 Soumia Benbrika
Soumia Benbrika Béatrice Desgranges
Béatrice Desgranges Francis Eustache
Francis Eustache Fausto Viader
Fausto Viader- Neuropsychology and Imaging of Human Memory, Normandy University-PSL Research University-EPHE-INSERM U1077, Caen University Hospital, Caen, France
It is now well recognized that, in addition to motor impairment, amyotrophic lateral sclerosis (ALS) may cause extra-motor clinical signs and symptoms. These can include the alteration of certain cognitive functions, impaired social cognition, and changes in the perception and processing of emotions. Where these extra-motor manifestations occur in ALS, they usually do so from disease onset. In about 10% of cases, the cognitive and behavioral changes meet the diagnostic criteria for frontotemporal dementia. The timecourse of behavioral and cognitive involvement in ALS is unclear. Whereas longitudinal studies have failed to show cognitive decline over time, some cross-sectional studies have demonstrated poorer cognitive performances in the advanced stages of the disease. Neuroimaging studies show that in ALS, extra-motor signs and symptoms are associated with specific brain lesions, but little is known about how they change over time. Finally, patients with ALS appear less depressed than might be expected, given the prognosis. Moreover, many patients achieve satisfactory psychosocial adjustment throughout the course of the disease, regardless of their degree of motor disability. There are scant longitudinal data on extra-motor impairment in ALS, and to our knowledge, no systematic review on this subject has yet been published. Even so, a better understanding of patients’ clinical trajectory is essential if they are to be provided with tailored care and given the best possible support. We therefore undertook to review the evidence for extra-motor changes and their time course in ALS, in both the cognitive, emotional and psychological domains, with a view to identifying mechanisms that may help these patients cope with their disease.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of upper and lower motor neurons. In all sporadic and most genetic cases, proteinaceous aggregates of TAR-DNA binding protein 43 (TDP43) are found in these upper and lower motor neurons, as well as in certain glial cells (Saberi et al., 2015). The disease worsens relentlessly, and death occurs after a median duration of 3 years after onset and 2 years after diagnosis (Couratier et al., 2016).
All patients experience extensive and progressive muscle paralysis that results in severe functional disability, but as many as 50% may also have extra-motor signs and symptoms, including cognitive impairment (Abrahams et al., 2000; Murphy J. M. et al., 2007; Murphy J. et al., 2007) mainly affecting executive functions, emotion processing and social cognition, and behavior.
Despite being given such a gloomy prognosis, not all patients display symptoms of depression. Indeed, depression rates remain low, considering the severity of the disease (Rabkin et al., 2016), and patients’ quality of life (QoL) is not as impaired as might be expected (Roach et al., 2009; Lulé et al., 2012; Jakobsson Larsson et al., 2017). This relatively good psychosocial adjustment raises important questions about the coping strategies used by patients.
While we now have substantial knowledge about extra-motor signs of ALS at baseline, data on their clinical course remain scarce. The results of cross-sectional studies attempting to correlate disease duration and/or physical symptom severity with cognitive/psychological status suggest that, unlike motor impairment, cognitive performances are not always significantly worse in the late stages of the disease than they were in the early stages (Ringholz et al., 2005). These observations suggest that the mechanisms of psychological involvement do not simply come down to degenerative changes in the neurons of the brain. Furthermore, the fact that some studies have described patients as retaining a relatively satisfactory level of wellbeing, despite the continuous worsening of their physical condition, may seem a little surprising and deserves further consideration.
We therefore set out to review the cognitive, emotional and psychological impairments that can emerge at the beginning of the disease, and then to address the question of how they change throughout the course of the disease. We focused on ALS cases that do not meet the clinical criteria for dementia, as ALS-dementia syndromes raise different issues, and may not be as similar to ALS without dementia as is usually claimed (Lulé et al., 2018). Longitudinal studies are the most appropriate way of assessing how these impairments change, but they have several drawbacks. First, the follow-up periods are often short, and a large proportion of patients are liable to drop out, owing to the rapid progression of ALS, making it difficult to detect significant changes in extra-motor signs and symptoms over time. Second, it is difficult to find a suitable control group that accurately matches the patient sample. Third and last, the tools used to assess cognitive functioning have to be carefully chosen in order to be both adequate and usable at all stages of the disease-even the most advanced-, in order to accommodate the motor impairments that invariably interfere with testing procedures.
To fulfill the objectives of our work, we conducted a bibliographical search on PUBMED prior to the 1st October 2018 with the following keywords: Cognition, emotion, psychological adjustment, neuroimaging, longitudinal, AND ALS. We retained the articles that met our keywords, and that were written in English or in French. 190 articles were finally included in our study.
Cognitive, Emotional, and Behavioral Changes in Als
Cognition in ALS (Table 1)
Risk Factors for Developing Cognitive Deficits
Female sex, older age at onset, and low education level seem to increase the risk of cognitive impairment (Irwin et al., 2007; Montuschi et al., 2015; Flaherty et al., 2017). The presence of a C9orf72 gene mutation is associated with more severe cognitive deficits, even in non-demented patients (Byrne et al., 2012; Montuschi et al., 2015). There are still conflicting data about the potential positive correlation between bulbar symptoms and cognitive decline, with some studies supporting this correlation (Giordana et al., 2011; Gordon et al., 2011; Strutt et al., 2012) while others don’t (Sterling et al., 2010; Zalonis et al., 2012).
