 Rachel Walters
Rachel Walters John Manion
John Manion G. Gregory Neely
G. Gregory Neely- Dr. John and Anne Chong Lab for Functional Genomics, Charles Perkins Centre, School of Life and Environmental Sciences, The University of Sydney, Sydney, NSW, Australia
Motor Neuron Disease (MND) typically affects patients during the later stages of life, and thus, MND is having an increasingly devastating impact on diagnosed individuals, their families and society. The umbrella term MND refers to diseases which cause the progressive loss of upper and/or lower motor neurons and a subsequent decrease in motor ability such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA). The study of these diseases is complex and has recently involved the use of genome-wide association studies (GWAS). However, in the case of MND, it has been difficult to identify the complex genetics involved in subtypes, and functional investigation of new candidate disease genes is warranted. Drosophila is a powerful model for addressing these complex diseases. The UAS/Gal4/Gal80 system allows for the upregulation of Drosophila genes, the “knockdown” of genes and the ectopic expression of human genes or mutations in a tissue-specific manner; often resulting in Drosophila models which exhibit typical MND disease pathologies. These can then be further interrogated to identify disease-modifying genes or mutations and disease pathways. This review will discuss two common MNDs and the current Drosophila models which are being used to research their genetic basis and the different pathologies of MND.
Introduction
As the global population ages, our societies face an increased prevalence of age-related diseases. A deeper understanding of the biology involved is required to develop new therapies to halt or even reverse disease progression. MND is characterized as the progressive loss of upper and/or lower motor neurons and a decrease in motor ability and function. It has been hypothesized that incorrect synaptic development and function could underlie MND progression (for full review see Murray et al., 2010). Typically, but not exclusively, symptoms begin to develop in the second half of a patient’s life, with incidence peaking between 75 and 79 years of age (Alonso et al., 2009), and leading to a rapid decrease in the quality of life and death (Tabrizi, 2006). The major forms of MND are ALS and SMA.
Genome-wide association studies have been used to establish the heritability and molecular etiology of MND. These studies identify SNPs that show statistical association with a specific phenotype. However, by design GWAS do not typically identify causative mutations (Auton et al., 2015). Instead, these studies flag common SNP variants within haplotype blocks. These regions may contain causative coding mutations, regulatory non-coding mutations, or complex elements that could influence disease susceptibility or progression. Moreover, these associations do not always predict directionality toward disease effect. Significant haplotype blocks are likely enriched for genes that modify disease, or regulatory elements that act in either cis or trans fashion, or even exert effects at long range or via unknown complex mechanisms. To pinpoint any disease-modifying genes within significant disease-associated haplotype blocks, large scale in vivo genetic screens are required. Drosophila is a well-established model organism through which to perform these screens for the study of MND.
The Drosophila UAS/Gal4/Gal80 system is used for tissue-specific targeted regulation of transgene expression (Brand and Perrimon, 1993; Figure 1A), or RNA interference (RNAi) inverted repeats designed to “knockdown” genes (Dietzl et al., 2007; Figure 1B). This tool makes Drosophila a potent system for the high throughput investigation of candidate disease genes in vivo. Many genetic diseases have a root cause of loss-of-function mutations, which is particularly easy to recapitulate using available whole genome in vivo RNAi libraries which allow for the targeted knockdown of all corresponding individual genes (Clark and Ding, 2006; Figure 1B). Moreover, disease genes which exert their disease-causing effects via gain-of-function mechanisms may be central to the development and function of the tissue of interest, and thus often will also exhibit loss-of-function phenotypes (Drenth and Waxman, 2007). Establishing an experimental interaction between loss of function phenotypes for candidate disease genes and the underlying disease process can be informative regardless of directionality. Drosophila is amenable to rapid and systematic genetic manipulation and is a cost-effective, ethical system to evaluate large gene sets for in vivo relevance with respect to organ dysfunction and disease. Another advantage of Drosophila models is the high degree of conservation between its genome and that of humans; with around 60% of all genes and 75% of human disease genes having a Drosophila ortholog (Adams et al., 2000; Fortini et al., 2000). Furthermore, 76% of human synaptic genes have a Drosophila ortholog (Lloyd et al., 2000) and, as it is hypothesized MND is, in part, due to synaptic dysfunction, candidate disease genes and synaptic regulators can be rapidly assessed for function in the well characterized and easily accessible Drosophila motor neuron. Drosophila in vivo screens allow for the study of morphological changes which occur during MND. The highly accessible Drosophila NMJ has been well developed as a model system and this knowledge can be used to study molecular and morphological aspects of synaptic dysfunction (Keshishian et al., 1996). The Drosophila glutamatergic NMJ also closely resembles vertebrate glutamatergic central synapses. It has been successfully used to study many aspects of synaptic transmission such as neurotransmitter release (Megighian et al., 2013; Valdez et al., 2015), signaling (Kamimura et al., 2013; Vonhoff and Keshishian, 2017) and homeostatic plasticity (for full review see Frank, 2014). For example, the Drosophila NMJ has aided in the discovery of important synaptic genes and proteins such as Bruchpilot (Kittel et al., 2006) and Synaptotagmin 4 (Yoshihara et al., 2005) (for full review see Hewitt and Whitworth, 2017). The use of Drosophila also allows for an in-depth study of disease progression; from the earliest signs to terminal stages. Finally, the large range of simple assay systems that can be undertaken rapidly using Drosophila, from lifespan and motor assays, to anatomical screens, would not be possible in other systems, and can each provide important functional information on the conserved machinery required for proper motor neuron function, and how these systems may be dysregulated during MND.
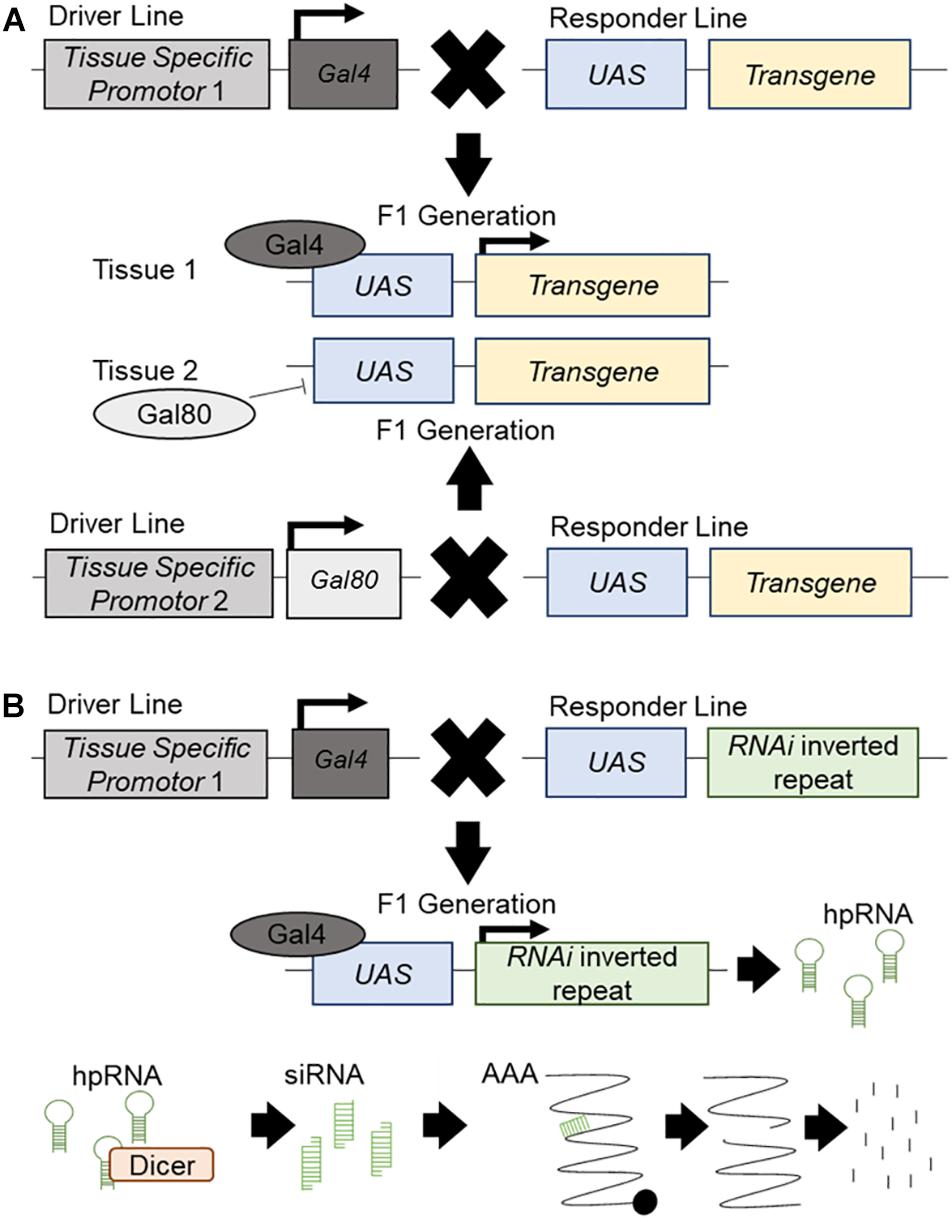
Figure 1. The UAS/Gal4/Gal80 system and RNAi knockdown. Schematic presentation of the UAS/Gal4/Gal80 system and its use in transgene expression and RNAi mediated knockdown. (A) The driver line contains a different tissue-specific promotor for both Gal4 and Gal80. It will drive the expression of Gal4 or Gal80 in the corresponding tissue. The responder line carries an upstream activator sequence (UAS) upstream of a transgene. In the F1 generation, each tissue specific promotor will drive the expression of Gal4 or Gal80 in the respective tissues. Gal80 will bind to the UAS and repress its expression in this tissue. Gal4 will bind to the UAS and promote the expression of the transgene. (B) The driver line contains a tissue-specific promotor which will drive the expression of Gal4 in the specific tissue. The responder line carries a UAS upstream of an inverted repeat of an RNAi construct. In the F1 generation, the tissue specific promotor will drive the expression of the Gal4 which will bind to the UAS and promote the expression of the RNAi construct, producing hairpin RNA (hpRNA). hpRNA is processed by Dicer into short interrupting RNA (siRNA) which will attach to endogenous mRNA and create a break in the strand. This broken mRNA will then be targeted for degradation and knockdown of the target gene will occur.
Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis, the most common form of MND, is a progressive neurodegenerative disease which can be categorized as either sporadic (SALS); cases where no immediate family member is affected, or familial (FALS); cases which have an inherited and often monogenic cause. FALS accounts for just 10% of ALS cases (Kiernan et al., 2011). The worldwide annual incidence is approximately 1.9 per 100,000 (Chiò et al., 2013) with a projected increase of approximately 70% between 2015 and 2040 (Arthur et al., 2016). ALS is ∼1.56 times more common in men than women (Mehta et al., 2014). However, this difference becomes less prevalent with an increase in age, with around the same number of male and female sufferers beyond the age of 70 (Worms, 2001). Symptoms can begin at any stage of life, however, middle age (40–49 years) to elderly (70+ years) are the most common ages at which the disease starts to develop. Following onset, patients suffer a progressive loss of motor function and after diagnosis have an average life expectancy of 5–6 years (Koppers et al., 2012). The pathology of ALS is varied, with patients suffering from symptoms at different time points after diagnostics, this may in part be due to slow prognosis, but, there are three main stages of disease progression; early, middle, and late. Through these stages, the patient will suffer from worsening symptoms and more increased physical weakness (Table 1; The ALS Association of Texas, 2018; Muscular Dystrophy Association, 2019). The leading cause of death in ALS sufferers is respiratory insufficiency. The disease progression of ALS has traditionally been evaluated through phenotypic assessments, however, the phenotypic variability of ALS results in inaccurate measurements and can delay diagnosis. Because of this, the use of multiple biomarkers is now being studied as a novel assessment method for disease progression (for full review see Simon et al., 2014; Huynh et al., 2018).
