 Kensuke Ikenaka
Kensuke Ikenaka Mari Suzuki
Mari Suzuki Hideki Mochizuki1
Hideki Mochizuki1 Yoshitaka Nagai
Yoshitaka Nagai- 1Department of Neurology, Graduate School of Medicine, Osaka University, Osaka, Japan
- 2Department of Neurotherapeutics, Graduate School of Medicine, Osaka University, Osaka, Japan
- 3Diabetic Neuropathy Project, Department of Sensory and Motor Systems, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan
Aggregation of α-synuclein (αSyn) plays a central role in the pathogenesis of Parkinson’s disease (PD) and dementia with Lewy bodies (DLB). Lewy bodies (LBs) and Lewy neurites, which consist mainly of aggregated αSyn, are widely observed in the affected regions of patient brains. Except for some familial forms of PD/DLB, most sporadic PD/DLB patients express the wild-type (WT) αSyn protein without any mutations, and the mechanisms as to how WT αSyn gains the propensity to pathologically aggregate still remains unclear. Furthermore, the mechanisms by which the same αSyn protein can cause different synucleinopathies with distinct phenotypes and pathologies, such as PD, DLB, and multiple system atrophy (MSA), still remain largely unknown. Recently, mutations in the GBA1 gene (encoding glucocerebrosidase), which are responsible for the lysosomal storage disorder Gaucher disease (GD), have been reported to be the strongest risk factor for developing sporadic PD/DLB. We previously demonstrated that glucosylceramide accumulated by GBA1 deficiency promotes the conversion of αSyn into a proteinase K-resistant conformation. Furthermore, decreased glucocerebrosidase activity has also been reported in the brains of patients with sporadic PD/DLB. Moreover, αSyn pathology has also been shown in the brains of lysosomal storage disorder patients, which show glycosphingolipid accumulation. These observations suggest the possibility that altered lipid metabolism and lipid accumulation play roles in αSyn aggregation and PD/DLB pathogenesis. Indeed, several previous studies have demonstrated that lipid interactions affect the conformation of αSyn and induces its oligomerization and aggregation. In this review, we will give an overview of the association between αSyn aggregation and lipid interactions from the viewpoints of the etiology, pathology, and genetics of PD/DLB. We also discuss the distinct species of αSyn aggregates and their association with specific types of synucleinopathies, and introduce our hypothesis that lipid interactions play a role as trans-acting effectors in producing distinct strains of αSyn fibrils.
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by preferential loss of dopaminergic neurons in the pars compacta of the substantia nigra (Scott and Netsky, 1961). PD is the second most common neurodegenerative disease after Alzheimer disease, and its prevalence is growing steadily. Clinical features of PD include motor symptoms, such as resting tremor, bradykinesia, and rigidity, as well as non-motor symptoms, such as dementia, depression, autonomic failure, and hallucinations (Marinus et al., 2018). Dopamine replacement therapy is widely used to improve the motor symptoms of PD patients, but does not attenuate disease progression. The exact mechanisms causing PD are still unknown, but the deposition of Lewy bodies (LBs) in the substantia nigra, which are composed mainly of fibrillar alpha-synuclein (αSyn), is a pathological hallmark of PD (Spillantini et al., 1997, 1998b). LBs are also widely observed in the cortex and other brain regions of patients with dementia with Lewy bodies (DLB) (Spillantini et al., 1998a), which predominantly exhibits dementia accompanied with frequent visual hallucinations and dopa-responsive Parkinsonism (Burkhardt et al., 1988; Byrne et al., 1989; Klatka et al., 1996). Since these diseases share pathological and clinical features, PD and DLB are considered to belong to the spectrum of disorders called Lewy body disease.
Genetic studies have demonstrated that missense or multiplication mutations in the αSyn gene cause familial forms of PD/DLB (Polymeropoulos et al., 1997), and that single nucleotide polymorphisms (SNPs) in the αSyn gene are major risk factors for sporadic PD/DLB (Satake et al., 2009; Simon-Sanchez et al., 2009). Considering the above pathological and genetic evidence, αSyn aggregation is a key target for developing disease-modifying therapies for PD/DLB and identifying the triggers of αSyn aggregation is expected to lead to new therapeutic strategies that attenuate or prevent PD/DLB.
Recently, there has been a large amount of discussion about the interaction of lipids with αSyn, which affects the structure of αSyn and accelerates its aggregation. In this review, we give an overview of the association between αSyn aggregation and lipid interactions from the viewpoints of the etiology, pathology, and genetics of PD/DLB patients and experimental models. We also propose our hypothesis that lipids interacting with αSyn may work as trans-acting effectors that induce variable conformational changes in the αSyn monomer, into structurally distinct αSyn fibrils, which may lead to clinically distinct synucleinopathies.
α-Synuclein
Physiological Characteristics of αSyn
αSyn is a small (14 kD) cytosolic protein that is enriched in presynaptic terminals. It consists of three major regions; the N-terminus (amino acid (aa) 1–60), the non-amyloid beta component (NAC, aa 61–95), and the C-terminus (aa 96–140) (Figure 1). In solution, αSyn is considered to be an intrinsically disordered protein, which lacks a single stable structure (Weinreb et al., 1996). Although the exact molecular function of αSyn has not yet been fully elucidated, it has been proposed to interact with biological membranes and membrane proteins, and to play roles in synaptic plasticity and neurotransmitter release (Chandra et al., 2004; Fortin et al., 2005; Nakata et al., 2012; Burre, 2015). In particular, αSyn was found to promote the assembly of SNARE complexes both in vivo and in vitro via the formation of multimers at the surface of synaptic vesicles (Burre et al., 2010).
