 Camille Pernègre
Camille Pernègre Antoine Duquette
Antoine Duquette Nicole Leclerc
Nicole Leclerc- 1Research Centre of the University of Montreal Hospital (CRCHUM), Montréal, QC, Canada
- 2Département de Neurosciences, Faculty of Medicine, Université de Montréal, Montréal, QC, Canada
In Alzheimer’s disease (AD), neurofibrillary tangles (NFTs), lesions composed of hyperphosphorylated and aggregated tau, spread from the transentorhinal cortex to the hippocampal formation and neocortex. Growing evidence indicates that tau pathology propagates trans-synaptically, implying that pathological tau released by pre-synaptic neurons is taken up by post-synaptic neurons where it accumulates and aggregates. Observations such as the presence of tau in the cerebrospinal fluid (CSF) from control individuals and in the CSF of transgenic mice overexpressing human tau before the detection of neuronal death indicate that tau can be secreted by neurons. The increase of tau in the CSF in pathological conditions such as AD suggests that tau secretion is enhanced and/or other secretory pathways take place when neuronal function is compromised. In physiological conditions, extracellular tau could exert beneficial effects as observed for other cytosolic proteins also released in the extracellular space. In such a case, blocking tau secretion could have negative effects on neurons unless the mechanism of tau secretion are different in physiological and pathological conditions allowing the prevention of pathological tau secretion without affecting the secretion of physiological tau. Furthermore, distinct extracellular tau species could be secreted in physiological and pathological conditions, species having the capacity to induce tau pathology being only secreted in the latter condition. In the present review, we will focus on the mechanisms and function of tau secretion in both physiological and pathological conditions and how this information can help to elaborate an efficient therapeutic strategy to prevent tau pathology and its propagation.
Observations Supporting Tau Secretion by Neurons
Tau protein, an axonal microtubule-associated protein, accumulates both intracellularly and extracellularly in AD. Extracellular tau is presently a promising therapeutic target to block the propagation of tau pathology in the brain of AD patients. Based on this, the elucidation of tau routes to the extracellular space could serve to elaborate therapies for prevention of the tau pathology spreading and the cognitive deficits linked to it. The presence of tau in CSF and the medium of neuronal cultures suggested that neurons can secrete it. However, its accumulation in the CSF during the progression of AD was believed to correlate with neuronal cell death (Hampel et al., 2010). The interneuronal transfer of human tau overexpressed in anterior bulbar neurons of the lamprey was the first observation indicating that tau could be secreted by neurons in vivo (Kim et al., 2010). More recently, tau release by neurons in vivo was demonstrated by its detection in the interstitial fluid in the absence of neurodegeneration using microdialysis in tau transgenic mouse brain (Yamada et al., 2011). Neuronal activity was shown to increase tau release by neurons both in vitro and in vivo (Pooler et al., 2013; Yamada et al., 2014). In particular, neuronal hyperexcitability observed at early stages of both sporadic and familial AD and in AD mouse models was shown to increase tau release (Busche et al., 2008, 2012; Palop and Mucke, 2009; Bakker et al., 2012; Pooler et al., 2013; Wu et al., 2016). Furthermore, we reported that the induction of autophagy and/or lysosomal dysfunction increased the release of tau by primary cortical neurons (Mohamed et al., 2014). Intriguingly, we recently reported that tau secretion induced by neuronal hyperactivity was associated with Golgi fragmentation indicating that Golgi membranes may play a role in tau secretion (Mohamed et al., 2017). Consistent with this, blocking the fragmentation of the Golgi upon the induction of hyperactivity reduced tau secretion. Lastly, the pattern of tau forms found in the medium upon cell damage differs from that resulting from active secretion (Plouffe et al., 2012; Kanmert et al., 2015). All the above observations demonstrate that neurons can secrete tau and that changes in its levels of secretion are linked to pathological conditions.
Mechanisms Involved in Membrane Free-Tau and Vesicle/Exosome-Tau Secretion
Two pools of extracellular tau are detected: a small pool found in vesicles and a large membrane-free pool (Saman et al., 2012; Pooler et al., 2013; Dujardin et al., 2014; Mohamed et al., 2014). Several groups, including ours, have demonstrated that neurons release tau by unconventional secretory pathways, rather than by the conventional endoplasmic reticulum (ER)-Golgi pathway, in both physiological and pathological conditions (ex. starvation, lysosomal dysfunction and hyperactivity) (Pooler et al., 2013; Mohamed et al., 2014, 2017). Both non-vesicular (free-tau) and vesicular (vesicle/exosome-tau) mechanisms were shown to contribute to tau secretion (Figure 1).
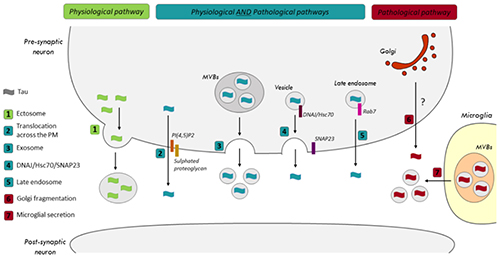
Figure 1. Secretory pathways of tau in physiological and pathological conditions. (1) The release of tau by ectosomes was only reported in physiological conditions. In both physiological and pathological conditions, tau could be released by (2) its translocation across the plasma membrane (PM), (3) exosomes, (4) the SNARE, SNAP-23, and the co-chaperone DNAJ/Hsc70 and (5) Rab7A, a Rab GTPase associated with late endosomes (LEs). (6) A correlation was noted between the fragmentation of the Golgi and an increase of tau secretion in pathological conditions. However, the pathway is not characterized yet. (7) Microglia were shown to release exosomes containing pathological tau in a tauopathy mouse model.
Translocation Across the Plasma Membrane
Two recent studies reported that both endogenous tau and overexpressed human tau can be secreted by a non-vesicular mechanism involving its direct translocation across the plasma membrane (PM) (Katsinelos et al., 2018; Merezhko et al., 2018). This mechanism was previously described for the fibroblast growth factor 2 (FGF2) (Steringer et al., 2017). Unconventional secretion of FGF2 occurs through a mechanism of self-sustained membrane translocation with FGF2 forming its own membrane translocation intermediates by reversible oligomerization, membrane insertion and disassembly in the extracellular space. As noted for FGF2, the interaction of tau with PI(4,5)P2 and sulphated proteoglycans mediates its translocation (Katsinelos et al., 2018; Merezhko et al., 2018). PI(4,5)P2 are involved in the binding of tau at the inner surface of the PM and sulphated proteoglycans found at the cell surface facilitate the export process. Auxiliary factors such as ATP1A1 and Tec kinase located at the PM are involved in FGF2 secretion (Ebert et al., 2010; Zacherl et al., 2015). Such factors remain to be identified for tau. As noted for FGF2, the formation of oligomer intermediates was shown to be necessary for tau secretion by this mechanism (Merezhko et al., 2018). The properties of the PM also modulate tau secretion. For example, the reduction of specific lipid species such as cholesterol and sphingomyelin impaired tau secretion (Merezhko et al., 2018). Interestingly, the secretion of phosphorylation-mimicking tau mutant through this mechanism was significantly more important than that of wild-type tau and a non-phosphorylatable tau mutant (Merezhko et al., 2018).
