 Joana S. Cristóvão
Joana S. Cristóvão Cláudio M. Gomes
Cláudio M. Gomes- 1Biosystems and Integrative Sciences Institute, Faculdade de Ciências, Universidade de Lisboa, Lisbon, Portugal
- 2Departamento de Química e Bioquímica, Universidade de Lisboa, Lisbon, Portugal
S100 proteins are calcium-binding proteins that regulate several processes associated with Alzheimer’s disease (AD) but whose contribution and direct involvement in disease pathophysiology remains to be fully established. Due to neuroinflammation in AD patients, the levels of several S100 proteins are increased in the brain and some S100s play roles related to the processing of the amyloid precursor protein, regulation of amyloid beta peptide (Aβ) levels and Tau phosphorylation. S100 proteins are found associated with protein inclusions, either within plaques or as isolated S100-positive puncta, which suggests an active role in the formation of amyloid aggregates. Indeed, interactions between S100 proteins and aggregating Aβ indicate regulatory roles over the aggregation process, which may either delay or aggravate aggregation, depending on disease stage and relative S100 and Aβ levels. Additionally, S100s are also known to influence AD-related signaling pathways and levels of other cytokines. Recent evidence also suggests that metal-ligation by S100 proteins influences trace metal homeostasis in the brain, particularly of zinc, which is also a major deregulated process in AD. Altogether, this evidence strongly suggests a role of S100 proteins as key players in several AD-linked physiopathological processes, which we discuss in this review.
Pathological Features of Alzheimer’s Disease
Alzheimer’s disease (AD) is a chronic and progressive neurodegenerative disorder that affects wide areas of the cerebral cortex and hippocampus. Most AD patients (>95%) are idiopathic and disease is characterized by late onset (80–90 years of age) with failure in the clearance of amyloid-β peptide (Aβ) from the brain (Masters et al., 2015). The main symptoms of the disease are progressive memory deficits, cognitive impairment, and personality changes. The neuropathological and neurochemical hallmarks of AD include selective neuronal death, synaptic loss and the presence of proteinaceous deposits in the extracellular space (known as diffuse and neuritic plaques) as well as inside neurons [known as neurofibrillary tangles (NFTs)]. Neuroinflammation, oxidative stress, and calcium dysregulation are also important features implicated in AD pathology (Wang X. et al., 2014).
Diffuse and neuritic plaques, most commonly known as amyloid plaques, are mainly constituted by Aβ deposits, surrounded by degenerative presynaptic endings, astrocytes and microglial cells (Weiner and Frenkel, 2006). Aβ peptides are formed from proteolytic cleavage of the amyloid precursor protein (APP) by the γ- and β-secretases (BACE1). Even though the normal function of APP is not known, it is possibly related to regulation of neurite outgrowth, cell adhesion, and neuron migration (Turner et al., 2003). APP processing can involve non-amyloidogenic or amyloidogenic pathways. When APP is cleaved by α-secretase and subsequently by γ-secretase, sAPPα is predominantly formed, which has an important role in neuronal plasticity and survival. However, in the amyloidogenic pathway, APP is cleaved by β- and γ-secretase producing sAPPβ, C-terminal fragments and Aβ peptides, which promote a range of detrimental effects in neurons and in the brain (Ling et al., 2003). Aβ40 and Aβ42 are the predominant accumulating peptides, whose aggregation into fibrillar cross-β structures is a central feature in AD pathogenesis. Aβ aggregation is naturally heterogeneous and monomers assemble and polymerize into structurally distinct forms, including protofibers, polymorphic oligomers and amyloid fibrils, all found within plaques. Extracellular accumulation of Aβ fibrils is not necessarily intrinsically cytotoxic and emerging evidence suggests precursor oligomers as the key toxic agents, also because of their seeding potential (Dahlgren et al., 2002; Walsh et al., 2002). Moreover, Aβ peptides can also be deposited intracellularly (Gouras et al., 2005).
The presence of neurofibrillar tangles, formed by neuronal intracellular deposition of hyperphosphorylated tau protein, is also a major AD hallmark. It has also been suggested that NFT may not be the major player in neurotoxicity, and that tau oligomers are in fact the major toxic forms promoting synaptic impairment (Tai et al., 2014; Fa et al., 2016). In agreement, it has been described that propagation of tau pathology occurs trans-synaptically (Liu et al., 2012). Other factors that contribute to tauophaties, besides tau hyperphosphorylation, are tau truncation, glycosylation, glycation, nitration, and ubiquitination (Chong et al., 2018).
Additionally, metal ion homeostasis and calcium signaling are also implicated in disease pathogenesis. In the early stages of AD, calcium imbalance promotes Aβ formation and tau hyperphosphorylation, as reviewed previously in LaFerla (2002). Aβ destabilizes neuronal calcium homeostasis generally leading to an increase in cytosolic calcium levels and formation of calcium-permeable pores. Calcium alterations lead to cytoskeletal modifications, triggering neuronal apoptosis and formation of free radicals through mitochondrial dysfunction. Moreover, familial AD mutations in presenilins are linked to altered synaptic Ca2+ signaling that imbalance the activities of Ca2+-calmodulin-dependent kinase II (CaMKII) and Ca2+-dependent phosphatase calcineurin (CaN), increasing the long-term depression and causing memory loss (Popugaeva et al., 2017). Transition metals such as Zn2+, Cu2+, and Fe2+ have well-established roles as chemical modulators of protein folding, amyloid aggregation and toxicity and are found to accumulate at protein deposits (Leal et al., 2012; Barnham and Bush, 2014; Cristovao et al., 2016).
Neuroinflammation is another cellular process linked to AD pathogenesis. Senile plaques are often closely associated with activated microglial cells and surrounded by activated astrocytes that have abundant filaments (Weiner and Frenkel, 2006). In response to Aβ deposition, activated microglia upregulate the expression of cell-surface proteins and cytokines such as the tumor-necrosis factor (TNF), interleukine-6 (IL-6), interleukine-1 (IL-1), S100 proteins, and chemokines. The presence of Aβ activates different cell receptors and intracellular signaling pathways, mainly those related to the receptor of advanced glycation end products (RAGE)/nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, that is responsible for the transcription of pro-inflammatory cytokines and chemokines in astrocytes (Gonzalez-Reyes et al., 2017). Additionally, astrogliosis appears as an early manifestation of AD. The migration of astrocytes into Aβ plaques is promoted by chemokines CCL2 and CCL3, that are released by activated microglial cells surrounding amyloid plaques. Astrocytes recruited to Aβ plaques have the potential to mediate both neurotoxicity and participate in the clearance of Aβ (Weiner and Frenkel, 2006). S100 proteins are among the alarmins that are upregulated and are highly secreted by astrocytes during this process (Venegas and Heneka, 2017), which results in their accumulation within Aβ deposits and brain tissues, as overviewed in the following sections.
The S100 Protein Family
S100 proteins are a family of low-molecular weight EF-hand Ca2+ proteins that are expressed in distinct organs and tissues. They are involved in multiple intracellular functions, including cell proliferation, differentiation, protein phosphorylation, cytoskeletal assembly, and disassembly and intracellular calcium homeostasis (Mrak and Griffinbc, 2001; Donato et al., 2009, 2013). In pathological conditions S100 proteins can be expressed in a cell type where they are not expressed under normal conditions. Additionally, some S100 proteins are secreted and regulate cell functions in an autocrine or paracrine manner by activation of surface receptors, such as the RAGE receptor, thereby promoting NF-κB signaling, an important trigger of inflammatory processes, recruiting and activating cellular pro-inflammatory effectors (Hofmann et al., 1999; Leclerc et al., 2009). Albeit S100 proteins are not cytokines in stricto sensu, in these cases, they have such functions, and act as extracellular alarmins or as damage-associated molecular pattern (DAMP) factors, that can either be beneficial or detrimental depending on concentration and molecular and cellular moiety (Donato et al., 2009, 2013). From the 25 S100 proteins described so far, several are present in the brain and from those, seven have been implicated in AD pathways: S100B, S100A1, S100A6, S100A7, S100A8, S100A9, and S100A12.
S100 proteins occur mainly as homodimers (Barger et al., 1992; Giannakopoulos et al., 1996; Matsui Lee et al., 2000; Cunden et al., 2017). Specifically, it is known that calcium binding to S100 proteins triggers conformational changes that expose a hydrophobic cleft that is crucial to interaction with partners to their activation, regulation and signaling functions. Several S100 proteins also bind zinc and copper, which interestingly are highly abundant in senile plaques (Heizmann et al., 2002; Senior et al., 2003; Maynard et al., 2005). A few S100 proteins are also found as heterodimers, including S100A8/A9 (Teigelkamp et al., 1991), S100B/A1 (Garbuglia et al., 1999) and S100A6/B (Yang et al., 1999). S100 proteins interconvert into functional oligomers, including tetramers, hexamers, and octamers and formation of these species can be promoted by Ca2+ or Zn2+ binding (Botelho et al., 2012). The functions described for these S100 oligomers involve a tighter interaction with RAGE, assistance in microtubule formation, neurite outgrowth, and tumor suppression.
