 Xu-Qiao Chen
Xu-Qiao Chen William C. Mobley
William C. Mobley- Department of Neurosciences, University of California, San Diego, La Jolla, CA, United States
Alzheimer disease (AD) represents an oncoming epidemic that without an effective treatment promises to exact extraordinary financial and emotional burdens (Apostolova, 2016). Studies of pathogenesis are essential for defining critical molecular and cellular events and for discovering therapies to prevent or mitigate their effects. Through studies of neuropathology, genetic and cellular, and molecular biology recent decades have provided many important insights. Several hypotheses have been suggested. Documentation in the 1980s of selective loss of cholinergic neurons of the basal forebrain, followed by clinical improvement in those treated with inhibitors of acetylycholinesterase, supported the “cholinergic hypothesis of age-related cognitive dysfunction” (Bartus et al., 1982). A second hypothesis, prompted by the selective loss of cholinergic neurons and the discovery of central nervous system (CNS) neurotrophic factors, including nerve growth factor (NGF), prompted the “deficient neurotrophic hypothesis” (Chen et al., 2018). The most persuasive hypothesis, the amyloid cascade hypothesis first proposed more than 25 years ago (Selkoe and Hardy, 2016), is supported by a wealth of observations. Genetic studies were exceptionally important, pointing to increased dose of the gene for the amyloid precursor protein (APP) in Down syndrome (DS) and a familial AD (FAD) due to duplication of APP and to mutations in APP and in the genes for Presenilin 1 and 2 (PSEN1, 2), which encode the γ-secretase enzyme that processes APP (Dorszewska et al., 2016). The “tau hypothesis” noted the prominence of tau-related pathology and its correlation with dementia (Kametani and Hasegawa, 2018). Recent interest in induction of microglial activation in the AD brain, as well as other manifestations of inflammation, supports the “inflammatory hypothesis” (Mcgeer et al., 2016). We place these findings in the context of the selective, but by no means unique, involvement of BFCNs and their trophic dependence on NGF signaling and speculate as to how pathogenesis in these neurons is initiated, amplified and ultimately results in their dysfunction and death. In so doing we attempt to show how the different hypotheses for AD may interact and reinforce one another. Finally, we address current attempts to prevent and/or treat AD in light of advances in understanding pathogenetic mechanisms and suggest that studies in the DS population may provide unique insights into AD pathogenesis and treatment.
Introduction
Alzheimer disease (AD) is the most common cause of dementia, accounting for up to 70% of cases (Apostolova, 2016). Clinical manifestations, which are insidious in onset, include memory loss and cognitive decline as well as behavioral dysfunction and failure to maintain function in activities of daily living (Scheltens et al., 2016). Patients progress from normal cognition to mild cognitive impairment (MCI) followed by increasing dementia severity (i.e., mild, moderate, and severe). While most cases are sporadic and occur late in life, other types of AD are recognized that present with earlier onset. Autosomal dominant forms of AD, which typically present in middle age, are due to mutations in the amyloid precursor protein (APP) (Bateman et al., 2011; Van Cauwenberghe et al., 2016), increased copy number for APP gene, and mutations in Presenilin 1 and 2, whose protein products regulate APP processing (Bateman et al., 2011; Van Cauwenberghe et al., 2016). APP, a type 1 transmembrane protein, is processed by sequential cleavage via either β- or α-secretase to produce the C-terminal fragments, β-CTF (or C99), or a-CTF (or C83), respectively. C99 is then cleaved by γ-secretase to yield the APP intracellular domain (AICD) and Aβ peptides of varying length; C83 cleavage yields the AICD and the P3 peptide (Thinakaran and Koo, 2008). A large body of evidence points to a pathogenic role for APP and its products in AD (Selkoe and Hardy, 2016). Down syndrome (DS), the most common genetic cause of AD, is due to trisomy for all or part of a third copy of chromosome 21. It is typically associated with the presence of a number of abnormal clinical phenotypes, including characteristic changes in craniofacial anatomy, and is universally marked by mild to moderate intellectual disability. The clinical presentation of dementia follows a course similar to AD, with problems with recall, explicit memory, and receptive language function before frank dementia. In those aged 30–40 the process may feature changes in behavior and personality (Ballard et al., 2016). Almost all adults with DS develop AD-like neuropathology by the age of 40. More than 50% display progressive cognitive impairment, leading to a diagnosis of dementia (Hithersay et al., 2017). The cause of death in DS is dementia in about 70% of cases (Hithersay et al., 2018). Given these findings this disorder is now termed AD in DS (AD-DS). The APP gene is present on chromosome 21 and is thus triplicated in DS (Megarbane et al., 2009; Ballard et al., 2016; Antonarakis, 2017). Correspondingly, the majority of adults with DS show increased expression of the gene with increases in the levels of the full length APP protein (Fl-APP) and all of its products (Prasher et al., 1998; Korbel et al., 2009; Wiseman et al., 2015; Ballard et al., 2016). Importantly, data are compelling that increased APP gene dose is necessary for AD-DS (Prasher et al., 1998; Korbel et al., 2009; Doran et al., 2017).
The neuropathology of AD manifests several cardinal features, including neuritic amyloid plaques and neurofibrillary tangles (NFTs) (Serrano-Pozo et al., 2011a). These markers are present in all types of AD, including AD-DS (Ballard et al., 2016). Amyloid plaques mark the extracellular accumulation and deposition of Aβ peptides, while aberrantly folded, and abnormally phosphorylated isoforms of the microtubule-associated protein tau serve as the principle constituent of NFTs. Both amyloid and tau pathologies show characteristic regional distributions and temporal patterns of evolution. Amyloid deposition begins in the frontal, temporal, and occipital lobes with later spread to hippocampus, the entorhinal, insular and cingulate cortices and amygdala; this is followed by involvement of several subcortical regions, including basal forebrain cholinergic nuclei, and later the substantia nigra, several brainstem nuclei, and the molecular layer of the cerebellum (Thal et al., 2002). While Aβ deposition is a prominent feature of AD pathology, studies do not support a role for plaque deposition in the temporal progression of dementia (Arriagada et al., 1992; Bierer et al., 1995; Giannakopoulos et al., 2003). Deposition of NFTs also follows a stereotypical spatiotemporal pattern, but one that is different than that for amyloid plaques. Braak and Braak defined six stages (i.e., Stages I-VI) in NFT involvement (Braak and Braak, 1991). The pattern features initial involvement of medial temporal cortex; later there is involvement of most of neocortex but with relative sparing of sensory, motor, and visual cortex. In contrast to plaque burden, the progressive involvement by NFTs is strongly linked to increasing cognitive deficits, with respect both the severity and duration of dementia (Arriagada et al., 1992; Bierer et al., 1995; Gomez-Isla et al., 1997; Giannakopoulos et al., 2003).
While for many years pathogenesis theories focused on amyloid plaques and tangles, an increasing body of evidence points to toxic Aβ and tau oligomers as having a primary causal role in the induction and spread of pathology and neuronal dysfunction and loss (Shafiei et al., 2017; Cline et al., 2018). In addition to plaques and tangles, other neuropathological hallmarks of AD include synapse loss and neuronal loss (Ball, 1977; Whitehouse et al., 1981; Dekosky and Scheff, 1990; Scheff et al., 1990, 2006, 2007; Scheff and Price, 1993; Gomez-Isla et al., 1996; Serrano-Pozo et al., 2011a; Andrade-Moraes et al., 2013; Calderon-Garciduenas and Duyckaerts, 2017) whose significance for disease manifestations is obvious. In addition, there is granulovacuolar degeneration (GVD), Hirano Bodies (Ball, 1977, 1978; Xu et al., 1992; Hirano, 1994), astrocytosis and microgliosis (Serrano-Pozo et al., 2011b). Importantly, changes in early endosomes are also manifestations in early stages of AD and AD-DS (Chen et al., 2018). Deficient endosomal transport of neurotrophic signals may play an important role in pathogenesis, as supported herein (Chen et al., 2018).
Selective Vulnerability in Ad: Lessons From Parkinson’s Disease
Deciphering the pathogenesis and discovering treatments for AD has benefitted greatly by defining pathological manifestations in selectively vulnerable neurons. Studies in Parkinson’s disease (PD) provided a model for pursuit of selective vulnerability of AD. PD is a movement disorder whose cardinal features include bradykinesia (slowness of movement), rigidity, tremor at rest, and impairment of gait. The evolution of the dopaminergic hypothesis of PD, and its impact on therapeutic drug discovery, is a fascinating lesson in disease neurobiology (Fahn, 2018). PD was first described clinically by James Parkinson in 1817. More than 100 years would intercede before several key discoveries would guide the elaboration of the dopaminergic hypothesis and development of effective PD treatments: (1) the synthesis of dopamine (DA) in the 1910s; (2) in the late 1930s and 1940s, recognition of DA as an intermediate in the catecholamine synthetic pathway leading to norepinephrine and epinephrine (Blaschko, 1942); (3) documentation of its presence in brain and mapping its presence in several neuronal populations, including nigro-striatal DA neurons by Carlsson, Hokfelt, Fuxe, Dahlstrom, and colleagues in the late 1950s and 1960s (Carlsson et al., 1958; Anden et al., 1964, 1966; Dahlstroem and Fuxe, 1964; Fuxe, 1965a,b) (4) evidence for reductions in DA in the brains of patients with PD, including the substantia nigra, in the early 1960s (Ehringer and Hornykiewicz, 1960); (5) the demonstration in 1965 that neurons of the substantia nigra pars compacta were the source of DA in striatum (Poirier and Sourkes, 1965); (6) in the same year, evidence that lesioning the substantia nigra resulted in loss of DA in striatum (Poirier and Sourkes, 1965); and (7) increasing recognition during the same and later years that selective degeneration of DA-synthesizing neurons of the substantia nigra pars compacta is a fundamental neuropathological feature of PD (Fahn, 2018). These and related events motivated treatments to restore DA in PD patients. Cotzias and colleagues published the first definitive report of the utility of L-3,4-dihydroxyphenylalanine (L-Dopa, the precursor to DA) therapy, documenting its value in improving akinesia, rigidity and tremor, with sustained improvement ranging from moderate to dramatic in most of the 28 patients treated (Cotzias et al., 1969). While there is much yet to learn about the pathogenesis of PD, L-Dopa, and DA-based treatments continue to serve as the gold-standard of symptomatic treatment (Fahn, 2018). Thus, successful treatment was built on an approach that linked the loss of a specific population of neurons and their neurotransmitter with efforts to restore neurotransmitter levels.
The Cholinergic Hypothesis: From Alzheimer’s Description to Treatment
Studies leading to the cholinergic hypothesis of age-related memory loss can be viewed in the context of those supporting the development of dopaminergic hypothesis. Key questions included: (1) is there a neurotransmitter whose actions critically support memory; (2) if so, are there changes in that neurotransmitter in disorders of age-related memory loss, most importantly in Alzheimer disease (AD); (3) if so, what neurons are responsible for producing the neurotransmitter; (4) do these neurons undergo degeneration in AD; and (5) will replacing the neurotransmitter confer symptomatic improvement. For AD, the first clinical description was that of Alois Alzheimer who described at a scientific conference in 1906 the case of a 51 year old woman, Auguste Deter, who presented in 1901 with reduced comprehension and memory, confusion, disorientation, changes in language including paraphasic errors, and perseveration, as well as psychiatric manifestations including paranoia and hallucinations (Maurer et al., 1997). She survived for 5 years. In 1907, in his full paper on the case Alzheimer noted that in addition to brain atrophy, silver staining methods showed “numerous small military foci,” now known to be amyloid β (Aβ)-containing plaques, and that within cells “there stands out one or several fibrils due to their characteristic thickness and peculiar impregnability,” the lesions we now refer to as neurofibrillary tangles. These findings were reconfirmed when the original slides were reexamined in the late 1990s (Maurer et al., 1997; Toodayan, 2016). The disorder was first named AD by Kraepelin in 1910 who indicated that the “anatomical findings suggest that we are dealing with a particularly serious form of senile dementia” but nevertheless noted that ‘this disease sometimes starts as early as the late forties” (Maurer et al., 1997). Subsequent years confirmed the existence of AD as a distinct entity and as the leading cause of dementia in the elderly (Reitz and Mayeux, 2014) with a worldwide prevalence of ∼4% of those greater than age 60 and an estimated number affected globally of 24 million (Calderon-Garciduenas and Duyckaerts, 2017).
As for PD, decades elapsed between the clinical description of AD, and the first effective treatment. But the focus on a possible role for acetylcholine in AD followed a somewhat different path than for DA. Indeed, already by the 1920s Otto Loewi had defined acetylcholine (ACh) as a neurotransmitter released by the vagus nerve to act on the heart to reduce its frequency of contractions, thus identifying ACh as a neurotransmitter (Loewi, 1921; Zimmer, 2006). Furthermore, by showing that physostigmine potentiated the actions of ACh, Loewi, and Navratil established that the heart contains an endogenous esterase for ACh, supporting the concept of a “cholinesterase” (Loewi and Navratil, 1926). In the next 15 years ACh was shown to serve as a neurotransmitter in autonomic ganglia and at the neuromuscular junction (Feldberg and Gaddum, 1934; Dale et al., 1936). The demonstration that ACh was released by simulation of motor nerves, that direct injection in muscle produced the kind of contraction elicited by nerve stimulation and that the response to ACh was increased by blocking cholinesterase activity strongly supported its role as a neurotransmitter for motor neurons (Feldberg and Gaddum, 1934; Brown et al., 1936; Bennett, 2000).
The case for ACh as a neurotransmitter in CNS built on insights from studies of its actions in the peripheral nervous system. Indeed, the use of drugs that increased the levels of ACh or blocked its actions played a key role in implicating ACh in memory. In memory studies on rodents, Deutsch and colleagues provided compelling evidence, demonstrating that effects of drugs that impact ACh levels are a function of the age of the memory, the dose of the drug, and the efficiency with which a task was initially learned (Deutsch, 1971; Squire, 1982). Importantly, while cholinergic neuronal systems were thus implicated in memory, the possible participation on non-cholinergic systems was considered possible. Studies in humans confirmed a role for cholinergic neurotransmission in memory. Those of Drachman and Levitt proved highly influential. Treatment with scopolamine, a competitive antagonist of acetylcholine at muscarinic receptors, induced in normal young human subjects impairment of memory storage, and retrieval as well as cognitive non-memory tasks with sparing of immediate memory (Drachman and Leavitt, 1974). The authors noted that the changes were strikingly similar to the changes seen in normal aged subjects (average age of ∼67 years) and speculated that deficits in cholinergic function might underlie age-related memory loss. That the latter was a common occurrence in humans and other mammals was widely appreciated by mid- to late 1970s, leading to the enunciation of the “cholinergic hypothesis of age-related memory dysfunction” and the consideration that such changes were present during both normal aging as well as, and to a greater extent in, AD (Bartus et al., 1982). An important development came from studies in AD brain noting selective reduction of the activity of the acetylcholine synthetic enzyme choline acetyltransferase (ChAT) in hippocampus, a region known to participate in memory functions, as well as in cortex and amygdala, with lesser reductions in other brain regions. A similar pattern of reduced activity was noted for acetylcholinesterase (AChE), but not for the activity of glutamic acid decarboxylase (GAD), the synthetic enzyme for g-aminobutyric acid (GABA). These data pointed to selective failure of the cholinergic system and raised the possibility that this change contributed to the pathogenesis of AD (Davies and Maloney, 1976). The reduction of ChAT activity was later confirmed and expanded to other brain regions of AD patients (Perry et al., 1977; Davies, 1979). Remarkably, when cases were stratified by age, a decrease in ChAT activity was apparent in older vs. younger controls, suggesting age-related changes in cholinergic function in asymptomatic aged people (Davies, 1979). A correlation between ChAT activity and mental test score in demented subjects suggested that deficits in ACh synthesis could underlie cognitive dysfunction (Perry et al., 1978).
