 Nnana Amakiri
Nnana Amakiri Aaron Kubosumi
Aaron Kubosumi James Tran
James Tran P. Hemachandra Reddy
P. Hemachandra Reddy- 1Department of Internal Medicine, Texas Tech University Health Sciences Center, Lubbock, TX, United States
- 2Garrison Institute on Aging, Texas Tech University Health Sciences Center, Lubbock, TX, United States
- 3Garrison Institute on Aging, South West Campus, Texas Tech University Health Sciences Center, Lubbock, TX, United States
- 4Department of Cell Biology and Biochemistry, Texas Tech University Health Sciences Center, Lubbock, TX, United States
- 5Department Pharmacology and Neuroscience, Texas Tech University Health Sciences Center, Lubbock, TX, United States
- 6Department of Neurology, Texas Tech University Health Sciences Center, Lubbock, TX, United States
- 7Department of Speech-Language and Hearing Clinics, Texas Tech University Health Sciences Center, Lubbock, TX, United States
- 8Department of Public Health, Graduate School of Biomedical Sciences, Texas Tech University Health Sciences Center, Lubbock, TX, United States
Alzheimer’s disease (AD) is a progressive mental illness characterized by memory loss and multiple cognitive impairments. In the last several decades, significant progress has been made in understanding basic biology, molecular mechanisms, and development of biomarkers and therapeutic drugs. Multiple cellular changes are implicated in the disease process including amyloid beta and phosphorylation of tau synaptic damage and mitochondrial dysfunction in AD. Among these, amyloid beta is considered a major player in the disease process. Recent advancements in molecular biology revealed that microRNAs (miRNAs) are considered potential biomarkers in AD with a focus on amyloid beta. In this article we discussed several aspects of AD including its prevalence, classifications, risk factors, and amyloid species and their accumulation in subcellular compartments. This article also discusses the discovery and biogenesis of miRNAs and their relevance to AD. Today’s research continues to add to the wealth of miRNA data that has been accumulated, however, there still lacks clear-cut understanding of the physiological relevance of miRNAs to AD. MiRNAs appear to regulate translation of gene products in AD and other human diseases. However, the mechanism of how many of these miRNAs regulate both the 5′ and 3′UTR of amyloid precursor protein (APP) processing is still being extrapolated. Hence, we still need more research on miRNAs and APP/amyloid beta formation in the progression and pathogenesis of AD.
Introduction
Alzheimer’s disease (AD) is a progressive multifactorial neurodegenerative form of dementia. AD was first discovered in the late 19th and early 20th centuries by pioneer Alois Alzheimer who documented many patients sharing similar symptoms with similar deteriorations seen in the patients’ brain anatomy (Alzheimer et al., 1991). Due to the progressive degeneracy of the brain seen in this disease, AD is characterized by memory loss, numerous cognitive impairments, and changes in personality, thought, and behavior (Reddy et al., 2017). Early in the disease, AD has a substantial impact on one’s daily routine by affecting areas of the brain that control memory, executive cognition and visuospatial awareness. Personality, behavior, and language impairments tend to occur much later in the progression of AD. While AD greatly impacts patients, it also poses a significant emotional and financial burden on the patients’ family members.
Alzheimer’s disease is recognized as a disease especially prevalent in the elderly. Of almost 5 million people diagnosed with AD in the United States in 2010, approximately 4.1 million were age 75 or older (Hebert et al., 2013). Due to advances in healthcare the average lifespan of human beings has rapidly increased from 57.3 to 64.2 years for males and 58.2 to 68.5 years for females between 1990 and 2013 (Abubakar et al., 2015). Because the average lifespan of human beings is increasing, AD’s impact on our world today is increasing as well. Though an estimated 5.5 million people in the United States currently suffer from AD, this number is expected to increase to 13.8 million by 2050. Due to the growing aging population, AD has become a major public health concern, with global costs in 2018 estimated at around $1 trillion (World Alzheimer Report, 2018). Unfortunately, there is a lack of effective prevention methods and no cure to combat this growing health concern. Because of its growing worldwide medical and financial burden it’s important to understand the different ways AD presents itself, the disease at the molecular level, and the modifiable and non-modifiable factors contributing to the characteristic features of AD. Currently, there are no early detectable biomarkers and drugs that can delay and/or prevent the disease process.
The purpose of this article is to highlight recent developments in AD including, (1) amyloid beta (Aβ) toxicity, (2) abnormal APP processing, (3) discovery of miRNAs and their biogenesis, and (4) involvement of miRNAs in aging and AD, particularly with abnormal APP processing and Aβ formation. This article also summarizes miRNAs as potential biomarkers for AD.
Types of Alzheimer’s Disease
Two forms of AD exist: sporadic AD and familial AD. Early-onset AD, known as familial AD, is an extremely uncommon form of the disease seen in one to two percent of all AD cases. It is defined as AD occurring before the age of 65. Late-onset AD, termed sporadic AD, is the overwhelmingly more common form of the disease affecting anyone at any age but usually occurring in those above the age of 65 (Dementia Australia, 2014). Familial AD is due to mutations in three major genes: amyloid beta precursor protein (APP) gene, presenilin1 (PSEN1) gene, and presenilin 2 (PSEN2) gene. Mutations in these genes induce the abnormal overproduction of amyloid-β, leading to familial AD. In sporadic AD, the cause is not as well understood. Sporadic AD is believed to be determined by a combination of genetic factors, environmental factors, and lifestyle (Piaceri et al., 2013).
Risk Factors of AD
Several risk factors affect the probability of acquiring sporadic Alzheimer’s disease. This includes both modifiable and non-modifiable risk factors. Modifiable risk factors for sporadic AD involve type 2 diabetes or obesity, environmental factors such as stress, vascular diseases, depression, stroke, hypertension, traumatic brain injury, alcohol, smoking, exercise, and other lifestyle habits. Sporadic AD non-modifiable risk factors include genetic mutations, age, sex, or genetic polymorphisms. Apolipoprotein E4 (ApoE4) genotype is believed to be a non-modifiable risk factor present in both familial and sporadic AD. Genetic polymorphisms that can serve as risk factors in sporadic AD include but are not limited to alterations in sortilin-related receptor 1, clusterin, complement component receptor 1, CD2-associated protein (CD2AP), Ephrin type-A receptor 1 protein (EPHA1), and Membrane-spanning 4-domains subfamily A (MS4A6A/MS4A4E genes) (Herrera-Rivero, 2013). This article investigates the role miRNAs play in AD. Specifically, it delves into the interaction between miRNAs and Amyloid-β (Aβ) in order to show how miRNAs might induce early-onset familial AD and late-onset sporadic AD.