Membranous Organelles-Based Tau Secretion
The contribution of membranous organelles to tau secretion was revealed by demonstrating that SNAREs and Rabs, two classes of proteins implicated in membrane trafficking are involved in tau secretion. In a recent study, it was observed that the suppression of SNAP-23, a SNARE protein implicated in neurotransmitter exocytosis, resulted in a significant reduction of tau secretion (Fontaine et al., 2016). The co-chaperone DNAJ/Hsc70 was shown to be necessary for tau secretion through this pathway. However, in this study, the membranous compartment involved in tau secretion was not characterized. This secretory pathway was shown to take place in both physiological and pathological conditions and to be used by wild-type and mutated human tau (P301L and R406W) (Fontaine et al., 2016).
We recently investigated tau secretory pathways (Plouffe et al., 2012; Mohamed et al., 2013, 2014, 2017; Rodriguez et al., 2017) and showed that late endosomes (LEs) are involved in the unconventional secretion of membrane-free tau (Rodriguez et al., 2017). Our data revealed that LEs could secrete membrane-free tau via Rab7A, a GTPase associated with these endosomes and involved in their trafficking (Rodriguez et al., 2017). Tau partially co-localized with Rab7A-positive structures and its secretion was significantly reduced by Rab7A depletion (Rodriguez et al., 2017). Interestingly, LEs were recently shown to be involved in the secretion of the huntingtin mutant (mHtt) linked to Huntington’s disease (Trajkovic et al., 2017). At early stages of AD when tau intracellular accumulation occurs, Rab7A is upregulated and there is an accumulation of endosomal structures (Nixon, 2005; Ginsberg et al., 2010a, b, 2011; Tiernan et al., 2016). Based on our above data, all these changes could favor tau secretion and its accumulation in the CSF of AD patients. In yeast, endosomes contribute to the formation of a compartment for unconventional protein secretion (CUPS), which arises following starvation (Cruz-Garcia et al., 2014; Curwin et al., 2016). We have reported that tau secretion is increased upon starvation (Mohamed et al., 2014). Thus, one can speculate that the accumulation of endosomes noted in AD may contribute to the production of CUPS yielding increased tau secretion.
We also reported that Rab1A was involved in tau secretion (Mohamed et al., 2017). Rab1A is associated with the membranes of the Golgi apparatus and its suppression induces their fragmentation (Shorter and Warren, 2002; Coune et al., 2011). In AD and tau transgenic mice, a fragmentation of the Golgi membranes was observed (Dal Canto, 1996; Stieber et al., 1996; Liazoghli et al., 2005; Anton-Fernandez et al., 2016). We showed that inducing Golgi fragmentation by suppression of Rab1A increased tau secretion (Mohamed et al., 2017).
Exosomes/Vesicles
Exosomes and ectosomes are the two main types of extracellular vesicles involved in the secretion of cytosolic proteins (Kalra et al., 2012). In the case of exosomes, cytosolic proteins are captured from the cytoplasm during the formation of internal endosomal vesicles resulting in the generation of multi-vesicular bodies (MVBs) (Hessvik and Llorente, 2018). Upon the fusion of MVBs with the PM, exosomes are released in the extracellular space. Ectosomes are larger than exosomes and result from the shedding of the PM (Muralidharan-Chari et al., 2010). Both exosomes and ectosomes were shown to be involved in tau secretion (Saman et al., 2012; Simon et al., 2012; Dujardin et al., 2014; Asai et al., 2015; Kanmert et al., 2015; Polanco et al., 2016; Wang et al., 2017; Guix et al., 2018). However, only a small pool of tau was found in these structures (1 to 10% of extracellular tau) (Kanmert et al., 2015). This mechanism was observed for endogenous tau and overexpressed human tau (Table 1). Vesicles containing tau leaving from anterior bulbar neurons in lamprey was the first observation indicating that tau could be released by exosomes/ectosomes (Lee et al., 2012). Several cell lines and primary neurons were reported to release tau by exosomes (Table 1). In pathological conditions, microglia were also shown to release tau by exosomes (Asai et al., 2015). In one study, tau was found in ectosomes (Dujardin et al., 2014). In this study, the authors observed the secretion of tau by ectosomes in physiological conditions whereas tau secretion by exosomes was observed in pathological conditions.
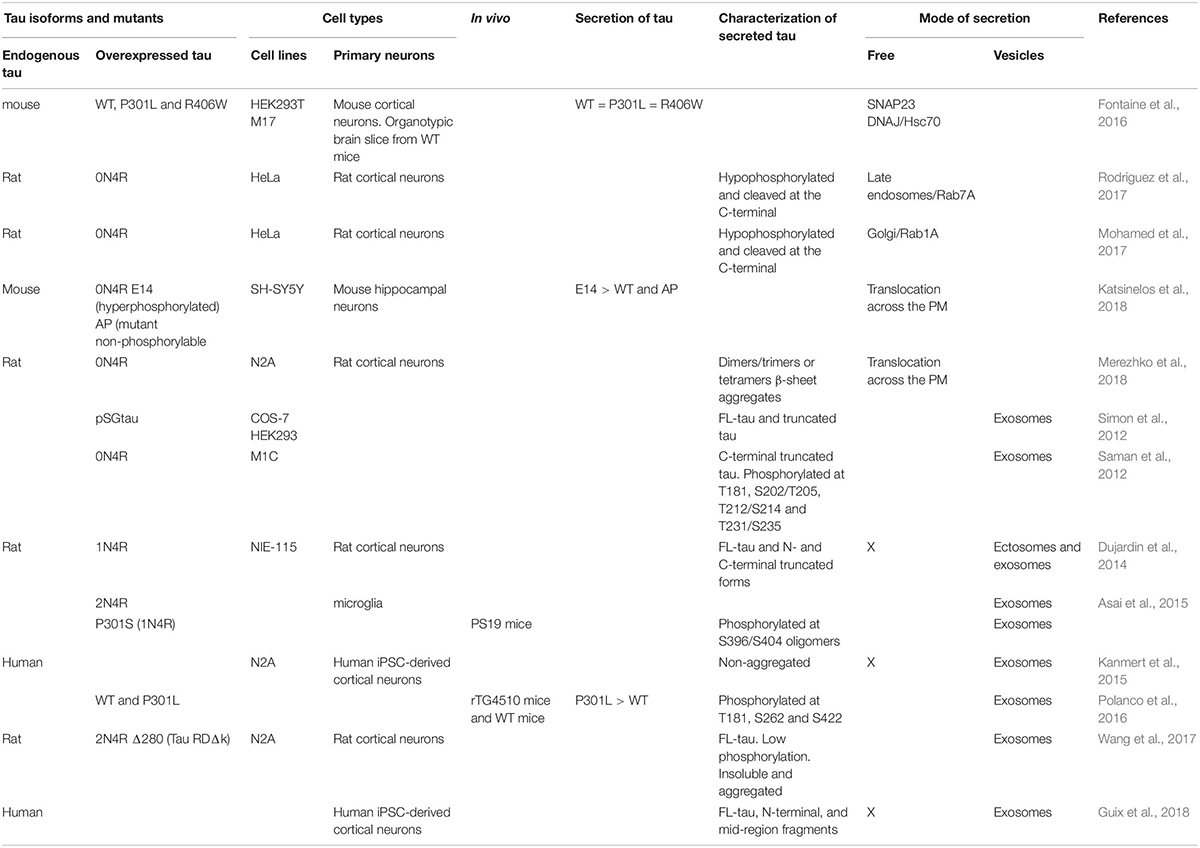
Table 1. Studies on tau secretory pathways.