With a role for cholinergic dysfunction posited for AD, the next step was to define the cholinergic neurons whose axons innervate cortex. The first documentation was provided by Johnston, McKinney, and Coyle in 1979 (Johnston et al., 1979). Studies in which kainic acid was used to selectively lesion neurons in the basal forebrain of rats resulted in striking reductions in ChAT, ACh, and the activity of synaptosomal high-affinity choline uptake, but not in neurochemical markers of GABAergic, noradrenergic, or serotoninergic neurons in ipsilateral neocortex. The locus of ChAT expressing neurons was defined as the nucleus basalis of Meynert (NBM) (Johnston et al., 1979) (see below for the anatomy of basal forebrain cholinergic neurons (BFCNs). With the locus of cortical cholinergic afferents now established, it remained to be described whether or not they were impacted in AD. This was evidenced by marked loss neurons of NBM neurons in the postmortem AD brain (Whitehouse et al., 1981, 1982). The role ascribed to hippocampus in memory pointed to cholinergic denervation of this region as likely contributing to memory loss, a suggestion consistent with evidence for marked neuron loss in the NBM in AD patient samples (Coyle et al., 1983).
Additional persuasive support for the “cholinergic hypothesis” came from clinical trials demonstrating efficacy for an AChE inhibitor in improving cognition in patients with AD. The initial effort, led by Summers and colleagues, evaluated tetrahydroaminoacridine (THA; Tacrine) treatment of subjects with AD using daily global assessment, the Names Learning Test, the Orientation Test, and the Alzheimer Deficit Scale. They showed improvement: (1) in an unblinded study (n = 17); and (2) in a blinded, placebo-controlled, crossover trial (n = 15 of the original group). Long term treatment (n = 12 of the original group) reported showing no serious side-affects and as providing continued symptomatic cognitive benefit of varying degree (Summers et al., 1986). The authors pointed to THA as a symptomatic, not curative, treatment and suggested that benefits would not be long-lasting. One follow-on study demonstrated statistically significant, dose-related improvements on clinician interview-based impression (CIBI), and the Alzheimer’s disease assessment scale-cognitive subscale (ADAS-Cog) (Knapp et al., 1994). But not all such studies confirmed benefits for cognition and behavior and concerns were raised due to evidence of hepatic dysfunction. Approved by the FDA in 1993, Tacrine was later withdrawn due to concern for hepatotoxicity (Anand and Singh, 2013). Nevertheless, robust efforts to build on the cholinergic hypothesis led to discovery and development of additional AChE inhibitors. Currently, three such agents are FDA-approved for treatment of AD-related cognitive dysfunction: donepezil, rivastigmine, and galantamine (Anand and Singh, 2013). Meta-analyses of their use demonstrate significant effects on improving cognition, typically as assessed using the ADAS-Cog, as well as clinician-rated global measures and measures of function and behavior. It is important to note that while such changes are significant, and long-term use is correlated with benefits for cognition and function, the benefits conferred by AChE inhibitors are modest (Deardorff et al., 2015).
Subsequent years have seen significant advances in understanding the anatomy of the basal forebrain cholinergic complex and the role BFCNs play in cognition. The cell bodies of BFCNs are found in a series of nuclei in the basal forebrain, including the medial septal nucleus (MSN), the diagonal band of Broca (DBB) (including both vertical and horizontal domains), the NBM, and the substantia innominate (SI) (Ballinger et al., 2016). In primates, the cholinergic nuclear groups are referred to as: Ch1 = MSN; Ch2 = vertical limb of the DBB; Ch3 = horizontal limb of the DBB; and Ch4 = the basal magnocellular complex that includes the SI, the NBM, the magnocellular preoptic nucleus and ventral pallidum (Mesulam et al., 1983). The axons emerging from these nuclei demonstrate distinct and characteristic pattern of innervation: the MSN and vertical limb of the DBB send axons to hippocampus, entorhinal cortex, and parahippocampus; the horizontal limb of the DBB, NBM, and SI send projections to neocortex and amygdala. A wealth of data now point to BFCNs, and their release of ACh, as supporting cognitive processes, including attention and memory (Ballinger et al., 2016). In earlier studies, selective lesions of BFCNs using saporin conjugated to an antibody against p75 neurotrophin receptor (p75NTR) (see below) impaired cognitive functions (Johnson et al., 2002; Conner et al., 2003; Knox, 2016). More recently, optogenetics has been used to explore BFCN contributions to learning and memory, demonstrating a role in a range of cognitive behaviors (Hangya et al., 2015). As expected, different portions of the forebrain cholinergic complex are implicated in different cognitive functions (Knox, 2016; Boskovic et al., 2018; Staib et al., 2018). Dysfunction and loss of BFCNs is now accepted as playing a significant role in cognitive dysfunction in AD (Ballinger et al., 2016).
The emergence and testing of the cholinergic hypothesis stands as an important milestone in the pursuit of the pathogenesis and treatment of AD. But it was evident that cholinergic hypothesis would not account fully for the pathophysiology of AD. Indeed, even as the cholinergic hypothesis emerged observations pointed to degeneration of other non-cholinergic populations in AD (Price et al., 1985). Nor did inhibition of AChE provide more than a modest benefit for cognition. Nevertheless the cholinergic hypothesis was highly influential. Some years later, Mesulam commented: “The cholinergic pathway emanating from the basal forebrain constitutes one of the most important modulatory afferents of the mammalian cortex. The initial expectation that the cholinergic deficiency would provide a unifying pathophysiological basis for Alzheimer’s disease and that cholinergic therapies would cure the dementia were clearly too optimistic. Nonetheless, the cortical cholinergic denervation remains one of the earliest, most severe, and most consistent transmitter changes in this disease. The cholinergic depletion may provide an important substrate for the neuropsychological features of Alzheimer’s disease, and may eventually yield important clues to its pathogenesis.” Indeed, this has proven to be the case as the involvement of BFCNs inspired studies of other approaches to pathogenesis. One such approach pursued a search for neurotrophic factors active on BFCNs to decipher whether deficient trophic support contributes to pathogenesis.
Ngf-Cholinergic Neuron Axis
The selective vulnerability of neuronal populations in AD and AD-DS should serve as a source of insights into pathogenesis. The early, selective loss of cholinergic neurons is yet to be fully understood, but an early suggested a link to deficient neurotrophic support. As suggested by Appel, neurodegeneration may be due to failure of innervated cells, including hippocampal and cortical neurons, to supply a relevant cholinergic neurotrophic factor (NTF), resulting in functional impairment of MSN and NBM neurons (Appel, 1981). If a deficient supply of a NTF contributes to pathogenesis it is rational to propose that treatment with an exogenous source of the NTF would restore the deficit and protect neurons from dysfunction and death. At the time of the Appel hypothesis, which might be called “the neurotrophic factor deficiency hypothesis,” relatively few NTFs had been identified and none were known to be active on CNS neurons. The first and best characterized was nerve growth factor (NGF) a small polypeptide discovered in the early 1950s by Levi-Montalcini and Hamburger with robust effects on sensory and sympathetic neurons (Levi-Montalcini and Hamburger, 1951). The first convincing suggestion that NGF might act in the brain was the demonstration by Seiler and Schwab that injecting radiolabeled NGF in the cortex of the rat was followed by selective retrograde transport to the basal forebrain cholinergic complex (Seiler and Schwab, 1984). This suggested that BFCNs expressed a receptor for NGF and may be responsive to NGF. Though the significance of this finding was little appreciated at the time it inspired studies over the next decade that provided a convincing demonstration that NGF does serve as a target-derived NTF for BFCNs. The key elements of such a demonstration include the following: (1) that the factor is produced in the target of innervation; (2) that it is released therein to bind to specific receptors on innervating axons; (3) that receptor-mediated signaling is registered in receptive neurons; and (4) that signaling events are linked to differentiation and/or maintenance of these neurons. Each of these criteria were satisfied for NGF and BFCNs. Critical steps in this line of investigation began with demonstrating that NGF acts on BFCNs and later to the demonstration of its expression in target territories and the expression of its receptors on BFCNs. Thus, the following was shown: (1) in vitro and in vivo, NGF acted in a dose-dependent fashion on the neurochemical differentiation of cholinergic neurons of the basal forebrain by significantly increasing the expression of activity of ChAT (Gnahn et al., 1983; Hefti et al., 1985; Mobley et al., 1985, 1986; Hagg et al., 1989; Li et al., 1995); (2) NGF intracerebroventricular (ICV) injection resulted in sparing of BFCNs in several lesion models, as measured by neuron size and number (Hefti, 1986; Williams et al., 1986; Kromer, 1987; Figueiredo et al., 1996); (3) NGF treatment reversed atrophy of BFCNs in a mouse model of DS (Holtzman et al., 1993); (4) NGF mRNA and protein were demonstrated in the target regions of BFCNs in hippocampus and cortex, with greater NGF expression in interneurons (Korsching et al., 1985; Large et al., 1986; Shelton and Reichardt, 1986; Whittemore et al., 1986; Ayer-Lelievre et al., 1988; Rocamora et al., 1996); (5) NGF present in target territories was selectively internalized and retrogradely transported in the axons of BFCNs (Seiler and Schwab, 1984); (6) cholinergic denervation of hippocampus induced a transient accumulation of NGF in hippocampus without a change in NGF mRNA (Korsching et al., 1986); (7) in developing rodents, disrupting NGF gene expression, or antibody-mediated sequestration of NGF, decreased expression of ChAT, and reduced the developmental increase in BFCN size and number (Crowley et al., 1994; Li et al., 1995; Debeir et al., 1999).
An interesting feature of the NGF-BFCN connection was evidence not just of the continued presence of NGF in target territories of mature BFCNs but that NGF acting on mature neurons robustly regulates trophic status. For example, prolonged infusion of NGF in adult mice deleted for one copy of NGF (i.e., NGF+/- mice) increased the cell size of BFCNs and the density of cholinergic innervation in hippocampus (Chen et al., 1997). In another example, in the elderly Ts65Dn mouse model of DS, in which there was reduction in the size and number of these neurons as well as decreased in cholinergic innervation of hippocampus, intraventricular delivery of NGF restored each of these measures to normal (Cooper et al., 2001). The latter findings were attributed to the ability of NGF to reverse the “phenotypic silence” of BFCNs – i.e., the downregulation of markers of BFCN differentiation in the setting of deficient supply of NGF signaling (Cooper et al., 2001). Moreover, NGF treatment of the wild type controls of aged Ts65Dn mice also increased the density of their cholinergic fibers in hippocampus, suggesting that augmenting normal levels of NGF serves to increase trophic status in normal neurons (Cooper et al., 2001). Continued NGF signaling in BFCNs may point to a role for NGF in dynamically modulating the synaptic function in hippocampal and cortical circuits. The robust responsiveness of BFCNs to increasing NGF levels mirrors a much earlier demonstration of the same phenomenon in sympathetic neurons in which it was speculated that endogenous NGF acting on adult neurons plays a regulatory role directed at maintenance of the adrenergic terminal network (Bjerre et al., 1975).
As work on NGF progressed a critical task was to identify the receptor(s) that mediated NGF signaling. Two receptors were discovered. The first was p75NTR (Johnson et al., 1986; Radeke et al., 1987). p75NTR is a transmembrane glycoprotein that binds with approximately nanomolar affinity to all members of the neurotrophin family, of which NGF is a member together with BDNF (brain-derived neurotrophic factor), NT3 (neurotrophin 3), and NT4 (Sofroniew et al., 2001; Chao, 2003). p75NTR also binds to the pro-forms of NGF and other neurotrophins with subnanomolar affinity. A member of the death domain-containing receptor subgroup of the tumor necrosis factor (TNF) receptor family, p75NTR signaling appears to serve a number of functions. Its signaling is cell context-specific, owing to the existence of co-receptors, signaling adaptors, and possibly other cell-specific features. p75NTR signaling shares features with other death receptors, including signaling through NF-κB and c-Jun N-terminal kinase (JNK) pathways as well as through RhoA. In perhaps the best studied neuronal population, p75NTR signaling in sympathetic neurons promotes axonal degeneration and cell death (Bothwell, 2016). Moreover, it appears that p75NTR regulates pruning of BFCN axonal arbors. Cre-mediated knockout of p75NTR in BFCNs significantly increased synaptic connectivity in medial prefrontal cortex, an effect that was prevented by co-expressing the p75NTR intracellular domain in these neurons. Thus, p75NTR appears to control BFCN synaptic arborization (Boskovic et al., 2018) and may do so in the mature CNS. What role(s) are played by retrograde axonal transport of p75NTR and its signaling properties in BFCNs is unknown, but recent data point to transport of the intracellular domain of p75NTR in mediating pro-apoptotic signaling in sympathetic neurons (Pathak et al., 2018). Consistent with these data, and in agreement with earlier studies pointing to increased differentiation and numbers of BFCNs in p75NTR knockout mice, a recent study showed that selective loss of p75NTR in BFCNs resulted in reduced cell death during the early postnatal period (Yeo et al., 1997; Naumann et al., 2002; Boskovic et al., 2014).
Another p75NTR function is to positively modulate trophic signaling through tropomyosin receptor kinase A (TrkA) (Davies et al., 1993; Bibel et al., 1999), the second NGF receptor discovered (Kaplan et al., 1991). TrkA, a member of the Trk receptor family which also includes TrkB (a receptor for BDNF and NT-4), and TrkC (a receptor for NT-3 receptor) (Huang and Reichardt, 2001; Chao, 2003) is activated by NGF alone among the neurotrophins (Belliveau et al., 1997). TrkA is a receptor tyrosine kinase whose activation by NGF leads to dimerization with tyrosine auto-phosphorylation resulting in the activation of classical signaling cascades of the mitogen-activated protein kinases (MAPK), phosphoinositide 3-kinase (PI3K), and phospholipase C-γ (PLC-γ) pathways (Bothwell, 2016). TrkA activation is responsible for the classical trophic effects of NGF on BFCNs. TrkA knockout mice show a marked decrease in the number of BFCNs and decreased cholinergic fiber density in hippocampus, indicating that NGF signaling through TrkA is essential for the normal development of BFCNs (Fagan et al., 1997). Enabling target-derived trophic support for BFCNs, retrograde axonal transport of activated TrkA delivers trophic signals to BFCN cell bodies (Xu et al., 2016). Both p75NTR and TrkA are robustly expressed in BFCNs and both are present on their axons (Holtzman et al., 1992, Kordower and Mufson, 1993; Holtzman et al., 1995; Fagan et al., 1997). Remarkably, as one facet of the response to NGF, expression of both TrkA and p75NTR were induced by NGF treatment in vivo (Miller et al., 1994; Li et al., 1995), pointing to the possibility that NGF signaling positively reinforces later signaling events.