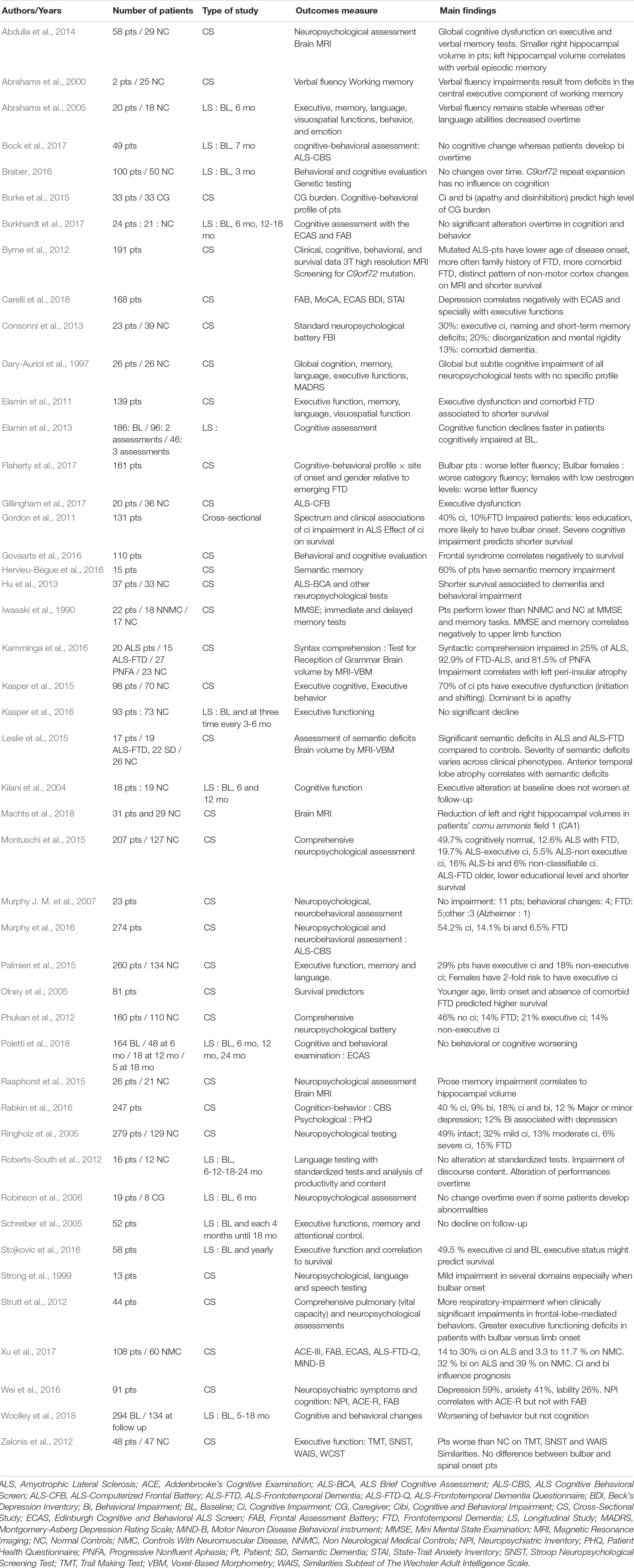
Table 1. Cognition in ALS.
The presence of depressive symptoms also seems to exacerbate the executive deficit in patients, with a negative correlation between scores on depression scales and those on cognitive tests (Wei et al., 2016; Carelli et al., 2018). These findings are not surprising, given the negative impact that depression is known to have on cognitive functions, in terms of attention and memory (Bortolato et al., 2016).
Baseline
Frequency and profile
Cognitive impairment occurs in 30–50% of patients with ALS, depending on the study and the neuropsychological tools used to assess cognitive functions (Giordana et al., 2011; Zago et al., 2011; Byrne et al., 2012; Kasper et al., 2015; Beeldman et al., 2016; Murphy et al., 2016). The cognitive deficit profile includes impairment of executive functions, verbal fluency, language, social cognition and verbal memory (Phukan et al., 2012; Beeldman et al., 2016). In 6–14% of patients, the cognitive impairment meets the criteria for a behavioral variant of frontotemporal dementia (FTD) (Consonni et al., 2013; Montuschi et al., 2015; Murphy et al., 2016). In the study by Ringholz et al. (2005), 51% of patients were cognitively impaired, compared with 5% of controls, and 14% met the diagnostic criteria for FTD. A cluster analysis indicated four patient subgroups: 49% with intact cognition, 32% with mild cognitive impairment, 13% with moderate impairment, and 6% with severe impairment.
Executive alterations affect verbal fluency, attention monitoring, switching, working memory, cognitive flexibility and mental control, and reasoning and coordinating rules (Phukan et al., 2012; Beeldman et al., 2016). Other executive functions are also impaired, such as initiation and shifting (Kasper et al., 2015). A study by Palmieri et al. (2015) found that female patients with ALS were twice as likely as males to have dysexecutive dysfunction-an intriguing finding that is not clearly explained to date.
Cognitive alterations observed in ALS also include language impairments (Iwasaki et al., 1990; Raaphorst et al., 2010; Giordana et al., 2011). According to Woolley and Rush (2017), language may be altered in 30–40% of patients without dementia, regardless of executive dysfunction, dysarthria or respiratory failure. Linguistic impairments may include deficits in syntactic processing, verb naming and action verb processing, semantic and verbal paraphasias, and syntactic comprehension deficits. Kamminga et al. (2016) found that syntactic comprehension was defective in 25% of patients with ALS without dementia. Leslie et al. (2015) observed semantic deficits in 35% of not demented ALS patients, and Hervieu-Bègue et al. (2016) in as many as 60% of such patients. Woolley and Rush (2017) suggested that language alterations observed in ALS reflect the fact that ALS and nonfluent/agrammatic primary progressive aphasia lie on a pathogenesis continuum.
Memory may also be altered to some extent in ALS (Dary-Auriol et al., 1997; Abrahams et al., 2000; Neary et al., 2000; Ringholz et al., 2005), but the nature of this impairment is subject to debate. It has been suggested that defective memory is mainly the consequence of executive dysfunction, especially since memory deficits very rarely occur in isolation in ALS (Strong, 2017). There is, however, some evidence in favor of hippocampal involvement. Abdulla et al. (2014) found reduced hippocampal volume in a series of patients with ALS relative to controls, and Machts et al. (2018) showed that this reduction mostly affects the anterior part, including the CA1 field-a critical structure for episodic memory. Furthermore, Raaphorst et al. (2015) found that immediate and delayed story recall scores were below normal in 23% of patients with ALS, and these performances were correlated with hippocampal gray-matter volume. Further studies are needed to try to determine the real status of memory in ALS.
