 Dario Valdinocci1
Dario Valdinocci1 Rui F. Simões2
Rui F. Simões2 Jaromira Kovarova3
Jaromira Kovarova3 Teresa Cunha-Oliveira2
Teresa Cunha-Oliveira2 Jiri Neuzil1,3
Jiri Neuzil1,3 Dean L. Pountney1*
Dean L. Pountney1*- 1School of Medical Science, Griffith University, Southport, QLD, Australia
- 2CNC - Center for Neuroscience and Cell Biology, University of Coimbra, Cantanhede, Portugal
- 3Institute of Biotechnology, Czech Academy of Sciences, Prague-West, Czechia
The appearance of alpha-synuclein-positive inclusion bodies (Lewy bodies) and the loss of catecholaminergic neurons are the primary pathological hallmarks of Parkinson’s disease (PD). However, the dysfunction of mitochondria has long been recognized as a key component in the progression of the disease. Dysfunctional mitochondria can in turn lead to dysregulation of calcium homeostasis and, especially in dopaminergic neurons, raised mean intracellular calcium concentration. As calcium binding to alpha-synuclein is one of the important triggers of alpha-synuclein aggregation, mitochondrial dysfunction will promote inclusion body formation and disease progression. Increased reactive oxygen species (ROS) resulting from inefficiencies in the electron transport chain also contribute to the formation of alpha-synuclein aggregates and neuronal loss. Recent studies have also highlighted defects in mitochondrial clearance that lead to the accumulation of depolarized mitochondria. Transaxonal and intracytoplasmic translocation of mitochondria along the microtubule cytoskeleton may also be affected in diseased neurons. Furthermore, nanotube-mediated intercellular transfer of mitochondria has recently been reported between different cell types and may have relevance to the spread of PD pathology between adjacent brain regions. In the current review, the contributions of both intracellular and intercellular mitochondrial dynamics to the etiology of PD will be discussed.
Introduction: PD, α-Synuclein and Mitochondria
The principal histopathological marker of Parkinson’s disease (PD) is the presence in neurons of α-synuclein (α-syn) protein aggregates that occur in inclusion bodies known as Lewy bodies (Shults, 2006; McCann et al., 2014). α-Syn is primarily expressed pre-synaptically and evidence exists of α-syn transfer from neurons to neuronal and non-neuronal cells in vitro, indicating that α-syn pathology spreads between anatomically adjacent brain regions by a cell-to-cell transfer mechanism (Valdinocci et al., 2017). α-Syn is a small (14 kDa), acidic protein expressed in the brain, peripheral nervous system and circulating erythrocytes (Thakur et al., 2019). Its pre-synaptic localization and high abundance implicate an important role in synaptic transmission (Burre et al., 2010) with specific functions implicated in synaptic vesicle recycling and regulating soluble NSF attachment protein receptor (SNARE) interactions and dopamine biosynthesis (Theillet et al., 2016; Sulzer and Edwards, 2019). In vitro α-syn is a dynamically unfolded protein, although in vivo the membrane-associated tetrameric form is proposed to be α-helical (Bartels et al., 2011). Various factors, such as raised copper or calcium concentration, oxidative stress and post-translational modifications can trigger intracellular α-syn aggregation (Fujiwara et al., 2002; El-Agnaf et al., 2006; Reynolds et al., 2008; Rcom-H’cheo-Gauthier et al., 2016). Whilst a definitive link between mitochondrial dysfunction and initiation of PD still does not exist, it is clear that dysfunctional mitochondria are omnipresent in PD (Chen et al., 2019). Moreover, α-Syn can be located at mitochondrial membranes, especially under stress conditions (Cole et al., 2008; Devi et al., 2008), and its aggregation can be linked to mitochondrial dysfunction in PD (Perfeito et al., 2013; Celardo et al., 2014). α-Syn aggregates may in turn cause deleterious alterations in mitochondrial function, including intracellular dynamics. This review focusses on α-syn interactions with mitochondria in PD.
Intracellular Mitochondrial Dynamics in PD
Mitochondria are highly dynamic and interconnected entities, with the ability to change their morphology in addition to mobilizing within the cell to help power critical functions (Burte et al., 2015). Mitochondrial dynamic processes include fusion/fission, transport and clearance, and are so interconnected and interdependent that they have been proposed to form an interactome that ultimately controls mitochondrial quality, quantity and metabolism (Dorn and Kitsis, 2015; Shirihai et al., 2015). Under normal conditions, mitochondria constantly undergo cycles of fusion and fission that affect their morphology and shape, processes that may be perturbed in PD. α-Syn has been shown to influence mitochondrial size both independently and dependent on fusion/fission proteins, with recent reports detailing these interactions as an attribute of the pathological variants such as oligomers and fibrils, ruling out any negative effects on mitochondrial dynamics as a normal function of the monomer (Wang et al., 2019). Factors such as the GTPases Mitofusin1 and 2 (Mfn1/2), and optic atrophy protein 1 (OPA1) are involved in fusing the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM), respectively, forming elongated structures that are more efficient in ATP generation (Chan, 2012; Tilokani et al., 2018). As illustrated in Figure 1, oligomeric α-syn can bind to lipids in the OMM and distress the membrane curvature, leading to a decrease in mitochondrial fusion rate (Pozo Devoto and Falzone, 2017). In addition, overexpression of α-Syn in transgenic mice led to reductions in Mfn1/2 protein levels, correlating with a decrease in mitochondrial fusion and smaller mitochondria (Xie and Chung, 2012). α-Syn knockdown was shown to trigger mitochondrial elongation (Kamp et al., 2010).