Cellular Changes in Alzheimer’s Disease
Histopathological and morphological examination of postmortem AD brains and transgenic mouse models of AD revealed that multiple cellular changes are involved with the disease process including, loss of neurons, synaptic loss/damage, mitochondrial fragmentation, increased free radical production, mitochondrial DNA damage, proliferation of astrocytes and microglia, hormonal imbalance and altered neurotransmitter levels (e.g., decreased acetylcholine), in addition to neurofibrillary tangles and senile plaques (Selkoe, 2001; Reddy et al., 2010, 2012; Zhu et al., 2013; Swerdlow et al., 2014). These changes primarily were observed in learning and memory regions of the brain, including entorhinal cortex and spreads to the hippocampus, temporal cortex, frontoparietal cortex, and finally to subcortical nuclei (Reddy and McWeeney, 2006).
Amyloid-β
The presence of extracellular amyloid plaques composed mainly of Aβ peptides in the brain is one of the hallmark features of AD. Identified in 1984 by scientists George Glenner and Caine Wong, Aβ are peptides of ∼40 amino acids long first described as a “novel cerebrovascular amyloid protein” (Glenner and Wong, 1984). Aβ is derived from a larger protein APP, one of the numerous mutated genes responsible for familial AD. In the non-AD pathway APP is predominantly cleaved by α-secretase, though the β-secretase pathway is part of normal physiology. In the disease-promoting pathway, the β-secretase pathway predominates. Cleavage by α- or β-secretase yields a protein fragment called secreted APP (sAPP) α or β, respectively. After β-secretase cleavage, sAPPβ is then cleaved by γ-secretase to yield Aβ. Cleavage by γ-secretase is inconsistent resulting in differences at the C-terminal end. These differences contribute to the existence of a wide array of Aβ species. The most numerous Aβ species include Aβ40 and Aβ42, or cleavage at the 40 and 42 positions, respectively (Murphy and LeVine, 2010).
Amyloid Beta Species
Extensive past research has revealed that multiple Aβ species are present in the postmortem AD brains and in AD mouse models – these are Aβ1–36, 1–37, 1–38, 1–39, 1–40, and Aβ1–41, Aβ1–42, and Aβ1–43 (Kakuda et al., 2017). Among these Aβ40 is the most abundant form of Aβ species (Mori et al., 1992). While similar in structure, Aβ40 is composed of two protofilaments whereas Aβ42 is composed of only one protofilament. Both also comprise of the same number of Aβ molecules per cross-β repeat. Aβ42 is a slightly longer and less abundant form of Aβ species (Schmidt et al., 2009). Aβ42 is hydrophobic, fibrillogenic, and the main Aβ variant that accumulates in the brain of AD patients (Murphy and LeVine, 2010).
Amyloid Beta – Pathogenic or Simply Bioproduct of Disease Process?
Though the exact pathogenic role of Aβ is unknown, the pathogenicity of Aβ is amplified when Aβ monomers become Aβ oligomers. Research shows that increasing Aβ plaque does not necessarily correlate with an amplified AD pathology. Aβ plaques can be present in both cognitively impaired individuals and those with normal cognitive function. Additionally, APP genetically modified mice have physiologic and behavioral abnormalities present before the appearance of Aβ plaque. This suggests that Aβ plaque buildup alone may not be responsible for the cognitive impairment associated with AD. Rather, smaller Aβ oligomers may play a more important role in the neurotoxicity seen in AD via its toxic effects on synapses in the brain (Mucke and Selkoe, 2012). Furthermore, it has also been shown that one can have very little plaque buildup but still demonstrate AD pathology (Erten-Lyons et al., 2009). This all is possible because the presence of small Aβ oligomers, not Aβ plaques, have been found to be primarily responsible for the impending cognitive impairment. Aβ oligomers are more likely to be pathogenic than Aβ plaques because they have a greater surface area to interact with the synapses of cells in the brain (Mucke and Selkoe, 2012). When analyzing mice with high Aβ plaque loads but deficient in oligomeric Aβ levels, researchers demonstrated that the mice did not experience any memory impairment (Lesné et al., 2008).
Evidence suggests that Aβ induces several different pathologies seen in AD. An example of said induction includes a role in stimulating tau protein tangles, the second hallmark feature of AD (Nussbaum et al., 2013). Another example of Aβ induced changes include the neuritic alterations and synaptic distortions seen greatest in cortical regions most proximal to Aβ plaques. The change from straight to coiled cortical dendrites is believed to decrease the efficiency of the transmission of signals along dendrites (Mucke and Selkoe, 2012).
Amyloid Beta Accumulation in Subcellular Compartments and Cellular Toxicity
Several lines of evidence suggest that Aβ accumulates in multiple cellular compartments. Under normal conditions, Aβ was found in the small cytoplasmic granules in both neurites and perikarya. Minor portions of Aβ are colocalized with trans-Golgi network, Golgi-derived vesicles, early and late endosomes, lysosomes, synaptic vesicles, and mitochondria (Reddy and Beal, 2008; Zheng et al., 2012). It is well documented that toxicity of Aβ is dependent on the size, aggregation state, and diffusion of Aβ in subcellular compartments and at neuronal terminals (Sengupta et al., 2016).
miRNAs and AD
Alzheimer’s disease research shows that miRNAs help regulate genes, protein expressions, and phenotypic changes in human diseases. Studies have shown miRNAs can contribute to the development of a variety of pathologic conditions. This includes a role in aging, cardiovascular disease, cancers, arthritis, cataracts, osteoporosis, diabetes/obesity, hypertension, and various neurodegenerative diseases such as AD, Huntington’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and schizophrenia (Kumar and Reddy, 2016; Reddy, 2017).
Discovery of MicroRNAs
miRNAs are a large family of small RNAs, approximately 18–25 nucleotides in length, that play an integral role in the regulation of gene expression. Discovered in 1993, a non-protein-coding transcript named lin-4 was identified as the first miRNA in the nematode Caenorhabditis elegans. Lin-4 was shown to negatively regulate lin-14 protein by complementary binding to its 3′UTR (Lee et al., 1993). In this species, the downregulation of lin-14 by lin-4 helps progress the embryo from the first to the second larval stage of development. Today thousands of miRNAs have been identified that anneal at the 3′ UTR of many human genes. Most recently, miRNAs have been shown to also have activity at the 5′ UTR of genes (Long et al., 2019). There are two classes of miRNAs: miRNAs associated with exons or the coding part of genes and those originating from introns, the non-coding part of genes. One-third of these miRNAs are found in the coding part of genes, and the remaining are in the non-coding regions (Abe and Bonini, 2013; Reddy, 2017).