Extracellular Tau Forms: Secreted Tau Vs. CSF-Tau
Although several studies indicate that tau found in CSF most likely results from its secretion by neurons, this remains to be experimentally demonstrated. Observations performed in lamprey where human tau was overexpressed in anterior bulbar neurons indicate that tau released by neurons could reach the CSF by periventricular and perimeningeal routes (Le et al., 2012). The fact that CSF-tau and secreted tau share similar post-translational modifications (PTMs) such as cleavage, phosphorylation and aggregation indicates that CSF-tau corresponds to secreted tau. In CSF obtained from control individuals and AD patients, both full-length tau (FL-tau) and truncated tau were detected (Johnson et al., 1997; Meredith et al., 2013; Wagshal et al., 2015; Guix et al., 2018). In all studies, the major form of tau present in CSF was cleaved at the C-terminal. In the medium from neuronal cultures, the amount of FL-tau varies from one study to another. In some studies, FL-tau was the main form of tau whereas in other studies it was the minor one (less than 1% of total secreted tau) (Plouffe et al., 2012; Pooler et al., 2013; Mohamed et al., 2014, 2017; Bright et al., 2015; Kanmert et al., 2015; Rodriguez et al., 2017). In transgenic mice, extracellular FL-tau was the main form of tau detected by microdialysis (Yamada et al., 2011). The differences in the amounts of FL-tau and truncated tau between CSF and culture medium could be explained by the three following reasons. First, all of the above observations are based on the use of antibodies to capture tau in the extracellular space. Because of this, one has to take into consideration that, depending on tau conformation and aggregation state in CSF and culture medium, the antibodies might have differential access to their epitopes. Consequently, this could affect the quantitation of FL-tau and cleaved tau. Secondly, the age and conditions of cultures could influence tau species released by neurons. For example, young cultures might release more FL-tau than old cultures, which might contain proteases able to cleave tau that would have been activated following an accumulation of insults. Thirdly, the turnover of extracellular FL-tau might differ in culture medium and CSF. For example, it might be higher in the CSF than in the culture medium. In such a case, a lower amount of FL-tau would be found in the CSF than in the culture medium. Phosphorylation of tau was also examined in both CSF and culture medium. The increase of phosphorylated tau in CSF is extensively used as a biomarker for AD (Hampel et al., 2010). However, in culture medium, most studies reported that tau presents either no phosphorylation or low phosphorylation levels (Mohamed et al., 2014; Rodriguez et al., 2017; Wang et al., 2017). More recently, two studies reported that an important amount of tau in the CSF of AD patients is not phosphorylated (Lewczuk et al., 2017; Ermann et al., 2018). In the case of tau aggregation, most studies did not report the presence of aggregated tau in culture medium and CSF. This could be explained by the lack of sensitivity of the techniques that were used. Indeed, tau aggregates were recently detected in both culture medium and CSF using a seeding assay (Takeda et al., 2016; Merezhko et al., 2018).
The PTMs of exosomal tau were also examined. Most studies reported that both FL-tau and C- and N-terminal truncated tau was found in exosomes, truncated forms being more abundant than FL-tau as noted for free-tau (Saman et al., 2012; Simon et al., 2012; Dujardin et al., 2014; Guix et al., 2018). In some studies, exosomal tau was found to be oligomeric or aggregated (Saman et al., 2012; Asai et al., 2015; Wang et al., 2017). Discrepancies are found in the literature concerning the phosphorylation levels of exosomal tau. Some studies reported that it was phosphorylated whereas in other studies, low phosphorylation levels were observed (Saman et al., 2012; Wang et al., 2017).
In adult human brain, six tau isoforms (0N3R, 1N3R, 2N3R, 0N4R, 1N4R, and 2N4R) are expressed (Buée et al., 2000). Very sparse information exists on the amount of each of these isoforms in the CSF and culture medium in both physiological and pathological conditions. In control CSF, the isoform 1N/3R was reported to be the most abundant (Barthelemy et al., 2016). A decrease of the 4R isoforms was observed in AD and other tauopathies such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), compared to controls (Luk et al., 2012). The preferential aggregation of the 4R isoforms in PSP and CBD could explain the decrease of these isoforms in the CSF (Lee et al., 2001). In the case of AD, where the 3R and 4R isoforms aggregate, the selective decrease of 4R remains unexplained.
Altogether, the above data reveal that several forms of tau co-exist in both the culture medium and CSF and strongly suggest that CSF-tau corresponds to secreted tau.
How the Elucidation of Tau Secretory Pathways and the Characterization of Extracellular Tau Can Help to Elaborate Therapeutic Strategies for Preventing the Propagation of Tau Pathology
Elucidation of Tau Secretory Pathways in Physiological Conditions: Understanding the Normal Function of Extracellular Tau
The fact that tau is released by neurons in physiological conditions strongly suggests that it has a role in the extracellular space. This was demonstrated for other cytosolic proteins. For example, annexin A4 and A5, calcium and phospholipids-binding proteins, act as anticoagulant factors via their Ca2+-regulated binding to phospholipids in the extracellular space (Gerke and Moss, 2002). Heat shock proteins are also secreted in the extracellular space (Santos et al., 2017). It was suggested that these proteins can trigger immunosuppression when they are released in the extracellular space under physiological conditions (Tanaka et al., 2007). No study has examined the normal function of extracellular tau. Previous studies indicate that tau could act as a signaling protein by binding to receptors such as muscarinic receptors. Indeed, extracellular tau was shown to bind and activate the M1 and M3 muscarinic receptors (Gomez-Ramos et al., 2008, 2009). However, it remains to be demonstrated whether the concentration of extracellular tau in physiological conditions could exert such effects.
Unconventional secretion of cytosolic proteins could also take place to prevent PTMs that can inactivate them. For example, the unconventional secretion of FGF2 prevents its O-glycosylation that renders its biologically inactive (Wegehingel et al., 2008). The PTMs of extracellular tau are still poorly characterized. It will be important to compare the PTMs of extracellular and intracellular tau to better understand the function of each of these pools. Unconventional secretion of protein could also aim at preventing inappropriate interactions. All these possibilities remain to be explored for tau. A better understanding of the distinct function of each extracellular tau form is crucial when designing anti-tau antibodies for immunotherapy as the sequestration of tau species having a physiological role by an antibody could accelerate neurodegeneration or be associated with significant side effects.