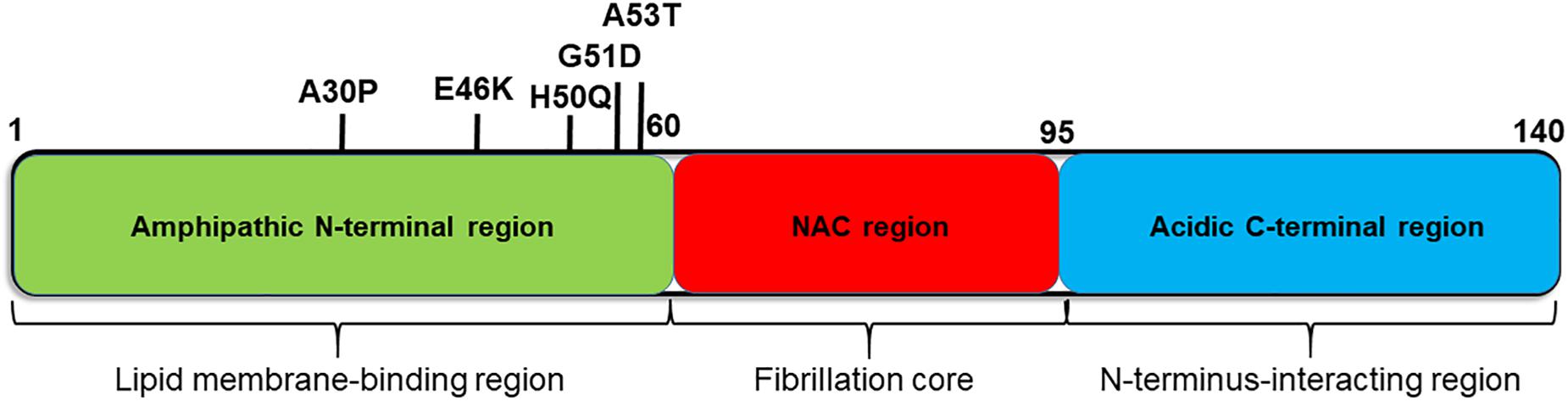
Figure 1. Diagram of the αSyn protein showing the characteristics of each region. αSyn can be divided into three regions; the N-terminal region (green), non-amyloid β-component (NAC) region (red), and C-terminal region (blue). The sites of causative mutations for familial PD are shown below, and functions and structural features are listed above each region.
Role of Lewy Bodies in the Pathogenesis of PD/DLB
Classical LBs are round and eosinophilic cytoplasmic inclusions that displace other cytoplasmic components. They consist of a dense core surrounded by a halo of radiating fibrils with a width of 10 nm (Roy and Wolman, 1969; Spillantini et al., 1998b). Cortical LB is mainly found in the cortex of DLB patients and advanced PD patients and their morphologies are slightly different from classical LB, which are less defined and lack halos (Kosaka et al., 1976; Spillantini et al., 1998b). The components of LB are mainly αSyn, together with many other molecules, including proteins, such as neurofilament (Trojanowski and Lee, 1998), microtubule-associated protein 1B (Jensen et al., 2000), and galectin-3 (Flavin et al., 2017), as well as various lipids (Araki et al., 2015). The mechanism as to how LB is formed still remains unclear.
Genetics of PD/DLB
Several point mutations in the αSyn gene are linked to autosomal-dominant PD/DLB (Polymeropoulos et al., 1997). Both pathogenic missense mutations (A53T, A30P, E46K, G51D, and H50Q) (PARK1) and multiplication of the entire gene (duplications and triplications) (PARK4) cause familial types of PD/DLB (Polymeropoulos et al., 1997; Kruger et al., 1998; Chartier-Harlin et al., 2004; Zarranz et al., 2004; Appel-Cresswell et al., 2013; Kiely et al., 2013). Moreover, in 2009, a European and a Japanese group both independently performed a genome-wide association study on sporadic PD and demonstrated strong associations of SNPs in the αSyn gene with PD (Satake et al., 2009; Simon-Sanchez et al., 2009), which have recently been shown to be also associated with DLB (Guerreiro et al., 2018). Taken together, these lines of genetic evidence for a causative role of αSyn as well as pathological evidence for the accumulation of αSyn in LBs strongly indicate the central role of αSyn in the pathogenesis of sporadic PD/DLB.
αSyn Aggregation
As fibrillar αSyn is a major component of LB, the mechanism of fibril formation of αSyn has been studied extensively. Although αSyn is an intrinsically disordered protein, it forms a β-sheet-rich structure when aggregated (Maiti et al., 2004). Amyloid-like fibril formation of αSyn has been experimentally reproduced in vitro, and the resulting fibrils are morphologically similar to those found in LBs of patient brains (Rochet et al., 2000; Serpell et al., 2000).
Previous studies demonstrated that three major regions of αSyn, namely, the C-terminus, NAC, and N-terminus, play different roles in the process of fibril formation (Figure 1). First, the NAC region plays a central role in fibril formation and aggregation, via its formation of cross β-sheet structures. Indeed, several studies have shown that the NAC is necessary and sufficient for the aggregation and toxicity of αSyn (Giasson et al., 2001; Periquet et al., 2007; Rodriguez et al., 2015).
The negative charges at the C-terminal region of αSyn play an important role in inhibiting fibril formation, by interacting with the NAC region and masking it (Hong et al., 2011). The C-terminal region is also known as a metal-binding site. Upon binding to metals, such as Al3+, the C-terminal domain loses its inhibitory effect on NAC, leading to αSyn aggregation (Uversky et al., 2001; Dedmon et al., 2005; Ly and Julian, 2008). The C-terminus was also shown to form transient, long-range interactions with the N-terminus resulting in the formation of multiple compact monomeric structures (Bertoncini et al., 2005). These compact structures of αSyn are resistant to aggregation. Moreover, C-terminally truncated forms of αSyn were found to aggregate faster than the full-length protein (Hoyer et al., 2004; Li et al., 2005; Anderson et al., 2006). Interestingly, truncation of the C-terminal domain of αSyn has been detected in the brains of PD/DLB patients and a specific antibody against truncated αSyn stained the core of LBs (Zhang et al., 2017).
The N-terminal region of αSyn (aa 1–60) is the amphipathic region, and is involved in its interaction with lipid membranes and the structural formation of natively unfolded αSyn (George et al., 1995; Uversky, 2007). Seven imperfect repeats containing a KTKEGV consensus motif are found in the N-terminal region and in the NAC region close to the N-terminal region (aa 61–85), which play an important role in membrane binding (Jakes et al., 1994; Zarbiv et al., 2014). Similar motifs are also found in apolipoprotein, which is known to cause systemic amyloidosis (George et al., 1995). Very interestingly, all the missense mutations of aSyn are located in the N-terminal region, suggesting the importance of membrane binding for its inherent toxicity. In the next section, we will introduce and discuss the interaction of lipids with αSyn and their roles in αSyn aggregation.
αSyn and Lipid Metabolism
Lipid Membrane Binding and Structural Changes of αSyn
Although αSyn aggregation is a central player in PD/DLB pathogenesis, the mechanisms as to how wild-type (WT) αSyn without any mutations gains pathological aggregation properties in sporadic PD patients remain unknown. To solve this important question, many recent studies have examined the association between the nature of the interaction of αSyn with lipids and the propensity of αSyn to aggregate.
There are two possible mechanisms by which lipid membranes promote the aggregation of αSyn. One possibility is that the membrane surface facilitates the local concentration of αSyn. Consistently, the magnitude by which lipids affect the aggregation of αSyn was found to correlate with the strength of binding of αSyn to lipids by the following two main parameters: the lipid-to-protein-ratio and the chemical properties of the lipids (Zhu et al., 2003; De Franceschi et al., 2011; Galvagnion et al., 2015, 2016). The other mechanism is that membrane binding induces the formation of an “αSyn seed,” by changing the protein conformation of αSyn (Lee et al., 2002). αSyn is a intrinsically disordered protein that does not adopt a stable structure; however, it has been shown that upon binding to small unilamellar lipid vesicles or membranes, a fraction of the first 95 residues of αSyn undergoes a structural change from random coil to a horseshoe-like two-helix antiparallel α-helix (Chandra et al., 2003; Drescher et al., 2008; Bodner et al., 2009).
