phenomena in other early-onset genetic forms of PD.
Here, we review the clinical evidence for early hyperkinetic symptoms in patients with PARKIN mutations and the recent evidence that parkin modulates glutamatergic neurotransmission. We discuss the hypothesis that early hyperkinetic symptoms of PD patients with PARKIN mutation may be considered manifestations of corticostriatal synaptopathy. These features can be linked to dopamine depletion, a well-recognized mechanism in the literature, but they also may be related to a potential direct effect of parkin on the corticostriatal synapse.
Clinical Evidence for Dystonia and Dyskinesia in Parkin Patients
Since the first description of juvenile parkinsonism caused by mutations of the PARKIN gene in two families (Kitada et al., 1998), dystonia has been proposed as a clinical hallmark of the disease (Schrag and Schott, 2006). Recent literature reviews have reported that an overall, clinically typical form of PD with early onset, slow progression, and excellent response to levodopa treatment is frequently associated with dystonia and dyskinesia in patients with PARKIN mutations (Grünewald et al., 2013; Kasten et al., 2018). Usually independent of levodopa intake, dystonia is described as the presenting symptom in a large percentage of PARKIN patients in whom it can be present in isolation for years before the appearance of parkinsonism (Khan et al., 2003). The reported phenotypic overlap between PD patients with PARKIN mutation and rare forms of dystonia-parkinsonism, such as levodopa-responsive dystonia (DRD), prompts screening for PARKIN mutations along with GTP cyclohydrolase 1 and tyrosine hydroxylase in clinically diagnosed DRD patients (Tassin et al., 2000; Wu et al., 2005, 2008; Schneider and Bhatia, 2010; Potulska-Chromik et al., 2017). To determine whether dystonia contributes to gait abnormalities in patients with PARKIN mutations, a recent study used a clinical computerized video motion analysis system to evaluate lower limb dystonia severity in 15 patients. Lower limb dystonia occurred in most PARKIN patients who displayed a specific, abnormal gait pattern that differed from the patterns observed in healthy controls independent of the OFF/ON state (Castagna et al., 2016).
In addition to dystonia, PARKIN patients show dyskinesia at exceedingly low dosages of levodopa as compared with late-onset PD patients. On average, the response to low doses of levodopa is reported as being excellent and sustained. However, the likelihood of developing LID is reportedly higher than in individuals with parkinsonism resulting from other aetiologies (Brüggemann and Klein, 1993; Paviour et al., 2004) and it is inducible by very-low-dose levodopa in some cases (Khan et al., 2003). While bradykinesia responds well even to low levodopa dosages in PARKIN patients, the same low doses seem to exacerbate dystonia already in very early stages of the disease. Although conflicting results have also been reported (Kasten et al., 2018), the present evidence suggests that the incidence of hyperkinetic disorders at earlier disease stages is higher in PARKIN patients than in those with iPD.
Potential Molecular Mechanisms Underlying Dystonia and Dyskinesia
The precise mechanisms underlying dystonia and LID in PD are unknown. Studies on PD animal models, mainly dopamine-depleted animals, and PD patients strongly suggest that dyskinesia results from dysfunction of glutamatergic transmission from the cortex to the striatal medium spiny neurons (MSNs) (Picconi et al., 2017). MSNs are characterized by radially projecting dendrites densely studded with spines and receiving excitatory glutamatergic inputs from the cerebral cortex which form in the order of 5000 contacts per neuron (Graveland et al., 1985; David Smith and Paul Bolam, 1990; Yelnik et al., 1991). The dendritic spines bear the asymmetrical glutamatergic synapse on the head and the symmetric dopaminergic synapses on the neck (Figure 1A; David Smith and Paul Bolam, 1990). This characteristic synaptic triad boosted the widely accepted theory that dopamine (DA) physiologically modulates the strength of cortical inputs to the MSNs (Gerfen and Surmeier, 2011) and that long-term DA depletion occurring over the course of idiopathic PD triggers prominent secondary changes in the corticostriatal synapses (Picconi et al., 2017). Indeed, DA depletion leads to strong sensitization of post-synaptic D1 receptors, which is critical for the development of dyskinesia (Darmopil et al., 2009), and induces a variety of molecular changes in the downstream signaling pathways, including an increase in the state of phosphorylation of the DA- and cAMP-regulated phosphoprotein of 32 KDa (DARPP-32) (Santini et al., 2007; Heumann et al., 2014; Ruiz-Dediego et al., 2015; Solís et al., 2017). DARPP-32 phosphorylation promotes a cascade of intracellular events in MSNs: activation of extracellular signal-regulated kinase 1 and 2 (ERK1 and 2) and, as a consequence, activation of the ERK and the mammalian target of rapamycin complex 1 (mTORC1) signaling pathways (Santini et al., 2012; Heumann et al., 2014).