Elucidation of Tau Secretory Pathways in Pathological Conditions: Preventing and/or Enhancing the Release of Pathological Tau
Several observations indicate that blocking tau secretion could be beneficial by preventing the propagation of tau pathology in the brain and the detrimental effects of extracellular tau on synaptic function. Tau pathology is believed to propagate trans-synaptically by the release of tau at the pre-synaptic terminal and its uptake by the post-synaptic neurons (Calafate et al., 2015; Goedert and Spillantini, 2017). This cascade of events was confirmed in a recent study (Katsinelos et al., 2018). When tau translocation across the PM was blocked in pre-synaptic neurons, transcellular spreading of tau to post-synaptic neurons was prevented. Extracellular tau could also exert detrimental effects on synaptic function (Figures 2A,B). Extracellular tau secreted from human-derived iPSCs differentiated neurons induced deficits in long-term potentiation (LTP), a synaptic event underlying memory, when it was injected in mouse brain (Hu et al., 2018). It was also reported that a soluble and unaggregated N-terminal fragment shown to accumulate in the CSF of AD patients induced a pre-synaptic deficit in K+-evoked glutamate release on hippocampal synaptosomes along with an alteration in local Ca2+ dynamics (Florenzano et al., 2017). Furthermore, prolonged exposure of this peptide induces neuritic dystrophy, microtubule breakdown, deregulation in pre-synaptic proteins and loss of mitochondria located at the nerve endings. In a recent study, tau fibrils composed of either 1N3R or 1N4R isoform were shown to form clusters on the PM of excitatory synapses (Shrivastava et al., 2019). This clustering of tau fibrils was mediated by their interaction with Na+/K+-ATPse (NKA) α3 subunit and the AMPA receptor subunit GLUA2. Upon tau-fibril clustering, a decrease of α3-NKA was observed resulting in a decrease capacity of restoring Na+ level after depolarization. An increase of GLUA2-AMPA was also observed, which induced higher Ca2+-influx following action potential. A higher activity of M1 and M3 muscarinic receptors induced by extracellular tau could also contribute to synaptic dysfunction. Interestingly, non-phosphorylated tau abundant in the extracellular space but not phosphorylated tau was shown to interact with these receptors (Gomez-Ramos et al., 2006, 2008; Diaz-Hernandez et al., 2010). Lastly, extracellular tau could affect synapses by altering the biophysical properties of lipid rafts (Flach et al., 2012).
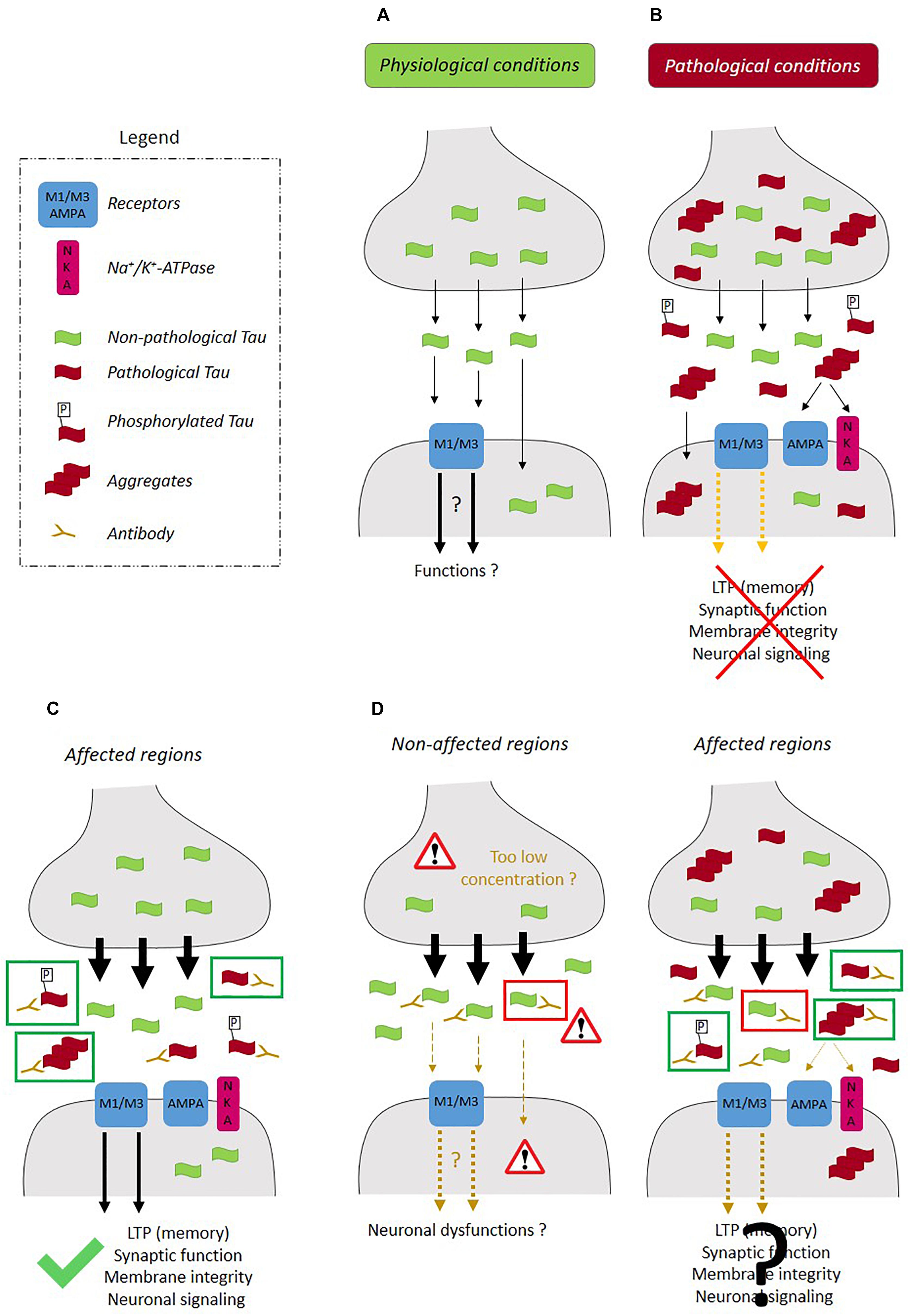
Figure 2. Therapeutic strategies for prevention of the propagation of tau pathology. (A) In physiological conditions, tau released at the pre-synaptic terminal presenting low phosphorylation levels, can interact with the receptors such as M1/M3 muscarinic receptors and activate signaling pathways involved in neuronal function. (B) In pathological conditions, extracellular pathological tau (phosphorylated and oligomeric) can interfere with synaptic function by inducing deficits in LTP, deficits in K+-evoked glutamate release, alterations in local Ca2+ dynamics and by altering the synaptic composition of NKA and AMPA receptors. The most (C) efficient therapeutic strategy would prevent the intracellular accumulation and aggregation of pathological tau by increasing its secretion (black arrows) and not that of physiological tau and would also involve the sequestration of pathological tau by an anti-tau antibody in the extracellular space (green rectangle). (D) Most pathways involved in tau secretion contribute to the release of tau in both physiological and pathological conditions. In such a case, the activation of tau secretion would not be confined to the affected regions and could also take place in non-affected regions leading to a decrease of intracellular physiological tau and an increase of extracellular physiological tau, which could be detrimental to healthy neurons. Furthermore, the capture of extracellular physiological tau by an anti-tau antibody could be detrimental in non-affected regions (red rectangle). In affected regions, both physiological (red rectangle) and pathological tau (green rectangle) could be released and captured by the anti-tau antibodies. If extracellular physiological tau plays a role in synaptic function and neuronal signaling, its capture could compromise neuronal recovery in the affected brain regions.
Conditions of stress might harm the degradative pathways such as proteasome and/or autophagy normally used to eliminate pathological tau. In such conditions, its secretion could be a very efficient clearance mechanism to prevent its intracellular accumulation and aggregation. This was recently demonstrated for TDP-43, a protein linked to ALS (Iguchi et al., 2016). Blocking TDP-43 secretion increased significantly its aggregation and toxicity. It is still unclear if preventing tau secretion will be detrimental to neurons.
