 Alberto Benussi1
Alberto Benussi1 Antonella Alberici1
Antonella Alberici1 Emanuele Buratti2
Emanuele Buratti2 Roberta Ghidoni3
Roberta Ghidoni3 Fabrizio Gardoni4
Fabrizio Gardoni4 Monica Di Luca4
Monica Di Luca4 Alessandro Padovani1
Alessandro Padovani1 Barbara Borroni1*
Barbara Borroni1*- 1Neurology Unit, Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy
- 2International Centre for Genetic Engineering and Biotechnology, ICGEB, Trieste, Italy
- 3IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italy
- 4Department of Pharmacological and Biomolecular Sciences, University of Milan, Milan, Italy
Frontotemporal dementia (FTD) is a heterogenous neurodegenerative disorder, characterized by diverse clinical presentations, neuropathological characteristics and underlying genetic causes. Emerging evidence has shown that FTD is characterized by a series of changes in several neurotransmitter systems, including serotonin, dopamine, GABA and, above all, glutamate. Indeed, several studies have now provided preclinical and clinical evidence that glutamate is key in the pathogenesis of FTD. Animal models of FTD have shown a selective hypofunction in N-methyl D-aspartate (NMDA) and α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, while in patients, glutamatergic pyramidal neurons are depleted in several areas, including the frontal and temporal cortices. Recently, a selective involvement of the AMPA GluA3 subunit has been observed in patients with autoimmune anti-GluA3 antibodies, which accounted for nearly 25% of FTD patients, leading to a decrease of the GluA3 subunit synaptic localization of the AMPA receptor and loss of dendritic spines. Other in vivo evidence of the involvement of the glutamatergic system in FTD derives from non-invasive brain stimulation studies using transcranial magnetic stimulation, in which specific stimulation protocols have indirectly identified a selective and prominent impairment in glutamatergic circuits in patients with both sporadic and genetic FTD. In view of limited disease modifying therapies to slow or revert disease progression in FTD, an important approach could consist in targeting the neurotransmitter deficits, similarly to what has been achieved in Parkinson’s disease with dopaminergic therapy or Alzheimer’s disease with cholinergic therapy. In this review, we summarize the current evidence concerning the involvement of the glutamatergic system in FTD, suggesting the development of new therapeutic strategies.
Introduction
Frontotemporal dementia (FTD) is one of the most common neurodegenerative conditions after Alzheimer’s Disease (AD), characterized by behavioral abnormalities, language impairment, and deficits of executive functions (Bang et al., 2015). The different clinical features have been grouped in different variants, represented by the behavioral variant of FTD (bvFTD) (Rascovsky et al., 2011), the agrammatic variant of Primary Progressive Aphasia (avPPA) and the semantic variant of PPA (svPPA) (Gorno-Tempini et al., 2011). Over the past ten years, for a common sharing of the same genetic and pathological determinants, atypical extrapyramidal conditions, including Corticobasal Syndrome (CBS) and Progressive Supranuclear Palsy (PSP), but also motor neuron disease (MND), were grouped under the same frontotemporal lobar degeneration (FTLD) disease spectrum (Litvan et al., 1996; Armstrong et al., 2013; Burrell et al., 2016). Concomitantly, the structural and functional brain correlates of each phenotype have been precisely reported (Rohrer et al., 2011). FTLD selectively affects the frontal and temporal regions, in which the main neuropathological hallmarks are constituted primarily by tau or TAR DNA-binding protein 43 (TDP-43) depositions (Spillantini and Goedert, 2013; Neumann and Mackenzie, 2019).
The identification of genetic mutations associated with FTLD helped to elucidate the underlying pathology, with mutations in Microtubule Associated Protein Tau (MAPT) causing tau accumulation, and Granulin (GRN) or the expansion on chromosome 9 open reading frame 72 (C9orf72) being associated with TDP-43 inclusions (Borroni and Padovani, 2013). Lastly, reappraisal of the pathological criteria for subtyping FTLD cases has benefited from some refinements, being updated with recent immunohistochemical, biochemical, and genetic advances (Cairns et al., 2007). In addition to FTLD-Tau or FTLD-TDP, several other neuropathological depositions have been defined, including FTLD-FET [with positivity for the FET family of DNA/RNA-binding proteins, comprising the fused in sarcoma (FUS), TATA-binding protein-associated factor 2N (TAF-15) and Ewing sarcoma protein (EWS)], FTLD-UPS (with inclusions of proteins of the ubiquitin-proteasome system) and FTLD-ni (with no inclusions observed) (Sieben et al., 2012; Van Mossevelde et al., 2018). Other uncommon genetic mutations have been described, including valosin containing protein (VCP) (Watts et al., 2004; van der Zee et al., 2009), sequestosome 1 (SQSTM1) (Rubino et al., 2012; Le Ber et al., 2013; van der Zee et al., 2014; Kovacs et al., 2016), and TANK-binding kinase 1 (TBK1) (Freischmidt et al., 2015; Gijselinck et al., 2015; Pottier et al., 2015), with an underlying TDP-43 pathology, charged multivesicular body protein 2B (CHMP2B) (Skibinski et al., 2005; Holm et al., 2009), associated with FTLD-UPS, and FUS mutations (Broustal et al., 2010; Van Langenhove et al., 2010) probably associated to FTLD-FET (no autopsy confirmation in patients with FTD to date but only in patients with amyotrophic lateral sclerosis) (Benussi et al., 2015a).
Despite the giant step forward in the knowledge of clinical, imaging, genetic and biological underpinnings of the disease, the absence of a reliable biomarker to predict the ongoing neuropathology represents a major limit to develop disease-modifying therapies that target tau or TDP-43 deposits, and that could be administered only to subjects with known pathogenetic mutations (Bang et al., 2015; Borroni et al., 2015). Moreover, it is still unknown whether tau and TDP-43 deposits represent the initial mechanism or simply the result of other trigger events.
Indeed, two different approaches might be pursued in the next future for treatment purposes: on one hand, there is urgent need to develop diagnostic markers able to identify the specific proteinopathies associated with FTLD, on the other, it might be possible to characterize neurotransmitter deficits shared by the entire FTLD spectrum (Rohrer et al., 2011).
Emerging evidence has now shown that FTD is characterized by a series of changes in several neurotransmitter systems, including serotonin, dopamine, GABA and, above all, glutamate (Murley and Rowe, 2018) (see Table 1).