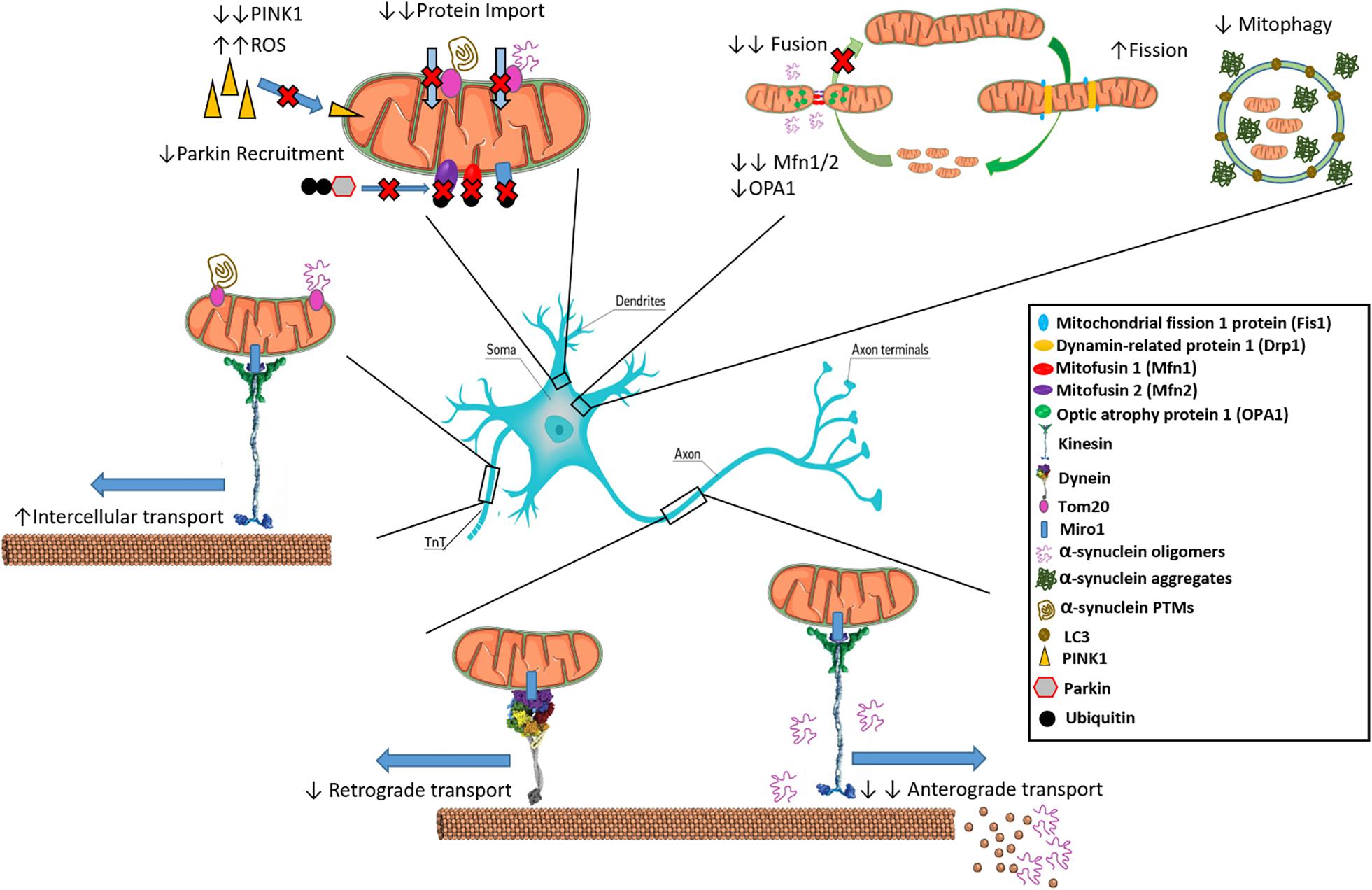
Figure 1. Interactions of α-synuclein with inter- and intra-cellular mitochondrial dynamics. α-syn inhibits mitochondrial fusion as over expression leads to reduced Mfn1/2 and OPA-1. Oligomeric α-syn can interact with kinesin to dislodge it from microtubules preventing anterograde transport along axons. Oligomeric α-Syn also promotes the depolymerization of microtubules, preventing transport of mitochondria. Increased aggregated α-syn is associated with reduced mitophagy. Certain α-Syn types such as oligomers and mutated variants like S129E bind to Tom20 resulting in inhibition of mitochondrial protein import. Reduction in PINK1 leads to inability to signal for mitophagy of α-syn bound mitochondria through Parkin recruitment, leading to ROS generation. Movement of pathological aggregates between cells via TnTs may occur if bound to Tom20 on actively transferring mitochondria.
Conversely, mitochondrial fission is dependent on dynamin-related protein 1 (Drp1), mitochondrial fission factor (Mff), mitochondrial fission protein 1 (Fis1) and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49/51) for shrinkage of structures typically promoted during cellular replication (Korobova et al., 2013; Pagliuso et al., 2018). Depending on the isoform, post-translational modification of Drp1 by the small ubiquitin-like modifier (SUMO) drastically alters the effect on mitochondrial fission. SUMO-2/3 for instance has been shown to inhibit Drp1-Mff interaction preventing mitochondrial fragmentation, however, SUMO-1 stabilizes Drp1 leading to enhanced fragmentation; with evidence establishing a link between SUMO and PD, this further implicates mitochondrial dynamics in the pathogenesis of the disease (Guo et al., 2013, 2017; Fu et al., 2014; Vijayakumaran et al., 2015; Henley et al., 2018; Vijayakumaran and Pountney, 2018). Mitochondrial complex I inhibitors, Rotenone and MPP+, inducers of parkinsonian phenotypes, were shown to promote mitochondrial fission (Barsoum et al., 2006; Thomas et al., 2011) and Drp1 inactivation prevented the fission phenotype (Wang et al., 2011a). In the substantia nigra of sporadic PD patients, the short form of OPA1 (OPA1-S) was decreased in the absence of changes in Mfn1, further suggesting mitochondrial fusion deficiency (Zilocchi et al., 2018). Thus, a net increase in mitochondrial fission over fusion may ultimately lead to a fragmented mitochondrial network negatively impacting the efficiency of neuronal signaling in PD. Worth noting are reductions in Drp1 in the later stages of degeneration in transgenic mice (Xie and Chung, 2012). Mitochondria are also a major source of intracellular calcium which is released upon electron transport chain dysfunction, such as that caused by rotenone-mediated inhibition, and interacts with α-syn to promote aggregation. Indeed, induction of the endogenous neuronal calcium buffering protein, calbindin-D28k (CB), was able to block rotenone-induced α-syn aggregation and neurons expressing high levels of CB excluded α-syn inclusion bodies in both human and mouse model tissues (Rcom-H’cheo-Gauthier et al., 2016, 2017; McLeary et al., 2019). Moreover, the Miro1 protein acts as a calcium sensor at the mitochondrial outer membrane, reacting to high calcium by augmenting mitophagy (Nemani et al., 2018).