Additional studies are needed to address the specific roles for p75NTR and TrkA in the development and maintenance of BFCNs and to address possible interactions between these receptors. In this context it is noteworthy that increased levels of pro-NGF were present in the AD and the adult DS brain, evidently due to reduced processing to the mature form of NGF. This raises the possibility that increased levels of pro-NGF, acting through p75NTR in BFCN axons, could compromise the integrity of axonal arbors, and synaptic function in AD (Bruno et al., 2009; Iulita and Cuello, 2014; Iulita et al., 2014). Moreover, p75NTR has been identified as one of the receptors for Aβ (Yaar et al., 1997; Yaar et al., 2002); robust p75NTR expression in BFCNs could thereby predispose them to Aβ-mediated signaling and endocytosis. Finally, speaking to the importance of continued NGF signaling in mature BFCNs, viral-mediated suppression of TrkA receptor expression in BFCNs acted selectively in aged rats to compromise attentional performance, reduce ACh release, and reduce ChAT immunostaining in BFCN cell bodies and cortical axons (Parikh et al., 2013). Taken together, the data are compelling that NGF and its receptors play critical roles in the development and maintenance of BFCNs and that studies of deficits in NGF and NGF signaling are germane to discussions of AD pathogenesis.
Ngf/Trk Signaling From Endosomes; the Signaling Endosome Hypothesis
For target-derived NTFs to impact the function of neurons they must communicate signals over the long distances between the ends of axons and the corresponding neuron cell bodies. The most well studied means by which this occurs begins with endocytosis of the NTF/receptor complex. The NTF/receptor complex first identified as playing a role in long distance signal transmission consisted of NGF bound to its activated TrkA receptor. Endocytosis of the NGF/activated TrkA complex was shown to form the “signaling endosome,” an organelle that can then be retrogradely transported to the soma via dynein-based transport (Howe and Mobley, 2005; Wu et al., 2009; Harrington and Ginty, 2013; Chen et al., 2018). Remarkably, and apropos its name, the NGF/TrkA signaling endosome contains not only the internalized NGF/active TrkA complex but also on its cytosolic surface the activated isoforms of many signaling molecules involved in the MAPK, Akt and PLCγ pathways (Howe et al., 2001; Delcroix et al., 2003; Zweifel et al., 2005; Chen et al., 2018). Excellent reviews speak to the formation, composition and regulation of signaling endosomes (Harrington and Ginty, 2013; Cosker and Segal, 2014; Scott-Solomon and Kuruvilla, 2018).
Studies in vivo and in vitro showed that Rab5-positive early endosomes were the first endocytic compartment in the retrograde pathway for transmitting the NGF/TrkA signaling endosomes (Wu et al., 2001; Delcroix et al., 2003; Cui et al., 2007; Liu et al., 2007). Recent studies point to the participation of other endocytic compartment(s) in this process and raise the possibility of neuron-specific differences in cargo management (Saxena et al., 2005; Deinhardt et al., 2006; Chen et al., 2018; Ye et al., 2018). It follows that retrograde signaling of NGF is impacted by factors that regulate the endocytic pathway and that dysregulation of such factors could disrupt the ability of signaling endosomes to support the long-distance communication needed to ensure normal neuronal structure and function. Rab5 belongs to the Ras superfamily of small Rab GTPases. Active Rab5 localizes to both the plasma membrane and early endosomes. Through its localization it is poised to regulate endocytosis of surface proteins and to regulate their trafficking through the early endosomal compartment (Zerial and Mcbride, 2001). It is noteworthy that AD risk variants include several genes whose proteins regulate endocytosis and transport, among which are Bin1, and Rin3 (Karch and Goate, 2015).
Ngf Signaling Dysregulation in Ad and Ad-Ds
The Appel hypothesis, pointing to a deficiency of NTF support as contributing to neurodegeneration, suggested that changes in the expression and release of such factors would be responsible. Given the robust harvest of data since that time, it is now possible to think about several levels at which an NTF deficiency might arise: NTF synthesis, NTF release, NTF binding to receptors, endocytosis of the NTF and its activated receptors into signaling endosomes, retrograde transport of signaling endosomes, and the robustness of signaling events catalyzed by signaling endosomes in the neuronal soma. There are data for many of these aspects for NGF and BFCNs in AD. NGF mRNA levels were not reduced in AD cortex or hippocampus in comparison to controls (Phillips et al., 1991; Jette et al., 1994; Fahnestock et al., 1996; Hock et al., 1998). In contrast, NGF protein levels were found to be increased in cortex and hippocampus (Crutcher et al., 1993; Scott et al., 1995; Fahnestock et al., 1996; Hock et al., 2000). Suggesting that decreased retrograde NGF transport contributed to this finding was that in the same brains increased NGF levels in cortex contrasted with decreased NGF levels in the cholinergic basal forebrain NBM (Scott et al., 1995). Supporting this interpretation, in the Ts65Dn mouse model of DS NGF transport from hippocampus to the basal forebrain was shown to be deficient, a change correlated with increased NGF in hippocampus and a trend to decreased levels in the MSN of the basal forebrain (Cooper et al., 2001). In contrast to the findings for NGF in the AD brain, BDNF protein was reduced in hippocampus and cortex as were BDNF mRNA levels (Siegel and Chauhan, 2000) suggesting a decrease in local synthesis for this NTF. There is little data for the temporal characteristics of NGF release in the targets of BFCNs, but one report speaks to long-lived increases in cortex following stimulation of the NBM (Hotta et al., 2007), suggesting that deficient innervation of cortex could reduce release.
Unlike NGF, there were decreases in the levels of both the mRNA and protein levels for TrkA in AD. Indeed, in surviving BFCNs in the NBM there was decreased TrkA mRNA (Boissiere et al., 1997; Ginsberg et al., 2006); the decreases were correlated with decreases in mini-mental state examination (MMSE) scores (Ginsberg et al., 2006). Reductions in mRNAs for TrkB and TrkC were also evident (Ginsberg et al., 2006). Corresponding to the reduction in mRNA, TrkA protein levels were also decreased in cell bodies of BFCNs as well as in AD cortex (Mufson et al., 1997). TrkA protein levels were also reduced in surviving neurons in NBM in DS (average age ∼50) (Sendera et al., 2000). In contrast to the findings for TrkA there was no change in the mRNA for p75NTR in basal forebrain, including the NBM (Goedert et al., 1989; Ginsberg et al., 2006). However, in one report remaining neurons and their processes were less immunoreactive for p75NTR (Salehi et al., 2000).
The studies just reviewed are evidence against a change in NGF gene expression as responsible for degeneration of BFCNs. The reduction in TrkA raises the possibility that this could contribute, but given evidence for decreased NGF retrograde transport and studies showing that NGF positively regulates TrkA gene expression, reduced retrograde NGF signaling could explain the decrease in TrkA gene expression. The question then is whether reduced retrograde transport of NGF signaling contributes to the degeneration of BFCNs in AD. This question has been addressed in a model of DS, the Ts65Dn mouse, whose genome is segmentally trisomic for approximately 100 mouse orthologs on human chromosome 21. The mouse App gene is present in three copies. In vivo increased App dose caused endosomal defects in retrograde trafficking of NGF in BFCNs in these mice (Salehi et al., 2006). This was manifest in BFCN axons in hippocampus by increased immunostaining for APP and its CTFs in enlarged Rab5-positive endosomes; thus, endosomes containing NGF and its receptors were significantly larger in cholinergic axons in Ts65Dn vs. control (2N) mice. Correspondingly, transport of NGF from hippocampus to basal forebrain was reduced to ∼10% of 2N controls (Salehi et al., 2006); synaptosomal studies showed that decreased transport was not due decreased NGF binding or internalization (Cooper et al., 2001). The defect in transport was largely restored by normalizing App dose. Significantly, reducing App dose also prevented the degeneration of BFCNs in the Ts65Dn mouse, pointing to defective retrograde signaling of NGF as responsible for BFCN loss (Salehi et al., 2006). Using the same methods to examine NGF transport, mice that overexpressed wild type human APP or a mutant of human APP, APPSwe also showed a modest but significant reduction in NGF transport (Salehi et al., 2006). That App overexpressing mice demonstrate a more general disruption of endosomal function is evidence that transport of BDNF signaling was also deficient in the Ts65Dn cortex. Thus, enlarged Rab5-positive endosomes containing activated TrkB receptors were seen in synapses and there was a corresponding decrease in TrkB-positive endosomes in cell bodies (Nosheny et al., 2015).
To explore the molecular basis by which increased APP gene dose compromises NGF retrograde transport studies were carried out on rat BFCNs in vitro. Overexpression of APP or C99, in BFCNs each induced increased activation of Rab5 and enlargement of early endosomes. These changes were correlated with decreased retrograde axonal transport of NGF, decreased NGF signaling, and atrophy of BFCNs (Jiang et al., 2010; Kim et al., 2016; Xu et al., 2016). Significantly, C99 mediated atrophy was mediated by increased activation of Rab5 because it was prevented when a dominant negative version of Rab5 was co-expressed (Xu et al., 2016). These data are evidence that increased APP gene dose, as reflected in increased levels of its products, including C99, acts to increase activation of Rab5 and reduce retrograde transport of NGF and NGF signaling in BFCNs. These findings may be directly relevant to dysfunction and loss of BFCNs in both AD and AD-DS.
Explaining the Selective Vulnerability of Bfcns: a Role for Deficient Ngf Signaling
Selective vulnerability of neurons is a well appreciated but poorly understood feature of many neurodegenerative disorders. As discussed above, selective loss of BFCNs characterizes AD. This question arises as to why BFCNs are more vulnerable than many other populations. Amyloid and tau pathologies, including NFTs and p-tau in pre-tangles, impact BFCNs relatively early in AD. It is likely that these markers significantly postdate the presence and toxic actions of oligomers of Aβ and tau. Such species are likely to have direct access to BFCNs. Addressing this suggestion, one can ask how oligomers of Aβ and tau might predelict BFCNs for degeneration. Below we speculate a series of events by which the biology of BFCNs and their dependence on NGF may feature in selective vulnerability. The stages of degeneration are assigned to distinct, but likely overlapping, temporal phases: initiation, amplification and termination (Figure 1).
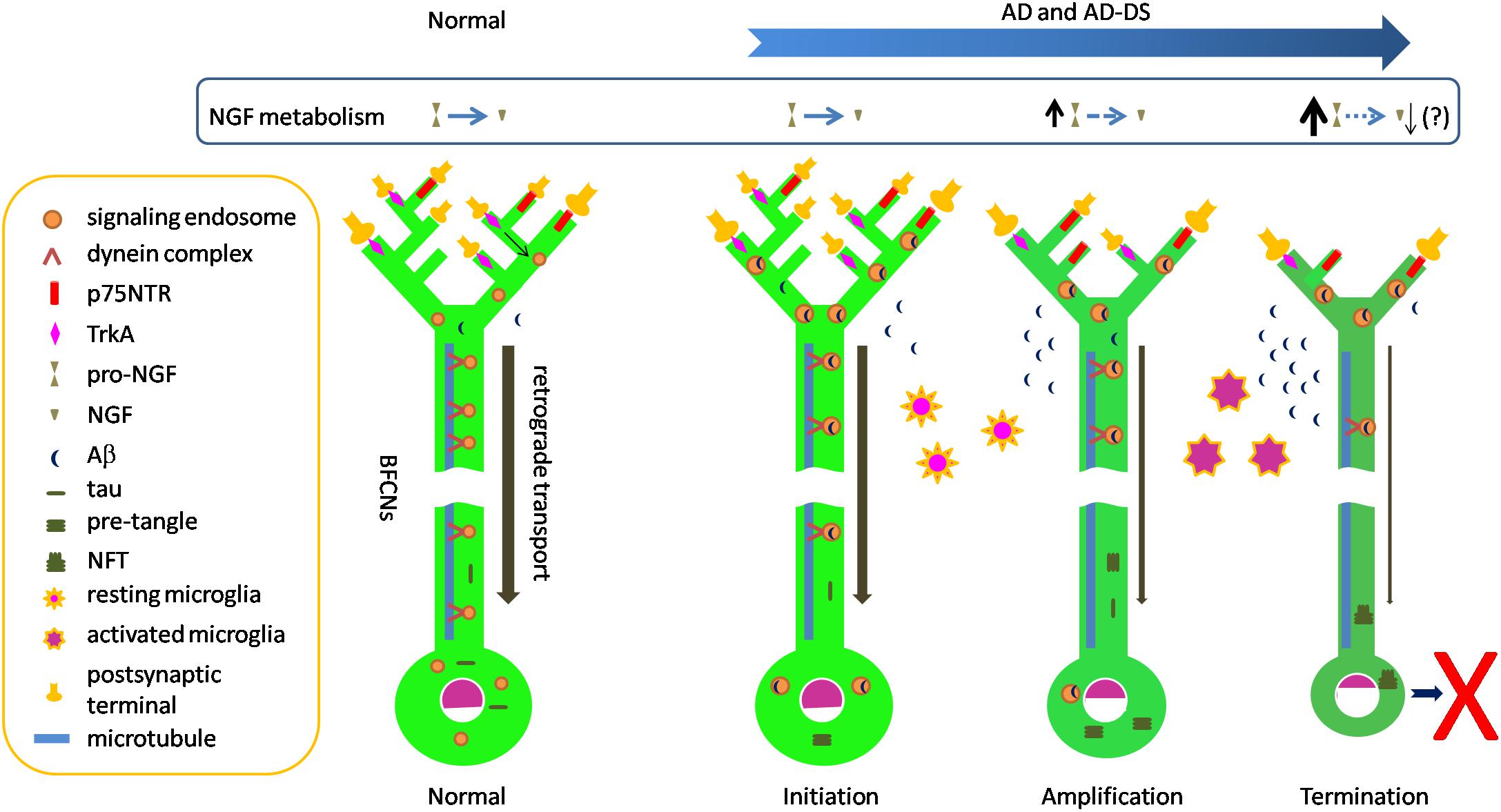
Figure 1. Speculative scheme to explain selective vulnerability of BFCNs in AD and AD-DS. Normal: dynein-mediated retrograde transport provides a steady flow of NGF/TrkA-containing Rab5-positive signaling endosomes from synapses to cell bodies. In this way target-derived NGF released by target neurons in hippocampus and cortex dynamically regulate the structure and function of BFCNs. The remarkable length of BFCN axons and their widespread arborization in target fields creates the need to move trophic signals over long distances. Initiation: Aβ42 and Aβ oligomeric species accumulate in BFCNs. Together with increased levels of C99 these species may contribute to the dysregulation of early endosomes seen at this stage. Dysregulation of endosomes reduces axonal transport of NGF/TrkA signaling endosomes, thus compromising trophic support of BFCNs. p75NTR binds and internalizes Aβ. Tau in pre-tangles is present, possibly reflecting increasing levels of tau oligomers which may also impact axonal transport. Meanwhile, resting microglial cells respond to Aβ and become activated and migrate to plaques to phagocytose Aβ. Amplification: Intracellular Aβ continues to impact BFCNs and failing retrograde transport of NGF signals results in BFCN dysfunction as measured by reduced expression of the genes for TrkA and ChAT. Reduced synthesis and anterograde transport of proteins needed for synaptic function and maintenance of axonal arbors reduces synapse number and function. p75NTR gene expression is relatively maintained, creating an imbalance in NGF signaling with excessive pruning of axonal arbors. Deficient activation of BFCN-responsive postsynaptic neurons may result in reduced processing of pro-NGF to mature NGF. Increased pro-NGF may increase p75NTR signaling. Termination: continuing compromise of retrograde axonal transport of NGF/TrkA signaling endosomes severely compromises the trophic status of BFCNs with marked changes in cell bodies and further shrinkage of axonal arbors and synaptic dysfunction. Sustained microglial activation results in defective Aβ phagocytosis and release of proinflammatory cytokines. The cumulative effect of these changes is atrophy and eventual death of BFCNs.