S100 proteins influence cognitive processes in the healthy brain and play roles in development and neuronal maintenance. Depending on the study, antisera to neurospecific S100 protein (Gromov et al., 1992; O’Dowd et al., 1997; Epstein et al., 2006) and antibodies directed against S100A1 and S100B (Gromov et al., 1992; O’Dowd et al., 1997; Epstein et al., 2006) either impair or improve learning and memory functions in rat brains (Gromov et al., 1992; O’Dowd et al., 1997; Epstein et al., 2006). Recent studies suggested that elevated S100B levels have deleterious effects during the neurodevelopmental period through RAGE-dependent processes (Santos et al., 2018). Additionally, S100B has been associated with Down Syndrome, a genetic variation where the most profound neurological features are mental retardation, seizures and early onset AD. Levels of S100B are increased in neuronal progenitor cells of patients with Down Syndrome (Esposito et al., 2008a) and in human induced pluripotent stem cells derived from Down Syndrome patients (Chen et al., 2014). S100B overexpression causes toxicity to neurons, reduces neurogenesis, and increases the production of reactive oxygen species (Esposito et al., 2008a; Lu et al., 2011; Chen et al., 2014).
There are several studies using clinical specimens and animal models implicating S100 proteins in AD pathophysiology (Marshak et al., 1992; Akiyama et al., 1994; Sheng et al., 1994; Gerlai et al., 1995; Mrak et al., 1996; Boom et al., 2004; Shepherd et al., 2006; Chaves et al., 2010; Ha et al., 2010; Mori et al., 2010; Roltsch et al., 2010; Chang et al., 2012; Afanador et al., 2014; Cirillo et al., 2015; Horvath et al., 2016; Gruden et al., 2017; Lodeiro et al., 2017; Iashchishyn et al., 2018; Wang et al., 2018). As overviewed in the following sections, different S100 proteins seem to be involved in several processes related to APP processing, influencing Aβ levels, tau post-translational modifications, formation of protein inclusions, and multiple signaling pathways. The ability of acting as Ca2+ sensors, regulating the activity of channels/pumps involved in Ca2+ release/uptake also provide feedback and feedforward mechanisms for sustaining aberrant Ca2+ signaling in AD. Therefore, involvement in all these processes makes a strong case for the importance of the S100 protein family in AD development (Table 1).
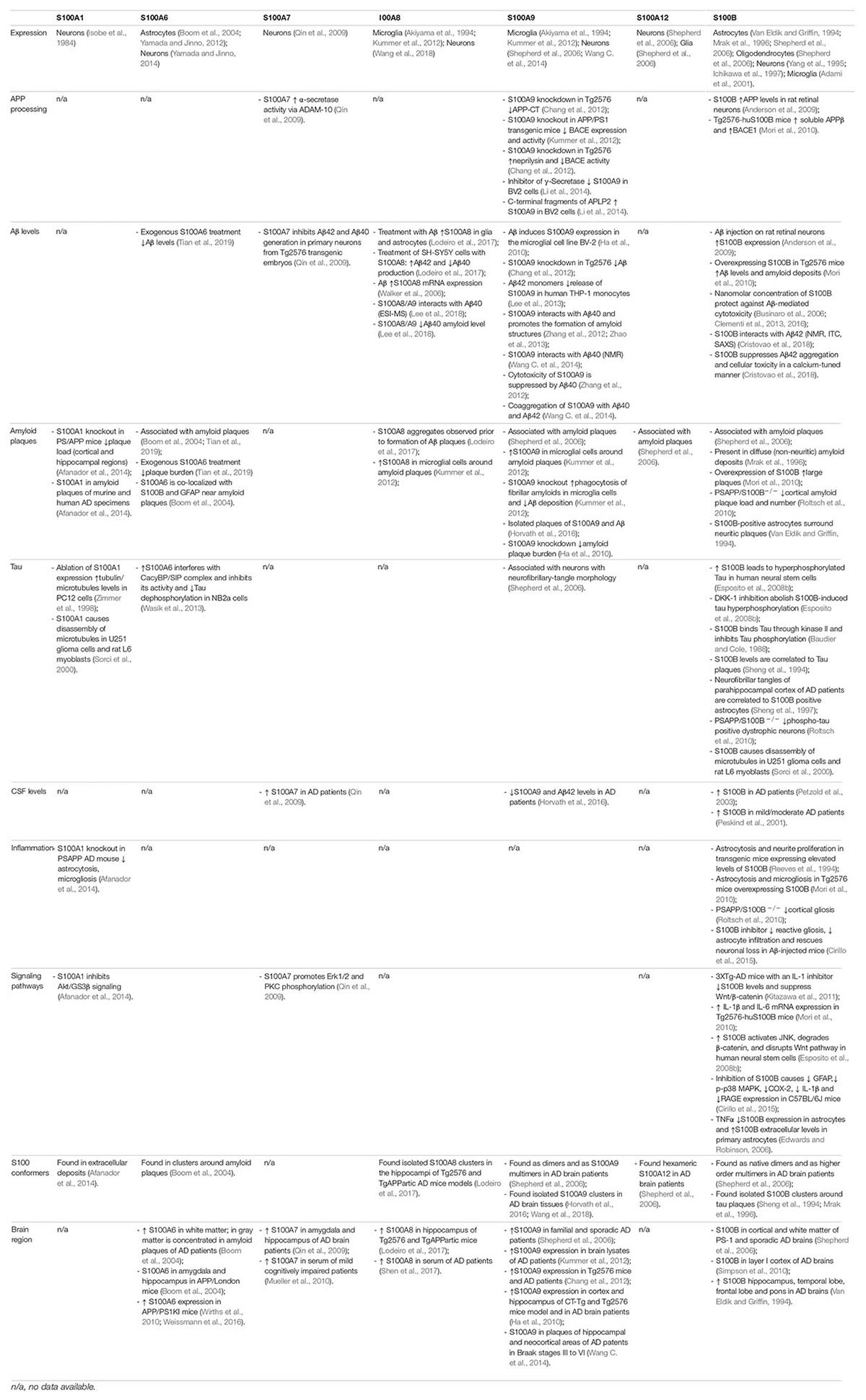
Table 1. Distribution, levels, and implication of brain S100 proteins in AD pathways.
S100B
S100B is the most studied S100 protein in the scope of AD, as reviewed in Steiner et al. (2011) and Yardan et al. (2011). S100B acts as a pro-inflammatory cytokine and a DAMP molecule depending on its concentration. S100B secreted from astrocytes can have trophic and toxic effect on neurons. At nanomolar concentration, S100B displays neurotrophic effects, leading to promotion of neuronal survival and neurite outgrowth. At micromolar concentrations S100B has deleterious effects inducing neuronal apoptosis (Mrak and Griffinbc, 2001). Upregulation of S100B leads to behavioral abnormalities and loss of dendritic density in mice (Gerlai et al., 1995). S100B also regulates the intracellular levels of free calcium in several central nervous system cell types, such as neurons and astrocytes. Recently we demonstrated that S100B acts as a sensor and regulator of elevated zinc levels in the brain and that this metal-buffering activity is tied to a neuroprotective role, through an indirect effect on calcium levels and in inhibition of excitotoxicity (Hagmeyer et al., 2017).
Several studies point to high levels of S100B in AD patients (Marshak et al., 1992; Van Eldik and Griffin, 1994; Peskind et al., 2001; Petzold et al., 2003; Chaves et al., 2010) and in AD mouse models (Yeh et al., 2015). The largest increase in S100B levels is observed in the hippocampus, temporal lobe (Van Eldik and Griffin, 1994) and in the layer I of the cortex (Simpson et al., 2010). It is demonstrated that sera of AD patients with moderate and severe dementia have a 60- and 37-fold increase in S100B, respectively. In AD patients with moderate dementia, the increase of S100B levels is followed by a 10-fold increase in auto-antibodies: however, in AD patients with severe dementia the levels of auto-antibodies remain identical to controls (Gruden et al., 2007), indicating that there is no immune-protection against elevated S100B levels in AD patients with severe dementia. S100B is also elevated in the cerebrospinal fluid (CSF) of AD patients (Peskind et al., 2001; Petzold et al., 2003). S100B is involved in APP cleavage processes: high levels of S100B increase BACE1 activity resulting in higher levels of toxic APPβ and C-terminal fragments, including the amyloidogenic β-CTF (C99) (Anderson et al., 2009; Mori et al., 2010).
S100B surrounding amyloid plaques is mostly produced by astrocytes (Van Eldik and Griffin, 1994; Mrak et al., 1996; Shepherd et al., 2006; Roltsch et al., 2010), but can also originate from oligodendrocytes (Simpson et al., 2010) and microglia (Roltsch et al., 2010). It was also observed that S100B positive astrocytes are present in the diffuse non-neuritic amyloid plaques (Mrak et al., 1996), suggesting an early, yet unclear, action of S100B in the formation of senile plaques. Evidence suggests that S100B may regulate plaque formation as the knockout of S100B in the PS/APP AD mouse model selectively decreases plaque load in the cortical region (Roltsch et al., 2010) and the overexpression of S100B increases Aβ levels and deposits, at early stages (Mori et al., 2010). Even though, it is established that elevated levels of S100B have deleterious effects that promote AD features, nanomolar concentrations of S100B effectively protect cells against Aβ-mediated cytotoxicity (Businaro et al., 2006; Clementi et al., 2013, 2016). Additionally, we have recently found that, in vitro, S100B binds to Aβ42 monomers, oligomers, and fibrils resulting in a calcium-tuned suppression of Aβ42 aggregation and cellular toxicity in SH-SY5Y cells (Cristovao et al., 2018). S100B was found both in normal and in AD brains in various oligomeric states (Shepherd et al., 2006); however, the protective or pathological functions of S100B oligomers are still unclear.
