 Francesco Gentile1
Francesco Gentile1 Stefania Scarlino1Yuri Matteo Falzone1,2
Stefania Scarlino1Yuri Matteo Falzone1,2 Christian Lunetta3
Christian Lunetta3 Lucio Tremolizzo4
Lucio Tremolizzo4 Angelo Quattrini1†
Angelo Quattrini1† Nilo Riva1,2*†
Nilo Riva1,2*†- 1Experimental Neuropathology Unit, Division of Neuroscience, Institute of Experimental Neurology – San Raffaele Scientific Institute, Milan, Italy
- 2Department of Neurology, San Raffaele Scientific Institute, Milan, Italy
- 3NeuroMuscular Omnicentre, Serena Onlus Foundation, Milan, Italy
- 4Neurology Unit, ALS Clinic, San Gerardo Hospital, University of Milano-Bicocca, Monza, Italy
Although amyotrophic lateral sclerosis (ALS) has been considered as a disorder of the motor neuron (MN) cell body, recent evidences show the non-cell-autonomous pathogenic nature of the disease. Axonal degeneration, loss of peripheral axons and destruction of nerve terminals are early events in the disease pathogenic cascade, anticipating MN degeneration, and the onset of clinical symptoms. Therefore, although ALS and peripheral axonal neuropathies should be differentiated in clinical practice, they also share damage to common molecular pathways, including axonal transport, RNA metabolism and proteostasis. Thus, an extensive evaluation of the molecular events occurring in the peripheral nervous system (PNS) could be fundamental to understand the pathogenic mechanisms of ALS, favoring the discovery of potential disease biomarkers, and new therapeutic targets.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common and severe form of motor neuron disease (MND), an heterogeneous group of disorders defined by prominent motor neuron (MN) degeneration (Saberi et al., 2015; Riva et al., 2016). Recent evidence has challenged such traditional view of selective neuronal loss demonstrating widespread extra-motor involvement in ALS and implying that neuronal populations other than MNs may also be affected (Geser et al., 2008; Brettschneider et al., 2013; McCluskey et al., 2014). Furthermore, an expanded clinical spectrum has now been recognized, as overlapping phenotypes with other neuromuscular disorders (Sabatelli et al., 2016), including peripheral neuropathies, have been described (Rajabally and Jacob, 2008; Bhatt et al., 2009; Sawa et al., 2012). Although in clinical practice MNDs should be differentiated from other peripheral nervous system (PNS) disorders, sensory and autonomic neurons in the dorsal root ganglia (DRG) and lower MNs in the ventral horns share important challenges, as proper stability and functioning of their long projections throughout the body requires a protective environment and efficient communication between the central nervous system (CNS) and the outermost areas of these cells. Nonetheless, differences in morphology and function may confer different patterns of resistance or vulnerability to specific stressors.
The aim of this review is to dissect PNS involvement in ALS, analyzing the evidence from clinical and pathological data to genetics in order to provide novel pathophysiological insights and potential implication for therapeutic strategies.
Diagnostic Challenges in ALS and ALS-Mimics
In the absence of pathognomonic diagnostic biomarkers, ALS diagnosis relies on clinical findings suggestive of selective upper, and lower MN (LMN) lesion and the exclusion of alternative causes (Ludolph et al., 2015). The wide clinical heterogeneity of the disease led to the development of several phenotypic classifications, mainly dependent on the distribution of UMN and LMN lesions. In patients presenting with isolated signs of LMN involvement (LMN Syndrome – LMNS), the differential diagnosis may be more challenging since the primary disease target may be the cell soma, the axon and its myelin, the neuromuscular junction (NMJ) or the muscle (Riva et al., 2011b; Garg et al., 2017; Muller et al., 2018). Such disorders not only share the motor unit as a common target of damage, but may be potentially underpinned by common pathogenic mechanisms. Within this context, ALS, with special reference to its restricted LMN phenotypes, such as flair legs and flail arm syndromes, shows high overlap in terms of clinical presentation, targets and mechanisms of damage with hereditary axonal neuropathies, such as axonal Charcot-Marie-Tooth neuropathy (CMT2) and distal hereditary motor neuropathy (dHMN) (Fischer and Glass, 2007). Besides the different rate of progression and spreading, distinction between these disorders could be difficult especially in the early phase, as clinical and neurophysiologic studies may be strikingly similar and may not able to discern the primary lesion site. Nonetheless, a recent study supported the value of the electroneurographic split hand index has recently been suggested for distinguishing ALS from mimic disorders (Menon et al., 2014). Moreover, pes cavus, traditionally considered a distinguishing clinical sign of CMT, has also been anecdotally reported in ALS patients (Ceroni et al., 2001), and is estimated to be as high as 2% our case series (data not published). Notably, the percentage of misdiagnosis in degenerative LMNS has been reported to be as high as 19%; moreover, up to 10% of patients initially diagnosed as ALS are ultimately re-diagnosed as having another disease, including peripheral neuropathy (Davenport et al., 1996; Traynor et al., 2000; Visser et al., 2002). Therefore, in some cases, MND diagnosis remains uncertain, and only follow up can lead to a satisfying level of diagnostic confidence. Of note, we previously showed that biopsy of obturator nerve may be useful in the distinction between ALS and motor neuropathies (Corbo et al., 1997; Riva et al., 2011b). Signs of acute axonal damage, focal/multifocal fiber loss and absent or scarce axonal regeneration are consistently observed in motor nerve biopsies from patients with ALS (Figure 1A), while neuropathies show a higher regeneration capacity and may present signs of myelin damage, inflammation, or pathologic deposits (Benedetti et al., 2010).
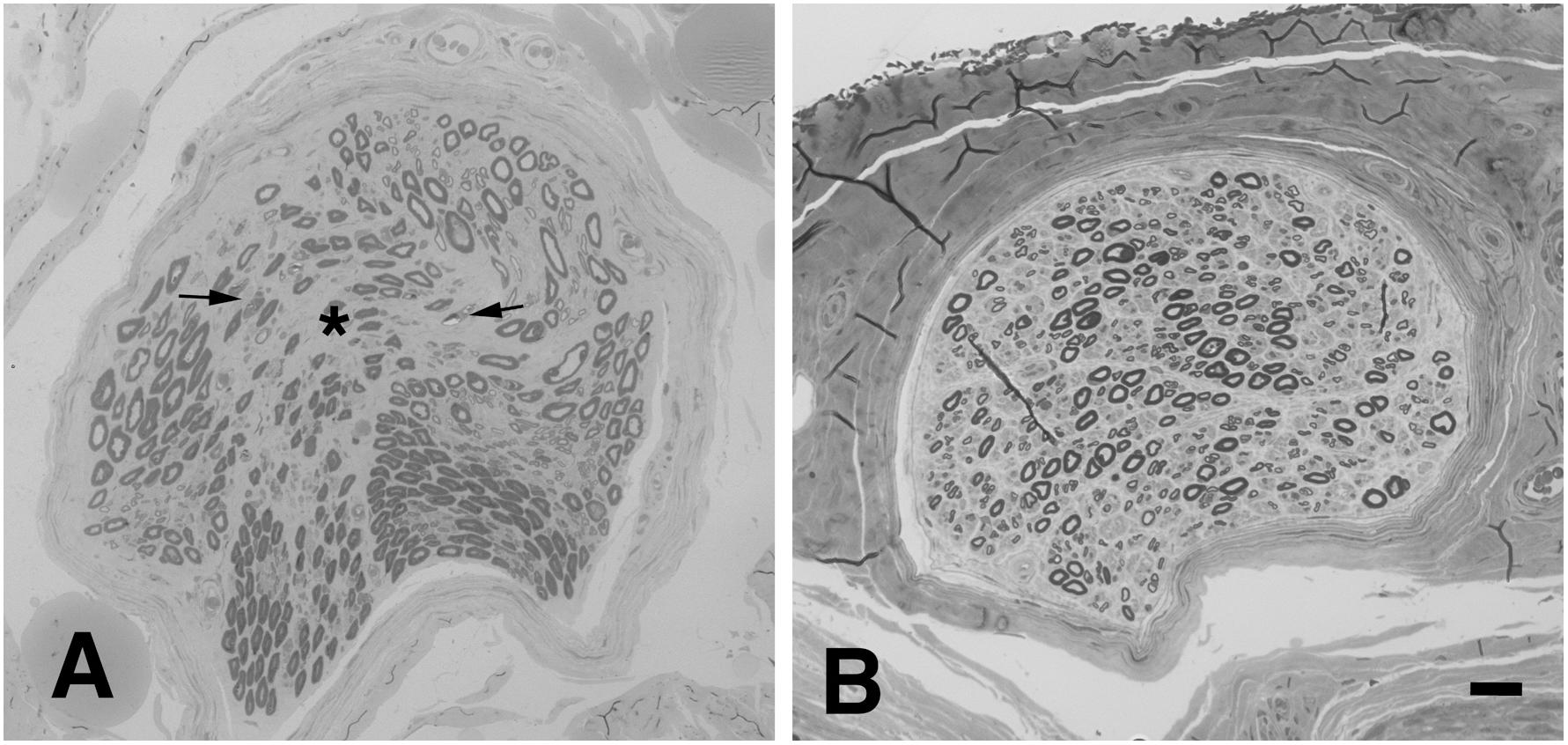
Figure 1. Representative neuropathological cases. (A) Transverse semi-thin sections of motor nerve biopsy (obturator nerve), and sural sensory nerve biopsy (B) from a patients with MND. (A) Focal loss of myelinated nerve fibers is evident (asterisk) in the endoneurium together with signs of acute axonal degeneration (myelin ovoids, arrows); no signs of regeneration are observed. (B) In sural sensory nerve biopsies a diffuse mild reduction of large nerve fibers is observed. Bar: 20 μm.
The PNS as a Converging Point in ALS
Evidence of Peripheral Nervous System Involvement in Human ALS
Although the circumstances leading to MN damage in ALS are still unknown, different mechanisms have been proposed to explain disease onset and spread. The dying-forward hypothesis states that the primary site of damage resides in the cell soma and subsequently spreads to the peripheral compartments. In the recent years, the dying-back pattern of degeneration has obtained a large attention in the context of ALS pathophysiology (Fischer et al., 2004), suggesting that ALS is a distal axonopathy, a pattern typically seen in peripheral neuropathies with a distal to proximal gradient of damage (England and Asbury, 2004). Therefore, considering that both motor and sensory nerves share mechanisms of axonal degeneration, it may be plausible that in ALS signs of PNS dysfunction may also extend to the sensory system. Notably, other neurodegenerative diseases such as Parkinson’s disease and Alzheimer’s disease show evidence of distal axonopathy (Arendt, 2009; Chung et al., 2009).
While diagnostic assessment in ALS relays on the assumption that non-motor components of the PNS are usually spared, increasing amount of evidence suggests the presence of sensory and autonomic dysfunction in ALS. Sensory symptoms are reported in about 2–30% of cases (Gubbay et al., 1985; Hammad et al., 2007). Numbness, tingling and pain are the most common symptoms reported. Decreased vibration sense and abnormal pain and temperature thresholds are rarely observed during standard examination, however quantitative sensory testing shows abnormal findings in about 10% of patients, rising to almost half of the cases in advanced stages of the disease (Truini et al., 2015; Isak et al., 2017).
Autonomic dysfunction has been reported in 5–30% of cases, while quantitative autonomic testing detected abnormal sudomotor and cardiovagal response in up to 50% of patients (McCluskey et al., 2014; Piccione et al., 2015). Abnormalities in either sensory nerve amplitudes or nerve conduction velocity may be apparent in about 20% of cases (Mondelli et al., 1993; Pugdahl et al., 2008), with higher frequencies reported in patients with longer disease duration (Pugdahl et al., 2007; Isak et al., 2016).
Pathologic studies of the sural nerve in ALS have demonstrated a mild reduction of the number of large nerve fibers (Figure 1B) in most of the examined cases (Bradley et al., 1983; Hammad et al., 2007; Devigili et al., 2011; Luigetti et al., 2012). Clustering of mitochondria, dilation of small vesicles and accumulation of neurofilaments (NF) have also been reported (Dyck et al., 1975; Di Trapani et al., 1987; Heads et al., 1991). Skin biopsy shows in about 80% of ALS patients a reduction of intraepidermal nerve fiber density (Weis et al., 2011; Truini et al., 2015; Dalla Bella et al., 2016; Nolano et al., 2017) and focal axonal swellings, suggesting axonal degeneration (Isak et al., 2017). Interestingly, small nerve fibers in the cornea are also reduced in ALS patients, correlating with bulbar disability scores (Ferrari et al., 2014). Finally, a significant neuronal loss has been observed in DRG of ALS patients, with preferential involvement of large-sized cells (Kawamura et al., 1981). Notably, evidence of pTDP-43 pathology has been shown in Clarke’s column, intermediolateral nucleus and dorsal root ganglia in a subset of cases, proving that these structures are also affected (Nishihira et al., 2008; Brettschneider et al., 2014).
The Role of Genetics in ALS: At the Borders of the Disease Spectrum
The knowledge about ALS genetics has had a relevant acceleration in the last decade, with more than 30 genes identified as pathogenic and more than 100 as ALS-related (Riva et al., 2016). The first gene identified as a cause of ALS was SOD1 in 1993 (Rosen et al., 1993) and together with TARDBP (Sreedharan et al., 2008), FUS (Kwiatkowski et al., 2009), and chromosome 9 open reading frame 72 (C9orf72) (DeJesus-Hernandez et al., 2011; Renton et al., 2011), represent the most common ALS-related genes, covering up to 70% of familial and 10% of sporadic cases (Riva et al., 2016; Chia et al., 2018). ALS-key genes have been associated with almost all clinical ALS phenotypes, although patients with SOD1, and FUS mutations may show preferential LMN involvement (Waibel et al., 2013; Picher-Martel et al., 2016).