Initiation
What events might initiate the process of BFCN degeneration? Increasing levels of soluble oligomeric Aβ42 species in BFCNs may represent the earliest molecular provocation. Absolute increases in Aβ42 are present in DS and FAD due to APP gene triplication. Pathogenic mutations in PSEN and APP converge on impairment of the sequential cleavage efficiency, leading to production of longer and more hydrophobic Aβ peptides (Szaruga et al., 2017). Indeed, due to γ-secretase dysfunction, reduced clearance of longer and more hydrophobic Aβ peptides, or both, Aβ42 accumulates in both FAD and SAD (Selkoe and Hardy, 2016). Focusing on BFCNs, Geula and colleagues showed that intraneuronal immunopositivity for Aβ42, including the amino terminus of Aβ, was present in these neurons in young brains (ages 20–66) as well as in older non-demented and AD cases. There was no quantitative difference between groups. Staining with one Aβ42 antibody revealed granular, punctate staining consistent with the presence of Aβ42 within a vesicular compartment. Virtually all BFCNs were immunopositive at all ages and immunoreactivity in BFCNs exceeded that in cortical regions and in another magnocellular group in globus pallidus. Interestingly, BFCN homogenates demonstrated oligomeric Aβ species of a variety of sizes, with increased levels of a cluster of bands between ∼35 and 44 kDa in non-demented elderly, and to a greater extent, in AD, in the absence of increases in either APP or total Aβ (Baker-Nigh et al., 2015). The presence of Aβ within early endosomes may be especially relevant to initiation of pathogenesis. Aβ is present in early endosomes in early stage AD and in the young DS brain. The finding that early endosomal enlargement is one of the earliest pathological manifestations of AD and AD in DS, raises the possibility that Aβ within endosomes acts to induce hyperactivation of Rab5 with dysregulation of this compartment and endosomal enlargement through increased fusion of early endosomes. Whether or not and how Aβ within endosomes induces activation of Rab5 is unknown, but if Aβ impacts processing of APP within endosomes, for example by compromising the activity of γ-secretase, this would lead to increased levels of C99. Importantly, C99 levels are increased in FAD and, more variably, in SAD (Pera et al., 2013). Due to increased APP dose C99 levels are also increased in the DS brain. With disruption of endosome size comes a reduction in endosomal trafficking, as has been demonstrated (Xu et al., 2016). While a variety of cargoes would be impacted, in BFCNs this would prominently impact retrograde transport of NGF signaling endosomes and thus compromise their trophic support of BFCNs, including the genetic and cellular programs that support structure and function of these neurons (Figure 1). Quite possibly this stage also features interaction of Aβ-mediated and tau-mediated toxic events; the latter may include compromised transport of TrkA (Vossel et al., 2010, 2015). Aβ-mediated dysregulation of tau homeostasis, which has been demonstrated (Zheng et al., 2002; Stoothoff and Johnson, 2005), could initiate increased levels of p-tau and tau oligomer formation. In this context it is noteworthy that tangle and p-tau pre-tangle cytopathology and immunoreactivity were present in the NBM in aged normals as well as in those with MCI, with increased frequency in the latter. Remarkably, in cognitively unimpaired subjects at the earliest stages of tau pathology (i.e., Braak Stage 1 and 2), apart from the medial temporal lobe, the NBM was the only forebrain structure to show tau cytopathology, pointing to an unusual propensity to tau accumulation in these cells (Mesulam et al., 2004). In this early AD stage microglia would respond to Aβ by becoming activated and migrating to plaques to phagocytose and clear Aβ (Kinney et al., 2018).
Amplification
What elements of BFCNs would amplify the effect of increasing levels of toxic Aβ and tau oligomers on pathogenesis? Several possibilities can be envisioned. First, the need for BFCNs to engage in both volume-based and point-to-point synaptic neurotransmission may impose considerable metabolic demands (Ballinger et al., 2016). Second, there is need for continued NGF signaling to maintain the differentiated status of BFCNs (Counts and Mufson, 2005). Third, the remarkable length of BFCN axons and their widespread arborization in target fields creates the need to move trophic signals over very long distances in relatively thin unmyelinated axons. A recent study notes that a single human BFCN may be ∼100 m long and that the highly arborized distal axon, with perhaps more than a 1000 branch points per arbor, covers a very large volume of cortex. Added to this, the arbors converge on a single relatively long (10 cm) proximal axon whose diameter is ∼0.3 μm (Wu et al., 2014). Fourth, given complex axonal arbors, as additional burden may attend trafficking of NGF signals within enlarged, abnormal endosomes. Fifth, the presence on BFCN axons of p75NTR may serve to bind and internalize Aβ (Yaar et al., 2002) and possibly AβOs. Indeed, there is evidence that soluble and aggregated Aβ bind to p75NTR (Yaar et al., 1997). Sixth, with failed retrograde transport of NGF signals there would be reduced expression of TrkA receptors, as has been demonstrated (Mufson et al., 1997), further compromising the ability to respond to NGF present in target territories. Given that p75NTR gene expression appears to be less severely affected (Ginsberg et al., 2006), persistence of p75NTR on BFCN axons may negatively impact axonal arbors, possibly through pruning (Schuldiner and Yaron, 2015). One consequence of deficient signaling to postsynaptic neurons could be reduced ability to properly process NGF in cholino-receptive target neurons, leading to the presence of increased levels of pro-NGF, a ligand for p75NTR that could signal to further disrupt axonal arbors and synaptic function. The progressive decrease in ChAT activity characteristic of later stages in AD (Davis et al., 1999) could be explained by increasing failure to transport NGF signaling to the cell bodies of BFCNs (Figure 1).
Termination
The events just outlined would lead to a significant and increasingly severe compromise of the ability of BFCNs to maintain key facets of its differentiated state with downregulation of genetic and cellular programs that support maintenance of and synaptic signaling from axonal arbors. Another feature at this stage of disease could be sustained activation of microglial cells, which might exacerbate AD through a combination of defective Aβ phagocytosis and release of proinflammatory cytokines (Kinney et al., 2018). Activated microglia could contribute to neuronal injury. The cumulative effect of these changes would be atrophy and eventual loss of BFCNs. Thus, the process leading to death of these cells would proceed over decades – from early events triggered by toxic Aβ and tau oligomers, to progressive compromise of retrograde NGF signaling, to increasing deficits in synaptic function, to dysregulation of secretion of NGF from postsynaptic targets and activation of nearby microglia, to eventual demise of BFCNs (Figure 1).
Therapeutic Strategies in Ad and Ad-Ds
Given its trophic role for BFCNs, NGF has been considered a potential therapy to prevent loss of these neurons in AD, and DS. An important question is how to deliver NGF. Due to its size, NGF does not cross the blood-brain barrier when administered peripherally (Granholm et al., 1998), thus precluding this method. ICV NGF administration has been practiced effectively in rodent models and was delivered in this fashion in a pilot clinical trial. Treatment of three patients was accompanied by constant back pain and weight reduction (Eriksdotter Jonhagen et al., 1998). Pointing to another possible complication, continuous ICV delivery of NGF in the rat resulted in marked, albeit reversible, Schwann cell hyperplasia, possibly in response to sprouting of NGF-responsive sensory and sympathetic neurites at the dorsolateral caudal medulla and upper cervical spinal cord (Winkler et al., 1997). Studies in primates also demonstrated Schwann cell hyperplasia (Day-Lollini et al., 1997). To avoid these complications an alternative method for NGF delivery, allowing for local delivery to the basal forebrain, was employed by Tuszynski and colleagues (Tuszynski et al., 2005). NGF gene therapy was effected via introduction into the basal forebrain of autologous fibroblasts induced to express NGF or via an NGF-expressing adeno-associated viral vector (serotype 2) (AAV2). Classic trophic responses were detected in the basal forebrain. Increased levels of NGF were present in basal forebrain up to one year after viral delivery. Patients did not report pain and there was no evidence of Schwann cell hyperplasia (Tuszynski et al., 2015). A Phase Ib study of NGF gene therapy used AAV2-mediated delivery to the basal forebrain bilaterally in 10 AD patients. AAV2-NGF was reportedly safe and well tolerated. At autopsy, performed in 5 patients up to 6 years following delivery of NGF-encoding virus, NGF immunostaining was present near the needle tract and nearby BFCNs stained positive for NGF, p75NTR, and ChAT. There was also evidence of increased size of BFCNs near the site of injection. Cognitive measures showed no change, but sample size was small (Rafii et al., 2014). In a recently reported Phase II trial, 49 subjects with mild to moderate AD were randomized to receive stereotactically guided injection of AAV2-NGF or sham surgery. AAV2-NGF therapy was safe and well tolerated but there was no significant difference in either the primary or secondary outcome measures for cognition or function (Rafii et al., 2018). In yet another approach to local delivery of NGF to basal forebrain, a recent study reports the neurosurgical introduction of a device encapsulating NGF secreting cells; two devices were implanted in each hemisphere. Four patients with mild to moderate AD were examined for 6 months in an open-label Phase Ib study; the treatment was well tolerated and there was evidence for continued secretion at the time the devices were retrieved (Eyjolfsdottir et al., 2016). While the rationale for CNS delivery of NGF-based treatments is clear, effecting this form of treatment entails invasive measures.
An alternative to NGF itself would be delivery of a small molecule (Bruno et al., 2004) or monoclonal antibody agonist of TrkA (Lesauteur et al., 1996). Use of these modalities would facilitate delivery to axons, rather than the cell bodies, of BFCNs and would thus more closely address the locus of NGF deficiency in AD. Having stated this, one still has to address the concern that disrupted transport of NGF or NGF signaling may compromise delivery to BFCN cell bodies. The possibility that treatment efficacy may require higher cortical and hippocampal levels of NGF or NGF-mimics is suggested by deficient trophic responses in cultured BFCNs in which C99 overexpression was used to induce hyperactivation of Rab5 resulting in reduced retrograde NGF transport. Under these conditions, BFCN soma size in C99 expressing neurons was far less responsive than controls even when their axons were bathed in much higher than physiological levels of NGF (Xu et al., 2016). Finally, a small molecule antagonist of p75NTR (LM11A-31-BHS) aimed at improving synaptic function is in a Phase II trial (NCT03069014).
Addressing more generally the use of treatments directed at the pathogenesis of AD and AD-DS, and in view of increased APP gene dose in DS as well as changes in the processing of APP with absolute or relative increases in Aβ42 in the brain in all forms of AD, a focus on reducing the levels of APP and its products is rational. In DS, reducing the levels of APP mRNA and protein may prove effective and could be delivered well before the onset of AD pathogenesis. One potential strategy for DS is the use of RNA-based treatments [e.g., antisense oligonucleotides (ASOs)] to reduce APP mRNA levels (Sawa et al., 2018). ASOs have been reported to modestly lower APP protein in AD mouse models (Kumar et al., 2000; Erickson et al., 2012; Farr et al., 2014). Given restricted CNS access of peripherally administered ASOs, intrathecal delivery of ASOs may be required; this mode of delivery has recently been shown to be effective in treating neurodegenerative disorders (Schoch and Miller, 2017). Another RNA-based approach is suggested by a study using nanoparticles containing siRNAs against APP (Shyam et al., 2015). Alternatively, small molecules that interfere with translation of APP mRNA may prove useful. Posiphen is one such molecule; it reduced the levels of APP and its products, including Aβ42, in a translation-dependent manner in AD mouse models (Lahiri et al., 2007; Teich et al., 2018). In studies on a small number of MCI patients, treatment with Posiphen for 10 days reduced in CSF the secreted amino terminal fragments of APP (i.e., sAPPα and sAPPβ), total tau, p-tau, and showed a trend toward reduced Aβ42 (Maccecchini et al., 2012). Posiphen was also shown to reduce App protein in the Ts65Dn mouse model of DS (Salehi et al., 2008).
At present the pipeline of AD treatments lists 112 different agents, of which 63% aim at disease modification, 22% address cognitive symptoms, and 12% focus on psychiatric and behavioral symptoms (Cummings et al., 2018). To date there is no convincing evidence of efficacy for disease-modifying approaches, many of which have targeted APP processing (Cummings et al., 2018). Inhibiting the synthesis of Aβ through use β-secretase inhibitors generated a great deal of interest resulting in development and clinical testing of several compounds. Recent Phase III trials of three such drugs (Verubecestat, Atabecestat, and Lanabecestat), examining patient cohorts ranging from those at risk for AD to those with MCI, demonstrated not only lack of efficacy but evidence of worsened cognition (Panza et al., 2019). Equally disappointing were studies of γ-secretase inhibitors, including Begacestat, Semagacestat, and Avagacestat. Clinical trials showed lack of efficacy and worsened cognition (Panza et al., 2019). A much more promising rationale focuses on enhancing the processing of APP to reduce the levels of Aβ42. Several well-tolerated small molecule modulators of γ-secretases (GSMs), which act to enhance processivity of the enzyme complex, significantly reduced Aβ42 as well as Aβ40 in vitro and in vivo and reduced amyloid burden in mouse models of AD (Kounnas et al., 2010; Wagner et al., 2017). The possibility that GSMs impact both the endopeptidase as well as the exopeptidase (i.e., carboxypeptidase-like) activity of γ-secretases suggests that GSMs might also reduce the levels of C99, a topic that deserves additional study to counter the C99-mediated increase in activation of Rab5 and endosomal dysfunction.