Overexpression of S100B in Tg2576 AD transgenic mice is also linked with neuroinflammation, promoting astrogliosis, microgliosis, and neurite proliferation (Reeves et al., 1994; Mori et al., 2010). However, knockout of S100B in PS/APP mouse model decreases cortical gliosis (Roltsch et al., 2010). IL-1 regulates the expression and secretion of S100B from astrocytes (de Souza et al., 2009). Treatment of the 3XTg-AD mice with an antibody against IL-1 reduces S100B levels and results in attenuation of tau pathology and in partial reduction of certain fibrillar and oligomeric forms of Aβ (Kitazawa et al., 2011) Therefore, S100B seems to be tied to different processes related to AD pathology as in addition to its ability to promote brain inflammatory response and tau pathology (Esposito et al., 2008b) it may play roles in directly promoting amyloidogenic APP processing, as proposed by Mori et al. (2010).
Inhibition of S100B using pentamidine in AD mouse models, lead to a reduction in the levels of proinflammatory mediators such as nitrite, MDA, PGE2 and IL-1, followed by an inhibition of Aβ-induced gliosis (Cirillo et al., 2015). Indeed, S100B-overexpressing mice that were infused with oligomeric Aβ exhibited enhanced glial activation. Neuroinflammation and loss of synaptic markers were noted, however there was no difference in the amyloid plaque burden, in comparison to controls (Craft et al., 2005). These results suggest a relationship between S100B and other cytokines that are also implicated in AD pathways. Indeed, the TNF-α cytokine, which is present at high levels in the AD brain, decreases both GFAP and S100B intracellular levels in astrocytes, while increasing their extracellular levels (Edwards and Robinson, 2006). This crosstalk suggests a relationship between TNF-α and the increase of these two proteins in CSF and sera of AD patients. Other reports showed that S100B enhances IL-6 mRNA (Mori et al., 2010; Yeh et al., 2015) and IL-1 mRNA levels in microglia and in neurons (Mori et al., 2010), via Sp1 and NF-κB signaling pathways (Liu et al., 2005). Additionally, knockout of S100B in the Tg2576 AD mouse model background results in a decrease in GFAP-positive astrocytes and in Iba-1 positive microglia (Roltsch et al., 2010), while its overexpression has opposite effects (Mori et al., 2010). Overall, these results suggest that S100B can influence and be influenced by the levels of other cytokines involved in AD pathogenesis.
Regarding tau pathology, high S100B levels in AD patients positively correlate to tau tangles with which S100B was found to be clustered (Sheng et al., 1994, 1997). Knockout of S100B in the PS/APP mouse model decreases phosphorylated-tau positive dystrophic neurons (Roltsch et al., 2010) and in mouse models expressing tau, S100B levels are upregulated (Sidoryk-Wegrzynowicz et al., 2017). Indeed, it has been demonstrated, in vitro, that S100B directly binds tau inhibiting its phosphorylation by yet unclear non-covalent interactions in a process that is Ca2+ or Zn2+-dependent (Baudier and Cole, 1988). However, contradictory results show that S100B promotes tau hyperphosphorylation by inducing GSK-3β activation and disrupting Wnt signaling (Esposito et al., 2008b), an important pathway to regulate synaptic transmission and plasticity. Indeed, S100B promotes the expression of the Dickkopf-related protein 1 (Dkk1), an antagonist of Wnt signaling that has previously been suggested to play a role in AD (Guo et al., 2016).
As previously mentioned, calcium dysregulation contributes to AD pathology and S100B is a key factor in the Ca2+ homeostasis of astrocytes. It was demonstrated that S100B knockout leads to a decrease of induced-Ca2+ transients (Xiong et al., 2000) such as those induced by Aβ. In what could be a potentially protective mechanism, S100B levels were found to be up-regulated in astrocytes upon Aβ induced Ca2+ intracellular waves (Chow et al., 2010).
S100A1
The investigation of the role of S100A1 in AD is encouraged by the fact that some of its targets are altered in the disease, such as the ryanodine receptor (RyR), an intracellular calcium release channel, tau and RAGE. S100A1 is primarily expressed in neurons and, as reviewed in Zimmer et al. (2005) is implicated in tau phosphorylation, neuronal cell sensitivity to Aβ and in the regulation of APP expression. In respect to the latter, available data indicates that βAPP steady-state mRNA and intracellular protein levels are down-regulated in response to ablation of S100A1 expression (Zimmer et al., 2005). In the PS/APP mouse model, knockout of S100A1 decreases inflammatory processes, such as astrocytosis and microgliosis, diminishing 3.7-fold the number of cortical plaques and 1.5-fold the number of hippocampal plaques (Afanador et al., 2014). Decreased S100A1 levels in PC12 cells increase tubulin levels and the number of neurites (Zimmer et al., 1998). Additionally, knockout of S100A1 in PC12 cells increases the resistance to Aβ-induced cell death (Zimmer et al., 2005).
S100A1 induces Glycogen synthase kinase 3 (GSK3) phosphorylation (Afanador et al., 2014), that is involved in several processes such as glycogen metabolism and gene transcription. GSK3 over-activation is also related to memory impairment and other AD related features (Hooper et al., 2008). In human and mouse AD brain tissue, S100A1:RyR complexes are present and their formation is Ca2+-dependent. RyR is a receptor with altered levels in AD that is associated with APP processing and Aβ production, however it is not known if it exerts a protective or pathogenic role in AD (Del Prete et al., 2014). S100A1 also binds to stress-inducible phosphoprotein 1 (STIP1) (Maciejewski et al., 2017), a Hsp90 cochaperone that is reported to be present in the vicinity of Aβ oligomers, preventing Aβ-induced synaptic loss and neuronal death in primary neurons (Ostapchenko et al., 2013). S100A1 and S100B also have the ability to cause microtubule disassembly in glioma cells and myoblasts in a Ca2+-dependent manner, suggesting a possible role of S100A1 in tau pathology (Sorci et al., 2000). Moreover, in human AD patients and in the PS/APP mouse model, extracellular S100A1 has been observed in plaque-like deposits (Afanador et al., 2014).
S100A6
S100A6 was identified in the AD gene signature as one of the most significantly positively correlated proteins with AD phenotype (Wruck et al., 2016). As other S100 proteins, S100A6 is upregulated in AD patients and in AD mouse models (Boom et al., 2004; Wirths et al., 2010; Weissmann et al., 2016) and is found in astrocyte-positive clusters that surround Aβ amyloid deposits in the brain’s gray matter (Boom et al., 2004). In PS/APP mouse brains, S100A6 localizes in the peripheral region of amyloid plaques and exogenous S100A6 treatment in mouse brain sections reduces Aβ levels and plaque burden (Tian et al., 2019).
Zinc ions are colocalized with senile plaques in AD patients and there is evidence that AD related-cognitive decline is dependent on extracellular zinc levels (Lovell et al., 1998; Takeda and Tamano, 2016). In particular, one study suggested that zinc-binding S100A6 exerts a zinc sequestering function, identical to what has been proposed for S100B (Hagmeyer et al., 2017), thus preventing zinc-induced toxicity in COS-7 cells (Tian et al., 2019). Additionally, PS/APP mice treated with a high-zinc diet have increased S100A6 levels and Aβ deposits. These studies point to a correlation between S100A6, zinc ions and decrease in Aβ plaque load.
The heterodimer S100A6/B is also implicated in pathological signal transduction in melanoma (Yang et al., 1999). It is possible that the formation of the heterodimer also occurs in AD since S100A6 is colocalized with S100B and astrocytic glial fibrillary acidic protein (GFAP), a marker of astrogliosis, near amyloid plaques (Boom et al., 2004). Additionally, it is reported that S100A6 binds to the CacyBP/SIP complex, a complex known to participate in the organization of microtubules. Overexpression of S100A6 in neuroblastoma NB2a cells inhibits CacyBP/SIP complex activity, and consequently lowers the rate of tau dephosphorylation (Wasik et al., 2013).
S100A7
There is scarce evidence regarding the role of S100A7 on AD pathways. It is reported that S100A7 is increased in mildly cognitively impaired patients (Mueller et al., 2010) and in the brain and CSF of AD dementia patients (Qin et al., 2009). S100A7 mRNA expression is regulated in the brain as a function of AD dementia and amyloid neuropathology (Qin et al., 2009). Exogenous S100A7 in primary hippocampal neurons of Tg2576 AD transgenic embryos inhibits the generation of Aβ42 and Aβ40 peptides and promotes the activity of “non- amyloidogenic” α-secretase, via upregulation of ADAM-10 (a disintegrin and metalloproteinase) and phosphorylation of Erk1/2 and PKC (Qin et al., 2009). Therefore, a beneficial role of S100A7 on APP processing is suggested, albeit other studies are required to more extensively support this possibility.
S100A8
S100A8 was found to be upregulated in the sera of AD patients (Shen et al., 2017) and in the hippocampus of Tg2576 and TgAPParc AD mice (Lodeiro et al., 2017). Indeed, several studies establish a correlation between Aβ and S100A8 expression. An increase in S100A8 mRNA levels was induced when aggregated Aβ was added to a microglia culture isolated from post-mortem AD brain tissues. Subsequent culture growth suggested that chronic secretion of S100A8 can lead to chronic activation of microglia (Walker et al., 2006). In rat primary astrocytes, Aβ42 treatment induces a significant increase in S100A8 mRNA levels. Treatment of SH-SY5Y neuroblastoma cells with recombinant S100A8 increased Aβ42 and decreased Aβ40 production (Lodeiro et al., 2017). In PS/APP mice, the S100A8/A9 heterodimer is found to be upregulated in microglial cells surrounding amyloid plaques (Kummer et al., 2012). Additionally, it is reported that S100A8/A9 binds directly to Aβ40 and that it interferes with amyloid formation but no effect was observed over Aβ42 aggregation (Lee et al., 2018). There is also a link between S100A8/A9 and the “non-amyloidogenic” α-secretase ADAM-10, since S100A8/A9 has lower expression in AD mice models overexpressing ADAM-10 (Prinzen et al., 2009).