Intracellular mitochondrial dynamics involves the transport of mitochondrial units from one area of the cell to another, and is of key importance in neuronal cells that are polarized with long axons and dendrites (Lin and Sheng, 2015). Mitochondria need to be transported to synaptic terminals, active growth cones and axonal branches, where they maintain energy and Ca2+ homeostasis (Sheng, 2017). Trafficking of mitochondria is microtubule (MT)-based, relying on ATP and motor proteins. MTs are polar α/β-tubulin polymers with the minus end within the cell body and the plus end to the cell extremity (Tas and Kapitein, 2018), and neurons can move mitochondria in both directions using independent motor proteins. Kinesins drive transport toward the cell extremities (anterograde transport) whereas dynein motors are responsible for retrograde transport (Hirokawa et al., 2010). Hydrolysis of ATP is essential to fuel mitochondrial movement in both directions (Zala et al., 2013). Bridging between mitochondria and MT-bound motor proteins are motor adaptor proteins such as Trafficking Kinesin Proteins 1 and 2 (TRAK1 and 2), which connect the OMM protein Mitochondrial Rho GTPase 1 (Miro1) to kinesin (Melkov and Abdu, 2018). Indeed, Miro1 also mediates Drp1/Fis1-independent mitochondrial shape transition (Mist), needed for mitophagy (Nemani et al., 2018). Syntabulin can also link mitochondria and kinesins whilst syntaphilin acts as an anchor stopping mitochondrial movement (Cai et al., 2005; Chen and Sheng, 2013).
An increase in α-syn concentration was shown to result in mitochondrial traffic arrest even before axonal degeneration, affecting both anterograde and retrograde transport (O’Donnell et al., 2014; Pozo Devoto and Falzone, 2017). Nigral dopaminergic neurons especially display a “dying back” pattern, wherein anterograde trafficking of mitochondria becomes disrupted in early stage sporadic PD followed by retrograde transport at late stage (Chu et al., 2012). Loss of this trafficking severely reduces nigral neuron ability to regulate the conditions necessary for axonal signaling and neurotransmitter release at terminals. Regarding anterograde trafficking, α-syn oligomers disrupt through direct binding interactions between kinesin and the MT in addition to increasing expression of tau, a MT structure disruptor (Prots et al., 2013, 2018). α-Syn appears to induce MT fragmentation directly as well, hindering mitochondrial movement from distal cell areas (Melo et al., 2017). Interestingly, this finding produced the opposite outcome to Chu et al. (2012) whereby retrograde transport was disrupted first. This may be due to differences in α-syn species interaction with trafficking complexes disrupting mitochondrial transport as Chu et al. (2012) brought about “dying back” through viral overexpression of α-Syn whilst Melo et al. (2017) utilized an A53T transgenic model. Moreover, the PD-linked protein leucine rich repeat kinase-2 (LRRK2) seems to alter MT polymerization/depolymerization cycles, affecting mitochondrial trafficking (Gillardon, 2009; Godena et al., 2014). Impairment of mitochondrial transport is also induced by the parkinsonian toxin MPP+, which inhibits kinesin-1-mediated anterograde transport leading to an increase in dynein-dependent retrograde transport (Morfini et al., 2007). LRRK2 and PTEN-induced kinase 1 (PINK1) mutant drosophila also exhibit disturbed mitochondrial calcium homeostasis with functional involvement of Miro1 (Lee et al., 2018).
Role of Mitophagy in PD
Mitochondrial dynamics also includes mitochondrial clearance by mitophagy, a mitochondrion-specific autophagic process that drives dysfunctional mitochondria to degradation in autophagosomes (Rodolfo et al., 2018). Several mitophagy mechanisms have been described, which can be dependent on or independent of mitochondrial receptors (Martinez-Vicente, 2017), including B-cell lymphoma 2 nineteen kilodalton interacting protein 3 (BNIP3), Nix, Bcl-2-like protein 13 (Bcl2-L-13) and Fun14 domain-containing protein 1 (FUNDC1), which interact with microtubule-associated proteins 1A/1B light chain 3B (LC3), recruiting the autophagosomal machinery (Chu, 2018). Cardiolipin may also mediate mitophagy, driving mitochondrial degradation when this phospholipid (that has a LC3-binding motif) moves from the IMM to the OMM (Chu et al., 2013). Receptor independent mitophagy involves the priming of mitochondria after PINK1 translocation from the cytosol to the mitochondria. The loss of mitochondrial membrane potential, and inhibition of PINK1-degrading proteases, leads to PINK1 accumulation in mitochondria, where it recruits the E3 ubiquitin ligase Parkin that initiates mitophagy by ubiquitinating OMM proteins, such as Mfn1 and Mfn2, Miro1, translocase of outer mitochondrial membrane 20 (TOM20), and voltage-dependent anion channel (VDAC) (Meissner et al., 2015). Poly-ubiquitinated OMM proteins act as mediators that ultimately cause mitochondrial engulfment by autophagosomes (Yoshii and Mizushima, 2015).
Evidence for a role of mitophagy in PD includes the observation that α-syn overexpression decreases the level of LC3-positive vesicles in human neuroblastoma cells (Winslow et al., 2010). Interestingly, mutations in PINK1 and Parkin genes cause autosomal recessive forms of familial early onset PD (Valente et al., 2001; Mata et al., 2004; Kumar et al., 2017), implicating a role of mitophagy in the aetiopathogenesis of PD (Dawson and Dawson, 2010). Mitochondrial morphology aberrations were found in a PINK1-mutant Drosophila model, and overexpression of Parkin was shown to rescue the phenotype (Clark et al., 2006). The role of Parkin mutations in mitophagy impairment was confirmed using iPSC-derived dopaminergic neurons with mutations in the Parkin gene (Suzuki et al., 2017). Additionally, Parkin has been demonstrated to be highly insoluble in PD, compromising autophagic systems (Lonskaya et al., 2013). Increased pathological α-syn leads to increased cytosolic Ca2+ and Miro1 upregulation, however, the adaptor function of Miro1 between mitochondria and motor transport complexes is abrogated at high Ca2+ concentration (Saotome et al., 2008; MacAskill et al., 2009; Wang and Schwarz, 2009). In addition to transport, increased Miro1 protects mitochondria from mitophagy. Thus, in PD it is likely that Miro1 upregulation in combination with PINK1 reduction serves to delay degradation allowing for unregulated ROS generation (Shaltouki et al., 2018). Interestingly, an increase in Miro1-dependent anterograde transport of mitochondria was found in a PINK1-knockout model (Liu et al., 2012). It is perhaps possible that in the early stages of PD, whilst cytosolic Ca2+ is low, that the transport function of Miro1 is unaffected, then changing to act as a protector especially in later stages. Phosphorylation of Miro1 by PINK1 is required for Miro1 degradation, however, mutations in PD likely affect this function (Wang et al., 2011b; Shlevkov et al., 2016). In combination with the protective capabilities of Miro1, this may explain why there is a frequent accumulation of dysfunctional mitochondria in distal axonal areas in PD models whilst still ubiquitinated by Parkin. Recent work by Grassi et al. (2018) suggests the possibility of differing α-syn variants affecting different cellular systems, with a non-fibrillar phosphorylated α-syn species described to induce mitophagy. However, understanding of mitophagy in PD is relatively limited due to the lack of amenable in vivo experimental approaches.