There is a great deal of interest in the immune approach to Aβ through vaccination and passive immunization. These efforts followed considerable success using the immune approach in animal models of AD (Schenk et al., 1999; Das et al., 2001; Spencer and Masliah, 2014). An early vaccine trial was complicated by sub-acute meningoencephalitis (Hock et al., 2003; Orgogozo et al., 2003). This led to changes in vaccine design, including use of the N-terminus of the Aβ peptide and different adjuvants or display platforms (Muhs et al., 2007; Van Dyck, 2018). Several vaccine trials are ongoing (Castro et al., 2017). Considerable effort has focused on the use of monoclonal antibodies against Aβ. No fewer than seven such antibodies have been evaluated; clinical trials continue for solanezumab, gantenerumab, crenezumab, aducanumab, and BAN2401. The different epitopes used to generate antibodies, and the different Aβ species targeted, are noteworthy. The lack of understanding of which Aβ species, and in particular which toxic species, should be targeted, may limit success using the immune approach. Indeed, given data for a role for Aβ oligomers in AD pathogenesis it is noteworthy that at least 2 of the antibodies currently being evaluated are stated to not interact with oligomers (Van Dyck, 2018). In spite of the excitement generated by these trials, immune-based trials are yet to demonstrate efficacy. Nevertheless, the hope remains that antibody and vaccine approaches will show promise.
To achieve therapeutic efficacy without compromising neuronal function immune-based approaches may need to be informed by a detailed evaluation of all the Aβ targets addressed by antibodies. Indeed, Aβ monomers may be important for normal neuronal function. Aβ1-42 monomers were shown to be critical in maintaining neuronal survival and glucose homeostasis (Giuffrida et al., 2009, 2015). It has been suggested that both the gain of toxicity of Aβ oligomers and loss of the normal physiological function of the Aβ monomer may contribute to the pathogenesis of AD (Copani, 2017). Aducanumab, a human monoclonal antibody that selectively targets aggregated Aβ without binding to soluble Aβ monomers, may represent an agent that selectively targets toxic species (Sevigny et al., 2016). Unfortunately, two Phase III trials of Aducanumab in AD were recently halted due to the conclusion by an independent data monitoring committee that the trials were unlikely to meet their primary endpoints, which included measures of cognition (Servick, 2019). Use of advanced trial designs and the conduct of trials in presymptomatic or early symptomatic AD may support further developments. Equally or more important may be immune approaches informed by defining the structure of toxic Aβ conformers. Given the shared Aβ-related pathology between AD and DS, successful trials of vaccines and monoclonal antibodies in AD trials would support their use in DS. A recent study of vaccine targeting Aβ showed relative preservation of BFCNs in Ts65Dn mice without obvious side effects (Belichenko et al., 2016). A vaccine trial is underway in adults with DS (NCT02738450).
Together with increased interest in a role for tau in AD pathogenesis, efforts have also been directed to tau-directed therapeutics. Diverse approaches have been suggested (Holtzman et al., 2016). Among them, multiple tau-based immunotherapy strategies have been successfully tested in AD animal models, pointing to this as an option for treating AD (Asuni et al., 2007; Boutajangout et al., 2011; Pedersen and Sigurdsson, 2015). Several tau-based immunotherapies have entered clinical trials in AD. Active immunotherapies, including AADvac-1 and ACI-35, target misfolded tau or p-tau epitopes in Phase II and Ib trials, respectively. Anti-tau antibodies, including ABBV-8E12 and RG6100, are in Phase II trials (Medina, 2018). As for Aβ, successfully targeting tau may benefit from defining the structure of toxic tau species. Future trials may also explore other facets of tau pathology, including the role that DYRK1A plays in priming tau for phosphorylation by other kinases (Wegiel et al., 2011).
The hyperactivation of Rab5 resulting in early endosomal enlargement serves as one of the earliest manifestations of AD and AD-DS. The link to decreased retrograde axonal transport and signaling of NGF, and atrophy of BFCNs (Xu et al., 2016) points to regulation of Rab5 activity as a rational treatment target. A Rab5-based strategy would benefit AD and AD-DS by restoring axonal transport of NGF-containing signaling endosomes as well as transport of other axonal cargoes impacted by excessive activation of Rab5. A number of approaches can be considered, including the discovery of small molecules that impact GTP loading of Rab5 as well as RNA-based approaches, including CNS delivery of specific ASOs.
Summary
The foregoing makes clear the complexity of AD. In spite of this, we are of the optimistic view that existing insights may be sufficient for launching a first wave of disease-modifying treatments. Nevertheless, advances in genetics and cellular biology will be needed to further enhance our understanding of pathogenesis and the mechanisms that must be targeted for the most robustly effective treatments. Especially important will be efforts to define the structure, function and location of toxic species of Aβ and tau, including biomarkers to announce their presence and interventions to combat their production, accumulation, or actions. A focus on detecting the earliest manifestations in selectively vulnerable populations, including BFCNs, would augment progress. In particular, we are intrigued by the intersection of Aβ- and tau-mediated pathology in dysregulation of the endosomal pathway and disordered transport of neurotrophic factor signaling as playing a defining role in neuron dysfunction and death. Finally, we anticipate continued benefit from asking how the various hypotheses for AD interact and reinforce one another.
Author Contributions
X-QC and WM wrote and edited the manuscript and approved it for publication.
Funding
Support for WM was provided by NIH: P01NS092525, R01AG055523; Alzheimer’s Association: DSAD-15-363207; Cure Alzheimer’s Fund: 2018-1301; DH Chen Foundation: R-86U55A; Ono Pharma Foundation: UCSD 2019-0742; and Lumind: Establishing a Clinical Trials Network for Down Syndrome.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our laboratory members and colleagues for their many helpful discussions. Special thanks go to Mariko Sawa, Chengbiao Wu, Larry Goldstein, Larry Squire, Ralph Nixon, Claudio Cuello, Elizabeth Head, Ira Lott, Michael Rafii, Andreas Muhs, Andrea Pfeifer, Douglas Galasko, Maria Maccecchini, Steve Wagner, Rudy Tanzi, Kevin Rynerson, Steve Dowdy, and Chris Glass.
References
Anand, P., and Singh, B. (2013). A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 36, 375–399.
Anden, N. E., Carlsson, A., Dahlstroem, A., Fuxe, K., Hillarp, N. A., and Larsson, K. (1964). Demonstration and mapping out of nigro-neostriatal dopamine neurons. Life Sci. 3, 523–530. doi: 10.1016/0024-3205(64)90161-4
Anden, N. E., Hfuxe, K., Hamberger, B., and Hokfelt, T. (1966). A quantitative study on the nigro-neostriatal dopamine neuron system in the rat. Acta Physiol. Scand. 67, 306–312. doi: 10.1111/j.1748-1716.1966.tb03317.x
Andrade-Moraes, C. H., Oliveira-Pinto, A. V., Castro-Fonseca, E., Da Silva, C. G., Guimaraes, D. M., Szczupak, D., et al. (2013). Cell number changes in Alzheimer’s disease relate to dementia, not to plaques and tangles. Brain 136, 3738–3752.
Antonarakis, S. E. (2017). Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 18, 147–163. doi: 10.1038/nrg.2016.154
Appel, S. H. (1981). A unifying hypothesis for the cause of amyotrophic lateral sclerosis, parkinsonism, and Alzheimer disease. Ann. Neurol. 10, 499–505. doi: 10.1002/ana.410100602
Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T., and Hyman, B. T. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639.
Asuni, A. A., Boutajangout, A., Quartermain, D., and Sigurdsson, E. M. (2007). Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 27, 9115–9129. doi: 10.1523/jneurosci.2361-07.2007
Ayer-Lelievre, C., Olson, L., Ebendal, T., Seiger, A., and Persson, H. (1988). Expression of the beta-nerve growth factor gene in hippocampal neurons. Science 240, 1339–1341. doi: 10.1126/science.2897715
Baker-Nigh, A., Vahedi, S., Davis, E. G., Weintraub, S., Bigio, E. H., Klein, W. L., et al. (2015). Neuronal amyloid-beta accumulation within cholinergic basal forebrain in ageing and Alzheimer’s disease. Brain 138, 1722–1737. doi: 10.1093/brain/awv024
Ball, M. J. (1977). Neuronal loss, neurofibrillary tangles and granulovacuolar degeneration in the hippocampus with ageing and dementia. A quantitative study. Acta Neuropathol. 37, 111–118. doi: 10.1007/bf00692056
Ball, M. J. (1978). Topographic distribution of neurofibrillary tangles and granulovacuolar degeneration in hippocampal cortex of aging and demented patients. A quantitative study. Acta Neuropathol. 42, 73–80. doi: 10.1007/bf00690970
Ballard, C., Mobley, W., Hardy, J., Williams, G., and Corbett, A. (2016). Dementia in Down’s syndrome. Lancet Neurol. 15, 622–636.
Ballinger, E. C., Ananth, M., Talmage, D. A., and Role, L. W. (2016). Basal forebrain cholinergic circuits and signaling in cognition and cognitive decline. Neuron 91, 1199–1218. doi: 10.1016/j.neuron.2016.09.006
Bartus, R. T., Dean, R. L. III, Beer, B., and Lippa, A. S. (1982). The cholinergic hypothesis of geriatric memory dysfunction. Science 217, 408–414. doi: 10.1126/science.7046051
Bateman, R. J., Aisen, P. S., De Strooper, B., Fox, N. C., Lemere, C. A., Ringman, J. M., et al. (2011). Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 3:1.
Belichenko, P. V., Madani, R., Rey-Bellet, L., Pihlgren, M., Becker, A., Plassard, A., et al. (2016). An anti-beta-amyloid vaccine for treating cognitive deficits in a mouse model of down syndrome. PLoS One 11:e0152471. doi: 10.1371/journal.pone.0152471
Belliveau, D. J., Krivko, I., Kohn, J., Lachance, C., Pozniak, C., Rusakov, D., et al. (1997). NGF and neurotrophin-3 both activate TrkA on sympathetic neurons but differentially regulate survival and neuritogenesis. J. Cell Biol. 136, 375–388. doi: 10.1083/jcb.136.2.375
Bennett, M. R. (2000). The concept of transmitter receptors: 100 years on. Neuropharmacology 39, 523–546. doi: 10.1016/s0028-3908(99)00137-9
Bibel, M., Hoppe, E., and Barde, Y. A. (1999). Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 18, 616–622. doi: 10.1093/emboj/18.3.616
Bierer, L. M., Hof, P. R., Purohit, D. P., Carlin, L., Schmeidler, J., Davis, K. L., et al. (1995). Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch. Neurol. 52, 81–88. doi: 10.1001/archneur.1995.00540250089017
Bjerre, B., Bjorklund, A., Mobley, W., and Rosengren, E. (1975). Short- and long-term effects of nerve growth factor on the sympathetic nervous system in the adult mouse. Brain Res. 94, 263–277. doi: 10.1016/0006-8993(75)90061-x
Blaschko, H. (1942). The activity of l(-)-dopa decarboxylase. J. Physiol. 101, 337–349. doi: 10.1113/jphysiol.1942.sp003988
Boissiere, F., Faucheux, B., Ruberg, M., Agid, Y., and Hirsch, E. C. (1997). Decreased TrkA gene expression in cholinergic neurons of the striatum and basal forebrain of patients with Alzheimer’s disease. Exp. Neurol. 145, 245–252. doi: 10.1006/exnr.1997.6443
Boskovic, Z., Alfonsi, F., Rumballe, B. A., Fonseka, S., Windels, F., and Coulson, E. J. (2014). The role of p75NTR in cholinergic basal forebrain structure and function. J. Neurosci. 34, 13033–13038. doi: 10.1523/jneurosci.2364-14.2014
Boskovic, Z., Milne, M. R., Qian, L., Clifton, H. D., Mcgovern, A. E., Turnbull, M. T., et al. (2018). Cholinergic basal forebrain neurons regulate fear extinction consolidation through p75 neurotrophin receptor signaling. Transl. Psychiatry 8:199.
Bothwell, M. (2016). Recent advances in understanding neurotrophin signaling. F1000Res. 5:F1000 Faculty Rev-1885.
Boutajangout, A., Ingadottir, J., Davies, P., and Sigurdsson, E. M. (2011). Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J. Neurochem. 118, 658–667. doi: 10.1111/j.1471-4159.2011.07337.x
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/bf00308809
Brown, G. L., Dale, H. H., and Feldberg, W. (1936). Reactions of the normal mammalian muscle to acetylcholine and to eserine. J. Physiol. 87, 394–424. doi: 10.1113/jphysiol.1936.sp003414
Bruno, M. A., Clarke, P. B., Seltzer, A., Quirion, R., Burgess, K., Cuello, A. C., et al. (2004). Long-lasting rescue of age-associated deficits in cognition and the CNS cholinergic phenotype by a partial agonist peptidomimetic ligand of TrkA. J. Neurosci. 24, 8009–8018. doi: 10.1523/jneurosci.1508-04.2004
Bruno, M. A., Leon, W. C., Fragoso, G., Mushynski, W. E., Almazan, G., and Cuello, A. C. (2009). Amyloid beta-induced nerve growth factor dysmetabolism in Alzheimer disease. J. Neuropathol. Exp. Neurol. 68, 857–869. doi: 10.1097/nen.0b013e3181aed9e6
Calderon-Garciduenas, A. L., and Duyckaerts, C. (2017). Alzheimer disease. Handb. Clin. Neurol. 145, 325–337.
Carlsson, A., Lindqvist, M., Magnusson, T., and Waldeck, B. (1958). On the presence of 3-hydroxytyramine in brain. Science 127:471. doi: 10.1126/science.127.3296.471
Castro, P., Zaman, S., and Holland, A. (2017). Alzheimer’s disease in people with Down’s syndrome: the prospects for and the challenges of developing preventative treatments. J. Neurol. 264, 804–813. doi: 10.1007/s00415-016-8308-8
Chao, M. V. (2003). Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 4, 299–309. doi: 10.1038/nrn1078
Chen, K. S., Nishimura, M. C., Armanini, M. P., Crowley, C., Spencer, S. D., and Phillips, H. S. (1997). Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J. Neurosci. 17, 7288–7296. doi: 10.1523/jneurosci.17-19-07288.1997
Chen, X. Q., Sawa, M., and Mobley, W. C. (2018). Dysregulation of neurotrophin signaling in the pathogenesis of Alzheimer disease and of Alzheimer disease in Down syndrome. Free Radic. Biol. Med. 114, 52–61. doi: 10.1016/j.freeradbiomed.2017.10.341
Cline, E. N., Bicca, M. A., Viola, K. L., and Klein, W. L. (2018). The amyloid-beta oligomer hypothesis: beginning of the third decade. J. Alzheimers Dis. 64, S567–S610.