S100A8 assemblies were found in the hippocampus of Tg2576 and TgAPParc AD mice brains, distinct from corpora amylacea, that are formed independently of Aβ plaques (Lodeiro et al., 2017). These S100A8 aggregates are likely not amyloidogenic as no staining was observed with thioflavin-S (Lodeiro et al., 2017).
S100A9
S100A9 was found to be strongly increased in brain lysates of AD patients and AD mice compared to healthy, age-matched controls (Ha et al., 2010; Chang et al., 2012; Kummer et al., 2012) and in familial PS-1 AD tissues (Shepherd et al., 2006). S100A9 is present in activated glia and neurons positive for tau neurofibrillary tangles (Shepherd et al., 2006). Additionally, there is a strong correlation between S100A9 and Aβ. In in vitro cell assays, Aβ42 reduces extracellular release of S100A9 in human THP-1 monocytes (Lee et al., 2013) and induces S100A9 expression in microglia BV2 cells (Ha et al., 2010). However, in CSF from AD patients with mild cognitive impairment and vascular dementia, the levels of S100A9 and Aβ42 are decreased (Horvath et al., 2016). Knockdown of S100A9 decreases cognition decline on Tg2576 mice and reduces amyloid plaque burden (Ha et al., 2010; Chang et al., 2012). S100A9 was found within amyloid plaques of sporadic and familial PS-1 AD brains (Shepherd et al., 2006; Wang et al., 2018) with distinct Braak stages from III to VI (Kummer et al., 2012; Wang C. et al., 2014). Indeed, in some studies it was possible to observe Aβ42 plaques and also isolated S100A9 plaques that are not colocalized, forming separate tissue deposits (Horvath et al., 2016; Wang et al., 2018).
Regarding the formation of S100A9 puncta in AD brain, a recent study reported that, in vitro, S100A9 is able to form polymeric structures that resemble amyloid structures and bind amyloid fluorophores. The polymerization reaction occurs through a two-step nucleation with initial misfolding of S100A9 and β-sheet formation (Iashchishyn et al., 2017). The impact of S100A9 oligomers on memory was studied through intranasal administration of S100A9 (dimers, oligomers, and fibrillar states) in aged mice. S100A9 oligomers and fibrils, but not dimers, evoked amnestic activity which is correlated with disruption of dopaminergic and glutamate neurochemistry in the prefrontal cortical and hippocampal regions (Gruden et al., 2016, 2018). Additionally, intranasal administration of S100A9 induces cellular stress in the frontal lobe, hippocampus, and cerebellum of aged mice, as well as impaired learning. However, co-treatment with S100A9 fibrillar species and glutamate antibodies increases locomotor activity (Gruden et al., 2017; Iashchishyn et al., 2018). In Tg2576 AD mice, knockdown or knockout of S100A9 significantly reduced the neuropathology and greatly improved learning and memory (Chang et al., 2012), suggesting a link between S100A9 and AD pathology. Indeed, knockout of S100A9 in the PS/APP AD mouse model led to increased phagocytosis of fibrillar Aβ and to decreased Aβ deposition (Kummer et al., 2012).
However, several studies focused on the relationship between S100A9 and Aβ42. In vitro biophysical approaches showed that S100A9 binds to Aβ40 through hydrophobic interactions (Zhang et al., 2012; Zhao et al., 2013; Wang C. et al., 2014). Kinetic assays suggested that S100A9 co-aggregates with Aβ40, promoting the formation of amyloid fibrils. The co-aggregation of S100A9 with Aβ42 was also referred to inhibit Aβ42 cytotoxicity (Wang C. et al., 2014).
Regarding APP processing, it was found that C-terminal fragments of amyloid precursor-like protein 2 (APLP2) upregulate S100A9 protein and mRNA expression in BV2 cell and that inhibitor of γ-secretase promotes downregulation of S100A9 protein levels (Li et al., 2014). AD mice deficient in S100A9 have decreased levels of key cytokines involved in APP processing and a reduction of BACE1 expression and activity (Kummer et al., 2012). S100A9 knockout also reduced overall levels of Aβ and APP C-terminal fragments in Tg2576 AD mice, due to increase in neprilysin levels and decreased BACE activity (Chang et al., 2012). Knockdown of S100A9 significantly attenuated the increase of Ca2+ levels provoked by C-terminals of APP or by Aβ treatment (Ha et al., 2010); however, others observed that a reduction of S100A9 extracellular release is followed by an increase in intracellular Ca2+ levels (Lee et al., 2013), evidencing a correlation between S100A9 and calcium dysregulation in AD. In the AD brain and in mouse models, S100A9 is present in its native form but also as large complexes ranging from 90 to 190 kDa (Shepherd et al., 2006). Indeed, after intranasal administration of S100A9 fibrils in aged mice, S100A9 plaques were observed in the brain which resulted in an exacerbation of cell stress (Iashchishyn et al., 2018). Overall there are solid evidences regarding S100A9 as a potential regulator of AD pathways.
S100A12
S100A12 is the least studied S100 protein in the context of AD. To date, a single study reported S100A12 hexamers associated with senile plaques, reactive glia and neurons in brains of sporadic and PS-1 AD patients (Shepherd et al., 2006).
Outlook
Considering the involvement of S100 proteins in multiple regulatory functions in the brain, the fact that they have age- and damage- related expression, and a direct involvement in neuroinflammation, it is not surprising that they are implicated in molecular processes associated with AD pathogenesis (Figure 1).
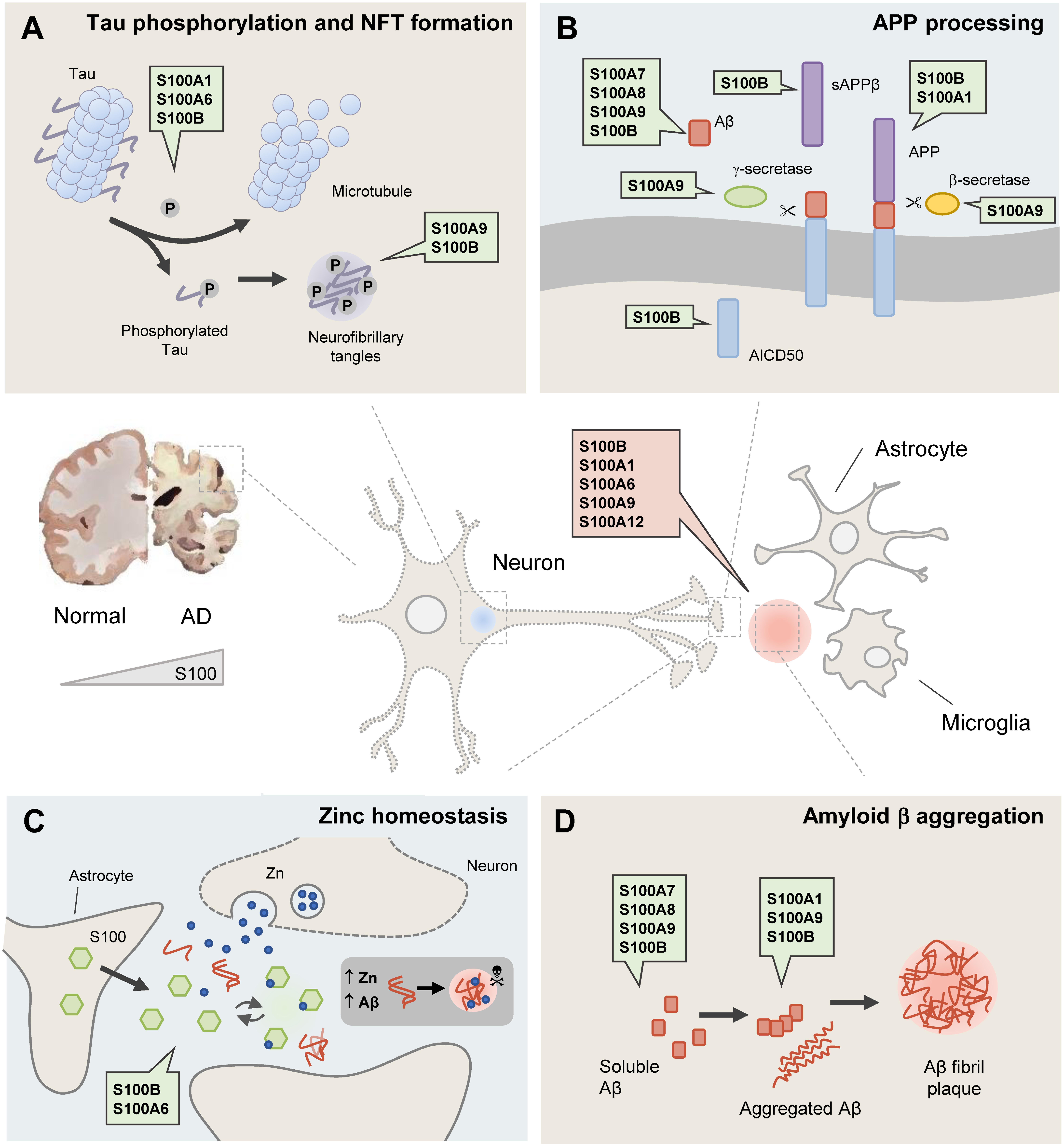
Figure 1. S100 proteins are involved in the main processes associated with Alzheimer’s disease (AD). In the AD brain, affected neurons (central panel) are damaged due to the formation of intracellular neurofibrillary tangles (represented by the blue dot) and extracellular amyloid species including an ensemble of low molecular weight aggregates, protofibrils and fibrils (represented by the red dot). As a result of astrocyte and microglia over-activation, some S100 proteins become upregulated, being implicated in several molecular processes altered in AD (A–D). (A) Tau phosphorylation and NFT formation. S100A1, S100A6 and S100B are involved in the disassembly of microtubules and Tau release, while S100A9 and S100B are found within neurofibrillary tangles. (B) APP processing. Several S100 proteins are implicated in APP cleavage and its amyloidogenic processing. S100A9 regulates γ- and β-secretase expression and activity and S100B and S100A1 regulate APP levels. Moreover, S100A7, S100A8, S100A9 and S100B influence Aβ levels (Table 1). (C) Zinc homeostasis. Due to their zinc-binding properties, S100B and S100A6 have zinc-buffering activities that are related to neuroprotective roles; and S100A6 reduces zinc levels and senile plaque load in PS/APP mouse brains. (D) Amyloid β aggregation. S100A1, S100A9, and S100B proteins can interact, modulate the aggregation and co-aggregate with the Aβ peptide. Several S100 proteins (S100B, S100A1, S100A6, S100A8, S100A9, and S100A12), are found within amyloid plaques and in astrocytes and/or microglia around amyloid deposits. Further details and references can be found in the text and in Table 1.