The frequency of gene variants in cases with extra-motor involvement is significantly higher compared to the pure ALS phenotype, and has been associated with poorer survival (McCluskey et al., 2014). Prominent sensory involvement had been described in rare mutated cases, with SOD1 and TARDBP genes being reported most frequently (Camdessanche et al., 2011).
Many of the genes involved in ALS are associated also with other neurological disorders sharing common targets of neurodegeneration, such as hereditary spastic paraplegia (HSP), axonal CMT neuropathy and dHMN. Among the genes involved in dHMN and axonal CMT there are Berardinelli-Seip Congenital Lipodystrophy 2 (BSCL2), neurofilament light (NEFL), transient receptor potential cation channel subfamily V member 4 (TRPV4), heat shock protein family B1, B3, and B8 (HSPB1, HSPB3, and HSPB8) (Martini et al., 2000; Rossor et al., 2012). Despite the discrepancy between the major genes mutated in hereditary neuropathies and ALS, each of them exerts pleiotropic functions in neuronal homeostasis, including RNA metabolism, protein quality control, axonal transport, stress response. Therefore, generation of a spectrum of clinical phenotypes from alteration in master genes, broadly involved in key neuronal metabolic pathways, could be expected (Table 1).
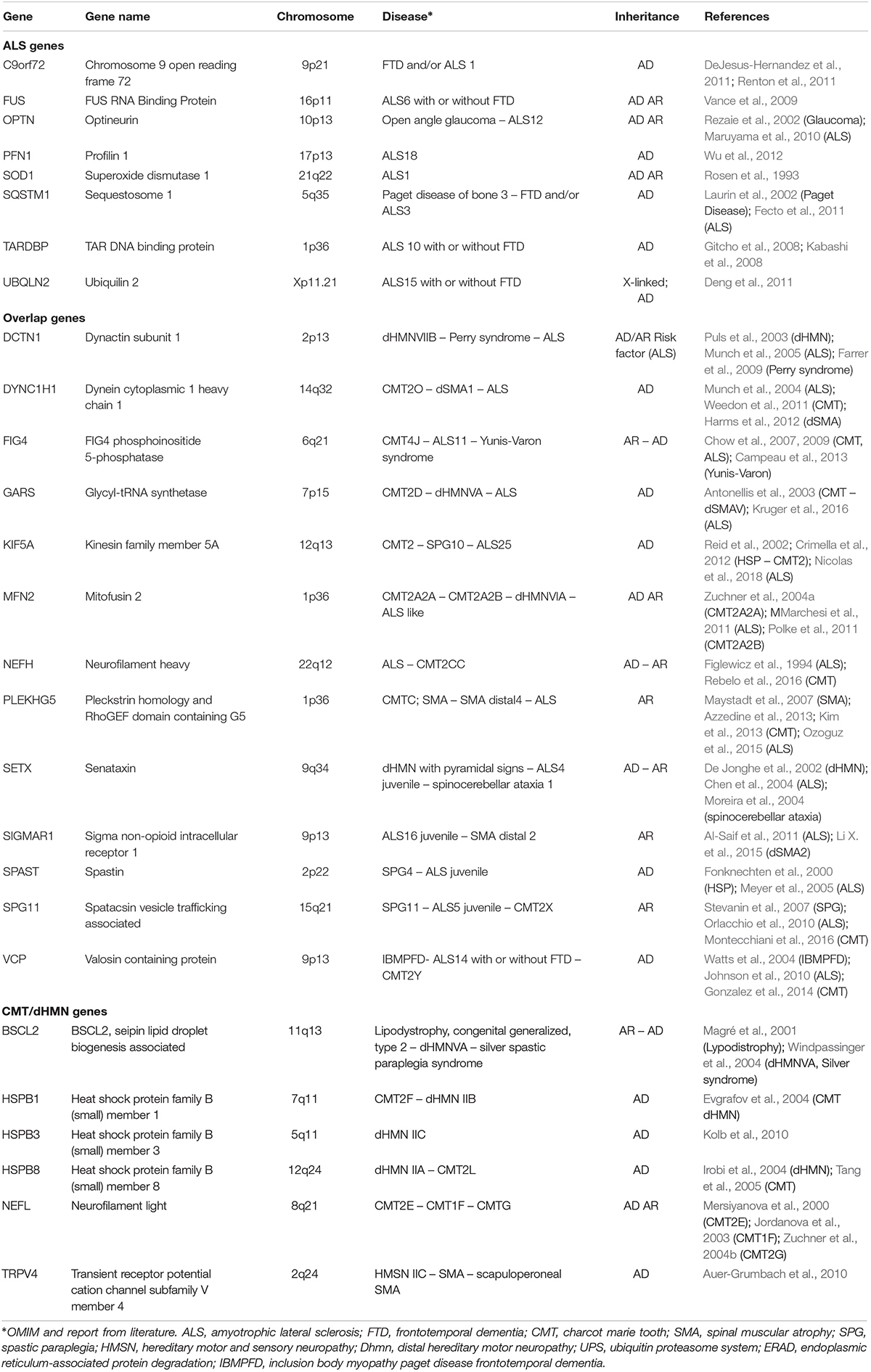
Table 1. Key genes associated with ALS, hereditary neuropathy, and overlapping phenotypes.
Neurofilaments (NF) are composed of three subunits defined by their molecular weight and encoded by the NF light (NEFL), medium (NEFM), and heavy (NEFH) genes (Lieberburg et al., 1989). NFs are specifically expressed in neurons and are the most abundant cytoskeletal components of myelinated axons, regulating their caliber, growth, and conduction rate (Hoffman et al., 1987). Mutations in NEFL are known to cause both axonal and demyelinating forms of CMT with different phenotypes, including pyramidal signs (Mersiyanova et al., 2000; Jordanova et al., 2003; Rebelo et al., 2016; Jacquier et al., 2017). Mutations in NEFH gene are involved in the pathogenesis of sporadic ALS (Figlewicz et al., 1994; Al-Chalabi et al., 1999) but also in CMT (Jacquier et al., 2017; Nam et al., 2017).
The cellular abundance of PI(3,5)P2, a phosphoinositide involved in the control of vesicles trafficking, is regulated by a phosphoinositide 5-phosphatase encoded by the FIG4 gene. CMT4J cases, clinically characterized by early onset and aggressive disease progression, have been associated with by an autosomal dominant pattern of transmission and by biallelic FIG4 mutations (Nicholson et al., 2011). Notably, heterozygous autosomal dominant FIG4 variants have been more recently associated with ALS and identified as ALS11 (Osmanovic et al., 2017).
The valosin containing protein (VCP) is member of the AAA ATPase family of proteins. This protein is ubiquitously expressed, and it is implicated in multiple cellular processes, such as cell survival (Vandermoere et al., 2006; Braun and Zischka, 2008), stress response and DNA and protein quality control (DeLaBarre et al., 2006; Ju et al., 2009; Weihl et al., 2009). Mutations in VCP have been described in patients with autosomal dominant inclusion body myopathy (IBM) associated with Paget disease and fronto-temporal dementia (FTD) (IBMPFD) (Watts et al., 2004), pure ALS patients (Johnson et al., 2010; Miller et al., 2012), and recently also CMT (Gonzalez et al., 2014; Jerath et al., 2015).
The exact function of senataxin (SETX) is unknown but it may be involved in RNA metabolism. Studies have shown a role in DNA transcription and repair (Suraweera et al., 2009; Skourti-Stathaki et al., 2011). Mutations in STX have been described in ataxia-ocular apraxia 2 (AOA2) (Moreira et al., 2004; Duquette et al., 2005; Fogel and Perlman, 2006;Arning et al., 2008; Airoldi et al., 2010; Fogel et al., 2014), autosomal dominant juvenile ALS (Chen et al., 2004; Zhao et al., 2009; Avemaria et al., 2011; Arning et al., 2013; Tripolszki et al., 2017), and in dHMN with pyramidal features (De Jonghe et al., 2002).
The Spastic Paraplegia 11 gene (SPG11) encodes the spatacsin protein, selectively expressed in neuron with a role in axonal growth, transport, and cytoskeletal stability (Perez-Branguli et al., 2014). SPG11 variants were first described in patients with autosomal recessive spastic paraplegia 11 with thin corpus callosum (Hehr et al., 2007). Then, descriptions in juvenile ALS (ALS5) and in CMT2X were also reported (Orlacchio et al., 2010; Daoud et al., 2012; Iskender et al., 2015; Montecchiani et al., 2016).
The dynactin subunit 1 (DCTN1) and kinesin family member 5A (KIF5A) genes encode for dynactin and kinesin subunits, involved in retrograde and anterograde axonal transport, respectively (Hirokawa et al., 2009; Kwinter et al., 2009). DCTN1 and KIF5A variants have been variably described in ALS (Munch et al., 2004, 2005; Liu et al., 2014, 2017; Brenner et al., 2018; Nicolas et al., 2018), CMT, and dHMN (Crimella et al., 2012; Lopez et al., 2015).
Finally, pleckstrin homology and RhoGEF domain containing G5 (PLEKHG5) and mitofusin 2 (MFN2) genes, whose mutations are known to cause CMT (Muglia et al., 2001; Zuchner et al., 2004a, 2006; Engelfried et al., 2006; Del Bo et al., 2008; Nicholson et al., 2008; Azzedine et al., 2013; Kim et al., 2013; Iapadre et al., 2018), have been also reported in patients with ALS or an “ALS-like” phenotype (Marchesi et al., 2011; Ozoguz et al., 2015).
Pathogenic Mechanisms of PNS Damage in ALS
Peripheral Motor and Sensory Dysfunction in ALS Models
The strongest evidence supporting the dying-back pattern of degeneration comes from transgenic mice overexpressing the mutated form of human SOD1 G93A (hSOD-1G93A), the most studied ALS model. Axonal pathology precedes spinal MN death and symptom onset, with the NMJ being the first site of morphological alteration (Fischer et al., 2004). Along with disease progression, the burden of axonal pathology overcomes the moderate MN loss in spinal cord, and suggesting that the motor phenotype in these mice models is mainly driven by the PNS damage (Gould et al., 2006; Hegedus et al., 2007). Indeed, interventions leading to complete rescue of MNs in the spinal cord of mutant SOD1 (mSOD1) ALS mice were not sufficient to halt axonal loss and motor phenotype, suggesting that mechanisms of central, and peripheral degeneration may be at least partially independent (Gould et al., 2006; Rouaux et al., 2007; Suzuki et al., 2007).
Despite the wealth amount of data generated from this model about MN degeneration, important concerns remain about the reproducibility of these findings in human ALS. The preferential involvement of spinal compared to cortical MNs, the anatomical differences in CNS organization between mice and humans and the confounding effects of artificial manipulation, such as transgenic overexpression, limit the translation of ALS preclinical research on patients (Kato, 2008; Philips and Rothstein, 2015). Nonetheless, some studies reported evidence of early NMJ alterations and axonal injury preceding MN loss also in ALS patients (Bradley et al., 1983; Dengler et al., 1990; Fischer et al., 2004; Bruneteau et al., 2015). Moreover, progressive NMJ and motor axon loss is a consistent finding also in other transgenic ALS mice with mutations in TARDBP, FUS, and C9orf72 repeat expansion (Picher-Martel et al., 2016). Overexpression of hTDP-43A315T and hTDP-43WT leads to NMJ denervation and loss of corticospinal axons, which in some cases predominate over MN al loss (Wegorzewska et al., 2009; Arnold et al., 2013; Herdewyn et al., 2014). In the hFUSP525L mouse lines, where the mutation is conditionally expressed in MNs, the progressive degeneration is preceded by early pre-symptomatic retraction of motor axons (Sharma et al., 2016).
Reflecting evidence of sensory dysfunction in human ALS, transgenic hSOD-1G93A mice also display neurodegeneration in sensory axon, DRG and proprioceptive sensory fibers of muscle spindles. Signs of axonal damage are detected since the pre-symptomatic stage, progressing with a distal-to-proximal gradient (Guo et al., 2009; Vaughan et al., 2015). Furthermore, loss of dorsal root axons has been detected in rodent models overexpressing mutant TDP-43 and FUS (Huang et al., 2011; Arnold et al., 2013), suggesting that ALS-specific proteins may also affect sensory neurons. However, it is still unclear whether the damage comes from nearby disease spreading or arise independently from MNs. Interestingly, transfection of mutant TDP-43 and SOD1 in cultured sensory neurons exert a negative impact on neurites length and arborization after prolonged culture, suggesting a direct effect (Vaughan et al., 2018).
Therefore, the reported evidence demonstrates that all components of the PNS are affected in ALS, although the differing kinetics of damage and progression still point to differences in vulnerability between the sensory and motor axons and neurons.
Unraveling Mechanisms of Vulnerability and Resistance of Peripheral Motor and Sensory Neurons in ALS
Studies addressing the mechanisms of PNS involvement in ALS may provide a unique opportunity for unraveling the determinants of the different patterns of vulnerability of motor and sensory neurons. This would allow the identification of specific protective or deleterious factors amenable of therapeutic intervention. Hereafter, we discuss key factors at the PNS level potentially implied in such differences.