Intercellular Transfer of Mitochondria and α-Synuclein
Genes move from progenitors to progeny, i.e., in a vertical manner, while horizontal gene transfer (HGT) is rare among eukaryotes (Keeling and Palmer, 2008; Davis et al., 2014; Davis and Xi, 2015). Recently, HGT has been reported for mitochondrial genes, via horizontal transfer of mitochondria between cells in vitro (Rustom et al., 2004; Spees et al., 2006; Rogers and Bhatacharya, 2013; Ahmad et al., 2014; Wang and Gerdes, 2015; Rustom, 2016; Sinha et al., 2016). Wang and Gerdes (2015) showed that organelles, including mitochondria, move between cells via so-called tunnelling nanotubes (TNTs), narrow inter-cellular bridges with actin as a structural protein and with tubulin fibers as “tracks” for movement of subcellular structures between cells (Rustom et al., 2004). Co-culture studies showed that transfer of mitochondria from mesenchymal stem cells (MSCs) into cancer cells with defects in mitochondrial DNA (mtDNA) resulted in recovery of mitochondrial respiration in cancer cells (Spees et al., 2006) and mitochondria, moving from healthy cells via TNTs, rescued cancer cells exposed to mitochondrial insults during early stages of apoptosis (Wang and Gerdes, 2015). Mice with experimental lung disease were grafted with allogenic MSCs with labeled mitochondria resulting in movement of the mitochondria to the diseased cells, recovering their respiration and alleviating the pathology (Islam et al., 2012). Inter-cellular transfer of mitochondria maintains balanced heteroplasmy of mtDNA in outbred individuals (Jayaprakash et al., 2015), pointing to mitochondrial HGT as a more frequent event than previously considered (Berridge et al., 2015, 2016).
In relation to disease, transfer of damaged mtDNA has been observed in various mouse models of engrafted tumor cells with other works such as that by Dong et al. (2018) revealing that damaged mtDNA is transported within the mitochondria (Tan et al., 2015). Such a process allows for the damaged mtDNA to affect the acceptor cell through the generation and leakage of ROS, especially so if mitophagy or systems regulating the prevention of damaged mitochondrial spread are faulty. Regarding PD, knowledge on mtDNA and mitochondrial spread in general is still unclear. mtDNA mutations are observed in neuronal cells in early stages of PD (Braak Stage 3 onward) due to damage attained via oxidative stress (Lin et al., 2012). This is not observed in late stage PD. However, this could potentially be due to the increased chances of neurons that previously hosted the damaged mtDNA of being destroyed, in addition to a lower population of neurons with damaged mtDNA prior to death. Various interactions of α-syn with mitochondria lead to the generation of ROS and thus increase risk of damaged mtDNA, including certain pathological α-syn species shown to bind with high affinity to the Tom20 mitochondrial outer membrane protein, thereby inhibiting mitochondrial protein uptake and promoting ROS generation (Di Maio et al., 2016; Grassi et al., 2018). Recent work by Wang et al. (2019) further reveals α-syn bound mitochondria only occurs with pathological α-syn aggregate species and not physiological monomers. This finding did not pertain only to PD but other synucleinopathies such as Dementia with Lewy Bodies and Multiple System Atrophy. In addition α-syn is also shown to inhibit Complex I directly, further compromising the energy production of the mitochondrion and increasing the generation of ROS (Reeve et al., 2015). No evidence has emerged to suggest that α-syn-affected mtDNA or mitochondria can spread to healthy cells, however, further investigation is required.
Mitochondrial transport is driven by motor complexes via binding through specific adaptor proteins, such as Miro1, suggesting that a potential mechanism of mitochondrial transfer between cells is mediated via the motor systems using TNTs (Ahmad et al., 2014; Sinha et al., 2016). This is based on experimental data (Ahmad et al., 2014) as well as analogy with the movement of mitochondria along axons in neuronal cells, where the kinesin and dynein motor systems include the motor protein plus two adaptor proteins Milton and Miro1, the latter with high affinity for mitochondria (Hase et al., 2009; Wang and Schwarz, 2009; MacAskill and Kittler, 2010; Kimura et al., 2012; Schiller et al., 2013). Indeed, mutations in the Miro1 gene, RHOT1, have recently been linked to PD cases, wherein the mutations led to fewer ER-mitochondria contact sites, calcium dyshomeostasis and exacerbation of calcium-dependent mitochondrial fragmentation and increased mitochondrial clearance (Grossmann et al., 2019). How the movement of mitochondria between cells is triggered and regulated is still not fully understood. Recent work has shown α-syn utilizing TNTs for spread. Thus, Dieriks et al. (2017) demonstrated the establishment of TNTs and transport of α-syn from neurons and pericytes in culture. Interestingly, Rostami et al. (2017) revealed TNT formation and α-syn transfer between astrocytic cells, transport of healthy mitochondria to rescue stressed mitochondria damaged by α-syn and transport of α-syn from α-syn affected astrocytes to healthy astrocytes. There were no reports of α-syn utilizing mitochondria for TNT-mediated migration, however, mitochondria may also represent an efficient carrier of α-syn between certain cell types. Furthermore, prion proteins, which have some similarities in mode of propagation to α-syn aggregates, have been established to hijack TNTs to mediate cell-to-cell transfer of the infectious protein (Gousset et al., 2009; Dorban et al., 2010; Prusiner et al., 2015; Steiner et al., 2018). Although there are many potential mechanisms of α-syn spread (Valdinocci et al., 2017), in view of findings that α-syn can migrate within TNTs bound to organelles such as lysosomal vesicles (Abounit et al., 2016) it is tempting to speculate that α-syn may transfer between neighboring cells bound to mitochondria that are translocated actively along TNTs (Figure 1).
Potential for Mitochondrially Targeted PD Therapeutics
The influences of pathological α-syn on mitochondrial dynamics in PD are potentially wide-ranging. Mitochondria-targeted drugs, such as Mito-Q and Mito-Apocynin showed therapeutic potential in experimental PD models whilst others such as Metformin are variable likely attributed to model utilized (Patil et al., 2014; Bayliss et al., 2016; Ismaiel et al., 2016; Lu et al., 2016; Langley et al., 2017; Xi et al., 2018). Although therapies that target calcium mobilization or oxidative stress tackle the effects of mitochondrial dysfunction, future innovative approaches could ameliorate mis-location of mitochondria or intercellular mitochondrial transfer. Further work is clearly needed to articulate the full significance of changed mitochondrial dynamics in PD etiology.