Biogenesis of MicroRNAs
Generation of miRNAs starts with the initial transcription by RNA polymerase II into a long primary transcript (pri-miRNA). In the nucleus, these pri-miRNAs are cleaved by two proteins Drosha and DiGeorge Syndrome Critical region 8 (DGCR8) proteins which dimerize together to form a functional microprocessor complex. These dimerized proteins cleave pri-miRNAs into precursor miRNAs (pre-miRNA) which are transported to the cytoplasm to be digested by proteins Dicer and TAR RNA binding protein (TRBP) to release a double-stranded miRNA duplex. A helicase unwinds the duplex assembly to form mature miRNA strands. One of these strands is usually degenerated while the other associates with the Ago2 protein to form miRNA-induced Silencing Complex (miRISC). Rarely, both can serve as mature functional miRNA. Functional miRNAs modulate gene activity by inhibiting miRNA cleavage or suppressing translation in the same way lin-4 was previously described as suppressing lin-14 translation in C. Elegans (Abe and Bonini, 2013).
Most reported miRNAs are found in the human brain. Many of these miRNAs are responsible for synaptic functions, neurotransmitter release, synapse formation and neurite outgrowth. In diseased states such as AD, expression levels of several miRNAs are either downregulated or upregulated (Abe and Bonini, 2013; see Figure 1).

Figure 1. Major microRNAs in Alzheimer’s Disease. Up arrow indicates an increased levels of the miRNAs in Alzheimer’s disease. Down arrow indicates downregulated miRNAs in Alzheimer’s disease.
Aging and MicroRNAs
Aging is the greatest risk factor for AD and many other neurodegenerative diseases. Within the aging process, cellular changes lead to increased disease vulnerability and mortality. Key to the aging process is cellular senescence, which miRNAs play a pivotal role in. Due to the critical role miRNAs play in cellular senescence and thus aging, they are pivotal in assessing one’s risk for AD. Some of the roles miRNAs largely influence include telomerase shortening, chronic inflammation, oxidative stress, mitochondrial dysfunction, DNA damage response, protein misfolding, stem cell impairment, and altered nutrient sensing (Reddy and Beal, 2008). Research is starting to show that miRNA’s have a wide array of functions that contribute to aging and cellular senescence (see Table 1).
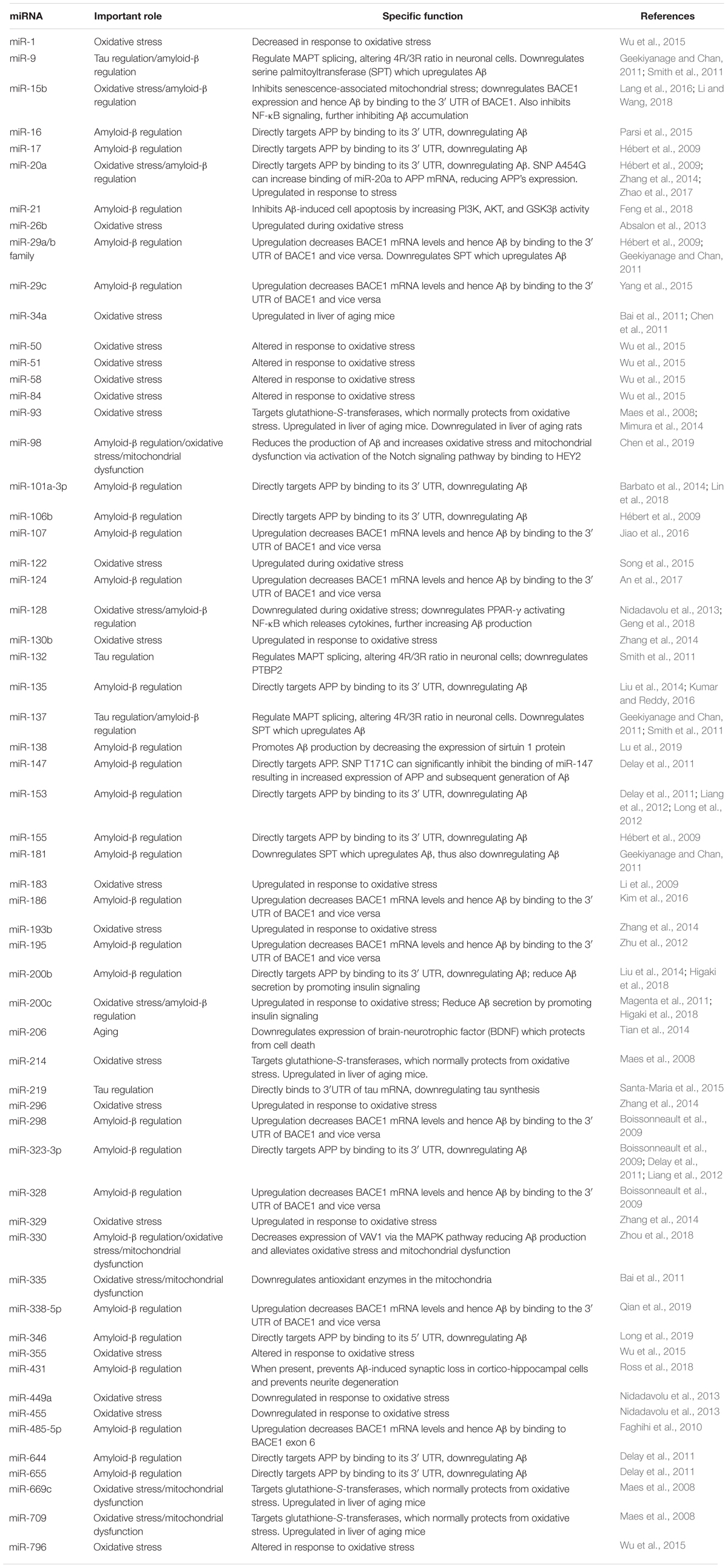
Table 1. Summary of miRNAs related to aging and Alzheimer’s disease.