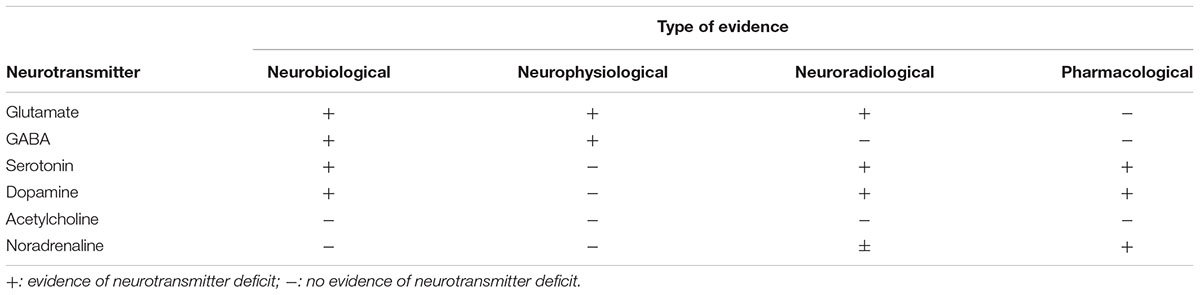
Table 1. Evidence for neurotransmitter deficits in frontotemporal dementia.
The recent identification of anti-AMPA GluA3 antibodies in the serum and in the cerebrospinal fluid (CSF) from FTLD patients (Borroni et al., 2017) has suggested that the impairment of glutamate neurotransmission through an autoimmune mechanism might be considered as a possible target to slow or revert the disease. In this framework, we can hypothesize that a restoration of the appropriate glutamatergic stimulation could be reached by modulating (i) the immune system or (ii) the glutamatergic receptors, developing the latter approach in analogy to what has been demonstrated effective for Parkinson or Alzheimer disease, with dopaminergic and cholinergic therapies, respectively (Murley and Rowe, 2018).
In this review, we summarize the current evidence concerning the involvement of the glutamatergic system in FTD, suggesting the development of new therapeutic strategies.
Molecular Biology
Glutamate, which represents the main excitatory neurotransmitter in the brain, largely contributes to memory and learning processes (Bliss and Collingridge, 1993), while being also involved in brain damage when abnormally activated in several conditions, including brain ischemia, epilepsy and neurodegeneration (Bowie, 2008). Glutamate exerts its functions at the synaptic level through both ionotropic (iGluR) and metabotropic glutamate receptors (mGluR).
iGluR are cation permeable tetramers, distinguished in N-methyl D-aspartate (NMDA), α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate (KA) on the basis of their affinity properties, the selectivity for different ions and the ability to generate rapid or slow electric kinetics. NMDA receptors (NMDAR) are known to mediate plasticity phenomena, as long-term potentiation (LTP) (Lescher et al., 2012), with a critical role of extra synaptic receptor subtype 2B (NR2B) subunit-containing ones (Paoletti et al., 2013). AMPA receptors are primarily involved in synaptic plasticity by modifications of subunits editing and composition, or interactions with different receptors and phosphorylation (Opazo et al., 2010; Huganir and Nicoll, 2013).
mGluR are a family of receptors coupled to G proteins, activating different transduction signals, mainly represented by phospholipase C and adenylate cyclase (Castillo et al., 2010). mGluRs contribute to neuronal plasticity and cognitive abilities, being able to mediate self-dependent forms of LTP and long-term depression (LTD) (Wang et al., 2015; Longhena et al., 2017).
Several evidences arise from both preclinical and clinical studies, showing the involvement of the glutamate neurotransmitter receptors, both iGluR and mGluR in the pathogenesis of FTLD.
In murine cortical neurons, silencing the FTD-associated gene granulin (GRN) decreases the expression of extra synaptic NR2B-containing NMDAR (Longhena et al., 2017); on the other hand, hyper-phosphorylated tau enhances glutamate release and produces an overactivation of the same receptor ending with neuron death, that can eventually be reduced by stimulating its reuptake through the astrocytic glutamate transporter 1 (GLT1)/excitatory amino acid transporter 2 (EAAT2) (Decker et al., 2016). Furthermore, FTLD has been related to the dysfunction in RNA pathways (Sephton and Yu, 2015), as corroborated by evidence that FUS depletion downregulates the transcription of GluA1, an essential AMPA-subunit involved in LTP phenomena (Udagawa et al., 2015). In that regard, also charged multivesicular body protein 2b (CHMP2B) FTD-related mutation increases GluA2 expression by disrupting microRNA levels (Gascon et al., 2014).
Knock out of the glutamate ionotropic receptor AMPA type subunit 3 gene (GRIA3) produces modifications in social behavior with an increase in aggressiveness (Adamczyk et al., 2012): in a recent study GluA3-containing AMPAR turned to be dormant receptors, triggered by a peculiar intracellular signaling pathway (Renner et al., 2017). Neuronal activity stimulated by AMPAR activation induces tau release from mature cortical neurons in a calcium-dependent way, suggesting the glutamatergic modulation as a further approach to prevent tau depositions (Pooler et al., 2013). Autoantibodies for the GluA3 subunit of AMPARs have been identified both in the serum and CSF of FTD patients (Borroni et al., 2017), characterized by a bvFTD phenotype with presenile onset, absence of an autosomal dominant pattern of inheritance, and greater bitemporal atrophy. These anti-GluA3 antibodies lead to a reduction of the synaptic levels of GluA3-containing AMPARs both in rat primary neurons and in human neurons differentiated from induced pluripotent stem cells (iPSCs). In addition, the presence of GluA3 antibodies in the CSF induced a loss of dendritic spine density, and increased levels of tau protein in vitro human neurons (Borroni et al., 2017).
Interestingly, Leuzy et al. (2016) reported a reduced availability of mGlur5 in bvFTD patients. Several observations argued for a link between autoimmunity and FTD (Alberici et al., 2018), and more recently, it was demonstrated a significant increase in frequency of anti-nuclear antibodies (ANA) observed in FTD patients, as compared to normal control subjects (Cavazzana et al., 2018). According to these findings, it might be hypothesized that an immune system dysregulation results into an abnormal production of autoantibodies directed against the GluA3 subunit, causing a deficit in glutamatergic transmission, eventually leading to FTLD.
The involvement of glutamatergic transmission has also been reported in amyotrophic lateral sclerosis (ALS), which is part of the FTLD-ALS spectrum disorder, in which a glutamate-induced excitotoxicity of motor neurons has been hypothesized (Blasco et al., 2014). Deficient editing of the GluR2 AMPA receptor subunit (Kawahara et al., 2004) and a diminished functional transport of glutamate and reduced EAAT2 immunoreactivity has been observed in motor neurons of patients with ALS (Rothstein et al., 1992, 1995). These findings further support the possible complex role of glutamatergic transmission abnormalities in the pathophysiology of FTD-ALS.