Most of the available studies report essentially deleterious effects of S100 proteins in AD pathological processes. Indeed, elevated levels of S100 proteins around amyloid plaques and neurofibrillary tangles, exacerbate neuroinflammation and interfere with APP processing and with several AD-related proteins and signaling pathways. This scenario is compatible with an aggravating role as part of downstream inflammatory processes in later stages of AD neurodegeneration. However, some protective roles of S100 proteins are emerging as equally important. Indeed, it is now proposed that early inflammatory responses start at very early stages of the AD neurodegenerative process, most likely before amyloid plaques are formed (Cuello, 2017). This process would involve an elevation of inflammatory molecules, including S100, at the earliest stages of amyloid pathology, even before the onset of the disease phenotypes. Interestingly, several recent studies suggest that low levels of S100B protect cells against Aβ42-mediated cytotoxicity and that low levels of S100A9 inhibit Aβ42 cytotoxicity. This has led to the recent proposal of a new chaperone-like function for S100B (Cristovao et al., 2018), which seems to be extensible to a metal buffering activity (Hagmeyer et al., 2017), both of which are certainly relevant in the context of AD. Therefore, also in AD, S100 proteins may exert different functions according to their (extra)cellular concentrations. At early inflammatory stages and relatively low concentrations they play protective roles, while later in pathology, at higher concentrations, they play essentially disease aggravating roles. This is in line with prior evidence that suggests that decreasing the levels of S100 proteins may be a strategy to mitigate AD progression. The development of S100 neutralizing antibodies and small molecules is already a current therapeutic approach in cancer, autoimmune diseases and chronic inflammatory disorders, as reviewed in Bresnick (2018), and could present a promising strategy for AD as well. Ongoing research in our laboratory will shed light into this subject in the near future.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by Fundação para a Ciência e a Tecnologia through grants UID/Multi/04046/2013 (to BioISI), PTDC/NEU-NMC/2138/2014 (to CG), IF/01046/2014 (to CG), Ph.D. fellowship SFRH/BD/101171/2014 (to JC) and by Bial Foundation through grant PT/FB/BL-2014-343 (to CG).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adami, C., Sorci, G., Blasi, E., Agneletti, A. L., Bistoni, F., and Donato, R. (2001). S100B expression in and effects on microglia. Glia 33, 131–142. doi: 10.1002/1098-1136(200102)33
Afanador, L., Roltsch, E. A., Holcomb, L., Campbell, K. S., Keeling, D. A., Zhang, Y., et al. (2014). The Ca2+ sensor S100A1 modulates neuroinflammation, histopathology and Akt activity in the PSAPP Alzheimer’s disease mouse model. Cell Calcium 56, 68–80. doi: 10.1016/j.ceca.2014.05.002
Akiyama, H., Ikeda, K., Katoh, M., Mcgeer, E. G., and Mcgeer, P. L. (1994). Expression of MRP14, 27E10, interferon-alpha and leukocyte common antigen by reactive microglia in postmortem human brain tissue. J. Neuroimmunol. 50, 195–201. doi: 10.1016/0165-5728(94)90046-9
Anderson, P. J., Watts, H. R., Jen, S., Gentleman, S. M., Moncaster, J. A., Walsh, D. T., et al. (2009). Differential effects of interleukin-1beta and S100B on amyloid precursor protein in rat retinal neurons. Clin. Ophthalmol. 3, 235–242.
Barger, S. W., Wolchok, S. R., and Van Eldik, L. J. (1992). Disulfide-linked S100 beta dimers and signal transduction. Biochim. Biophys. Acta 1160, 105–112. doi: 10.1016/0167-4838(92)90043-d
Barnham, K. J., and Bush, A. I. (2014). Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 43, 6727–6749. doi: 10.1039/c4cs00138a
Baudier, J., and Cole, R. D. (1988). Interactions between the microtubule-associated Pi-Proteins and S100b regulate Pi-Phosphorylation by the Ca-2+ calmodulin-dependent protein kinase-Ii. J. Biol. Chem. 263, 5876–5883.
Boom, A., Pochet, R., Authelet, M., Pradier, L., Borghgraef, P., Van Leuven, F., et al. (2004). Astrocytic calcium/zinc binding protein S100A6 over expression in Alzheimer’s disease and in PS1/APP transgenic mice models. Biochim. Biophys. Acta 1742, 161–168. doi: 10.1016/j.bbamcr.2004.09.011
Botelho, H. M., Fritz, G., and Gomes, C. M. (2012). Analysis of S100 oligomers and amyloids. Methods Mol. Biol. 849, 373–386. doi: 10.1007/978-1-61779-551-0_25
Bresnick, A. R. (2018). S100 proteins as therapeutic targets. Biophys. Rev 10, 1617–1629. doi: 10.1007/s12551-018-0471-y
Businaro, R., Leone, S., Fabrizi, C., Sorci, G., Donato, R., Lauro, G. M., et al. (2006). S100B protects LAN-5 neuroblastoma cells against Abeta amyloid-induced neurotoxicity via RAGE engagement at low doses but increases Abeta amyloid neurotoxicity at high doses. J. Neurosci. Res. 83, 897–906. doi: 10.1002/jnr.20785
Chang, K. A., Kim, H. J., and Suh, Y. H. (2012). The role of S100a9 in the pathogenesis of Alzheimer’s disease: the therapeutic effects of S100a9 knockdown or knockout. Neurodegener. Dis. 10, 27–29. doi: 10.1159/000333781
Chaves, M. L., Camozzato, A. L., Ferreira, E. D., Piazenski, I., Kochhann, R., Dall’igna, O., et al. (2010). Serum levels of S100B and NSE proteins in Alzheimer’s disease patients. J. Neuroinflam. 7:6. doi: 10.1186/1742-2094-7-6
Chen, C., Jiang, P., Xue, H., Peterson, S. E., Tran, H. T., Mccann, A. E., et al. (2014). Role of astroglia in down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 5:4430. doi: 10.1038/ncomms5430
Chong, F. P., Ng, K. Y., Koh, R. Y., and Chye, S. M. (2018). Tau Proteins and tauopathies in Alzheimer’s disease. Cell Mol. Neurobiol. 38, 965–980. doi: 10.1007/s10571-017-0574-1
Chow, S. K., Yu, D., Macdonald, C. L., Buibas, M., and Silva, G. A. (2010). Amyloid beta-peptide directly induces spontaneous calcium transients, delayed intercellular calcium waves and gliosis in rat cortical astrocytes. ASN Neuro 2:e00026. doi: 10.1042/AN20090035
Cirillo, C., Capoccia, E., Iuvone, T., Cuomo, R., Sarnelli, G., Steardo, L., et al. (2015). S100B inhibitor pentamidine attenuates reactive gliosis and reduces neuronal loss in a mouse model of alzheimer’s disease. Biomed. Res. Int. 2015:508342. doi: 10.1155/2015/508342
Clementi, M. E., Sampaolese, B., Triggiani, D., Tiezzi, A., and Giardia, B. (2013). S100b protects IMR-32 cells against Ab(1-42) induced neurotoxicity via modulation of apoptotic genes expression. Adv. Alzh. Dis. 2, 99–108. doi: 10.4236/aad.2013.23013
Clementi, M. E., Sampaolese, B., and Giardina, B. (2016). S100b induces expression of myoglobin in apbeta treated neuronal cells in vitro: a possible neuroprotective mechanism. Curr. Aging Sci. 9, 279–283. doi: 10.2174/1874609809666160222112850
Craft, J. M., Watterson, D. M., Marks, A., and Van Eldik, L. J. (2005). Enhanced susceptibility of S-100B transgenic mice to neuroinflammation and neuronal dysfunction induced by intracerebroventricular infusion of human beta-amyloid. Glia 51, 209–216. doi: 10.1002/glia.20194