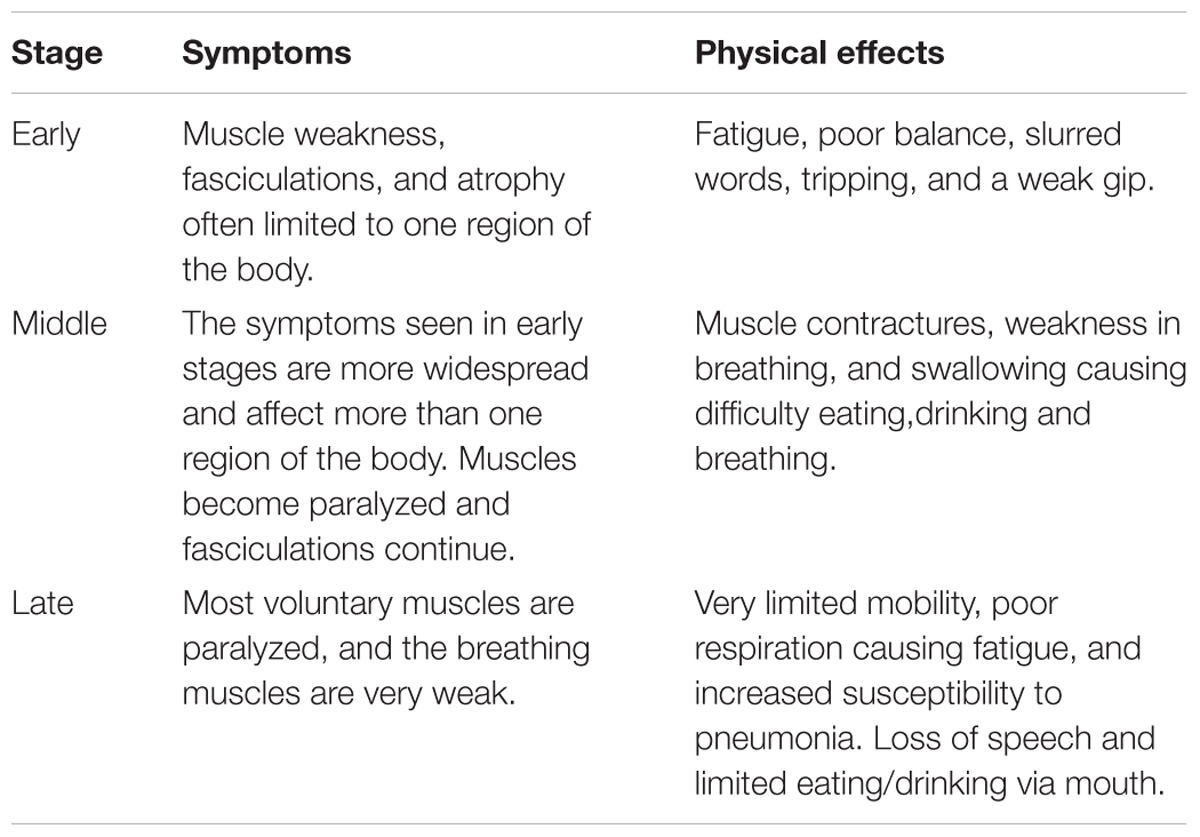
Table 1. The progression of ALS stages, symptoms, and physical effects (The ALS Association of Texas, 2018; Muscular Dystrophy Association, 2019).
The cause of ALS is thought to be a combination of multiple genetic risk variants and environmental factors including heavy metals (Kamel et al., 2002; Gait et al., 2003) or pesticides (Malek et al., 2012) (for full review see Bozzoni et al., 2016). From twin studies, heritability is estimated to be between 76% (Al-Chalabi et al., 2010)and 61% when known familial cases are excluded (SALS) (Wingo et al., 2011) (for a full review see Al-Chalabi and Visscher, 2014). However, more recent work using genome-wide complex trait analysis (GCTA) has placed the estimate much lower, at between approximately 21% (Keller et al., 2014) and 12% (Fogh et al., 2014). The main reason for the disparity in these estimates is due to the differing methods of obtaining the data, where twin studies often yield higher heritability values compared to GWAS for a variety of complex diseases (Tenesa and Haley, 2013; Feldman and Ramachandran, 2018).
Whilst familial studies have established that mutations in a number of genes can cause ALS, mutations in C9orf72 (DeJesus-Hernandez et al., 2011), Superoxide Dismutase 1 (SOD1) (Rosen et al., 1993), TAR Binding-Protein 43 (TARDBP) (Neumann et al., 2006), Ataxin 2 (ATXN2) (Elden et al., 2010), and FUS (Vance et al., 2009; Table 2) are the most common.
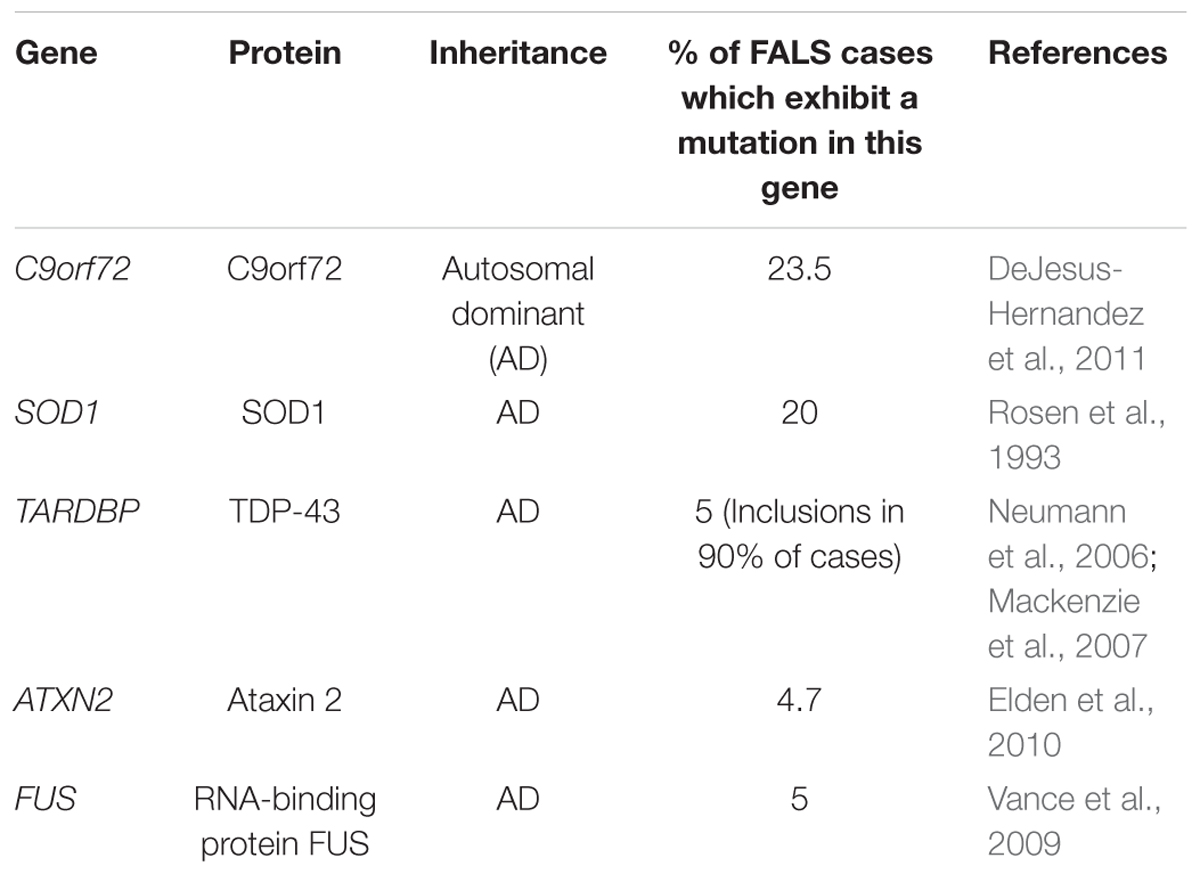
Table 2. The major genetic contributors to a patient’s risk of developing ALS.
To elucidate the contribution of genetic variation to sporadic ALS, population-wide studies have been performed. To date, 14 GWAS have been completed (for full review see Max-Planck-Gesellschaft, 2017) spanning a wide range of populations. These studies have linked many genes to an increased risk of ALS, including, with the exception of FUS, those genes discussed above. However, C9orf72 is the only gene that exhibits a genome-wide significant peak which has been confirmed in a second cohort (Ahmeti et al., 2013). As ALS is heterogeneous in nature, the mechanism by which these or other nearby genes may modify disease is not fully understood and requires further study. These genetic risk factors have well-established Drosophila disease models which have routinely been used in the study of ALS, its causes, and its effects on motor neurons.
C9orf72
The cellular function of C9orf72 is currently under contention. Similar to TDP-43 (discussed below), C9orf72 is thought to play a role in mRNA stability and transport (Figure 2). The first intron of human C9orf72 contains a hexanucleotide repeat expansion G4C2. Importantly, an increased number of G4C2 repeats has been linked to an increased risk of ALS (DeJesus-Hernandez et al., 2011). C9orf72 genetic mutations are the most frequent known cause of ALS (Majounie et al., 2012); with around 40% of FALS cases exhibiting various numbers of repeat expansions (Renton et al., 2011) and C9orf72 is also the only locus to show a genome-wide significance in meta-analysis studies (Malek et al., 2012; Al-Chalabi and Visscher, 2014).