Intrinsic Axonal Vulnerability
Hyperexcitability of the axolemma, due to altered Na+ and K+ conductance properties, has been demonstrated in motor but not sensory axons of ALS patients (Shibuya et al., 2011; Park et al., 2017; Matamala et al., 2018). The consequent increase in Ca2+ influx may be selectively harmful in the motor compartment, since motor axons show a low Ca2+-buffering capacity (Jaiswal, 2013; Leal and Gomes, 2015). In turn, Ca2+ overload leads to the activation of effector proteins such as calpain, a calcium-dependent protease, involved in TDP-43 fragmentation, which predisposes to aggregation, Wallerian degeneration and NMJ disassembly (Yamashita et al., 2012; Conforti et al., 2014; Campanari et al., 2016). Specific demise of MNs may also be driven by the involvement of ALS-key proteins, such as TDP-43 and FUS. They are mRNA-binding proteins, mainly localized in the nucleus, which participate in the metabolism of broad pools of mRNAs, by affecting splicing, stability, transport. Mutations in these proteins affect neurons by both loss and gain of function, leading to altered RNA processing, increasing cytoplasmic translocation and propensity to aggregation (Ratti and Buratti, 2016). They are essential during embryonic development, but only MNs in the cortex and anterior horns display sustained expression of both proteins during lifetime (Huang et al., 2010). Although most of the transcriptome is shared between motor and DRG axons, more than one third is unique (Rotem et al., 2017). It has been estimated that TDP43 binds roughly 30% of the whole transcriptome (Ling et al., 2013), including coding and non-coding RNAs, such as miRNA (Kawahara and Mieda-Sato, 2012). TDP-43 is actively recruited in RNA granules along motor axons for active transport (Fallini et al., 2012). Examples of TDP43-bound mRNAs include human neurofilaments, components of the endosomal trafficking and mitochondrial proteins (Stalekar et al., 2015; Schwenk et al., 2016; Wang et al., 2016). Over-expression of wild type and mutant TDP-43 alters the dynamics of its axonal transport (Alami et al., 2014), leading to mitochondrial toxicity and NMJ loss (Lin et al., 2011; Tripathi et al., 2014; Wang et al., 2016). Furthermore, a transient increase in TDP-43 localization along axonal routes is observed following axotomy, suggesting a role in peripheral nerve regeneration (Moisse et al., 2009). However, TDP-43G348C mice show abnormal persistence of cytoplasmic levels after peripheral injury, impairing axonal regeneration (Swarup et al., 2012). Such finding may provide a potential additional explanation to the paucity of regenerative clusters observed in ventral roots and motor nerves of human and animal ALS (Riva et al., 2011b, 2014; Luigetti et al., 2012). In contrast, sensory nerves still show some level of regeneration capacity in ALS patients, suggesting that efficient repair mechanisms may dampen progressive neurodegeneration (Hammad et al., 2007; Isaacs et al., 2007).
Neuromuscular Junction
The early susceptibility of the NMJ in ALS relies on peculiar features that distinguish it from other peripheral ending structures, including the sensory receptors. It has high energy demands and requires efficient mechanisms of plasticity and repair to cope with the challenges faced by muscle contraction. Oxidative stress, which contributes to ALS pathogenesis (D’Amico et al., 2013), has a great influence on NMJ function, as the early production of reactive oxygen species (ROS) in distal muscles inhibits transmitter release (Kraft et al., 2007; Naumenko et al., 2011) and leads to synaptic loss (Fischer et al., 2011, Fischer et al., 2012). High levels of oxidative stress have also been observed in muscles and spinal cord of ALS patients (D’Amico et al., 2013; Lanfranconi et al., 2017). Mitochondrial dysfunction further participates to NMJ dismantling, as its role in energy production and Ca2+ buffering is essential for proper function and maintenance of the synapse (Fischer-Hayes et al., 2013). Non-neuronal factors are also involved, as terminal schwann cells (TSCs) exert important functions in NMJ stability and repair, adapting their state according to local synaptic environment. Such decoding ability depends on the muscarinic acetylcholine receptor (mAchR), a G-protein-coupled receptor (GPCR) present on the membrane of TSCs, which detects the level of Ach released in the synaptic cleft. SOD1 mutants have been shown to alter the mAchR sensitivity of these cells to local damage, leading to deficient repair of denervated NMJ in ALS mice models (Arbour et al., 2015). Still, such findings need confirmation in humans and other models of ALS, although a relationship between TDP-43 function and TSC-related receptors has been suggested (Arbour et al., 2017).
PNS-Related Non-cell Autonomous Toxicity
Besides TSCs, other non-cell autonomous mechanisms in the PNS may further influence MNs vulnerability. The role of Schwann cells in the peripheral nerve has also been recently re-evaluated in ALS. Early deficits in axonal transport may be initially compensated by the transfer of polyribosomes from Schwann cells to the axonal compartment, boosting local protein synthesis as an adaptive response to mSOD1-induced injury (Verheijen et al., 2014). Moreover, Schwann cells may exert partial protection against the increased production of ROS related to axonal degeneration, as the loss of dismutase activity in these cells accelerated disease progression (Lobsiger et al., 2009). However, pathogenic gain-of-functions SOD1 mutants may affect Schwann cells functioning and protective effects as well (Wang et al., 2012). Neuroinflammatory response within the PNS and alterations of the blood-nerve barrier might offer additional insights in the pathogenesis of ALS (Kang and Rivest, 2007; Beers et al., 2008), as suggested by observations raised in mice models (Pupillo et al., 2014; Nardo et al., 2016, 2018) and in human ALS (Devigili et al., 2011; Riva et al., 2011a; Gerevini et al., 2016). Pathological studies of sural nerve biopsies of ALS patients have demonstrated, in a subset of cases, the presence of vasculitic-like inflammatory infiltrates associated with normal nerve morphology, suggesting a potential neuroprotective effect of neuroinflammation within the PNS (Devigili et al., 2011). Indeed, the recruitment of macrophages and stimulation of their phagocytic function in peripheral nerves is important to create a favorable milieu for axonal regeneration, as shown in mSOD1 ALS mice (Nardo et al., 2016).
Common Grounds for ALS and Hereditary Neuropathies
Despite the distinctive prognosis and disease course, ALS, related MNDs and hereditary neuropathies show a non-negligible degree of overlap in terms of both clinical presentations and genetics. Therefore, a better understanding of the key cellular pathways involved in both diseases may allow a broader comprehension of the molecular events leading to axonal and MN degeneration in ALS.
Neuronal Cytoskeleton
Microtubules, NFs and actin are the main components of axonal cytoskeleton, and impairment in either structure or functioning of any of these components may lead to axonal atrophy, retraction and degeneration or transport defects (Gentil et al., 2015; Kevenaar and Hoogenraad, 2015). Elevated levels of light NFs (NFL) and phosphorylated heavy NFs have been observed in the cerebrospinal fluid and serum of ALS, together with lower mRNA NFL levels in MNs (Volkening et al., 2009; Gaiani et al., 2017; Feneberg et al., 2018). Accumulation of NF components in axons is a prominent feature of both human and animal ALS (Leigh and Swash, 1991; Rouleau et al., 1996; Morrison et al., 2000), and has been observed along both motor and sensory axons in the hSOD-1G93A ALS mouse model (King et al., 2012; Gentil et al., 2015; Sassone et al., 2016). Alterations of ALS-key genes, such as SOD1 and TARDBP, have been shown to affect the stability of NFL mRNA, suggesting that altered stoichiometry between the different NF components may contribute to the disease (Volkening et al., 2009). Mutations in NEFL and NEFH, leading to axonal CMT, promote protein aggregation, aberrant mitochondrial morphology and shortening of axonal length (Yoshihara et al., 2002; Rebelo et al., 2016; Nam et al., 2017). Tubulin Alpha 4a (TUBA4A), encoding for α-tubulin, has been recently identified as a novel ALS gene (Smith et al., 2014). It has been shown that TUBA4A mutations are able to alter microtubule dynamics, with depolymerization and degradation of α-tubulin (Helferich et al., 2018). Although this gene has been described only in cohorts of ALS and FTD patients, a sensory and motor neuronopathy is observed in progressive motor neuronopathy mice mutated in tubulin binding cofactor E (TBCE), a known interactor of α-tubulin (Schafer et al., 2017). Profilin 1 (PFN1) has been found as a rare cause of familial ALS (Wu et al., 2012). It is a crucial protein for the conversion of monomeric (G)-actin to filamentous (F)-actin. Interestingly, although pathogenic variants do not disrupt actin dynamics (Freischmidt et al., 2015), alterations in microtubule growth and increased propensity to cytoplasmic TDP43 aggregation have been described (Matsukawa et al., 2016; Tanaka et al., 2016; Henty-Ridilla et al., 2017), revealing potential cell-type specific mechanisms of neurodegeneration.
Axonal Transport
Defects in axonal transport have been clearly observed both in ALS and dHMN. SOD1 and TDP43 mutants impair anterograde and retrograde axonal transport, leading to loss of essential synaptic components, mitochondrial abnormalities, and neurite shortening (Williamson and Cleveland, 1999; Perlson et al., 2009; Alami et al., 2014). Similarly abnormal transport of organelles, such as mitochondria and the endoplasmic reticulum (ER), is found in CMT models due to NEFL mutations (Tradewell et al., 2009). The failure of axon-cell body communication may dampen the activation of repair mechanisms, thus conferring an increase vulnerability of the peripheral compartment, more pronounced for the distal segments (Fischer et al., 2004; Dadon-Nachum et al., 2011). Strategies aiming at reinforcing axonal transport may thus confer protection to a broad range of pathogenic alterations, preserving the axonal routes and its endings.
Protein Folding
The highly differentiated and post-mitotic state of MNs exerts a relevant pressure over the protein quality control system of the cell, which is devoted to the correct folding, function and turnover of the whole proteome. HSPB1/27, HSPB8/22, and sigma non-opioid intracellular receptor 1 (SIGMAR1) are three chaperones with a significant expression levels in MNs (Mavlyutov et al., 2010). Mutations of the small heat shock proteins have been mostly associated with a dHMN or axonal CMT2 phenotype, while SIGMAR1 gene variants have been reported also in rare ALS patients (Al-Saif et al., 2011; Rossor et al., 2012). HSPB1 mutants alter both its chaperone activity in NF organization and preservation of axonal stability (Evgrafov et al., 2004; Ackerley et al., 2006). HSPB8 mutant mice develop a progressive motor neuropathy through specific neurite degeneration (Irobi et al., 2010; Bouhy et al., 2018). In addition, alterations in TDP-43 expression, and associated splicing is observed in the muscles of these mice (Cortese et al., 2018). Conversely, there is a significantly higher HSPB8 expression in surviving neurons in the hSOD-1G93A mouse model of ALS (Crippa et al., 2010). Finally, SIGMAR1, together with MFN2 and vesicle-associated membrane protein-associated protein B/C (VAPB), are able to modulate unfolded protein response (UPR) sensors, including protein kinase RNA-like endoplasmic reticulum kinase (PERK) (Nguyen et al., 2015).
Mitochondrial/Associated Membranes (MAMs)
The ER and mitochondria have evolved complex sites of interactions, defined as mitochondrial/associated membranes (MAMs), which are important for essential cellular functions, such as calcium homeostasis, autophagy, regulation of mitochondrial dynamics, and axon survival (Giorgi et al., 2015). Both ALS and hereditary neuropathies display dysfunction in one or more of MAM components. A huge number of cellular proteins participate in MAMs, including MFN2 and VAPB, which act as tethering sites between the two organelles (Bernard-Marissal et al., 2015). VAPB mutants affect the interaction with the mitochondrial protein tyrosine phosphatase-interacting protein 51 (PTPIP5) (de Brito and Scorrano, 2008; De Vos et al., 2012). Interestingly, TDP43 and FUS mutants have been shown to decrease ER-mitochondria contacts by disrupting VAPB-PTPIP5 binding (Stoica et al., 2014). MFN2 mutants alter other MAM functions, such as mitochondrial transport, fusion, and autophagosome assembly (Kim et al., 2015). Loss-of function mutations in SIGMAR1 display decreased ER-mitochondria association, resulting in MN death, and axonal degeneration due to impaired retrograde transport (Bernard-Marissal et al., 2015; Watanabe et al., 2016).
Therapeutic Perspectives
Due to the high similarity between these neuromuscular disorders and the intimate relationship observed in the molecular pathways involved, it may not be surprising if functional proteins involved in either of the two diseases may provide benefits on their respective counterpart.
Preservation of NMJ is an attractive target to delay muscle denervation and disability in ALS. A drug-screening platform in C. elegans and zebrafish models of ALS identified pimozide, an already approved neuroleptic, as a NMJ stabilizer by blocking T-type Ca2+ channels (Patten et al., 2017). Despite some benefits in neurophysiologic measures were reported in hSOD-1G93A mice and human patients in the first report, one study assessing long-term effects showed warned caution, as worsening of survival and muscle function were observed in mutant SOD1, and TDP-43 mice models (Pozzi et al., 2018). Notably, an agonist antibody directed toward muscle-specific kinase (MuSK), a post-synaptic tyrosine kinase receptor essential for NMJ maintenance, preserved motor synapses, delayed muscle denervation and extended lifespan in hSOD-1G93A ALS mice (Cantor et al., 2018). The promising findings of this study still awaits confirmation and replication in other disease models.
Reinforcing the action of factors involved in axonal stability and transport may restore soma-axon communication, delaying NMJ loss and axonal degeneration. Pharmacological inhibition of histone deacetylase 6 (HDAC6), a known client protein of HSPB1, was able to increase the stability of microtubules, rescuing axonal loss, and improving outcome in a CMT2F mouse model with mutant HSPB1 (d’Ydewalle et al., 2011). Interestingly, HDAC6 inhibition was also able to restore axonal transport defects in patient-derived MNs mutated in FUS (Guo et al., 2017). Pharmacologic agents targeting microtubule dynamics, such as noscapine and vinblastine, delayed disease onset and prolonged survival in hSOD-1G93A mice, by reducing microtubule turnover (Fanara et al., 2007).
Inhibition of p38/mitogen-associated protein kinase (MAPK) signaling in MNs was able to restore retrograde axonal transport defects in vivo (Gibbs et al., 2018), together with reduced microglia-induced neuroinflammation in the spinal cord (Kim and Choi, 2010; Zhan et al., 2015; Leal-Lasarte et al., 2017). However, it is remarkable that no significant improvement was observed on the clinical phenotype (Gibbs et al., 2018), suggesting that inhibition of this pathway alone is not sufficient to fully rescue MN function.