Interestingly, the time course of the propagation of tau pathology in the brain and the amount of tau in the CSF differ among tauopathies. In AD where the progression of the disease is slow, an increase of tau in the CSF is observed whereas in other tauopathies such as PSP and CBD, where the disease progression is fast, no change or a small decrease of tau is noted in the CSF (Lee et al., 2001; Jabbari et al., 2017). At first glance, these observations do not support that extracellular tau is involved in the propagation of tau pathology. In a recent study, PSP-tau and CBD-tau species were reported to be more potent than AD-tau species at inducing propagation, meaning that they induced it faster and earlier than AD-tau (Narasimhan et al., 2017). Three explanations can reconcile all of the above observations. Firstly, in PSP and CBD, it is possible that tau secretion is lower than in AD but the tau species that are released are more potent than AD-tau at inducing the propagation of tau pathology. Lower secretion levels of tau would fit with the fact that PSP- and CBD-tau have a higher capacity to aggregate than AD-tau. Indeed, in transgenic mice, a decrease of tau in the CSF was correlated to the increase of tau aggregation (Yamada et al., 2011). Secondly, the secretion levels of tau are similar in PSP, CBD and AD but the uptake of PSP- and CBD-tau species is increased when compared to AD-tau resulting in a lower CSF concentration but faster propagation than AD-tau. Thirdly, in both PSP and CBD, tau pathology is noted in glial cells and neurons whereas in AD, tau pathology is mainly neuronal (Lee et al., 2001). Tau released by glial cells might be less abundant but more potent to induce propagation in both neurons and glial cells. Microglia can facilitate tau pathology propagation between neurons by phagocytosing and exocytosing tau protein (Guix et al., 2018). In the case of astrocytes, it was reported that they could decrease the spreading of tau pathology by taken up extracellular tau and targeting it for degradation (Martini-Stoica et al., 2018). However, in PSP and CBD, the tau taken up by astrocytes might be exocytosed instead of degraded.
Based on the above observations, one could speculate that blocking tau secretion could prevent the propagation of tau pathology and the detrimental effects exerted by extracellular tau on synaptic function. On the other hand, this could accelerate the intracellular accumulation and aggregation of pathological tau in the somato-dendritic (SD) compartment as noted for TDP-43. In either case, the elucidation of tau secretory pathways is central for the elaboration of a therapeutic strategy to prevent the development of tau pathology and its propagation. An efficient therapeutic strategy should prevent the intracellular and extracellular accumulation of pathological tau. To achieve this, an increase of pathological tau secretion for prevention of its intracellular accumulation and its sequestration in the extracellular space by an antibody to prevent its propagation and synaptic detrimental effects are necessary (Figure 2C). Furthermore, it is important to take into consideration that most of the mechanisms involved in tau secretion are active in both physiological and pathological conditions indicating that blocking them could have detrimental effects in the areas of the brain not affected by tau pathology where extracellular tau could play a role in neuronal function (Figure 2D). An aspect of tau secretion that was neglected in most studies is the fact that it reaches a plateau. This was only reported by Bright et al. (2015). This phenomenon was observed with all the cell types that we used to examine tau secretory pathways (unpublished data). This observation indicates that a feedback mechanism regulates tau secretion. Taking this into account, the administration of an anti-tau antibody that would block this mechanism by binding to the region of tau involved in it, could lead to an increase of tau secretion. In such a case, the sequestration of pathological tau by the antibody would be limited because of the sustained release of tau compromising the beneficial effects of this antibody. All the above concerns and questions will need to be addressed before the elaboration of therapeutic strategies based on tau secretion. Furthermore, the identification of tau secretory pathways will help to determine whether other proteins are secreted by these pathways, which could contribute to neuronal dysfunction and/or could be used as novel biomarkers for tauopathies.
From recent studies, one could conclude that combined therapeutic strategies with tau secretion would be the most efficient way for preventing the propagation of tau pathology in the brain. Three combined strategies could be elaborated. Firstly, a recent study reported that impaired dural lymphatic function resulted in an increased retention of extracellular tau in the brain as well as a delayed clearance of tau to the periphery (Patel et al., 2019). A therapeutic strategy combining an increase of tau secretion and an increase of tau clearance by the dural lymphatic system could be very efficient in preventing intracellular and extracellular accumulation of tau in the brain. Secondly, intraneuronal transfer of tau was shown to occur by the formation of tunneling nanotubes (Tardivel et al., 2016). This implies that both tau secretion and formation of tunneling nanotubes would have to be blocked to prevent tau pathology propagation. Thirdly, extracellular tau can be taken up by astrocytes. Such an uptake was shown to reduce tau spreading (Martini-Stoica et al., 2018). A concomitant increase of tau secretion and tau uptake by astrocytes could prevent extracellular and intracellular tau accumulation.
Lastly, a recent study reported that END-binding proteins found at the tip of microtubules could modulate tau secretion in non-neuronal cells indicating that microtubules could play a determinant role in the release of tau by neurons (Sayas et al., 2019). Although this remains to be demonstrated in neurons, microtubule network could be modulated to increase tau secretion, which could be combined with the administration of an anti-tau antibody for sequestration of secreted tau.
Characterization of Extracellular Tau Species in Physiological and Pathological Conditions: An Essential Step for Improving the Efficacy of Tau Immunotherapy and Improving the Use of Tau as a Biomarker
The non-vesicular extracellular tau, the main target for immunotherapy, was shown to be able to transfer from neuron to neuron (Takeda et al., 2015; Katsinelos et al., 2018). Several studies have reported that tau aggregates are necessary to induce propagation. The major drawback of these studies is that aggregates composed of recombinant tau, which does not present the PTMs of secreted tau such as cleavage and phosphorylation, was used instead of tau purified either from culture medium or CSF (Wu et al., 2012; Mirbaha et al., 2015). Tau aggregates isolated from the brain of patients presenting a tauopathy were also used in these studies (Takeda et al., 2015; Narasimhan et al., 2017). However, it is not clear if these aggregates are released and/or formed in the extracellular space in AD brains. In recent studies where new sensitive techniques were used, aggregates of tau were detected in the culture medium and CSF indicating that if aggregates are needed to induce the propagation of tau pathology in the brain, extracellular tau could do it (Takeda et al., 2016; Merezhko et al., 2018). In the case of phosphorylation, it is less clear if it is necessary for inducing propagation. Most studies reported that extracellular tau presents low levels of phosphorylation, an observation recently confirmed in human CSF (Le et al., 2012; Mohamed et al., 2014; Lewczuk et al., 2017; Rodriguez et al., 2017; Wang et al., 2017; Ermann et al., 2018). Aggregates composed of non-phosphorylated recombinant tau were shown to induce tau propagation (Wu et al., 2012; Mirbaha et al., 2015). Cleavage of extracellular tau seems to increase its spreading and toxicity. In lamprey, the N-terminal domain of tau was sufficient for secretion and propagation (Le et al., 2012). However, the pattern of secretion differed between the N-terminal and FL-tau in this system. The N-terminal fragment presented a diffuse secretion occurring at the somata whereas FL- tau presented a focal secretion taking place in dendrites and the axon. Interestingly, extracellular N-terminal fragments of tau were shown to increase the production of Aβ in primary human cortical neurons (Bright et al., 2015). When these fragments were neutralized by an antibody, Aβ production was decreased.
The isoform composition of extracellular tau remains to be well characterized for most tauopathies. This point is important since it was recently reported that tau isoforms do not have the same propensity for spreading in the brain. Indeed, the 4R isoforms were found to propagate farther than 3R tau (Dujardin et al., 2018). In the same study, it was reported that misfolded tau composed of either P301L or P332S tau mutant linked to FTLD-tau presented a spreading more distant from the initiation site than hyperphosphorylated tau whereas the opposite was noted for wild-type tau (Dujardin et al., 2018). Based on these observations, antibodies targeting hyperphosphorylated tau might not be as efficient as antibodies targeting misfolded tau in blocking tau propagation in patients presenting these mutations. Altogether, the above observations indicate that extracellular tau could induce the propagation of tau pathology in the brain. However, a better characterization of secreted tau PTMs and isoforms is needed for a more complete understanding of its function and detrimental effects in the extracellular space. Most importantly, the analysis of tau PTMs and isoforms in physiological and pathological conditions will help to design antibodies that will sequester the pathological not the physiological species.