Additional support for miRNA as a causal factor includes the observation of altered miRNA levels in those suffering or at risk for AD. The role of miRNAs in aging and AD is further summarized in Reddy et al. (2017). MiRNAs are directly involved with aging and cellular senescence by disrupting protein regulation. In addition, as shown in Table 1, a large number of miRNAs are associated with aging and cellular senescence. Further research is needed to better understand the relationship between age-related miRNAs in inducing AD and other neurodegenerative diseases.
MicroRNAs and Abnormal APP Processing and Aβ Formation
Recent research has reinforced previously hypothesized mechanisms in which miRNAs induce Aβ production while highlighting more miRNAs that may work via these methods and perhaps revealing new mechanisms through which miRNAs operate. Table 2 shows altered miRNAs directly related to Aβ in AD while Figure 1 illustrates the ways in which these miRNAs are altered in the AD state compared to a normal physiologic state. Though their involvement is not well defined, miRNAs are thought to be a causal factor in Aβ formation.
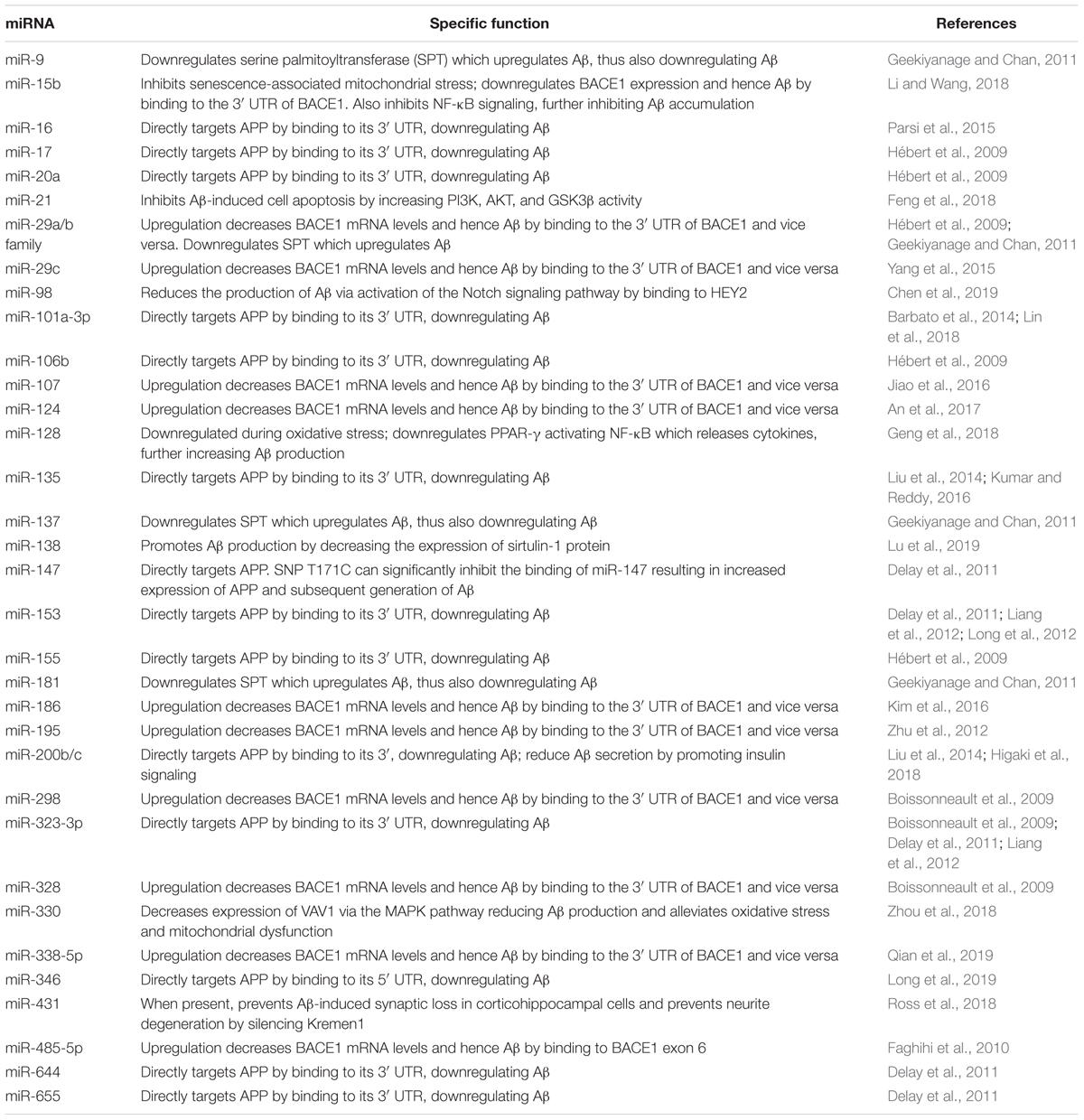
Table 2. Summary of miRNAs regulating amyloid-β.
Many miRNAs have been documented as directly regulating APP expression levels on its 3′ UTR. Recently, Lahiri’s group reported a novel activity of miR-346: specifically, that it targets the APP mRNA 5′-UTR to upregulate APP translation and Aβ production (Long et al., 2019). This upregulation is reduced but not eliminated by the knockdown of the argonaute 2 protein. The target site for miR-346 overlaps with active sites for an iron-responsive element (IRE) and an interleukin-1 (IL-1) acute box element. IREs interact with iron response protein1 (IRP1), an iron-dependent translational repressor. In primary human brain cultures, miR-346 activity required chelation of Fe. In addition, miR-346 levels are altered in late-Braak stage AD. Thus, miR-346 plays a role in upregulation of APP and increased formation of Aβ in AD brains (Long et al., 2019).
In addition, 3′ UTR of APP gene is also largely involved with APP regulation. Recently, a number of single nucleotide polymorphisms located in the 3′UTR of APP have been found in AD patients with family history of dementia. Delay et al. (2011) extensively studied the role of polymorphisms in 3′ UTR in regulating APP expression regulation and miRNA function. Their bioinformatics analysis identified 12 putative miRNA bindings sites located in or near the APP 3′UTR variants T117C, A454G, and A833C. Among those candidates, seven miRNAs, including miR-20a, miR-17, miR-147, miR-655, miR-323-3p, miR-644, and miR-153 could regulate APP expression in vitro and under physiological conditions in cells. Using luciferase assay, they demonstrated that the T117C variant inhibited miR-147 binding, whereas the A454G variant increased miR-20a binding, consequently having opposite effects on APP expression. Further, research is still needed to understand the roles of the APP gene’s 3′ UTR in relation to miRNAs.