Reducing ER stress through either a potentiation of the chaperone system or inhibition of stress-related factors may limit the production rate of misfolded proteins. Enhancing HSPB8 activity has been shown to be protective against accumulation of TDP43 aggregates in MNs (Crippa et al., 2016; Rusmini et al., 2017), and to extend survival of hSOD-1G93A mice (Aurelian et al., 2012). Trehalose, a chemical chaperone known to induce HSPB8, reduces ER stress levels and improve autophagy, delaying disease, and prolonging MN survival in a mouse model (Zhang et al., 2014; Li Y. et al., 2015). Upon UPR activation, cells switch to a state of translational repression and stimulation of chaperones’ synthesis, processes regulated by phosphorylation of the eukaryotic translation initiation factor 2A (eiF2α). Salubrinal and guanabenz, two eiF2α phosphatase inhibitors, protected against MN degeneration in ALS mice (Saxena et al., 2009;Wang et al., 2014), although one study reported that guanabenz accelerated disease progression in an ALS model (Vieira et al., 2015).
Considering their importance in cell physiology, MAMs could represent an important target for therapy in ALS and may prove beneficial for a wide array of downstream signaling cascades relying upon these structures. An agonist of SIGMAR1 was shown to improve muscle activity and motor performance in pre-symptomatic hSOD-1G93A ALS mice (Mancuso et al., 2012). Notably, overexpression of MFN2 prevented NMJ loss and delayed onset and progression in a murine model by raising calpastatin levels, a calpain inhibitor, essential for survival, and function of the NMJ (Wang et al., 2018).
Boosting axonal regeneration through either neuron- or Schwann-targeting factors may also be a valuable alternative strategy in order to preserve motor function (Gilley et al., 2017). Studies modulating Neuregulin 1 activity, a neurotrophic factor involved in peripheral nerve development and regeneration, in murine hSOD-1G93A ALS lead to increased re-innervation and MN survival (Mancuso et al., 2016; Modol-Caballero et al., 2017). However, other approaches aimed at slowing Wallerian degeneration, such as the mutations in the Slowed Wallerian degeneration (WldS) and Sterile Alpha and TIR Motif Containing 1 (Sarm1) proteins, gave only modest or no results in terms of progression and survival (Fischer et al., 2005; Peters et al., 2018), suggesting that alternative mechanisms may underlie the axonal loss observed in ALS.
Conclusion
Clinical and pathological studies in both human patients and rodent models suggest that the PNS is involved in ALS pathogenetic cascade. A better comprehension of the molecular events occurring into the PNS may prove essential for a better understanding of MND pathomechanisms and may pave the way to the discovery of novel therapeutic targets. Considering that in ALS neurodegeneration derives from the combination of several dysfunctional pathways, it might be possible that the development of combination therapies acting on multiple cascades located in both the central and the peripheral compartments may show synergistic neuroprotective effect in order to prevent axonal degeneration, MN cell death, und ultimately in prolonging patients’ survival.
Author Contributions
FG and SS contributed to the collection of available evidence and writing. YF, AQ, NR, CL, and LT contributed to the manuscript writing and review of intellectual content of the manuscript, and approved the final version of the manuscript. AQ and NR contributed to the manuscript design.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ackerley, S., James, P. A., Kalli, A., French, S., Davies, K. E., and Talbot, K. (2006). A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum. Mol. Genet. 15, 347–354. doi: 10.1093/hmg/ddi452
Airoldi, G., Guidarelli, A., Cantoni, O., Panzeri, C., Vantaggiato, C., Bonato, S., et al. (2010). Characterization of two novel SETX mutations in AOA2 patients reveals aspects of the pathophysiological role of senataxin. Neurogenetics 11, 91–100. doi: 10.1007/s10048-009-0206-0
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S. W., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Al-Chalabi, A., Andersen, P. M., Nilsson, P., Chioza, B., Andersson, J. L., Russ, C., et al. (1999). Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum. Mol. Genet. 8, 157–164. doi: 10.1093/hmg/8.2.157
Al-Saif, A., Al-Mohanna, F., and Bohlega, S. (2011). A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 70, 913–919. doi: 10.1002/ana.22534
Antonellis, A., Ellsworth, R. E., Sambuughin, N., Puls, I., Abel, A., Lee-Lin, S. Q., et al. (2003). Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 72, 1293–1299. doi: 10.1086/375039
Arbour, D., Tremblay, E., Martineau, E., Julien, J. P., and Robitaille, R. (2015). Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J. Neurosci. 35, 688–706. doi: 10.1523/JNEUROSCI.1379-14.2015
Arbour, D., Vande Velde, C., and Robitaille, R. (2017). New perspectives on amyotrophic lateral sclerosis: the role of glial cells at the neuromuscular junction. J. Physiol. 595, 647–661. doi: 10.1113/JP270213
Arning, L., Epplen, J. T., Rahikkala, E., Hendrich, C., Ludolph, A. C., and Sperfeld, A. D. (2013). The SETX missense variation spectrum as evaluated in patients with ALS4-like motor neuron diseases. Neurogenetics 14, 53–61. doi: 10.1007/s10048-012-0347-4
Arning, L., Schols, L., Cin, H., Souquet, M., Epplen, J. T., and Timmann, D. (2008). Identification and characterisation of a large senataxin (SETX) gene duplication in ataxia with ocular apraxia type 2 (AOA2). Neurogenetics 9, 295–299. doi: 10.1007/s10048-008-0139-z
Arnold, E. S., Ling, S. C., Huelga, S. C., Lagier-Tourenne, C., Polymenidou, M., Ditsworth, D., et al. (2013). ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U.S.A. 110, E736–E745. doi: 10.1073/pnas.1222809110
Auer-Grumbach, M., Olschewski, A., Papic, L., Kremer, H., Mcentagart, M. E., Uhrig, S., et al. (2010). Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat. Genet. 42, 160–164. doi: 10.1038/ng.508
Aurelian, L., Laing, J. M., and Lee, K. S. (2012). H11/HspB8 and its herpes simplex virus type 2 homologue ICP10PK share functions that regulate cell life/death decisions and human disease. Autoimmune Dis. 2012:395329. doi: 10.1155/2012/395329
Avemaria, F., Lunetta, C., Tarlarini, C., Mosca, L., Maestri, E., Marocchi, A., et al. (2011). Mutation in the senataxin gene found in a patient affected by familial ALS with juvenile onset and slow progression. Amyotroph. Lateral Scler. 12, 228–230. doi: 10.3109/17482968.2011.566930
Azzedine, H., Zavadakova, P., Plante-Bordeneuve, V., Vaz Pato, M., Pinto, N., Bartesaghi, L., et al. (2013). PLEKHG5 deficiency leads to an intermediate form of autosomal-recessive Charcot-Marie-Tooth disease. Hum. Mol. Genet. 22, 4224–4232. doi: 10.1093/hmg/ddt274
Beers, D. R., Henkel, J. S., Zhao, W., Wang, J., and Appel, S. H. (2008). CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. U.S.A. 105, 15558–15563. doi: 10.1073/pnas.0807419105
Benedetti, S., Previtali, S. C., Coviello, S., Scarlato, M., Cerri, F., Di Pierri, E., et al. (2010). Analyzing histopathological features of rare charcot-marie-tooth neuropathies to unravel their pathogenesis. Arch. Neurol. 67, 1498–1505. doi: 10.1001/archneurol.2010.303
Bernard-Marissal, N., Medard, J. J., Azzedine, H., and Chrast, R. (2015). Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138, 875–890. doi: 10.1093/brain/awv008
Bhatt, A., Farooq, M. U., Aburashed, R., Kassab, M. Y., Majid, A., Bhatt, S., et al. (2009). Hereditary neuropathy with liability to pressure palsies and amyotrophic lateral sclerosis. Neurol. Sci. 30, 241–245. doi: 10.1007/s10072-009-0034-x
Bouhy, D., Juneja, M., Katona, I., Holmgren, A., Asselbergh, B., De Winter, V., et al. (2018). A knock-in/knock-out mouse model of HSPB8-associated distal hereditary motor neuropathy and myopathy reveals toxic gain-of-function of mutant Hspb8. Acta Neuropathol. 135, 131–148. doi: 10.1007/s00401-017-1756-0
Bradley, W. G., Good, P., Rasool, C. G., and Adelman, L. S. (1983). Morphometric and biochemical studies of peripheral nerves in amyotrophic lateral sclerosis. Ann. Neurol. 14, 267–277. doi: 10.1002/ana.410140304
Braun, R. J., and Zischka, H. (2008). Mechanisms of Cdc48/VCP-mediated cell death: from yeast apoptosis to human disease. Biochim. Biophys. Acta 1783, 1418–1435. doi: 10.1016/j.bbamcr.2008.01.015
Brenner, D., Yilmaz, R., Muller, K., Grehl, T., Petri, S., Meyer, T., et al. (2018). Hot-spot KIF5A mutations cause familial ALS. Brain 141, 688–697. doi: 10.1093/brain/awx370
Brettschneider, J., Arai, K., Del Tredici, K., Toledo, J. B., Robinson, J. L., Lee, E. B., et al. (2014). TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 128, 423–437. doi: 10.1007/s00401-014-1299-6
Brettschneider, J., Del Tredici, K., Toledo, J. B., Robinson, J. L., Irwin, D. J., Grossman, M., et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. doi: 10.1002/ana.23937
Bruneteau, G., Bauche, S., Gonzalez De Aguilar, J. L., Brochier, G., Mandjee, N., Tanguy, M. L., et al. (2015). Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol. 2, 362–372. doi: 10.1002/acn3.179
Camdessanche, J. P., Belzil, V. V., Jousserand, G., Rouleau, G. A., Creac’h, C., Convers, P., et al. (2011). Sensory and motor neuronopathy in a patient with the A382P TDP-43 mutation. Orphanet J. Rare Dis. 6:4. doi: 10.1186/1750-1172-6-4
Campanari, M. L., Garcia-Ayllon, M. S., Ciura, S., Saez-Valero, J., and Kabashi, E. (2016). Neuromuscular junction impairment in amyotrophic lateral sclerosis: reassessing the role of acetylcholinesterase. Front. Mol. Neurosci. 9:160. doi: 10.3389/fnmol.2016.00160
Campeau, P. M., Lenk, G. M., Lu, J. T., Bae, Y., Burrage, L., Turnpenny, P., et al. (2013). Yunis-Varon syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am. J. Hum. Genet. 92, 781–791. doi: 10.1016/j.ajhg.2013.03.020
Cantor, S., Zhang, W., Delestree, N., Remedio, L., Mentis, G. Z., and Burden, S. J. (2018). Preserving neuromuscular synapses in ALS by stimulating MuSK with a therapeutic agonist antibody. eLife 7:e34375
Ceroni, M., Curti, D., and Alimonti, D. (2001). Amyotrophic lateral sclerosis and SOD1 gene: an overview. Funct. Neurol. 16, 171–180.
Chen, Y. Z., Bennett, C. L., Huynh, H. M., Blair, I. P., Puls, I., Irobi, J., et al. (2004). DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 74, 1128–1135. doi: 10.1086/421054
Chia, R., Chio, A., and Traynor, B. J. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102. doi: 10.1016/S1474-4422(17)30401-5
Chow, C. Y., Landers, J. E., Bergren, S. K., Sapp, P. C., Grant, A. E., Jones, J. M., et al. (2009). Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 84, 85–88. doi: 10.1016/j.ajhg.2008.12.010
Chow, C. Y., Zhang, Y., Dowling, J. J., Jin, N., Adamska, M., Shiga, K., et al. (2007). Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448, 68–72. doi: 10.1038/nature05876
Chung, C. Y., Koprich, J. B., Siddiqi, H., and Isacson, O. (2009). Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV α-synucleinopathy. J. Neurosci. 29, 3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009
Conforti, L., Gilley, J., and Coleman, M. P. (2014). Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 15, 394–409. doi: 10.1038/nrn3680
Corbo, M., Abouzahr, M. K., Latov, N., Iannaccone, S., Quattrini, A., Nemni, R., et al. (1997). Motor nerve biopsy studies in motor neuropathy and motor neuron disease. Muscle Nerve 20, 15–21. doi: 10.1002/(sici)1097-4598(199701)20:1<15::aid-mus2>3.0.co;2-k
Cortese, A., Laura, M., Casali, C., Nishino, I., Hayashi, Y. K., Magri, S., et al. (2018). Altered TDP-43-dependent splicing in HSPB8-related distal hereditary motor neuropathy and myofibrillar myopathy. Eur. J. Neurol. 25, 154–163. doi: 10.1111/ene.13478
Crimella, C., Baschirotto, C., Arnoldi, A., Tonelli, A., Tenderini, E., Airoldi, G., et al. (2012). Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin. Genet. 82, 157–164. doi: 10.1111/j.1399-0004.2011.01717.x
Crippa, V., Cicardi, M. E., Ramesh, N., Seguin, S. J., Ganassi, M., Bigi, I., et al. (2016). The chaperone HSPB8 reduces the accumulation of truncated TDP-43 species in cells and protects against TDP-43-mediated toxicity. Hum. Mol. Genet. 25, 3908–3924. doi: 10.1093/hmg/ddw232
Crippa, V., Sau, D., Rusmini, P., Boncoraglio, A., Onesto, E., Bolzoni, E., et al. (2010). The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 19, 3440–3456. doi: 10.1093/hmg/ddq257
Dadon-Nachum, M., Melamed, E., and Offen, D. (2011). The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 43, 470–477. doi: 10.1007/s12031-010-9467-1
Dalla Bella, E., Lombardi, R., Porretta-Serapiglia, C., Ciano, C., Gellera, C., Pensato, V., et al. (2016). Amyotrophic lateral sclerosis causes small fiber pathology. Eur. J. Neurol. 23, 416–420. doi: 10.1111/ene.12936
D’Amico, E., Factor-Litvak, P., Santella, R. M., and Mitsumoto, H. (2013). Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 65, 509–527. doi: 10.1016/j.freeradbiomed.2013.06.029
Daoud, H., Zhou, S., Noreau, A., Sabbagh, M., Belzil, V., Dionne-Laporte, A., et al. (2012). Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol. Aging 33:839.e5-9. doi: 10.1016/j.neurobiolaging.2011.11.012
Davenport, R. J., Swingler, R. J., Chancellor, A. M., and Warlow, C. P. (1996). Avoiding false positive diagnoses of motor neuron disease: lessons from the Scottish motor neuron disease register. J. Neurol. Neurosurg. Psychiatry 60, 147–151. doi: 10.1136/jnnp.60.2.147
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
De Jonghe, P., Auer-Grumbach, M., Irobi, J., Wagner, K., Plecko, B., Kennerson, M., et al. (2002). Autosomal dominant juvenile amyotrophic lateral sclerosis and distal hereditary motor neuronopathy with pyramidal tract signs: synonyms for the same disorder? Brain 125, 1320–1325. doi: 10.1093/brain/awf127
De Vos, K. J., Morotz, G. M., Stoica, R., Tudor, E. L., Lau, K. F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Del Bo, R., Moggio, M., Rango, M., Bonato, S., D’angelo, M. G., Ghezzi, S., et al. (2008). Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology 71, 1959–1966. doi: 10.1212/01.wnl.0000327095.32005.a4
DeLaBarre, B., Christianson, J. C., Kopito, R. R., and Brunger, A. T. (2006). Central pore residues mediate the p97/VCP activity required for ERAD. Mol. Cell 22, 451–462. doi: 10.1016/j.molcel.2006.03.036
Deng, H. X., Chen, W., Hong, S. T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. doi: 10.1038/nature10353
Dengler, R., Konstanzer, A., Kuther, G., Hesse, S., Wolf, W., and Struppler, A. (1990). Amyotrophic lateral sclerosis: macro-EMG and twitch forces of single motor units. Muscle Nerve 13, 545–550. doi: 10.1002/mus.880130612
Devigili, G., Uceyler, N., Beck, M., Reiners, K., Stoll, G., Toyka, K. V., et al. (2011). Vasculitis-like neuropathy in amyotrophic lateral sclerosis unresponsive to treatment. Acta Neuropathol. 122, 343–352. doi: 10.1007/s00401-011-0837-8
Di Trapani, G., David, P., La Cara, A., Tonali, P., and Laurienzo, P. (1987). Light and ultrastructural studies in sural biopsies of the pseudopolyneuropathic form of ALS. Adv. Exp. Med. Biol. 209, 111–119. doi: 10.1007/978-1-4684-5302-7_18
Duquette, A., Roddier, K., Mcnabb-Baltar, J., Gosselin, I., St-Denis, A., Dicaire, M. J., et al. (2005). Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann. Neurol. 57, 408–414. doi: 10.1002/ana.20408
Dyck, P. J., Stevens, J. C., Mulder, D. W., and Espinosa, R. E. (1975). Frequency of nerve fiber degeneration of peripheral motor and sensory neurons in amyotrophic lateral sclerosis. Morphometry of deep and superficial peroneal nerves. Neurology 25, 781–785.