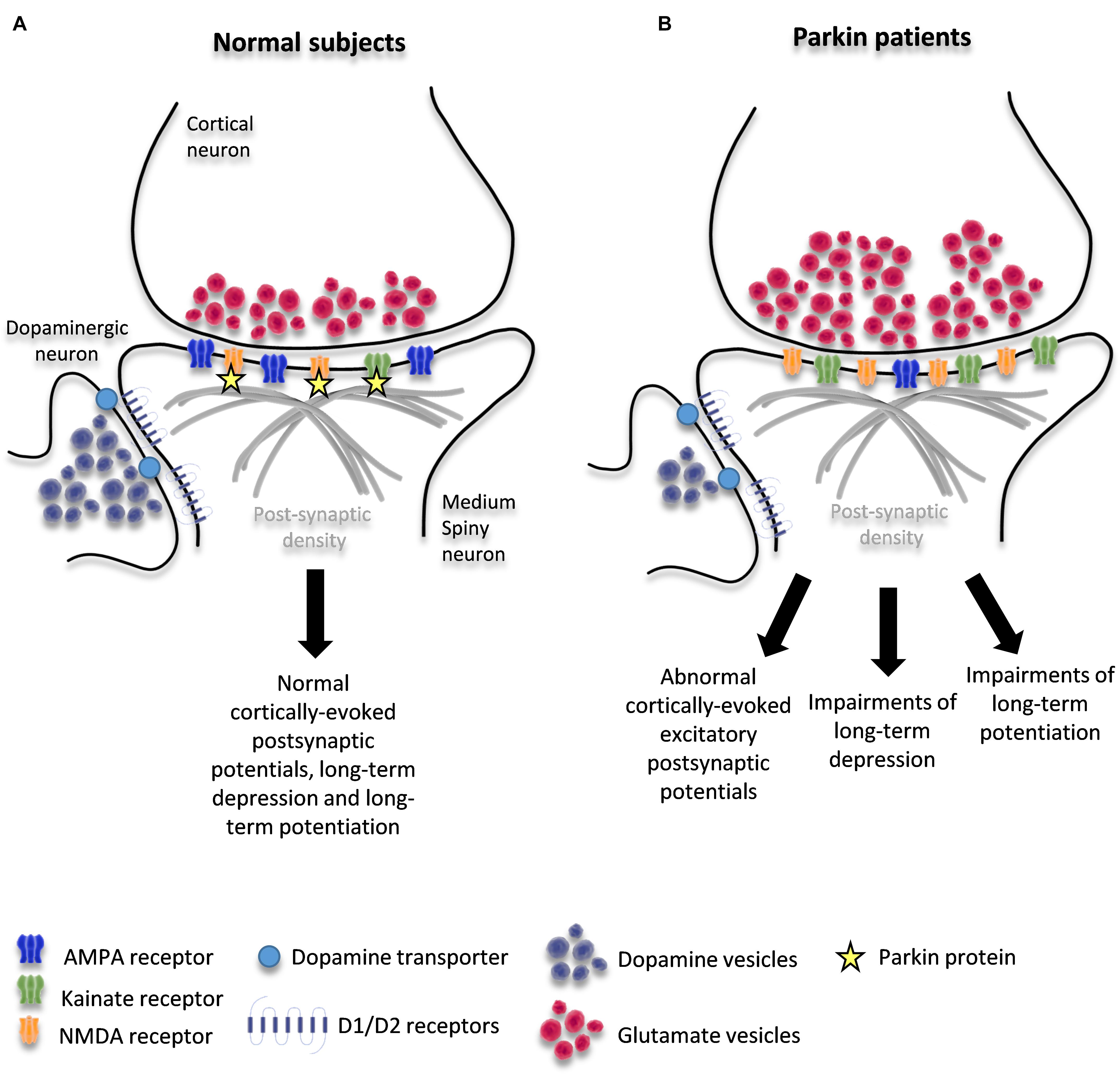
Figure 1. Schematic diagram of corticostriatal synapse in healthy condition (A) and in patients with PARKIN mutations (B). The figure shows the interaction of parkin with KAR and NMDAR and potential changes induced by loss of PARKIN function.