Other possible modulators of glutamatergic transmission which have been shown to be impaired in FTD are serotonin (5-HT) and GABA. 5-HT has been shown to differently modify glutamate mediated effects, acting on distinct 5-HT receptor subtypes both at the pre-synaptic and post-synaptic site and in different brain regions: in the frontal cortex glutamate release is inhibited by serotonin whereas in the prefrontal cortex serotonin enhances glutamatergic transmission (Dawson et al., 2001; Ciranna, 2006). In FTD, a dysfunction of the serotoninergic system has been frequently observed (Bowen et al., 2008; Vermeiren et al., 2016), possibly opening an avenue for glutamatergic modulation through serotonin regulation (Huey et al., 2006).
Furthermore, GABA, which is the predominant inhibitory neurotransmitter in the brain with different functions other that merely counteracting excitatory glutamatergic neurons, has been shown to be impaired in FTD patients. Initial studies have shown that a subgroup of GABAergic neurons that bind calbinidin-D28k are reduced in the upper neocortical layers of the frontal and temporal cortices in FTD (Ferrer, 1999), while gamma oscillations and coherence, which reflect GABA inhibition, are reduced between the frontal lobes of patients with behavioral variant FTD (Hughes et al., 2018). These findings are corroborated by reports of the toxic effects mediated by tau and TDP-43 on GABAergic interneurons, leading to a loss of GABAergic function in animal models (Levenga et al., 2013; Yamashita and Kwak, 2014).
Neurophysiology
Indirect evidence of the involvement of the glutamatergic system in FTD also comes from neurophysiological studies using both in vitro and in vivo techniques.
In vitro studies in transgenic mice expressing pathological human tau (V337M mutation), which is one of the main pathological hallmarks of FTD, have shown both AMPA and NMDA receptor hypofunction in the ventral striatum and insular cortex, which were reversible after the administration of cycloserine, an NMDA receptor co-agonist (Warmus et al., 2014). Further in vitro studies in transgenic mice carrying a CHMP2B mutation, which is another gene associated with FTD, have also shown altered AMPA receptor composition and function in the medial prefrontal cortex (Gascon et al., 2014).
In vivo neurophysiological evidence of the involvement of glutamatergic circuits in FTD mainly comes from non-invasive brain stimulation studies using transcranial magnetic stimulation (TMS) (Borroni et al., 2018). In this context, different paired-pulse TMS paradigms have been implemented to assess intracortical inhibitory and excitatory interneuronal circuits (Benussi et al., 2015b; Rossini et al., 2015). In particular, intracortical facilitation (ICF), which consists in a physiological facilitation elicited by applying a subthreshold conditioning magnetic stimulus followed by a suprathreshold test stimulus at an inter stimulus interval of 6–30 ms, has shown to depend mainly on glutamatergic circuits in the primary motor cortex (Ziemann et al., 2015), with NMDA receptor antagonists decreasing ICF (Ziemann et al., 1998; Schwenkreis et al., 1999).
Reduced ICF has been observed in patients with genetic FTD, carrying a GRN or C9orf72 mutation, even in the presymptomatic phases of disease, compared to non-carrier first degree relatives (Benussi et al., 2016). These dysfunctions correlated with reduced cortical thickness and surface area of the right insula in presymptomatic GRN carriers, suggesting that glutamatergic impairment in the presymptomatic phases of GRN-related FTD could reflect the beginning of insular dysfunction, even in absence of cognitive or behavioral abnormalities (Gazzina et al., 2018).
Recently, the reduction of ICF has been observed up to 30 years before expected symptom onset in a very large cohort of GRN and C9orf72 mutation carriers compared to non-carriers, long before the onset of clinical and neuroimaging abnormalities (Benussi et al., 2019).
An impairment of ICF has also been observed in sporadic FTD (Burrell et al., 2011; Benussi et al., 2017), confirming how this biomarker may be useful not only to track disease progression, but also to distinguish FTD from other forms of dementias, even in the early disease stages (Benussi et al., 2018a; Padovani et al., 2018).
Regarding other syndromes in the FTLD spectrum, a reduced ICF has been also observed in patients with CBS and PSP, highlighting how this technique may also be used to distinguish other atypical parkinsonian disorders, including dementia with Lewy bodies (Benussi et al., 2018b).
Alterations in ICF have been observed also in patients with both sporadic and familial ALS; however, contrary to what has been observed in FTD, an increase in ICF seems to be predominant (Geevasinga et al., 2015; Van den Bos et al., 2018). It is still debated if cortical hyperexcitability might act as an adaptive process in response to peripheral neurodegeneration and could serve as a neuroprotective strategy, or if cortical hyperexcitability may serve as a final common pathway in ALS, mediating neuronal degeneration via a trans-synaptic glutamate process (Geevasinga et al., 2015).
Treatment Approaches: Targeting Glutamatergic Neurotransmission
Currently there are no approved treatments for FTD, and there are no therapies able to stop or alter the disease course. Pharmacological treatments to date have mostly concerned the off-label use of medications for symptomatic management. Recent advancements in understanding the molecular and genetic basis of FTD, and several clinical trials based on these insights are underway and have been reviewed elsewhere (Tsai and Boxer, 2016).
Glutamate neurotransmission has been considered a possible target for FTD symptomatic treatment. Memantine, a NMDA receptor antagonist with an indication for the treatment of moderate to severe AD (Tariot et al., 2004), was studied in two randomized, placebo-controlled trials over 52 and 26 weeks in FTD (Vercelletto et al., 2011; Boxer et al., 2013). Both studies failed to demonstrate significant benefits on behavioral disturbances or clinical global impression of change.
The recent observations of an effect exerted by the AMPARs activation on tau aggregation renewed the interest of glutamatergic modulation as a further approach to prevent tau depositions (Pooler et al., 2013; Borroni et al., 2017). Moreover, the identification of autoantibodies directed against GluA3 subunits provided evidence for an autoimmune dysregulation as a possible pathogenetic mechanism in FTD (Alberici et al., 2018). The link between autoimmune antibodies and neurodegeneration has been previously shown in the anti-IgLON5-related tauopathy, in which extensive neuropathological tau and TDP-43 inclusions have been observed (Sabater et al., 2014), placing these disorders at the convergence of neurodegenerative and autoimmune mechanisms. However, further research is necessary to validate these findings and elucidate the mechanisms by which these, or still other unidentified auto-antibodies, induce pathologic protein aggregates and neurodegeneration.