d’Ydewalle, C., Krishnan, J., Chiheb, D. M., Van Damme, P., Irobi, J., Kozikowski, A. P., et al. (2011). HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med. 17, 968–974. doi: 10.1038/nm.2396
Engelfried, K., Vorgerd, M., Hagedorn, M., Haas, G., Gilles, J., Epplen, J. T., et al. (2006). Charcot-Marie-Tooth neuropathy type 2A: novel mutations in the mitofusin 2 gene (MFN2). BMC Med. Genet. 7:53. doi: 10.1186/1471-2350-7-53
Evgrafov, O. V., Mersiyanova, I., Irobi, J., Van Den Bosch, L., Dierick, I., Leung, C. L., et al. (2004). Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 36, 602–606. doi: 10.1038/ng1354
Fallini, C., Bassell, G. J., and Rossoll, W. (2012). The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 21, 3703–3718. doi: 10.1093/hmg/dds205
Fanara, P., Banerjee, J., Hueck, R. V., Harper, M. R., Awada, M., Turner, H., et al. (2007). Stabilization of hyperdynamic microtubules is neuroprotective in amyotrophic lateral sclerosis. J. Biol. Chem. 282, 23465–23472. doi: 10.1074/jbc.m703434200
Farrer, M. J., Hulihan, M. M., Kachergus, J. M., Dachsel, J. C., Stoessl, A. J., Grantier, L. L., et al. (2009). DCTN1 mutations in Perry syndrome. Nat. Genet. 41, 163–165. doi: 10.1038/ng.293
Fecto, F., Yan, J., Vemula, S. P., Liu, E., Yang, Y., Chen, W., et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. doi: 10.1001/archneurol.2011.250
Feneberg, E., Oeckl, P., Steinacker, P., Verde, F., Barro, C., Van Damme, P., et al. (2018). Multicenter evaluation of neurofilaments in early symptom onset amyotrophic lateral sclerosis. Neurology 90, e22–e30. doi: 10.1212/WNL.0000000000004761
Ferrari, G., Grisan, E., Scarpa, F., Fazio, R., Comola, M., Quattrini, A., et al. (2014). Corneal confocal microscopy reveals trigeminal small sensory fiber neuropathy in amyotrophic lateral sclerosis. Front. Aging Neurosci. 6:278. doi: 10.3389/fnagi.2014.00278
Figlewicz, D. A., Krizus, A., Martinoli, M. G., Meininger, V., Dib, M., Rouleau, G. A., et al. (1994). Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 3, 1757–1761. doi: 10.1093/hmg/3.10.1757
Fischer, L. R., Culver, D. G., Davis, A. A., Tennant, P., Wang, M., Coleman, M., et al. (2005). The WldS gene modestly prolongs survival in the SOD1G93A fALS mouse. Neurobiol. Dis. 19, 293–300. doi: 10.1016/j.nbd.2005.01.008
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Fischer, L. R., and Glass, J. D. (2007). Axonal degeneration in motor neuron disease. Neurodegener. Dis. 4, 431–442. doi: 10.1159/000107704
Fischer, L. R., Igoudjil, A., Magrane, J., Li, Y., Hansen, J. M., Manfredi, G., et al. (2011). SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain 134, 196–209. doi: 10.1093/brain/awq314
Fischer, L. R., Li, Y., Asress, S. A., Jones, D. P., and Glass, J. D. (2012). Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp. Neurol. 233, 163–171. doi: 10.1016/j.expneurol.2011.09.020
Fischer-Hayes, L. R., Brotherton, T., and Glass, J. D. (2013). Axonal degeneration in the peripheral nervous system: implications for the pathogenesis of amyotrophic lateral sclerosis. Exp. Neurol. 246, 6–13. doi: 10.1016/j.expneurol.2013.05.001
Fogel, B. L., Cho, E., Wahnich, A., Gao, F., Becherel, O. J., Wang, X., et al. (2014). Mutation of senataxin alters disease-specific transcriptional networks in patients with ataxia with oculomotor apraxia type 2. Hum. Mol. Genet. 23, 4758–4769. doi: 10.1093/hmg/ddu190
Fogel, B. L., and Perlman, S. (2006). Novel mutations in the senataxin DNA/RNA helicase domain in ataxia with oculomotor apraxia 2. Neurology 67, 2083–2084. doi: 10.1212/01.wnl.0000247661.19601.28
Fonknechten, N., Mavel, D., Byrne, P., Davoine, C. S., Cruaud, C., Bonsch, D., et al. (2000). Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum. Mol. Genet. 9, 637–644. doi: 10.1093/hmg/9.4.637
Freischmidt, A., Schopflin, M., Feiler, M. S., Fleck, A. K., Ludolph, A. C., and Weishaupt, J. H. (2015). Profilin 1 with the amyotrophic lateral sclerosis associated mutation T109M displays unaltered actin binding and does not affect the actin cytoskeleton. BMC Neurosci. 16:77. doi: 10.1186/s12868-015-0214-y
Gaiani, A., Martinelli, I., Bello, L., Querin, G., Puthenparampil, M., Ruggero, S., et al. (2017). Diagnostic and prognostic biomarkers in amyotrophic lateral sclerosis: neurofilament light chain levels in definite subtypes of disease. JAMA Neurol. 74, 525–532. doi: 10.1001/jamaneurol.2016.5398
Garg, N., Park, S. B., Vucic, S., Yiannikas, C., Spies, J., Howells, J., et al. (2017). Differentiating lower motor neuron syndromes. J. Neurol. Neurosurg. Psychiatry 88, 474–483. doi: 10.1136/jnnp-2016-313526
Gentil, B. J., Tibshirani, M., and Durham, H. D. (2015). Neurofilament dynamics and involvement in neurological disorders. Cell Tissue Res. 360, 609–620. doi: 10.1007/s00441-014-2082-7
Gerevini, S., Agosta, F., Riva, N., Spinelli, E. G., Pagani, E., Caliendo, G., et al. (2016). MR imaging of brachial plexus and limb-girdle muscles in patients with amyotrophic lateral sclerosis. Radiology 279, 553–561. doi: 10.1148/radiol.2015150559
Geser, F., Brandmeir, N. J., Kwong, L. K., Martinez-Lage, M., Elman, L., Mccluskey, L., et al. (2008). Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch. Neurol. 65, 636–641. doi: 10.1001/archneur.65.5.636
Gibbs, K. L., Kalmar, B., Rhymes, E. R., Fellows, A. D., Ahmed, M., Whiting, P., et al. (2018). Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis. 9:596.
Gilley, J., Ribchester, R. R., and Coleman, M. P. (2017). Sarm1 deletion, but not Wld(S), confers lifelong rescue in a mouse model of severe axonopathy. Cell Rep. 21, 10–16. doi: 10.1016/j.celrep.2017.09.027
Giorgi, C., Missiroli, S., Patergnani, S., Duszynski, J., Wieckowski, M. R., and Pinton, P. (2015). Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 22, 995–1019. doi: 10.1089/ars.2014.6223
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D., et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538.
Gonzalez, M. A., Feely, S. M., Speziani, F., Strickland, A. V., Danzi, M., Bacon, C., et al. (2014). A novel mutation in VCP causes Charcot-Marie-Tooth Type 2 disease. Brain 137, 2897–2902. doi: 10.1093/brain/awu224
Gould, T. W., Buss, R. R., Vinsant, S., Prevette, D., Sun, W., Knudson, C. M., et al. (2006). Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J. Neurosci. 26, 8774–8786. doi: 10.1523/jneurosci.2315-06.2006
Gubbay, S. S., Kahana, E., Zilber, N., Cooper, G., Pintov, S., and Leibowitz, Y. (1985). Amyotrophic lateral sclerosis. A study of its presentation and prognosis. J. Neurol. 232, 295–300. doi: 10.1007/bf00313868
Guo, W., Naujock, M., Fumagalli, L., Vandoorne, T., Baatsen, P., Boon, R., et al. (2017). HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 8:861. doi: 10.1038/s41467-017-00911-y
Guo, Y. S., Wu, D. X., Wu, H. R., Wu, S. Y., Yang, C., Li, B., et al. (2009). Sensory involvement in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Exp. Mol. Med. 41, 140–150.
Hammad, M., Silva, A., Glass, J., Sladky, J. T., and Benatar, M. (2007). Clinical, electrophysiologic, and pathologic evidence for sensory abnormalities in ALS. Neurology 69, 2236–2242. doi: 10.1212/01.wnl.0000286948.99150.16
Harms, M. B., Ori-Mckenney, K. M., Scoto, M., Tuck, E. P., Bell, S., Ma, D., et al. (2012). Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 78, 1714–1720. doi: 10.1212/WNL.0b013e3182556c05
Heads, T., Pollock, M., Robertson, A., Sutherland, W. H., and Allpress, S. (1991). Sensory nerve pathology in amyotrophic lateral sclerosis. Acta Neuropathol. 82, 316–320.