It remains to be demonstrated whether CSF-tau could be used for the differential diagnosis of tauopathies at early stages of the disease and/or for following the progression of a tauopathy. The amount of total and phosphorylated tau is increased in the CSF of AD patients (Hampel et al., 2010). More recently, it was demonstrated that non-phosphorylated tau was also increased in the CSF of these patients (Lewczuk et al., 2017). The use of CSF-tau as a biomarker for tauopathies other than AD such as PSP is less clear. Some studies reported an increase of CSF-tau in PSP whereas others noted either no change or a decrease (Jabbari et al., 2017). In the PSP studies, the analysis of CSF-tau was performed with the anti-tau antibodies used for AD. The lower detection of tau in PSP than in AD could indicate that extracellular tau in PSP and AD differs and therefore that antibodies used in AD may not be able to detect tau in PSP. A better characterization of extracellular tau in PSP and AD is needed to demonstrate this. Another explanation could be that tau is less secreted in PSP, leading to a faster accumulation and aggregation of intracellular pathological tau, which would explain the faster progression of the disease compared to AD. A better characterization of tau secretion pathways will help to demonstrate whether these pathways are less active in PSP.
Conclusion
In conclusion, the elucidation of tau secretory pathways is central to further investigate the physiological function of extracellular tau and its contribution to tau pathology. Extracellular tau is the main target of therapies involving the administration of an anti-tau antibody. A better characterization of all tau forms released in the extracellular space and their function is needed for the design of more efficient antibodies that will solely sequester toxic tau species. This characterization will require the use of more sensitive techniques for the detection of extracellular tau PTMs and aggregation state in the CSF.
Author Contributions
CP, AD, and NL wrote the manuscript. CP prepared the figures.
Funding
This work was supported by the Pfizer-FRQS innovative grant (27443) to NL and the Canadian Institutes of Health Research grants (MOP-114874 and PJT-155993) to NL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anton-Fernandez, A., Aparicio-Torres, G., Tapia, S., DeFelipe, J., and Munoz, A. (2016). Morphometric alterations of Golgi apparatus in Alzheimer’s disease are related to tau hyperphosphorylation. Neurobiol. Dis. 97(Pt A), 11–23. doi: 10.1016/j.nbd.2016.10.005
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Bakker, A., Krauss, G. L., Albert, M. S., Speck, C. L., Jones, L. R., Stark, C. E., et al. (2012). Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74, 467–474. doi: 10.1016/j.neuron.2012.03.023
Barthelemy, N. R., Gabelle, A., Hirtz, C., Fenaille, F., Sergeant, N., Schraen-Maschke, S., et al. (2016). Differential mass spectrometry profiles of tau protein in the cerebrospinal fluid of patients with Alzheimer’s disease, progressive supranuclear palsy, and dementia with lewy bodies. J. Alzheimers Dis. 51, 1033–1043. doi: 10.3233/jad-150962
Bright, J., Hussain, S., Dang, V., Wright, S., Cooper, B., Byun, T., et al. (2015). Human secreted tau increases amyloid-beta production. Neurobiol. Aging 36, 693–709. doi: 10.1016/j.neurobiolaging.2014.09.007
Buée, L., Bussiere, T., Buee-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 33, 95–130. doi: 10.1016/s0165-0173(00)00019-9
Busche, M. A., Chen, X., Henning, H. A., Reichwald, J., Staufenbiel, M., Sakmann, B., et al. (2012). Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 109, 8740–8745. doi: 10.1073/pnas.1206171109
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K. H., Haass, C., et al. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science (New York, NY) 321, 1686–1689. doi: 10.1126/science.1162844
Calafate, S., Buist, A., Miskiewicz, K., Vijayan, V., Daneels, G., de Strooper, B., et al. (2015). Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep. 11, 1176–1183. doi: 10.1016/j.celrep.2015.04.043
Coune, P. G., Bensadoun, J. C., Aebischer, P., and Schneider, B. L. (2011). Rab1A over-expression prevents Golgi apparatus fragmentation and partially corrects motor deficits in an alpha-synuclein based rat model of Parkinson’s disease. J. Parkinsons Dis. 1, 373–387.
Cruz-Garcia, D., Curwin, A. J., Popoff, J. F., Bruns, C., Duran, J. M., and Malhotra, V. (2014). Remodeling of secretory compartments creates CUPS during nutrient starvation. J. Cell Biol. 207, 695–703. doi: 10.1083/jcb.201407119
Curwin, A. J., Brouwers, N., Alonso, Y. A. M., Teis, D., Turacchio, G., Parashuraman, S., et al. (2016). ESCRT-III drives the final stages of CUPS maturation for unconventional protein secretion. Elife 5:e16299.
Dal Canto, M. C. (1996). The Golgi apparatus and the pathogenesis of Alzheimer’s disease. Am. J. Pathol. 148, 355–360.
Diaz-Hernandez, M., Gomez-Ramos, A., Rubio, A., Gomez-Villafuertes, R., Naranjo, J. R., Miras-Portugal, M. T., et al. (2010). Tissue non-specific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J. Biol. Chem. 285, 32539–32548. doi: 10.1074/jbc.m110.145003
Dujardin, S., Begard, S., Caillierez, R., Lachaud, C., Carrier, S., Lieger, S., et al. (2018). Different tau species lead to heterogeneous tau pathology propagation and misfolding. Acta Neuropathol. Commun. 6:132.
Dujardin, S., Begard, S., Caillierez, R., Lachaud, C., Delattre, L., Carrier, S., et al. (2014). Ectosomes: a new mechanism for non-exosomal secretion of tau protein. PLoS One 9:e100760. doi: 10.1371/journal.pone.0100760
Ebert, A. D., Laussmann, M., Wegehingel, S., Kaderali, L., Erfle, H., Reichert, J., et al. (2010). Tec-kinase-mediated phosphorylation of fibroblast growth factor 2 is essential for unconventional secretion. Traffic (Copenhagen, Denmark) 11, 813–826. doi: 10.1111/j.1600-0854.2010.01059.x
Ermann, N., Lewczuk, P., Schmitz, M., Lange, P., Knipper, T., Goebel, S., et al. (2018). CSF nonphosphorylated Tau as a biomarker for the discrimination of AD from CJD. Ann. Clin. Transl. Neurol. 5, 883–887. doi: 10.1002/acn3.584
Flach, K., Hilbrich, I., Schiffmann, A., Gartner, U., Kruger, M., Leonhardt, M., et al. (2012). Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 287, 43223–43233. doi: 10.1074/jbc.m112.396176
Florenzano, F., Veronica, C., Ciasca, G., Ciotti, M. T., Pittaluga, A., Olivero, G., et al. (2017). Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 8, 64745–64778.