Author Contributions
All authors have contributed to the preparation of the manuscript, with DP taking responsibility for editing the final version of the manuscript.
Funding
DV and DP were funded by Griffith University. JK and JN were funded by the Czech Academy of Sciences. RS and TC-O were funded by the ERDF through COMPETE 2020/FCT under research grants PD/BD/128254/2016 (RS), POCI-01-0145-FEDER-029297 (TC-O), and UID/NEU/04539/2019.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abounit, S., Bousset, L., Loria, F., Zhu, S., de Chaumont, F., Pieri, L., et al. (2016). Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 35, 2120–2138. doi: 10.15252/embj.201593411
Ahmad, T., Mukherjee, S., Pattnaik, B., Kumar, M., Singh, S., Kumar, M., et al. (2014). Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 33, 994–1010. doi: 10.1002/embj.201386030
Barsoum, M. J., Yuan, H., Gerencser, A. A., Liot, G., Kushnareva, Y., Graber, S., et al. (2006). Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25, 3900–3911. doi: 10.1038/sj.emboj.7601253
Bartels, T., Choi, J., and Selkoe, D. (2011). α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Bayliss, J., Lemus, M., Santos, V., Deo, M., Davies, J., Kemp, B., et al. (2016). Metformin prevents nigrostriatal dopamine degeneration independent of AMPK activation in dopamine neurons. PLoS One 11:e0159381. doi: 10.1371/journal.pone.0159381
Berridge, M., Dong, L., and Neuzil, J. (2015). Mitochondrial DNA in tumor initiation, progression, and metastasis: role of horizontal mtDNA transfer. Cancer Res. 75, 3203–3208. doi: 10.1158/0008-5472.CAN-15-0859
Berridge, M., McConnell, M., Grasso, C., Bajzikova, M., Kovarova, J., and Neuzil, J. (2016). Horizontal transfer of mitochondria between mammalian cells: beyond co-culture approaches. Curr. Opin. Genet. Dev. 38, 75–82. doi: 10.1016/j.gde.2016.04.003
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M., and Sudhof, T. (2010). α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Burte, F., Carelli, V., Chinnery, P. F., and Yu-Wai-Man, P. (2015). Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 11, 11–24. doi: 10.1038/nrneurol.2014.228
Cai, Q., Gerwin, C., and Sheng, Z. H. (2005). Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol. 170, 959–969. doi: 10.1083/jcb.200506042
Celardo, I., Martins, L. M., and Gandhi, S. (2014). Unravelling mitochondrial pathways to Parkinson’s disease. Br. J. Pharmacol. 171, 1943–1957. doi: 10.1111/bph.12433
Chan, D. C. (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 46, 265–287. doi: 10.1146/annurev-genet-110410-132529
Chen, C., Turnbull, D. M., and Reeve, A. K. (2019). Mitochondrial dysfunction in Parkinson’s disease-cause or consequence? Biology 8:E38.
Chen, Y., and Sheng, Z. H. (2013). Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J. Cell Biol. 202, 351–364. doi: 10.1083/jcb.201302040
Chu, C. T. (2018). Multiple pathways for mitophagy: a neurodegenerative conundrum for Parkinson’ disease. Neurosci. Lett. 697, 66–71. doi: 10.1016/j.neulet.2018.04.004
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. doi: 10.1038/ncb2837
Chu, Y., Morfini, G., Langhamer, L., He, Y., Brady, S., and Kordower, J. (2012). Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 135, 2058–2073. doi: 10.1093/brain/aws133
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. doi: 10.1038/nature04779
Cole, N. B., Dieuliis, D., Leo, P., Mitchell, D. C., and Nussbaum, R. L. (2008). Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp. Cell Res. 314, 2076–2089. doi: 10.1016/j.yexcr.2008.03.012
Davis, C., Kim, K., Bushong, E., Mills, E., Boassa, D., Shih, T., et al. (2014). Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. U.S.A. 111, 9633–9638. doi: 10.1073/pnas.1404651111
Davis, C., and Xi, Z. (2015). Horizontal gene transfer in parasitic plants. Curr. Opin. Plant Biol. 26, 14–19. doi: 10.1016/j.pbi.2015.05.008
Dawson, T. M., and Dawson, V. L. (2010). The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 25(Suppl. 1), S32–S39. doi: 10.1002/mds.22798
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Di Maio, R., Barrett, P., Hoffman, E., Barrett, C., Zharikov, A., Borah, A., et al. (2016). α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Trans. Med. 8:342ra78. doi: 10.1126/scitranslmed.aaf3634
Dieriks, B., Park, T., Fourie, C., Faull, R., Dragunow, M., and Curtis, M. (2017). α-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci. Rep. 7:42984.
Dong, L., Kovarova, J., Bajzikova, M., Coelho, A., Boukalova, S., Rohlena, J., et al. (2018). Horizontal transfer of mitochondria and dihydroorotate dehydrogenase function in respiration recovery of mtDNA deficient cancer cells. Free Radic. Biol. Med. 120, S53–S54.
Dorban, G., Defaweux, V., Heinen, E., and Antoine, N. (2010). Spreading of prions from the immune to the peripheral nervous system: a potential implication of dendritic cells. Histochem. Cell Biol. 133, 493–504. doi: 10.1007/s00418-010-0687-9
Dorn, G. W. II, and Kitsis, R. N. (2015). The mitochondrial dynamism-mitophagy-cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circ. Res. 116, 167–182. doi: 10.1161/CIRCRESAHA.116.303554
El-Agnaf, O., Salem, S., Paleologou, K., Curran, M., Gibson, M., Court, J., et al. (2006). Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 20, 419–425. doi: 10.1096/fj.03-1449com
Fu, J., Yu, H., Chiu, S., Mirando, A., Maruyama, E., Cheng, J., et al. (2014). Disruption of SUMO-Specific Protease 2 induces mitochondria mediated neurodegeneration. PLoS Genet. 10:e1004579. doi: 10.1371/journal.pgen.1004579
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M., et al. (2002). α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Gillardon, F. (2009). Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability–a point of convergence in parkinsonian neurodegeneration? J. Neurochem. 110, 1514–1522. doi: 10.1111/j.1471-4159.2009.06235.x
Godena, V. K., Brookes-Hocking, N., Moller, A., Shaw, G., Oswald, M., Sancho, R. M., et al. (2014). Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 5:5245. doi: 10.1038/ncomms6245
Gousset, K., Schiff, E., Langevin, C., Marijanovic, Z., Caputo, A., Browman, D., et al. (2009). Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 11, 328–336. doi: 10.1038/ncb1841
Grassi, D., Howard, S., Zhou, M., Diaz-Perez, N., Urban, N., Guerrero-Given, D., et al. (2018). Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 115, E2634–E2643.