The feasibility of targeting an autoimmune response is an attractive potential therapeutic approach, suggesting immunomodulatory therapies as an evidence-based approach to treat FTLD. In the absence of prospective and randomized clinical trials for the treatment of autoimmune encephalitis, literature data are based on case reports with anti-NMDA, or more rarely, anti-AMPA receptor encephalitis (Dalmau and Graus, 2018). We can hypothesize that scavenging anti-GluA3 antibodies by using immunomodulation might restore glutamatergic transmission, thus slowing or reverting FTLD neurodegenerative process. Alternatively, in agreement with the glutamatergic hypothesis, and in analogy to what has been proposed for schizophrenia, positive allosteric modulators of AMPA receptors as well as orthosteric ligands and modulators of metabotropic glutamatergic receptors in particular ligands acting on mGlu receptors might be considered promising potential medications in FTLD (Menniti et al., 2013).
The modulation of glutamatergic transmission via 5-HT regulation may also be a promising approach to seek. Favorable evidence with selective serotonin reuptake inhibitors (SSRIs) has been observed in FTD patients, with several open label and placebo-controlled studies with SSRIs showing an improvement of several behavioral symptoms, as disinhibition, irritability and depression (Moretti et al., 2003; Lebert et al., 2004; Anneser et al., 2007; Herrmann et al., 2012; Hughes et al., 2015). However, it is still not known if this is a direct effect on serotoninergic transmission or possibly an indirect downstream effect on glutamatergic systems.
Regarding GABAergic therapies, evidence is currently lacking for a clinical efficacy in FTD patients.
Conclusion
We have observed how the involvement of the glutamatergic system may play a key role in the pathogenesis of FTD both from a biological and neurophysiological perspective. This implication may open several avenues regarding treatment options which will have to be verified experimentally, both from a symptomatic but also possibly disease modifying approach.
The involvement of glutamate in FTLD may answer some of the open issues in this field, yet we caution that FTD symptoms almost certainly do not flow from a single neurotransmitter abnormality. Indeed, this proposed model does not negate the involvement of other neurotransmitters, which have already been observed in FTLD, including GABA, serotonin and dopamine, and all of these may be ultimately brought together in a unified and interconnected framework (Murley and Rowe, 2018) (see Figure 1). Restoring these deficits, individually or in combination, has the potential to improve cognitive, behavioral and motor symptoms. More realistically, in fact the ultimate phenotypic expression probably arises from combinations of neurotransmitter abnormalities, genetic mutations, and environmental factors; combinations that may vary considerably from patient to patient.
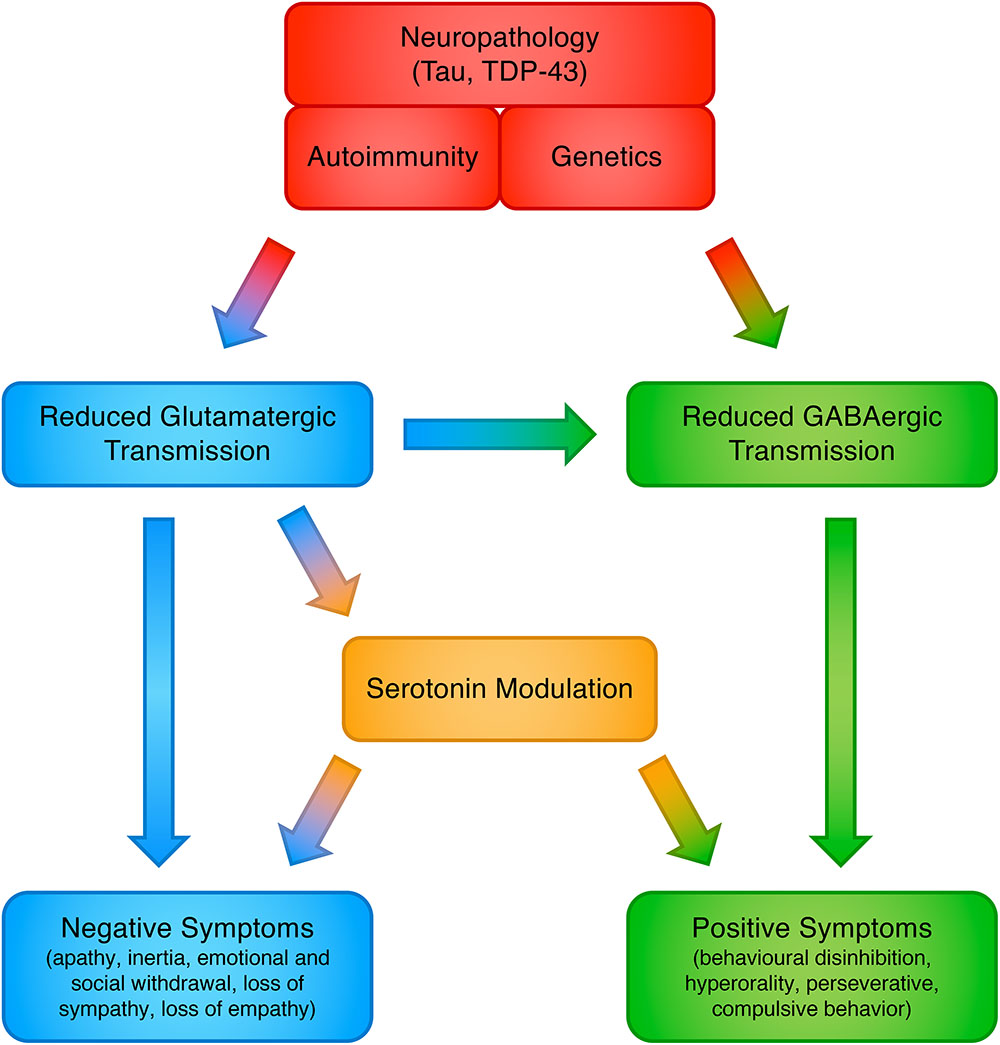
Figure 1. Proposed model for the involvement of neurotransmitter systems in frontotemporal dementia.
Another interesting avenue worth pursuing is the potential for this amino-acid to act as a biomarker, either in establishing the diagnosis or as a measure of disease progression. Direct measurements in the CSF have shown a negative correlation between glutamate levels with verbally agitated behavior in FTD patients (Vermeiren et al., 2013). On the other hand, indirect measurements come from magnetic resonance spectroscopy of FTD patients in which glutamate/glutamine levels have been found to be reduced in the frontal and temporal lobes (Ernst et al., 1997; Sarac et al., 2008) and from neurophysiological studies with TMS, showing in both sporadic and genetic FTD a reduced ICF, which is partially mediated by glutamatergic transmission. In future, glutamate levels could also be indirectly assessed with electroencephalography (EEG) (Lally et al., 2014) or by TMS-EEG evoked potentials (Cash et al., 2016). To define which direct or indirect biomarker of glutamatergic neurotransmission might be the most useful and informative has still to be elucidated, considering the lack of studies on the subject, with different biomarkers perhaps providing distinct information from both a physiopathological and topographical perspective.