Lu’s lab highlighted a mechanism used by miR-138 involving the miRNA, sirtulin-1 protein, and circular RNA. They describe how miR-138 downregulates the Sirtulin-1 protein which induces Aβ production. This can then be reversed by inhibition of miR-138 via circular RNA, specifically circular HDAC9 (Lu et al., 2019).
Past research has shown β-site amyloid precursor protein cleaving enzyme, (BACE)-1, to be required for the cleavage of amyloid precursor protein (APP) to generate Aβ. This cleavage stimulates the nuclear transcription factor (NF-κB) leading to the secretion of inflammatory cytokines (Li and Wang, 2018). Many miRNAs, as seen in Table 2, inhibit BACE1 mRNA levels so in the AD state they are downregulated, contributing to elevated Aβ production. Geng’s lab illuminated how some miRNAs can activate PPAR-gamma which also leads to the activation of NF-κB further leading to the release of cytokines and Aβ production (Geng et al., 2018).
Several miRNAs also act to increase Aβ production via the Notch signaling pathway. Specifically, miRNAs are shown to inhibit HEY2 protein levels which inactivates the Notch signaling pathway. This Notch pathway is responsible for suppressing Aβ production among many other functions in the non-AD state (Chen et al., 2019).
Another of the many mechanisms by which miRNAs are proposed to regulate amyloid-beta production is through insulin signaling (Higaki et al., 2018). Higaki’s research detailed how the miR-200 family inhibit the expression of ribosomal protein S6 kinase B1 (S6K1), a downstream effector of mammalian target of rapamycin (mTOR). The protein m-TOR normally would suppress insulin release which leads to insulin resistance, typically seen in AD brains.
In Feng’s lab, they highlighted another mechanism through which miRNAs can alter amyloid-β levels. They show how miR-21 can inhibit Aβ-induced apoptosis (Feng et al., 2018). Normally, Aβ would be able to increase Bax and inhibit Bcl-2 protein levels. Both actions would compromise the cell membrane of cells and lead to eventual cell death. MiR-21 is upregulated in AD to invoke its protective effect by increasing the activity of the phosphatidylinositol 3-kinase/Protein Kinase B (also known as PI3K/AKT) pathway and increase glycogen synthase kinase-3β (GSK-3β) levels. These signaling pathways diminish Aβ-induced cell death.
MiRNA dysregulation is present in both tau and Aβ pathologies. Though both pathologies can lead to the AD state, it’s not clear which changes miRNA levels and what results from these changes. Interestingly, four miRNAs (miR142a-5p, miR146a-5p, miR155-5p, and miR-455-5p) have been shown to be commonly upregulated in transgenic tau and APP mice in the AD state (Sierksma et al., 2018). Through Sierksma’s investigation, it is shown that these miRNAs are not responsible for inducing cognitive disturbances because when they are upregulated in healthy mice the miRNAs do not induce AD-related cognitive disturbances. Henceforth, these miRNAs may be upregulated as part of a protective mechanism in the AD brain. Previous literature has shown several of these miRNAs to be downregulated in the AD brain, further supporting this hypothesized protecting role in the AD brain (see Table 1).
MicroRNAs as Potential Biomarkers
A potential biomarker for early diagnosis of AD are miRNAs found in circulatory biofluids. Circulatory biofluids include cerebrospinal fluid (CSF), extracellular fluid (ECF), peritoneal fluid, pleural fluid, seminal fluid, bronchial secretions, breast milk, serum, plasma, and various other bodily fluids. The stability and abundance of circulatory miRNAs contribute to their ability to be a potential diagnostic biomarker for disease. The levels of miRNAs in these fluids change with changes in body physiology naturally seen in aging, changing diets, and pathologic states. It is believed that in addition to brain imaging, miRNA analysis of a blood sample might serve as a promising tool when attempting to diagnose whether a person has or is likely to contract cognitive impairment due to AD (Kumar and Reddy, 2016).
Various tests have revealed that disrupting miRNA biogenesis causes neurodegeneration. For example, a neurodegenerative disease is likely to result if one disrupts the Dicer protein which cleaves pre-miRNA into a double-stranded miRNA duplex. Studies like this show that miRNAs strongly impact long-term brain integrity (Abe and Bonini, 2013). Though a disruption in miRNA biogenesis is thought to be linked to the onset of many neurodegenerative diseases, outside of AD, very few studies have shown miRNAs use as a potential biomarker (Reddy, 2017).
While still much can be ascertained on the identity of miRNAs as peripheral biomarkers for Alzheimer’s disease in general, their presence and identity might help clarify what stage of AD the patient is in. Age is the biggest risk factor for AD so recently Ryan et al. (2018) tracked how the expression of miRNAs changed over time. Eight miRNAs were significantly affected by age either prior to or during Aβ plaque deposition. There experiments helped reinforce the notion that biological processes that lead to AD change over time and highlights potential pre-symptomatic biomarkers that are found to be distinct from the miRNAs highlighted in Table 2 which generally highlight the dysregulation of miRNA in the symptomatic AD state.
Conclusion and Future Directions
Alzheimer’s disease is a progressive neurodegenerative disease characterized by memory loss and multiple cognitive impairments. With growing aging population, the prevalence of AD is high and is a major health concern in the society. Although tremendous progress has been made in understanding basic biology, molecular mechanisms of disease and therapeutics, we still do not have non-invasive early detectable biomarkers and drugs that can delay and/or prevent disease process. Several years of intense research revealed that multiple cellular changes are implicated in disease progression and pathogenesis, including synaptic damage, mitochondrial dysfunction, increased proliferation of astrocytes and microglia, in addition to the presence of core amyloid beta and phosphorylation of tau in AD affected regions of the brain. Amyloid beta is considered a major player in the disease process because of its relevance to both early-onset familial and late-onset sporadic AD. Recent research also revealed that microRNAs are considered as potential, non-invasive peripheral biomarkers in aging and other age-related neurodegenerative diseases such as AD. In this article, we discussed several aspects, including altered levels of miRNAs with aging, AD, particularly related to amyloid beta in disease process. In recent years, a wealth of miRNA data has been accumulated, but there is no single microRNA that regulate APP processing and amyloid beta formation and cognitive decline. It is possible that several miRNAs are involved in APP processing – some regulate 5′ UTR and others 3′ UTR of APP gene. To understand the biology of miRNAs and how they regulate both 5′- and 3′-UTR of APP processing, we still need more research on miRNAs and APP/amyloid beta formation in the progression and pathogenesis of AD.