The precise mechanisms as to how membrane-bound α-helical αSyn changes its structure into a β-sheet and starts to aggregate is still unknown. However, it is well known that membrane lipids play an important role in this process, and therefore, alterations in the lipid composition of neuronal membranes may be a key step in the pathogenesis of PD.
Glucocerebrosidase 1 (GBA1) Mutations and PD/DLB
One proof of concept that explains the involvement of lipid dysregulation in the pathogenesis of PD/DLB is that GBA1 mutations increase the risk of PD/DLB (Tayebi et al., 2003; Goker-Alpan et al., 2004; Sidransky and Lopez, 2012). The GBA1 gene encodes the lysosomal enzyme glucocerebrosidase (GCase), an enzyme involved in sphingolipid metabolism, catalyzing its conversion to glucose and ceramides. Homozygous mutations in the GBA1 gene cause Gaucher disease (GD), which is the most common lysosomal storage disorder. The accumulation of glucosylceramide (GlcCer) in macrophages is observed as “Gaucher cells,” which serve as the hallmark of GD.
Interestingly, a subset of type 1 GD patients was reported to demonstrate typical PD symptoms (Neudorfer et al., 1996). A multicenter genetic analysis confirmed that heterozygous mutations in the GBA1 gene are significant risk factors for PD (Sidransky et al., 2009) and also for DLB (Nalls et al., 2013; Gamez-Valero et al., 2016). Clinical studies reported that GBA1-linked PD/DLB is virtually indistinguishable from idiopathic PD/DLB, with a slightly earlier age of onset (Nichols et al., 2009; Gamez-Valero et al., 2016) and higher prevalence of cognitive impairment (Sidransky et al., 2009). Regarding pathology, no differences in LB pathology have been reported between GBA1-linked PD and idiopathic PD (Neumann et al., 2009).
Importantly, not only a rare mutation in the GBA1 gene increases the risk of developing PD/DLB, but also a decrease in GCase activity has been reported in the biofluid or brains of sporadic PD patients (Balducci et al., 2007; Gegg et al., 2012; Parnetti et al., 2014; Chiasserini et al., 2015; Rocha et al., 2015; Moors et al., 2019). Furthermore, a decrease in GCase activity in the brains of sporadic PD/DLB patients was reported to be associated with an increase in the amounts of insoluble αSyn (Murphy et al., 2014). These findings indicate the possibility that decreased GCase activity is a common pathway leading to αSyn aggregation in the brain (Gegg and Schapira, 2018).
Glucosylceramide and αSyn Aggregation
How does decreased GCase activity lead to αSyn aggregation? Several in vitro studies have shown that both GlcCer and glucosylsphingosine can directly cause monomeric αSyn to aggregate (Mazzulli et al., 2011; Taguchi et al., 2017; Maor et al., 2019). We showed a significant increase in αSyn dimers upon incubation with GlcCer-containing liposomes, which is consistent with the finding that the amount of αSyn dimers was significantly increased in erythrocytes of GD patients (Argyriou et al., 2012; Suzuki et al., 2015; Moraitou et al., 2016).
From the view of a loss-of GCase function model, Mazzulli et al. (2011) showed that GCase deficiency leads to the accumulation of GlcCer in neurons, which in turn promotes the formation of toxic αSyn aggregates in cultured neuronal cells. Interestingly, not only GlcCer directly affects the amyloid formation of purified αSyn by stabilizing soluble oligomeric intermediates, but αSyn also inhibits the lysosomal activity of normal GCase, suggesting a bidirectional association between αSyn and GCase in the pathogenesis of the synucleinopathies (Mazzulli et al., 2011). We hence examined the effects of GCase deficiency on the neurotoxicity of αSyn in a Drosophila model (Suzuki et al., 2015). Behavioral and histological analyses showed that knockdown of the Drosophila homologue of GBA1 exacerbates locomotor dysfunction and loss of dopaminergic neurons of αSyn-expressing flies. We showed that GlcCer accumulated owing to GCase deficiency interacts with αSyn and confers proteinase K (PK)-resistance to αSyn, suggesting that GlcCer promotes the toxic conformational conversion of αSyn.
Lipid Dysregulation in Sporadic PD/DLB
What is the status of lipid metabolism in sporadic PD/DLB patients? As discussed above, decreased activity of the lysosomal enzyme GCase has been reported in the brains of sporadic PD/DLB patients, suggesting the accumulation of glycosphingolipids and αSyn (Gegg et al., 2012; Moors et al., 2018). Indeed, several studies reported altered lipid metabolism in PD/DLB patients. For example, plasma ceramides and monohexosylceramides were increased in DLB patients (Savica et al., 2016) and polyunsaturated fatty acids were increased in PD/DLB patient brains (Sharon et al., 2003).
Interestingly, the level of lysosomal enzyme activity was reported to be higher in the substantia nigra, and selectively decreased in older people as well as in PD/DLB patients, suggesting a possible explanation as to how PD/DLB pathology can selectively affect specific regions of the brain (Murphy et al., 2014; Chiasserini et al., 2015). Considering that lipid accumulation leads to αSyn aggregation, lipids might be accumulated in LBs in the brains of PD/DLB patients. Indeed, we examined LBs in the brains of PD patients by Fourier transform infrared microscopy, and found that lipids are accumulated in the core of LBs and are surrounded by a halo that is rich in fibril-like β-sheet structures (Araki et al., 2015). These results are consistent with a report that the core of LB was stained with the lipid-soluble fluorescent dye rhodamine B (Issidorides et al., 1991).
Structural Variants of αSyn and Lipids
Clinical and Pathological Diversity of Synucleinopathies
αSyn is aggregated and accumulated in a group of neurodegenerative diseases, including PD, DLB, and multiple system atrophy (MSA), which are collectively known as the synucleinopathies. Although they share the same disease-causing protein, each synucleinopathy demonstrates distinct clinical and pathological phenotypes, which may result from diverse pathological αSyn strains in patients (Guo et al., 2013; Prusiner et al., 2015). Recent studies have indicated that αSyn may act in a way similar to prions and that structural variants of αSyn aggregates may behave as strains with distinct biochemical and functional properties, inducing specific phenotypic traits, which might finally provide an explanation for the clinical heterogeneity observed among PD, DLB, and MSA patients (Peng et al., 2018b). In this section, we summarize the characteristics of different αSyn strains that are experimentally generated from recombinant αSyn monomers. We also discuss αSyn strains that may potentially exist in synucleinopathy patients. Finally, we describe the possible roles of lipids in determining the structural diversity of αSyn fibrils.