The molecular signatures that have recently emerged as key signals in LIDs are ERK1/2 phosphorylation (Pavón et al., 2006), abnormal activation of mitogen- and stress-activated protein kinase-1 (MSK-1), and phosphorylation of histone H3 (Santini et al., 2007; Heumann et al., 2014). The final result of this chain of events is a change in the molecular make-up of MSNs due to alterations in gene expression and protein synthesis that might account for maladaptive morphological plasticity at the corticostriatal synapse (Picconi et al., 2017). Morphological examination of MSNs in dopamine-depleted animals and in autopsy specimens from iPD patients has revealed dendrite atrophy, truncated dendrites, and decreased spine density (Stephens et al., 2005; Zaja-Milatovic et al., 2005; Suarez et al., 2016). These functional and morphological changes in the corticostriatal synapses are believed to result in dyskinesias (Picconi et al., 2017).
The question arises as to whether the same molecular mechanisms underlie the early and atypical dyskinesia displayed by PARKIN patients. Clinical studies with [18F] Dopa as a marker of the functional integrity of pre-synaptic dopamine nerve terminals have shown that Parkin mutated patients have widespread pre-synaptic dopaminergic deficits (Scherfler et al., 2004). So while the early hyperkinetic symptoms in patients with parkin mutations can be linked to DA depletion, the appearance of dyskinesia in early disease stages, before the onset of typical and cardinal signs of parkinsonism, suggests that additional mechanisms may be involved. In particular, dyskinesia-related molecular changes in patients with PARKIN mutation may partially stem from dysfunctions of the corticostriatal synapses due to the loss of parkin function in these synapses.
The Potential Role of Parkin at the Corticostriatal Synapse
In vitro studies have revealed that parkin can localize at the pre-synapse, where it associates with the cytoplasmic surface of synaptic vesicles (Kubo et al., 2001; Mouatt-Prigent et al., 2004) and binds to synaptotagmin-11, a pre-synaptic protein involved in synaptic vesicle formation, docking, and recycling (Huynh et al., 2003; Wang et al., 2018). The loss of parkin function may inhibit endocytosis and the processes of vesicle replenishment and recycling, leading to as yet undefined changes in neurotransmitter release. Interestingly, the pre-synaptic functions of parkin resemble the function of synuclein, another key protein involved in PD pathogenesis and a regulator of pre-synapse size and synaptic vesicle pool organization (Anwar et al., 2011; Vargas et al., 2017), additionally, the roles of other PD genes such as DNAJC6, SYNJ1, SH3GL2, LRRK2, and VPS35 in the regulation of synaptic vesicle trafficking SVE are beginning to emerge (Nguyen et al., 2018).
In vitro studies on glutamatergic neurons showed that parkin can also localize at the post-synapse, where it directly modulates ionotropic glutamate receptor function. Parkin directly interacts with and ubiquitinates the GluK2 subunit of kainate receptor (KAR) and thus regulates its function: loss of parkin increases KAR levels in the human cortex and KAR function in glutamatergic hippocampal neurons in vitro (Maraschi et al., 2014). At the post-synapse, loss of parkin function probably leads to dysregulation of other neurotransmitter receptors as well: a study on brain tissues from parkin knockout mice showed up-regulation of KAR density, increased NMDA receptor density, and reduced AMPA receptor density throughout various brain regions (Cremer et al., 2015). The role of parkin in modulating AMPA receptors was further confirmed in an in vitro study showing that parkin-deficient hippocampal neurons exhibit significantly reduced AMPA receptor-mediated currents (Cortese et al., 2016). The suggested underlying mechanism is the decrease in the post-synaptic expression of the adaptor protein Homer1, which is necessary for coupling AMPAR endocytic zones with post-synaptic density. A more recent study evaluated the impact of four parkin point mutations on glutamatergic synaptic function in hippocampal neurons and found that these mutants alter NMDA and AMPA receptor-mediated currents and cell-surface levels and that they prevent the induction of long-term depression. The study demonstrated that parkin regulates NMDA receptor trafficking through ubiquitination of the subunit GluN1 and that the mutations impair ubiquitination activity (Zhu et al., 2018). Hence, the bulk of evidence strongly suggests that PARKIN mutations disturb glutamatergic synaptic transmission by directly changing NMDAR, AMPAR, and KAR receptor function. Since parkin is widely expressed in various brain regions and neuron types, including cortical neurons and MSNs, (Kitada et al., 1998; Stichel et al., 2000) it is conceivable that the loss of parkin directly induces pathological effects on these neurons and contributes to the pathophysiology of dyskinesia.