Hegedus, J., Putman, C. T., and Gordon, T. (2007). Time course of preferential motor unit loss in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 28, 154–164. doi: 10.1016/j.nbd.2007.07.003
Hehr, U., Bauer, P., Winner, B., Schule, R., Olmez, A., Koehler, W., et al. (2007). Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann. Neurol. 62, 656–665. doi: 10.1002/ana.21310
Helferich, A. M., Brockmann, S. J., Reinders, J., Deshpande, D., Holzmann, K., Brenner, D., et al. (2018). Dysregulation of a novel miR-1825/TBCB/TUBA4A pathway in sporadic and familial ALS. Cell Mol. Life Sci. 75, 4301–4319. doi: 10.1007/s00018-018-2873-1
Henty-Ridilla, J. L., Juanes, M. A., and Goode, B. L. (2017). Profilin directly promotes microtubule growth through residues mutated in amyotrophic lateral sclerosis. Curr. Biol. 27, 3535.e4–3543.e4. doi: 10.1016/j.cub.2017.10.002
Herdewyn, S., Cirillo, C., Van Den Bosch, L., Robberecht, W., Vanden Berghe, P., and Van Damme, P. (2014). Prevention of intestinal obstruction reveals progressive neurodegeneration in mutant TDP-43 (A315T) mice. Mol. Neurodegener. 9:24. doi: 10.1186/1750-1326-9-24
Hirokawa, N., Noda, Y., Tanaka, Y., and Niwa, S. (2009). Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682–696. doi: 10.1038/nrm2774
Hoffman, P. N., Cleveland, D. W., Griffin, J. W., Landes, P. W., Cowan, N. J., and Price, D. L. (1987). Neurofilament gene expression: a major determinant of axonal caliber. Proc. Natl. Acad. Sci. U.S.A. 84, 3472–3476. doi: 10.1073/pnas.84.10.3472
Huang, C., Xia, P. Y., and Zhou, H. (2010). Sustained expression of TDP-43 and FUS in motor neurons in rodent’s lifetime. Int. J. Biol. Sci. 6, 396–406. doi: 10.7150/ijbs.6.396
Huang, C., Zhou, H., Tong, J., Chen, H., Liu, Y. J., Wang, D., et al. (2011). FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 7:e1002011. doi: 10.1371/journal.pgen.1002011
Iapadre, G., Morana, G., Vari, M. S., Pinto, F., Lanteri, P., Tessa, A., et al. (2018). A novel homozygous MFN2 mutation associated with severe and atypical CMT2 phenotype. Eur. J. Paediatr. Neurol. 22, 563–567. doi: 10.1016/j.ejpn.2017.12.020
Irobi, J., Almeida-Souza, L., Asselbergh, B., De Winter, V., Goethals, S., Dierick, I., et al. (2010). Mutant HSPB8 causes motor neuron-specific neurite degeneration. Hum. Mol. Genet. 19, 3254–3265. doi: 10.1093/hmg/ddq234
Irobi, J., Van Impe, K., Seeman, P., Jordanova, A., Dierick, I., Verpoorten, N., et al. (2004). Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat. Genet. 36, 597–601. doi: 10.1038/ng1328
Isaacs, J. D., Dean, A. F., Shaw, C. E., Al-Chalabi, A., Mills, K. R., and Leigh, P. N. (2007). Amyotrophic lateral sclerosis with sensory neuropathy: part of a multisystem disorder? J. Neurol. Neurosurg. Psychiatry 78, 750–753. doi: 10.1136/jnnp.2006.098798
Isak, B., Pugdahl, K., Karlsson, P., Tankisi, H., Finnerup, N. B., Furtula, J., et al. (2017). Quantitative sensory testing and structural assessment of sensory nerve fibres in amyotrophic lateral sclerosis. J. Neurol. Sci. 373, 329–334. doi: 10.1016/j.jns.2017.01.005
Isak, B., Tankisi, H., Johnsen, B., Pugdahl, K., Torvin, M. A., Finnerup, N. B., et al. (2016). Involvement of distal sensory nerves in amyotrophic lateral sclerosis. Muscle Nerve 54, 1086–1092. doi: 10.1002/mus.25157
Iskender, C., Kartal, E., Akcimen, F., Kocoglu, C., Ozoguz, A., Kotan, D., et al. (2015). Turkish families with juvenile motor neuron disease broaden the phenotypic spectrum of SPG11. Neurol. Genet. 1:e25. doi: 10.1212/NXG.0000000000000025
Jacquier, A., Delorme, C., Belotti, E., Juntas-Morales, R., Sole, G., Dubourg, O., et al. (2017). Cryptic amyloidogenic elements in mutant NEFH causing Charcot-Marie-Tooth 2 trigger aggresome formation and neuronal death. Acta Neuropathol. Commun. 5:55. doi: 10.1186/s40478-017-0457-1
Jaiswal, M. K. (2013). Calcium, mitochondria, and the pathogenesis of ALS: the good, the bad, and the ugly. Front. Cell Neurosci. 7:199. doi: 10.3389/fncel.2013.00199
Jerath, N. U., Crockett, C. D., Moore, S. A., Shy, M. E., Weihl, C. C., Chou, T. F., et al. (2015). Rare Manifestation of a c.290 C > T, p.Gly97Glu VCP Mutation. Case Rep. Genet. 2015:239167. doi: 10.1155/2015/239167
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
Jordanova, A., De Jonghe, P., Boerkoel, C. F., Takashima, H., De Vriendt, E., Ceuterick, C., et al. (2003). Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain 126, 590–597. doi: 10.1093/brain/awg059
Ju, J. S., Fuentealba, R. A., Miller, S. E., Jackson, E., Piwnica-Worms, D., Baloh, R. H., et al. (2009). Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875–888. doi: 10.1083/jcb.200908115
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., Mcconkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kang, J., and Rivest, S. (2007). MyD88-deficient bone marrow cells accelerate onset and reduce survival in a mouse model of amyotrophic lateral sclerosis. J. Cell Biol. 179, 1219–1230. doi: 10.1083/jcb.200705046
Kato, S. (2008). Amyotrophic lateral sclerosis models and human neuropathology: similarities and differences. Acta Neuropathol. 115, 97–114. doi: 10.1007/s00401-007-0308-4
Kawahara, Y., and Mieda-Sato, A. (2012). TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. U.S.A. 109, 3347–3352. doi: 10.1073/pnas.1112427109
Kawamura, Y., Dyck, P. J., Shimono, M., Okazaki, H., Tateishi, J., and Doi, H. (1981). Morphometric comparison of the vulnerability of peripheral motor and sensory neurons in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 40, 667–675. doi: 10.1097/00005072-198111000-00008
Kevenaar, J. T., and Hoogenraad, C. C. (2015). The axonal cytoskeleton: from organization to function. Front. Mol. Neurosci. 8:44. doi: 10.3389/fnmol.2015.00044
Kim, E. K., and Choi, E. J. (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405. doi: 10.1016/j.bbadis.2009.12.009
Kim, H. J., Hong, Y. B., Park, J. M., Choi, Y. R., Kim, Y. J., Yoon, B. R., et al. (2013). Mutations in the PLEKHG5 gene is relevant with autosomal recessive intermediate Charcot-Marie-Tooth disease. Orphanet J. Rare Dis. 8:104. doi: 10.1186/1750-1172-8-104
Kim, H. J., Nagano, Y., Choi, S. J., Park, S. Y., Kim, H., Yao, T. P., et al. (2015). HDAC6 maintains mitochondrial connectivity under hypoxic stress by suppressing MARCH5/MITOL dependent MFN2 degradation. Biochem. Biophys. Res. Commun. 464, 1235–1240. doi: 10.1016/j.bbrc.2015.07.111
King, A. E., Blizzard, C. A., Southam, K. A., Vickers, J. C., and Dickson, T. C. (2012). Degeneration of axons in spinal white matter in G93A mSOD1 mouse characterized by NFL and alpha-internexin immunoreactivity. Brain Res. 1465, 90–100. doi: 10.1016/j.brainres.2012.05.018
Kolb, S. J., Snyder, P. J., Poi, E. J., Renard, E. A., Bartlett, A., Gu, S., et al. (2010). Mutant small heat shock protein B3 causes motor neuropathy: utility of a candidate gene approach. Neurology 74, 502–506. doi: 10.1212/WNL.0b013e3181cef84a
Kraft, A. D., Resch, J. M., Johnson, D. A., and Johnson, J. A. (2007). Activation of the Nrf2–ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp. Neurol. 207, 107–117. doi: 10.1016/j.expneurol.2007.05.026
Kruger, S., Battke, F., Sprecher, A., Munz, M., Synofzik, M., Schols, L., et al. (2016). Rare variants in neurodegeneration associated genes revealed by targeted panel sequencing in a German ALS cohort. Front. Mol. Neurosci. 9:92. doi: 10.3389/fnmol.2016.00092
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Kwinter, D. M., Lo, K., Mafi, P., and Silverman, M. A. (2009). Dynactin regulates bidirectional transport of dense-core vesicles in the axon and dendrites of cultured hippocampal neurons. Neuroscience 162, 1001–1010. doi: 10.1016/j.neuroscience.2009.05.038
Lanfranconi, F., Ferri, A., Corna, G., Bonazzi, R., Lunetta, C., Silani, V., et al. (2017). Inefficient skeletal muscle oxidative function flanks impaired motor neuron recruitment in amyotrophic lateral sclerosis during exercise. Sci. Rep. 7:2951. doi: 10.1038/s41598-017-02811-z
Laurin, N., Brown, J. P., Morissette, J., and Raymond, V. (2002). Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70, 1582–1588. doi: 10.1086/340731
Leal, S. S., and Gomes, C. M. (2015). Calcium dysregulation links ALS defective proteins and motor neuron selective vulnerability. Front. Cell. Neurosci. 9:225. doi: 10.3389/fncel.2015.00225
Leal-Lasarte, M. M., Franco, J. M., Labrador-Garrido, A., Pozo, D., and Roodveldt, C. (2017). Extracellular TDP-43 aggregates target MAPK/MAK/MRK overlapping kinase (MOK) and trigger caspase-3/IL-18 signaling in microglia. FASEB J. 31, 2797–2816. doi: 10.1096/fj.201601163R
Leigh, P. N., and Swash, M. (1991). Cytoskeletal pathology in motor neuron diseases. Adv. Neurol. 56, 115–124.
Li, X., Hu, Z., Liu, L., Xie, Y., Zhan, Y., Zi, X., et al. (2015). A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology 84, 2430–2437. doi: 10.1212/WNL.0000000000001680
Li, Y., Guo, Y., Wang, X., Yu, X., Duan, W., Hong, K., et al. (2015). Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience 298, 12–25. doi: 10.1016/j.neuroscience.2015.03.061
Lieberburg, I., Spinner, N., Snyder, S., Anderson, J., Goldgaber, D., Smulowitz, M., et al. (1989). Cloning of a cDNA encoding the rat high molecular weight neurofilament peptide (NF-H): developmental and tissue expression in the rat, and mapping of its human homologue to chromosomes 1 and 22. Proc. Natl. Acad. Sci. U.S.A. 86, 2463–2467. doi: 10.1073/pnas.86.7.2463
Lin, M. J., Cheng, C. W., and Shen, C. K. (2011). Neuronal function and dysfunction of Drosophila dTDP. PLoS One 6:e20371. doi: 10.1371/journal.pone.0020371
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu, X., Yang, L., Tang, L., Chen, L., and Fan, D. (2017). DCTN1 gene analysis in Chinese patients with sporadic amyotrophic lateral sclerosis. PLoS One 12:e0182572. doi: 10.1371/journal.pone.0182572
Liu, Z. J., Li, H. F., Tan, G. H., Tao, Q. Q., Ni, W., Cheng, X. W., et al. (2014). Identify mutation in amyotrophic lateral sclerosis cases using HaloPlex target enrichment system. Neurobiol Aging 35, 2881.e11-2881.e15. doi: 10.1016/j.neurobiolaging.2014.07.003
Lobsiger, C. S., Boillee, S., Mcalonis-Downes, M., Khan, A. M., Feltri, M. L., Yamanaka, K., et al. (2009). Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc. Natl. Acad. Sci. U.S.A. 106, 4465–4470. doi: 10.1073/pnas.0813339106
Lopez, E., Casasnovas, C., Gimenez, J., Santamaria, R., Terrazas, J. M., and Volpini, V. (2015). Identification of two novel KIF5A mutations in hereditary spastic paraplegia associated with mild peripheral neuropathy. J. Neurol. Sci. 358, 422–427. doi: 10.1016/j.jns.2015.08.1529
Ludolph, A., Drory, V., Hardiman, O., Nakano, I., Ravits, J., Robberecht, W., et al. (2015). A revision of the El Escorial criteria - 2015. Amyotroph. Lateral Scler. Frontotemporal Degener. 16, 291–292. doi: 10.3109/21678421.2015.1049183
Luigetti, M., Conte, A., Del Grande, A., Bisogni, G., Romano, A., and Sabatelli, M. (2012). Sural nerve pathology in ALS patients: a single-centre experience. Neurol. Sci. 33, 1095–1099. doi: 10.1007/s10072-011-0909-5
Magré, J., Delépine, M., Khallouf, E., Gedde-Dahl, T. Jr., Van Maldergem, L., Sobel, E., Papp, J. (2001). Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet. 28, 365–370. doi: 10.1038/ng585
Mancuso, R., Martinez-Muriana, A., Leiva, T., Gregorio, D., Ariza, L., Morell, M., et al. (2016). Neuregulin-1 promotes functional improvement by enhancing collateral sprouting in SOD1(G93A) ALS mice and after partial muscle denervation. Neurobiol. Dis. 95, 168–178. doi: 10.1016/j.nbd.2016.07.023
Mancuso, R., Olivan, S., Rando, A., Casas, C., Osta, R., and Navarro, X. (2012). Sigma-1R agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics 9, 814–826. doi: 10.1007/s13311-012-0140-y
Marchesi, C., Ciano, C., Salsano, E., Nanetti, L., Milani, M., Gellera, C., et al. (2011). Co-occurrence of amyotrophic lateral sclerosis and Charcot-Marie-Tooth disease type 2A in a patient with a novel mutation in the mitofusin-2 gene. Neuromuscul. Disord. 21, 129–131. doi: 10.1016/j.nmd.2010.09.009
Martini, R., Berciano, J., and Van Broeckhoven, C. (2000). 5th Workshop of the European CMT Consortium, 69th ENMC International Workshop: therapeutic approaches in CMT neuropathies and related disorders 23-25 April 1999, Soestduinen, The Netherlands. Neuromuscul. Disord. 10, 69–74. doi: 10.1016/s0960-8966(99)00095-4
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
Matamala, J. M., Howells, J., Dharmadasa, T., Huynh, W., Park, S. B., Burke, D., et al. (2018). Excitability of sensory axons in amyotrophic lateral sclerosis. Clin. Neurophysiol. 129, 1472–1478. doi: 10.1016/j.clinph.2018.03.014