Grossmann, D., Berenguer-Escuder, C., Bellet, M. E., Scheibner, D., Bohler, J., and Massart, F. (2019). Mutations in RHOT1 disrupt ER-mitochondria contact sites interfering with calcium homeostasis and mitochondrial dynamics in Parkinson’s disease. Antioxid. Redox Signal. doi: 10.1089/ars.2018.7718 [Epub ahead of print].
Guo, C., Hildick, K. L., Luo, J., Dearden, L., Wilkinson, K. A., and Henley, J. M. (2013). SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. 32, 1514–1528. doi: 10.1038/emboj.2013.65
Guo, C., Wilkinson, K. A., Evans, A. J., Rubin, P. P., and Henley, J. M. (2017). SENP3-mediated deSUMOylation of Drp1 facilitates interaction with Mff to promote cell death. Sci. Rep. 7:43811. doi: 10.1038/srep43811
Hase, K., Kimura, S., Takatsu, H., Ohmae, M., Kawano, S., Kitamura, H., et al. (2009). M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 11, 1427–1432. doi: 10.1038/ncb1990
Henley, J. M., Carmichael, R. E., and Wilkinson, K. A. (2018). Extranuclear SUMOylation in Neurons. Trends Neurosci. 41, 198–210. doi: 10.1016/j.tins.2018.02.004
Hirokawa, N., Niwa, S., and Tanaka, Y. (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–638. doi: 10.1016/j.neuron.2010.09.039
Islam, M. N., Das, S. R., Emin, M. T., Wei, M., Sun, L., Westphalen, K., et al. (2012). Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 18, 759–765. doi: 10.1038/nm.2736
Ismaiel, A. A., Espinosa-Oliva, A. M., Santiago, M., García-Quintanilla, A., Oliva-Martín, M. J., Herrera, A. J., et al. (2016). Metformin, besides exhibiting strong in vivo anti-inflammatory properties, increases mptp-induced damage to the nigrostriatal dopaminergic system. Toxicol. Appl. Pharmacol. 298, 19–30. doi: 10.1016/j.taap.2016.03.004
Jayaprakash, A. D., Benson, E. K., Gone, S., Liang, R., Shim, J., Lambertini, L., et al. (2015). Stable heteroplasmy at the single-cell level is facilitated by intercellular exchange of mtDNA. Nucleic. Acids Res. 43, 2177–2187. doi: 10.1093/nar/gkv052
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., et al. (2010). Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1. Parkin and DJ-1. EMBO J. 29, 3571–3589. doi: 10.1038/emboj.2010.223
Keeling, P., and Palmer, J. (2008). Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 9, 605–618. doi: 10.1038/nrg2386
Kimura, S., Hase, K., and Ohno, H. (2012). Tunneling nanotubes: Emerging view of their molecular components and formation mechanisms. Exp. Cell Res. 318, 1699–1706. doi: 10.1016/j.yexcr.2012.05.013
Korobova, F., Ramabhadran, V., and Higgs, H. N. (2013). An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339, 464–467. doi: 10.1126/science.1228360
Kumar, A., Tamjar, J., Waddell, A., Woodroof, H., Raimi, O., Shaw, A., et al. (2017). Structure of PINK1 and mechanisms of Parkinson’ disease-associated mutations. eLife. 6: e29985
Langley, M., Ghosh, A., Charli, A., Sarkar, S., Ay, M., Luo, J., et al. (2017). Mito-apocynin prevents mitochondrial dysfunction, microglial activation, oxidative damage, and progressive neurodegeneration in mitopark transgenic mice. Antioxid. Redox Signal. 27, 1048–1066. doi: 10.1089/ars.2016.6905
Lee, K. S., Huh, S., Lee, S., Wu, Z., Kim, A. K., Kang, H. Y., et al. (2018). Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. U.S.A. 115, E8844–E8853. doi: 10.1073/pnas.1721136115
Lin, M., Cantuti-Castelvetri, I., Zheng, K., Jackson, K., Tan, Y., Arzberger, T., et al. (2012). Somatic mitochondrial DNA mutations in early parkinson and incidental lewy body disease. Ann. Neurol. 71, 850–854. doi: 10.1002/ana.23568
Lin, M. Y., and Sheng, Z. H. (2015). Regulation of mitochondrial transport in neurons. Exp. Cell Res. 334, 35–44. doi: 10.1016/j.yexcr.2015.01.004
Liu, S., Sawada, T., Lee, S., Yu, W., Silverio, G., Alapatt, P., et al. (2012). Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 8:e1002537. doi: 10.1371/journal.pgen.1002537
Lonskaya, I., Hebron, M., Algarzae, N., Desforges, N., and Moussa, C. (2013). Decreased parkin solubility is associated with impairment of autophagy in the nigrostriatum of sporadic Parkinson’s disease. Neuroscience 232, 90–105. doi: 10.1016/j.neuroscience.2012.12.018
Lu, M., Su, C., Qiao, C., Bian, Y., Ding, J., and Hu, G. (2016). Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of parkinson’s disease via autophagy and mitochondrial ROS clearance. Int. J. Neuropsychopharmacol. 19:yw047.
MacAskill, A., and Kittler, J. (2010). Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 20, 102–112. doi: 10.1016/j.tcb.2009.11.002
MacAskill, A., Rinholm, J., Twelvetrees, A., Arancibia-Carcamo, I., Muir, J., Fransson, A., et al. (2009). Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61, 541–555. doi: 10.1016/j.neuron.2009.01.030
Martinez-Vicente, M. (2017). Neuronal mitophagy in neurodegenerative diseases. Front. Mol. Neurosci. 10:64. doi: 10.3389/fnmol.2017.00064
Mata, I. F., Lockhart, P. J., and Farrer, M. J. (2004). Parkin genetics: one model for Parkinson’s disease. Hum. Mol. Genet. 13, R127–R133.
McCann, H., Stevens, C., Cartwright, H., and Halliday, G. (2014). α-Synucleinopathy phenotypes. Parkinsonism Relat. Disord. 20, S62–S67.