These studies on the localization of parkin and its capacity to directly regulate excitatory transmission constitute an important step toward understanding the functions of parkin at the synapses (Sassone et al., 2017). The complementary approach was to investigate the function of nigrostriatal and corticostriatal synapses in murine models knockout for Parkin gene. Unfortunately, studies on the nigrostriatal pathway revealed many discrepancies probably deriving from different experimental conditions and/or from in vivo age-dependent compensatory mechanisms. Increased spontaneous DA release from SNc DA neurons was found in Parkin-knockout mice aged 8 to 9 months (Goldberg et al., 2003). Decreased evoked DA release was found in 3-to-9-month-old Parkin knockout mice (Oyama et al., 2010). In the striatum of 22-month-old wild-type and Parkin-knockout mice, DA levels were reported to be indistinguishable (Perez and Palmiter, 2005). DA overflow in striatal slices from 6-to-8-week-old mice was found to be equal between wild-type and Parkin-knockout mice (Sanchez et al., 2014). Indeed, not all the evidence supports the idea that DA release is impaired in the nigrostriatal pathway of Parkin-knockout mice. Studies on corticostriatal synapses performed by intracellular recordings of MSNs revealed impairment in long-term depression and long-term potentiation in Parkin-knockout mice (Kitada et al., 2009). These results suggest that parkin is involved in the regulation of striatal synaptic plasticity. Unfortunately, however, Parkin-knockout mice do not show DA neuron loss, motor impairment, dystonic phenotype or hypersensitivity to levodopa (Perez and Palmiter, 2005). The authors suggest that this murine model may represent a tool to study the early stages of the disease (Dawson et al., 2010); nonetheless, its usefulness as a disease model is limited and complicates the interpretation of the results obtained on these mice. More promising results come from studies on dopaminergic neurons differentiated from induced human pluripotent stem cells. When glutamatergic transmission was studied in a mixed population of GABAergic, glutamatergic, and dopaminergic neurons differentiated from induced human pluripotent stem cells, PARKIN mutations were observed to potentiate glutamatergic transmission by modifying synaptic vesicle function at the pre-synaptic site of the corticostriatal synapse (Zhong et al., 2017). Moreover, the actions of dopamine on D1-class receptors were noted to be markedly altered in dopaminergic neurons differentiated from induced human pluripotent stem cells of PD patients with PARKIN mutations. In particular, as compared with control neurons, the parkin-deficient neurons had a stronger response to the activation of D1-class dopamine receptors (Zhong et al., 2017).
This raises the possibility that parkin could be involved in D1 receptor hypersensitization after dopaminergic denervation, an important mechanism underlying the dyskinesia in iPD (Heumann et al., 2014). Accordingly, a recent paper showed that parkin ubiquitinates and regulates the levels of STEP61, the striatal enriched protein tyrosine phosphatase, whereas clinically relevant parkin mutants fail to do so. Because STEP61 substrates include ERK1/2, it is conceivable that a parkin-induced increase in STEP61 might influence D1 signaling in MSNs (Kurup et al., 2015).
Taken together, these findings suggest that parkin orchestrates a complex remodeling of corticostriatal synapses through multiple mechanisms. These mechanisms are likely to include changes due to DA depletion as well as those due to loss of parkin function at these synapses: alterations in the trafficking of glutamate vesicles at the glutamatergic pre-synapse and/or dysregulation of glutamate receptor levels on MSNs and/or dysregulation of D1-signaling. These mechanisms may be responsible for the precocious maladaptive plasticity of the corticostriatal synapses that leads to early dyskinesia (Figure 1B).