In conclusion, it is therefore now clear that the role of glutamate in FTD can represent an interesting and innovative approach to better understand the underlying ongoing neurodegenerative process in this pathology, although further investigations will be needed in order to increase our biological understanding of the disease, which will probably be contingent to the development of appropriate models and biomarkers for glutamatergic drug development.
Author Contributions
All authors gave their substantial contribution to conception and design of the manuscript and drafting the manuscript and revising it critically for important intellectual content, approved the manuscript in its present form for publication, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This work was supported by the Italian Ministry of Health (Ricerca Corrente).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Marcello Giunta for his significant contributions to the manuscript.
References
Adamczyk, A., Mejias, R., Takamiya, K., Yocum, J., Krasnova, I. N., Calderon, J., et al. (2012). GluA3-deficiency in mice is associated with increased social and aggressive behavior and elevated dopamine in striatum. Behav. Brain Res. 229, 265–272. doi: 10.1016/j.bbr.2012.01.007
Alberici, A., Cristillo, V., Gazzina, S., Benussi, A., Padovani, A., and Borroni, B. (2018). Autoimmunity and frontotemporal dementia. Curr. Alzheimer Res. 15, 602–609. doi: 10.2174/1567205015666180119104825
Anneser, J. M. H., Jox, R. J., and Borasio, G. D. (2007). Inappropriate sexual behaviour in a case of ALS and FTD: successful treatment with sertraline. Amyotroph. Lateral Scler. 8, 189–190. doi: 10.1080/17482960601073543
Armstrong, M. J., Litvan, I., Lang, A. E., Bak, T. H., Bhatia, K. P., Borroni, B., et al. (2013). Criteria for the diagnosis of corticobasal degeneration. Neurology 80, 496–503. doi: 10.1212/WNL.0b013e31827f0fd1
Bang, J., Spina, S., and Miller, B. L. (2015). Frontotemporal dementia. Lancet 386, 1672–1682. doi: 10.1016/S0140-6736(15)00461-4
Benussi, A., Alberici, A., Ferrari, C., Cantoni, V., Dell’Era, V., Turrone, R., et al. (2018a). The impact of transcranial magnetic stimulation on diagnostic confidence in patients with Alzheimer disease. Alzheimers Res. Ther. 10:94. doi: 10.1186/s13195-018-0423-6
Benussi, A., Dell’Era, V., Cantoni, V., Ferrari, C., Caratozzolo, S., Rozzini, L., et al. (2018b). Discrimination of atypical parkinsonisms with transcranial magnetic stimulation. Brain Stimul. 11, 366–373. doi: 10.1016/j.brs.2017.11.013
Benussi, A., Cosseddu, M., Filareto, I., Dell’Era, V., Archetti, S., Sofia Cotelli, M., et al. (2016). Impaired long-term potentiation-like cortical plasticity in presymptomatic genetic frontotemporal dementia. Ann. Neurol. 80, 472–476. doi: 10.1002/ana.24731
Benussi, A., Di Lorenzo, F., Dell’Era, V., Cosseddu, M., Alberici, A., Caratozzolo, S., et al. (2017). Transcranial magnetic stimulation distinguishes Alzheimer disease from frontotemporal dementia. Neurology 89, 665–672. doi: 10.1212/WNL.0000000000004232
Benussi, A., Gazzina, S., Premi, E., Cosseddu, M., Archetti, S., Dell’Era, V., et al. (2019). Clinical and biomarker changes in presymptomatic genetic frontotemporal dementia. Neurobiol. Aging 76, 133–140. doi: 10.1016/j.neurobiolaging.2018.12.018
Benussi, A., Padovani, A., and Borroni, B. (2015a). Phenotypic heterogeneity of monogenic frontotemporal dementia. Front. Aging Neurosci. 7:171. doi: 10.3389/fnagi.2015.00171
Benussi, A., Padovani, A., and Borroni, B. (2015b). Transcranial magnetic stimulation in Alzheimer’s Disease and cortical dementias. J. Alzheimers Dis. Parkinsonism 5:197.
Blasco, H., Mavel, S., Corcia, P., and Gordon, P. H. (2014). The glutamate hypothesis in ALS: pathophysiology and drug development. Curr. Med. Chem. 21, 3551–3575. doi: 10.2174/0929867321666140916120118
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Borroni, B., Benussi, A., Archetti, S., Galimberti, D., Parnetti, L., Nacmias, B., et al. (2015). Csf p-tau181/tau ratio as biomarker for TDP pathology in frontotemporal dementia. Amyotroph. Lateral Scler. Front. Degener. 16, 86–91. doi: 10.3109/21678421.2014.971812
Borroni, B., Benussi, A., Premi, E., Alberici, A., Marcello, E., Gardoni, F., et al. (2018). Biological, neuroimaging, and neurophysiological markers in frontotemporal dementia: three faces of the same coin. J. Alzheimers Dis. 62, 1113–1123. doi: 10.3233/JAD-170584
Borroni, B., and Padovani, A. (2013). Dementia: a new algorithm for molecular diagnostics in FTLD. Nat. Rev. Neurol. 9, 241–242. doi: 10.1038/nrneurol.2013.72
Borroni, B., Stanic, J., Verpelli, C., Mellone, M., Bonomi, E., Alberici, A., et al. (2017). Anti-AMPA GluA3 antibodies in frontotemporal dementia: a new molecular target. Sci. Rep. 7:1006. doi: 10.1038/s41598-017-06117-y
Bowen, D. M., Procter, A. W., Mann, D. M. A., Snowden, J. S., Esiri, M. M., Neary, D., et al. (2008). Imbalance of a serotonergic system in frontotemporal dementia: implication for pharmacotherapy. Psychopharmacology 196, 603–610. doi: 10.1007/s00213-007-0992-8
Bowie, D. (2008). Ionotropic glutamate receptors & CNS disorders. CNS Neurol. Disord. Drug Targets 7, 129–143. doi: 10.2174/187152708784083821
Boxer, A. L., Knopman, D. S., Kaufer, D. I., Grossman, M., Onyike, C., Graf-Radford, N., et al. (2013). Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 12, 149–156. doi: 10.1016/S1474-4422(12)70320-4
Broustal, O., Camuzat, A., Guillot-Noël, L., Guy, N., Millecamps, S., Deffond, D., et al. (2010). FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J. Alzheimers. Dis. 22, 765–769.