Cristovao, J. S., Morris, V. K., Cardoso, I., Leal, S. S., Martinez, J., Botelho, H. M., et al. (2018). The neuronal S100B protein is a calcium-tuned suppressor of amyloid-beta aggregation. Sci. Adv. 4:eaaq1702. doi: 10.1126/sciadv.aaq1702
Cristovao, J. S., Santos, R., and Gomes, C. M. (2016). Metals and neuronal metal binding proteins implicated in alzheimer’s disease. Oxid. Med. Cell Longev. 2016:9812178. doi: 10.1155/2016/9812178
Cuello, A. C. (2017). Early and late CNS inflammation in alzheimer’s disease: two extremes of a continuum? Trends Pharmacol. Sci. 38, 956–966. doi: 10.1016/j.tips.2017.07.005
Cunden, L. S., Brophy, M. B., Rodriguez, G. E., Flaxman, H. A., and Nolan, E. M. (2017). Biochemical and functional evaluation of the intramolecular disulfide bonds in the zinc-chelating antimicrobial protein human S100A7 (Psoriasin). Biochemistry 56, 5726–5738. doi: 10.1021/acs.biochem.7b00781
Dahlgren, K. N., Manelli, A. M., Stine, W. B., Baker, L. K., Krafft, G. A., and Ladu, M. J. (2002). Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053. doi: 10.1074/jbc.m201750200
de Souza, D. F., Leite, M. C., Quincozes-Santos, A., Nardin, P., Tortorelli, L. S., Rigo, M. M., et al. (2009). S100B secretion is stimulated by IL-1beta in glial cultures and hippocampal slices of rats: likely involvement of MAPK pathway. J. Neuroimmunol. 206, 52–57. doi: 10.1016/j.jneuroim.2008.10.012
Del Prete, D., Checler, F., and Chami, M. (2014). Ryanodine receptors: physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 9:21. doi: 10.1186/1750-1326-9-21
Donato, R., Cannon, B. R., Sorci, G., Riuzzi, F., Hsu, K., Weber, D. J., et al. (2013). Functions of S100 proteins. Curr. Mol. Med. 13, 24–57. doi: 10.2174/1566524011307010024
Donato, R., Sorci, G., Riuzzi, F., Arcuri, C., Bianchi, R., Brozzi, F., et al. (2009). S100B’s double life: Intracellular regulator and extracellular signal. Biochim. Biophys. Acta Mol. Cell Res. 1793, 1008–1022. doi: 10.1016/j.bbamcr.2008.11.009
Edwards, M. M., and Robinson, S. R. (2006). TNF alpha affects the expression of GFAP and S100B: implications for Alzheimer’s disease. J. Neural Transm. 113, 1709–1715. doi: 10.1007/s00702-006-0479-5
Epstein, O. I., Pavlov, I. F., and Shtark, M. B. (2006). Improvement of memory by means of ultra-low doses of antibodies to S-100B antigen. Evid. Based Compl. Alternat. Med. 3, 541–545. doi: 10.1093/ecam/nel073
Esposito, G., Imitola, J., Lu, J., De Filippis, D., Scuderi, C., Ganesh, V. S., et al. (2008a). Genomic and functional profiling of human down syndrome neural progenitors implicates S100B and aquaporin 4 in cell injury. Hum. Mol. Genet. 17, 440–457. doi: 10.1093/hmg/ddm322
Esposito, G., Scuderi, C., Lu, J., Savani, C., De Filippis, D., Iuvone, T., et al. (2008b). S100B induces tau protein hyperphosphorylation via Dickopff-1 up-regulation and disrupts the Wnt pathway in human neural stem cells. J. Cell Mol. Med. 12, 914–927. doi: 10.1111/j.1582-4934.2008.00159.x
Fa, M., Puzzo, D., Piacentini, R., Staniszewski, A., Zhang, H., Baltrons, M. A., et al. (2016). Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci. Rep. 6:19393. doi: 10.1038/srep19393
Garbuglia, M., Verzini, M., Sorci, G., Bianchi, R., Giambanco, I., Agneletti, A. L., et al. (1999). The calcium-modulated proteins, S100A1 and S100B, as potential regulators of the dynamics of type III intermediate filaments. Braz. J. Med. Biol. Res. 32, 1177–1185. doi: 10.1590/s0100-879x1999001000001
Gerlai, R., Wojtowicz, J. M., Marks, A., and Roder, J. (1995). Overexpression of a calcium-binding protein, S100 beta, in astrocytes alters synaptic plasticity and impairs spatial learning in transgenic mice. Learn. Mem. 2, 26–39. doi: 10.1101/lm.2.1.26
Giannakopoulos, P., Hof, P. R., Kovari, E., Vallet, P. G., Herrmann, F. R., and Bouras, C. (1996). Distinct patterns of neuronal loss and alzheimer’s disease lesion distribution in elderly individuals older than 90 years. J. Neuropathol. Exp. Neurol. 55, 1210–1220. doi: 10.1097/00005072-199612000-00004
Gonzalez-Reyes, R. E., Nava-Mesa, M. O., Vargas-Sanchez, K., Ariza-Salamanca, D., and Mora-Munoz, L. (2017). Involvement of astrocytes in alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 10:427. doi: 10.3389/fnmol.2017.00427
Gouras, G. K., Almeida, C. G., and Takahashi, R. H. (2005). Intraneuronal abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol. Aging 26, 1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022
Gromov, L. A., Syrovatskaya, L. P., and Ovinova, G. V. (1992). Functional role of the neurospecific S-100 protein in the processes of memory. Neurosci. Behav. Physiol. 22, 25–29. doi: 10.1007/bf01186664
Gruden, M. A., Davidova, T. B., Malisauskas, M., Sewell, R. D., Voskresenskaya, N. I., Wilhelm, K., et al. (2007). Differential neuroimmune markers to the onset of Alzheimer’s disease neurodegeneration and dementia: autoantibodies to Abeta(25-35) oligomers, S100b and neurotransmitters. J. Neuroimmunol. 186, 181–192. doi: 10.1016/j.jneuroim.2007.03.023
Gruden, M. A., Davydova, T. V., Fomina, V. G., Vetrile, L. A., Morozova-Roche, L. A., and Sewell, R. D. E. (2017). Antibodies to glutamate reversed the amnesic effects of proinflammatory S100A9 protein fibrils in aged C57Bl/6 Mice. Bull. Exp. Biol. Med. 162, 430–432. doi: 10.1007/s10517-017-3632-2
Gruden, M. A., Davydova, T. V., Kudrin, V. S., Wang, C., Narkevich, V. B., Morozova-Roche, L. A., et al. (2018). S100A9 protein aggregates boost hippocampal glutamate modifying monoaminergic neurochemistry: a glutamate antibody sensitive outcome on alzheimer-like memory decline. ACS Chem. Neurosci. 9, 568–577. doi: 10.1021/acschemneuro.7b00379
Gruden, M. A., Davydova, T. V., Wang, C., Narkevich, V. B., Fomina, V. G., Kudrin, V. S., et al. (2016). The misfolded pro-inflammatory protein S100A9 disrupts memory via neurochemical remodelling instigating an Alzheimer’s disease-like cognitive deficit. Behav. Brain Res. 306, 106–116. doi: 10.1016/j.bbr.2016.03.016
Guo, X., Tang, P., Liu, P., Liu, Y., Chong, L., and Li, R. (2016). Dkk1: a promising molecule to connect Alzheimer’s disease and osteoporosis. Med. Hypoth. 88, 30–32. doi: 10.1016/j.mehy.2015.12.023
Ha, T. Y., Chang, K. A., Kim, J., Kim, H. S., Kim, S., Chong, Y. H., et al. (2010). S100a9 knockdown decreases the memory impairment and the neuropathology in Tg2576 mice, AD animal model. PLoS One 5:e8840. doi: 10.1371/journal.pone.0008840
Hagmeyer, S., Cristóvão, J. S., Mulvihill, J. J., Boeckers, T. M., Gomes, C. M., and Grabrucker, A. M. (2017). Zinc binding to S100B affords regulation of trace metal homeostasis and excitotoxicity in the brain. Front. Mol. Neurosci. 10:456. doi: 10.3389/fnmol.2017.00456
Heizmann, C. W., Fritz, G., and Schafer, B. W. (2002). S100 proteins: structure, functions and pathology. Front. Biosci. 7, d1356–d1368.