Conclusion
Since the parkin protein modulates glutamatergic receptor function and possibly glutamate release as well, it is conceivable that PARKIN mutation affects corticostriatal transmission, leading to early maladaptive changes in this glutamatergic synapse. This alteration could produce changes due to corticostriatal-induced depletion of DA at the synapses. What does this scenario imply for the development of specific and effective treatments? The only currently available drug for symptomatic treatment of dyskinesia is amantadine, an antiviral agent introduced in the early 1970s that also displays NMDA receptor antagonism (Hubsher et al., 2012). With a better understanding of the mechanisms by which parkin causes early dystonia and dyskinesia, researchers may be able to identify novel therapeutic targets and develop strategies that include drugs that act on the glutamatergic system. Such drugs are already under investigation for treating dyskinesias in iPD (Müller, 2017). Moreover, if our hypothesis proves true, the distinct molecular mechanisms underlying dyskinesia between PARKIN patients and late-onset PD patients could be relevant for devising precision medicine strategies for parkin patients. The recent discovery that LID is caused by a distinct and stable group of striatal neurons (Girasole et al., 2018) paves the way to further studies aimed at investigating the role of parkin in these neurons and at their corticostriatal synapse.
In conclusion, published data support the hypothesis of an early corticostriatal synaptopathy in parkin patients caused by parkin modulating the glutamatergic synapse. This hypothesis could provide a biological basis for studying the disease phenotype and lay the groundwork for the pharmacological treatment of PD patients with PARKIN mutation.
Author Contributions
JS wrote first draft of the manuscript. FV and AC reviewed and critically revised the manuscript.
Funding
This work was supported by the Telethon Foundation (Grant GGP15167 to AC) and the Italian Ministry of Health (GR-2016-02361366 to JS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Floriano Girotti for critical revision and reading of the manuscript. The Michael J. Fox Foundation for Parkinson’s Research (Grant MJFF14110 to JS).
References
Anwar, S., Peters, O., Millership, S., Ninkina, N., Doig, N., Connor-Robson, N., et al. (2011). Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J. Neurosci. 31, 7264–7274. doi: 10.1523/JNEUROSCI.6194-10.2011
Brüggemann, N., and Klein, C. (1993). “Parkin type of early-onset Parkinson disease,” in GeneReviews® [Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Castagna, A., Frittoli, S., Ferrarin, M., Del Sorbo, F., Romito, L. M., Elia, A. E., et al. (2016). Quantitative gait analysis in parkin disease: possible role of dystonia. Mov. Disord. 31, 1720–1728. doi: 10.1002/mds.26672
Cortese, G. P., Zhu, M., Williams, D., Heath, S., and Waites, C. L. (2016). Parkin deficiency reduces hippocampal glutamatergic neurotransmission by impairing AMPA receptor endocytosis. J. Neurosci. 36, 12243–12258. doi: 10.1523/JNEUROSCI.1473-16.2016
Cremer, J. N., Amunts, K., Schleicher, A., Palomero-Gallagher, N., Piel, M., Rösch, F., et al. (2015). Changes in the expression of neurotransmitter receptors in Parkin and DJ-1 knockout mice. quantitative multireceptor study. Neuroscience 311, 539–551. doi: 10.1016/j.neuroscience.2015.10.054
Darmopil, S., Martín, A. B., De Diego, I. R., Ares, S., and Moratalla, R. (2009). Genetic inactivation of dopamine D1 but Not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 66, 603–613. doi: 10.1016/j.biopsych.2009.04.025
David Smith, A., and Paul Bolam, J. (1990). The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurones. Trends Neurosci. 13, 259–265. doi: 10.1016/0166-2236(90)90106-K
Dawson, T. M., Ko, H. S., and Dawson, V. L. (2010). Genetic animal models of Parkinson’s disease. Neuron 66, 646–661. doi: 10.1016/j.neuron.2010.04.034