Burrell, J. R., Halliday, G. M., Kril, J. J., Ittner, L. M., Götz, J., Kiernan, M. C., et al. (2016). The frontotemporal dementia-motor neuron disease continuum. Lancet 388, 919–931. doi: 10.1016/S0140-6736(16)00737-6
Burrell, J. R., Kiernan, M. C., Vucic, S., and Hodges, J. R. (2011). Motor neuron dysfunction in frontotemporal dementia. Brain 134, 2582–2594. doi: 10.1093/brain/awr195
Cairns, N. J., Bigio, E. H., Mackenzie, I. R. A., Neumann, M., Lee, V. M. Y., Hatanpaa, K. J., et al. (2007). Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol. 114, 5–22. doi: 10.1007/s00401-007-0237-2
Cash, R. F. H., Noda, Y., Zomorrodi, R., Radhu, N., Farzan, F., Rajji, T. K., et al. (2016). Characterization of glutamatergic and GABAA-Mediated neurotransmission in motor and dorsolateral prefrontal cortex using paired-pulse TMS–EEG. Neuropsychopharmacology 42, 502–511. doi: 10.1038/npp.2016.133
Castillo, C. A., León, D. A., Ballesteros-Yáñez, I., Iglesias, I., Martín, M., and Albasanz, J. L. (2010). Glutamate differently modulates metabotropic glutamate receptors in neuronal and glial cells. Neurochem. Res. 35, 1050–1063. doi: 10.1007/s11064-010-0154-y
Cavazzana, I., Alberici, A., Bonomi, E., Ottaviani, R., Kumar, R., Archetti, S., et al. (2018). Antinuclear antibodies in Frontotemporal Dementia: the tip’s of autoimmunity iceberg? J. Neuroimmunol. 325, 61–63. doi: 10.1016/j.jneuroim.2018.10.006
Ciranna, L. (2006). Serotonin as a modulator of glutamate- and GABA-mediated neurotransmission: implications in physiological functions and in pathology. Curr. Neuropharmacol. 4, 101–114. doi: 10.2174/157015906776359540
Dalmau, J., and Graus, F. (2018). Antibody-mediated encephalitis. N. Engl. J. Med. 378, 840–851. doi: 10.1056/NEJMra1708712
Dawson, L. A., Nguyen, H. Q., and Li, P. (2001). The 5-HT6 receptor antagonist SB-271046 selectively enhances excitatory neurotransmission in the rat frontal cortex and hippocampus. Neuropsychopharmacology 25, 662–668. doi: 10.1016/S0893-133X(01)00265-2
Decker, J. M., Krüger, L., Sydow, A., Dennissen, F. J., Siskova, Z., Mandelkow, E., et al. (2016). The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 17, 552–569. doi: 10.15252/embr.201541439
Ernst, T., Chang, L., Melchor, R., and Mehringer, C. M. (1997). Frontotemporal dementia and early Alzheimer disease: differentiation with frontal lobe H-1 MR spectroscopy. Radiology 203, 829–836. doi: 10.1148/radiology.203.3.9169712
Ferrer, I. (1999). Neurons and their dendrites in frontotemporal dementia. Dement. Geriatr. Cogn. Disord. 10, 55–60. doi: 10.1159/000051214
Freischmidt, A., Wieland, T., Richter, B., Ruf, W., Schaeffer, V., Müller, K., et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636. doi: 10.1038/nn.4000
Gascon, E., Lynch, K., Ruan, H., Almeida, S., Verheyden, J. M., Seeley, W. W., et al. (2014). Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 20, 1444–1451. doi: 10.1038/nm.3717
Gazzina, S., Benussi, A., Premi, E., Paternicò, D., Cristillo, V., Dell’Era, V., et al. (2018). Neuroanatomical correlates of transcranial magnetic stimulation in presymptomatic granulin mutation carriers. Brain Topogr. 31, 488–497. doi: 10.1007/s10548-017-0612-9
Geevasinga, N., Menon, P., Nicholson, G. A., Ng, K., Howells, J., Kril, J. J., et al. (2015). Cortical Function in asymptomatic carriers and patients with C9orf72Amyotrophic lateral sclerosis. JAMA Neurol. 72, 1268–1274. doi: 10.1001/jamaneurol.2015.1872
Gijselinck, I., Van Mossevelde, S., van der Zee, J., Sieben, A., Philtjens, S., Heeman, B., et al. (2015). Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology 85, 2116–2125. doi: 10.1212/WNL.0000000000002220
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014. doi: 10.1212/WNL.0b013e31821103e6
Herrmann, N., Black, S. E., Chow, T., Cappell, J., Tang-Wai, D. F., and Lanctôt, K. L. (2012). Serotonergic function and treatment of behavioral and psychological symptoms of frontotemporal dementia. Am. J. Geriatr. Psychiatry 20, 789–797. doi: 10.1097/JGP.0b013e31823033f3
Holm, I. E., Isaacs, A. M., and Mackenzie, I. R. A. (2009). Absence of FUS-immunoreactive pathology in frontotemporal dementia linked to chromosome 3 (FTD-3) caused by mutation in the CHMP2B gene. Acta Neuropathol. 118, 719–720. doi: 10.1007/s00401-009-0593-1
Huey, E. D., Putnam, K. T., and Grafman, J. (2006). A systematic review of neurotransmitter deficits and treatments in frontotemporal dementia. Neurology 66, 17–22. doi: 10.1212/01.wnl.0000191304.55196.4d
Huganir, R. L., and Nicoll, R. A. (2013). AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717. doi: 10.1016/j.neuron.2013.10.025
Hughes, L. E., Rittman, T., Regenthal, R., Robbins, T. W., and Rowe, J. B. (2015). Improving response inhibition systems in frontotemporal dementia with citalopram. Brain 138, 1961–1975. doi: 10.1093/brain/awv133
Hughes, L. E., Rittman, T., Robbins, T. W., and Rowe, J. B. (2018). Reorganization of cortical oscillatory dynamics underlying disinhibition in frontotemporal dementia. Brain 141, 2486–2499. doi: 10.1093/brain/awy176
Kawahara, Y., Ito, K., Sun, H., Ito, M., Kanazawa, I., and Kwak, S. (2004). GluR4c, an alternative splicing isoform of GluR4, is abundantly expressed in the adult human brain. Brain Res. Mol. Brain Res. 127, 150–155. doi: 10.1016/j.molbrainres.2004.05.020
Kovacs, G. G., van der Zee, J., Hort, J., Kristoferitsch, W., Leitha, T., Höftberger, R., et al. (2016). Clinicopathological description of two cases with SQSTM1 gene mutation associated with frontotemporal dementia. Neuropathology 36, 27–38. doi: 10.1111/neup.12233
