 Hannah Rostalski1
Hannah Rostalski1 Stina Leskelä1
Stina Leskelä1 Nadine Huber1
Nadine Huber1 Kasper Katisko2
Kasper Katisko2 Antti Cajanus2
Antti Cajanus2 Eino Solje2,3
Eino Solje2,3 Mikael Marttinen4
Mikael Marttinen4 Teemu Natunen4
Teemu Natunen4 Anne M. Remes5,6
Anne M. Remes5,6 Mikko Hiltunen4
Mikko Hiltunen4 Annakaisa Haapasalo1*
Annakaisa Haapasalo1*- 1A.I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, Kuopio, Finland
- 2Institute of Clinical Medicine – Neurology, University of Eastern Finland, Kuopio, Finland
- 3Neuro Center, Neurology, Kuopio University Hospital, Kuopio, Finland
- 4Institute of Biomedicine, University of Eastern Finland, Kuopio, Finland
- 5Medical Research Center, Oulu University Hospital, Oulu, Finland
- 6Research Unit of Clinical Neuroscience, Neurology, University of Oulu, Oulu, Finland
Frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) are neurodegenerative diseases with a complex, but often overlapping, genetic and pathobiological background and thus they are considered to form a disease spectrum. Although neurons are the principal cells affected in FTLD and ALS, increasing amount of evidence has recently proposed that other central nervous system-resident cells, including microglia and astrocytes, may also play roles in neurodegeneration in these diseases. Therefore, deciphering the mechanisms underlying the disease pathogenesis in different types of brain cells is fundamental in order to understand the etiology of these disorders. The major genetic cause of FTLD and ALS is a hexanucleotide repeat expansion (HRE) in the intronic region of the C9orf72 gene. In neurons, specific pathological hallmarks, including decreased expression of the C9orf72 RNA and proteins and generation of toxic RNA and protein species, and their downstream effects have been linked to C9orf72 HRE-associated FTLD and ALS. In contrast, it is still poorly known to which extent these pathological changes are presented in other brain cells. Here, we summarize the current literature on the potential role of astrocytes and microglia in C9orf72 HRE-linked FTLD and ALS and discuss their possible phenotypic alterations and neurotoxic mechanisms that may contribute to neurodegeneration in these diseases.
Introduction
Frontotemporal lobar degeneration (FTLD) is a group of neurodegenerative disorders affecting predominantly the frontal and temporal lobes of the brain (Gorno-Tempini et al., 2011; Rascovsky et al., 2011) and the second most prevalent early-onset dementia (Onyike and Diehl-Schmid, 2013). FTLD clinical syndromes are characterized by changes in behavior, personality, and executive functions or deterioration of language functions. GGGGCC hexanucleotide repeat expansion (HRE) in the non-coding region of chromosome 9 open reading frame 72 (C9orf72) (C9-HRE) is the major genetic cause of familial FTLD (12–48%) and amyotrophic lateral sclerosis (ALS) (24–46%) cases and 6–20% of sporadic cases for both diseases (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Majounie et al., 2012). C9-HRE may also lead to concomitant FTLD and ALS. The pathobiology of C9-HRE-associated FTLD and ALS (C9-FTLD or C9-ALS) is complex. Neurons in the C9-HRE carriers display specific pathological hallmarks, including toxic RNA and proteins derived from the expanded C9-HRE and decreased expression of C9orf72 due to haploinsufficiency (see reviews Gitler and Tsuiji, 2016; Freibaum and Taylor, 2017; Balendra and Isaacs, 2018). However, recently the potential role of other central nervous system (CNS)-resident cells, especially astrocytes and microglia, has also started to gain attention.
Glial cells are essential for brain homeostasis, but they also may mediate neuroinflammation (Jang et al., 2013; Franco and Fernández-Suárez, 2015; Shinozaki et al., 2017). Chronic changes in their physiological functions may contribute to neurodegeneration via both cell autonomous and non-cell autonomous mechanisms in neurodegenerative diseases, including ALS and FTLD. Whereas, most of the early findings on glial involvement in ALS pathogenesis derived from studies on mutant superoxide dismutase 1 (SOD1), there is accumulating evidence for glial contribution in other subtypes of ALS as well (Broe et al., 2004; Haidet-Phillips et al., 2011; Minami et al., 2015; Radford et al., 2015; Chen et al., 2016; Lee et al., 2016; Taylor et al., 2016; Cooper-Knock et al., 2017; Hallmann et al., 2017; Krabbe et al., 2017; Bachiller et al., 2018; Deczkowska et al., 2018); for a recent review on the role of neuroinflammation and complement system in ALS see also (Parker et al., 2019). Communication between neurons and glia via secreted factors and membrane-bound receptors is crucial for e.g., regulation of synaptic pruning and detection and clearance of apoptotic cells, and alterations in this crosstalk are suggested to contribute to the pathogenesis of neurodegenerative diseases (Rama Rao and Kielian, 2015; Szepesi et al., 2018). For example, complement 3 (C3) levels are increased, whereas the levels of signal regulatory protein (SIRP) α, a protein negatively regulating phagocytosis, and its corresponding receptor on microglia, cluster of differentiation (CD) 47, are reduced in the frontal cortex of FTLD patients compared to healthy controls or ALS and FTLD/ALS patients (Gitik et al., 2011; Umoh et al., 2018). Also, levels of CD200, expressed on neurons and restricting microglial activation (Barclay et al., 2002), are reduced in the frontal cortex of FTLD compared to FTLD/ALS patients (Umoh et al., 2018). These findings imply that microglia-mediated synaptic pruning and phagocytosis might be enhanced in FTLD brains. RNA expression analyses have indicated that pathways including the complement system, antigen presentation, and interferon (IFN) γ and interleukin (IL) 1-β signaling are significantly upregulated in the brains of C9-ALS patients compared to sporadic ALS patients (Prudencio et al., 2015; O'Rourke et al., 2016). Also transcription factors of the nuclear factor kappa B (NFκB) pathway were differentially expressed in C9-ALS and non-C9-ALS patients compared to healthy subjects (Ismail et al., 2013), but it is unclear in which cells these alterations occur. Lower levels of C-X-C motif chemokine ligand 10 protein, a microglial chemoattractant, in the cerebrospinal fluid (CSF) of C9-ALS patients were observed compared to non-C9-ALS cases (Ismail et al., 2013), but the physiological consequences of this are unknown. Astrocytes from C9-ALS patients show glucose hypermetabolism as compared to non-C9-ALS cases and controls, possibly as a consequence of astrogliosis (Cistaro et al., 2014). Decreased levels of excitatory amino acid transporter (EAAT) 2 in the frontal cortex of FTLD patients compared to controls (Umoh et al., 2018) and in C9-ALS compared to sporadic ALS patients (Fomin et al., 2018) suggest that astrocytes in both C9-HRE carriers and non-carriers might show defective uptake of glutamate, which could lead to excitotoxicity. Thus, mounting evidence points to a potentially altered physiology of microglia and astrocytes in FTLD and ALS associated with C9-HRE.