Conner, J. M., Culberson, A., Packowski, C., Chiba, A. A., and Tuszynski, M. H. (2003). Lesions of the Basal forebrain cholinergic system impair task acquisition and abolish cortical plasticity associated with motor skill learning. Neuron 38, 819–829. doi: 10.1016/s0896-6273(03)00288-5
Cooper, J. D., Salehi, A., Delcroix, J. D., Howe, C. L., Belichenko, P. V., Chua-Couzens, J., et al. (2001). Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc. Natl. Acad. Sci. U.S.A. 98, 10439–10444. doi: 10.1073/pnas.181219298
Copani, A. (2017). The underexplored question of beta-amyloid monomers. Eur. J. Pharmacol. 817, 71–75. doi: 10.1016/j.ejphar.2017.05.057
Cosker, K. E., and Segal, R. A. (2014). Neuronal signaling through endocytosis. Cold Spring Harb. Perspect. Biol. 6:a020669.
Cotzias, G. C., Papavasiliou, P. S., and Gellene, R. (1969). Modification of Parkinsonism–chronic treatment with L-dopa. N. Engl. J. Med. 280, 337–345. doi: 10.1056/nejm196902132800701
Counts, S. E., and Mufson, E. J. (2005). The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J. Neuropathol. Exp. Neurol. 64, 263–272. doi: 10.1093/jnen/64.4.263
Coyle, J. T., Price, D. L., and Delong, M. R. (1983). Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 219, 1184–1190. doi: 10.1126/science.6338589
Crowley, C., Spencer, S. D., Nishimura, M. C., Chen, K. S., Pitts-Meek, S., Armanini, M. P., et al. (1994). Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 76, 1001–1011. doi: 10.1016/0092-8674(94)90378-6
Crutcher, K. A., Scott, S. A., Liang, S., Everson, W. V., and Weingartner, J. (1993). Detection of NGF-like activity in human brain tissue: increased levels in Alzheimer’s disease. J. Neurosci. 13, 2540–2550. doi: 10.1523/jneurosci.13-06-02540.1993
Cui, B., Wu, C., Chen, L., Ramirez, A., Bearer, E. L., Li, W. P., et al. (2007). One at a time, live tracking of NGF axonal transport using quantum dots. Proc. Natl. Acad. Sci. U.S.A. 104, 13666–13671. doi: 10.1073/pnas.0706192104
Cummings, J., Lee, G., Ritter, A., and Zhong, K. (2018). Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement. 4, 195–214.
Dahlstroem, A., and Fuxe, K. (1964). Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol. Scand. Suppl. 232, 231–255.
Dale, H. H., Feldberg, W., and Vogt, M. (1936). Release of acetylcholine at voluntary motor nerve endings. J. Physiol. 86, 353–380. doi: 10.1113/jphysiol.1936.sp003371
Das, P., Murphy, M. P., Younkin, L. H., Younkin, S. G., and Golde, T. E. (2001). Reduced effectiveness of Abeta1-42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol. Aging 22, 721–727. doi: 10.1016/s0197-4580(01)00245-7
Davies, A. M., Lee, K. F., and Jaenisch, R. (1993). p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron 11, 565–574. doi: 10.1016/0896-6273(93)90069-4
Davies, P. (1979). Neurotransmitter-related enzymes in senile dementia of the Alzheimer type. Brain Res. 171, 319–327. doi: 10.1016/0006-8993(79)90336-6
Davies, P., and Maloney, A. J. (1976). Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2:1403. doi: 10.1016/s0140-6736(76)91936-x
Davis, K. L., Mohs, R. C., Marin, D., Purohit, D. P., Perl, D. P., Lantz, M., et al. (1999). Cholinergic markers in elderly patients with early signs of Alzheimer disease. JAMA 281, 1401–1406.
Day-Lollini, P. A., Stewart, G. R., Taylor, M. J., Johnson, R. M., and Chellman, G. J. (1997). Hyperplastic changes within the leptomeninges of the rat and monkey in response to chronic intracerebroventricular infusion of nerve growth factor. Exp. Neurol. 145, 24–37. doi: 10.1006/exnr.1997.6448
Deardorff, W. J., Feen, E., and Grossberg, G. T. (2015). The use of cholinesterase inhibitors across all stages of Alzheimer’s disease. Drugs Aging 32, 537–547. doi: 10.1007/s40266-015-0273-x
Debeir, T., Saragovi, H. U., and Cuello, A. C. (1999). A nerve growth factor mimetic TrkA antagonist causes withdrawal of cortical cholinergic boutons in the adult rat. Proc. Natl. Acad. Sci. U.S.A. 96, 4067–4072. doi: 10.1073/pnas.96.7.4067
Deinhardt, K., Salinas, S., Verastegui, C., Watson, R., Worth, D., Hanrahan, S., et al. (2006). Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 52, 293–305. doi: 10.1016/j.neuron.2006.08.018
Dekosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464. doi: 10.1002/ana.410270502
Delcroix, J. D., Valletta, J. S., Wu, C., Hunt, S. J., Kowal, A. S., and Mobley, W. C. (2003). NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron 39, 69–84.
Deutsch, J. A. (1971). The cholinergic synapse and the site of memory. Science 174, 788–794. doi: 10.1126/science.174.4011.788
Doran, E., Keator, D., Head, E., Phelan, M. J., Kim, R., Totoiu, M., et al. (2017). Down syndrome, partial trisomy 21, and absence of Alzheimer’s disease: the role of APP. J. Alzheimers Dis. 56, 459–470. doi: 10.3233/jad-160836
Dorszewska, J., Prendecki, M., Oczkowska, A., Dezor, M., and Kozubski, W. (2016). Molecular basis of familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 13, 952–963. doi: 10.2174/1567205013666160314150501
Drachman, D. A., and Leavitt, J. (1974). Human memory and the cholinergic system. A relationship to aging? Arch. Neurol. 30, 113–121.
Ehringer, H., and Hornykiewicz, O. (1960). [Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system]. Klin. Wochenschr. 38, 1236–1239.
Erickson, M. A., Niehoff, M. L., Farr, S. A., Morley, J. E., Dillman, L. A., Lynch, K. M., et al. (2012). Peripheral administration of antisense oligonucleotides targeting the amyloid-beta protein precursor reverses AbetaPP and LRP-1 overexpression in the aged SAMP8 mouse brain. J. Alzheimers Dis. 28, 951–960. doi: 10.3233/jad-2011-111517
Eriksdotter Jonhagen, M., Nordberg, A., Amberla, K., Backman, L., Ebendal, T., Meyerson, B., et al. (1998). Intracerebroventricular infusion of nerve growth factor in three patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 9, 246–257. doi: 10.1159/000017069
Eyjolfsdottir, H., Eriksdotter, M., Linderoth, B., Lind, G., Juliusson, B., Kusk, P., et al. (2016). Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: application of a second-generation encapsulated cell biodelivery device. Alzheimers Res. Ther. 8:30.
Fagan, A. M., Garber, M., Barbacid, M., Silos-Santiago, I., and Holtzman, D. M. (1997). A role for TrkA during maturation of striatal and basal forebrain cholinergic neurons in vivo. J. Neurosci. 17, 7644–7654. doi: 10.1523/jneurosci.17-20-07644.1997
Fahn, S. (2018). The 200-year journey of Parkinson disease: reflecting on the past and looking towards the future. Parkinsonism Relat. Disord. 46(Suppl. 1), S1–S5.
Fahnestock, M., Scott, S. A., Jette, N., Weingartner, J. A., and Crutcher, K. A. (1996). Nerve growth factor mRNA and protein levels measured in the same tissue from normal and Alzheimer’s disease parietal cortex. Brain Res. Mol. Brain Res. 42, 175–178. doi: 10.1016/s0169-328x(96)00193-3
Farr, S. A., Erickson, M. A., Niehoff, M. L., Banks, W. A., and Morley, J. E. (2014). Central and peripheral administration of antisense oligonucleotide targeting amyloid-beta protein precursor improves learning and memory and reduces neuroinflammatory cytokines in Tg2576 (AbetaPPswe) mice. J. Alzheimers Dis. 40, 1005–1016. doi: 10.3233/jad-131883
Feldberg, W., and Gaddum, J. H. (1934). The chemical transmitter at synapses in a sympathetic ganglion. J. Physiol. 81, 305–319. doi: 10.1113/jphysiol.1934.sp003137
Figueiredo, B. C., Hu, L., Bedard, A. M., Tetzlaff, W., and Cuello, A. C. (1996). NGF prevents further atrophy of cholinergic cells of the nucleus basalis due to cortical infarction in adult post-hypothyroid rats but does not restore cell size compared to euthyroid [correction of euthroid] rats. J. Chem. Neuroanat. 12, 15–27. doi: 10.1016/s0891-0618(96)00171-8
Fuxe, K. (1965a). Evidence for the existence of monoamine neurons in the central nervous system. 3. the monoamine nerve terminal. Z. Zellforsch. Mikrosk. Anat. 65, 573–596. doi: 10.1007/bf00337069
Fuxe, K. (1965b). Evidence for the existence of monoamine neurons in the central nervous system. IV. Distribution of monoamine nerve terminals in the central nervous system. Acta Physiol. Scand. Suppl. 247, 237.
Giannakopoulos, P., Herrmann, F. R., Bussiere, T., Bouras, C., Kovari, E., Perl, D. P., et al. (2003). Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01
Ginsberg, S. D., Che, S., Wuu, J., Counts, S. E., and Mufson, E. J. (2006). Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 97, 475–487. doi: 10.1111/j.1471-4159.2006.03764.x
Giuffrida, M. L., Caraci, F., Pignataro, B., Cataldo, S., De Bona, P., Bruno, V., et al. (2009). Beta-amyloid monomers are neuroprotective. J. Neurosci. 29, 10582–10587.
Giuffrida, M. L., Tomasello, M. F., Pandini, G., Caraci, F., Battaglia, G., Busceti, C., et al. (2015). Monomeric ss-amyloid interacts with type-1 insulin-like growth factor receptors to provide energy supply to neurons. Front. Cell. Neurosci. 9:297. doi: 10.3389/fncel.2015.00297
Gnahn, H., Hefti, F., Heumann, R., Schwab, M. E., and Thoenen, H. (1983). NGF-mediated increase of choline acetyltransferase (ChAT) in the neonatal rat forebrain: evidence for a physiological role of NGF in the brain? Brain Res. 285, 45–52. doi: 10.1016/0165-3806(83)90107-4
Goedert, M., Fine, A., Dawbarn, D., Wilcock, G. K., and Chao, M. V. (1989). Nerve growth factor receptor mRNA distribution in human brain: normal levels in basal forebrain in Alzheimer’s disease. Brain Res. Mol. Brain Res. 5, 1–7. doi: 10.1016/0169-328x(89)90011-9
Gomez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., et al. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 41, 17–24. doi: 10.1002/ana.410410106
Gomez-Isla, T., Price, J. L., Mckeel, D. W. Jr., Morris, J. C., Growdon, J. H., et al. (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci. 16, 4491–4500. doi: 10.1523/jneurosci.16-14-04491.1996
Granholm, A. C., Albeck, D., Backman, C., Curtis, M., Ebendal, T., Friden, P., et al. (1998). A non-invasive system for delivering neural growth factors across the blood-brain barrier: a review. Rev. Neurosci. 9, 31–55.
Hagg, T., Fass-Holmes, B., Vahlsing, H. L., Manthorpe, M., Conner, J. M., and Varon, S. (1989). Nerve growth factor (NGF) reverses axotomy-induced decreases in choline acetyltransferase, NGF receptor and size of medial septum cholinergic neurons. Brain Res. 505, 29–38. doi: 10.1016/0006-8993(89)90112-1
Hangya, B., Ranade, S. P., Lorenc, M., and Kepecs, A. (2015). Central cholinergic neurons are rapidly recruited by reinforcement feedback. Cell 162, 1155–1168. doi: 10.1016/j.cell.2015.07.057
Harrington, A. W., and Ginty, D. D. (2013). Long-distance retrograde neurotrophic factor signalling in neurons. Nat. Rev. Neurosci. 14, 177–187. doi: 10.1038/nrn3253
Hefti, F. (1986). Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J. Neurosci. 6, 2155–2162. doi: 10.1523/jneurosci.06-08-02155.1986
Hefti, F., Hartikka, J., Eckenstein, F., Gnahn, H., Heumann, R., and Schwab, M. (1985). Nerve growth factor increases choline acetyltransferase but not survival or fiber outgrowth of cultured fetal septal cholinergic neurons. Neuroscience 14, 55–68. doi: 10.1016/0306-4522(85)90163-0
Hirano, A. (1994). Hirano bodies and related neuronal inclusions. Neuropathol. Appl. Neurobiol. 20, 3–11. doi: 10.1111/j.1365-2990.1994.tb00951.x
Hithersay, R., Hamburg, S., Knight, B., and Strydom, A. (2017). Cognitive decline and dementia in Down syndrome. Curr. Opin. Psychiatry 30, 102–107. doi: 10.1097/yco.0000000000000307
Hithersay, R., Startin, C. M., Hamburg, S., Mok, K. Y., Hardy, J., Fisher, E. M. C., et al. (2018). Association of dementia with mortality among adults with down syndrome older than 35 years. JAMA Neurol. doi: 10.1001/jamaneurol.2018.3616
Hock, C., Heese, K., Hulette, C., Rosenberg, C., and Otten, U. (2000). Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch. Neurol. 57, 846–851.
Hock, C., Heese, K., Muller-Spahn, F., Hulette, C., Rosenberg, C., and Otten, U. (1998). Decreased trkA neurotrophin receptor expression in the parietal cortex of patients with Alzheimer’s disease. Neurosci. Lett. 241, 151–154. doi: 10.1016/s0304-3940(98)00019-6
Hock, C., Konietzko, U., Streffer, J. R., Tracy, J., Signorell, A., Muller-Tillmanns, B., et al. (2003). Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron 38, 547–554. doi: 10.1016/s0896-6273(03)00294-0
Holtzman, D. M., Carrillo, M. C., Hendrix, J. A., Bain, L. J., Catafau, A. M., Gault, L. M., et al. (2016). Tau: from research to clinical development. Alzheimers Dement. 12, 1033–1039.
Holtzman, D. M., Kilbridge, J., Li, Y., Cunningham, E. T. Jr., Lenn, N. J., et al. (1995). TrkA expression in the CNS: evidence for the existence of several novel NGF-responsive CNS neurons. J. Neurosci. 15, 1567–1576. doi: 10.1523/jneurosci.15-02-01567.1995
Holtzman, D. M., Li, Y., Chen, K., Gage, F. H., Epstein, C. J., and Mobley, W. C. (1993). Nerve growth factor reverses neuronal atrophy in a Down syndrome model of age-related neurodegeneration. Neurology 43, 2668–2673.