Hofmann, M. A., Drury, S., Fu, C., Qu, W., Taguchi, A., Lu, Y., et al. (1999). RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97, 889–901. doi: 10.1016/s0092-8674(00)80801-6
Hooper, C., Killick, R., and Lovestone, S. (2008). The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 104, 1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x
Horvath, I., Jia, X., Johansson, P., Wang, C., Moskalenko, R., Steinau, A., et al. (2016). Pro-inflammatory S100A9 protein as a robust biomarker differentiating early stages of cognitive impairment in alzheimer’s disease. ACS Chem. Neurosci. 7, 34–39. doi: 10.1021/acschemneuro.5b00265
Iashchishyn, I. A., Gruden, M. A., Moskalenko, R. A., Davydova, T. V., Wang, C., Sewell, R. D. E., et al. (2018). Intranasally Administered S100A9 Amyloids Induced Cellular Stress, Amyloid Seeding, and Behavioral Impairment in Aged Mice. ACS Chem Neurosci. 9, 1338–1348 doi: 10.1021/acschemneuro.7b00512
Iashchishyn, I. A., Sulskis, D., Nguyen Ngoc, M., Smirnovas, V., and Morozova-Roche, L. A. (2017). Finke-Watzky Two-Step Nucleation-Autocatalysis Model of S100A9 Amyloid Formation: Protein Misfolding as “Nucleation” Event. ACS Chem Neurosci 8, 2152–2158. doi: 10.1021/acschemneuro.7b00251
Ichikawa, H., Jacobowitz, D. M., and Sugimoto, T. (1997). S100 protein-immunoreactive primary sensory neurons in the trigeminal and dorsal root ganglia of the rat. Brain Res 748, 253–257. doi: 10.1016/s0006-8993(96)01364-9
Isobe, T., Takahashi, K., and Okuyama, T. (1984). S100a0 (alpha alpha) protein is present in neurons of the central and peripheral nervous system. J. Neurochem. 43, 1494–1496. doi: 10.1111/j.1471-4159.1984.tb05415.x
Kitazawa, M., Cheng, D., Tsukamoto, M. R., Koike, M. A., Wes, P. D., Vasilevko, V., et al. (2011). Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 187, 6539–6549. doi: 10.4049/jimmunol.1100620
Kummer, M. P., Vogl, T., Axt, D., Griep, A., Vieira-Saecker, A., Jessen, F., et al. (2012). Mrp14 deficiency ameliorates amyloid beta burden by increasing microglial phagocytosis and modulation of amyloid precursor protein processing. J. Neurosci. 32, 17824–17829. doi: 10.1523/JNEUROSCI.1504-12.2012
LaFerla, F. M. (2002). Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 3, 862–872. doi: 10.1038/nrn960
Leal, S. S., Botelho, H. M., and Gomes, C. M. (2012). Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev. 256, 2253–2270. doi: 10.1016/j.ccr.2012.04.004
Leclerc, E., Fritz, G., Vetter, S. W., and Heizmann, C. W. (2009). Binding of S100 proteins to RAGE: an update. Biochim. Biophys. Acta Mol. Cell Res. 1793, 993–1007. doi: 10.1016/j.bbamcr.2008.11.016
Lee, E. O., Yang, J. H., Chang, K. A., Suh, Y. H., and Chong, Y. H. (2013). Amyloid-beta peptide-induced extracellular S100A9 depletion is associated with decrease of antimicrobial peptide activity in human THP-1 monocytes. J. Neuroinflam. 10:68. doi: 10.1186/1742-2094-10-68
Lee, H. J., Savelieff, M. G., Kang, J., Brophy, M. B., Nakashige, T. G., Lee, S. J. C., et al. (2018). Calprotectin influences the aggregation of metal-free and metal-bound amyloid-beta by direct interaction. Metallomics 10, 1116–1127. doi: 10.1039/c8mt00091c
Li, G., Chen, H., Cheng, L., Zhao, R., Zhao, J., and Xu, Y. (2014). Amyloid precursor-like protein 2 C-terminal fragments upregulate S100A9 gene and protein expression in BV2 cells. Neural Regen. Res. 9, 1923–1928. doi: 10.4103/1673-5374.145362
Ling, Y., Morgan, K., and Kalsheker, N. (2003). Amyloid precursor protein (APP) and the biology of proteolytic processing: relevance to Alzheimer’s disease. Int. J. Biochem. Cell Biol. 35, 1505–1535. doi: 10.1016/s1357-2725(03)00133-x
Liu, L., Drouet, V., Wu, J. W., Witter, M. P., Small, S. A., Clelland, C., et al. (2012). Trans-synaptic spread of tau pathology in vivo. PLoS One 7:e31302. doi: 10.1371/journal.pone.0031302
Liu, L., Li, Y. K., Van Eldik, L. J., Griffin, W. S. T., and Barger, S. W. (2005). S100B-induced microglial and neuronal IL-1 expression is mediated by cell type-specific transcription factors. J. Neurochem. 92, 546–553. doi: 10.1111/j.1471-4159.2004.02909.x
Lodeiro, M., Puerta, E., Ismail, M. A., Rodriguez-Rodriguez, P., Ronnback, A., Codita, A., et al. (2017). Aggregation of the inflammatory S100A8 precedes a beta plaque formation in transgenic APP mice: positive feedback for S100A8 and a beta productions. J. Gerontol. Ser. Biol. Sci. Med. Sci. 72, 319–328. doi: 10.1093/gerona/glw073
Lovell, M. A., Robertson, J. D., Teesdale, W. J., Campbell, J. L., and Markesbery, W. R. (1998). Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 158, 47–52. doi: 10.1016/s0022-510x(98)00092-6
Lu, J., Esposito, G., Scuderi, C., Steardo, L., Delli-Bovi, L. C., Hecht, J. L., et al. (2011). S100B and APP promote a gliocentric shift and impaired neurogenesis in down syndrome neural progenitors. PLoS One 6:e22126. doi: 10.1371/journal.pone.0022126
Maciejewski, A., Prado, V. F., Prado, M. A. M., and Choy, W. Y. (2017). Molecular basis for the interaction between stress-inducible phosphoprotein 1 (STIP1) and S100A1. Biochem. J. 474, 1853–1866. doi: 10.1042/BCJ20161055
Marshak, D. R., Pesce, S. A., Stanley, L. C., and Griffin, W. S. (1992). Increased S100 beta neurotrophic activity in Alzheimer’s disease temporal lobe. Neurobiol. Aging 13, 1–7. doi: 10.1016/0197-4580(92)90002-f
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer’s disease. Nat. Rev. Dis. Primers 1:15056. doi: 10.1038/nrdp.2015.56
Matsui Lee, I. S., Suzuki, M., Hayashi, N., Hu, J., Van Eldik, L. J., Titani, K., et al. (2000). Copper-dependent formation of disulfide-linked dimer of S100B protein. Arch. Biochem. Biophys. 374, 137–141. doi: 10.1006/abbi.1999.1595
Maynard, C. J., Bush, A. I., Masters, C. L., Cappai, R., and Li, Q. X. (2005). Metals and amyloid-beta in Alzheimer’s disease. Int. J. Exp. Pathol. 86, 147–159.
Mori, T., Koyama, N., Arendash, G. W., Horikoshi-Sakuraba, Y., Tan, J., and Town, T. (2010). Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer’s disease. Glia 58, 300–314. doi: 10.1002/glia.20924
Mrak, R. E., and Griffinbc, W. S. (2001). The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 22, 915–922. doi: 10.1016/s0197-4580(01)00293-7
Mrak, R. E., Sheng, J. G., and Griffin, W. S. (1996). Correlation of astrocytic S100 beta expression with dystrophic neurites in amyloid plaques of Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 55, 273–279. doi: 10.1097/00005072-199603000-00002
Mueller, C., Zhou, W. D., Vanmeter, A., Heiby, M., Magaki, S., Ross, M. M., et al. (2010). The heme degradation pathway is a promising serum biomarker source for the early detection of alzheimer’s disease. J. Alzh. Dis. 19, 1081–1091. doi: 10.3233/jad-2010-1303
O’Dowd, B. S., Zhao, W. Q., Ng, K. T., and Robinson, S. R. (1997). Chicks injected with antisera to either S-100 alpha or S-100 beta protein develop amnesia for a passive avoidance task. Neurobiol. Learn. Mem. 67, 197–206. doi: 10.1006/nlme.1997.3766
Ostapchenko, V. G., Beraldo, F. H., Mohammad, A. H., Xie, Y. F., Hirata, P. H., Magalhaes, A. C., et al. (2013). The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-beta oligomer toxicity. J. Neurosci. 33, 16552–16564. doi: 10.1523/JNEUROSCI.3214-13.2013
Peskind, E. R., Griffin, W. S. T., Akama, K. T., Raskind, M. A., and Van Eldik, L. J. (2001). Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer’s disease. Neurochem. Int. 39, 409–413. doi: 10.1016/s0197-0186(01)00048-1
Petzold, A., Jenkins, R., Watt, H. C., Green, A. J., Thompson, E. J., Keir, G., et al. (2003). Cerebrospinal fluid S100B correlates with brain atrophy in Alzheimer’s disease. Neurosci. Lett. 336, 167–170. doi: 10.1016/s0304-3940(02)01257-0
Popugaeva, E., Pchitskaya, E., and Bezprozvanny, I. (2017). Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease – A therapeutic opportunity? Biochem. Biophys. Res. Commun. 483, 998–1004. doi: 10.1016/j.bbrc.2016.09.053
Prinzen, C., Trumbach, D., Wurst, W., Endres, K., Postina, R., and Fahrenholz, F. (2009). Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice. BMC Genomics 10:66. doi: 10.1186/1471-2164-10-66
Qin, W., Ho, L., Wang, J., Peskind, E., and Pasinetti, G. M. (2009). S100A7, a novel alzheimer’s disease biomarker with non-amyloidogenic alpha-secretase activity acts via selective promotion of ADAM-10. PLoS One 4:e4183. doi: 10.1371/journal.pone.0004183
Reeves, R. H., Yao, J., Crowley, M. R., Buck, S., Zhang, X., Yarowsky, P., et al. (1994). Astrocytosis and axonal proliferation in the hippocampus of S100b transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 91, 5359–5363. doi: 10.1073/pnas.91.12.5359
Roltsch, E., Holcomb, L., Young, K. A., Marks, A., and Zimmer, D. B. (2010). PSAPP mice exhibit regionally selective reductions in gliosis and plaque deposition in response to S100B ablation. J. Neuroinflam. 7:78. doi: 10.1186/1742-2094-7-78
Santos, G., Barateiro, A., Gomes, C. M., Brites, D., and Fernandes, A. (2018). Impaired oligodendrogenesis and myelination by elevated S100B levels during neurodevelopment. Neuropharmacology 129, 69–83. doi: 10.1016/j.neuropharm.2017.11.002
Senior, S. Z., Mans, L. L., Vanguilder, H. D., Kelly, K. A., Hendrich, M. P., and Elgren, T. E. (2003). Catecholase activity associated with copper-S100B. Biochemistry 42, 4392–4397. doi: 10.1021/bi0205799
Shen, L. M., Liao, L. P., Chen, C., Guo, Y., Song, D. L., Wang, Y., et al. (2017). Proteomics analysis of blood serums from alzheimer’s disease patients using iTRAQ labeling technology. J. Alzh. Dis. 56, 361–378. doi: 10.3233/JAD-160913
Sheng, J. G., Mrak, R. E., and Griffin, W. S. (1994). S100 beta protein expression in Alzheimer disease: potential role in the pathogenesis of neuritic plaques. J. Neurosci. Res. 39, 398–404. doi: 10.1002/jnr.490390406
Sheng, J. G., Mrak, R. E., and Griffin, W. S. (1997). Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J. Neuropathol. Exp. Neurol. 56, 285–290. doi: 10.1097/00005072-199703000-00007
Shepherd, C. E., Goyette, J., Utter, V., Rahimi, F., Yang, Z., Geczy, C. L., et al. (2006). Inflammatory S100A9 and S100A12 proteins in Alzheimer’s disease. Neurobiol. Aging 27, 1554–1563. doi: 10.1016/j.neurobiolaging.2005.09.033
Sidoryk-Wegrzynowicz, M., Gerber, Y. N., Ries, M., Sastre, M., Tolkovsky, A. M., and Spillantini, M. G. (2017). Astrocytes in mouse models of tauopathies acquire early deficits and lose neurosupportive functions. Acta Neuropathol. Commun. 5:89. doi: 10.1186/s40478-017-0478-9
Simpson, J. E., Ince, P. G., Lace, G., Forster, G., Shaw, P. J., Matthews, F., et al. (2010). Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 31, 578–590. doi: 10.1016/j.neurobiolaging.2008.05.015
Sorci, G., Agneletti, A. L., and Donato, R. (2000). Effects of S100A1 and S100B on microtubule stability. An in vitro study using triton-cytoskeletons from astrocyte and myoblast cell lines. Neuroscience 99, 773–783. doi: 10.1016/s0306-4522(00)00238-4
Steiner, J., Bogerts, B., Schroeter, M. L., and Bernstein, H. G. (2011). S100B protein in neurodegenerative disorders. Clin. Chem. Lab. Med. 49, 409–424. doi: 10.1515/CCLM.2011.083
Tai, H. C., Wang, B. Y., Serrano-Pozo, A., Frosch, M. P., Spires-Jones, T. L., and Hyman, B. T. (2014). Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s disease. Acta Neuropathol. Commun. 2:146. doi: 10.1186/s40478-014-0146-2
Takeda, A., and Tamano, H. (2016). Insight into cognitive decline from Zn(2+) dynamics through extracellular signaling of glutamate and glucocorticoids. Arch. Biochem. Biophys. 611, 93–99. doi: 10.1016/j.abb.2016.06.021
Teigelkamp, S., Bhardwaj, R. S., Roth, J., Meinardus-Hager, G., Karas, M., and Sorg, C. (1991). Calcium-dependent complex assembly of the myeloic differentiation proteins MRP-8 and MRP-14. J. Biol. Chem. 266, 13462–13467.