Pathological Hallmarks of C9-HRE in Glial Cells
RNA Foci Are Less Abundant in Glial Cells Compared to Neurons
Sense and antisense RNA foci, formed by aggregated C9-HRE-containing RNA, represent a unique pathological feature of C9-FTLD and C9-ALS. RNA foci have mainly been reported in neurons (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Gendron et al., 2013; Mizielinska et al., 2013; Dafinca et al., 2016), but also in non-neuronal cells, such as fibroblasts and lymphoblasts (Donnelly et al., 2013; Lagier-Tourenne et al., 2013; Cooper-Knock et al., 2014). Interestingly, glial fibrillary acidic protein (GFAP)-positive and negative glial cells in the cerebellum of C9-ALS and C9-FTLD cases (Gendron et al., 2013) and induced pluripotent stem cell (iPSC)-derived astroglia have also been confirmed to contain sense foci (Sareen et al., 2013). However, compared to neurons, RNA foci have only been detected in a small fraction of microglia and astrocytes in post-mortem C9-ALS frontal cortex (Mizielinska et al., 2013) and/or cerebellum of C9-HRE carrying FTLD, ALS and FTLD/ALS patients (DeJesus-Hernandez et al., 2017). Furthermore, the number of RNA foci per cell was lower in microglia and astrocytes compared to neurons (Mizielinska et al., 2013). Whereas, neurons may exhibit nuclear and, to a lower extent, cytoplasmic RNA foci, microglia, and astrocytes showed only intranuclear RNA foci (Lagier-Tourenne et al., 2013; Mizielinska et al., 2013). This may suggest that (i) glial cells might not express C9-HRE to the same extent as neurons; (ii) expression of RNA-binding proteins, known to stabilize RNA foci, and/or proteins involved in cytoplasmic translocation of C9-HRE RNA are less abundant; (iii) glial cells can better clear C9-HRE-containing RNA (Peters et al., 2015); or (iv) somatic heterogeneity of the C9-HRE length, which can occur in different cells within the same tissue of C9-HRE carriers (DeJesus-Hernandez et al., 2011; Almeida et al., 2013; van Blitterswijk et al., 2013; Nordin et al., 2015), might underlie the lower prevalence of RNA foci in glial cells compared to neurons. The RNA foci in neurons are suggested to cause disturbances in RNA metabolism through sequestration of RNA-binding proteins (Donnelly et al., 2013; Mori et al., 2013a; Sareen et al., 2013; Cooper-Knock et al., 2014). Similar effects might occur in glial cells exhibiting RNA foci, but further investigations are required to ascertain this.
Dipeptide Repeat Proteins Appear Less Frequent in Glial Cells Than Neurons
In addition to the RNA foci, five dipeptide repeat protein (DRP) species, namely poly-glycine-alanine (poly-GA), poly-glycine-proline (poly-GP), poly-glycine-arginine (poly-GR), poly-proline-arginine (poly-PR), and poly-proline-alanine (poly-PA) are directly derived via repeat-associated non-AUG (RAN) translation from the C9-HRE-containing RNA and represent additional pathological hallmarks unique to C9-FTLD and C9-ALS (Zu et al., 2011, 2013; Ash et al., 2013; Gendron et al., 2013; Mori et al., 2013b). Consistent with the low prevalence of RNA foci in glia, poly-GA inclusions were not detected in microglia or astrocytes of post-mortem C9-FTLD or C9-ALS brains (Mackenzie et al., 2013) or in astrocytes in the hippocampus of a C9-ALS patient (Ash et al., 2013). Also, poly-GP inclusions were undetectable in glial cells of C9-ALS and C9-FTLD patients showing sense RNA foci in cerebellar astrocytes (Gendron et al., 2013). Another study on C9-ALS post-mortem brains could not detect any of the DRPs in glial cells in subcortical white matter, hippocampus or white matter in the spinal cord, where glial cells are abundant (Saberi et al., 2018). However, poly-GA, poly-GP and poly-GR inclusions were detected in the ependymal cells of the spinal cord central canal of C9-FTLD and C9-FTLD/ALS cases. Poly-GA inclusions were also observed in ependymal and subependymal cells of the lateral ventricular wall (Schludi et al., 2015). These results suggest that DRP inclusions might be present in glial cells, but to a lower extent than in neurons and in defined CNS areas. Use of cell type-specific antibodies may help to decipher which glial cell types exhibit DRP inclusions and whether they, if present, compromise glial cell function. The lower prevalence of DRPs in glia as compared to neurons could mean that (i) less C9-HRE-containing RNA is translocated into the cytosol; ii) it undergoes RAN translation less efficiently; (iii) and/or DRPs are degraded before they can accumulate. These might be supported by the finding that adeno-associated virus (AAV)-mediated expression of DRPs in mice leads to DRP accumulation in neurons but not glial cells (Chew et al., 2015). However, it cannot be excluded that glial DRPs might in fact derive from DRPs secreted by neurons, since neuron-to-glia transmission has been shown to occur in vitro (Westergard et al., 2016) (Figures 1, 2).
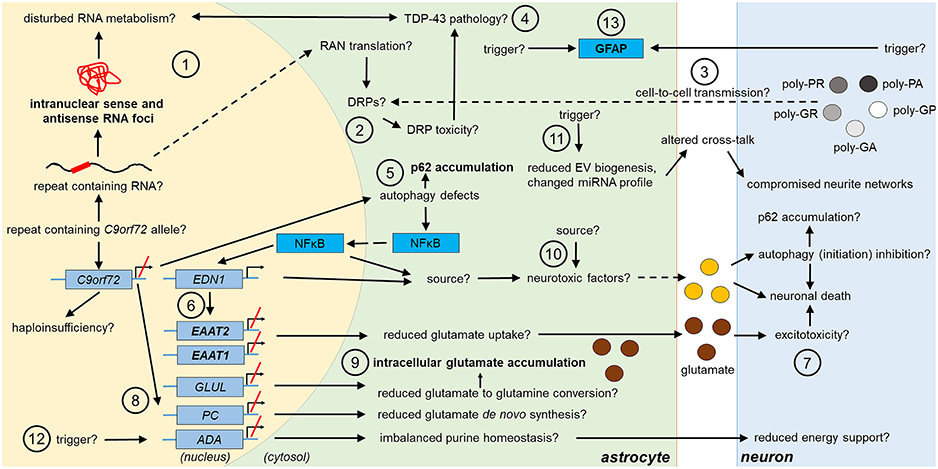
Figure 1. Potential and confirmed phenotypic features of C9orf72 HRE-associated astrocytes in FTLD/ALS. Features detected in the astrocytes of FTLD or ALS patient post-mortem brain are indicated in bold text. The presence of the typical C9-HRE-associated pathological hallmarks, which have previously been observed mainly in neurons, as well as other potential mechanisms, which still need to be confirmed in human patient astrocytes, are indicated with a question mark. Directions of sequential events are visualized with arrows. Steps requiring intracellular or intercellular translocation of molecules are indicated by dashed arrows. The different events are indicated by numbers as follows: (1) C9-HRE-containing RNA might be transcribed in astrocytes, forming intranuclear sense and antisense RNA foci, which may disturb RNA metabolism. (2) C9-HRE-containing RNA might be translocated into the cytosol of astrocytes, where it could undergo RAN translation creating potentially toxic DRPs. (3) DRPs might also be transmitted from other cell types, such as neurons, to astrocytes. (4) Potential disturbances in RNA metabolism as well as DRP toxicity might lead to TDP-43 pathology, which in turn could lead to defects in RNA processing in astrocytes. (5) Reduced C9orf72 levels in astrocytes might cause defects in autophagy, resulting in p62 accumulation and increase of NFκB levels in the cytosol and nucleus. (6) Enhanced NFκB levels with simultaneously decreased C9orf72 levels might enhance EDN1 expression, which suppresses EAAT1 and 2 expression in astrocytes. (7) As a result, astrocytic uptake of extracellular glutamate might be diminished, which might lead to excitotoxicity. (8) Reduced C9orf72 levels might lead to decreased expression of genes involved in glutamate de novo synthesis (e.g., PC) and glutamate to glutamine conversion (e.g., GLUL). (9) Reduced conversion of glutamate to glutamine might underlie intracellular glutamate accumulation. (10) Neurotoxic factors, which partly might be created through the altered NFκB signaling, cause neuronal death. Neurotoxic factors might cause autophagy inhibition in neurons, which might lead to p62 accumulation. (11) Reduced EV synthesis, which might lead to decreased EV secretion, and an altered miRNA expression profile, might influence the crosstalk between astrocytes and neurons, which could lead to disturbed neurite growth and networks. (12) Decreased expression of ADA might disrupt purine metabolism and lead to decreased energy support of neurons by astrocytes. (13) Enhanced GFAP expression might derive from endogenous or exogenous triggers. ADA, adenosine deaminase; DRP, dipeptide repeat protein; EAAT1/2, excitatory amino acid transporter1/2; EDN1, endothelin 1; EV, extracellular vesicle; GFAP, glial fibrillary acidic protein; GLUL, glutamate-ammonia ligase; HRE, hexanucleotide repeat expansion; miRNA, micro ribonucleic acid; NFκB, nuclear factor “kappa-light-chain-enhancer” of activated B-cells; PC, pyruvate carboxylase; RAN, repeat-associated non-AUG; TDP-43, Transactive response DNA-binding protein 43.