The Strong (2017) revised diagnostic criteria of ALS-FTSD classify ALS into several categories according to the type and severity of neuropsychological impairments: pure ALS (no impairment), ALSci (cognitive impairment), ALSbi (behavioral impairment), ALScibi (cognitive and behavioral impairment), all three without dementia, and ALS-FTD. According to those criteria, the diagnosis of ALS-ci requires either executive dysfunction or language impairment or both, and the diagnosis of ALS-bi requires either apathy or two of the Rascovsky criteria for FTD.
Over Time
Most longitudinal studies of cognitive changes in patients with ALS have a follow-up period of 6 months. A number of authors have reported an absence of decline in cognitive functions during the course of the disease (Kilani et al., 2004; Schreiber et al., 2005; Braber, 2016; Kasper et al., 2016; Burkhardt et al., 2017; Poletti et al., 2018; Woolley et al., 2018). Although patients may initially have lower performances than controls, their deficits remain stable over time. It has been argued, however, that unlike normal controls, patients with ALS do not display a practice effect in repeated assessments with the Edinburgh Cognitive and Behavioural ALS Screen (ECAS) battery, which has been interpreted as evidence of a pre-symptomatic cognitive decline (Burkhardt et al., 2017). Abrahams et al. (2005) found that while most cognitive scores remained stable over time (including written and spoken verbal fluency) patients with ALS performed a single word retrieval test increasingly slowly whereas controls performed it faster. Furthermore, the caregivers of patients report increasing cognitive dysfunction in daily life over time, unlike controls’ partners. Robinson et al. (2006) found that cognitive changes occurred in 36% of patients with ALS over a 6-month period. Cognitive status seems to have a heterogeneous outcome in ALS. Behavioral symptoms may appear in patients with stable cognitive performances (Bock et al., 2017). The presence of even mild cognitive or behavioral impairment (as defined by the 2017 Strong criteria) at baseline seems to be a significant risk factor for the later appearance of a full-blown frontotemporal syndrome (Elamin et al., 2013). The choice of the neuropsychological tests used to assess cognitive status is of primary importance, as Roberts-South et al. (2012) suggested in their longitudinal study of discourse changes in a group of patients with ALS compared with healthy participants over 24 months. Subtle cognitive language deficits affecting discourse (content rather than productivity) were found to emerge early in ALS and worsen as the disease progressed. The authors concluded that language deficits are more thoroughly detected by the discourse analysis method than by standard language tasks. Gillingham et al. (2017) assessed patients with ALS at baseline and 9 months later using both standardized non-specific cognitive tests and the ALS-Computerized Frontal Battery (ALS-CFB), which was specifically designed for patients with ALS. While the basic cognitive tests failed to reveal any change over time, the ALS-CFB showed a significant decrease in cognitive performances in patients compared with controls (e.g., for verbal fluency). Finally, patients with bulbar onset seem to exhibit a progressive decline in cognitive functions (Strong et al., 1999).
It is therefore difficult to give a simple answer to the question “do cognitive functions decline over time in ALS?” owing to the heterogeneity of the patients and the difficulty of differentiating genuine cognitive deficits from the consequences of the steadily worsening motor impairment. A tentative, preliminary answer could be that when ALS is associated with a cognitive impairment at baseline, this impairment is likely to progress, and when dementia is present at diagnosis, decline is faster (Woolley and Katz, 2008). Normal cognition at baseline was associated with tendency to remain cognitively intact overtime (Elamin et al., 2013).
Consequences for Patients and Caregivers
Cognitive impairment in ALS is associated with a more rapid progression of the disease and a poorer prognosis, with reduced survival (Elamin et al., 2011; Giordana et al., 2011; Gordon et al., 2011; Hu et al., 2013; Xu et al., 2017). Stojkovic et al. (2016) found that the death risk was increased threefold by the presence of executive dysfunction in ALS. Poorer survival could be explained by patients’ difficulty weighing up the benefits of non-invasive ventilation (Govaarts et al., 2016), or their reduced compliance in the use of medical devices (Olney et al., 2005). The correlation between the severity of bulbar symptoms and the cognitive deficit may also influence survival figures (Olney et al., 2005). Elamin et al. (2013) evaluated the clinical impact of cognitive deficit in patients with ALS in a longitudinal study. Executive impairment at the initial consultation was associated with significantly higher rates of attrition due to disability or death, and faster rates of motor functional decline, particularly bulbar function. QoL in patients has been found to be worse in the case of cognitive impairment (Hu et al., 2013). Caregivers are the pillars of patient care. They may be the spouse, children, brothers or sisters. It is cognitive and behavioral impairment, rather than the patients’ physical disability, that increases caregivers’ burden and anxiety (Rabkin et al., 2009; Burke et al., 2015). This underscores the importance of screening patients with ALS for cognitive dysfunction, in order to predict disease progression and provide more adequate care.