McLeary, F. A., Rcom-H’cheo-Gauthier, A. N., Goulding, M., Radford, R. A. W., Okita, Y., Faller, P., et al. (2019). Switching on Endogenous metal binding proteins in Parkinson’s Disease. Cells 8:E179. doi: 10.3390/cells8020179
Meissner, C., Lorenz, H., Hehn, B., and Lemberg, M. K. (2015). Intramembrane protease PARL defines a negative regulator of PINK1- and PARK2/Parkin-dependent mitophagy. Autophagy 11, 1484–1498. doi: 10.1080/15548627.2015.1063763
Melkov, A., and Abdu, U. (2018). Regulation of long-distance transport of mitochondria along microtubules. Cell. Mol. Life Sci. 75, 163–176. doi: 10.1007/s00018-017-2590-1
Melo, T. Q., Van Zomeren, K. C., Ferrari, M. F., Boddeke, H. W., and Copray, J. C. (2017). Impairment of mitochondria dynamics by human A53T alpha-synuclein and rescue by NAP (davunetide) in a cell model for Parkinson’s disease. Exp. Brain Res. 235, 731–742. doi: 10.1007/s00221-016-4836-9
Morfini, G., Pigino, G., Opalach, K., Serulle, Y., Moreira, J. E., Sugimori, M., et al. (2007). 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 104, 2442–2447. doi: 10.1073/pnas.0611231104
Nemani, N., Carvalho, E., Tomar, D., Dong, Z., Ketschek, A., and Breves, S. L. (2018). MIRO-1 determines mitochondrial shape transition upon GPCR activation and Ca2+ Stress. Cell Rep. 24, 1005–1019. doi: 10.1016/j.celrep.2018.03.098
O’Donnell, K. C., Lulla, A., Stahl, M. C., Wheat, N. D., Bronstein, J. M., and Sagasti, A. (2014). Axon degeneration and PGC-1alpha-mediated protection in a zebrafish model of alpha-synuclein toxicity. Dis. Mod. Mech. 7, 571–582. doi: 10.1242/dmm.013185
Pagliuso, A., Cossart, P., and Stavru, F. (2018). The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 75, 355–374. doi: 10.1007/s00018-017-2603-0
Patil, S., Jain, P., Ghumatkar, P., Tambe, R., and Sathaye, S. (2014). Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 277, 747–754. doi: 10.1016/j.neuroscience.2014.07.046
Perfeito, R., Cunha-Oliveira, T., and Rego, A. C. (2013). Reprint of: revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease-resemblance to the effect of amphetamine drugs of abuse. Free Radic. Biol. Med. 62, 186–201. doi: 10.1016/j.freeradbiomed.2013.05.042
Pozo Devoto, V. M., and Falzone, T. L. (2017). Mitochondrial dynamics in Parkinson’s disease: a role for alpha-synuclein? Dis. Mod. Mech. 10, 1075–1087. doi: 10.1242/dmm.026294
Prots, I., Grosch, J., Brazdis, R., Simmnacher, K., Veber, V., Havlicek, S., et al. (2018). α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. U.S.A. 115, 7813–7818. doi: 10.1073/pnas.1713129115
Prots, I., Veber, V., Brey, S., Campioni, S., Buder, K., Riek, R., et al. (2013). Alpha-synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 288, 21742–21754. doi: 10.1074/jbc.M113.451815
Prusiner, S., Woerman, A., Mordes, D., Watts, J., Rampersaud, R., Berry, D., et al. (2015). Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 112, E5308–E5317.
Rcom-H’cheo-Gauthier, A. N., Meedeniya, A. C., and Pountney, D. L. (2017). Calcipotriol inhibits α-synuclein aggregation in SH-SY5Y neuroblastoma cells by a Calbindin-D28k-dependent mechanism. J. Neurochem. 141, 263–274. doi: 10.1111/jnc.13971
Rcom-H’cheo-Gauthier, A. N., Osborne, S., Meedeniya, A., and Pountney, D. (2016). Calcium: alpha-synuclein interactions in alpha-synucleinopathies. Front. Neurosci. 10:570. doi: 10.3389/fnins.2016.00570
Reeve, A., Ludtmann, M., Angelova, P., Simcox, E., Horrocks, M., Klenerman, D., et al. (2015). Aggregated α-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis. 6, e1820–e1820. doi: 10.1038/cddis.2015.166
Reynolds, A., Glanzer, J., Kadiu, I., Ricardo-Dukelow, M., Chaudhuri, A., Ciborowski, P., et al. (2008). Nitrated alpha-synuclein-activated microglial profiling for Parkinson’s disease. J. Neurochem. 104, 1504–1525. doi: 10.1111/j.1471-4159.2007.05087.x
Rodolfo, C., Campello, S., and Cecconi, F. (2018). Mitophagy in neurodegenerative diseases. Neurochem. Int. 117, 156–166. doi: 10.1016/j.neuint.2017.08.004
Rogers, R., and Bhatacharya, J. (2013). When cells become organelle donors. Physiology 28, 414–422. doi: 10.1152/physiol.00032.2013
Rostami, J., Holmqvist, S., Lindström, V., Sigvardson, J., Westermark, G., Ingelsson, M., et al. (2017). Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J. Neurosci. 37, 11835–11853. doi: 10.1523/JNEUROSCI.0983-17.2017
Rustom, A. (2016). The missing link: does tunnelling nanotube-based supercellularity provide a new understanding of chronic and lifestyle diseases? Open Biol. 6:160057. doi: 10.1098/rsob.160057
Rustom, A., Saffrich, R., Markovic, I., Walther, P., and Gerdes, H. H. (2004). Nanotubular highways for intercellular organelle transport. Science 303, 1007–1010. doi: 10.1126/science.1093133
Saotome, M., Safiulina, D., Szabadkai, G., Das, S., Fransson, A., Aspenstrom, P., et al. (2008). Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. U.S.A. 105, 20728–20733. doi: 10.1073/pnas.0808953105
Schiller, C., Diakopoulos, K., Rohwedder, I., Kremmer, E., von Toerne, C., Ueffing, M., et al. (2013). LST1 promotes the assembly of a molecular machinery responsible for tunneling nanotube formation. J. Cell Sci. 126, 767–777. doi: 10.1242/jcs.114033
Shaltouki, A., Hsieh, C., Kim, M., and Wang, X. (2018). Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 136, 607–620. doi: 10.1007/s00401-018-1873-4
Sheng, Z. H. (2017). The Interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol. 27, 403–416. doi: 10.1016/j.tcb.2017.01.005
Shirihai, O. S., Song, M., and Dorn, G. W. II (2015). How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 116, 1835–1849. doi: 10.1161/CIRCRESAHA.116.306374
Shlevkov, E., Kramer, T., Schapansky, J., LaVoie, M., and Schwarz, T. (2016). Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. U.S.A. 113, E6097–E6106.