Holtzman, D. M., Li, Y., Parada, L. F., Kinsman, S., Chen, C. K., Valletta, J. S., et al. (1992). p140trk mRNA marks NGF-responsive forebrain neurons: evidence that trk gene expression is induced by NGF. Neuron 9, 465–478. doi: 10.1016/0896-6273(92)90184-f
Hotta, H., Uchida, S., and Kagitani, F. (2007). Stimulation of the nucleus basalis of Meynert produces an increase in the extracellular release of nerve growth factor in the rat cerebral cortex. J. Physiol. Sci. 57, 383–387. doi: 10.2170/physiolsci.sc008107
Howe, C. L., and Mobley, W. C. (2005). Long-distance retrograde neurotrophic signaling. Curr. Opin. Neurobiol. 15, 40–48. doi: 10.1016/j.conb.2005.01.010
Howe, C. L., Valletta, J. S., Rusnak, A. S., and Mobley, W. C. (2001). NGF signaling from clathrin-coated vesicles: evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron 32, 801–814. doi: 10.1016/s0896-6273(01)00526-8
Huang, E. J., and Reichardt, L. F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736. doi: 10.1146/annurev.neuro.24.1.677
Iulita, M. F., and Cuello, A. C. (2014). Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol. Sci. 35, 338–348. doi: 10.1016/j.tips.2014.04.010
Iulita, M. F., Do Carmo, S., Ower, A. K., Fortress, A. M., Flores Aguilar, L., Hanna, M., et al. (2014). Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain 137, 860–872. doi: 10.1093/brain/awt372
Jette, N., Cole, M. S., and Fahnestock, M. (1994). NGF mRNA is not decreased in frontal cortex from Alzheimer’s disease patients. Brain Res. Mol. Brain Res. 25, 242–250. doi: 10.1016/0169-328x(94)90159-7
Jiang, Y., Mullaney, K. A., Peterhoff, C. M., Che, S., Schmidt, S. D., Boyer-Boiteau, A., et al. (2010). Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. U.S.A. 107, 1630–1635. doi: 10.1073/pnas.0908953107
Johnson, D., Lanahan, A., Buck, C. R., Sehgal, A., Morgan, C., Mercer, E., et al. (1986). Expression and structure of the human NGF receptor. Cell 47, 545–554. doi: 10.1016/0092-8674(86)90619-7
Johnson, D. A., Zambon, N. J., and Gibbs, R. B. (2002). Selective lesion of cholinergic neurons in the medial septum by 192 IgG-saporin impairs learning in a delayed matching to position T-maze paradigm. Brain Res. 943, 132–141. doi: 10.1016/s0006-8993(02)02623-9
Johnston, M. V., Mckinney, M., and Coyle, J. T. (1979). Evidence for a cholinergic projection to neocortex from neurons in basal forebrain. Proc. Natl. Acad. Sci. U.S.A. 76, 5392–5396. doi: 10.1073/pnas.76.10.5392
Kametani, F., and Hasegawa, M. (2018). Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 12:25. doi: 10.3389/fnins.2018.00025
Kaplan, D. R., Hempstead, B. L., Martin-Zanca, D., Chao, M. V., and Parada, L. F. (1991). The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science 252, 554–558. doi: 10.1126/science.1850549
Karch, C. M., and Goate, A. M. (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. doi: 10.1016/j.biopsych.2014.05.006
Kim, S., Sato, Y., Mohan, P. S., Peterhoff, C., Pensalfini, A., Rigoglioso, A., et al. (2016). Evidence that the rab5 effector APPL1 mediates APP-betaCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol. Psychiatry 21, 707–716. doi: 10.1038/mp.2015.97
Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., and Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 4, 575–590.
Knapp, M. J., Knopman, D. S., Solomon, P. R., Pendlebury, W. W., Davis, C. S., and Gracon, S. I. (1994). A 30-week randomized controlled trial of high-dose tacrine in patients with Alzheimer’s disease. The Tacrine Study Group. JAMA 271, 985–991. doi: 10.1001/jama.271.13.985
Knox, D. (2016). The role of basal forebrain cholinergic neurons in fear and extinction memory. Neurobiol. Learn. Mem. 133, 39–52. doi: 10.1016/j.nlm.2016.06.001
Korbel, J. O., Tirosh-Wagner, T., Urban, A. E., Chen, X. N., Kasowski, M., Dai, L., et al. (2009). The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. U.S.A. 106, 12031–12036. doi: 10.1073/pnas.0813248106
Kordower, J. H., and Mufson, E. J. (1993). NGF receptor (p75)-immunoreactivity in the developing primate basal ganglia. J. Comp. Neurol. 327, 359–375. doi: 10.1002/cne.903270305
Korsching, S., Auburger, G., Heumann, R., Scott, J., and Thoenen, H. (1985). Levels of nerve growth factor and its mRNA in the central nervous system of the rat correlate with cholinergic innervation. EMBO J. 4, 1389–1393. doi: 10.1002/j.1460-2075.1985.tb03791.x
Korsching, S., Heumann, R., Thoenen, H., and Hefti, F. (1986). Cholinergic denervation of the rat hippocampus by fimbrial transection leads to a transient accumulation of nerve growth factor (NGF) without change in mRNANGF content. Neurosci. Lett. 66, 175–180. doi: 10.1016/0304-3940(86)90186-2
Kounnas, M. Z., Danks, A. M., Cheng, S., Tyree, C., Ackerman, E., Zhang, X., et al. (2010). Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron 67, 769–780.
Kromer, L. F. (1987). Nerve growth factor treatment after brain injury prevents neuronal death. Science 235, 214–216. doi: 10.1126/science.3798108
Kumar, V. B., Farr, S. A., Flood, J. F., Kamlesh, V., Franko, M., Banks, W. A., et al. (2000). Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides 21, 1769–1775. doi: 10.1016/s0196-9781(00)00339-9
Lahiri, D. K., Chen, D., Maloney, B., Holloway, H. W., Yu, Q. S., Utsuki, T., et al. (2007). The experimental Alzheimer’s disease drug posiphen [(+)-phenserine] lowers amyloid-beta peptide levels in cell culture and mice. J. Pharmacol. Exp. Ther. 320, 386–396. doi: 10.1124/jpet.106.112102
Large, T. H., Bodary, S. C., Clegg, D. O., Weskamp, G., Otten, U., and Reichardt, L. F. (1986). Nerve growth factor gene expression in the developing rat brain. Science 234, 352–355. doi: 10.1126/science.3764415
Lesauteur, L., Maliartchouk, S., Le Jeune, H., Quirion, R., and Saragovi, H. U. (1996). Potent human p140-TrkA agonists derived from an anti-receptor monoclonal antibody. J. Neurosci. 16, 1308–1316. doi: 10.1523/jneurosci.16-04-01308.1996
Levi-Montalcini, R., and Hamburger, V. (1951). Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J. Exp. Zool. 116, 321–361. doi: 10.1002/jez.1401160206
Li, Y., Holtzman, D. M., Kromer, L. F., Kaplan, D. R., Chua-Couzens, J., Clary, D. O., et al. (1995). Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J. Neurosci. 15, 2888–2905. doi: 10.1523/jneurosci.15-04-02888.1995
Liu, J., Lamb, D., Chou, M. M., Liu, Y. J., and Li, G. (2007). Nerve growth factor-mediated neurite outgrowth via regulation of Rab5. Mol. Biol. Cell 18, 1375–1384. doi: 10.1091/mbc.e06-08-0725
Loewi, O. (1921). Über humorale übertragbarkeit der herznervenwirkung. I.Mitteilung. Pflügers Arch. Gesamte Physiol. Menschen Tiere 189, 239–242.
Loewi, O., and Navratil, E. (1926). Über humorale übertragbarkeit der herznerven-wirkung. X.Mitteilung. Über das schicksal des vagusstoffes. Pflügers Arch. Ges. Physiol. 214, 678–688. doi: 10.1007/bf01741946
Maccecchini, M. L., Chang, M. Y., Pan, C., John, V., Zetterberg, H., and Greig, N. H. (2012). Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-beta peptide and tau levels: target engagement, tolerability and pharmacokinetics in humans. J. Neurol. Neurosurg. Psychiatry 83, 894–902. doi: 10.1136/jnnp-2012-302589
Maurer, K., Volk, S., and Gerbaldo, H. (1997). Auguste D and Alzheimer’s disease. Lancet 349, 1546–1549.
Mcgeer, P. L., Rogers, J., and Mcgeer, E. G. (2016). Inflammation, antiinflammatory agents, and Alzheimer’s disease: the last 22 years. J. Alzheimers Dis. 54, 853–857. doi: 10.3233/jad-160488
Medina, M. (2018). An overview on the clinical development of tau-based therapeutics. Int. J. Mol. Sci. 19:E1160.
Megarbane, A., Ravel, A., Mircher, C., Sturtz, F., Grattau, Y., Rethore, M. O., et al. (2009). The 50th anniversary of the discovery of trisomy 21: the past, present, and future of research and treatment of Down syndrome. Genet. Med. 11, 611–616. doi: 10.1097/gim.0b013e3181b2e34c
Mesulam, M., Shaw, P., Mash, D., and Weintraub, S. (2004). Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann. Neurol. 55, 815–828. doi: 10.1002/ana.20100
Mesulam, M. M., Mufson, E. J., Levey, A. I., and Wainer, B. H. (1983). Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J. Comp. Neurol. 214, 170–197. doi: 10.1002/cne.902140206
Miller, F. D., Speelman, A., Mathew, T. C., Fabian, J., Chang, E., Pozniak, C., et al. (1994). Nerve growth factor derived from terminals selectively increases the ratio of p75 to trkA NGF receptors on mature sympathetic neurons. Dev. Biol. 161, 206–217. doi: 10.1006/dbio.1994.1021
Mobley, W. C., Rutkowski, J. L., Tennekoon, G. I., Buchanan, K., and Johnston, M. V. (1985). Choline acetyltransferase activity in striatum of neonatal rats increased by nerve growth factor. Science 229, 284–287. doi: 10.1126/science.2861660
Mobley, W. C., Rutkowski, J. L., Tennekoon, G. I., Gemski, J., Buchanan, K., and Johnston, M. V. (1986). Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Brain Res. 387, 53–62. doi: 10.1016/0169-328x(86)90020-3
Mufson, E. J., Lavine, N., Jaffar, S., Kordower, J. H., Quirion, R., and Saragovi, H. U. (1997). Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer’s disease. Exp. Neurol. 146, 91–103. doi: 10.1006/exnr.1997.6504
Muhs, A., Hickman, D. T., Pihlgren, M., Chuard, N., Giriens, V., Meerschman, C., et al. (2007). Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 104, 9810–9815. doi: 10.1073/pnas.0703137104
Naumann, T., Casademunt, E., Hollerbach, E., Hofmann, J., Dechant, G., Frotscher, M., et al. (2002). Complete deletion of the neurotrophin receptor p75NTR leads to long-lasting increases in the number of basal forebrain cholinergic neurons. J. Neurosci. 22, 2409–2418. doi: 10.1523/jneurosci.22-07-02409.2002
Nosheny, R. L., Belichenko, P. V., Busse, B. L., Weissmiller, A. M., Dang, V., Das, D., et al. (2015). Increased cortical synaptic activation of TrkB and downstream signaling markers in a mouse model of Down Syndrome. Neurobiol. Dis. 77, 173–190. doi: 10.1016/j.nbd.2015.02.022
Orgogozo, J. M., Gilman, S., Dartigues, J. F., Laurent, B., Puel, M., Kirby, L. C., et al. (2003). Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61, 46–54. doi: 10.1212/01.wnl.0000073623.84147.a8
Panza, F., Lozupone, M., Seripa, D., and Imbimbo, B. P. (2019). Amyloid-beta immunotherapy for Alzheimer’s disease - is it now a long shot? Ann. Neurol. 85, 303–315.
Parikh, V., Howe, W. M., Welchko, R. M., Naughton, S. X., D’amore, D. E., Han, D. H., et al. (2013). Diminished trkA receptor signaling reveals cholinergic-attentional vulnerability of aging. Eur. J. Neurosci. 37, 278–293. doi: 10.1111/ejn.12090
Pathak, A., Stanley, E. M., Hickman, F. E., Wallace, N., Brewer, B., Li, D., et al. (2018). Retrograde degenerative signaling mediated by the p75 neurotrophin receptor requires p150(glued) deacetylation by axonal HDAC1. Dev. Cell 46, 376–387.e7. doi: 10.1016/j.devcel.2018.07.001
Pedersen, J. T., and Sigurdsson, E. M. (2015). Tau immunotherapy for Alzheimer’s disease. Trends Mol. Med. 21, 394–402.
Pera, M., Alcolea, D., Sanchez-Valle, R., Guardia-Laguarta, C., Colom-Cadena, M., Badiola, N., et al. (2013). Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease. Acta Neuropathol. 125, 201–213. doi: 10.1007/s00401-012-1062-9
Perry, E. K., Perry, R. H., Blessed, G., and Tomlinson, B. E. (1977). Necropsy evidence of central cholinergic deficits in senile dementia. Lancet 1:189. doi: 10.1016/s0140-6736(77)91780-9
Perry, E. K., Tomlinson, B. E., Blessed, G., Bergmann, K., Gibson, P. H., and Perry, R. H. (1978). Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br. Med. J. 2, 1457–1459. doi: 10.1136/bmj.2.6150.1457
Phillips, H. S., Hains, J. M., Armanini, M., Laramee, G. R., Johnson, S. A., and Winslow, J. W. (1991). BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 7, 695–702. doi: 10.1016/0896-6273(91)90273-3
Poirier, L. J., and Sourkes, T. L. (1965). Influence of the substantia nigra on the catecholamine content of the striatum. Brain 88, 181–192. doi: 10.1093/brain/88.1.181
Prasher, V. P., Farrer, M. J., Kessling, A. M., Fisher, E. M., West, R. J., Barber, P. C., et al. (1998). Molecular mapping of Alzheimer-type dementia in Down’s syndrome. Ann. Neurol. 43, 380–383.
Price, D. L., Whitehouse, P. J., and Struble, R. G. (1985). Alzheimer’s disease. Annu. Rev. Med. 36, 349–356.
Radeke, M. J., Misko, T. P., Hsu, C., Herzenberg, L. A., and Shooter, E. M. (1987). Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature 325, 593–597. doi: 10.1038/325593a0
Rafii, M. S., Baumann, T. L., Bakay, R. A., Ostrove, J. M., Siffert, J., Fleisher, A. S., et al. (2014). A phase1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer’s disease. Alzheimers Dement. 10, 571–581. doi: 10.1016/j.jalz.2013.09.004
Rafii, M. S., Tuszynski, M. H., Thomas, R. G., Barba, D., Brewer, J. B., Rissman, R. A., et al. (2018). Adeno-associated viral vector (serotype 2)-nerve growth factor for patients with alzheimer disease: a randomized clinical trial. JAMA Neurol. 75, 834–841.