Distinct Species of αSyn Fibrils
The diversity of αSyn fibril structures have been well studied using αSyn mutants that cause familial forms of PD (Serpell et al., 2000; Choi et al., 2004; Sahay et al., 2015). The mutations A53T and E46K have been shown to significantly accelerate fibrillation in vitro when compared with WT αSyn, and the morphologies are also distinct from WT fibrils; A53T and E46K αSyn form twisted fibrils 5–14 nm in width, whereas WT αSyn forms non-twisted fibrils. The morphologies of A30P and G51D αSyn fibrils appears to be similar to those formed from WT fibrils, with a filament width of 6–9 nm (Serpell et al., 2000). In another study using single-molecule fluorescence, Sierecki et al. (2016) reported that pathogenic αSyn mutants surprisingly segregated into two groups: one group (E46K, H50Q, and A53T), formed large aggregates and fibrils whereas the other group (WT, G51D, and A30P) tended to form mostly oligomers. These findings suggest that cis-acting effects of mutations on αSyn itself lead to the distinct species of fibrils (Figure 2).
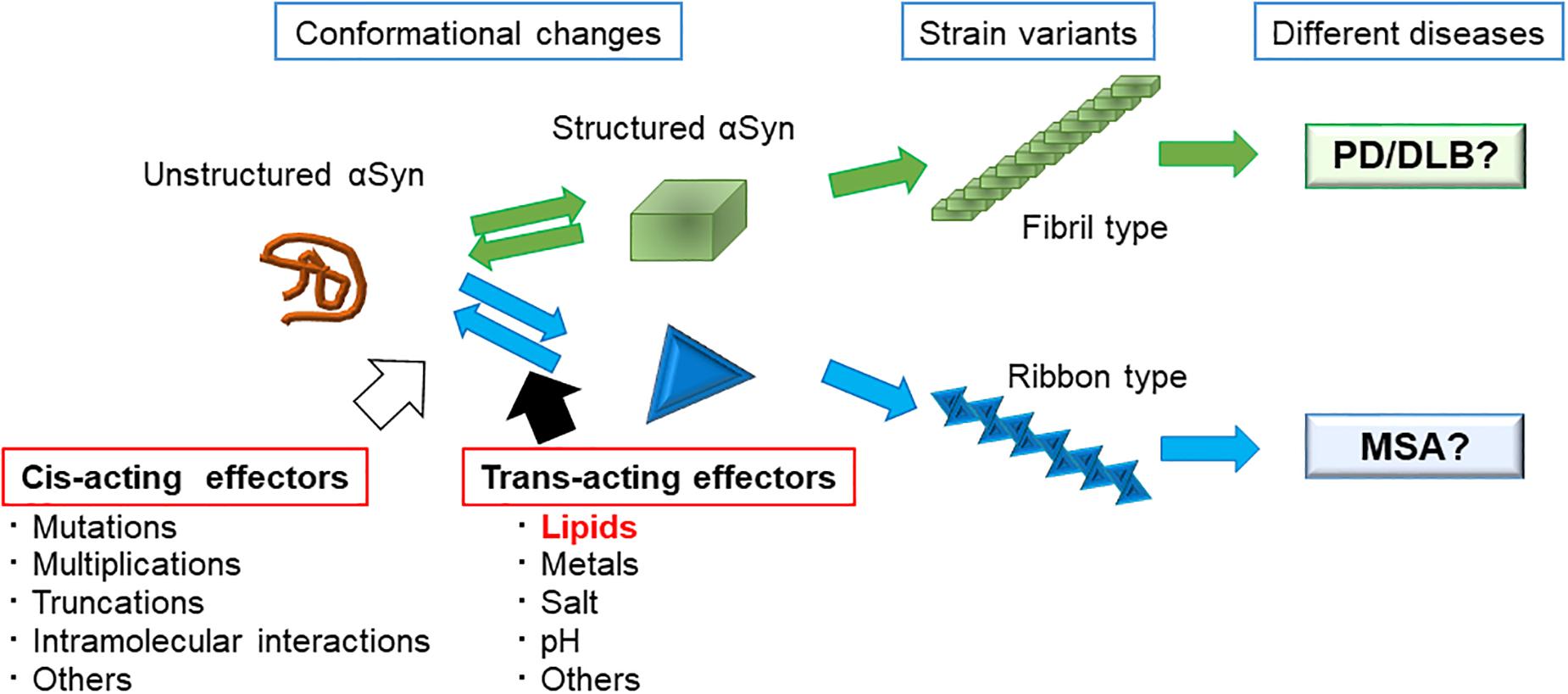
Figure 2. Strain variants of αSyn leading to different clinical phenotypes and possible factors that affect αSyn assembly. We propose the possible mechanisms as to how different αSyn species cause the different types of synucleinopathies. Unstructured αSyn can adopt several structural conformations. Once αSyn forms a structure that favors assembly with each other, it forms fibrils. The final morphology and characters of the fibrils are strongly associated with the initial conformational change. As changing the buffer conditions, such as adding salt or desalting leads to the formation of different types of fibrils, many environmental factors (trans-acting effectors) and some autologous factors (cis-acting effectors) are thought to affect the type of fibril that is formed.
How about the trans-acting effectors that affect the fibrillation of WT αSyn? In 2013, Bousset et al. (2013) assessed the effects of different buffer conditions, i.e., with or without salt, on the formation of αSyn fibrils, and showed that different conditions created two strains with different structures, levels of toxicity, and in vitro and in vivo seeding and propagation properties. Later, in 2015, they showed that distinct αSyn strains demonstrated different seeding capacities, inducing strain-specific pathologies and neurotoxic phenotypes in vivo (Peelaerts et al., 2015). These discoveries led to the hypothesis that trans-acting effectors affect αSyn fibrillation processes and that distinct strains may account for different clinicopathological traits among the synucleinopathies (Figure 2).
Cellular Environment and Synuclein Species
To date, there have been no direct lines of evidence clearly showing the detailed conformational differences among αSyn species in PD, DLB, and MSA. However, several reports strongly indicate the existence of different αSyn species in the different synucleinopathies. For example, the solubility of αSyn in the brains of MSA patients is very different from that in the brains of PD/DLB patients. αSyn extracted from MSA patient brains mainly appears in the sodium dodecyl sulfate (SDS)-soluble fraction, whereas αSyn extracted from PD/DLB patient brains appears in the insoluble fraction (Campbell et al., 2001). Moreover, Hirohata et al. (2011) reported that αSyn fibrils amplified from the cerebrospinal fluid of MSA patients by protein misfolding cyclic amplification using recombinant αSyn showed different widths compared with fibrils amplified from PD and DLB patients. In 2015, Prusiner et al. (2015) reported that brain lysates from MSA patients containing an abnormal form of αSyn could transmit this abnormal conformation to transgenic mice expressing αSyn 140∗A53T; however, neither αSyn from normal control brain lysates nor PD brain samples were able to transmit an abnormal conformation.