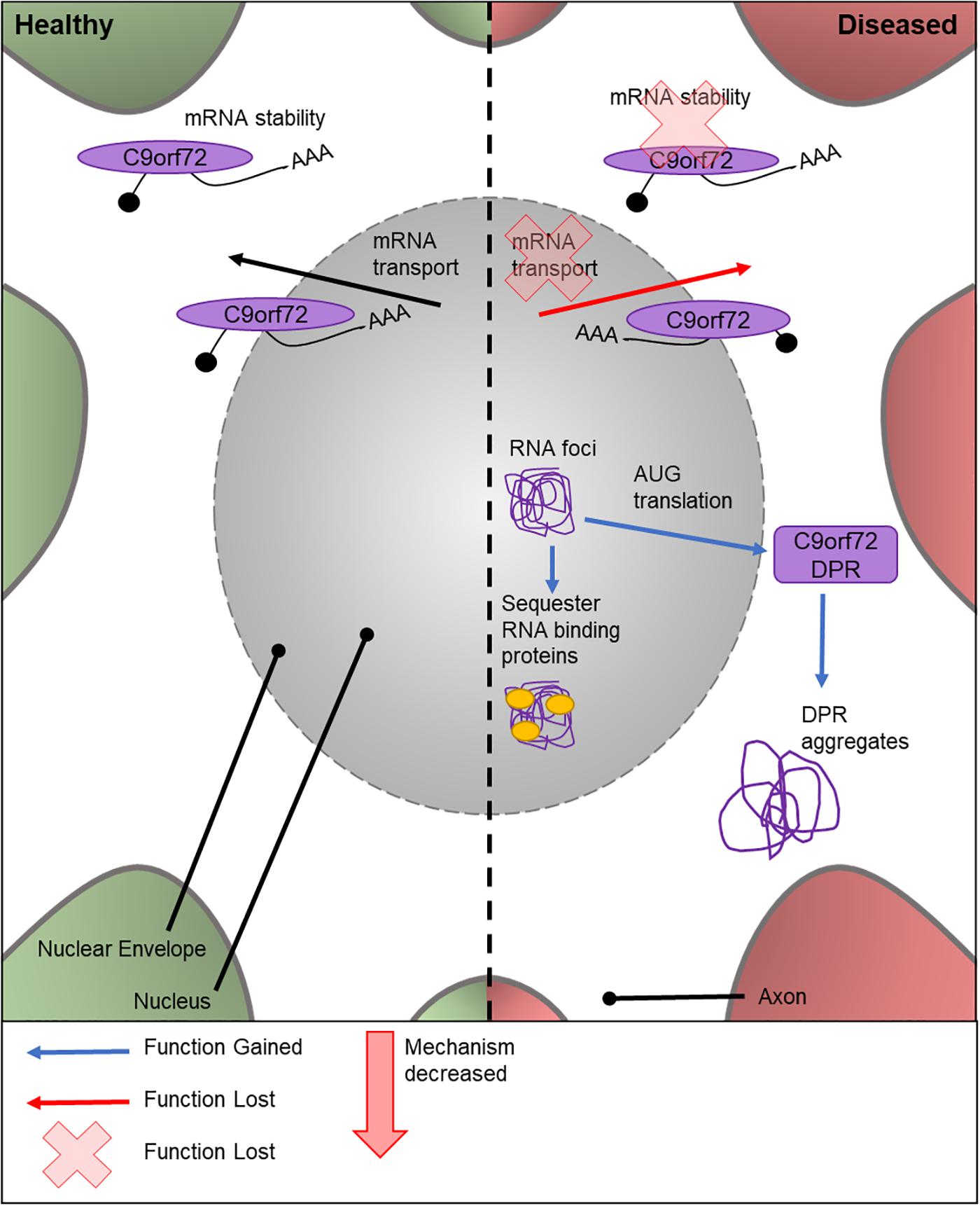
Figure 2. Cellular features of C9orf72 in a healthy and diseased neuron. Schematic presentation of a neuronal cell body with normal and disease-state C9orf72 cellular location and functions. In healthy neurons, C9orf72 is thought to function in mRNA stability and transport. In the diseased state, it is thought to form nuclear RNA foci which sequester RNA binding proteins. Through RAN translation of the mutated gene, DPR proteins are created. These form cytoplasmic protein aggregates. It is also thought mutations could cause the loss of C9orf72’s role in mRNA stability and transport.
There are multiple potential mechanisms by which the G4C2 repeat expansions can cause disease. These include a loss of C9orf72 normal cellular function, sequestering and altering the function of RNA binding proteins by RNA foci (Lill et al., 2011), or by AUG translated di-peptide repeat proteins (DPRs) (Ash et al., 2013; Figure 2). There is no known Drosophila C9orf72 ortholog. Therefore, to study the relative importance and contribution of C9orf72 in MND, transgenic Drosophila lines expressing either C9orf72 pure or RNA-only human G4C2 repeat variants, or animals which express both, have been generated (Mizielinska et al., 2014; Lee et al., 2016). C9orf72 pure lines will express both RNA and DPRs. Ectopic expression of human C9orf72 DPRs in fly motor neurons causes lethality at 25°C, while these models are semi-lethal at 18°C (Yang et al., 2015). The lack of endogenous Drosophila C9orf72 has proven to be a benefit to these studies, as it provides a control model for experimental use which contains no C9orf72.
Studies suggest that the C9orf72 RNA species form nuclear RNA foci, which could have a role in sequestering RNA-binding proteins in the nucleus (Ash et al., 2013). Furthermore, the G4C2 repeat expansions, but not the RNA foci (Mizielinska et al., 2014), are translated via repeat-association non-ATG translation to form DPR proteins, which form aggregates in the cytoplasm of the cell body (Mackenzie et al., 2013; Mori et al., 2013; Zu et al., 2013; Cooper-Knock et al., 2014). A further model has been developed, using a UAS-(G4C2)48 construct, to study the effect of G4C2 repeats on translation (Burguete et al., 2015). This highlighted that these repeats localize in neurites and can negatively impact the branching of the cell, ultimately affecting the neuron’s function. Mizielinska et al.’s (2014) study also suggests the pathogenicity of C9orf72 DPRs is highly associated with arginine containing DPR proteins, adding to the growing body of knowledge on DPR-specific pathology. The effects of RNA foci in the neuron has also been investigated (Burguete et al., 2015). However, the link between RNA and disease progression is widely debated. Drosophila lines expressing only the RNA foci have recently been created (Moens et al., 2018) for the study into the role of repeat RNA on disease progression without the input from the DPR proteins. Depending on the genomic location of the RNA repeats, lines can be produced with cytoplasmic or nuclear foci, allowing the elucidation of these different pathologies. These early RNA-only models have suggested that neither cytoplasmic or nuclear RNA are toxic and therefore has a limited role in ALS (Moens et al., 2018). Drosophila models have also shown that changes to transport through the nuclear pore, via the nuclear-pore complex, contribute to neurodegeneration in C9orf72 pathogenesis. This was validated in vitro using patient neurons. However, the cause of the defective transport has yet to be found. Zhang et al. (2015) attributes it to sense RNAs, whereas Freibaum et al. (2015) suggests it could be due to a combination of DPRs and toxic RNAs. As shown here, there have been many attempts to elucidate the contribution of different possible mechanisms responsible for C9orf72-caused disease progression. These studies highlight the ongoing effort to establish a timeline and mechanism for C9orf72 toxicity using genetically tractable systems.
SOD1
The human SOD1 gene encodes one of three members of the SOD enzyme family. As shown in Figure 3, SOD1 proteins bind to Cu2+ ions via a specific binding site and catalyze the dismutation of free radical species in the cell. SOD1-mediated removal of harmful superoxides is hypothesized to suppress apoptosis and prevent cellular damage by free radicals (Danial and Korsmeyer, 2004; Bunton-Stasyshyn et al., 2015). It was the first gene to be linked to familial ALS, with 11 missense SOD1 mutations showing an association with ALS (Redler and Dokholyan, 2012). Currently, over 90 disease-modifying mutations have been found in SOD1 (Redler and Dokholyan, 2012; Bunton-Stasyshyn et al., 2015) and around 20% of FALS cases carry a SOD1 mutation (Shaw et al., 1997). Although the mechanism(s) through which the mutated enzyme contributes to ALS are unknown, there is evidence that the accumulation of misfolded proteins and a gain of toxic function of the mutated enzyme is involved (Clement et al., 2003; Beers et al., 2006; Nagai et al., 2007).
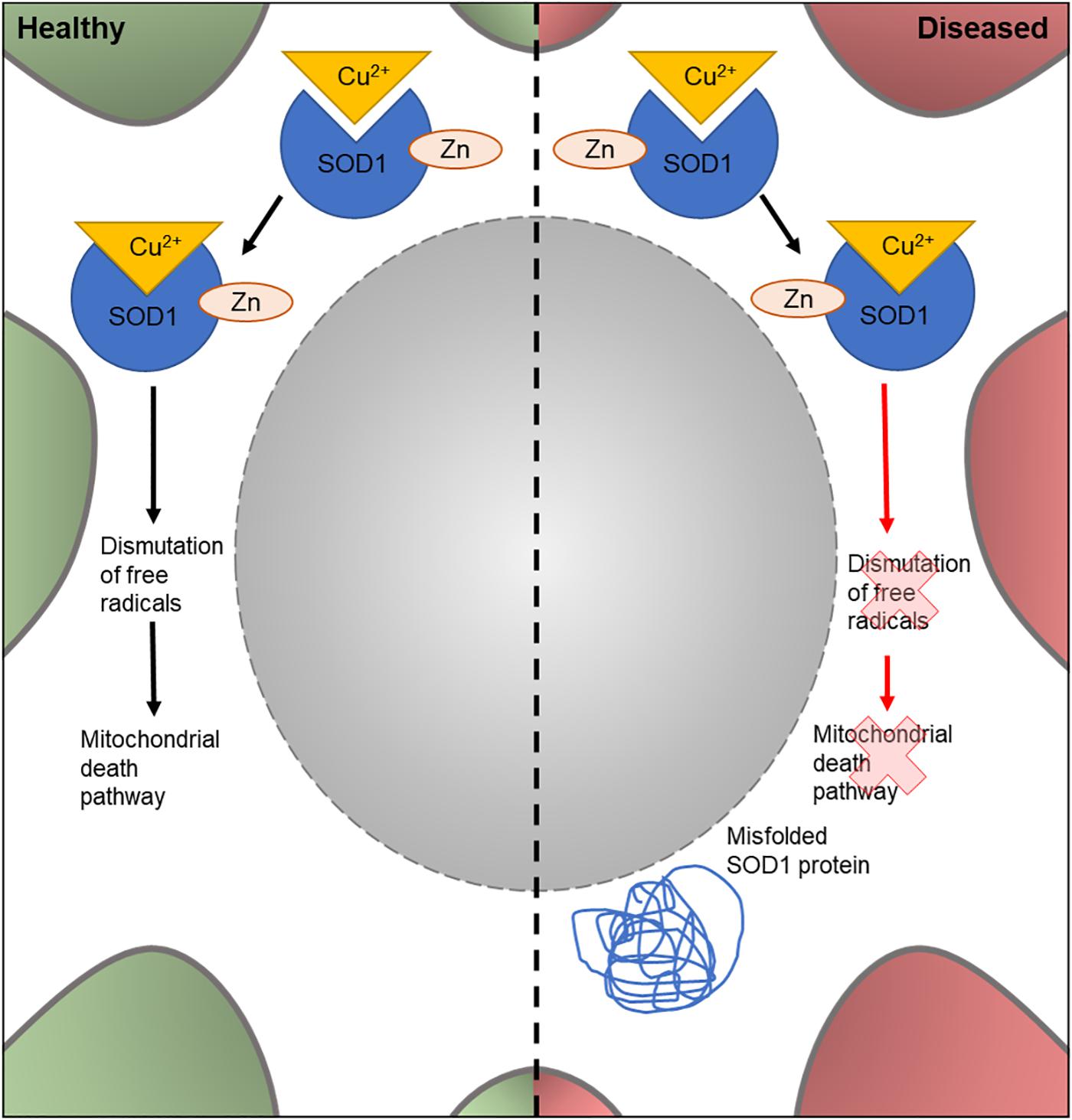
Figure 3. Cellular features of SOD1 in a healthy and diseased neuron. Schematic presentation of a neuronal cell body with normal and disease-state SOD1 cellular location and function. In healthy neurons, SOD1 is located in the cytoplasm. Here, it binds to Cu2+ and Zn to form a complex involved in dismutation of free radicals; part of the mitochondrial death pathway. In diseased neurons, mutated SOD1 is also found in the cytoplasm. However, it loses its dismutation function and has an unknown gain of function. It can also form misfolded protein aggregates in the cytoplasm.