Neuroimaging Correlates of Cognitive Deficits
Anatomical brain changes are more pronounced in patients with ALS who exhibit cognitive impairment than in those who do not. Gray matter volume in the frontal and temporal lobes is reduced when cognitive impairment is present (Abe et al., 1997; Giordana et al., 2011; Mioshi et al., 2013), as is that of the cerebellar cortex and basal ganglia (Christidi et al., 2018). Reduced cortical thickness is observed in the bilateral precentral gyrus, insular and cingulate cortices, and frontotemporal regions in the case of cognitive deficit (Schuster et al., 2014a; Agosta et al., 2016). Left peri-insular atrophy was found to correlate with scores on a syntactic comprehension task (Kamminga et al., 2016). There are greater white-matter changes in the corticospinal and corpus callosum tracts when cognitive functions are altered (Sarro et al., 2011), and these extend to extra-motor tracts, particularly within the frontal lobes and associative areas including the cingulum and the inferior longitudinal, inferior fronto-occipital, and uncinate fasciculi (Sarro et al., 2011; Kasper et al., 2014; Christidi et al., 2018). Verbal learning and memory test scores are correlated with white-matter values in the fornix (Sarro et al., 2011). Cerebral regional metabolism has been repeatedly studied with fluorodeoxyglucose positron emission tomography (18FDG-PET) in patients both with and without cognitive dysfunction. One of the most recent studies shows that cognitively impaired (but not demented) patients with ALS have relative hypometabolism in the right cingulate and frontal cortex, and bilaterally in the prefrontal cortex, compared with patients with no cognitive impairment (Canosa et al., 2016). These patients also have relative hypermetabolism in parts of the midbrain and corticospinal tracts (Canosa et al., 2016). In a combined MRI and PET study in patients with ALS, Buhour et al. (2017) found gray-matter atrophy, predominantly in the temporal poles, and hypometabolism in the left superior medial cortex. Hypermetabolism was also found in parts of the temporal lobes and the cerebellum. A series of negative correlations between cognitive performance and regional cerebral metabolism in functionally relevant areas suggest that hypermetabolism is more likely to reflect deleterious processes such as neuro-inflammation rather than compensatory neuronal activity.
Longitudinal studies of anatomical cerebral changes in ALS with cognitive modifications remain rare, for the same reasons as those dealing with purely clinical aspects of the disease. Floeter et al. (2016) assessed brain volume changes in patients who were either sporadic or carriers of the genetic mutation C9orf72 (C9+), which is often associated with FTD or cognitive impairment. These authors found that over a 6-month period, ventricular volume increased in C9+ versus sporadic cases, suggesting that subcortical involvement influences cognitive performances in this particular group of patients. Other microstructural changes over time in ALS have been documented. A significant decline in the cortical thickness of frontal, temporal and parietal regions is observed over time, whereas the reduced cortical thickness of the precentral gyrus at the beginning of the disease remains stable (Verstraete et al., 2012; Schuster et al., 2014b). Menke et al. (2018) assessed brain changes in patients with ALS over 2 years and reported widespread changes in both white and gray matter in the cingulate gyrus, thalami, caudate nuclei, pallidum, hippocampi and parahippocampal gyri, and insula. These results indicate that over time, cerebral changes extend into extra-motor areas, but it remains difficult to draw a link between these changes and clinical cognitive implications.
Changes in Emotion Perception and Social Cognition (Table 2)
Baseline
Perception of emotions
A number of studies have shown that emotion perception is impaired in ALS (Lulé et al., 2005; Zimmerman et al., 2007; Palmieri et al., 2010; Girardi et al., 2011), with patients exhibiting deficits in emotion recognition (facial or prosodic) and emotional valence attribution, and decreased excitability when emotional material is presented. By contrast, Cavallo et al. (2011) and Papps et al. (2005) found no deterioration in either facial emotional recognition or judgments of emotional valence. Clinical features such as type of onset and disease severity may explain the heterogeneity of patients’ emotional deficit profiles, as suggested by Sedda (2014). Oh et al. (2016) confirmed the presence of facial emotion recognition deficits in ALS. Bora (2017) carried out a meta-analysis of 15 studies of emotion recognition in ALS and concluded that ALS is associated with significant impairments in facial emotion recognition, especially for disgust and surprise.
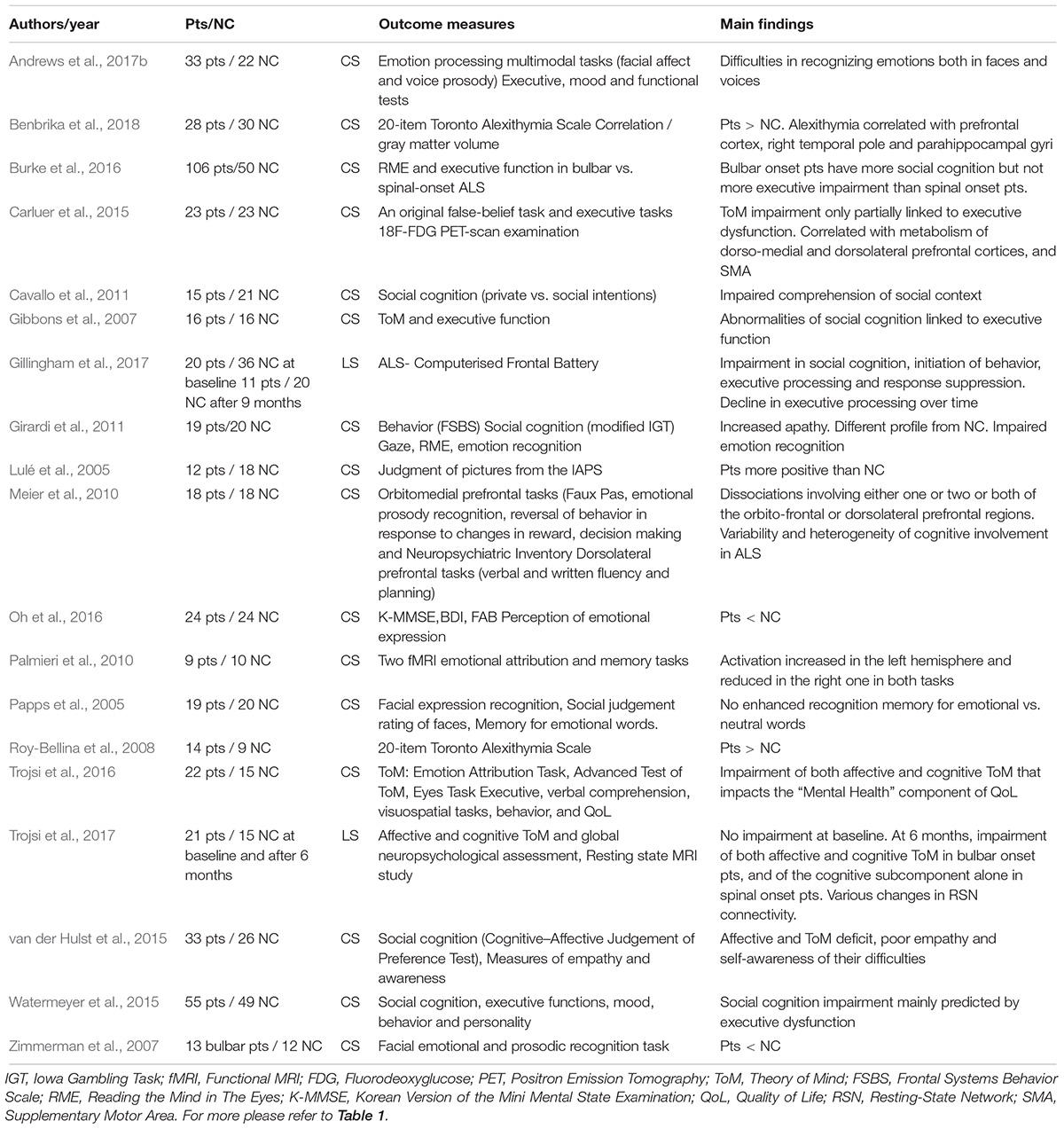
Table 2. Social cognition and emotion perception in ALS.