Matsukawa, K., Hashimoto, T., Matsumoto, T., Ihara, R., Chihara, T., Miura, M., et al. (2016). Familial amyotrophic lateral sclerosis-linked mutations in profilin 1 exacerbate TDP-43-induced degeneration in the retina of Drosophila melanogaster through an increase in the cytoplasmic localization of TDP-43. J. Biol. Chem. 291, 23464–23476. doi: 10.1074/jbc.m116.729152
Mavlyutov, T. A., Epstein, M. L., Andersen, K. A., Ziskind-Conhaim, L., and Ruoho, A. E. (2010). The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience 167, 247–255. doi: 10.1016/j.neuroscience.2010.02.022
Maystadt, I., Rezsohazy, R., Barkats, M., Duque, S., Vannuffel, P., Remacle, S., et al. (2007). The nuclear factor kappaB-activator gene PLEKHG5 is mutated in a form of autosomal recessive lower motor neuron disease with childhood onset. Am. J. Hum. Genet. 81, 67–76. doi: 10.1086/518900
McCluskey, L., Vandriel, S., Elman, L., Van Deerlin, V. M., Powers, J., Boller, A., et al. (2014). ALS-Plus syndrome: non-pyramidal features in a large ALS cohort. J. Neurol. Sci. 345, 118–124. doi: 10.1016/j.jns.2014.07.022
Menon, P., Kiernan, M. C., and Vucic, S. (2014). Cortical dysfunction underlies the development of the split-hand in amyotrophic lateral sclerosis. PLoS One 9:e87124. doi: 10.1371/journal.pone.0087124
Mersiyanova, I. V., Perepelov, A. V., Polyakov, A. V., Sitnikov, V. F., Dadali, E. L., Oparin, R. B., et al. (2000). A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am. J. Hum. Genet. 67, 37–46. doi: 10.1086/302962
Meyer, T., Schwan, A., Dullinger, J. S., Brocke, J., Hoffmann, K. T., Nolte, C. H., et al. (2005). Early-onset ALS with long-term survival associated with spastin gene mutation. Neurology 65, 141–143. doi: 10.1212/01.wnl.0000167130.31618.0a
Miller, J. W., Smith, B. N., Topp, S. D., Al-Chalabi, A., Shaw, C. E., and Vance, C. (2012). Mutation analysis of VCP in British familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging 33:2721.e1-2. doi: 10.1016/j.neurobiolaging.2012.06.003
Modol-Caballero, G., Santos, D., Navarro, X., and Herrando-Grabulosa, M. (2017). Neuregulin 1 reduces motoneuron cell death and promotes neurite growth in an in vitro model of motoneuron degeneration. Front. Cell. Neurosci. 11:431. doi: 10.3389/fncel.2017.00431
Moisse, K., Volkening, K., Leystra-Lantz, C., Welch, I., Hill, T., and Strong, M. J. (2009). Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 1249, 202–211. doi: 10.1016/j.brainres.2008.10.021
Mondelli, M., Rossi, A., Passero, S., and Guazzi, G. C. (1993). Involvement of peripheral sensory fibers in amyotrophic lateral sclerosis: electrophysiological study of 64 cases. Muscle Nerve 16, 166–172. doi: 10.1002/mus.880160208
Montecchiani, C., Pedace, L., Lo Giudice, T., Casella, A., Mearini, M., Gaudiello, F., et al. (2016). ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease. Brain 139, 73–85. doi: 10.1093/brain/awv320
Moreira, M. C., Klur, S., Watanabe, M., Nemeth, A. H., Le Ber, I., Moniz, J. C., et al. (2004). Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 36, 225–227. doi: 10.1038/ng1303
Morrison, B. M., Shu, I. W., Wilcox, A. L., Gordon, J. W., and Morrison, J. H. (2000). Early and selective pathology of light chain neurofilament in the spinal cord and sciatic nerve of G86R mutant superoxide dismutase transgenic mice. Exp. Neurol. 165, 207–220. doi: 10.1006/exnr.2000.7457
Muglia, M., Zappia, M., Timmerman, V., Valentino, P., Gabriele, A. L., Conforti, F. L., et al. (2001). Clinical and genetic study of a large Charcot-Marie-Tooth type 2A family from southern Italy. Neurology 56, 100–103. doi: 10.1212/wnl.56.1.100
Muller, H. P., Agosta, F., Riva, N., Spinelli, E. G., Comi, G., Ludolph, A. C., et al. (2018). Fast progressive lower motor neuron disease is an ALS variant: a two-centre tract of interest-based MRI data analysis. Neuroimage Clin. 17, 145–152. doi: 10.1016/j.nicl.2017.10.008
Munch, C., Rosenbohm, A., Sperfeld, A. D., Uttner, I., Reske, S., Krause, B. J., et al. (2005). Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 58, 777–780. doi: 10.1002/ana.20631
Munch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. doi: 10.1212/01.wnl.0000134608.83927.b1
Nam, D. E., Jung, S. C., Yoo, D. H., Choi, S. S., Seo, S. Y., Kim, G. H., et al. (2017). Axonal Charcot-Marie-Tooth neuropathy concurrent with distal and proximal weakness by translational elongation of the 3’ UTR in NEFH. J. Peripher. Nerv. Syst. 22, 200–207. doi: 10.1111/jns.12223
Nardo, G., Trolese, M. C., De Vito, G., Cecchi, R., Riva, N., Dina, G., et al. (2016). Immune response in peripheral axons delays disease progression in SOD1(G93A) mice. J. Neuroinflamm. 13:261.
Nardo, G., Trolese, M. C., Verderio, M., Mariani, A., De Paola, M., Riva, N., et al. (2018). Counteracting roles of MHCI and CD8(+) T cells in the peripheral and central nervous system of ALS SOD1(G93A) mice. Mol. Neurodegener. 13:42. doi: 10.1186/s13024-018-0271-7
Naumenko, N., Pollari, E., Kurronen, A., Giniatullina, R., Shakirzyanova, A., Magga, J., et al. (2011). Gender-specific mechanism of synaptic impairment and its prevention by GCSF in a mouse model of ALS. Front. Cell. Neurosci. 5:26. doi: 10.3389/fncel.2011.00026
Nguyen, L., Lucke-Wold, B. P., Mookerjee, S. A., Cavendish, J. Z., Robson, M. J., Scandinaro, A. L., et al. (2015). Role of sigma-1 receptors in neurodegenerative diseases. J. Pharmacol. Sci. 127, 17–29.
Nicholson, G., Lenk, G. M., Reddel, S. W., Grant, A. E., Towne, C. F., Ferguson, C. J., et al. (2011). Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P(2) phosphatase FIG4. Brain 134, 1959–1971. doi: 10.1093/brain/awr148
Nicholson, G. A., Magdelaine, C., Zhu, D., Grew, S., Ryan, M. M., Sturtz, F., et al. (2008). Severe early-onset axonal neuropathy with homozygous and compound heterozygous MFN2 mutations. Neurology 70, 1678–1681. doi: 10.1212/01.wnl.0000311275.89032.22
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268.e6–1283.e6. doi: 10.1016/j.neuron.2018.02.027
Nishihira, Y., Tan, C. -F., Onodera, O., Toyoshima, Y., Yamada, M., Morita, T., et al. (2008). Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol. 116:169. doi: 10.1007/s00401-008-0385-z
Nolano, M., Provitera, V., Manganelli, F., Iodice, R., Caporaso, G., Stancanelli, A., et al. (2017). Non-motor involvement in amyotrophic lateral sclerosis: new insight from nerve and vessel analysis in skin biopsy. Neuropathol. Appl. Neurobiol. 43, 119–132. doi: 10.1111/nan.12332
Orlacchio, A., Babalini, C., Borreca, A., Patrono, C., Massa, R., Basaran, S., et al. (2010). SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 133, 591–598. doi: 10.1093/brain/awp325
Osmanovic, A., Rangnau, I., Kosfeld, A., Abdulla, S., Janssen, C., Auber, B., et al. (2017). FIG4 variants in central European patients with amyotrophic lateral sclerosis: a whole-exome and targeted sequencing study. Eur. J. Hum. Genet. 25, 324–331. doi: 10.1038/ejhg.2016.186
Ozoguz, A., Uyan, O., Birdal, G., Iskender, C., Kartal, E., Lahut, S., et al. (2015). The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol. Aging 36, 1764.e9–1764.e18. doi: 10.1016/j.neurobiolaging.2014.12.032
Park, S. B., Kiernan, M. C., and Vucic, S. (2017). Axonal excitability in amyotrophic lateral sclerosis : axonal excitability in ALS. Neurotherapeutics 14, 78–90. doi: 10.1007/s13311-016-0492-9
Patten, S. A., Aggad, D., Martinez, J., Tremblay, E., Petrillo, J., Armstrong, G. A., et al. (2017). Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight 2: 97152
Perez-Branguli, F., Mishra, H. K., Prots, I., Havlicek, S., Kohl, Z., Saul, D., et al. (2014). Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum. Mol. Genet. 23, 4859–4874. doi: 10.1093/hmg/ddu200
Perlson, E., Jeong, G. B., Ross, J. L., Dixit, R., Wallace, K. E., Kalb, R. G., et al. (2009). A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J. Neurosci. 29, 9903–9917. doi: 10.1523/JNEUROSCI.0813-09.2009
Peters, O. M., Lewis, E. A., Osterloh, J. M., Weiss, A., Salameh, J. S., Metterville, J., et al. (2018). Loss of Sarm1 does not suppress motor neuron degeneration in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 27, 3761–3771. doi: 10.1093/hmg/ddy260
Philips, T., and Rothstein, J. D. (2015). Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 69, 67.1–67.21.
Piccione, E. A., Sletten, D. M., Staff, N. P., and Low, P. A. (2015). Autonomic system and amyotrophic lateral sclerosis. Muscle Nerve 51, 676–679. doi: 10.1002/mus.24457
Picher-Martel, V., Valdmanis, P. N., Gould, P. V., Julien, J. P., and Dupre, N. (2016). From animal models to human disease: a genetic approach for personalized medicine in ALS. Acta Neuropathol. Commun. 4:70. doi: 10.1186/s40478-016-0340-5
Polke, J. M., Laura, M., Pareyson, D., Taroni, F., Milani, M., Bergamin, G., et al. (2011). Recessive axonal Charcot-Marie-Tooth disease due to compound heterozygous mitofusin 2 mutations. Neurology 77, 168–173. doi: 10.1212/WNL.0b013e3182242d4d
Pozzi, S., Thammisetty, S. S., and Julien, J. P. (2018). Chronic administration of pimozide fails to attenuate motor and pathological deficits in two mouse models of amyotrophic lateral sclerosis. Neurotherapeutics 15, 715–727. doi: 10.1007/s13311-018-0634-3
Pugdahl, K., Fuglsang-Frederiksen, A., De Carvalho, M., Johnsen, B., Fawcett, P. R., Labarre-Vila, A., et al. (2007). Generalised sensory system abnormalities in amyotrophic lateral sclerosis: a European multicentre study. J. Neurol. Neurosurg. Psychiatry 78, 746–749. doi: 10.1136/jnnp.2006.098533
Pugdahl, K., Fuglsang-Frederiksen, A., Johnsen, B., De Carvalho, M., Fawcett, P. R., Labarre-Vila, A., et al. (2008). A prospective multicentre study on sural nerve action potentials in ALS. Clin. Neurophysiol. 119, 1106–1110. doi: 10.1016/j.clinph.2008.01.010
Puls, I., Jonnakuty, C., Lamonte, B. H., Holzbaur, E. L., Tokito, M., Mann, E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456.
Pupillo, E., Messina, P., Giussani, G., Logroscino, G., Zoccolella, S., Chio, A., et al. (2014). Physical activity and amyotrophic lateral sclerosis: a European population-based case-control study. Ann. Neurol. 75, 708–716. doi: 10.1002/ana.24150
Rajabally, Y. A., and Jacob, S. (2008). Chronic inflammatory demyelinating polyneuropathy-like disorder associated with amyotrophic lateral sclerosis. Muscle Nerve 38, 855–860. doi: 10.1002/mus.21010
Ratti, A., and Buratti, E. (2016). Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 138(Suppl. 1), 95–111. doi: 10.1111/jnc.13625
Rebelo, A. P., Abrams, A. J., Cottenie, E., Horga, A., Gonzalez, M., Bis, D. M., et al. (2016). Cryptic amyloidogenic elements in the 3’ UTRs of neurofilament genes trigger axonal neuropathy. Am. J. Hum. Genet. 98, 597–614. doi: 10.1016/j.ajhg.2016.02.022
Reid, E., Kloos, M., Ashley-Koch, A., Hughes, L., Bevan, S., Svenson, I. K., et al. (2002). A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J. Hum. Genet. 71, 1189–1194. doi: 10.1086/344210
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rezaie, T., Child, A., Hitchings, R., Brice, G., Miller, L., Coca-Prados, M., et al. (2002). Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 295, 1077–1079. doi: 10.1126/science.1066901
Riva, N., Agosta, F., Lunetta, C., Filippi, M., and Quattrini, A. (2016). Recent advances in amyotrophic lateral sclerosis. J. Neurol. 263, 1241–1254. doi: 10.1007/s00415-016-8091-6
Riva, N., Chaabane, L., Peviani, M., Ungaro, D., Domi, T., Dina, G., et al. (2014). Defining peripheral nervous system dysfunction in the SOD-1G93A transgenic rat model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 73, 658–670. doi: 10.1097/NEN.0000000000000081
Riva, N., Gallia, F., Iannaccone, S., Corbo, M., Terenghi, F., Lazzerini, A., et al. (2011a). Chronic motor axonal neuropathy. J. Peripher. Nerv. Syst. 16, 341–346.
Riva, N., Iannaccone, S., Corbo, M., Casellato, C., Sferrazza, B., Lazzerini, A., et al. (2011b). Motor nerve biopsy: clinical usefulness and histopathological criteria. Ann. Neurol. 69, 197–201. doi: 10.1002/ana.22110
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62.
Rossor, A. M., Kalmar, B., Greensmith, L., and Reilly, M. M. (2012). The distal hereditary motor neuropathies. J. Neurol. Neurosurg. Psychiatry 83, 6–14. doi: 10.1136/jnnp-2011-300952
Rotem, N., Magen, I., Ionescu, A., Gershoni-Emek, N., Altman, T., Costa, C. J., et al. (2017). ALS along the axons - expression of coding and noncoding RNA differs in axons of ALS models. Sci. Rep. 7:44500. doi: 10.1038/srep44500
Rouaux, C., Panteleeva, I., René, F., Gonzalez De Aguilar, J.-L., Echaniz-Laguna, A., Dupuis, L., et al. (2007). Sodium valproate exerts neuroprotective effects In Vivo through CREB-Binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J. Neurosci. 27, 5535–5545. doi: 10.1523/jneurosci.1139-07.2007
Rouleau, G. A., Clark, A. W., Rooke, K., Pramatarova, A., Krizus, A., Suchowersky, O., et al. (1996). SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann. Neurol. 39, 128–131.