Tian, Z.-Y., Wang, C.-Y., Wang, T., Li, Y.-C., and Wang, Z. Y. (2019). Glial S100A6 Degrades beta-amyloid aggregation through targeting competition with zinc ions. Aging Dis. 10:14.
Turner, P. R., O’connor, K., Tate, W. P., and Abraham, W. C. (2003). Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 70, 1–32. doi: 10.1016/s0301-0082(03)00089-3
Van Eldik, L. J., and Griffin, W. S. (1994). S100 beta expression in Alzheimer’s disease: relation to neuropathology in brain regions. Biochim. Biophys. Acta 1223, 398–403.
Venegas, C., and Heneka, M. T. (2017). Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 101, 87–98. doi: 10.1189/jlb.3MR0416-204R
Walker, D. G., Link, J., Lue, L. F., Dalsing-Hernandez, J. E., and Boyes, B. E. (2006). Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J. Leukoc. Biol. 79, 596–610. doi: 10.1189/jlb.0705377
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Wang, C., Iashchishyn, I. A., Pansieri, J., Nystrom, S., Klementieva, O., Kara, J., et al. (2018). S100A9-Driven amyloid-neuroinflammatory cascade in traumatic brain injury as a precursor state for alzheimer’s disease. Sci. Rep. 8:12836. doi: 10.1038/s41598-018-31141-x
Wang, C., Klechikov, A. G., Gharibyan, A. L., Warmlander, S. K., Jarvet, J., Zhao, L., et al. (2014). The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 127, 507–522. doi: 10.1007/s00401-013-1208-4
Wang, X., Wang, W., Li, L., Perry, G., Lee, H. G., and Zhu, X. (2014). Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 1842, 1240–1247.
Wasik, U., Schneider, G., Mietelska-Porowska, A., Mazurkiewicz, M., Fabczak, H., Weis, S., et al. (2013). Calcyclin binding protein and Siah-1 interacting protein in Alzheimer’s disease pathology: neuronal localization and possible function. Neurobiol. Aging 34, 1380–1388. doi: 10.1016/j.neurobiolaging.2012.11.007
Weiner, H. L., and Frenkel, D. (2006). Immunology and immunotherapy of Alzheimer’s disease. Nat. Rev. Immunol. 6, 404–416.
Weissmann, R., Huttenrauch, M., Kacprowski, T., Bouter, Y., Pradier, L., Bayer, T. A., et al. (2016). Gene expression profiling in the APP/PS1KI mouse model of familial alzheimer’s disease. J. Alzh. Dis. 50, 397–409. doi: 10.3233/JAD-150745
Wirths, O., Breyhan, H., Marcello, A., Cotel, M. C., Bruck, W., and Bayer, T. A. (2010). Inflammatory changes are tightly associated with neurodegeneration in the brain and spinal cord of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol. Aging 31, 747–757. doi: 10.1016/j.neurobiolaging.2008.06.011
Wruck, W., Schroter, F., and Adjaye, J. (2016). Meta-analysis of transcriptome data related to hippocampus biopsies and iPSC-derived neuronal cells from alzheimer’s disease patients reveals an association with FOXA1 and FOXA2 gene regulatory networks. J. Alzh. Dis. 50, 1065–1082. doi: 10.3233/JAD-150733
Xiong, Z., O’hanlon, D., Becker, L. E., Roder, J., Macdonald, J. F., and Marks, A. (2000). Enhanced calcium transients in glial cells in neonatal cerebellar cultures derived from S100B null mice. Exp. Cell Res. 257, 281–289. doi: 10.1006/excr.2000.4902
Yamada, J., and Jinno, S. (2012). Upregulation of calcium binding protein, S100A6, in activated astrocytes is linked to glutamate toxicity. Neuroscience 226, 119–129. doi: 10.1016/j.neuroscience.2012.08.068
Yamada, J., and Jinno, S. (2014). S100A6 (Calcyclin) is a novel marker of neural stem cells and astrocyte precursors in the subgranular zone of the adult mouse hippocampus. Hippocampus 24, 89–101. doi: 10.1002/hipo.22207
Yang, Q., Hamberger, A., Hyden, H., Wang, S., Stigbrand, T., and Haglid, K. G. (1995). S-100 beta has a neuronal localisation in the rat hindbrain revealed by an antigen retrieval method. Brain Res. 696, 49–61. doi: 10.1016/0006-8993(95)00755-f
Yang, Q., O’hanlon, D., Heizmann, C. W., and Marks, A. (1999). Demonstration of heterodimer formation between S100B and S100A6 in the yeast two-hybrid system and human melanoma. Exp. Cell Res. 246, 501–509. doi: 10.1006/excr.1998.4314
Yardan, T., Erenler, A. K., Baydin, A., Aydin, K., and Cokluk, C. (2011). Usefulness of S100B protein in neurological disorders. J. Pak. Med. Assoc. 61, 276–281.
Yeh, C. W., Yeh, S. H., Shie, F. S., Lai, W. S., Liu, H. K., Tzeng, T. T., et al. (2015). Impaired cognition and cerebral glucose regulation are associated with astrocyte activation in the parenchyma of metabolically stressed APPswe/PS1dE9 mice. Neurobiol. Aging 36, 2984–2994. doi: 10.1016/j.neurobiolaging.2015.07.022
Zhang, C., Liu, Y., Gilthorpe, J., and Van Der Maarel, J. R. (2012). MRP14 (S100A9) protein interacts with Alzheimer beta-amyloid peptide and induces its fibrillization. PLoS One 7:e32953. doi: 10.1371/journal.pone.0032953
Zhao, L. N., Zhang, T., Zhang, C., Wang, C., Morozova-Roche, L. A., Chew, L. Y., et al. (2013). S100A9 induces aggregation-prone conformation in abeta peptides: a combined experimental and simulation study. RSC Adv. 3, 24081–24089.
Zimmer, D. B., Chaplin, J., Baldwin, A., and Rast, M. (2005). S100-mediated signal transduction in the nervous system and neurological diseases. Cell Mol Biol. 51, 201–214.
Keywords: neuroinflammation and neurodegeneration, amyloid-β, tau, metal ions, protein misfolding, aggregation
Citation: Cristóvão JS and Gomes CM (2019) S100 Proteins in Alzheimer’s Disease. Front. Neurosci. 13:463. doi: 10.3389/fnins.2019.00463
Received: 07 January 2019; Accepted: 24 April 2019;
Published: 16 May 2019.
Edited by:
Natalia N. Nalivaeva, University of Leeds, United KingdomReviewed by:
Estelle Leclerc, North Dakota State University, United StatesWieslawa Lesniak, Nencki Institute of Experimental Biology (PAS), Poland
Fabrizio Michetti, Catholic University of the Sacred Heart, Italy
Copyright © 2019 Cristóvão and Gomes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cláudio M. Gomes, Y21nb21lc0BmYy51bC5wdA==