Author Contributions
PR contributed in designing the project. NA, PR, JT, and AK contributed in writing and checking and finalizing the manuscript.
Funding
Work presented in this manuscript was supported by NIH grants R01AG042178, R01AG047812, R01NS105473, and R41AG060836, the Garrison Family Foundation and The CH Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abe, M., and Bonini, N. M. (2013). MicroRNAs and neurodegeneration: role and impact. Trends Cell Biol. 23, 30–36. doi: 10.1016/j.tcb.2012.08.013
Absalon, S., Kochanek, D. M., Raghavan, V., and Krichevsky, A. M. (2013). MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci. 33, 14645–14659. doi: 10.1523/JNEUROSCI.1327-13.2013
Abubakar, I. I., Tillmann, T., and Banerjee, A. (2015). Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the global burden of disease study 2013. Lancet 385, 117–171. doi: 10.1016/S0140-6736(14)61682-2
Alzheimer, A., Förstl, H., and Levy, R. (1991). On certain peculiar diseases of old age. Hist. Psychiatry 2, 71–73.
An, F., Gong, G., Wang, Y., Bian, M., Yu, L., and Wei, C. (2017). MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget 8:114065. doi: 10.18632/oncotarget.23119
Bai, X. Y., Ma, Y., Ding, R., Fu, B., Shi, S., and Chen, X. M. (2011). miR-335 and miR-34a promote renal senescence by suppressing mitochondrial antioxidative enzymes. J. Am. Soc. Nephrol. 22, 1252–1261. doi: 10.1681/ASN.2010040367
Barbato, C., Pezzola, S., Caggiano, C., Antonelli, M., Frisone, P., Ciotti, M. T., et al. (2014). A lentiviral sponge for miR-101 regulates RanBP9 expression and amyloid precursor protein metabolism in hippocampal neurons. Front. Cell. Neurosci. 8:37. doi: 10.3389/fncel.2014.00037
Boissonneault, V., Plante, I., Rivest, S., and Provost, P. (2009). MicroRNA-298 and microRNA-328 regulate expression of mouse β-amyloid precursor protein-converting enzyme 1. J. Biol. Chem. 284, 1971–1981. doi: 10.1074/jbc.M807530200
Chen, F. Z., Zhao, Y., and Chen, H. Z. (2019). MicroRNA-98 reduces amyloid β-protein production and improves oxidative stress and mitochondrial dysfunction through the notch signaling pathway via HEY2 in Alzheimer’s disease mice. Int. J. Mol. Med. 43, 91–102. doi: 10.3892/ijmm.2018.3957
Chen, H., Sun, Y., Dong, R., Yang, S., Pan, C., Xiang, D., et al. (2011). Mir-34a is upregulated during liver regeneration in rats and is associated with the suppression of hepatocyte proliferation. PLoS One 6:e20238. doi: 10.1371/journal.pone.0020238
Delay, C., Calon, F., Mathews, P., and Hébert, S. S. (2011). Alzheimer-specific variants in the 3′ UTR of Amyloid precursor protein affect microRNA function. Mol. Neurodegener. 6:70. doi: 10.1186/1750-1326-6-70
Dementia Australia (2014). Alzheimer’s Disease. Available at: https://www.dementia.org.au/about-dementia/types-of-dementia/alzheimers-disease (accessed January 10, 2019).
Erten-Lyons, D., Woltjer, R. L., Dodge, H., Nixon, R., Vorobik, R., Calvert, J. F., et al. (2009). Factors associated with resistance to dementia despite high Alzheimer disease pathology. Neurology 72, 354–360. doi: 10.1212/01.wnl.0000341273.18141.64
Faghihi, M. A., Zhang, M., Huang, J., Modarresi, F., Van der Brug, M. P., Nalls, M. A., et al. (2010). Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol. 11:R56. doi: 10.1186/gb-2010-11-5-r56
Feng, M. G., Liu, C. F., Chen, L., Feng, W. B., Liu, M., Hai, H., et al. (2018). MiR-21 attenuates apoptosis-triggered by amyloid-β via modulating PDCD4/PI3K/AKT/GSK-3β pathway in SH-SY5Y cells. Biomed. Pharmacother. 101, 1003–1007. doi: 10.1016/j.biopha.2018.02.043
Geekiyanage, H., and Chan, C. (2011). MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer’s disease. J. Neurosci. 31, 14820–14830. doi: 10.1523/JNEUROSCI.3883-11.2011
Geng, L., Zhang, T., Liu, W., and Chen, Y. (2018). Inhibition of miR-128 abates Aβ-mediated cytotoxicity by targeting PPAR-γ via NF-κB inactivation in primary mouse cortical neurons and neuro2a cells. Yonsei Med. J. 59, 1096–1106. doi: 10.3349/ymj.2018.59.9.1096
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890.
Hebert, L. E., Weuve, J., Scherr, P. A., and Evans, D. L. (2013). Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 1778–1783. doi: 10.1212/WNL.0b013e31828726f5
Hébert, S. S., Horré, K., Nicolaï, L., Bergmans, B., Papadopoulou, A. S., Delacourte, A., et al. (2009). MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 33, 422–428. doi: 10.1016/j.nbd.2008.11.009
Herrera-Rivero, M. (2013). “Late-onset Alzheimer’s disease: risk factors, clinical diagnosis and the search for biomarkers,” in Neurodegenerative Diseases, ed. U. Kishore (Research Triangle Park: InTech).
Higaki, S., Muramatsu, M., Matsuda, A., Matsumoto, K., Satoh, J. I., Michikawa, M., et al. (2018). Defensive effect of microRNA-200b/c against amyloid-beta peptide-induced toxicity in Alzheimer’s disease models. PLoS One 13:e0196929. doi: 10.1371/journal.pone.0196929
Jiao, Y., Kong, L., Yao, Y., Li, S., Tao, Z., Yan, Y., et al. (2016). Osthole decreases beta amyloid levels through up-regulation of miR-107 in Alzheimer’s disease. Neuropharmacology 108, 332–344. doi: 10.1016/j.neuropharm.2016.04.046
Kakuda, N., Miyasaka, T., Iwasaki, N., Nirasawa, T., Wada-Kakuda, S., Takahashi-Fujigasaki, J., et al. (2017). Distinct deposition of amyloid-β species in brains with Alzheimer’s disease pathology visualized with MALDI imaging mass spectrometry. Acta Neuropathol. Commun. 5:73.