Patients’ processing of their own emotions (alexithymia) also seems to be altered, although there have been very few studies so far in this area. Roy-Bellina et al. (2008) reported that patients with ALS are more often alexithymic than controls. Benbrika et al. (2018) assessed a group of patients with ALS and a matched control group on the three dimensions of alexithymia: Difficulty Identifying Feelings, Difficulty Describing Feelings, and Externally-Oriented Thinking. Patients were more often alexithymic than controls and had a higher level of alexithymia, especially on the Difficulty Identifying Feelings dimension, suggesting that they have difficulty with the first stage of own emotion processing, namely recognizing one’s own emotions.
Social cognition
Social cognition is a set of cognitive processes used to encode, decode, store, retrieve and use information about people in social relationships. It has several dimensions, such as theory of mind (ToM), empathy, and moral reasoning. ToM refers to the ability to infer the mental and emotional states (i.e., beliefs, preferences and intentions) of oneself and others, and contributes to the understanding of other people’s behavior. It can be divided into cognitive and affective ToM. Studies of ToM abilities in ALS have sometimes yielded conflicting results, owing to the heterogeneity of the tasks used and the patients’ cognitive status, or the presence of depressive symptoms.
Early studies in this domain attested to a deficit in ToM, showing that patients with ALS perform more poorly than healthy individuals on the faux pas task (task assessing cognitive ToM through stories featuring appropriate and inappropriate social behavior), story comprehension task (strip stories presented to participants who have to choose the most appropriate picture) and decision making task (task evaluating decision making abilities under ambiguity in a card game) (Gibbons et al., 2007; Meier et al., 2010; Cavallo et al., 2011; Girardi et al., 2011).
More recently, van der Hulst et al. (2015) investigated whether the ToM deficit described in ALS could be further delineated as one of either affective or cognitive ToM, and explored the relationship between this social cognition deficit and the behavioral manifestations of empathy and self-awareness. Patients were evaluated on a neuropsychological battery that included a cognitive–affective judgment of preference test, a questionnaire to evaluate their self-awareness, and the Neuropsychiatric Inventory–Questionnaire (NPI-Q) to pick up any behavioral changes. The authors found deficits in both cognitive and affective ToM (affective + cognitive ToM deficit in 36% of patients, affective ToM deficit in 12%, and cognitive ToM deficit in 3%). Patients with a ToM deficit were more likely to display behavioral changes such as apathy, lack of empathy, and low self-awareness. When Carluer et al. (2015) compared 23 non-demented patients with controls matched for age, sex and educational level on cognitive ToM, by administering a false-belief task, they found a cognitive ToM deficit in patients with impaired executive functions. Trojsi et al. (2016) assessed ToM in patients with ALS using a comprehensive battery of specific tasks: the Advanced Task of ToM (ATM), in which participants listen to stories and then have to explain the protagonists’ actions; the Emotional Attribution Task (EAT), in which participants have to identify the emotions experienced by the protagonists in a story; and the Reading the Mind in the Eyes task (Eyes test or RME). Patients also underwent a cognitive battery and a QoL assessment. Patients scored lower than controls on the EAT but not on the ATM, and EAT and RME correlated positively with the education, prose memory and mental health items of the QoL questionnaire. Patients were not very cognitively impaired. By contrast, Andrews et al. (2017b) reported impaired complex facial and prosodic emotion recognition, but intact simple facial affect recognition, in a group of non-demented patients with ALS. Finally, Woolley and Rush (2017) showed that the performances of patients without dementia may be impaired on tasks of complex facial affect recognition or affective prosody recognition, and may have difficulty interpreting the gaze direction of others.
To summarize findings on social cognition in ALS, even though results are quite divergent, affective ToM seems to be systematically affected, whereas scores on cognitive ToM seem to be underpinned by patients’ cognitive profile. Studies including patients with severe cognitive impairment have reported cognitive ToM deficits (Carluer et al., 2015), whereas those among patients with only mild or moderate cognitive impairment point to preserved cognitive ToM (Trojsi et al., 2016). This prompts the question of a link between social cognition and other cognitive processes.
Interactions between cognitive status, social cognition, and emotion recognition
Most studies investigating the pattern of interaction between social cognition impairment and other cognitive functions support the idea of an interaction between executive functions and cognitive ToM, with positive correlations between executive functions (verbal fluency, composite executive score, executive score of the Frontal System Behavior Scale, etc.) and ToM (decision making task, faux pas task, etc.) (Gibbons et al., 2007; Meier et al., 2010; Watermeyer et al., 2015). In Carluer et al. (2015) study, cognitive ToM scores were only partially associated with executive performances, notably shifting, inhibition, and the ability to manipulate items in working memory. This led the authors to propose that the ToM deficit observed in some cases of ALS is not simply the consequence of an executive function impairment. Nevertheless, as studies do not generally exclude patients who have only a mild cognitive impairment, it is still unclear whether any ToM deficit is associated with executive deficit in ALS or whether ToM per se is disturbed.