A recent Drosophila SOD1 model (Gallart-Palau et al., 2016) was found to exhibit non-functioning mitochondria, a cellular phenotype of ALS. In this model, the human wildtype (hSOD) and mutant SOD1G93A was expressed in the thoracic muscles of Drosophila. This targeted expression lead to phenotypes which are stereotypical of ALS, such as impairment of normal motor behavior and mitochondrial pathology. This study builds on previous work, where transgenic motor neuron specific expression of hSOD1 exhibited progressive negative motor effects accompanied with defective neural circuit electrophysiology, accumulation of SOD1 aggregates and an increased stress response in ventral nerve cord glial cells (Watson et al., 2008). These studies suggest that overexpression of mutated SOD1 protein can cause both cell autonomous and non-cell-autonomous damage, as in the case of the effect on glial cells (Watson et al., 2008), and is a possible cause of SOD1-mediated ALS. However, in these studies, it is not clear whether the disease-phenotypes observed are due to overexpression of the mutant SOD1, or from the mutated protein itself and therefore would be caused by low-levels of the mutated protein. To address this issue, a recent study has investigated the effect of dosage of transgenes on subsequent disease pathology in Drosophila models of SOD1-associated ALS (Şahin et al., 2017). Through the mutation of endogenous Drosophila SOD1 and comparing with multiple copy number insertions, the study demonstrated a predominance of gain of function mutations. Moreover, this new model allows the study of SOD1 in ALS pathology at typical expression levels by utilizing the endogenous genetic machinery. Together this highlights the diversity of Drosophila disease models and their importance in investigating disease-linked mutations and their effect at the protein level.
TDP-43
TDP-43 (encoded by TARDBP) is a DNA- and RNA-binding protein which acts as a nuclear transcription factor and is thought to bind a large proportion of the transcriptome. It has been implicated in the transport of mRNAs to dendritic granules (Wang et al., 2007) and RNA metabolic processes (Buratti and Baralle, 2010; Figure 4). Homozygous TDP-43 knock-out causes peri-implantation lethality in mouse embryos (Wu et al., 2009), and utilizing the Cre/Lox system to induce TDP-43 knockout later in life is also lethal (Chiang et al., 2010). The Cre/Lox system allows for time- and/or tissue-specific activation or repression of target genes in mouse models and can therefore be used to study mutations in adult animals and their developed organs and tissues. In this system, tamoxifen acts as an inducer at a mutated estrogen receptor (Cre/ERT2), triggering recombination and deletion (Feil et al., 2009). This allows for temporal-specific inactivation of target genes. This demonstrates that TDP-43 is essential for development, as well as throughout adult life. In 2006, TDP-43 was found to be the characteristic ubiquitinated protein found in neuronal cytoplasmic inclusions, a stereotypical neuropathological feature of ALS (Neumann et al., 2006; Figure 4). TDP-43 pathology likely stems from the translocation of this protein from the nucleus into the cytoplasm, where it forms ubiquitinated aggregates (Arai et al., 2006). This has led to two possible pathogenic mechanisms being explored; a gain-of-function of the protein once translocated to the cytoplasm, possibly due to fragmentation and phosphorylation (Arai et al., 2006), and a loss-of-function mechanism in the nucleus due to mutation of the gene (Giordana et al., 2010). However, there is additional evidence that supports a combination of these pathways to be responsible for the proteinopathy (Lee et al., 2012).
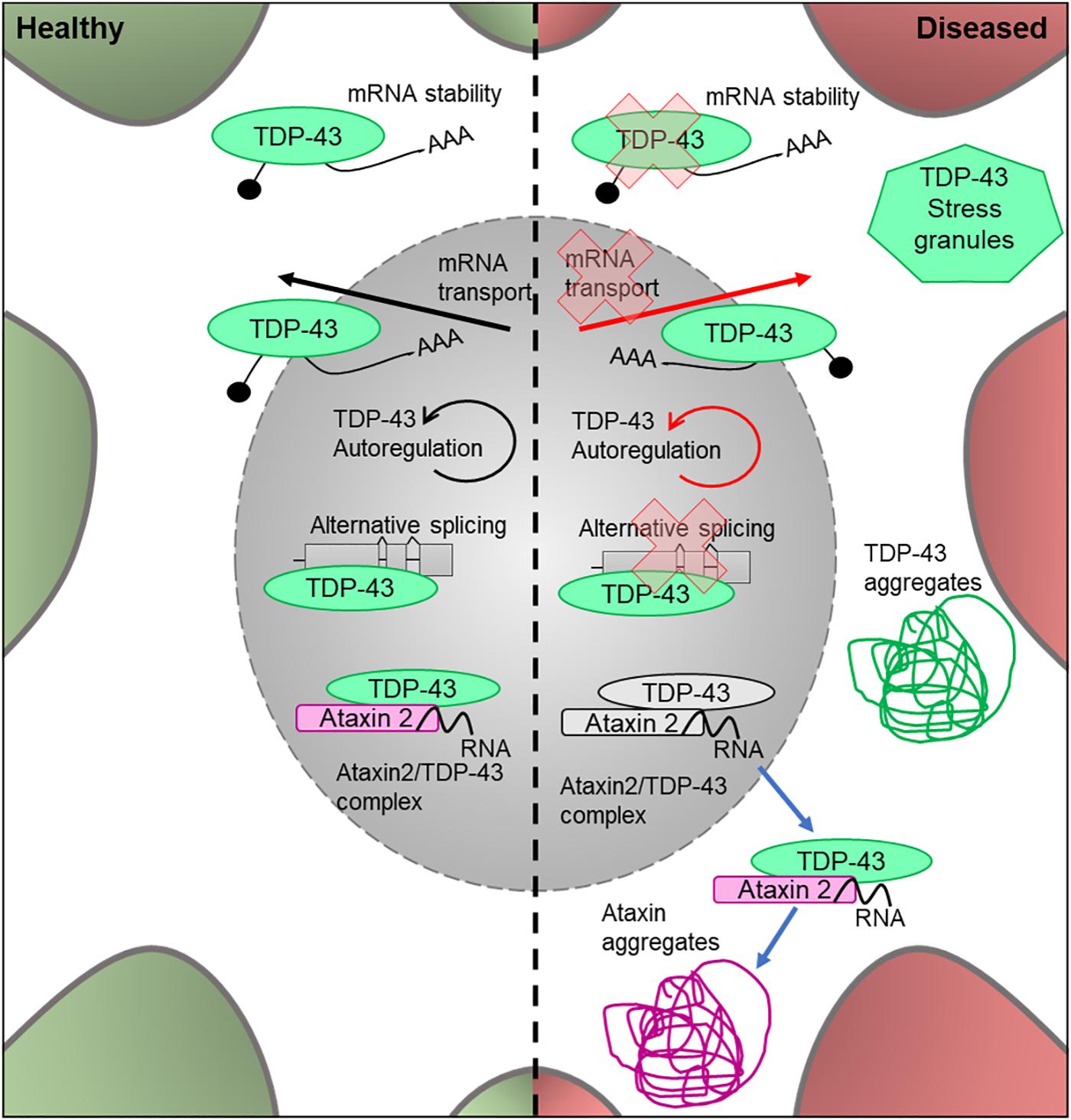
Figure 4. Cellular features of TDP-43 and Ataxin 2 in a healthy and diseased neuron. Schematic presentation of a neuronal cell body with normal and disease-state TDP-43 and Ataxin 2 cellular location and functions. In healthy neurons, TDP-43 is located in both the cytoplasm and the nucleus, where it undergoes autoregulation. It is an RNA-binding protein which is involved in alternative splicing, mRNA transport and cytoplasmic stability of the mRNA. In healthy neurons, Ataxin 2 is located in the nucleus and modifies TDP-43 activity by forming a complex in the presence of RNA. In the disease state, the protein loses its ability to autoregulate as well as its function in alternative splicing, mRNA transport and mRNA stability. It forms ubiqitinated TDP-43 aggregates in the cytoplasm and is known to associate in stress granules. In the disease state, the Ataxin 2/TDP-43 complex translocates to the cytoplasm where it forms TDP-43 and Ataxin 2 aggregates.
TARDBP is highly conserved between Drosophila and humans, and the fly homolog is called TBPH (Lukacsovich et al., 1999). This similarity allows for in-depth study of the role TDP-43 mutations play in ALS etiology. Both loss and gain of function TDP-43 models have been shown to negatively affect lifespan, motor function and synaptic transmission in Drosophila (Feiguin et al., 2009), all of which are common characteristics of ALS pathology (Diaper et al., 2013) showing that Drosophila can be successfully used to model this disease-associated gene. Moreover, these models have shown that manipulation of TDP-43 levels by either loss (mutant) or gain of function (overexpression of wildtype TBPH) leads to toxicity. A recent loss-of-function (TBPH null mutant) fly larval model suggests that TBPH binds to the pre-mRNA form of the gene and regulates its levels by preventing degradation of the transcripts (Chang et al., 2014). Drosophila models have been successfully utilized to suggest a mechanism by which TBPH proteins form aggregates in the cytoplasm of the cell. Other disease mechanisms have been discovered through the use of Drosophila disease models, for example, that synaptic transmission is an early event in the onset of ALS (Diaper et al., 2013). Both loss- and gain-of-function models have been shown to have impaired synaptic transmission in larval and adult models at the pre-synaptic bouton and a progressive loss of function phenotype (Chang et al., 2014). Moreover, a loss of TBPH can also lead to reduced cacophony protein levels. Cacophony encodes the alpha 1 subunit of the presynaptic calcium channel responsible for the presynaptic voltage gated Cav2 current. It is localized at the active zones of the NMJ and other synapses (Kawasaki et al., 2004; Peng and Wu, 2007) and is required for neurotransmitter release at the NMJ and for synaptic growth (Rieckhof et al., 2003). Due to its importance in NMJ function, cacophony dysregulation following TDP-43 loss may be an important disease mechanism in ALS and warrants further investigation. The discoveries highlighted here show the power of using in vivo fly models to study TDP-43 and its role in MND.
Ataxin 2
Ataxin 2 is thought to act as a modifier of TDP-43 toxicity, binding to TDP-43 in the presence of RNA and forming a nuclear complex of unknown function. In ALS sufferers, the complex is translocated into the cytoplasm where it contributes to the formation of TDP-43 protein aggregates (Elden et al., 2010; Figure 5). Thus, it is important to understand the interaction between these proteins, and whether mutations in ATXN2 play a causal role in TDP-43 toxicity in ALS.