Gerfen, C. R., and Surmeier, D. J. (2011). Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 34, 441–466. doi: 10.1146/annurev-neuro-061010-113641
Girasole, A. E., Lum, M. Y., Nathaniel, D., Bair-Marshall, C. J., Guenthner, C. J., Luo, L., et al. (2018). A subpopulation of striatal neurons mediates levodopa-induced dyskinesia. Neuron 97, 787–795. doi: 10.1016/j.neuron.2018.01.017
Goldberg, M. S., Fleming, S. M., Palacino, J. J., Cepeda, C., Lam, H. A., Bhatnagar, A., et al. (2003). Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 278, 43628–43635. doi: 10.1074/jbc.M308947200
Graveland, G. A., Williams, R. S., and Difiglia, M. (1985). A golgi study of the human neostriatum: neurons and afferent fibers. J. Comp. Neurol. 234, 317–333. doi: 10.1002/cne.902340304
Grünewald, A., Kasten, M., Ziegler, A., and Klein, C. (2013). Next-generation phenotyping using the Parkin example: time to catch up with genetics. JAMA Neurol. 70, 1186–1191. doi: 10.1001/jamaneurol.2013.488
Heumann, R., Moratalla, R., Herrero, M. T., Chakrabarty, K., Drucker-Colín, R., Garcia-Montes, J. R., et al. (2014). Dyskinesia in Parkinson’s disease: mechanisms and current non-pharmacological interventions. J. Neurochem. 130, 472–489. doi: 10.1111/jnc.12751
Hubsher, G., Haider, M., and Okun, M. S. (2012). Amantadine: the journey from fighting flu to treating Parkinson disease. Neurology 78, 1096–1099. doi: 10.1212/WNL.0b013e31824e8f0d
Huynh, D. P., Scoles, D. R., Nguyen, D., and Pulst, S. M. (2003). The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum. Mol. Genet. 12, 2587–2597. doi: 10.1093/hmg/ddg269
Kasten, M., Hartmann, C., Hampf, J., Schaake, S., Westenberger, A., Vollstedt, E. J., et al. (2018). Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov. Disord. 33, 730–741. doi: 10.1002/mds.27352
Kasten, M., Marras, C., and Klein, C. (2017). Nonmotor signs in genetic forms of Parkinson’s disease. Int. Rev. Neurobiol. 133, 129–178. doi: 10.1016/bs.irn.2017.05.030
Khan, N. L., Graham, E., Critchley, P., Schrag, A. E., Wood, N. W., Lees, A. J., et al. (2003). Parkin disease: a phenotypic study of a large case series. Brain 126, 1279–1292. doi: 10.1093/brain/awg142
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Kitada, T., Pisani, A., Karouani, M., Haburcak, M., Martella, G., Tscherter, A., et al. (2009). Impaired dopamine release and synaptic plasticity in the striatum of Parkin-/- mice. J. Neurochem. 110, 613–621. doi: 10.1111/j.1471-4159.2009.06152.x
Kubo, S., Kitami, T., Noda, S., Shimura, H., Uchiyama, Y., Asakawa, S., et al. (2001). Parkin is associated with cellular vesicles. J. Neurochem. 78, 42–54. doi: 10.1046/j.1471-4159.2001.00364.x
Kurup, P. K., Xu, J., Videira, R. A., Ononenyi, C., Baltazar, G., Lombroso, P. J., et al. (2015). STEP 61 is a substrate of the E3 ligase parkin and is upregulated in Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 112, 1202–1207. doi: 10.1073/pnas.1417423112
Lohmann, E., Thobois, S., Lesage, S., Broussolle, E., Du Montcel, S. T., Ribeiro, M. J., et al. (2009). A multidisciplinary study of patients with early-onset PD with and without parkin mutations. Neurology 72, 110–116. doi: 10.1212/01.wnl.0000327098.86861.d4
Maraschi, A. M., Ciammola, A., Folci, A., Sassone, F., Ronzitti, G., Cappelletti, G., et al. (2014). Parkin regulates kainate receptors by interacting with the GluK2 subunit. Nat. Commun. 5:5182. doi: 10.1038/ncomms6182
Mouatt-Prigent, A., Muriel, M. P., Gu, W. J., El Hachimi, K. H., Lücking, C. B., Brice, A., et al. (2004). Ultrastructural localization of parkin in the rat brainstem, thalamus and basal ganglia. J. Neural Transm. 111, 1209–1218. doi: 10.1007/s00702-004-0144-9
Müller, T. (2017). Current and investigational non-dopaminergic agents for management of motor symptoms (including motor complications) in Parkinson’s disease. Expert Opin. Pharmacother. 18, 1457–1465. doi: 10.1080/14656566.2017.1373089
Nguyen, M., Wong, Y. C., Ysselstein, D., Severino, A., and Krainc, D. (2018). Synaptic, mitochondrial, and lysosomal dysfunction in parkinson’s disease. Trends Neurosci. 42, 140–149. doi: 10.1016/j.tins.2018.11.001