Kim, W., Noh, H., Lee, Y., Jeon, J., Shanmugavadivu, A., McPhie, D. L., et al. (2016). MiR-126 regulates growth factor activities and vulnerability to toxic insult in neurons. Mol. Neurobiol. 53, 95–108. doi: 10.1007/s12035-014-8989-x
Kumar, S., and Reddy, P. H. (2016). Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim. Biophys. Acta 1862, 1617–1627. doi: 10.1016/j.bbadis.2016.06.001
Lang, A., Grether-Beck, S., Singh, M., Kuck, F., Jakob, S., Kefalas, A., et al. (2016). MicroRNA-15b regulates mitochondrial ROS production and the senescence-associated secretory phenotype through sirtuin 4/SIRT4. Aging 8:484.
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854.
Lesné, S., Kotilinek, L., and Ashe, K. H. (2008). Plaque-bearing mice with reduced levels of oligomeric amyloid-β assemblies have intact memory function. Neuroscience 151, 745–749.
Li, G., Luna, C., Qiu, J., Epstein, D. L., and Gonzalez, P. (2009). Alterations in microRNA expression in stress-induced cellular senescence. Mech. Ageing Dev. 130, 731–741. doi: 10.1016/j.mad.2009.09.002
Li, J., and Wang, H. (2018). miR-15b reduces amyloid-β accumulation in SH-SY5Y cell line through targetting NF-κB signaling and BACE1. Biosci. Rep. 38:BSR20180051. doi: 10.1042/BSR20180051
Liang, C., Zhu, H., Xu, Y., Huang, L., Ma, C., Deng, W., et al. (2012). MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 1455, 103–113. doi: 10.1016/j.brainres.2011.10.051
Lin, Y., Yao, Y., Liang, X., Shi, Y., Kong, L., Xiao, H., et al. (2018). Osthole suppresses amyloid precursor protein expression by up-regulating miRNA-101a-3p in Alzheimer’s disease cell model. Zhejiang Da Xue Xue Bao Yi Xue Ban 47, 473–479.
Liu, C. G., Wang, J. L., Li, L., Xue, L. X., Zhang, Y. Q., and Wang, P. C. (2014). MicroRNA-135a and-200b, potential Biomarkers for Alzheimer× s disease, regulate β secretase and amyloid precursor protein. Brain Res. 1583, 55–64. doi: 10.1016/j.brainres.2014.04.026
Long, J. M., Maloney, B., Rogers, J. T., and Lahiri, D. K. (2019). Novel upregulation of amyloid-β precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5′-untranslated region: implications in Alzheimer’s disease. Mol. Psychiatry 24, 345–363.
Long, J. M., Ray, B., and Lahiri, D. K. (2012). MicroRNA-153 physiologically inhibits expression of amyloid-β precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J. Biol. Chem. 287, 31298–31310. doi: 10.1074/jbc.M112.366336
Lu, Y., Tan, L., and Wang, X. (2019). Circular HDAC9/microRNA-138/Sirtuin-1 pathway mediates synaptic and amyloid precursor protein processing deficits in Alzheimer’s disease. Neurosci. Bull.
Maes, O. C., An, J., Sarojini, H., and Wang, E. (2008). Murine microRNAs implicated in liver functions and aging process. Mech. Ageing Dev. 129, 534–541. doi: 10.1016/j.mad.2008.05.004
Magenta, A., Cencioni, C., Fasanaro, P., Zaccagnini, G., Greco, S., Sarra-Ferraris, G., et al. (2011). miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 18:1628. doi: 10.1038/cdd.2011.42
Mimura, S., Iwama, H., Kato, K., Nomura, K., Kobayashi, M., Yoneyama, H., et al. (2014). Profile of microRNAs associated with aging in rat liver. Int. J. Mol. Med. 34, 1065–1072. doi: 10.3892/ijmm.2014.1892
Mori, H., Takio, K., Ogawara, M., and Selkoe, D. J. (1992). Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J. Biol. Chem. 267, 17082–17086.
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2:a006338. doi: 10.1101/cshperspect.a006338
Murphy, M. P., and LeVine, H. (2010). Alzheimer’s disease and the β-amyloid peptide. J. Alzheimers Dis. 19:311. doi: 10.3233/JAD-2010-1221
Nidadavolu, L. S., Niedernhofer, L. J., and Khan, S. A. (2013). Identification of microRNAs dysregulated in cellular senescence driven by endogenous genotoxic stress. Aging 5:460.
Nussbaum, J. M., Seward, M. E., and Bloom, G. S. (2013). Alzheimer disease: a tale of two prions. Prion 7, 14–19. doi: 10.4161/pri.22118
Parsi, S., Smith, P. Y., Goupil, C., Dorval, V., and Hébert, S. S. (2015). Preclinical evaluation of miR-15/107 family members as multifactorial drug targets for Alzheimer’s disease. Mol. Ther. Nucleic Acids 4:e256. doi: 10.1038/mtna.2015.33
Piaceri, I., Nacmias, B., and Sorbi, S. (2013). Genetics of familial and sporadic Alzheimer’s disease. Front. Biosci. 5:167–177. doi: 10.2741/E605
Qian, Q., Zhang, J., He, F. P., Bao, W. X., Zheng, T. T., Zhou, D. M., et al. (2019). Down-regulated expression of microRNA-338-5p contributes to neuropathology in Alzheimer’s disease. FASEB J. 33, 4404–4417. doi: 10.1096/fj.201801846R
Reddy, P. H. (2017). Molecular Biology of Aging, 1st Edn, Vol. 146. Cambridge, MA: Elsevier Academic Press.
Reddy, P. H., and Beal, M. F. (2008). Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14, 45–53.
Reddy, P. H., Manczak, M., Mao, P., Calkins, M. J., Reddy, A. P., and Shirendeb, U. (2010). Amyloid-beta and mitochondria in aging and Alzheimer’s disease: implications for synaptic damage and cognitive decline. J. Alzheimers. Dis. 20(Suppl. 2), S499–S512. doi: 10.3233/JAD-2010-100504
Reddy, P. H., and McWeeney, S. (2006). Mapping cellular transcriptosomes in autopsied Alzheimer’s disease subjects and relevant animal models. Neurobiol. Aging 27, 1060–1077.