Results on the interaction between emotion recognition and affective ToM performances and cognitive abilities have been somewhat conflicting. In a subset of patients with bulbar ALS, Zimmerman et al. (2007), found that 62% of them had emotion recognition defects, with no correlation with cognitive symptoms. However, the cognitive assessment was done using the Mini-Mental State Examination (MMSE), which is not well suited to patients with ALS. When they calculated a composite executive score, Watermeyer et al. (2015) also found no correlation between cognitive performances and RME test scores. By contrast, Girardi et al. (2011) reported a negative correlation between RME performances and z scores for verbal fluency. Burke et al. (2016) compared the RME test performances of three groups of patients: “no cognitive impairment,” “defect in only one cognitive domain,” and “multi-executive deficits/cognitively impaired.” The patients with no cognitive impairment performed better than those with a defect in only one cognitive domain, who in turn performed better than those in the third group. Finally, Andrews et al. (2017b) found that crossmodal (facial and prosodic integration) emotion recognition correlated with executive functions, whereas the separate modalities of emotion recognition did not. No specific cognitive function seems to be related to either affective ToM or emotion recognition, and as Trojsi et al. (2016) study suggests, the link between ToM performances and cognitive impairment may only concern the cognitive aspect of ToM.
Over Time
To our knowledge, only two studies have addressed changes in emotional and ToM abilities and their neural correlates in the course of ALS. Gillingham et al. (2017) assessed cognitive and emotional changes in patients with ALS at baseline and 9 months later using a specific cognitive and emotional battery (ALS-CFB) and other standardized cognitive tests, including cognitive first-order and cognitive and affective second-order ToM tasks. At baseline, patients showed a deficit in emotion perception for happy emotions, and scored significantly lower than controls on the first-order cognitive ToM task. At 9 months, in the emotion perception task, patients were better at recognizing angry faces than controls were, but there were no changes over time in ToM. Trojsi et al. (2017) assessed cognitive and affective ToM at baseline and 6 months later. At baseline, no ToM abnormalities were found in patients, whereas at follow up, patients with bulbar onset exhibited a decline in both affective and cognitive ToM, whereas those with limb onset only displayed impairment of cognitive ToM over time. This study also included an fMRI investigation, only at baseline, that will be discussed in the next section about neuroimaging. This result is in line with the more extensive prefrontal hypometabolism observed in patients with bulbar versus limb onset by Cistaro et al. (2012).
Neuroimaging Correlates of Emotional and Social Cognition Changes
Emotion recognition impairment is associated with alteration of white-matter integrity along the right inferior longitudinal fasciculus and inferior fronto-occipital fasciculus (Crespi et al., 2014). Defective emotional empathy attribution is correlated with reduced gray-matter density in the anterior cingulate cortex and right inferior frontal gyrus (Cerami et al., 2014). Palmieri et al. (2010) found a general increase in left-hemisphere activation and reduced right-hemisphere activation in patients when they were asked to attribute an emotional valence or remember an emotion. Aho-Özhan et al. (2016) assessed emotion recognition and its functional neural correlates in a group of patients with ALS and a matched control group. Patients recognized disgust and fear less accurately, and had lower activity in the hippocampus on both sides (brain regions involved in negative emotion processing) and more activity in the right inferior frontal gyrus.
Using a composite executive function score as a covariate, Carluer et al. (2015) found positive correlations between cognitive ToM performances and the metabolism of the bilateral superior frontal gyrus, bilateral middle frontal gyrus, and bilateral supplementary motor area. Buhour et al. (2017) sought to understand the metabolic dysfunction and its neurobehavioral consequences better by subjecting a sample of 37 patients with ALS to a comprehensive neuropsychological assessment and PET imaging. Significant negative correlations were found between metabolic activity within the left fusiform gyrus and performance on a false-belief task.
Longitudinal studies are very few and far between. Lulé et al. (2007) assessed emotional valence attribution, arousal, association of movement, and brain functioning when emotional material was presented to patients with ALS and healthy controls at baseline and 6 months later. Patients had an increased brain response in the right supramarginal area and a reduced brain response in extrastriatal visual areas at both measurement points, compared with healthy controls. In the patient group, a reduced brain response in the anterior insula at follow up was correlated with subjective arousal. This reduced response was tentatively interpreted as indicating reduced arousal during the course of the disease at the neural and behavioral levels. The reduced activity in extrastriatal visual areas could be similarly interpreted. The increased brain response in the right supramarginal area could represent altered sensitivity to social-emotional cues.
In the aforementioned study of ToM, Trojsi et al. (2017) assessed, only at baseline, the resting state functional connectivity with fMRI in 21 ALS patients compared to a matched group control. Subjects also underwent affective and cognitive ToM tasks both at baseline and after 6 months. Compared to controls, ALS patients exhibited abnormalities (1) within the DMN (Default mode network) with decreased connectivity in anterior node and increased connectivity in posterior node, (2) within the right FPN (Fronto-parietal network) with decreased connectivity in the supramarginal gyri and (3) within the left FPN and SLN (salient network) with decreased connectivity on the medial and dorsolateral prefrontal cortices. Positive correlations were found between affective ToM performances and functional connectivity of the posterior node of the DMN and the supramarginal gyri. ToM alterations were associated to decreased connectivity in the posterior cingulate cortex (PCC) and the occipital gyri of DMN. The authors suggest that these results support the hypothesis of the potential role of frontotemporoparietal network structures, such as the PCC and the supramarginal gyri in reasoning about the contents of another person’s mind particularly in affective mentalizing and in empathetic face processing.