Oyama, G., Yoshimi, K., Natori, S., Chikaoka, Y., Ren, Y. R., Funayama, M., et al. (2010). Impaired in vivo dopamine release in parkin knockout mice. Brain Res. 1352, 214–222. doi: 10.1016/j.brainres.2010.06.065
Paviour, D. C., Surtees, R. A. H., and Lees, A. J. (2004). Diagnostics considerations in juvenile parkinsonism. Mov. Disord. 19, 123–135. doi: 10.1002/mds.10644
Pavón, N., Martín, A. B., Mendialdua, A., and Moratalla, R. (2006). ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol. Psychiatry 59, 64–74. doi: 10.1016/j.biopsych.2005.05.044
Perez, F. A., and Palmiter, R. D. (2005). Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 102, 2174–2179. doi: 10.1073/pnas.0409598102
Picconi, B., Hernández, L. F., Obeso, J. A., and Calabresi, P. (2017). Motor complications in Parkinson’s disease: striatal molecular and electrophysiological mechanisms of dyskinesias. Mov. Disord. 33, 867–876. doi: 10.1002/mds.27261
Potulska-Chromik, A., Hoffman-Zacharska, D., Łukawska, M., and Kostera-Pruszczyk, A. (2017). Dopa-responsive dystonia or early-onset Parkinson disease – Genotype–phenotype correlation. Neurol. Neurochir. Pol. 51, 1–6. doi: 10.1016/j.pjnns.2016.07.013
Ruiz-Dediego, I., Mellstrom, B., Vallejo, M., Naranjo, J. R., and Moratalla, R. (2015). Activation of DREAM (Downstream Regulatory Element Antagonistic Modulator), a calcium-binding protein, reduces L-DOPA-induced dyskinesias in mice. Biol. Psychiatry 77, 95–105. doi: 10.1016/j.biopsych.2014.03.023
Sanchez, G., Varaschin, R. K., Büeler, H., Marcogliese, P. C., Park, D. S., and Trudeau, L. E. (2014). Unaltered striatal dopamine release levels in young Parkin knockout, Pink1 knockout, DJ-1 knockout and LRRK2 R1441G transgenic mice. PLoS One 9:e0094826. doi: 10.1371/journal.pone.0094826
Santini, E., Feyder, M., Gangarossa, G., Bateup, H. S., Greengard, P., and Fisone, G. (2012). Dopamine- and cAMP-regulated phosphoprotein of 32-kDa (DARPP-32)-dependent activation of extracellular signal-regulated kinase (ERK) and mammalian target of rapamycin complex 1 (mTORC1) signaling in experimental parkinsonism. J. Biol. Chem. 287, 27806–27812. doi: 10.1074/jbc.M112.388413
Santini, E., Valjent, E., Usiello, A., Carta, M., Borgkvist, A., Girault, J. A., et al. (2007). Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J. Neurosci. 27, 6995–7005. doi: 10.1523/JNEUROSCI.0852-07.2007
Sassone, J., Serratto, G., Valtorta, F., Silani, V., Passafaro, M., and Ciammola, A. (2017). The synaptic function of parkin. Brain 140, 2265–2272. doi: 10.1093/brain/awx006
Scherfler, C., Khan, N. L., Pavese, N., Eunson, L., Graham, E., Lees, A. J., et al. (2004). Striatal and cortical pre- and postsynaptic dopaminergic dysfunction in sporadic parkin-linked parkinsonism. Brain 127, 1332–1342. doi: 10.1093/brain/awh150
Schneider, S. A., and Alcalay, R. N. (2017). Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov. Disord. 32, 1504–1523. doi: 10.1002/mds.27193
Schneider, S. A., and Bhatia, K. P. (2010). Rare causes of dystonia parkinsonism. Curr. Neurol. Neurosci. Rep. 10, 431–439. doi: 10.1007/s11910-010-0136-0
Schrag, A., and Schott, J. M. (2006). Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol. 5, 355–363. doi: 10.1016/S1474-4422(06)70411-2
Solís, O., Garcia-Montes, J. R., González-Granillo, A., Xu, M., and Moratalla, R. (2017). Dopamine D3 receptor Modulates l-DOPA-Induced Dyskinesia by Targeting D1 Receptor-Mediated Striatal Signaling. Cereb. Cortex 27, 435–446. doi: 10.1093/cercor/bhv231
Stephens, B., Mueller, A. J., Shering, A. F., Hood, S. H., Taggart, P., Arbuthnott, G. W., et al. (2005). Evidence of a breakdown of corticostriatal connections in Parkinson’s disease. Neuroscience 132, 741–754. doi: 10.1016/j.neuroscience.2005.01.007
Stichel, C. C., Augustin, M., Kühn, K., Zhu, X. R., Engels, P., Ullmer, C., et al. (2000). Parkin expression in the adult mouse brain. Eur. J. Neurosci. 12, 4181–4194.