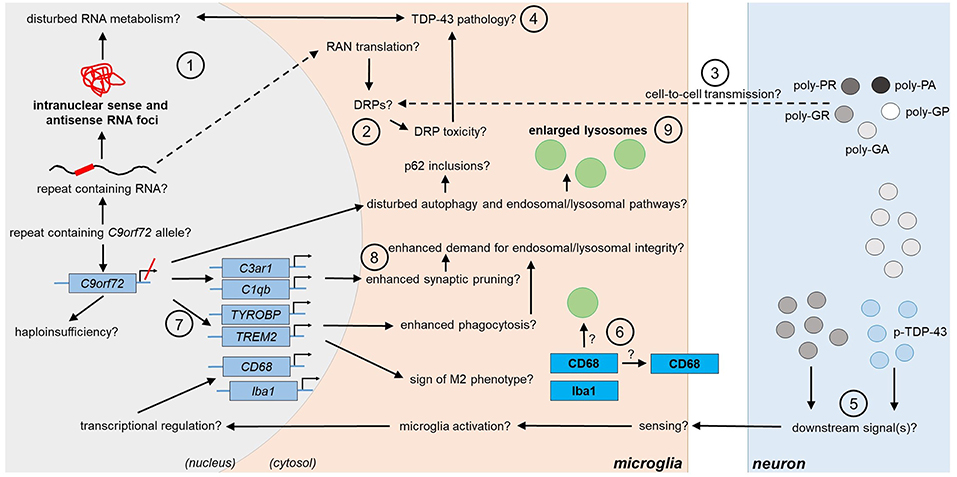
Figure 2. Potential and confirmed phenotypic features of C9orf72 HRE-associated microglia in FTLD/ALS. Features detected in the microglia of FTLD or ALS patient post-mortem brain are indicated in bold text. The presence of the typical C9-HRE-associated pathological hallmarks, which have previously been observed mainly in neurons, as well as other potential mechanisms, which still need to be confirmed in human patient microglia, are indicated with a question mark. Directions of sequential events are visualized with arrows. Steps requiring intracellular or intercellular translocation of molecules are indicated by dashed arrows. The different events are indicated by numbers as follows: (1) C9-HRE-containing RNA might be transcribed in microglia, forming intranuclear sense and antisense RNA foci, which may disturb RNA metabolism. (2) C9-HRE-containing RNA might be translocated into the cytosol of microglia, where it could undergo RAN translation creating potentially toxic DRPs. (3) DRPs might also be transmitted from other cell types, such as neurons, to microglia. (4) Potential disturbances in RNA metabolism as well as DRP toxicity might lead to TDP-43 pathology, which in turn could lead to defects in RNA processing in microglia. (5) Expression of poly-GA in combination with enhanced TDP-43 phosphorylation (p-TDP-43) or poly-GR expression alone in neurons might lead to downstream signals, which are recognized by adjacent microglia, leading first to enhanced CD68 and later Iba1 expression. (6) CD68 could serve as a receptor on the cell surface or localize in lysosomes. (7) Downregulation of C9orf72 might increase TREM2 and TYROBP expression, which might be a sign of M2 microglial phenotype switching and result in increased phagocytic activity. (8) Microglial C3ar1 and C1qb expression might be increased through decreased expression of C9orf72 and lead to enhanced synaptic pruning. (9) Decreased C9orf72 expression might disturb autophagy pathway, resulting in p62 accumulation and enlarged lysosomes in microglia. C1qb, complement subcomponent C1q chain B; C3ar1, complement C3a Receptor 1; CD68, cluster of differentiation 68; DRP, dipeptide repeat protein; HRE, hexanucleotide repeat expansion; Iba1, ionized calcium-binding adapter molecule 1; RAN, repeat-associated non-AUG; TDP-43, Transactive response DNA binding protein 43; TREM2, Triggering receptor expressed on myeloid cells 2; TYROBP, tyrosine kinase binding protein.
Glial Cells of C9-ALS and C9-FTLD Brain Present TDP-43 Pathology and p62 Inclusions
Transactive response DNA binding protein 43 (TDP-43) phosphorylation, cytoplasmic translocation, and truncation are pathological hallmarks of FTLD and ALS, including C9-FTLD and C9-ALS (Cooper-Knock et al., 2012), and potential contributors to disturbed RNA metabolism (Gendron et al., 2010). Neuronal TDP-43-negative, but p62-positive inclusions containing DRPs, represent another unique hallmark of C9-HRE (Mahoney et al., 2012). It has been suggested that TDP-43 aggregation can be caused by C9orf72 haploinsufficiency, formation of RNA foci, or DRP toxicity (Cooper-Knock et al., 2015; Sellier et al., 2016; Nonaka et al., 2018), whereas p62 accumulation might result from C9orf72 haploinsufficiency via impairment of autophagy (Sellier et al., 2016) or DRP expression (May et al., 2014). Cytoplasmic p62 and (phospho)-TDP-43-positive inclusions have been reported in glia in frontal, parietal, temporal, and motor cortex, hippocampus, brainstem, cerebellum, and spinal cord of C9-FTLD, C9-FTLD/ALS, and C9-ALS cases (Al-Sarraj et al., 2011; Cooper-Knock et al., 2012; Schipper et al., 2016), but it was not specified in which glial cell types the inclusions were detected. Some studies have reported phospho-TDP-43-positive inclusions in oligodendrocytes (Murray et al., 2011; Brettschneider et al., 2014; Fatima et al., 2015) and p62-positive inclusions in astrocytes (Simón-Sánchez et al., 2012). Co-immunostaining with cell type-specific antibodies would provide clarification to which extent astrocytes and microglia display p62 and TDP-43 inclusions. Understanding how the accumulation and aggregation of these proteins affect glial cell function could yield mechanistic insights into their potential contribution to disease pathogenesis. In conclusion, so far C9-HRE-associated pathological hallmarks have been detected to a lower extent in glial cells than in neurons in human post-mortem brains. In addition to the abovementioned potential reasons underlying the lower prevalence of these hallmarks in glial cells, yet one other option might be that glial cells can undergo extensive proliferation (Michell-Robinson et al., 2015; Verkhratsky and Nedergaard, 2018), which could either prevent the formation or dilute the amount of already existing RNA foci, DRPs, protein aggregates, or inclusions.
Astrocytes in Model Systems of C9-FTLD and C9-ALS
C9-HRE Might Cause Astrogliosis
Increased chitinase-3-like protein 1 (CHI3L1) and GFAP expression are considered as indicators of astrogliosis (Sofroniew and Vinters, 2010; Zamanian et al., 2012), and both proteins show increased levels in the frontal cortex and CSF of FTLD patients (Umoh et al., 2018; Oeckl et al., 2019). Interestingly, elevated GFAP expression was detected in mice upon AAV-mediated C9-HRE expression and in one bacterial artificial chromosome (BAC) C9-HRE mouse model (Liu et al., 2016). However, in two other BAC mice, signs of astrogliosis or microgliosis were not detected (Peters et al., 2015; Jiang et al., 2016). It should be noted that these mice did not concomitantly model haploinsufficiency. Astrocytes of C9orf72−/− mice did not show enhanced GFAP immunoreactivity, indicating that lack of C9orf72 does not cause astrogliosis (Koppers et al., 2015). Increased GFAP levels do not always correlate with enhanced ionized calcium-binding adapter molecule (Iba)1 levels (Zhang et al., 2016; Schludi et al., 2017), suggesting that trigger(s) for astro- and microgliosis are different. Identification of such triggers might be essential for elucidating pathogenic disease mechanisms.