Then, what are the trans-acting effectors and how are different strains created in the brains of patients? Recently, Peng et al. proposed the hypothesis that different cellular environments of neurons and oligodendrocytes promote the formation of distinct αSyn strains (Peng et al., 2018a). They showed that oligodendrocytes but not neurons transform misfolded αSyn into a strain that has more resistance to PK and stronger transmissibility, which is similar to fibrils obtained from MSA patients, thus highlighting the fact that distinct αSyn strains are generated by different intracellular milieus.
Possible Roles of Lipid Diversity in Synucleinopathies
Considering the types of cellular milieus that may differ between neurons and oligodendrocytes, we believe that lipid components are a possible candidate (Figure 2). The brain is primarily comprised of lipid, where lipids represent up to 50% of its dry weight. Some lipids are found abundantly in the brain, such as galactosylceramide (GalCer) and sulfatide in myelin, and ganglioside GM1 in neurons, and lipids are not homogeneously distributed in the brain. In fact, glial cells and neurons have specific glycosphingolipid compositions (van Echten-Deckert and Herget, 2006). Oligodendrocytes mainly express GalCer, sulfatide, and GM3, whereas astrocytes highly express ganglioside GM3. Neurons contain the widest variety of gangliosides, including GM1, GD1a, GD1b, and GT1b (van Echten-Deckert and Herget, 2006). Taking these observations into consideration, the high content of glycosphingolipids in brain membranes have a considerable effect on the aggregation process of the αSyn in each neuronal cell type. Indeed, our in vitro results suggest the possibility that distinct prion-like αSyn strains are generated by GlcCer and GM1 (Suzuki et al., 2015, 2018). The PK-digested band pattern of αSyn, which indicates structural differences of various amyloid fibrils, was different between αSyn incubated with GlcCer-containing liposomes and αSyn incubated with GM1-containing liposomes (Suzuki et al., 2015). We therefore propose that lipid components act as trans-acting effectors for αSyn in the pathogenesis of PD (Figure 2).
Few studies to date have investigated the changes in lipid metabolism of MSA patients. Don et al. (2014) analyzed the amount of galactosphingolipids in the white matter of the brains of MSA patients. They showed that total sphingomyelin, sulfatide, and GalCer levels were selectively decreased in disease-affected white matter (primary motor cortex), whereas no significant differences were detected in unaffected white matter (visual cortex). Interestingly, the physiological expression of these lipids are higher in the motor cortex than in the visual cortex, suggesting a possible explanation as to how αSyn selectively accumulates in specific regions of the brain in MSA patients.
Conclusion
In this review, we gave an overview of αSyn aggregation in PD/DLB and the possible roles of lipid dysregulation in the PD/DLB pathogenesis. Many genetic and pathological studies have shown that lipid metabolism is affected in the brains of PD patients, and many in vitro and in vivo studies have suggested that this might initiate conformational changes in αSyn, leading to its aggregation. Some preliminary data suggest the possibility that the accumulated lipids not only accelerate αSyn aggregation but also determine the different species of αSyn fibrils. If our hypothesis can be proven, we will be able to extend our understanding of the distinct pathologies among the synucleinopathies, such as PD, DLB, and MSA, and why they show different clinicopathological features despite showing accumulation of the same WT αSyn. Further studies, such as a comparison of lipid metabolism in the brains or biosamples of PD/DLB and MSA patients and investigations on how such altered lipid metabolism affects αSyn aggregation, are required to show the association between altered lipid metabolism and different αSyn species in PD/DLB and MSA.
Author Contributions
KI wrote the first draft of the manuscript. All authors contributed to manuscript revision, read it, and approved the submitted version.
Funding
This work was supported in part by the Grants-in-Aid for Challenging Exploratory Research (17K19658 to YN), for the Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control 17H05699 to YN and 17H05700 to HM), for the Scientific Research (B) (18H02741 to HM), and for the Young Scientists (B) (T17K161220 to KI and 15K19502 and 17K16134 to MS) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan; by grants for Practical Research Projects for Rare/Intractable Diseases (JP16ek0109018 and JP18ek0109222 to YN) from the Japan Agency for Medical Research and Development; by the Intramural Research Grants for Neurological and Psychiatric Disorders (27-7, 27-9, 30-3, and 30-9 to YN) from the National Center of Neurology and Psychiatry; by a grant from the Japan Foundation for Neuroscience and Mental Health, Japan (to YN); and by a grant provided by The Ichiro Kanehara Foundation (to MS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. H. Akiko Popiel for critical reading of the manuscript and lab members for their helpful discussions.
References
Anderson, J. P., Walker, D. E., Goldstein, J. M., de Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Appel-Cresswell, S., Vilarino-Guell, C., Encarnacion, M., Sherman, H., Yu, I., Shah, B., et al. (2013). Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 28, 811–813. doi: 10.1002/mds.25421
Araki, K., Yagi, N., Ikemoto, Y., Yagi, H., Choong, C. J., Hayakawa, H., et al. (2015). Synchrotron FTIR micro-spectroscopy for structural analysis of Lewy bodies in the brain of Parkinson’s disease patients. Sci. Rep. 5:17625. doi: 10.1038/srep17625
Argyriou, A., Dermentzaki, G., Papasilekas, T., Moraitou, M., Stamboulis, E., Vekrellis, K., et al. (2012). Increased dimerization of alpha-synuclein in erythrocytes in Gaucher disease and aging. Neurosci. Lett. 24, 205–209. doi: 10.1016/j.neulet.2012.08.069
Balducci, C., Pierguidi, L., Persichetti, E., Parnetti, L., Sbaragli, M., Tassi, C., et al. (2007). Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson’s disease. Mov. Disord. 22, 1481–1484. doi: 10.1002/mds.21399
Bertoncini, C. W., Jung, Y. S., Fernandez, C. O., Hoyer, W., Griesinger, C., Jovin, T. M., et al. (2005). Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 102, 1430–1435. doi: 10.1073/pnas.0407146102
Bodner, C. R., Dobson, C. M., and Bax, A. (2009). Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J. Mol. Biol. 390, 775–790. doi: 10.1016/j.jmb.2009.05.066
Bousset, L., Pieri, L., Ruiz-Arlandis, G., Gath, J., Jensen, P. H., Habenstein, B., et al. (2013). Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 4:2575. doi: 10.1038/ncomms3575
Burkhardt, C. R., Filley, C. M., Kleinschmidt-DeMasters, B. K., de la Monte, S., Norenberg, M. D., and Schneck, S. A. (1988). Diffuse Lewy body disease and progressive dementia. Neurology 38, 1520–1528.