Suarez, L. M., Solis, O., Aguado, C., Lujan, R., and Moratalla, R. (2016). L-DOPA oppositely regulates synaptic strength and spine morphology in D1 and D2 striatal projection neurons in dyskinesia. Cereb. Cortex 26, 4253–4264. doi: 10.1093/cercor/bhw263
Tassin, J., Dürr, A., Bonnet, A. M., Gil, R., Vidailhet, M., Lücking, C. B., et al. (2000). Levodopa-responsive dystonia. GTP cyclohydrolase I or parkin mutations? Brain 123, 1112–1121. doi: 10.1093/brain/123.6.1112
Vargas, K. J., Schrod, N., Davis, T., Fernandez-Busnadiego, R., Taguchi, Y. V., Laugks, U., et al. (2017). Synucleins Have Multiple Effects on Presynaptic Architecture. Cell Rep. 18, 161–173. doi: 10.1016/j.celrep.2016.12.023
Wang, C., Kang, X., Zhou, L., Chai, Z., Wu, Q., Huang, R., et al. (2018). Synaptotagmin-11 is a critical mediator of parkin-linked neurotoxicity and Parkinson’s disease-like pathology. Nat. Commun. 9:81. doi: 10.1038/s41467-017-02593-y
Wickremaratchi, M. M., Knipe, M. D. W., Sastry, B. S. D., Morgan, E., Jones, A., Salmon, R., et al. (2011). The motor phenotype of Parkinson’s disease in relation to age at onset. Mov. Disord. 26, 457–463. doi: 10.1002/mds.23469
Wu, R. M., Bounds, R., Lincoln, S., Hulihan, M., Lin, C. H., Hwu, W. L., et al. (2005). Parkin mutations and early-onset parkinsonism in a Taiwanese cohort. Arch. Neurol. 62, 82–87. doi: 10.1001/archneur.62.1.82
Wu, Z. Y., Lin, B., Chen, B., Zhao, G. X., Xie, H., Murong, S. X., et al. (2008). Molecular analyses of GCH-1, TH and parkin genes in Chinese dopa-responsive dystonia families. Clin. Genet. 74, 513–521. doi: 10.1111/j.1399-0004.2008.01039.x
Yelnik, J., Francis, C., Percheron, G., and Tandéa, D. (1991). Morphological taxonomy of the neurons of the primate striatum. J. Comp. Neurol. 313, 273–294. doi: 10.1002/cne.903130207
Zaja-Milatovic, S., Milatovic, D., Schantz, A. M., Zhang, J., Montine, K. S., Samii, A., et al. (2005). Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology 64, 545–547. doi: 10.1212/01.WNL.0000150591.33787.A4
Zhong, P., Hu, Z., Jiang, H., Yan, Z., and Feng, J. (2017). Dopamine induces oscillatory activities in human midbrain neurons with parkin mutations. Cell Rep. 19, 1033–1044. doi: 10.1016/j.celrep.2017.04.023
Keywords: synaptopathy, glutamatergic transmission, parkin, dyskinesia, striatum
Citation: Sassone J, Valtorta F and Ciammola A (2019) Early Dyskinesias in Parkinson’s Disease Patients With Parkin Mutation: A Primary Corticostriatal Synaptopathy? Front. Neurosci. 13:273. doi: 10.3389/fnins.2019.00273
Received: 27 November 2018; Accepted: 07 March 2019;
Published: 26 March 2019.
Edited by:
Gregory Jaye Bix, University of Kentucky, United StatesReviewed by:
Mattia Volta, EURAC Research, ItalySandeep Kumar Barodia, The University of Alabama at Birmingham, United States
Copyright © 2019 Sassone, Valtorta and Ciammola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jenny Sassone, c2Fzc29uZS5qZW5ueUBoc3IuaXQ=