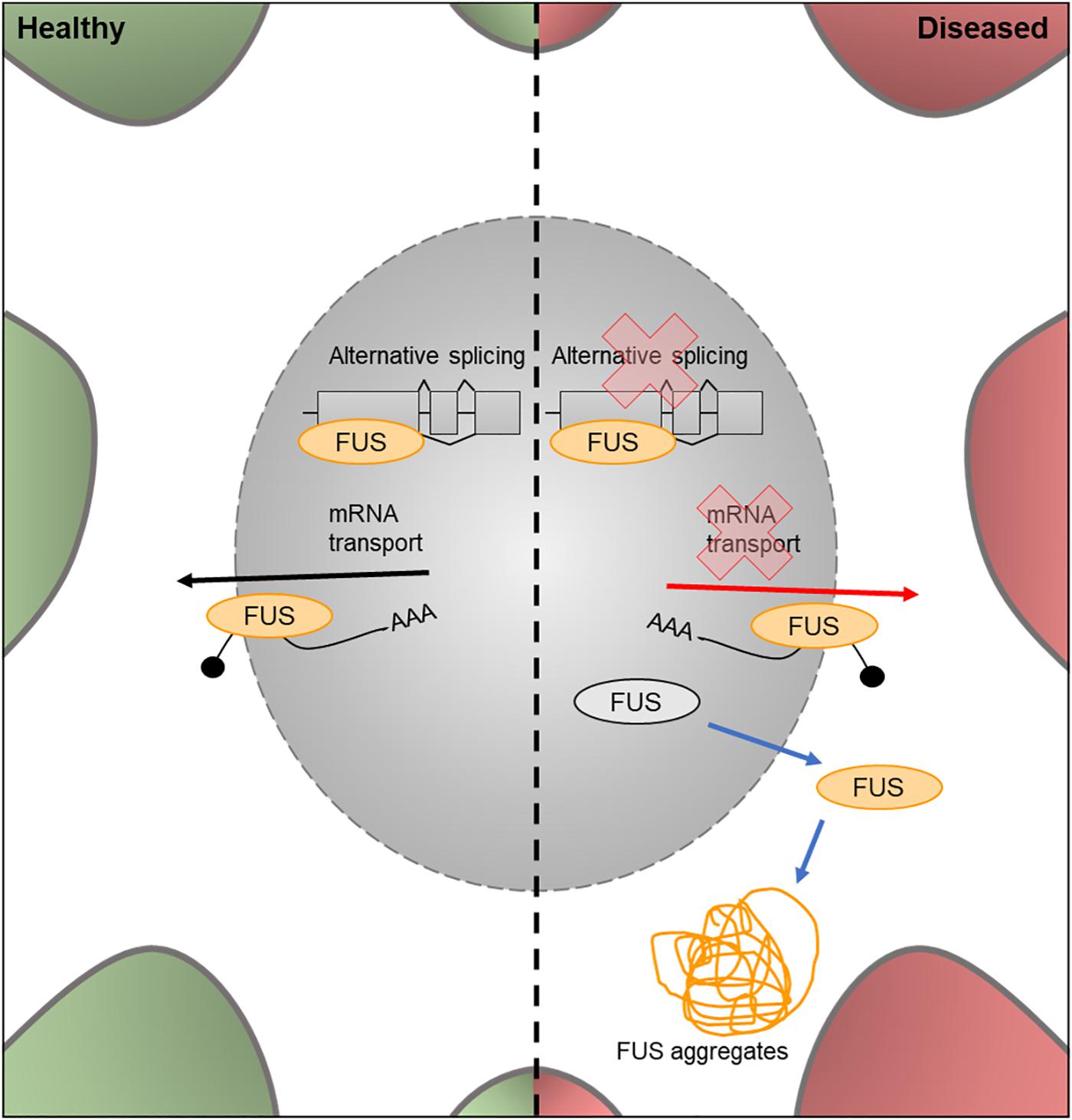
Figure 5. Cellular features of FUS in a healthy and diseased neuron. Schematic presentation of a neuronal cell body with normal and disease-state FUS cellular location and functions. In healthy neurons, FUS is located in both the nucleus and the cytoplasm and is involved in alternative splicing of pre-mRNA and mRNA transport out of the nucleus. In the disease state, FUS translocates into the cytoplasm and loses its function in splicing and mRNA transport. It can also form FUS aggregates due to phase transitioning.
Mutations which cause intermediate-length polyQ expansions (30Q) in ATXN2 are significantly associated with ALS. It has been shown that the upregulation of the Drosophila homolog of ATXN2 increased the toxicity of TDP-43 via increased accumulation of TDP-43 aggregates (Elden et al., 2010; Figure 5). These effects were observed in retinal structures and lead to decreased mobility and an overall reduction in lifespan. These results showed that the ability of ATXN2 to modulate TDP-43 toxicity is conserved through to Drosophila, again showing the value of this system to help understand the molecular events leading to MND.
However, TDP-43 is not the only ALS-associated gene which ATXN2 has been found to interact with. When ATXN2 (30Q) intermediate expansions are co-expressed with a depletion in C9orf72, there is an increase in ATXN2 aggregates and subsequent cell death (Ciura et al., 2016). Further, evaluation of ATXN2 as a disease modifier in patients carrying a C9orf72 expansion mutation suggests that patients with both intermediate ATXN2 repeat lengths and C9orf72 expansions are more susceptible to ALS development (van Blitterswijk et al., 2014). Together these studies support a complex mechanism for the onset and development of ALS, involving many genes and their interactions. With the power of the fruit fly genetic toolbox, these interactions can be rapidly characterized in vivo.
FUS
The FUS gene encodes an RNA binding protein, a component of the hnRNP complex. This protein is involved in the splicing of pre-mRNA and the export of processed mRNA into the cytoplasm from the nucleus (Vance et al., 2009; Figure 6). Mutations in the nuclear import factor signal of the FUS gene results in neuronal cytoplasmic mislocalization of FUS protein (Darovic et al., 2015) and these mutations have been observed in approximately 3% of FALS cases (Kwiatkowski et al., 2009).
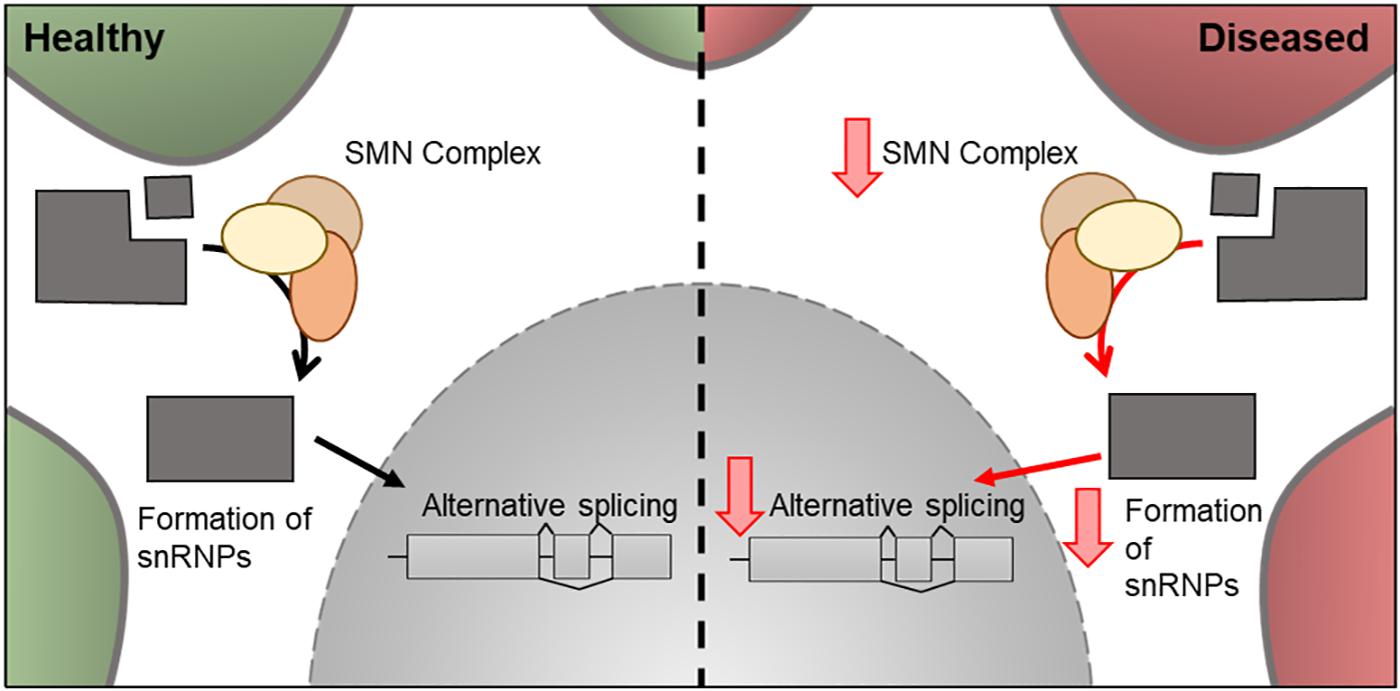
Figure 6. Cellular features of SMN1 in a healthy and diseased neuron. Schematic presentation of a neuronal cell body with normal and disease-state SMN cellular location and functions. In healthy neurons, SMN is located in the cytoplasm where it forms a complex involved in the assembly of snRNPs. These are essential for pre-mRNA splicing in the nucleus. In the disease state, the levels of SMN decreases and therefore there is a decrease in its downstream cellular functions.
The Drosophila ortholog of FUS is Caz and the mutants Caz P398L and Q349X are currently used as disease models for FUS-related ALS research. For example, expression of fly Caz, human wild-type or disease-relevant variants of FUS promote cytotoxicity (Jäckel et al., 2015). To study the function of Caz at the NMJ Machamer et al. (2014) used overexpression of wildtype Caz and mutant human FUS. It was suggested that Caz/FUS modulates the structure and function of the NMJ. Additionally, the overexpression of human FUS resulted in a decrease in presynaptic active zones and impaired synaptic transmission; possibly through a dominant-negative mechanism or by the downregulation of endogenous Caz. However, this finding was not repeated in wildtype Caz experiments, highlighting that overexpression of human genes in the fly may cause toxicity which is independent of disease-associated mutations. This finding was supported by another study, which showed disease-independent toxicity due to overexpression of human FUS (Xia et al., 2012). Drosophila has also been used to assess the role of FUS in axonal transport (Baldwin et al., 2016), which was affected in both FUS and Caz loss and gain-of-function models through perturbed vesicle and mitochondrial transport. It has also been shown that Caz mutants have severe developmental and locomotor defects which, in overexpression models, became more prominent with age, a stereotypical ALS phenotype. As both the loss and gain-of-function models affected the structure and function of the axon, it can be suggested that Caz is critical, and either loss or gain may result in a neurodegenerative phenotype.
Recent work has highlighted the mechanism of phase-transition in RNA-binding proteins and it is believed to play a role in RNA-binding protein-linked neurodegeneration (for full review see Harrison and Shorter, 2017). A Drosophila model of FUS-linked ALS has shown that this RNA-binding protein undergoes phase-transitioning into solid aggregates due to its prion-like binding domain and interactions with arginine-rich domains (Bogaert et al., 2018) and the effect of this on MND phenotypes and possible reversal of the transition has been further studied. Guo et al. (2018) have shown that in ALS-linked FUS-mutation overexpression models there is a higher level of neurodegeneration and a decrease in lifespan. However, these disease phenotypes can be rescued by overexpression of Kapβ2, a nuclear-import factor which reverses the production of FUS aggregates via phase-transitioning through prevention and disassociation of the formed aggregates.
Spinal Muscular Atrophy (SMA)
Spinal muscular atrophy is one of the leading genetic causes of infant mortality in the United States (Miniño et al., 2010) and results in the degeneration of the anterior motor neurons, progressive atrophy of the muscles and eventually respiratory failure and death (Lefebvre et al., 1995). It has an estimated incidence of between 1 in 6000 and 1 in 10,000 live births (Ogino et al., 2002) and a carrier frequency of 1/40 to 1/60 (Prior et al., 2010). There are five main forms of SMA, SMA0 (Pre-natal SMA), SMA1 (Werdnig-Hoffmann disease), SMA2 (Dubowitz disease), SMA3 (Kugelberg-Welander disease), and SMA4. These sub-types manifest themselves at different stages of life; prenatal, 0–6 months, 6–18 months, over 12 months and in adulthood, respectively (Pearn, 1980). The most severe form of SMA is SMA0 where death usually occurs rapidly after birth (Grotto et al., 2016). SMA1 is the next most severe, where patients cannot sit upright unaided and often do not survive past their second year. SMA2 and SMA3 are less severe and SMA4 is the least severe of the subgroups, with patients being able to walk unaided and only having mild symptoms (Munsat and Davies, 1992). Although they have different severities and ages of onset, the subgroup of an SMA sufferer is not considered when selecting patients for clinical trials as often endpoint and interventions-used are a more important factor. To date, the only drug which has been approved to treat SMA in the United States is SpinrazaTM (Nusinersen). In clinical trials, administering SpinrazaTM every 4 months resulted in an improvement in motor function in ∼60% of infants (FDA, 2016).