Rusmini, P., Cristofani, R., Galbiati, M., Cicardi, M. E., Meroni, M., Ferrari, V., et al. (2017). The role of the heat shock protein B8 (HSPB8) in motoneuron diseases. Front. Mol. Neurosci. 10:176. doi: 10.3389/fnmol.2017.00176
Sabatelli, M., Marangi, G., Conte, A., Tasca, G., Zollino, M., and Lattante, S. (2016). New ALS-Related genes expand the spectrum paradigm of amyotrophic lateral sclerosis. Brain Pathol. 26, 266–275. doi: 10.1111/bpa.12354
Saberi, S., Stauffer, J. E., Schulte, D. J., and Ravits, J. (2015). Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol. Clin. 33, 855–876. doi: 10.1016/j.ncl.2015.07.012
Sassone, J., Taiana, M., Lombardi, R., Porretta-Serapiglia, C., Freschi, M., Bonanno, S., et al. (2016). ALS mouse model SOD1G93A displays early pathology of sensory small fibers associated to accumulation of a neurotoxic splice variant of peripherin. Hum. Mol. Genet. 25, 1588–1599. doi: 10.1093/hmg/ddw035
Sawa, N., Kataoka, H., Sugie, K., Kawahara, M., Horikawa, H., Kusunoki, S., et al. (2012). Clinical analysis and outcomes of amyotrophic lateral sclerosis with demyelinating polyneuropathy. Amyotroph. Lateral Scler. 13, 125–131. doi: 10.3109/17482968.2011.627590
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Schafer, M. K., Bellouze, S., Jacquier, A., Schaller, S., Richard, L., Mathis, S., et al. (2017). Sensory neuropathy in progressive motor neuronopathy (pmn) mice is associated with defects in microtubule polymerization and axonal transport. Brain Pathol. 27, 459–471. doi: 10.1111/bpa.12422
Schwenk, B. M., Hartmann, H., Serdaroglu, A., Schludi, M. H., Hornburg, D., Meissner, F., et al. (2016). TDP-43 loss of function inhibits endosomal trafficking and alters trophic signaling in neurons. EMBO J. 35, 2350–2370. doi: 10.15252/embj.201694221
Sharma, A., Lyashchenko, A. K., Lu, L., Nasrabady, S. E., Elmaleh, M., Mendelsohn, M., et al. (2016). ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 7:10465. doi: 10.1038/ncomms10465
Shibuya, K., Misawa, S., Arai, K., Nakata, M., Kanai, K., Yoshiyama, Y., et al. (2011). Markedly reduced axonal potassium channel expression in human sporadic amyotrophic lateral sclerosis: an immunohistochemical study. Exp. Neurol. 232, 149–153. doi: 10.1016/j.expneurol.2011.08.015
Skourti-Stathaki, K., Proudfoot, N. J., and Gromak, N. (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell. 42, 794–805. doi: 10.1016/j.molcel.2011.04.026
Smith, B. N., Ticozzi, N., Fallini, C., Gkazi, A. S., Topp, S., Kenna, K. P., et al. (2014). Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. doi: 10.1016/j.neuron.2014.09.027
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Stalekar, M., Yin, X., Rebolj, K., Darovic, S., Troakes, C., Mayr, M., et al. (2015). Proteomic analyses reveal that loss of TDP-43 affects RNA processing and intracellular transport. Neuroscience 293, 157–170. doi: 10.1016/j.neuroscience.2015.02.046
Stevanin, G., Santorelli, F. M., Azzedine, H., Coutinho, P., Chomilier, J., Denora, P. S., et al. (2007). Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat. Genet. 39, 366–372. doi: 10.1038/ng1980
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K. F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5:3996. doi: 10.1038/ncomms4996
Suraweera, A., Lim, Y., Woods, R., Birrell, G. W., Nasim, T., Becherel, O. J., et al. (2009). Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum. Mol. Genet. 18, 3384–3396. doi: 10.1093/hmg/ddp278
Suzuki, M., Mchugh, J., Tork, C., Shelley, B., Klein, S. M., Aebischer, P., et al. (2007). GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS One 2:e689. doi: 10.1371/journal.pone.0000689
Swarup, V., Audet, J. N., Phaneuf, D., Kriz, J., and Julien, J. P. (2012). Abnormal regenerative responses and impaired axonal outgrowth after nerve crush in TDP-43 transgenic mouse models of amyotrophic lateral sclerosis. J. Neurosci. 32, 18186–18195. doi: 10.1523/JNEUROSCI.2267-12.2012
Tanaka, Y., Nonaka, T., Suzuki, G., Kametani, F., and Hasegawa, M. (2016). Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum. Mol. Genet. 25, 1420–1433. doi: 10.1093/hmg/ddw024
Tang, B. S., Zhao, G. H., Luo, W., Xia, K., Cai, F., Pan, Q., et al. (2005). Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum. Genet. 116, 222–224. doi: 10.1007/s00439-004-1218-3
Tradewell, M. L., Durham, H. D., Mushynski, W. E., and Gentil, B. J. (2009). Mitochondrial and axonal abnormalities precede disruption of the neurofilament network in a model of charcot-marie-tooth disease type 2E and are prevented by heat shock proteins in a mutant-specific fashion. J. Neuropathol. Exp. Neurol. 68, 642–652. doi: 10.1097/NEN.0b013e3181a5deeb
Traynor, B. J., Codd, M. B., Corr, B., Forde, C., Frost, E., and Hardiman, O. M. (2000). Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: a population-based study. Arch. Neurol. 57, 1171–1176.
Tripathi, V. B., Baskaran, P., Shaw, C. E., and Guthrie, S. (2014). Tar DNA-binding protein-43 (TDP-43) regulates axon growth in vitro and in vivo. Neurobiol. Dis. 65, 25–34. doi: 10.1016/j.nbd.2014.01.004
Tripolszki, K., Torok, D., Goudenege, D., Farkas, K., Sulak, A., Torok, N., et al. (2017). High-throughput sequencing revealed a novel SETX mutation in a Hungarian patient with amyotrophic lateral sclerosis. Brain Behav. 7:e00669. doi: 10.1002/brb3.669
Truini, A., Biasiotta, A., Onesti, E., Di Stefano, G., Ceccanti, M., La Cesa, S., et al. (2015). Small-fibre neuropathy related to bulbar and spinal-onset in patients with ALS. J. Neurol. 262, 1014–1018. doi: 10.1007/s00415-015-7672-0
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vandermoere, F., El Yazidi-Belkoura, I., Slomianny, C., Demont, Y., Bidaux, G., Adriaenssens, E., et al. (2006). The valosin-containing protein (VCP) is a target of Akt signaling required for cell survival. J. Biol. Chem. 281, 14307–14313. doi: 10.1074/jbc.m510003200
Vaughan, S. K., Kemp, Z., Hatzipetros, T., Vieira, F., and Valdez, G. (2015). Degeneration of proprioceptive sensory nerve endings in mice harboring amyotrophic lateral sclerosis-causing mutations. J. Comp. Neurol. 523, 2477–2494. doi: 10.1002/cne.23848
Vaughan, S. K., Sutherland, N. M., Zhang, S., Hatzipetros, T., Vieira, F., and Valdez, G. (2018). The ALS-inducing factors, TDP43(A315T) and SOD1(G93A), directly affect and sensitize sensory neurons to stress. Sci. Rep. 8:16582. doi: 10.1038/s41598-018-34510-8
Verheijen, M. H., Peviani, M., Hendricusdottir, R., Bell, E. M., Lammens, M., Smit, A. B., et al. (2014). Increased axonal ribosome numbers is an early event in the pathogenesis of amyotrophic lateral sclerosis. PLoS One 9:e87255. doi: 10.1371/journal.pone.0087255
Vieira, F. G., Ping, Q., Moreno, A. J., Kidd, J. D., Thompson, K., Jiang, B., et al. (2015). Guanabenz treatment accelerates disease in a mutant SOD1 mouse model of ALS. PLoS One 10:e0135570. doi: 10.1371/journal.pone.0135570
Visser, J., Van Den Berg-Vos, R. M., Franssen, H., Van Den Berg, L. H., Vogels, O. J., Wokke, J. H., et al. (2002). Mimic syndromes in sporadic cases of progressive spinal muscular atrophy. Neurology 58, 1593–1596. doi: 10.1212/wnl.58.11.1593
Volkening, K., Leystra-Lantz, C., Yang, W., Jaffee, H., and Strong, M. J. (2009). Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 1305, 168–182. doi: 10.1016/j.brainres.2009.09.105
Waibel, S., Neumann, M., Rosenbohm, A., Birve, A., Volk, A. E., Weishaupt, J. H., et al. (2013). Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: a clinico-genetic study in Germany. Eur. J. Neurol. 20, 540–546. doi: 10.1111/ene.12031
Wang, L., Gao, J., Liu, J., Siedlak, S. L., Torres, S., Fujioka, H., et al. (2018). Mitofusin 2 regulates axonal transport of calpastatin to prevent neuromuscular synaptic elimination in skeletal muscles. Cell Metab. 28, 400.e8–414.e8. doi: 10.1016/j.cmet.2018.06.011
Wang, L., Popko, B., Tixier, E., and Roos, R. P. (2014). Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis. 71, 317–324. doi: 10.1016/j.nbd.2014.08.010
Wang, L., Pytel, P., Feltri, M. L., Wrabetz, L., and Roos, R. P. (2012). Selective knockdown of mutant SOD1 in Schwann cells ameliorates disease in G85R mutant SOD1 transgenic mice. Neurobiol. Dis. 48, 52–57. doi: 10.1016/j.nbd.2012.05.014
Wang, W., Wang, L., Lu, J., Siedlak, S. L., Fujioka, H., Liang, J., et al. (2016). The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 22, 869–878. doi: 10.1038/nm.4130
Watanabe, S., Ilieva, H., Tamada, H., Nomura, H., Komine, O., Endo, F., et al. (2016). Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 8, 1421–1437. doi: 10.15252/emmm.201606403
Watts, G. D., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., et al. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381. doi: 10.1038/ng1332
Weedon, M. N., Hastings, R., Caswell, R., Xie, W., Paszkiewicz, K., Antoniadi, T., et al. (2011). Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 89, 308–312. doi: 10.1016/j.ajhg.2011.07.002
Wegorzewska, I., Bell, S., Cairns, N. J., Miller, T. M., and Baloh, R. H. (2009). TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 18809–18814. doi: 10.1073/pnas.0908767106
Weihl, C. C., Pestronk, A., and Kimonis, V. E. (2009). Valosin-containing protein disease: inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul. Disord. 19, 308–315. doi: 10.1016/j.nmd.2009.01.009
Weis, J., Katona, I., Muller-Newen, G., Sommer, C., Necula, G., Hendrich, C., et al. (2011). Small-fiber neuropathy in patients with ALS. Neurology 76, 2024–2029. doi: 10.1212/WNL.0b013e31821e553a
Williamson, T. L., and Cleveland, D. W. (1999). Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 2, 50–56. doi: 10.1038/4553
Windpassinger, C., Auer-Grumbach, M., Irobi, J., Patel, H., Petek, E., Horl, G., et al. (2004). Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet. 36, 271–276. doi: 10.1038/ng1313
Wu, C. H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280
Yamashita, T., Hideyama, T., Hachiga, K., Teramoto, S., Takano, J., Iwata, N., et al. (2012). A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 3:1307. doi: 10.1038/ncomms2303
Yoshihara, T., Yamamoto, M., Hattori, N., Misu, K., Mori, K., Koike, H., et al. (2002). Identification of novel sequence variants in the neurofilament-light gene in a Japanese population: analysis of Charcot-Marie-Tooth disease patients and normal individuals. J. Peripher. Nerv. Syst. 7, 221–224. doi: 10.1046/j.1529-8027.2002.02028.x
Zhan, L., Xie, Q., and Tibbetts, R. S. (2015). Opposing roles of p38 and JNK in a Drosophila model of TDP-43 proteinopathy reveal oxidative stress and innate immunity as pathogenic components of neurodegeneration. Hum. Mol. Genet. 24, 757–772. doi: 10.1093/hmg/ddu493
Zhang, X., Chen, S., Song, L., Tang, Y., Shen, Y., Jia, L., et al. (2014). MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 10, 588–602. doi: 10.4161/auto.27710
Zhao, Z. H., Chen, W. Z., Wu, Z. Y., Wang, N., Zhao, G. X., Chen, W. J., et al. (2009). A novel mutation in the senataxin gene identified in a Chinese patient with sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 118–122. doi: 10.1080/17482960802572673
Zuchner, S., De Jonghe, P., Jordanova, A., Claeys, K. G., Guergueltcheva, V., Cherninkova, S., et al. (2006). Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann. Neurol. 59, 276–281. doi: 10.1002/ana.20797
Zuchner, S., Mersiyanova, I. V., Muglia, M., Bissar-Tadmouri, N., Rochelle, J., Dadali, E. L., et al. (2004a). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36, 449–451. doi: 10.1038/ng1341
Keywords: motor neuron disease, lower motor neuron syndrome, nerve, neuropathy, CMT, distal SMA, hereditary neuropathy, genetics
Citation: Gentile F, Scarlino S, Falzone YM, Lunetta C, Tremolizzo L, Quattrini A and Riva N (2019) The Peripheral Nervous System in Amyotrophic Lateral Sclerosis: Opportunities for Translational Research. Front. Neurosci. 13:601. doi: 10.3389/fnins.2019.00601
Received: 15 February 2019; Accepted: 27 May 2019;
Published: 25 June 2019.
Edited by:
Foteini Christidi, National and Kapodistrian University of Athens, GreeceReviewed by:
Chiara F. Valori, German Center for Neurodegenerative Diseases (DZNE), GermanyYasushi Kitaoka, St. Marianna University School of Medicine, Japan
Copyright © 2019 Gentile, Scarlino, Falzone, Lunetta, Tremolizzo, Quattrini and Riva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nilo Riva, cml2YS5uaWxvQGhzci5pdA==
†These authors share last senior authorship