Fontaine, S. N., Zheng, D., Sabbagh, J. J., Martin, M. D., Chaput, D., Darling, A., et al. (2016). DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 35, 1537–1549. doi: 10.15252/embj.201593489
Gerke, V., and Moss, S. E. (2002). Annexins: from structure to function. Physiol. Rev. 82, 331–371. doi: 10.1152/physrev.00030.2001
Ginsberg, S. D., Alldred, M. J., Counts, S. E., Cataldo, A. M., Neve, R. L., Jiang, Y., et al. (2010a). Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry 68, 885–893. doi: 10.1016/j.biopsych.2010.05.030
Ginsberg, S. D., Mufson, E. J., Counts, S. E., Wuu, J., Alldred, M. J., Nixon, R. A., et al. (2010b). Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 22, 631–639. doi: 10.3233/jad-2010-101080
Ginsberg, S. D., Mufson, E. J., Alldred, M. J., Counts, S. E., Wuu, J., Nixon, R. A., et al. (2011). Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J. Chem. Neuroanat. 42, 102–110. doi: 10.1016/j.jchemneu.2011.05.012
Gomez-Ramos, A., Diaz-Hernandez, M., Cuadros, R., Hernandez, F., and Avila, J. (2006). Extracellular tau is toxic to neuronal cells. FEBS Lett. 580, 4842–4850. doi: 10.1016/j.febslet.2006.07.078
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Diaz-Hernandez, J. I., Miras-Portugal, M. T., and Avila, J. (2009). Characteristics and consequences of muscarinic receptor activation by tau protein. Eur. Neuropsychopharmacol. 19, 708–717. doi: 10.1016/j.euroneuro.2009.04.006
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Miras-Portugal, M. T., and Avila, J. (2008). Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 37, 673–681. doi: 10.1016/j.mcn.2007.12.010
Guix, F. X., Corbett, G. T., Cha, D. J., Mustapic, M., Liu, W., Mengel, D., et al. (2018). Detection of aggregation-competent tau in neuron-derived extracellular vesicles. Int. J. Mol. Sci. 19:E663.
Hampel, H., Blennow, K., Shaw, L. M., Hoessler, Y. C., Zetterberg, H., and Trojanowski, J. Q. (2010). Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp. Gerontol. 45, 30–40. doi: 10.1016/j.exger.2009.10.010
Hessvik, N. P., and Llorente, A. (2018). Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 75, 193–208. doi: 10.1007/s00018-017-2595-9
Hu, N. W., Corbett, G. T., Moore, S., Klyubin, I., O’Malley, T. T., Walsh, D. M., et al. (2018). Extracellular forms of abeta and tau from iPSC models of Alzheimer’s disease disrupt synaptic plasticity. Cell Rep. 23, 1932–1938. doi: 10.1016/j.celrep.2018.04.040
Iguchi, Y., Eid, L., Parent, M., Soucy, G., Bareil, C., Riku, Y., et al. (2016). Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 139(Pt 12), 3187–3201. doi: 10.1093/brain/aww237
Jabbari, E., Zetterberg, H., and Morris, H. R. (2017). Tracking and predicting disease progression in progressive supranuclear palsy: CSF and blood biomarkers. J. Neurol. Neurosurg. Psychiatry 88, 883–888. doi: 10.1136/jnnp-2017-315857
Johnson, G. V., Seubert, P., Cox, T. M., Motter, R., Brown, J. P., and Galasko, D. (1997). The tau protein in human cerebrospinal fluid in Alzheimer’s disease consists of proteolytically derived fragments. J. Neurochem. 68, 430–433. doi: 10.1046/j.1471-4159.1997.68010430.x
Kalra, H., Simpson, R. J., Ji, H., Aikawa, E., Altevogt, P., Askenase, P., et al. (2012). Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 10:e1001450. doi: 10.1371/journal.pbio.1001450
Kanmert, D., Cantlon, A., Muratore, C. R., Jin, M., O’Malley, T. T., Lee, G., et al. (2015). C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J. Neurosci. 35, 10851–10865. doi: 10.1523/jneurosci.0387-15.2015
Katsinelos, T., Zeitler, M., Dimou, E., Karakatsani, A., Muller, H. M., Nachman, E., et al. (2018). Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep. 23, 2039–2055. doi: 10.1016/j.celrep.2018.04.056
Kim, W., Lee, S., Jung, C., Ahmed, A., Lee, G., and Hall, G. F. (2010). Interneuronal transfer of human tau between Lamprey central neurons in situ. J. Alzheimers Dis. 19, 647–664. doi: 10.3233/jad-2010-1273
Le, M. N., Kim, W., Lee, S., McKee, A. C., and Hall, G. F. (2012). Multiple mechanisms of extracellular tau spreading in a non-transgenic tauopathy model. Am. J. Neurodegener. Dis. 1, 316–333.
Lee, S., Kim, W., Li, Z., and Hall, G. F. (2012). Accumulation of vesicle-associated human tau in distal dendrites drives degeneration and tau secretion in an in situ cellular tauopathy model. Int. J. Alzheimers Dis. 2012:172837.
Lee, V., Goedert, M., and Trojanowski, J. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159. doi: 10.1146/annurev.neuro.24.1.1121
Lewczuk, P., Lelental, N., Lachmann, I., Holzer, M., Flach, K., Brandner, S., et al. (2017). Non-phosphorylated tau as a potential biomarker of Alzheimer’s disease: analytical and Diagnostic Characterization. J. Alzheimers Dis. 55, 159–170. doi: 10.3233/jad-160448
Liazoghli, D., Perreault, S., Micheva, K., Desjardins, M., and Leclerc, N. (2005). Fragmentation of the Golgi apparatus induced by the overexpression of wild-type and mutant human tau forms in neurons. Am. J. Pathol. 166, 1499–1514. doi: 10.1016/s0002-9440(10)62366-8
Luk, C., Compta, Y., Magdalinou, N., Marti, M. J., Hondhamuni, G., Zetterberg, H., et al. (2012). Development and assessment of sensitive immuno-PCR assays for the quantification of cerebrospinal fluid three- and four-repeat tau isoforms in tauopathies. J. Neurochem. 123, 396–405. doi: 10.1111/j.1471-4159.2012.07911.x
Martini-Stoica, H., Cole, A. L., Swartzlander, D. B., Chen, F., Wan, Y. W., Bajaj, L., et al. (2018). TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J. Exp. Med. 215, 2355–2377. doi: 10.1084/jem.20172158
Meredith, J. E. Jr., Sankaranarayanan, S., Guss, V., Lanzetti, A. J., Berisha, F., Neely, R. J., et al. (2013). Characterization of novel CSF Tau and ptau biomarkers for Alzheimer’s disease. PLoS One 8:e76523. doi: 10.1371/journal.pone.0076523
Merezhko, M., Brunello, C. A., Yan, X., Vihinen, H., Jokitalo, E., Uronen, R. L., et al. (2018). Secretion of tau via an unconventional non-vesicular mechanism. Cell Rep. 25, 2027–2035.e4. doi: 10.1016/j.celrep.2018.10.078
Mirbaha, H., Holmes, B. B., Sanders, D. W., Bieschke, J., and Diamond, M. I. (2015). Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J. Biol. Chem. 290, 14893–14903. doi: 10.1074/jbc.m115.652693
Mohamed, N.-V., Desjardins, A., and Leclerc, N. (2017). Tau secretion is correlated to an increase of Golgi dynamics. PLoS One 12:e0178288. doi: 10.1371/journal.pone.0178288
Mohamed, N. V., Herrou, T., Plouffe, V., Piperno, N., and Leclerc, N. (2013). Spreading of tau pathology in Alzheimer’s disease by cell-to-cell transmission. Eur. J. Neurosci. 37, 1939–1948. doi: 10.1111/ejn.12229
Mohamed, N. V., Plouffe, V., Remillard-Labrosse, G., Planel, E., and Leclerc, N. (2014). Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci. Rep. 4:5715.