In 1995, mutations in exon 7 of the SMN1 gene and resulting decrease in SMN protein levels was associated with SMA (Lefebvre et al., 1995). Around 95% of SMA cases are caused by the homozygous absence of SMN1 (Sugarman et al., 2012). SMN1 is located near the telomere of chromosome 5, whereas SMN2, a nearly identical copy of SMN1 is located closer to the centromere and, due to a silent C-to-T transition (C840T), undergoes alternative splicing to create a truncated protein, which is rapidly degraded in the cell (Burnett et al., 2009). Although both SMN1 and SMN2 produce SMN protein, only 10% of SMN2 transcripts produce functional protein (Monani et al., 1999). A decrease of SMN protein is directly correlated with the classification of SMA and severity of the disease, with reductions in levels of ∼70, ∼50, and ∼30% in types 1, 2, and 3, respectively (Crawford et al., 2012). The copy number of the SMN2 gene, also increases with sub-group classification with SMN0 patients having only one copy of the gene (Butchbach, 2016). This suggests SMN2 can play a disease-modifying role in the severity of SMA (Crawford et al., 2012). The mechanism of actions of SpinrazaTM is to increase the levels of SMA and it contains an anti-sense oligonucleotide directed to SMN2, used to increase the levels of functional SMN protein produced in the cell (Zanetta et al., 2014). SMN is a cytoplasmic protein involved in the assembly of snRNP, which are essential for pre-mRNA splicing (Figure 6). It has been hypothesized that the mechanism which leads to SMA is either a loss-of-function of SMN-mediated snRNP assembly role, resulting in alternative splicing of target genes (Gabanella et al., 2007) or a loss-of-function of mRNA transport in neurons (Fan and Simard, 2002; Eggert et al., 2006). However, other functions related to the actin-cytoskeleton, such as neurite outgrowth, NMJ formation, and profilin binding, have also been implicated in SMA pathology (Giesemann et al., 1999; Chan et al., 2003; Rossoll et al., 2003).
The Drosophila genome has one ortholog of SMN1 (DmSMN) (Miguel-Aliaga et al., 2000; Chan et al., 2003). Drosophila models utilizing a mutated DmSMN73Ao have been used to show that reduced levels and activity of SMN protein causes the inability to fly or jump, morphological defects at the NMJ, and lethality (Chan et al., 2003; Chang et al., 2008). This shows that SMA pathology can be successfully modeled in Drosophila. Null mutants and RNAi lines have been used to reduce SMN levels by different degrees, mimicking the levels of SMN found in different SMA classifications and recapitulating the disease more thoroughly. These models have been successfully used to further classify the role of the SMN protein (Chan et al., 2003) and its specific binding capability to snRNPs (Garcia et al., 2016). Recent work suggests an additional role for SMN in regulating the actin cytoskeleton (Braun et al., 1995; Rajendra et al., 2007). By showing that DmSMN mutants require protein rescue in both nerves and muscle tissue to save normal motor function, these studies suggest a muscle-specific role and a novel disease-causing pathway in the muscle tissue. SMN may act as a sarcomeric protein which is required for the expression of muscle-specific actin, its organization and the subsequent formation of muscle tissue, something which is lacking in SMA patients. However, it has also been suggested that muscle degeneration can also occur due to lack of innervation (Ling et al., 2012), highlighting the important role SMN has in the nervous system. Further research is required to discern whether incorrect muscle formation is due to a motor neuron-specific role of SMN and a subsequent lack of innervation, or whether a muscle-specific novel function of SMN also contributes to SMA pathology. This highlights the importance of the overall view of SMA pathology to establish a timeline of the disease and the role of correct muscle cell function in the severity of SMA. The use of Drosophila in the study of SMA will allow for future study into tissue-specific rescue models and the analysis of the timeline of SMA pathology to elucidate the pathology and molecular etiology of this disease.
Conclusion
With the completion of the Drosophila genome sequence in 2000 (Adams et al., 2000), and the publishing of the first draft of the human genome in 2001 (International Human Genome Sequencing Consortium [IHGS], 2001; Venter et al., 2001) the high level of conservation of genes between fruit flies and humans was established, and the benefit of systematic genetic dissection of molecular pathways in the fruit fly was reinforced. The use of Drosophila genetics has a long and successful history in the study of various human genetic diseases, ranging from cancer biology (Simon et al., 1991; Olivier et al., 1993) to cardiovascular disease (Kim et al., 2010; Neely et al., 2010), from Parkinson’s (Feany and Bender, 2000; Song et al., 2017) to Alzheimer’s (Kilian et al., 2017). The studies discussed here highlight the successful research efforts into understanding MND pathologies but also show possible future directions of research efforts; which are needed to increase our understanding of MND and to find novel therapeutics. As a model organism, Drosophila provides a fast, cheap, and powerful platform to define the genetic underpinnings of complex human diseases and should be continually used in the investigation into MND.
Author Contributions
All authors contributed to the writing of the manuscript and read and approved the final manuscript.
Funding
This work was funded by the Motor Neuron Disease Research Institute of Australia under grants: GIA 1640 and GIA 1718.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ALS, amyotrophic lateral sclerosis; C9orf72, chromosome 9 open reading frame 72; Caz, Cabeza; DPR, di-peptide repeat(s); FALS, familial amyotrophic lateral sclerosis; FUS, fused-in-sarcoma; G4C2, GGGGCC; GWAS, genome-wide association study(ies); hnRNP, heterogeneous nuclear ribonucleoprotein(s); hSOD, human SOD; MND, Motor Neuron Disease(s); NMJ, neuromuscular junction; snRNP, small nuclear ribonucleoprotein(s); SALS, sporadic amyotrophic lateral sclerosis; SMA, spinal muscular atrophy; SMN1, survival of motor neuron 1; SNP, single nucleotide polymorphism(s); SOD, superoxide dismutase; TBPH, TAR DNA binding homolog.
References
Adams, M. D., Celniker, S. E., Holt, R. A., Evans, C. A., Gocayne, J. D., Amanatides, P. G., et al. (2000). The genome sequence of Drosophila melanogaster. Science 287, 2185–2195. doi: 10.1126/science.287.5461.2185
Ahmeti, K. B., Ajroud-Driss, S., Al-Chalabi, A., Andersen, P. M., Armstrong, J., Birve, A., et al. (2013). Age of onset of amyotrophic lateral sclerosis is modulated by a locus on 1p34.1. Neurobiol. Aging 34, 357.e7–357.e19. doi: 10.1016/j.neurobiolaging.2012.07.017
Al-Chalabi, A., Fang, F., Hanby, M. F., Leigh, P. N., Shaw, C. E., Ye, W., et al. (2010). An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 81, 1324–1326. doi: 10.1136/jnnp.2010.207464
Al-Chalabi, A., and Visscher, P. M. (2014). Motor neuron disease: common genetic variants and the heritability of ALS. Nat. Rev. Neurol. 10, 549–550. doi: 10.1038/nrneurol.2014.166
Alonso, A., Logroscino, G., Jick, S. S., and Hernán, M. A. (2009). Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. Eur. J. Neurol. 16, 745–751. doi: 10.1111/j.1468-1331.2009.02586.x
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Arthur, K. C., Calvo, A., Price, T. R., Geiger, J. T., Chiò, A., and Traynor, B. J. (2016). Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 7:12408. doi: 10.1038/ncomms12408
Ash, P. E. A., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W.-L., Dejesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., Korbel, J. O., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Baldwin, K. R., Godena, V. K., Hewitt, V. L., and Whitworth, A. J. (2016). Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet. 25, 2378–2392.
Beers, D. R., Henkel, J. S., Xiao, Q., Zhao, W., Wang, J., Yen, A. A., et al. (2006). Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 103, 16021–16026. doi: 10.1073/pnas.0607423103
Bogaert, E., Boeynaems, S., Kato, M., Guo, L., Caulfield, T. R., Steyaert, J., et al. (2018). Molecular dissection of FUS points at synergistic effect of low-complexity domains in toxicity. Cell Rep. 24, 529.e4–537.e4. doi: 10.1016/j.celrep.2018.06.070
Bozzoni, V., Pansarasa, O., Diamanti, L., Nosari, G., Cereda, C., and Ceroni, M. (2016). Amyotrophic lateral sclerosis and environmental factors. Funct. Neurol. 31, 7–19. doi: 10.11138/FNeur/2016.31.1.007
Brand, A. H., and Perrimon, N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415.
Braun, S., Warter, J. M., Poindron, P., Croizat, B., and Lagrange, M. C. (1995). Constitutive muscular abnormalities in culture in spinal muscular atrophy. Lancet 345, 694–695. doi: 10.1016/S0140-6736(95)90869-2
Bunton-Stasyshyn, R. K. A., Saccon, R. A., Fratta, P., and Fisher, E. M. C. (2015). SOD1 function and its implications for amyotrophic lateral sclerosis pathology. Neuroscience 21, 519–529. doi: 10.1177/1073858414561795
Buratti, E., and Baralle, F. E. (2010). The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 7, 420–429. doi: 10.4161/rna.7.4.12205
Burguete, A. S., Almeida, S., Gao, F.-B., Kalb, R., Akins, M. R., and Bonini, N. M. (2015). GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. eLife 4:e08881. doi: 10.7554/eLife.08881
Burnett, B. G., Muñoz, E., Tandon, A., Kwon, D. Y., Sumner, C. J., and Fischbeck, K. H. (2009). Regulation of SMN protein stability. Mol. Cell Biol. 29, 1107–1115. doi: 10.1128/MCB.01262-08
Butchbach, M. E. R. (2016). Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front. Mol. Biosci. 3:7. doi: 10.3389/fmolb.2016.00007
Chan, Y. B., Miguel-Aliaga, I., Franks, C., Thomas, N., Trülzsch, B., Sattelle, D. B., et al. (2003). Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum. Mol. Genet. 12, 1367–1376. doi: 10.1093/hmg/ddg157
Chang, H. C.-H., Dimlich, D. N., Yokokura, T., Mukherjee, A., Kankel, M. W., Sen, A., et al. (2008). Modeling spinal muscular atrophy in Drosophila. PLoS One. 3:3209. doi: 10.1371/journal.pone.0003209
Chang, J.-C., Hazelett, D. J., Stewart, J. A., and Morton, D. B. (2014). Motor neuron expression of the voltage-gated calcium channel cacophony restores locomotion defects in a Drosophila, TDP-43 loss of function model of ALS. Brain Res. 1584, 39–51. doi: 10.1016/j.brainres.2013.11.019
Chiang, P.-M., Ling, J., Jeong, Y. H., Price, D. L., Aja, S. M., and Wong, P. C. (2010). Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc. Natl. Acad. Sci. U.S.A. 107, 16320–16324. doi: 10.1073/pnas.1002176107
Chiò, A., Logroscino, G., Traynor, B. J., Collins, J., Simeone, J. C., Goldstein, L. A., et al. (2013). Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41, 118–130. doi: 10.1159/000351153
Ciura, S., Sellier, C., Campanari, M.-L., Charlet-Berguerand, N., and Kabashi, E. (2016). The most prevalent genetic cause of ALS-FTD, C9orf72 synergizes the toxicity of ATXN2 intermediate polyglutamine repeats through the autophagy pathway. Autophagy 12, 1406–1408. doi: 10.1080/15548627.2016.1189070
Clark, J., and Ding, S. (2006). Generation of RNAi libraries for high-throughput screens. J. Biomed. Biotechnol. 2006:45716. doi: 10.1155/JBB/2006/45716
Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillée, S., Rule, M., et al. (2003). Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302, 113–117. doi: 10.1126/science.1086071
Cooper-Knock, J., Walsh, M. J., Higginbottom, A., Robin Highley, J., Dickman, M. J., Edbauer, D., et al. (2014). Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 137, 2040–2051. doi: 10.1093/brain/awu120
Crawford, T. O., Paushkin, S. V., Kobayashi, D. T., Forrest, S. J., Joyce, C. L., Finkel, R. S., et al. (2012). Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS One 7:33572. doi: 10.1371/journal.pone.0033572
Danial, N. N., and Korsmeyer, S. J. (2004). Cell death: critical control points. Cell 116, 205–219. doi: 10.1016/S0092-8674(04)00046-7
Darovic, S., Prpar Mihevc, S., Župunski, V., Gunčar, G., Štalekar, M., Lee, Y.-B., et al. (2015). Phosphorylation of C-terminal tyrosine residue 526 in FUS impairs its nuclear import. J. Cell Sci. 128, 4151–4159. doi: 10.1242/jcs.176602
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Diaper, D. C., Adachi, Y., Sutcliffe, B., Humphrey, D. M., Elliott, C. J. H., Stepto, A., et al. (2013). Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum. Mol. Genet. 22, 1539–1557. doi: 10.1093/hmg/ddt005
Dietzl, G., Chen, D., Schnorrer, F., Su, K.-C., Barinova, Y., Fellner, M., et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156. doi: 10.1038/nature05954
Drenth, J. P. H., and Waxman, S. G. (2007). Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J. Clin. Invest. 117, 3603–3609. doi: 10.1172/JCI33297
Eggert, C., Chari, A., Laggerbauer, B., and Fischer, U. (2006). Spinal muscular atrophy: the RNP connection. Trends Mol. Med. 12, 113–121. doi: 10.1016/j.molmed.2006.01.005
Elden, A. C., Kim, H.-J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320
Fan, L., and Simard, L. R. (2002). Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum. Mol. Genet. 11, 1605–1614. doi: 10.1093/hmg/11.14.1605
FDA (2016). SPINRAZA (Nusinersen) Injection, for Intrathecal Use Initial U.S. Approval: 2016. Silver Spring, MD: FDA.