Lally, N., Mullins, P. G., Roberts, M. V., Price, D., Gruber, T., and Haenschel, C. (2014). Glutamatergic correlates of gamma-band oscillatory activity during cognition: a concurrent ER-MRS and EEG study. Neuroimage 85(Pt 2), 823–833. doi: 10.1016/j.neuroimage.2013.07.049
Le Ber, I., Camuzat, A., Guerreiro, R., Bouya-Ahmed, K., Bras, J., Nicolas, G., et al. (2013). SQSTM1 mutations in french patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 70, 1403–1410. doi: 10.1001/jamaneurol.2013.3849
Lebert, F., Stekke, W., Hasenbroekx, C., and Pasquier, F. (2004). Frontotemporal dementia: a randomised, controlled trial with trazodone. Dement. Geriatr. Cogn. Disord. 17, 355–359. doi: 10.1159/000077171
Lescher, J., Paap, F., Schultz, V., Redenbach, L., Scheidt, U., Rosewich, H., et al. (2012). MicroRNA regulation in experimental autoimmune encephalomyelitis in mice and marmosets resembles regulation in human multiple sclerosis lesions. J. Neuroimmunol. 246, 27–33. doi: 10.1016/j.jneuroim.2012.02.012
Leuzy, A., Zimmer, E. R., Dubois, J., Pruessner, J., Cooperman, C., Soucy, J.-P., et al. (2016). In vivo characterization of metabotropic glutamate receptor type 5 abnormalities in behavioral variant FTD. Brain Struct. Funct. 221, 1387–1402. doi: 10.1007/s00429-014-0978-3
Levenga, J., Krishnamurthy, P., Rajamohamedsait, H., Wong, H., Franke, T. F., Cain, P., et al. (2013). Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol. Commun. 1:34. doi: 10.1186/2051-5960-1-34
Litvan, I., Agid, Y., Calne, D., Campbell, G., Dubois, B., Duvoisin, R. C., et al. (1996). Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 47, 1–9. doi: 10.1212/WNL.47.1.1
Longhena, F., Zaltieri, M., Grigoletto, J., Faustini, G., La Via, L., Ghidoni, R., et al. (2017). Depletion of progranulin reduces GluN2B-containing NMDA receptor density, tau phosphorylation and dendritic arborization in mouse primary cortical neurons. J. Pharmacol. Exp. Ther. 363, 164–175. doi: 10.1124/jpet.117.242164
Menniti, F. S., Lindsley, W. C., Conn, P. J., Pandit, J., Zagouras, P., and Volkmann, R. A. (2013). Allosteric modulators for the treatment of schizophrenia: targeting glutamatergic networks. Curr. Top. Med. Chem. 13, 26–54. doi: 10.2174/1568026611313010005
Moretti, R., Torre, P., Antonello, R. M., Cazzato, G., and Bava, A. (2003). Frontotemporal dementia: paroxetine as a possible treatment of behavior symptoms: a randomized, controlled, open 14-month study. Eur. Neurol. 49, 13–19. doi: 10.1159/000067021
Murley, A. G., and Rowe, J. B. (2018). Neurotransmitter deficits from fronto temporal lobar degeneration. Brain 141, 1263–1285. doi: 10.1093/brain/awx327
Neumann, M., and Mackenzie, I. R. A. (2019). Review: neuropathology of non-tau frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 45, 19–40. doi: 10.1111/nan.12526
Opazo, P., Labrecque, S., Tigaret, C. M., Frouin, A., Wiseman, P. W., De Koninck, P., et al. (2010). CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 67, 239–252. doi: 10.1016/j.neuron.2010.06.007
Padovani, A., Benussi, A., Cantoni, V., Dell’Era, V., Cotelli, M. S., Caratozzolo, S., et al. (2018). Diagnosis of mild cognitive impairment due to Alzheimer’s disease with transcranial magnetic stimulation. J. Alzheimers Dis. 65, 221–230. doi: 10.3233/JAD-180293
Paoletti, P., Bellone, C., and Zhou, Q. (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. doi: 10.1038/nrn3504
Pooler, A. M., Polydoro, M., Wegmann, S., Nicholls, S. B., Spires-Jones, T. L., and Hyman, B. T. (2013). Propagation of tau pathology in Alzheimer’s disease: identification of novel therapeutic targets. Alzheimers Res. Ther. 5, 1–8. doi: 10.1186/alzrt214
Pottier, C., Bieniek, K. F., Finch, N., van de Vorst, M., Baker, M., Perkersen, R., et al. (2015). Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 130, 77–92. doi: 10.1007/s00401-015-1436-x
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477. doi: 10.1093/Brain/Awr179
Renner, M. C., Albers, E. H., Gutierrez-Castellanos, N., Reinders, N. R., van Huijstee, A. N., Xiong, H., et al. (2017). Synaptic plasticity through activation of GluA3-containing AMPA-receptors. eLife 6:e25462. doi: 10.7554/eLife.25462
Rohrer, J. D., Lashley, T., Schott, J. M., Warren, J. E., Mead, S., Isaacs, A. M., et al. (2011). Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 134, 2565–2581. doi: 10.1093/brain/awr198
Rossini, P. M., Burke, D., Chen, R., Cohen, L. G., Daskalakis, Z., Di Iorio, R., et al. (2015). Non-invasive electrical and magnetic stimulation of the brain, spinal cord, roots and peripheral nerves: basic principles and procedures for routine clinical and research application. An updated report from an I.F.C.N. Committee. Clin. Neurophysiol. 126, 1071–1107. doi: 10.1016/j.clinph.2015.02.001
Rothstein, J. D., Martin, L. J., and Kuncl, R. W. (1992). Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 326, 1464–1468. doi: 10.1056/NEJM199205283262204
Rothstein, J. D., Van Kammen, M., Levey, A. I., Martin, L. J., and Kuncl, R. W. (1995). Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84. doi: 10.1002/ana.410380114
Rubino, E., Rainero, I., Chio, A., Rogaeva, E., Galimberti, D., Fenoglio, P., et al. (2012). SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 79, 1556–1562. doi: 10.1212/WNL.0b013e31826e25df
Sabater, L., Gaig, C., Gelpi, E., Bataller, L., Lewerenz, J., Torres-Vega, E., et al. (2014). A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 13, 575–586. doi: 10.1016/S1474-4422(14)70051-1
Sarac, H., Zagar, M., Vranjes, D., Henigsberg, N., Bilić, E., and Pavlisa, G. (2008). Magnetic resonance imaging and magnetic resonance spectroscopy in a patient with amyotrophic lateral sclerosis and frontotemporal dementia. Coll. Antropol. 32(Suppl. 1), 205–210.