Reddy, P. H., Tonk, S., Kumar, S., Vijayan, M., Kandimalla, R., Kuruva, C. S., et al. (2017). A critical evaluation of neuroprotective and neurodegenerative MicroRNAs in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 483, 1156–1165. doi: 10.1016/j.bbrc.2016.08.067
Reddy, P. H., Tripathi, R., Troung, Q., Tirumala, K., Reddy, T. P., Anekonda, V., et al. (2012). Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 1822, 639–649. doi: 10.1016/j.bbadis.2011.10.011
Ross, S. P., Baker, K. E., Fisher, A., Hoff, L., Pak, E. S., and Murashov, A. K. (2018). miRNA-431 prevents amyloid-β-induced synapse loss in neuronal cell culture model of alzheimer’s disease by silencing kremen1. Front. Cell. Neurosci. 12:87. doi: 10.3389/fncel.2018.00087
Ryan, M. M., Guévremont, D., Mockett, B. G., Abraham, W. C., and Williams, J. M. (2018). Circulating plasma microRNAs are altered with amyloidosis in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 66, 835–852. doi: 10.3233/JAD-180385
Santa-Maria, I., Alaniz, M. E., Renwick, N., Cela, C., Fulga, T. A., Van Vactor, D., et al. (2015). Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J. Clin. Investig. 125, 681–686. doi: 10.1172/JCI78421
Schmidt, M., Sachse, C., Richter, W., Xu, C., Fändrich, M., and Grigorieff, N. (2009). Comparison of Alzheimer Aβ (1–40) and Aβ (1–42) amyloid fibrils reveals similar protofilament structures. Proc. Natl. Acad. Sci. U.S.A. 106, 19813–19818.
Sengupta, U., Nilson, A. N., and Kayed, R. (2016). The role of amyloid-beta oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 6, 42–49. doi: 10.1016/j.ebiom.2016.03.035
Sierksma, A., Lu, A., Salta, E., Eynden, E. V., Callaerts-Vegh, Z., D’hooge, R., et al. (2018). Deregulation of neuronal miRNAs induced by amyloid-β or TAU pathology. Mol. Neurodegener. 13:54. doi: 10.1186/s13024-018-0285-1
Smith, P. Y., Delay, C., Girard, J., Papon, M. A., Planel, E., Sergeant, N., et al. (2011). MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum. Mol. Genet. 20, 4016–4024. doi: 10.1093/hmg/ddr330
Song, K., Kwon, H., Han, C., Zhang, J., Dash, S., Lim, K., et al. (2015). Active glycolytic metabolism in CD133 (+) hepatocellular cancer stem cells: regulation by MIR-122. Oncotarget 6:40822. doi: 10.18632/oncotarget.5812
Swerdlow, R. H., Burns, J. M., and Khan, S. M. (2014). The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim. Biophys. Acta 1842, 1219–1231. doi: 10.1016/j.bbadis.2013.09.010
Tian, N., Cao, Z., and Zhang, Y. (2014). MiR-206 decreases brain-derived neurotrophic factor levels in a transgenic mouse model of Alzheimer’s disease. Neurosci. Bull. 30, 191–197.
Wu, H., Huang, C., Taki, F. A., Zhang, Y., Dobbins, D. L., Li, L., et al. (2015). Benzo-α-pyrene induced oxidative stress in Caenorhabditis elegans and the potential involvements of microRNA. Chemosphere 139, 496–503. doi: 10.1016/j.chemosphere.2015.08.031
Yang, G., Song, Y., Zhou, X., Deng, Y., Liu, T., Weng, G., et al. (2015). MicroRNA-29c targets β-site amyloid precursor protein-cleaving enzyme 1 and has a neuroprotective role in vitro and in vivo. Mol. Med. Rep. 12, 3081–3088. doi: 10.3892/mmr.2015.3728
Zhang, R., Zhang, Q., Niu, J., Lu, K., Xie, B., Cui, D., et al. (2014). Screening of microRNAs associated with Alzheimer’s disease using oxidative stress cell model and different strains of senescence accelerated mice. J. Neurol. Sci. 338, 57–64. doi: 10.1016/j.jns.2013.12.017
Zhao, J., Yue, D., Zhou, Y., Jia, L., Wang, H., Guo, M., et al. (2017). The role of MicroRNAs in Aβ deposition and tau phosphorylation in Alzheimer’s disease. Front. Neurol. 8:342. doi: 10.3389/fneur.2017.00342
Zheng, L., Cedazo-Minguez, A., Hallbeck, M., Jerhammar, F., Marcusson, J., and Terman, A. (2012). Intracellular distribution of amyloid beta peptide and its relationship to the lysosomal system. Trans. Neurodegener. 1:19. doi: 10.1186/2047-9158-1-19
Zhou, Y., Wang, Z.-F., Li, W., Hong, H., Chen, J., Tian, Y., and Liu, Z.-Y. (2018). Protective effects of microRNA-330 on amyloid β-protein production, oxidative stress, and mitochondrial dysfunction in Alzheimer’s disease by targeting VAV1 via the MAPK signaling pathway. J. Cell. Biochem. 119, 5437–5448. doi: 10.1002/jcb.26700
Zhu, H. C., Wang, L. M., Wang, M., Song, B., Tan, S., Teng, J. F., et al. (2012). MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res. Bull. 88, 596–601. doi: 10.1016/j.brainresbull.2012.05.018
Keywords: microRNA, Alzheimer’s disease, amyloid beta, mitochondria, aging
Citation: Amakiri N, Kubosumi A, Tran J and Reddy PH (2019) Amyloid Beta and MicroRNAs in Alzheimer’s Disease. Front. Neurosci. 13:430. doi: 10.3389/fnins.2019.00430
Received: 23 January 2019; Accepted: 15 April 2019;
Published: 03 May 2019.
Edited by:
Illana Gozes, Tel Aviv University, IsraelReviewed by:
Anthony J. Turner, University of Leeds, United KingdomZareen Amtul, Independent Researcher, London, Canada
Copyright © 2019 Amakiri, Kubosumi, Tran and Reddy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: P. Hemachandra Reddy, aGVtYWNoYW5kcmEucmVkZHlAdHR1aHNjLmVkdQ==