Feany, M. B., and Bender, W. W. (2000). A Drosophila model of Parkinson’s disease. Nature 404, 394–398. doi: 10.1038/35006074
Feiguin, F., Godena, V. K., Romano, G., D’Ambrogio, A., Klima, R., and Baralle, F. E. (2009). Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 583, 1586–1592. doi: 10.1016/j.febslet.2009.04.019
Feil, S., Valtcheva, N., and Feil, R. (2009). “Inducible Cre Mice,” in Gene Knockout Protocols, 2nd Edn, eds W. Wurst and R. Kühn (New York, NY: Humana Press), 343–363. doi: 10.1007/978-1-59745-471-1_18
Feldman, M. W., and Ramachandran, S. (2018). Missing compared to what? Revisiting heritability, genes and culture. Philos. Trans. R. Soc. B Biol. Sci. 373:20170064. doi: 10.1098/rstb.2017.0064
Fogh, I., Ratti, A., Gellera, C., Lin, K., Tiloca, C., Moskvina, V., et al. (2014). A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 23, 2220–2231. doi: 10.1093/hmg/ddt587
Fortini, M. E., Skupski, M. P., Boguski, M. S., and Hariharan, I. K. A. (2000). survey of human disease gene counterparts in the Drosophila genome. J. Cell Biol. 150, 23–30. doi: 10.1083/jcb.150.2.F23
Frank, C. A. (2014). Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology 78, 63–74. doi: 10.1016/j.neuropharm.2013.06.015
Freibaum, B. D., Lu, Y., Lopez-Gonzalez, R., Kim, N. C., Almeida, S., Lee, K.-H., et al. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. doi: 10.1038/nature14974
Gabanella, F., Butchbach, M. E. R., Saieva, L., Carissimi, C., Burghes, A. H. M., and Pellizzoni, L. (2007). Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One 2:921. doi: 10.1371/journal.pone.0000921
Gait, R., Maginnis, C., Lewis, S., Pickering, N., Antoniak, M., Hubbard, R., et al. (2003). Occupational exposure to metals and solvents and the risk of motor neuron disease. Neuroepidemiology 22, 353–356. doi: 10.1159/000072925
Gallart-Palau, X., Ng, C.-H., Ribera, J., Sze, S. K., and Lim, K.-L. (2016). Drosophila expressing human SOD1 successfully recapitulates mitochondrial phenotypic features of familial amyotrophic lateral sclerosis. Neurosci. Lett. 624, 47–52. doi: 10.1016/j.neulet.2016.05.006
Garcia, E. L., Wen, Y., Praveen, K., and Matera, A. G. (2016). Transcriptomic comparison of Drosophila snRNP biogenesis mutants reveals mutant-specific changes in pre-mRNA processing: implications for spinal muscular atrophy. RNA 22, 1215–1227. doi: 10.1261/rna.057208.116
Giesemann, T., Rathke-Hartlieb, S., Rothkegel, M., Bartsch, J. W., Buchmeier, S., Jockusch, B. M., et al. (1999). A role for polyproline motifs in the spinal muscular atrophy protein SMN. Profilins bind to and colocalize with smn in nuclear gems. J. Biol. Chem. 274, 37908–37914. doi: 10.1074/jbc.274.53.37908
Giordana, M. T., Piccinini, M., Grifoni, S., De Marco, G., Vercellino, M., Magistrello, M., et al. (2010). TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol. 20, 351–360. doi: 10.1111/j.1750-3639.2009.00284.x
Grotto, S., Cuisset, J.-M., Marret, S., Drunat, S., Faure, P., Audebert-Bellanger, S., et al. (2016). Type 0 spinal muscular atrophy: further delineation of prenatal and postnatal features in 16 patients. J. Neuromuscul. Dis. 3, 487–495. doi: 10.3233/JND-160177
Guo, L., Kim, H. J., Wang, H., Monaghan, J., Freyermuth, F., Sung, J. C., et al. (2018). Nuclear-import receptors reverse aberrant phase transitions of rna-binding proteins with prion-like domains. Cell 173, 677.e20–692.e20. doi: 10.1016/j.cell.2018.03.002
Harrison, A. F., and Shorter, J. (2017). RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 474, 1417–1438. doi: 10.1042/BCJ20160499
Hewitt, V. L., and Whitworth, A. J. (2017). Mechanisms of Parkinson’s disease: lessons from Drosophila. Curr. Top. Dev. Biol. 121, 173–200. doi: 10.1016/bs.ctdb.2016.07.005
Huynh, W., Dharmadasa, T., Vucic, S., and Kiernan, M. C. (2018). Functional biomarkers for amyotrophic lateral sclerosis. Front. Neurol. 9:1141. doi: 10.3389/fneur.2018.01141
International Human Genome Sequencing Consortium [IHGS]. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921.
Jäckel, S., Summerer, A. K., Thömmes, C. M., Pan, X., Voigt, A., Schulz, J. B., et al. (2015). Nuclear import factor transportin and arginine methyltransferase 1 modify FUS neurotoxicity in Drosophila. Neurobiol. Dis. 74, 76–88. doi: 10.1016/j.nbd.2014.11.003
Kamel, F., Umbach, D. M., Munsat, T. L., Shefner, J. M., Hu, H., and Sandler, D. P. (2002). Lead exposure and amyotrophic lateral sclerosis. Epidemiology 13, 311–319. doi: 10.1097/00001648-200205000-00012
Kamimura, K., Ueno, K., Nakagawa, J., Hamada, R., Saitoe, M., and Maeda, N. (2013). Perlecan regulates bidirectional Wnt signaling at the Drosophila neuromuscular junction. J. Cell Biol. 200, 219–233. doi: 10.1083/jcb.201207036
Kawasaki, F., Zou, B., Xu, X., and Ordway, R. W. (2004). Active zone localization of presynaptic calcium channels encoded by the cacophony locus of Drosophila. J. Neurosci. 24, 282–285. doi: 10.1523/JNEUROSCI.3553-03.2004
Keller, M. F., Ferrucci, L., Singleton, A. B., Tienari, P. J., Laaksovirta, H., Restagno, G., et al. (2014). Genome-wide analysis of the heritability of amyotrophic lateral sclerosis. JAMA Neurol. 71, 1123–1134. doi: 10.1001/jamaneurol.2014.1184
Keshishian, H., Broadie, K., Chiba, A., and Bate, M. (1996). The Drosophila neuromuscular junction: a model system for studying synaptic development and function. Annu. Rev. Neurosci. 19, 545–575. doi: 10.1146/annurev.ne.19.030196.002553
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kilian, J. G., Hsu, H.-W., Mata, K., Wolf, F. W., and Kitazawa, M. (2017). Astrocyte transport of glutamate and neuronal activity reciprocally modulate tau pathology in Drosophila. Neuroscience 348, 191–200. doi: 10.1016/j.neuroscience.2017.02.011
Kim, I.-M., Wolf, M. J., and Rockman, H. A. (2010). Gene deletion screen for cardiomyopathy in adult Drosophila identifies a new notch ligand. Circ. Res. 106, 1233–1243. doi: 10.1161/CIRCRESAHA.109.213785
Kittel, R. J., Wichmann, C., Rasse, T. M., Fouquet, W., Schmidt, M., Schmid, A., et al. (2006). Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science 312, 1051–1054. doi: 10.1126/science.1126308
Koppers, M., van Blitterswijk, M. M., Vlam, L., Rowicka, P. A., van Vught, P. W. J., Groen, E. J. N., et al. (2012). VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 33, 837.e7–837.e13. doi: 10.1016/j.neurobiolaging.2011.10.006
Kwiatkowski, T. J., Bosco, D. A., LeClerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Lee, E. B., Lee, V. M.-Y., and Trojanowski, J. Q. (2012). Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50. doi: 10.1038/nrn3121
Lee, K.-H., Zhang, P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774–788. doi: 10.1016/j.cell.2016.10.002
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. doi: 10.1016/0092-8674(95)90460-3
Lill, C. M., Abel, O., Bertram, L., and Al-Chalabi, A. (2011). Keeping up with genetic discoveries in amyotrophic lateral sclerosis: the ALSoD and ALSGene databases. Amyotroph. Lateral Scler. 12, 238–249. doi: 10.3109/17482968.2011.584629
Ling, K. K. Y., Gibbs, R. M., Feng, Z., and Ko, C.-P. (2012). Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 21, 185–195. doi: 10.1093/hmg/ddr453
Lloyd, T. E., Verstreken, P., Ostrin, E. J., Phillippi, A., Lichtarge, O., and Bellen, H. J. (2000). A genome-wide search for synaptic vesicle cycle proteins in Drosophila. Neuron 26, 45–50. doi: 10.1016/S0896-6273(00)81136-8
Lukacsovich, T., Asztalos, Z., Juni, N., Awano, W., and Yamamoto, D. (1999). The Drosophila melanogaster 60A chromosomal division is extremely dense with functional genes: their sequences, genomic organization, and expression. Genomics 57, 43–56. doi: 10.1006/geno.1999.5746
Machamer, J. B., Collins, S. E., and Lloyd, T. E. (2014). The ALS gene FUS regulates synaptic transmission at the Drosophila neuromuscular junction. Hum. Mol. Genet. 23, 3810–3822. doi: 10.1093/hmg/ddu094
Mackenzie, I. R., Arzberger, T., Kremmer, E., Troost, D., Lorenzl, S., Mori, K., et al. (2013). Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 126, 859–879. doi: 10.1007/s00401-013-1181-y
Mackenzie, I. R. A., Bigio, E. H., Ince, P. G., Geser, F., Neumann, M., Cairns, N. J., et al. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis withSOD1 mutations. Ann. Neurol. 61, 427–434. doi: 10.1002/ana.21147
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G. P., Waite, A., Rollinson, S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330. doi: 10.1016/S1474-4422(12)70043-1
Malek, A. M., Barchowsky, A., Bowser, R., Youk, A., and Talbott, E. O. (2012). Pesticide exposure as a risk factor for amyotrophic lateral sclerosis: a meta-analysis of epidemiological studies: pesticide exposure as a risk factor for ALS. Environ. Res. 117, 112–119. doi: 10.1016/j.envres.2012.06.007
Max-Planck-Gesellschaft (2017). ). The Max Planck Society for the Advancement of Science. ALSGene. Munich: Max-Planck-Gesellschaft.