Neurotoxicity Mediated by Astrocytes Containing C9-HRE
Recent studies suggest that astrocytes from C9-HRE carriers with ALS can mediate neurotoxicity. Murine embryonic stem cell-derived motor neurons co-cultured with fibroblast-derived astrocytes from sporadic ALS or C9-ALS patients underwent extensive cell death within 4 days. Partial replacement of the culture medium by control astrocyte conditioned medium (ACM) did not prevent cell death, suggesting involvement of a gain-of-toxic-function mechanism rather than insufficient trophic support by the C9-HRE astrocytes (Meyer et al., 2014). Moreover, iPSC-derived motor neurons from control subjects or C9-ALS patients cultured in C9-ALS ACM showed dramatically decreased viability after 5 days (Madill et al., 2017). These studies indicate that direct physical contact between C9-HRE astrocytes and neurons as well as secretion of neurotoxicants might mediate neuronal cell death in a non-cell autonomous manner. Varcianna et al. showed that extracellular vesicles (EVs) secreted by induced astrocytes from C9-ALS patients decreased motor neuron survival. Also, C9-HRE astrocytes revealed a profile of increased or decreased expression of certain microRNAs compared to healthy controls, of which many are involved in axonal guidance and maintenance. Among these, miRNA-494-3p, which was recently shown to protect against lipopolysaccharide-induced cell death by targeting IL-13 expression (Geng and Liu, 2019), was significantly reduced in EVs secreted by C9-HRE astrocytes. Treatment with miRNA-494-3p mimic restored neurite length and number of nodes, and promoted motor neuron survival, suggesting that C9-HRE astrocytes might have impaired capacity to support neurons. This lack of support might also partially involve impaired EV biogenesis in astrocytes, which has also been shown to take place in fibroblasts and motor neurons derived from C9-HRE carriers (Aoki et al., 2017; Varcianna et al., 2019). Addition of the ACM from the same C9-HRE astrocytes led to slightly decreased cell survival compared to EVs only, indicating that in addition to EVs, also other astrocyte-derived soluble factors might cause neurotoxicity (Varcianna et al., 2019). In addition, impaired autophagy initiation and increased levels of SOD1 in neurons have been proposed as possible underlying mechanisms of the neurotoxicity mediated by C9-HRE-containing astrocytes through secreted factors (Madill et al., 2017), but require further investigations. Since SOD1 is regulated by transcription factors, such as NFκB (Milani et al., 2011), which in turn are controlled by environmental stimuli, investigating the mechanism behind increased SOD1 levels might yield better understanding on mechanisms of astrocyte-mediated neurotoxicity. Since autophagy can be regulated via microRNAs (Shah et al., 2018), investigating whether microRNAs secreted by C9-HRE astrocytes are the underlying mechanism of the inhibition of autophagy initiation in co-cultured cells would be interesting in order to find therapeutic targets.
Notably, under stress conditions, such as increased extracellular adenosine levels, neurotoxicity might be even enhanced. This idea is supported by the finding that induced sporadic as well as C9-HRE astrocytes and neurons harbor lower levels of adenosine deaminase (ADA), which normally deaminates adenosine to inosine. This predisposition has been shown to lead to enhanced death of the induced astrocytes themselves as well as motor neurons when cultured together with C9-HRE or sporadic induced astrocytes. Several conditions can lead to elevated adenosine triphosphate (ATP) levels in the brain and these can be sensed by and activate microglia and astrocytes. Under normal conditions, excessive ATP can be metabolized. However, defective ATP metabolism, as e.g., during ADA deficiency, could lead to excessive glial activation and subsequent neuroinflammation. In addition, decreased ADA levels could disturb energy metabolism in astrocytes and result in impaired nutritional support for neurons by C9-HRE astrocytes (Allen et al., 2019).
Haploinsufficiency as a Potential Mechanism of Astrocyte-Mediated Neurotoxicity
In humans, two C9orf72 protein isoforms, long (~50 kDa) and short (~25 kDa), have been described. The levels of both protein isoforms are reduced in C9-HRE carrier tissues, including brain areas affected by neurodegeneration (Saberi et al., 2018). siRNA-mediated knockdown of both C9orf72 isoforms in U87 glioblastoma cells or normal human astrocytes has been shown to lead to the accumulation of p62 inclusions (Fomin et al., 2018), supporting the idea that loss of C9orf72 may lead to their formation. Also, reduced expression of pyruvate carboxylase (PC), EAAT1 and 2, and glutamine synthetase (GLUL) together with intracellular glutamate accumulation was observed, implying that disturbed glutamate de novo synthesis, uptake and conversion into glutamine, all crucial functions in astrocytes, may take place upon C9orf72 knockdown (Fomin et al., 2018). Decreased EAAT1 and EAAT2 levels suggest that C9-HRE astrocytes may not be able to efficiently take up excessive glutamate from synaptic cleft, which might lead to glutamate excitotoxicity, especially as induced motor neurons of C9-HRE ALS patients and C9orf72-deficient motor neurons have higher levels of glutamate receptors in neurites and dendritic spines (Shi et al., 2018). It was also observed that expression of endothelin (EDN) 1 as well as the levels of cytosolic and nuclear NFκB p65 were increased. The authors showed that the short C9orf72 isoform can bind to the predicted NFκB promoter binding site in EDN1 and further suggested that in C9orf72 knockdown cells, increased NFκB expression could lead to increased expression of EDN1, a negative regulator of EAAT2 expression (Fomin et al., 2018). Therefore, the mechanisms underlying C9-HRE astrocyte-mediated neurotoxicity might involve C9orf72 haploinsufficiency and p62 accumulation, and enhanced EDN1 expression and NFκB signaling, known to induce the expression of nitric oxide synthase (Fomin et al., 2018).
Knockdown of both C9orf72 isoforms in U251 human astroglioma cells increased transmembrane protein 106b (TMEM106b) and progranulin but not lysosomal-associated membrane protein (LAMP) 1 and cathepsin D protein levels (similar to microglia in C9orf72-deficient mice Sullivan et al., 2016, see below). Similar effect was not detected when the cells were only expressing C9-HRE (Nicholson et al., 2018). These findings implicate that C9orf72 haploinsufficiency may cause changes in astrocytic lysosomal pathways. Whether such effects can be observed in C9-HRE carriers and how they might affect astrocytic function still remains unknown. Taken together, these studies suggest that C9orf72 haploinsufficiency, resulting from the C9-HRE, may lead to p62 accumulation, which could also propagate to other cells (Madill et al., 2017). Additionally, changes in NFκB and EDN1 signaling, disturbed glutamate metabolism, and lysosomal alterations might be phenotypic features of C9-HRE astrocytes with potentially altered physiological functions (Figure 1).
Microglia in C9-FTLD and C9-ALS Models
Features of C9-HRE-Expressing Microglia
Depending on environmental cues, microglia can switch from resting to activated phenotype, characterized by elevated expression of CD68 (Choi et al., 2017; Hendrickx et al., 2017). Studies on post-mortem brains have shown increased microglial activation based on cell morphology and Iba1 and CD68 immunoreactivity in C9-FTLD and C9-ALS vs. sporadic FTLD and ALS cases. Significantly increased microglial activation according to number and morphology (ramified vs. ameoboid) of CD68-positive cells in frontal and temporal gray and white matter was detected in FTLD cases compared to controls, although there were no differences between C9-FTLD and sporadic FTLD cases (Lant et al., 2014). Also, augmented microglial activation based on CD68 immunoreactivity and cell morphology in the white matter of medulla and motor cortex of C9-ALS patients compared to non-C9-ALS cases has been reported. Iba1 immunoreactivity was also increased, indicating potentially increased number of microglia (Brettschneider et al., 2012). Higher CD68 immunoreactivity in the white matter of motor cortex, medulla, mid-crus cerebri, and lateral and anterior corticospinal tracts of C9-ALS patients (Cooper-Knock et al., 2012) and in the body and genu of corpus callosum of C9-HRE-carrying vs. non-carrying ALS patients has been detected (Cardenas et al., 2017), suggesting that microglial activation and infiltration in the brain might take place at least in late stages of C9-FTLD and C9-ALS. It would be crucial to assess at different stages of the disease process whether the phenotype of C9-HRE-carrying activated microglia is pro- or anti-inflammatory.