Sinha, P., Islam, M., Bhattacharya, S., and Bhattacharya, J. (2016). Intercellular mitochondrial transfer: bioenergetic crosstalk between cells. Curr. Opin. Genet. Dev. 38, 97–101. doi: 10.1016/j.gde.2016.05.002
Spees, J., Olson, S., Whitney, M., and Prockop, D. (2006). Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. U.S.A. 103, 1283–1288. doi: 10.1073/pnas.0510511103
Steiner, J., Quansah, E., and Brundin, P. (2018). The concept of alpha-synuclein as a prion-like protein: ten years after. Cell Tissue Res. 373, 161–173. doi: 10.1007/s00441-018-2814-1
Sulzer, D., and Edwards, R. H. (2019). The physiological role of α-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 150, 475–486. doi: 10.1111/jnc.14810
Suzuki, S., Akamatsu, W., Kisa, F., Sone, T., Ishikawa, K. I., Kuzumaki, N., et al. (2017). Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem. Biophys. Res. Commun. 483, 88–93. doi: 10.1016/j.bbrc.2016.12.188
Tan, A., Baty, J., Dong, L., Bezawork-Geleta, A., Endaya, B., Goodwin, J., et al. (2015). Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 21, 81–94. doi: 10.1016/j.cmet.2014.12.003
Tas, R. P., and Kapitein, L. C. (2018). Exploring cytoskeletal diversity in neurons. Science 361, 231–232. doi: 10.1126/science.aat5992
Thakur, P., Chiu, W. H., Roeper, J., and Goldberg, J. A. (2019). α-Synuclein 2.0 — Moving towards Cell Type Specific Pathophysiology. Neuroscience 412, 248–256. doi: 10.1016/j.neuroscience.2019.06.005
Theillet, F., Binolfi, A., Bekei, B., Martorana, A., Rose, H., Stuiver, M., et al. (2016). Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50. doi: 10.1038/nature16531
Thomas, K. J., Mccoy, M. K., Blackinton, J., Beilina, A., Van Der Brug, M., Sandebring, A., et al. (2011). DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 20, 40–50. doi: 10.1093/hmg/ddq430
Tilokani, L., Nagashima, S., Paupe, V., and Prudent, J. (2018). Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 62, 341–360. doi: 10.1042/EBC20170104
Valdinocci, D., Radford, R., Siow, S., Chung, R., and Pountney, D. (2017). Potential modes of intercellular α-synuclein transmission. Int. J. Mol. Sci. 18, E469. doi: 10.3390/ijms18020469
Valente, E. M., Bentivoglio, A. R., Dixon, P. H., Ferraris, A., Ialongo, T., Frontali, M., et al. (2001). Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet. 68, 895–900. doi: 10.1086/319522
Vijayakumaran, S., and Pountney, D. L. (2018). SUMOylation, aging and autophagy in neurodegeneration. Neurotoxicology. 66, 53–57. doi: 10.1016/j.neuro.2018.02.015
Vijayakumaran, S., Wong, M. B., Antony, H., and Pountney, D. L. (2015). Direct and/or indirect roles for SUMO in modulating alpha-synuclein toxicity. Biomolecules. 5, 1697–1716. doi: 10.3390/biom5031697
Wang, X., Becker, K., Levine, N., Zhang, M., Lieberman, A., Moore, D., et al. (2019). Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 7:41. doi: 10.1186/s40478-019-0696-4
Wang, X., and Gerdes, H. H. (2015). Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death. Differ. 22, 1181–1191. doi: 10.1038/cdd.2014.211
Wang, X., and Schwarz, T. (2009). The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. doi: 10.1016/j.cell.2008.11.046
Wang, X., Su, B., Liu, W., He, X., Gao, Y., Castellani, R. J., et al. (2011a). DLP1-dependent mitochondrial fragmentation mediates 1-methyl-4-phenylpyridinium toxicity in neurons: implications for Parkinson’s disease. Aging Cell 10, 807–823. doi: 10.1111/j.1474-9726.2011.00721.x
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y., Selkoe, D., et al. (2011b). PINK1 and parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. doi: 10.1016/j.cell.2011.10.018
Winslow, A. R., Chen, C. W., Corrochano, S., Acevedo-Arozena, A., Gordon, D. E., Peden, A. A., et al. (2010). Alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol. 190, 1023–1037. doi: 10.1083/jcb.201003122
Xi, Y., Feng, D., Tao, K., Wang, R., Shi, Y., Qin, H., et al. (2018). MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1α. Biochim. Biophys. Acta BBA Mol. Basis Dis. 1864, 2859–2870. doi: 10.1016/j.bbadis.2018.05.018
Xie, W., and Chung, K. K. (2012). Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson’s disease. J. Neurochem. 122, 404–414. doi: 10.1111/j.1471-4159.2012.07769.x
Yoshii, S. R., and Mizushima, N. (2015). Autophagy machinery in the context of mammalian mitophagy. Biochim. Biophys. Acta 853, 2797–2801. doi: 10.1016/j.bbamcr.2015.01.013
Zala, D., Hinckelmann, M. V., Yu, H., Lyra Da Cunha, M. M., Liot, G., Cordelieres, F. P., et al. (2013). Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 152, 479–491. doi: 10.1016/j.cell.2012.12.029
Keywords: alpha-synuclein, tunneling nanotube, Parkinson’s, mitophagy, mitochondria
Citation: Valdinocci D, Simões RF, Kovarova J, Cunha-Oliveira T, Neuzil J and Pountney DL (2019) Intracellular and Intercellular Mitochondrial Dynamics in Parkinson’s Disease. Front. Neurosci. 13:930. doi: 10.3389/fnins.2019.00930
Received: 01 March 2019; Accepted: 19 August 2019;
Published: 18 September 2019.
Edited by:
Sandeep Kumar Barodia, The University of Alabama at Birmingham, United StatesReviewed by:
Karina Ckless, State University of New York Plattsburgh, United StatesDilshan Shanaka Harischandra, Covance, United States
Tatiana Rosado Rosenstock, Faculty of Medical Sciences of Santa Casa de São Paulo, Brazil
Copyright © 2019 Valdinocci, Simões, Kovarova, Cunha-Oliveira, Neuzil and Pountney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dean L. Pountney, ZC5wb3VudG5leUBncmlmZml0aC5lZHUuYXU=