Muralidharan-Chari, V., Clancy, J. W., Sedgwick, A., and D’Souza-Schorey, C. (2010). Microvesicles: mediators of extracellular communication during cancer progression. J. Cell Sci. 123(Pt 10), 1603–1611. doi: 10.1242/jcs.064386
Narasimhan, S., Guo, J. L., Changolkar, L., Stieber, A., McBride, J. D., Silva, L. V., et al. (2017). Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 37, 11406–11423. doi: 10.1523/jneurosci.1230-17.2017
Nixon, R. A. (2005). Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging. 26, 373–382. doi: 10.1016/j.neurobiolaging.2004.09.018
Palop, J. J., and Mucke, L. (2009). Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol. 66, 435–440.
Patel, T. K., Habimana-Griffin, L., Gao, X., Xu, B., Achilefu, S., Alitalo, K., et al. (2019). Dural lymphatics regulate clearance of extracellular tau from the CNS. Mol. Neurodegener. 14:11.
Plouffe, V., Mohamed, N. V., Rivest-McGraw, J., Bertrand, J., Lauzon, M., and Leclerc, N. (2012). Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One 7:e36873. doi: 10.1371/journal.pone.0036873
Polanco, J. C., Scicluna, B. J., Hill, A. F., and Gotz, J. (2016). Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J. Biol. Chem. 291, 12445–12466. doi: 10.1074/jbc.m115.709485
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Rodriguez, L., Mohamed, N.-V., Desjardins, A., Lippe, R., Fon, E. A., and Leclerc, N. (2017). Rab7A regulates tau secretion. J. Neurochem. 141, 592–605. doi: 10.1111/jnc.13994
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Jackson, B., et al. (2012). Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.m111.277061
Santos, T. G., Martins, V. R., and Hajj, G. N. M. (2017). Unconventional secretion of heat shock proteins in cancer. Int. J. Mol. Sci. 18:E946.
Sayas, C. L., Medina, M., Cuadros, R., Olla, I., Garcia, E., Perez, M., et al. (2019). Role of tau N-terminal motif in the secretion of human tau by End Binding proteins. PLoS One 14:e0210864. doi: 10.1371/journal.pone.0210864
Shorter, J., and Warren, G. (2002). Golgi architecture and inheritance. Annu. Rev. Cell Dev. Biol. 18, 379–420. doi: 10.1146/annurev.cellbio.18.030602.133733
Shrivastava, A. N., Redeker, V., Pieri, L., Bousset, L., Renner, M., Madiona, K., et al. (2019). Clustering of Tau fibrils impairs the synaptic composition of alpha3-Na(+)/K(+)-ATPase and AMPA receptors. EMBO J. 38:e99871. doi: 10.15252/embj.201899871
Simon, D., Garcia-Garcia, E., Gomez-Ramos, A., Falcon-Perez, J. M., Diaz-Hernandez, M., Hernandez, F., et al. (2012). Tau overexpression results in its secretion via membrane vesicles. Neurodegener. Dis. 10, 73–75. doi: 10.1159/000334915
Steringer, J. P., Lange, S., Cujova, S., Sachl, R., Poojari, C., Lolicato, F., et al. (2017). Key steps in unconventional secretion of fibroblast growth factor 2 reconstituted with purified components. Elife 6:e28985.
Stieber, A., Mourelatos, Z., and Gonatas, N. K. (1996). In Alzheimer’s disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. Am. J. Pathol. 148, 415–426.
Takeda, S., Commins, C., DeVos, S. L., Nobuhara, C. K., Wegmann, S., Roe, A. D., et al. (2016). Seed-competent high-molecular-weight tau species accumulates in the cerebrospinal fluid of Alzheimer’s disease mouse model and human patients. Ann. Neurol. 80, 355–367. doi: 10.1002/ana.24716
Takeda, S., Wegmann, S., Cho, H., DeVos, S. L., Commins, C., Roe, A. D., et al. (2015). Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 6:8490.
Tanaka, K., Namba, T., Arai, Y., Fujimoto, M., Adachi, H., Sobue, G., et al. (2007). Genetic evidence for a protective role for heat shock factor 1 and heat shock protein 70 against colitis. J. Biol. Chem. 282, 23240–23252. doi: 10.1074/jbc.m704081200
Tardivel, M., Begard, S., Bousset, L., Dujardin, S., Coens, A., Melki, R., et al. (2016). Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 4:117.
Tiernan, C. T., Ginsberg, S. D., Guillozet-Bongaarts, A. L., Ward, S. M., He, B., Kanaan, N. M., et al. (2016). Protein homeostasis gene dysregulation in pretangle-bearing nucleus basalis neurons during the progression of Alzheimer’s disease. Neurobiol. Aging 42, 80–90. doi: 10.1016/j.neurobiolaging.2016.02.031
Trajkovic, K., Jeong, H., and Krainc, D. (2017). Mutant huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J. Neurosci. 37, 9000–9012. doi: 10.1523/jneurosci.0118-17.2017
Wagshal, D., Sankaranarayanan, S., Guss, V., Hall, T., Berisha, F., Lobach, I., et al. (2015). Divergent CSF tau alterations in two common tauopathies: Alzheimer’s disease and progressive supranuclear palsy. J. Neurol. Neurosurg. Psychiatry 86, 244–250. doi: 10.1136/jnnp-2014-308004
Wang, Y., Balaji, V., Kaniyappan, S., Kruger, L., Irsen, S., Tepper, K., et al. (2017). The release and trans-synaptic transmission of tau via exosomes. Mol. Neurodegener. 12:5.
Wegehingel, S., Zehe, C., and Nickel, W. (2008). Rerouting of fibroblast growth factor 2 to the classical secretory pathway results in post-translational modifications that block binding to heparan sulfate proteoglycans. FEBS Lett. 582, 2387–2392. doi: 10.1016/j.febslet.2008.05.042
Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., et al. (2012). Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. doi: 10.1074/jbc.m112.394528
Wu, J. W., Hussaini, S. A., Bastille, I. M., Rodriguez, G. A., Mrejeru, A., Rilett, K., et al. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092. doi: 10.1038/nn.4328
Yamada, K., Cirrito, J. R., Stewart, F. R., Jiang, H., Finn, M. B., Holmes, B. B., et al. (2011). In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J. Neurosci. 31, 13110–13117. doi: 10.1523/jneurosci.2569-11.2011
Yamada, K., Holth, J. K., Liao, F., Stewart, F. R., Mahan, T. E., Jiang, H., et al. (2014). Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. doi: 10.1084/jem.20131685
Keywords: tau protein, secretion, tauopathies, extracellular tau, propagation
Citation: Pernègre C, Duquette A and Leclerc N (2019) Tau Secretion: Good and Bad for Neurons. Front. Neurosci. 13:649. doi: 10.3389/fnins.2019.00649
Received: 25 January 2019; Accepted: 06 June 2019;
Published: 26 June 2019.
Edited by:
Diane Hanger, King’s College London, United KingdomReviewed by:
Luc Buee, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceFelix Hernandez, Spanish National Research Council (CSIC), Spain
Copyright © 2019 Pernègre, Duquette and Leclerc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole Leclerc, Tmljb2xlLmxlY2xlcmNAdW1vbnRyZWFsLmNh