Schwenkreis, P., Witscher, K., Janssen, F., Addo, A., Dertwinkel, R., Zenz, M., et al. (1999). Influence of the N-methyl-d-aspartate antagonist memantine on human motor cortex excitability. Neurosci. Lett. 270, 137–140. doi: 10.1016/S0304-3940(99)00492-9
Sephton, C. F., and Yu, G. (2015). The function of RNA-binding proteins at the synapse: implications for neurodegeneration. Cell. Mol. Life Sci. 72, 3621–3635. doi: 10.1007/s00018-015-1943-x
Sieben, A., Van Langenhove, T., Engelborghs, S., Martin, J.-J., Boon, P., Cras, P., et al. (2012). The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 124, 353–372. doi: 10.1007/s00401-012-1029-x
Skibinski, G., Parkinson, N. J., Brown, J. M., Chakrabarti, L., Lloyd, S. L., Hummerich, H., et al. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808. doi: 10.1038/ng1609
Spillantini, M. G., and Goedert, M. (2013). Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. doi: 10.1016/S1474-4422(13)70090-5
Tariot, P. N., Farlow, M. R., Grossberg, G. T., Graham, S. M., McDonald, S., Gergel, I., et al. (2004). Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 291, 317–324. doi: 10.1001/jama.291.3.317
Tsai, R. M., and Boxer, A. L. (2016). Therapy and clinical trials in frontotemporal dementia: past, present, and future. J. Neurochem. 138(Suppl.), 211–221. doi: 10.1111/jnc.13640
Udagawa, T., Fujioka, Y., Tanaka, M., Honda, D., Yokoi, S., Riku, Y., et al. (2015). FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun. 6:7098. doi: 10.1038/ncomms8098
Van den Bos, M. A. J., Higashihara, M., Geevasinga, N., Menon, P., Kiernan, M. C., and Vucic, S. (2018). Imbalance of cortical facilitatory and inhibitory circuits underlies hyperexcitability in ALS. Neurology 91, e1669–e1676. doi: 10.1212/WNL.0000000000006438
van der Zee, J., Pirici, D., Van Langenhove, T., Engelborghs, S., Vandenberghe, R., Hoffmann, M., et al. (2009). Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology 73, 626–632. doi: 10.1212/WNL.0b013e3181b389d9
van der Zee, J., Van Langenhove, T., Kovacs, G. G., Dillen, L., Deschamps, W., Engelborghs, S., et al. (2014). Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 128, 397–410. doi: 10.1007/s00401-014-1298-7
Van Langenhove, T., van der Zee, J., Sleegers, K., Engelborghs, S., Vandenberghe, R., Gijselinck, I., et al. (2010). Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74, 366–371. doi: 10.1212/WNL.0b013e3181ccc732
Van Mossevelde, S., Engelborghs, S., van der Zee, J., and Van Broeckhoven, C. (2018). Genotype–phenotype links in frontotemporal lobar degeneration. Nat. Rev. Neurol. 14, 363–378. doi: 10.1038/s41582-018-0009-8
Vercelletto, M., Boutoleau-Bretonnière, C., Volteau, C., Puel, M., Auriacombe, S., Sarazin, M., et al. (2011). Memantine in behavioral variant frontotemporal dementia: negative results. J. Alzheimers. Dis. 23, 749–759. doi: 10.3233/JAD-2010-101632
Vermeiren, Y., Janssens, J., Aerts, T., Martin, J. J., Sieben, A., Van Dam, D., et al. (2016). Brain serotonergic and noradrenergic deficiencies in behavioral variant frontotemporal dementia compared to early-onset Alzheimer’s Disease. J. Alzheimers Dis. 53, 1079–1096. doi: 10.3233/JAD-160320
Vermeiren, Y., Le Bastard, N., Van Hemelrijck, A., Drinkenburg, W. H., Engelborghs, S., and De Deyn, P. P. (2013). Behavioral correlates of cerebrospinal fluid amino acid and biogenic amine neurotransmitter alterations in dementia. Alzheimers Dement. 9, 488–498. doi: 10.1016/j.jalz.2012.06.010
Wang, Z., Neely, R., and Landisman, C. E. (2015). Activation of Group I and Group II metabotropic glutamate receptors causes LTD and LTP of electrical synapses in the rat thalamic reticular nucleus. J. Neurosci. 35, 7616–7625. doi: 10.1523/JNEUROSCI.3688-14.2015
Warmus, B. A., Sekar, D. R., McCutchen, E., Schellenberg, G. D., Roberts, R. C., McMahon, L. L., et al. (2014). Tau-mediated NMDA receptor impairment underlies dysfunction of a selectively vulnerable network in a mouse model of frontotemporal dementia. J. Neurosci. 34, 16482–16495. doi: 10.1523/JNEUROSCI.3418-14.2014
Watts, G. D. J., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., et al. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381. doi: 10.1038/ng1332
Yamashita, T., and Kwak, S. (2014). The molecular link between inefficient GluA2 Q/R site-RNA editing and TDP-43 pathology in motor neurons of sporadic amyotrophic lateral sclerosis patients. Brain Res. 1584, 28–38. doi: 10.1016/j.brainres.2013.12.011
Ziemann, U., Chen, R., Cohen, L. G., and Hallett, M. (1998). Dextromethorphan decreases the excitability of the human motor cortex. Neurology 51, 1320–1324. doi: 10.1212/WNL.51.5.1320
Keywords: frontotemporal dementia, frontotemporal lobar degeneration, glutamate, neurotransmitter, autoimmunity, transcranial magnetic stimulation
Citation: Benussi A, Alberici A, Buratti E, Ghidoni R, Gardoni F, Di Luca M, Padovani A and Borroni B (2019) Toward a Glutamate Hypothesis of Frontotemporal Dementia. Front. Neurosci. 13:304. doi: 10.3389/fnins.2019.00304
Received: 04 February 2019; Accepted: 18 March 2019;
Published: 29 March 2019.
Edited by:
Annakaisa Haapasalo, University of Eastern Finland, FinlandReviewed by:
Yannick Vermeiren, University of Antwerp, BelgiumRafael Linden, Federal University of Rio de Janeiro, Brazil
Copyright © 2019 Benussi, Alberici, Buratti, Ghidoni, Gardoni, Di Luca, Padovani and Borroni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Borroni, YmFyYmFyYS5ib3Jyb25pQHVuaWJzLml0