Iba1-positive microglia of C9-ALS patient post-mortem motor cortex and spinal cord contained enlarged lysosomes based on LAMP1 immunoreactivity compared to sporadic ALS cases (O'Rourke et al., 2016), suggesting lysosomal alterations in C9-HRE carriers. Total LAMP1 levels were not significantly changed in the frontal cortex of C9-ALS cases compared to sporadic ALS nor between FTLD and ALS and control samples (Umoh et al., 2018). This might suggest that (i) altered lysosomal morphology does not correlate with total LAMP1 levels or (ii) that microglia from spinal cord and frontal cortex show different lysosomal features. Enlarged microglial lysosomes may emerge under different conditions, such as cathepsin B and L inhibition and subsequent prevention of autophagosome-lysosome fusion (Jung et al., 2015), defects in lysosomal fission (Durchfort et al., 2012), progranulin deficiency (Evers et al., 2017) or TMEM106b overexpression (Nicholson and Rademakers, 2016), resulting in decreased lysosomal degradation capacity (Jung et al., 2015; Nicholson and Rademakers, 2016), and upon phagocytosis of fibrillar amyloid β, resulting in cathepsin B release (Halle et al., 2008). Further investigations are warranted related to the number of microglia with enlarged lysosomes and factors triggering such a phenotype, as well as understanding whether the enlarged lysosomes reflect dysfunction or improved function. Nevertheless, enhanced Iba1 and CD68 immunoreactivity and enlarged lysosomes may be considered as typical features of late stage C9-FTLD or C9-ALS microglia (Figure 2).
Neuron-Microglia Crosstalk Might Contribute to C9-HRE Microglia Phenotype
Expression of pathological C9-HRE in mouse cortex has been shown to upregulate Iba1 expression, but it is unclear whether this is due to cell-autonomous or non-cell autonomous effects (Nicholson et al., 2018). Significantly enhanced CD68 and Iba1 expression was detected at 6 months of age in the spinal cord of transgenic mice expressing poly-GA149 specifically in neurons. Increased microglial activation was absent in brain areas where the neurons did not exhibit poly-GA pathology. At this time point, no significant neuronal loss could be detected. However, the mice demonstrated enhanced TDP-43 phosphorylation, but no translocation or inclusions, and mild behavioral deficits, indicating that microglial activation might precede severe neuronal dysfunction. Interestingly, 1 month-old mice did not exhibit elevated Iba1 but already elevated CD68 expression (Schludi et al., 2017), implicating that enhanced CD68 expression in microglia may precede increased Iba1 expression. In contrast, expression of poly-GA50 did not increase Iba1 levels or cause TDP-43 pathology at 6 months of age, but the mice showed behavioral impairments and neurodegeneration (Zhang et al., 2016). Expression of poly-GR100 led to elevated Iba1 levels in mouse cortex and hippocampus, which peaked at 1.5 months of age. Notably, at this time, neuronal loss and brain atrophy, but no TDP-43 pathology, were already detectable in hippocampus and cortex (Zhang et al., 2018). These studies suggest that DRP length or type and/or concomitant additional factors, such as TDP-43 phosphorylation, might differentially regulate microglial Iba1 levels and activation. Also, neuron-microglia crosstalk might contribute to the activation of microglia.
Decreased C9orf72 Levels May Influence C9-HRE Microglia Phenotype
Human C9orf72 and its mouse ortholog 3110043O21Rik are strongly expressed in myeloid cells, including microglia (O'Rourke et al., 2016; Rizzu et al., 2016; Iyer et al., 2018). C9orf72−/− mice show severe autoimmune phenotypes, elevated levels of inflammatory cytokines [IL-12, IL-10, tumor necrosis factor (TNF) α, IL-17] and monocyte chemoattractant protein 1 in serum and pro-inflammatory macrophage polarization (Atanasio et al., 2016; O'Rourke et al., 2016; Sullivan et al., 2016). Microglia of C9orf72−/− mice showed phenotypes ranging from accumulation of enlarged lysosomes and enhanced LAMP1 immunoreactivity, strongly increased expression of IL-6 and IL-1β under basal conditions (O'Rourke et al., 2016) to no changes in LAMP1 or cathepsin D immunoreactivity (Sullivan et al., 2016). However, microglia from hemizygous mice were not reported to show significant increases in pro-inflammatory cytokine levels or lysosomal changes (O'Rourke et al., 2016). Furthermore, even in C9orf72−/− mice, no increase in Iba1 immunoreactivity or changes in the morphology, number, or distribution of microglia were detected in contrast to human post-mortem brain as described above (Koppers et al., 2015; Sullivan et al., 2016). On the other hand, antisense oligonucleotides targeting C9orf72 transcripts induced the mRNA levels of triggering receptor expressed on myeloid cells (TREM) 2, tyrosine kinase binding protein (TYROBP), C1q B chain (C1qb) and C3a receptor 1 (C3ar1), all predominantly expressed in microglia and regulating central microglial functions, including activation, pruning, and phagocytosis (Lagier-Tourenne et al., 2013), suggesting that reduced C9orf72 levels associate with alterations in these microglial functions. Altogether, the current data suggest that C9orf72 haploinsufficiency might not underlie the C9-ALS and C9-FTLD microglial phenotypes as assessed by Iba1 and CD68 immunoreactivity. Whether lysosomal changes resulting from reduced C9orf72 levels occur in microglia needs further investigation. C9orf72 knockout in HEK293T cells leads to enlarged lysosomes, suggesting that similar effects might also occur in other cell types (Amick et al., 2016). However, enhanced TREM2, TYROBP, C1qb, and C3ar1 expression might represent additional phenotypic features of C9-HRE microglia (Figure 2). Interestingly, also progranulin-deficient mice show enlarged lysosomes, increased CD68, TYROBP, TREM2 and complement factor expression (Evers et al., 2017) and Iba1 immunoreactivity (Tanaka et al., 2014).
Conclusions and Future Perspectives
Here, we have discussed the potential phenotypes of C9-HRE astrocytes and microglia based on the current literature. In general, FTLD and ALS are complex multifactorial diseases involving multi-cellular components. Thus, dysfunction of one cell type only might not be enough to trigger neurodegeneration but different cell types and their crosstalk are likely to contribute. Regarding C9-HRE-associated FTLD and ALS, astrocytes are suggested to mediate neurotoxic effects, but so far there is no conclusive experimental evidence of direct contribution of microglia to neurodegeneration in C9-FTLD and C9-ALS models. The reason for this might be that the current approaches have focused mostly on C9orf72 haploinsufficiency when modeling the C9-HRE-related effects in microglia. These studies have suggested that C9orf72 depletion, which leads to severe dysfunction of the immune system, might affect macrophages and microglia differently. Since neurons with C9-HRE-associated pathological features rather than C9orf72 haploinsufficiency might cause microglia activation, concomitant modeling of the loss-of-function and gain-of-toxic function mechanisms might help to decipher whether and how microglia may take part in C9-FTLD and C9-ALS pathogenesis. Also, since loss of C9orf72 has been linked to autoimmune phenotypes, it would be important to also investigate the crosstalk between the peripheral immune system and CNS-resident cells in the future. While the C9-HRE microglial phenotypes might result, at least partially, from neuron-microglia crosstalk, astrocytic neurotoxicity appears to derive from intrinsic factors, which then display non-cell autonomous deleterious effects on neurons. Since C9-HRE astrocytes show defects in EV-based communication with neurons, investigating whether the crosstalk between astrocytes and microglia of C9-HRE carriers is changed as well, would reveal further insights into mechanisms potentially underlying C9-HRE-associated ALS and FTLD. Future investigations are warranted to uncover potential spatial and temporal contributions of glia to onset and progression of C9-FTLD and C9-ALS. Also, even though C9-FTLD and C9-ALS share overlapping genetic background and pathological features, it will be interesting to find out the similarities and potential differences in the effects of microglia and astrocytes on the pathogenesis of these two diseases. As the presently available literature on glia in the context of C9-HRE is still limited, we hope that this summary of the current knowledge and the hypotheses presented will help to design future experiments for deciphering the so far poorly understood role of astrocytes and microglia in disease pathogenesis and for identifying potential novel biomarker and/or therapeutic target candidates for FTLD and ALS.
Author Contributions
HR and AH outlined and drafted the manuscript. HR, SL, NH, KK, AC, ES, MM, TN, AR, MH, and AH participated in the writing and editing the manuscript. All authors have accepted the contents of the submitted manuscript.