Burre, J. (2015). The synaptic function of alpha-synuclein. J. Parkinsons Dis. 5, 699–713. doi: 10.3233/JPD-150642
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Byrne, E. J., Lennox, G., Lowe, J., and Godwin-Austen, R. B. (1989). Diffuse Lewy body disease: clinical features in 15 cases. J. Neurol. Neurosurg. Psychiatry 52, 709–717. doi: 10.1136/jnnp.52.6.709
Campbell, B. C., McLean, C. A., Culvenor, J. G., Gai, W. P., Blumbergs, P. C., Jakala, P., et al. (2001). The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J. Neurochem. 76, 87–96. doi: 10.1046/j.1471-4159.2001.00021.x
Chandra, S., Chen, X., Rizo, J., Jahn, R., and Sudhof, T. C. (2003). A broken alpha -helix in folded alpha -Synuclein. J. Biol. Chem. 278, 15313–15318. doi: 10.1074/jbc.M213128200
Chandra, S., Fornai, F., Kwon, H. B., Yazdani, U., Atasoy, D., Liu, X., et al. (2004). Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc. Natl. Acad. Sci. U.S.A. 101, 14966–14971. doi: 10.1073/pnas.0406283101
Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., et al. (2004). Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. doi: 10.1016/S0140-6736(04)17103-1
Chiasserini, D., Paciotti, S., Eusebi, P., Persichetti, E., Tasegian, A., Kurzawa-Akanbi, M., et al. (2015). Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 10:15. doi: 10.1186/s13024-015-0010-2
Choi, W., Zibaee, S., Jakes, R., Serpell, L. C., Davletov, B., Crowther, R. A., et al. (2004). Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 576, 363–368. doi: 10.1016/j.febslet.2004.09.038
De Franceschi, G., Frare, E., Pivato, M., Relini, A., Penco, A., Greggio, E., et al. (2011). Structural and morphological characterization of aggregated species of alpha-synuclein induced by docosahexaenoic acid. J. Biol. Chem. 286, 22262–22274. doi: 10.1074/jbc.M110.202937
Dedmon, M. M., Lindorff-Larsen, K., Christodoulou, J., Vendruscolo, M., and Dobson, C. M. (2005). Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 127, 476–477. doi: 10.1021/ja044834j
Don, A. S., Hsiao, J. H., Bleasel, J. M., Couttas, T. A., Halliday, G. M., and Kim, W. S. (2014). Altered lipid levels provide evidence for myelin dysfunction in multiple system atrophy. Acta Neuropathol. Commun. 2:150. doi: 10.1186/s40478-014-0150-6
Drescher, M., Veldhuis, G., van Rooijen, B. D., Milikisyants, S., Subramaniam, V., and Huber, M. (2008). Antiparallel arrangement of the helices of vesicle-bound alpha-synuclein. J. Am. Chem. Soc. 130, 7796–7797. doi: 10.1021/ja801594s
Flavin, W. P., Bousset, L., Green, Z. C., Chu, Y., Skarpathiotis, S., Chaney, M. J., et al. (2017). Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 134, 629–653. doi: 10.1007/s00401-017-1722-x
Fortin, D. L., Nemani, V. M., Voglmaier, S. M., Anthony, M. D., Ryan, T. A., and Edwards, R. H. (2005). Neural activity controls the synaptic accumulation of alpha-synuclein. J. Neurosci. 25, 10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005
Galvagnion, C., Brown, J. W., Ouberai, M. M., Flagmeier, P., Vendruscolo, M., Buell, A. K., et al. (2016). Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 113, 7065–7070. doi: 10.1073/pnas.1601899113
Galvagnion, C., Buell, A. K., Meisl, G., Michaels, T. C., Vendruscolo, M., Knowles, T. P., et al. (2015). Lipid vesicles trigger alpha-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 11, 229–234. doi: 10.1038/nchembio.1750
Gamez-Valero, A., Prada-Dacasa, P., Santos, C., Adame-Castillo, C., Campdelacreu, J., Rene, R., et al. (2016). GBA mutations are associated with earlier onset and male sex in dementia with lewy bodies. Mov. Disord. 31, 1066–1070. doi: 10.1002/mds.26593
Gegg, M. E., Burke, D., Heales, S. J., Cooper, J. M., Hardy, J., Wood, N. W., et al. (2012). Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 72, 455–463. doi: 10.1002/ana.23614
Gegg, M. E., and Schapira, A. H. V. (2018). The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 285, 3591–3603. doi: 10.1111/febs.14393
George, J. M., Jin, H., Woods, W. S., and Clayton, D. F. (1995). Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15, 361–372. doi: 10.1016/0896-6273(95)90040-3
Giasson, B. I., Murray, I. V., Trojanowski, J. Q., and Lee, V. M. (2001). A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 276, 2380–2386. doi: 10.1074/jbc.M008919200
Goker-Alpan, O., Schiffmann, R., LaMarca, M. E., Nussbaum, R. L., McInerney-Leo, A., and Sidransky, E. (2004). Parkinsonism among gaucher disease carriers. J. Med. Genet. 41, 937–940. doi: 10.1136/jmg.2004.024455
Guerreiro, R., Ross, O. A., Kun-Rodrigues, C., Hernandez, D. G., Orme, T., Eicher, J. D., et al. (2018). Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol. 17, 64–74. doi: 10.1016/S1474-4422(17)30400-3
Guo, J. L., Covell, D. J., Daniels, J. P., Iba, M., Stieber, A., Zhang, B., et al. (2013). Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117. doi: 10.1016/j.cell.2013.05.057
Hirohata, M., Ono, K., Morinaga, A., Ikeda, T., and Yamada, M. (2011). Cerebrospinal fluid from patients with multiple system atrophy promotes in vitro alpha-synuclein fibril formation. Neurosci. Lett. 491, 48–52. doi: 10.1016/j.neulet.2011.01.005
Hong, D. P., Xiong, W., Chang, J. Y., and Jiang, C. (2011). The role of the C-terminus of human alpha-synuclein: intra-disulfide bonds between the C-terminus and other regions stabilize non-fibrillar monomeric isomers. FEBS Lett. 585, 561–566. doi: 10.1016/j.febslet.2011.01.009
Hoyer, W., Cherny, D., Subramaniam, V., and Jovin, T. M. (2004). Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry 43, 16233–16242. doi: 10.1021/bi048453u
Issidorides, M. R., Mytilineou, C., Panayotacopoulou, M. T., and Yahr, M. D. (1991). Lewy bodies in parkinsonism share components with intraneuronal protein bodies of normal brains. J. Neural Transm. Park Dis. Dement. Sect. 3, 49–61. doi: 10.1007/bf02251136
Jakes, R., Spillantini, M. G., and Goedert, M. (1994). Identification of two distinct synucleins from human brain. FEBS Lett. 345, 27–32. doi: 10.1016/0014-5793(94)00395-5
Jensen, P. H., Islam, K., Kenney, J., Nielsen, M. S., Power, J., and Gai, W. P. (2000). Microtubule-associated protein 1B is a component of cortical Lewy bodies and binds alpha-synuclein filaments. J. Biol. Chem. 275, 21500–21507. doi: 10.1074/jbc.M000099200
Kiely, A. P., Asi, Y. T., Kara, E., Limousin, P., Ling, H., Lewis, P., et al. (2013). Alpha-synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 125, 753–769. doi: 10.1007/s00401-013-1096-7