Behavioral Changes (Table 3)
Baseline
Frequency and typology
Behavioral changes are increasingly being recognized as a common feature in ALS, and may be similar to those observed in FTD. They occur in 24–69% of patients with ALS (Murphy J. M. et al., 2007; Gibbons et al., 2008; Witgert et al., 2010; Lillo et al., 2011; Bock et al., 2016; Burke et al., 2017), 6–25% of whom meet the criteria for FTD (Murphy J. et al., 2007; Lillo et al., 2011; Bock et al., 2016; Burke et al., 2017). In some patients, they may appear as early features, even prior to the development of the motor symptoms (Mioshi et al., 2014).
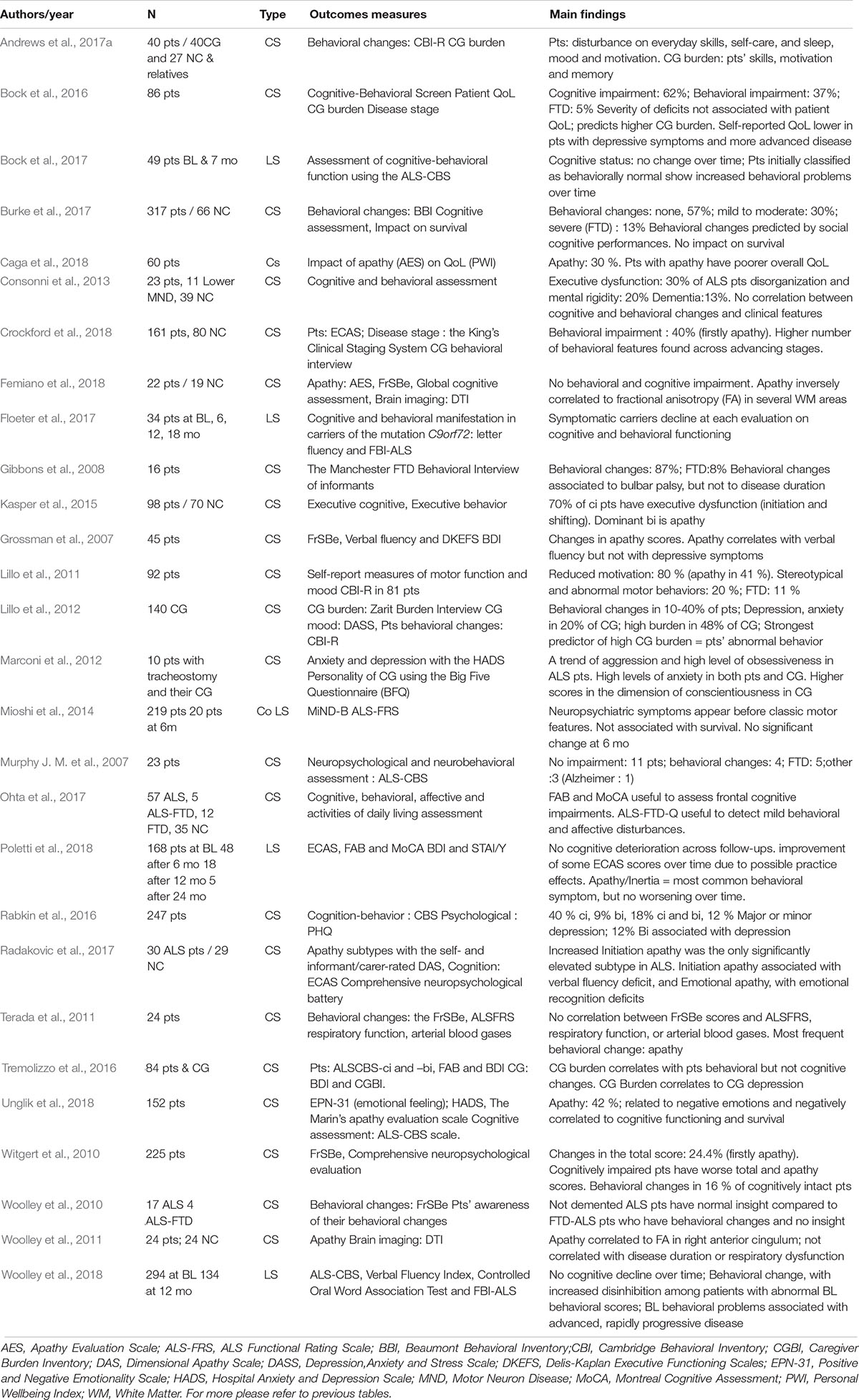
Table 3. Behavioral changes in ALS.
Apathy seems to be the most common behavioral change (Grossman et al., 2007; Witgert et al., 2010; Lillo et al., 2011; Kasper et al., 2015). The most frequently encountered subtype of apathy in ALS is lack of initiation, that is, a lack of motivation to self-generate thoughts (Radakovic et al., 2017). Patients in the advanced stage may display aggressiveness and obsessiveness (Marconi et al., 2012). Disinhibition, impulsivity, lack of foresight and planning, distractibility, reduced concern for hygiene, irritability, increased self-centeredness and reduced concern for the feelings and needs of others, new unusual habits, loss of insight, and blunting of the primary emotions of happiness, sadness, fear and anger have also been reported (Gibbons et al., 2008; Giordana et al., 2011; Burke et al., 2017). Finally, changes such as aspontaneity, disorganization, and mental rigidity have also been observed (Consonni et al., 2013). Insight on behavioral changes is altered in ALS-FTD, but not in ALS without dementia (Woolley et al., 2010).
Implications for the disease course
The presence of behavioral symptoms has practical implications, as it impacts the patients’ psychological state (and that of their caregivers), with consequences for their QoL and indeed their prognosis.