Megighian, A., Zordan, M., Pantano, S., Scorzeto, M., Rigoni, M., Zanini, D., et al. (2013). Evidence for a radial SNARE super-complex mediating neurotransmitter release at the Drosophila neuromuscular junction. J. Cell Sci. 126, 3134–3140. doi: 10.1242/jcs.123802
Mehta, P., Antao, V., Kaye, W., Sanchez, M., Williamson, D., Bryan, L., et al. (2014). Prevalence of amyotrophic lateral sclerosis - United States, 2010-2011. MMWR Suppl. 63, 1–14.
Miguel-Aliaga, I., Chan, Y. B., Davies, K. E., and van den Heuvel, M. (2000). Disruption of SMN function by ectopic expression of the human SMN gene in Drosophila. FEBS Lett. 486, 99–102. doi: 10.1016/S0014-5793(00)02243-2
Miniño, A. M., Xu, J., and Kochanek, K. D. (2010). Deaths: preliminary data for 2008. Natl. Vital. Stat. Rep. 59, 1–52.
Mizielinska, S., Grönke, S., Niccoli, T., Ridler, C. E., Clayton, E. L., Devoy, A., et al. (2014). C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194. doi: 10.1126/science.1256800
Moens, T. G., Mizielinska, S., Niccoli, T., Mitchell, J. S., Thoeng, A., Ridler, C. E., et al. (2018). Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta Neuropathol. 135, 445–457. doi: 10.1007/s00401-017-1798-3
Monani, U. R., Lorson, C. L., Parsons, D. W., Prior, T. W., Androphy, E. J., Burghes, A. H. M., et al. (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183. doi: 10.1093/hmg/8.7.1177
Mori, K., Weng, S.-M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Munsat, T. L., and Davies, K. E. (1992). International SMA consortium meeting. Neuromuscul. Disord. 2, 423–428. doi: 10.1016/S0960-8966(06)80015-5
Murray, L. M., Talbot, K., and Gillingwater, T. H. (2010). Neuromuscular synaptic vulnerability in motor neurone disease: amyotrophic lateral sclerosis and spinal muscular atrophy. Neuropathol. Appl. Neurobiol. 36, 133–156. doi: 10.1111/j.1365-2990.2010.01061.x
Muscular Dystrophy Association (2019). ALS: Amyotrophic Lateral Sclerosis - Stages of ALS. Chicago, IL: Muscular Dystrophy Association National Office.
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Neely, G. G., Kuba, K., Cammarato, A., Isobe, K., Amann, S., Zhang, L., et al. (2010). A global in vivo Drosophila RNAi screen identifies NOT3 as a conserved regulator of heart function. Cell 141, 142–153. doi: 10.1016/j.cell.2010.02.023
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Ogino, S., Leonard, D. G. B., Rennert, H., Ewens, W. J., and Wilson, R. B. (2002). Genetic risk assessment in carrier testing for spinal muscular atrophy. Am. J. Med. Genet. 110, 301–307. doi: 10.1002/ajmg.10425
Olivier, J. P., Raabe, T., Henkemeyer, M., Dickson, B., Mbamalu, G., Margolis, B., et al. (1993). A Drosophila SH2-SH3 adaptor protein implicated in coupling the sevenless tyrosine kinase to an activator of Ras guanine nucleotide exchange, Sos. Cell 73, 179–191. doi: 10.1016/0092-8674(93)90170-U
Pearn, J. (1980). Classification of spinal muscular atrophies. Lancet 315, 919–922. doi: 10.1016/S0140-6736(80)90847-8
Peng, I.-F., and Wu, C.-F. (2007). Drosophila cacophony channels: a major mediator of neuronal Ca2+ currents and a trigger for K+ channel homeostatic regulation. J. Neurosci. 27, 1072–1081. doi: 10.1523/JNEUROSCI.4746-06.2007
Prior, T. W., Snyder, P. J., Rink, B. D., Pearl, D. K., Pyatt, R. E., Mihal, D. C., et al. (2010). Newborn and carrier screening for spinal muscular atrophy. Am. J. Med. Genet. Part A 152, 1608–1616. doi: 10.1002/ajmg.a.33474
Rajendra, T. K., Gonsalvez, G. B., Walker, M. P., Shpargel, K. B., Salz, H. K., and Matera, A. G. (2007). A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J. Cell Biol. 176, 831–841. doi: 10.1083/jcb.200610053
Redler, R. L., and Dokholyan, N. V. (2012). The complex molecular biology of amyotrophic lateral sclerosis (ALS). Prog. Mol. Biol. Transl. Sci. 107, 215–262. doi: 10.1016/B978-0-12-385883-2.00002-3
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rieckhof, G. E., Yoshihara, M., Guan, Z., and Littleton, J. T. (2003). Presynaptic N-type calcium channels regulate synaptic growth. J. Biol. Chem. 278, 41099–41108. doi: 10.1074/jbc.M306417200
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rossoll, W., Jablonka, S., Andreassi, C., Kröning, A.-K., Karle, K., Monani, U. R., et al. (2003). Smn, the spinal muscular atrophy–determining gene product, modulates axon growth and localization of β-actin mRNA in growth cones of motoneurons. J. Cell Biol. 163, 801–812. doi: 10.1083/jcb.200304128
Şahin, A., Held, A., Bredvik, K., Major, P., Achilli, T.-M., Kerson, A. G., et al. (2017). Human SOD1 ALS mutations in a drosophila knock-in model cause severe phenotypes and reveal dosage-sensitive gain- and loss-of-function components. Genetics 205, 707–723. doi: 10.1534/genetics.116.190850
Shaw, C. E., Enayat, Z. E., Powell, J. F., Anderson, V. E., Radunovic, A., al-Sarraj, S., et al. (1997). Familial amyotrophic lateral sclerosis. Molecular pathology of a patient with a SOD1 mutation. Neurology 49, 1612–1616. doi: 10.1212/WNL.49.6.1612
Simon, M. A., Bowtell, D. D. L., Dodson, G. S., Laverty, T. R., and Rubin, G. M. (1991). Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 67, 701–716. doi: 10.1016/0092-8674(91)90065-7
Simon, N. G., Turner, M. R., Vucic, S., Al-Chalabi, A., Shefner, J., Lomen-Hoerth, C., et al. (2014). Quantifying disease progression in amyotrophic lateral sclerosis. Ann. Neurol. 76, 643–657. doi: 10.1002/ana.24273
Song, L., He, Y., Ou, J., Zhao, Y., Li, R., Cheng, J., et al. (2017). Auxilin underlies progressive locomotor deficits and dopaminergic neuron loss in a drosophila model of parkinson’s disease. Cell Rep. 18, 1132–1143. doi: 10.1016/j.celrep.2017.01.005
Sugarman, E. A., Nagan, N., Zhu, H., Akmaev, V. R., Zhou, Z., Rohlfs, E. M., et al. (2012). Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur. J. Hum. Genet. 20, 27–32. doi: 10.1038/ejhg.2011.134
Tabrizi, S. (2006). Neurodegenerative diseases neurobiology pathogenesis and therapeutics. J. Neurol. Neurosurg. Psychiatry 77, 284–284. doi: 10.1136/jnnp.2005.072710
Tenesa, A., and Haley, C. S. (2013). The heritability of human disease: estimation, uses and abuses. Nat. Rev. Genet. 14, 139–149. doi: 10.1038/nrg3377
Valdez, C., Scroggs, R., Chassen, R., and Reiter, L. T. (2015). Variation in Dube3a expression affects neurotransmission at the Drosophila neuromuscular junction. Biol. Open 4, 776–782. doi: 10.1242/bio.20148045
van Blitterswijk, M., Mullen, B., Heckman, M. G., Baker, M. C., DeJesus-Hernandez, M., Brown, P. H., et al. (2014). Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol. Aging 35, 2421.e13–2421.e17. doi: 10.1016/j.neurobiolaging.2014.04.016
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., et al. (2001). The sequence of the human genome. Science 291, 1304–1351. doi: 10.1126/science.1058040
Vonhoff, F., and Keshishian, H. (2017). Cyclic nucleotide signaling is required during synaptic refinement at the Drosophila neuromuscular junction. Dev. Neurobiol. 77, 39–60. doi: 10.1002/dneu.22407
Wang, I.-F., Wu, L.-S., Chang, H.-Y., and Shen, C.-K. J. (2007). TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem. 105, 797–806. doi: 10.1111/j.1471-4159.2007.05190.x
Watson, M. R., Lagow, R. D., Xu, K., Zhang, B., and Bonini, N. M. (2008). A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J. Biol. Chem. 283, 24972–24981. doi: 10.1074/jbc.M804817200
Wingo, T. S., Cutler, D. J., Yarab, N., Kelly, C. M., and Glass, J. D. (2011). The heritability of amyotrophic lateral sclerosis in a clinically ascertained United States research registry. PLoS One 6:27985. doi: 10.1371/journal.pone.0027985
Worms, P. M. (2001). The epidemiology of motor neuron diseases: a review of recent studies. J. Neurol. Sci. 191, 3–9. doi: 10.1016/S0022-510X(01)00630-X
Wu, L.-S., Cheng, W.-C., Hou, S.-C., Yan, Y.-T., Jiang, S.-T., and Shen, C.-K. J. (2009). TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis 48, 56–62. doi: 10.1002/dvg.20584
Xia, R., Liu, Y., Yang, L., Gal, J., Zhu, H., and Jia, J. (2012). Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS. Mol. Neurodegener. 7:10. doi: 10.1186/1750-1326-7-10
Yang, D., Abdallah, A., Li, Z., Lu, Y., Almeida, S., and Gao, F.-B. (2015). FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol. 130, 525–535. doi: 10.1007/s00401-015-1448-6
Yoshihara, M., Adolfsen, B., Galle, K. T., and Littleton, J. T. (2005). Retrograde signaling by Syt 4 induces presynaptic release and synapse-specific growth. Science 310, 858–863. doi: 10.1126/science.1117541
Zanetta, C., Nizzardo, M., Simone, C., Monguzzi, E., Bresolin, N., Comi, G. P., et al. (2014). Molecular therapeutic strategies for spinal muscular atrophies: current and future clinical trials. Clin. Ther. 36, 128–140. doi: 10.1016/j.clinthera.2013.11.006
Zhang, K., Donnelly, C. J., Haeusler, A. R., Grima, J. C., Machamer, J. B., Steinwald, P., et al. (2015). The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61. doi: 10.1038/nature14973
Keywords: Motor Neuron Disease, Drosophila melanogaster, UAS/Gal4/Gal80, amyotrophic lateral sclerosis, spinal muscular atrophy
Citation: Walters R, Manion J and Neely GG (2019) Dissecting Motor Neuron Disease With Drosophila melanogaster. Front. Neurosci. 13:331. doi: 10.3389/fnins.2019.00331
Received: 02 November 2018; Accepted: 21 March 2019;
Published: 12 April 2019.
Edited by:
Efthimios M. C. Skoulakis, Alexander Fleming Biomedical Sciences Research Center, GreeceReviewed by:
Federico Herrera, Instituto de Tecnologia Química e Biológica António Xavier (ITQB NOVA), PortugalDimitrios John Stravopodis, National and Kapodistrian University of Athens, Greece
Copyright © 2019 Walters, Manion and Neely. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John Manion, am9obi5tYW5pb25Ac3lkbmV5LmVkdS5hdQ== G. Gregory Neely, Z3JlZy5uZWVseUBzeWRuZXkuZWR1LmF1