Reitz, C., and Mayeux, R. (2014). Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 88, 640–651. doi: 10.1016/j.bcp.2013.12.024
Rocamora, N., Pascual, M., Acsady, L., De Lecea, L., Freund, T. F., and Soriano, E. (1996). Expression of NGF and NT3 mRNAs in hippocampal interneurons innervated by the GABAergic septohippocampal pathway. J. Neurosci. 16, 3991–4004. doi: 10.1523/jneurosci.16-12-03991.1996
Salehi, A., Delcroix, J. D., Belichenko, P. V., Zhan, K., Wu, C., Valletta, J. S., et al. (2006). Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51, 29–42. doi: 10.1016/j.neuron.2006.05.022
Salehi, A., Faizi, M., Takimoto, R., Valletta, J., Danks, A., and Mobley, W. C. (2008). “Posiphen treatment is able to reduce APP levels in Ts65Dn mouse model of Down syndrome,” in Proceedings of the Alzheimer’s Association International Conference on Alzheimer’s Disease, Chicago, IL.
Salehi, A., Ocampo, M., Verhaagen, J., and Swaab, D. F. (2000). P75 neurotrophin receptor in the nucleus basalis of meynert in relation to age, sex, and Alzheimer’s disease. Exp. Neurol. 161, 245–258. doi: 10.1006/exnr.1999.7252
Sawa, M., Zhao, H., Kordasiewicz, H., Fitzsimmons, B., Derse, D., Becker, A., et al. (2018). “APP-directed antisense oligonucleotides reduced APP gene expression in mouse models of Down Syndrome,” In Proceedings of the 48th annual meeting Society for Neuroscience’s, San Diego, CA.
Saxena, S., Bucci, C., Weis, J., and Kruttgen, A. (2005). The small GTPase Rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor TrkA. J. Neurosci. 25, 10930–10940. doi: 10.1523/jneurosci.2029-05.2005
Scheff, S. W., Dekosky, S. T., and Price, D. A. (1990). Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol. Aging 11, 29–37. doi: 10.1016/0197-4580(90)90059-9
Scheff, S. W., and Price, D. A. (1993). Synapse loss in the temporal lobe in Alzheimer’s disease. Ann. Neurol. 33, 190–199. doi: 10.1002/ana.410330209
Scheff, S. W., Price, D. A., Schmitt, F. A., Dekosky, S. T., and Mufson, E. J. (2007). Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68, 1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f
Scheff, S. W., Price, D. A., Schmitt, F. A., and Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012
Scheltens, P., Blennow, K., Breteler, M. M., De Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517.
Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., et al. (1999). Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. doi: 10.1038/22124
Schoch, K. M., and Miller, T. M. (2017). Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron 94, 1056–1070. doi: 10.1016/j.neuron.2017.04.010
Schuldiner, O., and Yaron, A. (2015). Mechanisms of developmental neurite pruning. Cell. Mol. Life Sci. 72, 101–119. doi: 10.1007/s00018-014-1729-6
Scott, S. A., Mufson, E. J., Weingartner, J. A., Skau, K. A., and Crutcher, K. A. (1995). Nerve growth factor in Alzheimer’s disease: increased levels throughout the brain coupled with declines in nucleus basalis. J. Neurosci. 15, 6213–6221. doi: 10.1523/jneurosci.15-09-06213.1995
Scott-Solomon, E., and Kuruvilla, R. (2018). Mechanisms of neurotrophin trafficking via Trk receptors. Mol. Cell. Neurosci. 91, 25–33. doi: 10.1016/j.mcn.2018.03.013
Seiler, M., and Schwab, M. E. (1984). Specific retrograde transport of nerve growth factor (NGF) from neocortex to nucleus basalis in the rat. Brain Res. 300, 33–39. doi: 10.1016/0006-8993(84)91338-6
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608.
Sendera, T. J., Ma, S. Y., Jaffar, S., Kozlowski, P. B., Kordower, J. H., Mawal, Y., et al. (2000). Reduction in TrkA-immunoreactive neurons is not associated with an overexpression of galaninergic fibers within the nucleus basalis in Down’s syndrome. J. Neurochem. 74, 1185–1196. doi: 10.1046/j.1471-4159.2000.741185.x
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011a). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Serrano-Pozo, A., Mielke, M. L., Gomez-Isla, T., Betensky, R. A., Growdon, J. H., Frosch, M. P., et al. (2011b). Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 179, 1373–1384. doi: 10.1016/j.ajpath.2011.05.047
Servick, K. (2019) Another major drug candidate targeting the brain plaques of Alzheimer’s disease has failed. What’s left? Science doi: 10.1126/science.aax4236
Sevigny, J., Chiao, P., Bussiere, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537, 50–56.
Shafiei, S. S., Guerrero-Munoz, M. J., and Castillo-Carranza, D. L. (2017). Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 9:83. doi: 10.3389/fnagi.2017.00083
Shelton, D. L., and Reichardt, L. F. (1986). Studies on the expression of the beta nerve growth factor (NGF) gene in the central nervous system: level and regional distribution of NGF mRNA suggest that NGF functions as a trophic factor for several distinct populations of neurons. Proc. Natl. Acad. Sci. U.S.A. 83, 2714–2718. doi: 10.1073/pnas.83.8.2714
Shyam, R., Ren, Y., Lee, J., Braunstein, K. E., Mao, H. Q., and Wong, P. C. (2015). Intraventricular delivery of siRNA nanoparticles to the central nervous system. Mol. Ther. Nucleic Acids 4:e242. doi: 10.1038/mtna.2015.15
Siegel, G. J., and Chauhan, N. B. (2000). Neurotrophic factors in Alzheimer’s and Parkinson’s disease brain. Brain Res. Brain Res. Rev. 33, 199–227.
Sofroniew, M. V., Howe, C. L., and Mobley, W. C. (2001). Nerve growth factor signaling, neuroprotection, and neural repair. Annu. Rev. Neurosci. 24, 1217–1281. doi: 10.1146/annurev.neuro.24.1.1217
Spencer, B., and Masliah, E. (2014). Immunotherapy for Alzheimer’s disease: past, present and future. Front. Aging Neurosci. 6:114. doi: 10.3389/fnagi.2014.00114
Staib, J. M., Della Valle, R., and Knox, D. K. (2018). Disruption of medial septum and diagonal bands of Broca cholinergic projections to the ventral hippocampus disrupt auditory fear memory. Neurobiol. Learn. Mem. 152, 71–79. doi: 10.1016/j.nlm.2018.05.009
Stoothoff, W. H., and Johnson, G. V. (2005). Tau phosphorylation: physiological and pathological consequences. Biochim. Biophys. Acta 1739, 280–297. doi: 10.1016/j.bbadis.2004.06.017
Summers, W. K., Majovski, L. V., Marsh, G. M., Tachiki, K., and Kling, A. (1986). Oral tetrahydroaminoacridine in long-term treatment of senile dementia, Alzheimer type. N. Engl. J. Med. 315, 1241–1245. doi: 10.1056/nejm198611133152001
Szaruga, M., Munteanu, B., Lismont, S., Veugelen, S., Horre, K., Mercken, M., et al. (2017). Alzheimer’s-causing mutations shift abeta length by destabilizing gamma-secretase-abetan interactions. Cell 170, 443-456.e14.
Teich, A. F., Sharma, E., Barnwell, E., Zhang, H., Staniszewksi, A., Utsuki, T., et al. (2018). Translational inhibition of APP by Posiphen: efficacy, pharmacodynamics, and pharmacokinetics in the APP/PS1 mouse. Alzheimers Dement. Transl. Res. Clin. Interv. 4, 37–45. doi: 10.1016/j.trci.2017.12.001
Thal, D. R., Rub, U., Orantes, M., and Braak, H. (2002). Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/wnl.58.12.1791
Thinakaran, G., and Koo, E. H. (2008). Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619. doi: 10.1074/jbc.r800019200
Toodayan, N. (2016). Professor alois Alzheimer (1864-1915): lest we forget. J. Clin. Neurosci. 31, 47–55. doi: 10.1016/j.jocn.2015.12.032
Tuszynski, M. H., Thal, L., Pay, M., Salmon, D. P., Bakay, R., Patel, P., et al. (2005). A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 11, 551–555. doi: 10.1038/nm1239
Tuszynski, M. H., Yang, J. H., Barba, D., Bakay, R. A., Pay, M. M., Masliah, E., et al. (2015). Nerve growth factor gene therapy: activation of neuronal responses in Alzheimer disease. JAMA Neurol. 72, 1139–1147.
Van Cauwenberghe, C., Van Broeckhoven, C., and Sleegers, K. (2016). The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18, 421–430. doi: 10.1038/gim.2015.117
Van Dyck, C. H. (2018). Anti-amyloid-beta monoclonal antibodies for alzheimer’s disease: pitfalls and promise. Biol. Psychiatry 83, 311–319. doi: 10.1016/j.biopsych.2017.08.010
Vossel, K. A., Xu, J. C., Fomenko, V., Miyamoto, T., Suberbielle, E., Knox, J. A., et al. (2015). Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J. Cell Biol. 209, 419–433. doi: 10.1083/jcb.201407065
Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A. C., Sharma, P., Finkbeiner, S., et al. (2010). Tau reduction prevents Abeta-induced defects in axonal transport. Science 330:198. doi: 10.1126/science.1194653
Wagner, S. L., Rynearson, K. D., Duddy, S. K., Zhang, C., Nguyen, P. D., Becker, A., et al. (2017). Pharmacological and toxicological properties of the potent oral gamma-secretase modulator BPN-15606. J. Pharmacol. Exp. Ther. 362, 31–44. doi: 10.1124/jpet.117.240861
Wegiel, J., Gong, C. X., and Hwang, Y. W. (2011). The role of DYRK1A in neurodegenerative diseases. FEBS J. 278, 236–245. doi: 10.1111/j.1742-4658.2010.07955.x
Whitehouse, P. J., Price, D. L., Clark, A. W., Coyle, J. T., and Delong, M. R. (1981). Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 10, 122–126. doi: 10.1002/ana.410100203
Whitehouse, P. J., Price, D. L., Struble, R. G., Clark, A. W., Coyle, J. T., and Delon, M. R. (1982). Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science 215, 1237–1239. doi: 10.1126/science.7058341
Whittemore, S. R., Ebendal, T., Larkfors, L., Olson, L., Seiger, A., Stromberg, I., et al. (1986). Development and regional expression of beta nerve growth factor messenger RNA and protein in the rat central nervous system. Proc. Natl. Acad. Sci. U.S.A. 83, 817–821. doi: 10.1073/pnas.83.3.817
Williams, L. R., Varon, S., Peterson, G. M., Wictorin, K., Fischer, W., Bjorklund, A., et al. (1986). Continuous infusion of nerve growth factor prevents basal forebrain neuronal death after fimbria fornix transection. Proc. Natl. Acad. Sci. U.S.A. 83, 9231–9235. doi: 10.1073/pnas.83.23.9231
Winkler, J., Ramirez, G. A., Kuhn, H. G., Peterson, D. A., Day-Lollini, P. A., Stewart, G. R., et al. (1997). Reversible Schwann cell hyperplasia and sprouting of sensory and sympathetic neurites after intraventricular administration of nerve growth factor. Ann. Neurol. 41, 82–93. doi: 10.1002/ana.410410114
Wiseman, F. K., Al-Janabi, T., Hardy, J., Karmiloff-Smith, A., Nizetic, D., Tybulewicz, V. L., et al. (2015). A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 16, 564–574. doi: 10.1038/nrn3983
Wu, C., Cui, B., He, L., Chen, L., and Mobley, W. C. (2009). The coming of age of axonal neurotrophin signaling endosomes. J. Proteomics 72, 46–55. doi: 10.1016/j.jprot.2008.10.007
Wu, C., Lai, C. F., and Mobley, W. C. (2001). Nerve growth factor activates persistent Rap1 signaling in endosomes. J. Neurosci. 21, 5406–5416. doi: 10.1523/jneurosci.21-15-05406.2001
Wu, H., Williams, J., and Nathans, J. (2014). Complete morphologies of basal forebrain cholinergic neurons in the mouse. eLife 3:e02444.
Xu, M., Shibayama, H., Kobayashi, H., Yamada, K., Ishihara, R., Zhao, P., et al. (1992). Granulovacuolar degeneration in the hippocampal cortex of aging and demented patients–a quantitative study. Acta Neuropathol. 85, 1–9. doi: 10.1007/bf00304627
Xu, W., Weissmiller, A. M., White, J. A. II, Fang, F., Wang, X., Wu, Y., et al. (2016). Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Invest. 126, 1815–1833. doi: 10.1172/jci82409
Yaar, M., Zhai, S., Fine, R. E., Eisenhauer, P. B., Arble, B. L., Stewart, K. B., et al. (2002). Amyloid beta binds trimers as well as monomers of the 75-kDa neurotrophin receptor and activates receptor signaling. J. Biol. Chem. 277, 7720–7725. doi: 10.1074/jbc.m110929200
Yaar, M., Zhai, S., Pilch, P. F., Doyle, S. M., Eisenhauer, P. B., Fine, R. E., et al. (1997). Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer’s disease. J. Clin. Invest. 100, 2333–2340. doi: 10.1172/jci119772
Ye, M., Lehigh, K. M., and Ginty, D. D. (2018). Multivesicular bodies mediate long-range retrograde NGF-TrkA signaling. eLife 7:e33012.
Yeo, T. T., Chua-Couzens, J., Butcher, L. L., Bredesen, D. E., Cooper, J. D., Valletta, J. S., et al. (1997). Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J. Neurosci. 17, 7594–7605. doi: 10.1523/jneurosci.17-20-07594.1997
Zerial, M., and Mcbride, H. (2001). Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117. doi: 10.1038/35052055
Zheng, W. H., Bastianetto, S., Mennicken, F., Ma, W., and Kar, S. (2002). Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 115, 201–211. doi: 10.1016/s0306-4522(02)00404-9
Zimmer, H. G. (2006). Otto Loewi and the chemical transmission of vagus stimulation in the heart. Clin. Cardiol. 29, 135–136. doi: 10.1002/clc.4960290313
Keywords: Alzheimer disease, Down syndrome, BFCNs, Aβ, NGF, signaling endosome, Rab5
Citation: Chen X-Q and Mobley WC (2019) Exploring the Pathogenesis of Alzheimer Disease in Basal Forebrain Cholinergic Neurons: Converging Insights From Alternative Hypotheses. Front. Neurosci. 13:446. doi: 10.3389/fnins.2019.00446
Received: 30 January 2019; Accepted: 18 April 2019;
Published: 07 May 2019.
Edited by:
A. Claudio Cuello, McGill University, CanadaReviewed by:
Filippo Caraci, Università degli Studi di Catania, ItalyLotta Granholm, University of Denver, United States
Copyright © 2019 Chen and Mobley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu-Qiao Chen, cTBjaGVuQHVjc2QuZWR1 William C. Mobley, d21vYmxleUB1Y3NkLmVkdQ==