Funding
This study was supported by grants from the Academy of Finland, grant nos. 315459 (AH), 315460 (AR), 288659 (TN), and 307866 (MH); Yrjö Jahnsson Foundation, grant no. 20187070 (AH); ALS tutkimuksen tuki ry. registered association (HR, SL, NH); Sigrid Jusélius Foundation (MH); Maire Taponen Foundation (AC); Finnish Cultural Foundation (AC); Finnish Brain Foundation (KK); Finnish Medical Foundation (KK); Päivikki and Sakari Sohlberg Foundation (KK); the Strategic Neuroscience Funding of the University of Eastern Finland (AH, MH); and Neurocenter Finland—AlzTrans pilot project (MH).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
HR, SL, and NH are supported by the University of Eastern Finland Doctoral Program in Molecular Medicine (DPMM) and GenomMed. This publication is part of a project that has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 740264.
References
Allen, S. P., Hall, B., Castelli, L. M., Francis, L., Woof, R., Siskos, A. P., et al. (2019). Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 142, 586–605. doi: 10.1093/brain/awy353
Almeida, S., Gascon, E., Tran, H., Chou, H. J., Gendron, T. F., DeGroot, S., et al. (2013). Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 126, 385–399. doi: 10.1007/s00401-013-1149-y
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. doi: 10.1007/s00401-011-0911-2
Amick, J., Roczniak-Ferguson, A., and Ferguson, S. M. (2016). C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol. Biol. Cell 27, 3040–3051. doi: 10.1091/mbc.e16-01-0003
Aoki, Y., Manzano, R., Lee, Y., Dafinca, R., Aoki, M., Douglas, A. G. L., et al. (2017). C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140, 887–897. doi: 10.1093/brain/awx024
Ash, P. E., Bieniek, K. F, Gendron, T. F., Caulfield, T., Lin, W. L., DeJesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Atanasio, A., Decman, V., White, D., Ramos, M., Ikiz, B., Lee, H., et al. (2016). C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep. 6:23204. doi: 10.1038/srep23204
Bachiller, S., Jiménez-Ferrer, I., Paulus, A., Yang, Y., Swanberg, M., Deierborg, T., et al. (2018). Microglia in neurological diseases: a road map to brain-disease dependent-inflammatory response. Front. Cell. Neurosci. 12:488. doi: 10.3389/fncel.2018.00488
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi: 10.1038/s41582-018-0047-2
Barclay, A. N., Wright, G. J., Brooke, G., and Brown, M. H. (2002). CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 23, 285–290. doi: 10.1016/S.1471-4906(02)02223-8
Brettschneider, J., Arai, K., Del Tredici, K., Toledo, J. B., Robinson, J. L., Lee, E. B., et al. (2014). TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 128, 423–437. doi: 10.1007/s00401-014-1299-6
Brettschneider, J., Toledo, J. B., Van Deerlin, V. M., Elman, L., McCluskey, L., Lee, V. M., et al. (2012). Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS ONE 7:e39216. doi: 10.1371/journal.pone.0039216
Broe, M., Kril, J., and Halliday, G. M. (2004). Astrocytic degeneration relates to the severity of disease in frontotemporal dementia. Brain J. Neurol. 127, 2214–2220. doi: 10.1093/brain/awh250
Cardenas, A. M., Sarlls, J. E., Kwan, J. Y., Bageac, D., Gala, Z. S., Danielian, L. E., et al. (2017). Pathology of callosal damage in ALS: an ex-vivo, 7T diffusion tensor MRI study. NeuroImage Clin. 15, 200–208. doi: 10.1016/j.nicl.2017.04.024
Chen, W. W., Zhang, X., and Huang, W. J. (2016). Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 13, 3391–3396. doi: 10.3892/mmr.2016.4948
Chew, J., Gendron, T. F., Prudencio, M., Sasaguri, H., Zhang, Y. J., Castanedes-Casey, M., et al. (2015). Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348, 1151–1154. doi: 10.1126/science.aaa9344
Choi, J. Y., Kim, J. Y., Kim, J. Y., Park, J., Lee, W. T., and Lee, J. E. (2017). M2 phenotype microglia-derived cytokine stimulates proliferation and neuronal differentiation of endogenous stem cells in ischemic brain. Exp. Neurobiol. 26, 33–41. doi: 10.5607/en.2017.26.1.33
Cistaro, A., Pagani, M., Montuschi, A., Calvo, A., Moglia, C., Canosa, A., et al. (2014). The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur. J. Nucl. Med. Mol. Imaging 41, 844–852. doi: 10.1007/s00259-013-2667-5
Cooper-Knock, J., Green, C., Altschuler, G., Wei, W., Bury, J. J., Heath, P. R., et al. (2017). A data-driven approach links microglia to pathology and prognosis in amyotrophic lateral sclerosis. Acta Neuropathol Commun 5:23. doi: 10.1186/s40478-017-0424-x
Cooper-Knock, J., Hewitt, C., Highley, J. R., Brockington, A., Milano, A., Man, S., et al. (2012). Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain J. Neurol. 135, 751–764. doi: 10.1093/brain/awr365
Cooper-Knock, J., Higginbottom, A., Stopford, M. J., Highley, J. R., Ince, P. G., Wharton, S. B., et al. (2015). Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol. 130, 63–75. doi: 10.1007/s00401-015-1429-9
Cooper-Knock, J., Walsh, M. J., Higginbottom, A., Robin Highley, J., Dickman, M. J., Edbauer, D., et al. (2014). Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 137, 2040–2051. doi: 10.1093/brain/awu120
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
Deczkowska, A., Keren-Shaul, H., Weiner, A., Colonna, M., Schwartz, M., and Amit, I. (2018). Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell 173, 1073–1081. doi: 10.1016/j.cell.2018.05.003
DeJesus-Hernandez, M., Finch, N. A., Wang, X., Gendron, T. F., Bieniek, K. F., Heckman, M. G., et al. (2017). In-depth clinico-pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol. 134, 255–269. doi: 10.1007/s00401-017-1725-7
DeJesus-Hernandez, M., Mackenzie, I., Boeve, B., Boxer, A., Baker, M., Rutherford, N., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Donnelly, C. J., Zhang, P. W., Pham, J. T., Haeusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. doi: 10.1016/j.neuron.2013.10.015
Durchfort, N., Verhoef, S., Vaughn, M. B., Shrestha, R., Adam, D., Kaplan, J., et al. (2012). The enlarged lysosomes in beigej cells result from decreased lysosome fission and not increased lysosome fusion. Traffic 13, 108–119. doi: 10.1111/j.1600-0854.2011.01300.x
Evers, B. M., Rodriguez-Navas, C., Tesla, R. J., Prange-Kiel, J., Wasser, C. R., Yoo, K. S., et al. (2017). Lipidomic and transcriptomic basis of lysosomal dysfunction in progranulin deficiency. Cell Rep. 20, 2565–2574. doi: 10.1016/j.celrep.2017.08.056
Fatima, M., Tan, R., Halliday, G. M., and Kril, J. J. (2015). Spread of pathology in amyotrophic lateral sclerosis: assessment of phosphorylated TDP-43 along axonal pathways. Acta Neuropathol. Commun. 3:47. doi: 10.1186/s40478-015-0226-y
Fomin, V., Richard, P., Hoque, M., Li, C., Gu, Z., Fissore-O'Leary, M., et al. (2018). The C9ORF72 gene, implicated in amyotrophic lateral sclerosis and frontotemporal dementia, encodes a protein that functions in control of endothelin and glutamate signaling. Mol. Cell. Biol. 38:e00155–18. doi: 10.1128/MCB.00155-18
Franco, R., and Fernández-Suárez, D. (2015). Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 131, 65–86. doi: 10.1016/j.pneurobio.2015.05.003
Freibaum, B. D., and Taylor, J. P. (2017). The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front. Mol. Neurosci. 10:35. doi: 10.3389/fnmol.2017.00035
Gendron, T. F., Bieniek, K. F., Zhang, Y. J., Jansen-West, K., Ash, P. E., Caulfield, T., et al. (2013). Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 126, 829–844. doi: 10.1007/s00401-013-1192-8