Klatka, L. A., Louis, E. D., and Schiffer, R. B. (1996). Psychiatric features in diffuse Lewy body disease: a clinicopathologic study using Alzheimer’s disease and Parkinson’s disease comparison groups. Neurology 47, 1148–1152. doi: 10.1212/wnl.47.5.1148
Kosaka, K., Oyanagi, S., Matsushita, M., and Hori, A. (1976). Presenile dementia with Alzheimer-, Pick- and Lewy-body changes. Acta Neuropathol. 36, 221–233. doi: 10.1007/bf00685366
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Lee, H. J., Choi, C., and Lee, S. J. (2002). Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 277, 671–678. doi: 10.1074/jbc.M107045200
Li, W., West, N., Colla, E., Pletnikova, O., Troncoso, J. C., Marsh, L., et al. (2005). Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. U.S.A. 102, 2162–2167. doi: 10.1073/pnas.0406976102
Ly, T., and Julian, R. R. (2008). Protein-metal interactions of calmodulin and alpha-synuclein monitored by selective noncovalent adduct protein probing mass spectrometry. J. Am. Soc. Mass Spectrom. 19, 1663–1672. doi: 10.1016/j.jasms.2008.07.006
Maiti, N. C., Apetri, M. M., Zagorski, M. G., Carey, P. R., and Anderson, V. E. (2004). Raman spectroscopic characterization of secondary structure in natively unfolded proteins: alpha-synuclein. J. Am. Chem. Soc. 126, 2399–2408. doi: 10.1021/ja0356176
Maor, G., Rapaport, D., and Horowitz, M. (2019). The effect of mutant GBA1 on accumulation and aggregation of alpha-synuclein. Hum. Mol. Genet. 28, 1768–1781. doi: 10.1093/hmg/ddz005
Marinus, J., Zhu, K., Marras, C., Aarsland, D., and van Hilten, J. J. (2018). Risk factors for non-motor symptoms in Parkinson’s disease. Lancet Neurol. 17, 559–568. doi: 10.1016/S1474-4422(18)30127-3
Mazzulli, J. R., Xu, Y. H., Sun, Y., Knight, A. L., McLean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
Moors, T. E., Paciotti, S., Ingrassia, A., Quadri, M., Breedveld, G., Tasegian, A., et al. (2018). Characterization of brain lysosomal activities in GBA-related and sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurobiol. 56, 1344–1355. doi: 10.1007/s12035-018-1090-0
Moors, T. E., Paciotti, S., Ingrassia, A., Quadri, M., Breedveld, G., Tasegian, A., et al. (2019). Characterization of brain lysosomal activities in GBA-related and sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurobiol. 56, 1344–1355. doi: 10.1007/s12035-018-1090-0
Moraitou, M., Dermentzaki, G., Dimitriou, E., Monopolis, I., Dekker, N., Aerts, H., et al. (2016). α-Synuclein dimerization in erythrocytes of Gaucher disease patients: correlation with lipid abnormalities and oxidative stress. Neurosci. Lett. 2, 1–5. doi: 10.1016/j.neulet.2015.12.013
Murphy, K. E., Gysbers, A. M., Abbott, S. K., Tayebi, N., Kim, W. S., Sidransky, E., et al. (2014). Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 137(Pt 3), 834–848. doi: 10.1093/brain/awt367
Nakata, Y., Yasuda, T., Fukaya, M., Yamamori, S., Itakura, M., Nihira, T., et al. (2012). Accumulation of alpha-synuclein triggered by presynaptic dysfunction. J. Neurosci. 32, 17186–17196. doi: 10.1523/JNEUROSCI.2220-12.2012
Nalls, M. A., Duran, R., Lopez, G., Kurzawa-Akanbi, M., McKeith, I. G., Chinnery, P. F., et al. (2013). A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 70, 727–735. doi: 10.1001/jamaneurol.2013.1925
Neudorfer, O., Giladi, N., Elstein, D., Abrahamov, A., Turezkite, T., Aghai, E., et al. (1996). Occurrence of Parkinson’s syndrome in type I gaucher disease. QJM 89, 691–694. doi: 10.1093/qjmed/89.9.691
Neumann, J., Bras, J., Deas, E., O’Sullivan, S. S., Parkkinen, L., Lachmann, R. H., et al. (2009). Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132(Pt 7), 1783–1794. doi: 10.1093/brain/awp044
Nichols, W. C., Pankratz, N., Marek, D. K., Pauciulo, M. W., Elsaesser, V. E., Halter, C. A., et al. (2009). Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 72, 310–316. doi: 10.1212/01.wnl.0000327823.81237.d1
Parnetti, L., Chiasserini, D., Persichetti, E., Eusebi, P., Varghese, S., Qureshi, M. M., et al. (2014). Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 29, 1019–1027. doi: 10.1002/mds.25772
Peelaerts, W., Bousset, L., Van der Perren, A., Moskalyuk, A., Pulizzi, R., Giugliano, M., et al. (2015). alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344. doi: 10.1038/nature14547
Peng, C., Gathagan, R. J., Covell, D. J., Medellin, C., Stieber, A., Robinson, J. L., et al. (2018a). Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 557, 558–563. doi: 10.1038/s41586-018-0104-4
Peng, C., Gathagan, R. J., and Lee, V. M. (2018b). Distinct alpha-synuclein strains and implications for heterogeneity among alpha-synucleinopathies. Neurobiol. Dis. 109(Pt B), 209–218. doi: 10.1016/j.nbd.2017.07.018
Periquet, M., Fulga, T., Myllykangas, L., Schlossmacher, M. G., and Feany, M. B. (2007). Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 27, 3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Prusiner, S. B., Woerman, A. L., Mordes, D. A., Watts, J. C., Rampersaud, R., Berry, D. B., et al. (2015). Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 112, E5308–E5317. doi: 10.1073/pnas.1514475112
Rocha, E. M., Smith, G. A., Park, E., Cao, H., Brown, E., Hallett, P., et al. (2015). Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2, 433–438. doi: 10.1002/acn3.177
Rochet, J. C., Conway, K. A., and Lansbury, P. T. Jr. (2000). Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse alpha-synuclein. Biochemistry 39, 10619–10626. doi: 10.1021/bi001315u
Rodriguez, J. A., Ivanova, M. I., Sawaya, M. R., Cascio, D., Reyes, F. E., Shi, D., et al. (2015). Structure of the toxic core of alpha-synuclein from invisible crystals. Nature 525, 486–490. doi: 10.1038/nature15368
Roy, S., and Wolman, L. (1969). Ultrastructural observations in Parkinsonism. J. Pathol. 99, 39–44. doi: 10.1002/path.1710990106
Sahay, S., Ghosh, D., Dwivedi, S., Anoop, A., Mohite, G. M., Kombrabail, M., et al. (2015). Familial parkinson disease-associated mutations alter the site-specific microenvironment and dynamics of alpha-synuclein. J. Biol. Chem. 290, 7804–7822. doi: 10.1074/jbc.M114.598607
Satake, W., Nakabayashi, Y., Mizuta, I., Hirota, Y., Ito, C., Kubo, M., et al. (2009). Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 41, 1303–1307. doi: 10.1038/ng.485
Savica, R., Murray, M. E., Persson, X. M., Kantarci, K., Parisi, J. E., Dickson, D. W., et al. (2016). Plasma sphingolipid changes with autopsy-confirmed Lewy Body or Alzheimer’s pathology. Alzheimers Dement 3, 43–50. doi: 10.1016/j.dadm.2016.02.005
Scott, T. R., and Netsky, M. G. (1961). The pathology of Parkinson’s syndrome: a critical review. Int. J. Neurol. 2, 51–60.