Gendron, T. F., Josephs, K. A., and Petrucelli, L. (2010). Review: transactive response DNA-binding protein 43 (TDP-43): mechanisms of neurodegeneration. Neuropathol. Appl. Neurobiol. 36, 97–112. doi: 10.1111/j.1365-2990.2010.01060.x
Geng, W., and Liu, L. (2019). MiR-494 alleviates lipopolysaccharide (LPS)-induced autophagy and apoptosis in PC-12 cells by targeting IL-13. Adv. Clin. Exp. Med. 28, 85–94. doi: 10.17219/acem/76749
Gitik, M., Liraz-Zaltsman, S., Oldenborg, P., Reichert, F., and Rotshenker, S. (2011). Myelin down-regulates myelin phagocytosis by microglia and macrophages through interactions between CD47 on myelin and SIRPα (signal regulatory protein-α) on phagocytes. J. Neuroinflammation 8:24. doi: 10.1186/1742-2094-8-24
Gitler, A. D., and Tsuiji, H. (2016). There has been an awakening: emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res. 1647, 19–29. doi: 10.1016/j.brainres.2016.04.004
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014. doi: 10.1212/WNL.0b013e31821103e6
Haidet-Phillips, A. M., Hester, M. E., Miranda, C. J., Meyer, K., Braun, L., Frakes, A., et al. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828. doi: 10.1038/nbt.1957
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-b. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
Hallmann, A. L., Araúzo-Bravo, M. J., Mavrommatis, L., Ehrlich, M., Röpke, A., Brockhaus, J., et al. (2017). Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci. Rep. 7:42991. doi: 10.1038/srep42991
Hendrickx, D. A. E., van Eden, C. G., Schuurman, K. G., Hamann, J., and Huitinga, I. (2017). Staining of HLA-DR, Iba1 and CD68 in human microglia reveals partially overlapping expression depending on cellular morphology and pathology. J. Neuroimmunol. 309, 12–22. doi: 10.1016/j.jneuroim.2017.04.007
Ismail, A., Cooper-Knock, J., Highley, J. R., Milano, A., Kirby, J., Goodall, E., et al. (2013). Concurrence of multiple sclerosis and amyotrophic lateral sclerosis in patients with hexanucleotide repeat expansions of C9ORF72. J. Neurol. Neurosurg. Psychiatr. 84, 79–87. doi: 10.1136/jnnp-2012-303326
Iyer, S., Acharya, K. R., and Subramanian, V. (2018). A comparative bioinformatic analysis of C9orf72. Peer J. 6:e4391. doi: 10.7717/peerj.4391
Jang, E., Kim, J. H., Lee, S., Kim, J. H., Seo, J. W., Jin, M., et al. (2013). Phenotypic polarization of activated astrocytes: the critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. J. Immunol. 191, 5204–5219. doi: 10.4049/jimmunol.1301637
Jiang, J., Zhu, Q., Gendron, T. F., Saberi, S., McAlonis-Downes, M., Seelman, A., et al. (2016). Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 Is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550. doi: 10.1016/j.neuron.2016.04.006
Jung, M., Lee, J., Seo, H. Y., Lim, J. S., and Kim, E. K. (2015). Cathepsin inhibition-induced lysosomal dysfunction enhances pancreatic beta-cell apoptosis in high glucose. PLoS ONE 10:e0116972. doi: 10.1371/journal.pone.0116972
Koppers, M., Blokhuis, A. M., Westeneng, H. J., Terpstra, M. L., Zundel, C. A., Vieira de Sa, R., et al. (2015). C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 78, 426–438. doi: 10.1002/ana.24453
Krabbe, G., Minami, S. S., Etchegaray, J. I., Taneja, P., Djukic, B., Davalos, D., et al. (2017). Microglial NFκB-TNFα hyperactivation induces obsessive–compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc. Natl. Acad. Sci. U S A. 114, 5029–5034. doi: 10.1073/pnas.1700477114
Lagier-Tourenne, C., Baughn, M., Rigo, F., Sun, S., Liu, P., Li, H. R., et al. (2013). Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U S A. 110, E4530–E4539. doi: 10.1073/pnas.1318835110
Lant, S. B., Robinson, A. C., Thompson, J. C., Rollinson, S., Pickering-Brown, S., Snowden, J. S., et al. (2014). Patterns of microglial cell activation in frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 40, 686–696. doi: 10.1111/nan.12092
Lee, J., Hyeon, S. J., Im, H., Ryu, H., Kim, Y., and Ryu, H. (2016). Astrocytes and microglia as non-cell autonomous players in the pathogenesis of ALS. Exp. Neurobiol. 25, 233–240. doi: 10.5607/en.2016.25.5.233
Liu, Y., Pattamatta, A., Zu, T., Reid, T., Bardhi, O., Borchelt, D., et al. (2016). C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 90, 521–534. doi: 10.1016/j.neuron.2016.04.005
Mackenzie, I. R., Arzberger, T., Kremmer, E., Troost, D., Lorenzl, S., Mori, K., et al. (2013). Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 126, 859–879. doi: 10.1007/s00401-013-1181-y
Madill, M., McDonagh, K., Ma, J., Vajda, A., McLoughlin, P., O'Brien, T., et al. (2017). Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 10, 22–12. doi: 10.1186/s13041-017-0300-4
Mahoney, C. J., Beck, J., Rohrer, J. D., Lashley, T., Mok, K., Shakespeare, T., et al. (2012). Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain 135, 736–750. doi: 10.1093/brain/awr361
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G., Waite, A., Rollinson, S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330. doi: 10.1016/S1474-4422(12)70043-1
May, S., Hornburg, D., Schludi, M. H., Arzberger, T., Rentzsch, K., Schwenk, B. M., et al. (2014). C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 128, 485–503. doi: 10.1007/s00401-014-1329-4
Meyer, K., Ferraiuolo, L., Miranda, C. J., Likhite, S., McElroy, S., Renusch, S., et al. (2014). Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. U S A. 111, 829–832. doi: 10.1073/pnas.1314085111
Michell-Robinson, M. A., Touil, H., Healy, L. M., Owen, D. R., Durafourt, B. A., Bar-Or, A., et al. (2015). Roles of microglia in brain development, tissue maintenance and repair. Brain 138, 1138–1159. doi: 10.1093/brain/awv066
Milani, P., Gagliardi, S., Cova, E., and Cereda, C. (2011). SOD1 transcriptional and posttranscriptional regulation and its potential implications in ALS. Neurol. Res. Int. 2011:458427. doi: 10.1155/2011/458427
Minami, S. S., Shen, V., Le, D., Krabbe, G., Asgarov, R., Perez-Celajes, L., et al. (2015). Reducing inflammation and rescuing FTD-related behavioral deficits in progranulin-deficient mice with α7 nicotinic acetylcholine receptor agonists. Biochem. Pharmacol. 97, 454–462. doi: 10.1016/j.bcp.2015.07.016
Mizielinska, S., Lashley, T., Norona, F. E., Clayton, E. L., Ridler, C. E., Fratta, P., et al. (2013). C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 126, 845–857. doi: 10.1007/s00401-013-1200-z
Mori, K., Arzberger, T., Grässer, F. A., Gijselinck, I., May, S., Rentzsch, K., et al. (2013b). Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893. doi: 10.1007/s00401-013-1189-3
Mori, K., Lammich, S., Mackenzie, I. R., Forné, I., Zilow, S., Kretzschmar, H., et al. (2013a). hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 125, 413–423. doi: 10.1007/s00401-013-1088-7
Murray, M. E., DeJesus-Hernandez, M., Rutherford, N. J., Baker, M., Duara, R., Graff-Radford, N. R., et al. (2011). Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 122, 673–690. doi: 10.1007/s00401-011-0907-y
Nicholson, A. M., and Rademakers, R. (2016). What we know about TMEM106B in neurodegeneration. Acta Neuropathol. 132, 639–651. doi: 10.1007/s00401-016-1610-9
Nicholson, A. M., Zhou, X., Perkerson, R. B., Parsons, T. M., Chew, J., Brooks, M., et al. (2018). Loss of Tmem106b is unable to ameliorate frontotemporal dementia-like phenotypes in an AAV mouse model of C9ORF72-repeat induced toxicity. Acta Neuropathol. Commun. 6:42. doi: 10.1186/s40478-018-0545-x
Nonaka, T., Masuda-Suzukake, M., Hosokawa, M., Shimozawa, A., Hirai, S., Okado, H., et al. (2018). C9ORF72 dipeptide repeat poly-GA inclusions promote intracellular aggregation of phosphorylated TDP-43. Hum. Mol. Genet. 27, 2658–2670. doi: 10.1093/hmg/ddy174
Nordin, A., Akimoto, C., Wuolikainen, A., Alstermark, H., Jonsson, P., Birve, A., et al. (2015). Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum. Mol. Genet. 24, 3133–3142. doi: 10.1093/hmg/ddv064
Oeckl, P., Weydt, P., Steinacker, P., Anderl-Straub, S., Nordin, F., Volk, A. E., et al. (2019). Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J. Neurol. Neurosurg. Psychiatr. 90, 4–10. doi: 10.1136/jnnp-2018-318868
Onyike, C. U., and Diehl-Schmid, J. (2013). The epidemiology of frontotemporal dementia. Int. Rev. Psychiatry 25, 130–137. doi: 10.3109/09540261.2013.776523
O'Rourke, J. G., Bogdanik, L., Yáñez, A., Lall, D., Wolf, A. J., Muhammad, A. K., et al. (2016). C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329. doi: 10.1126/science.aaf1064
Parker, S. E., Hanton, A. M., Stefanou, S. N., Noakes, P. G., Woodruff, T. M., and Lee, J. D. (2019). Revisiting the role of the innate immune complement system in ALS. Neurobiol. Dis. 127, 223–232. doi: 10.1016/j.nbd.2019.03.003
Peters, O., Cabrera, G., Tran, H., Gendron, T., McKeon, J., Metterville, J., et al. (2015). Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 88, 902–909. doi: 10.1016/j.neuron.2015.11.018
Prudencio, M., Belzil, V. V., Batra, R., Ross, C. A., Gendron, T. F., Pregent, L. J., et al. (2015). Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18, 1175–1182. doi: 10.1038/nn.4065
Radford, R. A., Morsch, M., Rayner, S. L., Cole, N. J., Pountney, D. L., and Chung, R. S. (2015). The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front. Cell. Neurosci. 9:414. doi: 10.3389/fncel.2015.00414
Rama Rao, K. V., and Kielian, T. (2015). Neuron–astrocyte interactions in neurodegenerative diseases: role of neuroinflammation. Clin. Exp. Neuroimmunol. 6, 245–263. doi: 10.1111/cen3.12237
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477. doi: 10.1093/brain/awr179
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rizzu, P., Blauwendraat, C., Heetveld, S., Lynes, E. M., Castillo-Lizardo, M., Dhingra, A., et al. (2016). C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol. Commun. 4:37. doi: 10.1186/s40478-016-0306-7
Saberi, S., Stauffer, J. E., Jiang, J., Garcia, S. D., Taylor, A. E., Schulte, D., et al. (2018). Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 135, 459–474. doi: 10.1007/s00401-017-1793-8
Sareen, D., O'Rourke, J. G., Meera, P., Muhammad, A. K., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5:208ra149. doi: 10.1126/scitranslmed.3007529
Schipper, L. J., Raaphorst, J., Aronica, E., Baas, F., de Haan, R., de Visser, M., et al. (2016). Prevalence of brain and spinal cord inclusions, including dipeptide repeat proteins, in patients with the C9ORF72 hexanucleotide repeat expansion: a systematic neuropathological review. Neuropathol. Appl. Neurobiol. 42, 547–560. doi: 10.1111/nan.12284
Schludi, M., May, S., Grässer, F., Rentzsch, K., Kremmer, E., Küpper, C., et al. (2015). Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 130, 537–555. doi: 10.1007/s00401-015-1450-z
Schludi, M. H., Becker, L., Garrett, L., Gendron, T. F., Zhou, Q., Schreiber, F., et al. (2017). Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol. 134, 241–254. doi: 10.1007/s00401-017-1711-0
Sellier, C., Campanari, M. L., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Shah, S. Z. A., Zhao, D., Hussain, T., Sabir, N., and Yang, L. (2018). Regulation of MicroRNAs-mediated autophagic flux: a new regulatory avenue for neurodegenerative diseases with focus on prion diseases. Front. Aging Neurosci. 10:139. doi: 10.3389/fnagi.2018.00139
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W. H., Hung, S. T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Shinozaki, Y., Shibata, K., Yoshida, K., Shigetomi, E., Gachet, C., Ikenaka, K., et al. (2017). Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y1 receptor downregulation. Cell Rep. 19, 1151–1164. doi: 10.1016/j.celrep.2017.04.047
Simón-Sánchez, J., Dopper, E. G., Cohn-Hokke, P. E., Hukema, R. K., Nicolaou, N., Seelaar, H., et al. (2012). The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 135, 723–35. doi: 10.1093/brain/awr353
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4:51. doi: 10.1186/s40478-016-0324-5
Szepesi, Z., Manouchehrian, O., Bachiller, S., and Deierborg, T. (2018). Bidirectional microglia-neuron communication in health and disease. Front. Cell. Neurosci. 12:323. doi: 10.3389/fncel.2018.00323
Tanaka, Y., Chambers, J. K., Matsuwaki, T., Yamanouchi, K., and Nishihara, M. (2014). Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol. Commun. 2:78. doi: 10.1186/s40478-014-0078-x
Taylor, J. P., Brown, R. H. Jr., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Umoh, M. E., Dammer, E. B., Dai, J., Duong, D. M., Lah, J. J., Levey, A. I., et al. (2018). A proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 10, 48–62. doi: 10.15252/emmm.201708202
van Blitterswijk, M., DeJesus-Hernandez, M., Niemantsverdriet, E., Murray, M. E., Heckman, M. G., Diehl, N. N., et al. (2013). Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 12, 978–988. doi: 10.1016/S1474-4422(13)70210-2
Varcianna, A., Myszczynska, M. A., Castelli, L. M., O'Neill, B., Kim, Y., Talbot, J., et al. (2019). Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. EBioMedicine 40, 626–635. doi: 10.1016/j.ebiom.2018.11.067
Verkhratsky, A., and Nedergaard, M. (2018). Physiology of astroglia. Physiol. Rev. 98, 239–389. doi: 10.1152/physrev.00042.2016
Westergard, T., Jensen, B. K., Wen, X., Cai, J., Kropf, E., Iacovitti, L., et al. (2016). Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 17, 645–652. doi: 10.1016/j.celrep.2016.09.032
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zhang, Y. J, Gendron, T. F., Grima, J. C., Sasaguri, H., Jansen-West, K., Xu, Y. F., et al. (2016). C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 19, 668– 677. doi: 10.1038/nn.4272
Zhang, Y. J., Gendron, T. F., Ebbert, M. T. W., O'Raw, A. D., Yue, M., Jansen-West, K., et al. (2018). Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 24, 1136–1142. doi: 10.1038/s41591-018-0071-1
Zu, T., Gibbens, B., Doty, N. S., Gomes-Pereira, M., Huguet, A., Stone, M. D., et al. (2011). Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U S A. 108, 260–265. doi: 10.1073/pnas.1013343108
Keywords: amyotrophic lateral sclerosis, astrocyte, C9orf72, C9orf72 expansion, frontotemporal lobar degeneration, microglia, neurodegeneration
Citation: Rostalski H, Leskelä S, Huber N, Katisko K, Cajanus A, Solje E, Marttinen M, Natunen T, Remes AM, Hiltunen M and Haapasalo A (2019) Astrocytes and Microglia as Potential Contributors to the Pathogenesis of C9orf72 Repeat Expansion-Associated FTLD and ALS. Front. Neurosci. 13:486. doi: 10.3389/fnins.2019.00486
Received: 28 February 2019; Accepted: 29 April 2019;
Published: 15 May 2019.
Edited by:
Jean-Marc Gallo, King's College London, United KingdomReviewed by:
Laura Ferraiuolo, University of Sheffield, United KingdomChiara F. Valori, German Center for Neurodegenerative Diseases (DZNE), Germany
Copyright © 2019 Rostalski, Leskelä, Huber, Katisko, Cajanus, Solje, Marttinen, Natunen, Remes, Hiltunen and Haapasalo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annakaisa Haapasalo, YW5uYWthaXNhLmhhYXBhc2Fsb0B1ZWYuZmk=