Serpell, L. C., Berriman, J., Jakes, R., Goedert, M., and Crowther, R. A. (2000). Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. U.S.A. 97, 4897–4902. doi: 10.1073/pnas.97.9.4897
Sharon, R., Bar-Joseph, I., Mirick, G. E., Serhan, C. N., and Selkoe, D. J. (2003). Altered fatty acid composition of dopaminergic neurons expressing alpha-synuclein and human brains with alpha-synucleinopathies. J. Biol. Chem. 278, 49874–49881. doi: 10.1074/jbc.M309127200
Sidransky, E., and Lopez, G. (2012). The link between the GBA gene and parkinsonism. Lancet Neurol. 11, 986–998. doi: 10.1016/S1474-4422(12)70190-4
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R., et al. (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Sierecki, E., Giles, N., Bowden, Q., Polinkovsky, M. E., Steinbeck, J., Arrioti, N., et al. (2016). Nanomolar oligomerization and selective co-aggregation of alpha-synuclein pathogenic mutants revealed by single-molecule fluorescence. Sci. Rep. 6:37630. doi: 10.1038/srep37630
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312. doi: 10.1038/ng.487
Spillantini, M. G., Crowther, R. A., Jakes, R., Cairns, N. J., Lantos, P. L., and Goedert, M. (1998a). Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 251, 205–208. doi: 10.1016/s0304-3940(98)00504-7
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998b). alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Suzuki, M., Fujikake, N., Takeuchi, T., Kohyama-Koganeya, A., Nakajima, K., Hirabayashi, Y., et al. (2015). Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant alpha-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 24, 6675–6686. doi: 10.1093/hmg/ddv372
Suzuki, M., Sango, K., Wada, K., and Nagai, Y. (2018). Pathological role of lipid interaction with alpha-synuclein in Parkinson’s disease. Neurochem. Int. 119, 97–106. doi: 10.1016/j.neuint.2017.12.014
Taguchi, Y. V., Liu, J., Ruan, J., Pacheco, J., Zhang, X., Abbasi, J., et al. (2017). Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson’s disease. J. Neurosci. 37, 9617–9631. doi: 10.1523/JNEUROSCI.1525-17.2017
Tayebi, N., Walker, J., Stubblefield, B., Orvisky, E., LaMarca, M. E., Wong, K., et al. (2003). Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 79, 104–109. doi: 10.1016/s1096-7192(03)00071-4
Trojanowski, J. Q., and Lee, V. M. (1998). Aggregation of neurofilament and alpha-synuclein proteins in Lewy bodies: implications for the pathogenesis of Parkinson disease and Lewy body dementia. Arch. Neurol. 55,151–152.
Uversky, V. N. (2007). Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 103, 17–37. doi: 10.1111/j.1471-4159.2007.04764.x
Uversky, V. N., Li, J., and Fink, A. L. (2001). Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296. doi: 10.1074/jbc.M105343200
van Echten-Deckert, G., and Herget, T. (2006). Sphingolipid metabolism in neural cells. Biochim. Biophys. Acta 1758, 1978–1994. doi: 10.1016/j.bbamem.2006.06.009
Weinreb, P. H., Zhen, W., Poon, A. W., Conway, K. A., and Lansbury, P. T. Jr. (1996). NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35, 13709–13715. doi: 10.1021/bi961799n
Zarbiv, Y., Simhi-Haham, D., Israeli, E., Elhadi, S. A., Grigoletto, J., and Sharon, R. (2014). Lysine residues at the first and second KTKEGV repeats mediate alpha-Synuclein binding to membrane phospholipids. Neurobiol. Dis. 70, 90–98. doi: 10.1016/j.nbd.2014.05.031
Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Zhang, Z., Kang, S. S., Liu, X., Ahn, E. H., He, L., Iuvone, P. M., et al. (2017). Asparagine endopeptidase cleaves alpha-synuclein and mediates pathologic activities in Parkinson’s disease. Nat. Struct. Mol. Biol. 24, 632–642. doi: 10.1038/nsmb.3433
Keywords: Parkinson’s disease, α-synuclein, lipid, fibril, synucleinopathy
Citation: Ikenaka K, Suzuki M, Mochizuki H and Nagai Y (2019) Lipids as Trans-Acting Effectors for α-Synuclein in the Pathogenesis of Parkinson’s Disease. Front. Neurosci. 13:693. doi: 10.3389/fnins.2019.00693
Received: 31 January 2019; Accepted: 18 June 2019;
Published: 03 July 2019.
Edited by:
Veerle Baekelandt, KU Leuven, BelgiumReviewed by:
Patrícia Maciel, Universidade do Minho, PortugalKatrin Beyer, Fundación Instituto de Investigación Biomédica “Germans Trias i Pujol”, Portugal
Copyright © 2019 Ikenaka, Suzuki, Mochizuki and Nagai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshitaka Nagai, bmFnYWlAbmV1cm90aGVyLm1lZC5vc2FrYS11LmFjLmpw