Rabkin et al. (2016) showed that whereas cognitive status does not correlate with mood, patients with behavioral impairment report more depressive symptoms, greater hopelessness, negative mood, and more negative feedback from spouses or caregivers. Caga et al. (2018) found that apathy was associated with more depressive symptoms and a poorer QoL, especially for achievement in life and community connectedness. Aggressiveness and obsession in the advanced stages of the disease correlate with a high level of anxiety in both patients and caregivers (Marconi et al., 2012). Unglik et al. (2018) reported that both apathetic and nonapathetic patients reported anxious and depressive symptoms, and the only significant difference between the two groups was that apathetic and anxious patients experienced more negative emotions, including sadness, shame and anger, than anxious patients without apathy.
The strongest predictor of high caregiver burden is patients’ abnormal behavior (e.g., apathy and disinhibition), rather than physical disability (Lillo et al., 2012; Watermeyer et al., 2015; Tremolizzo et al., 2016). The level of depressive and anxious symptoms in caregivers is also correlated with behavioral changes (Watermeyer et al., 2015; Tremolizzo et al., 2016). Caregiver burden is further influenced by patients’ everyday skills, motivation and memory, mostly because poor motivation, memory dysfunction, and difficulty performing activities of daily living require more support in the shape of direct supervision, prompting, or hands-on care (Andrews et al., 2017b).
Survival in ALS is highly influenced by the presence or absence of apathy, with a median survival time of 21.7 months in the case of moderate-to-severe apathy, 46.9 months in the case of mild apathy, and 51.9 months when apathy is absent (Caga et al., 2018). Apathy correlates negatively with survival time (Unglik et al., 2018).
Associated factors
Behavioral changes can occur either on their own or in the presence of a cognitive deficit (Murphy J. M. et al., 2007; Witgert et al., 2010). When cognitive impairment is present, behavioral symptoms seem to be greater (Witgert et al., 2010; Consonni et al., 2013). Apathy has been found to be associated with verbal fluency, leading some authors to suggest that apathy in ALS is underpinned by the medial prefrontal cortex (Grossman et al., 2007; Radakovic et al., 2017). Although Burke et al. (2017) found that social cognitive performances predicted behavioral changes, others have failed to do so (Terada et al., 2011).
Studies assessing relationships between behavioral changes and physical parameters have yielded somewhat conflicting results. Terada et al. (2011) found that apathy was correlated with the ALS condition per se, and not with the physical disability. Their population, however, consisted of only mildly disabled patients, who needed no assistance with activities of daily living, and all had normal blood gases. By contrast, in a larger sample of patients, with a wider range of physical impairments (although no details were given about their respiratory status), Ohta et al. (2017) did find a correlation between scores on the ALS-FTD Questionnaire and ALS Functional Rating Scale. Whereas the motor deficit may not directly impact behavior in mildly impaired patients, it is obviously mandatory to control for physical parameters, and first and foremost for blood gases, to avoid erroneously ascribing the consequences of hypercapnic encephalopathy to the cerebral neuronal involvement of ALS in the advanced stage.
Bock et al. (2017) found no association between bulbar or spinal onset and behavioral changes at baseline, whereas other authors (Gibbons et al., 2008; Crockford et al., 2018) have reported that bulbar palsy is associated with a higher rate of behavioral change.
Over Time
The way behavioral symptoms change as the disease progresses is still a matter of debate.
Most cross-sectional studies have shown no correlation between the severity of behavioral changes and time elapsed between disease onset and the time of study, suggesting that there is no significant decline over time (Woolley et al., 2011; Bock et al., 2017; Femiano et al., 2018). Crockford et al. (2018) assessed behavior in a sample of 149 patients using the ECAS caregiver behavioral interview and examined whether behavior was related to disease stages according to King’s Clinical Staging System (stage 1 from stage 4 depending on the number of affected bodily regions). Almost 40% of patients were behaviorally impaired (most of them with apathy) and a higher number of behavioral features was found across advancing stages. This result suggests that, contrary to what had been reported previously, behavioral and cognitive impairments are more severe in more severe disease stages.
Similarly, most of the longitudinal studies support the idea that behavioral manifestations over time may either increase (Bock et al., 2017) or appear as the illness progresses, with the emergence of frustration tolerance, reduced insight, mental rigidity, and lack of interest (Bock et al., 2017; Woolley et al., 2018). The severity of these disturbances increases faster over time when dementia is present in patients with the C9+ mutation (Floeter et al., 2017). Poletti et al. (2018) found no change over time in the prevalence of behavioral changes as measured with the ECAS, but did find an increase in behavioral disturbances as measured with the Frontal Behavioral Inventory, which is possibly a more sensitive tool, as it quantifies patients’ performances, whereas the ECAS only indicates whether or not there are changes.
Neuroimaging Correlates
Most studies addressing the issue of the neural correlates of behavioral changes in ALS point to a significant correlation between apathy scores and prefrontal cortex atrophy, especially in the orbitofrontal and dorsolateral areas (Tsujimoto et al., 2011; Consonni et al., 2018), whereas disinhibition is negatively correlated with thickness of the right frontotemporal and cingulate cortices (Consonni et al., 2018).
Diffusion tensor imaging studies have shown a significant negative correlation between apathy scores and fractional anisotropy in the right anterior cingulate region, corpus callosum, bilateral amygdalae, left thalamus, and fornix, with atrophy of these brain regions (Woolley et al., 2011; Branco et al., 2018; Femiano et al., 2018). Correlations between apathy and the prefrontal cortex have also been found in other neurological diseases.
On the whole, the relationships between the anterior cingulate (and possibly some subcortical structures) and apathy, and between the anterior temporal lobe and disinhibition, appear quite consistent across studies.
Psychological Adjustment (Table 4)
Psychological Reactions and Wellbeing
Baseline
Chronic diseases induce a wide range of psychological responses, such as uncertainty about the future, anxiety, and depression. These psychological responses can have a major impact on health, through for example the perceived somatic symptom burden, adherence to treatment and compliance with care, malnutrition, and mortality (Cukor et al., 2006; Katon et al., 2007), and therefore need to be detected and supported.
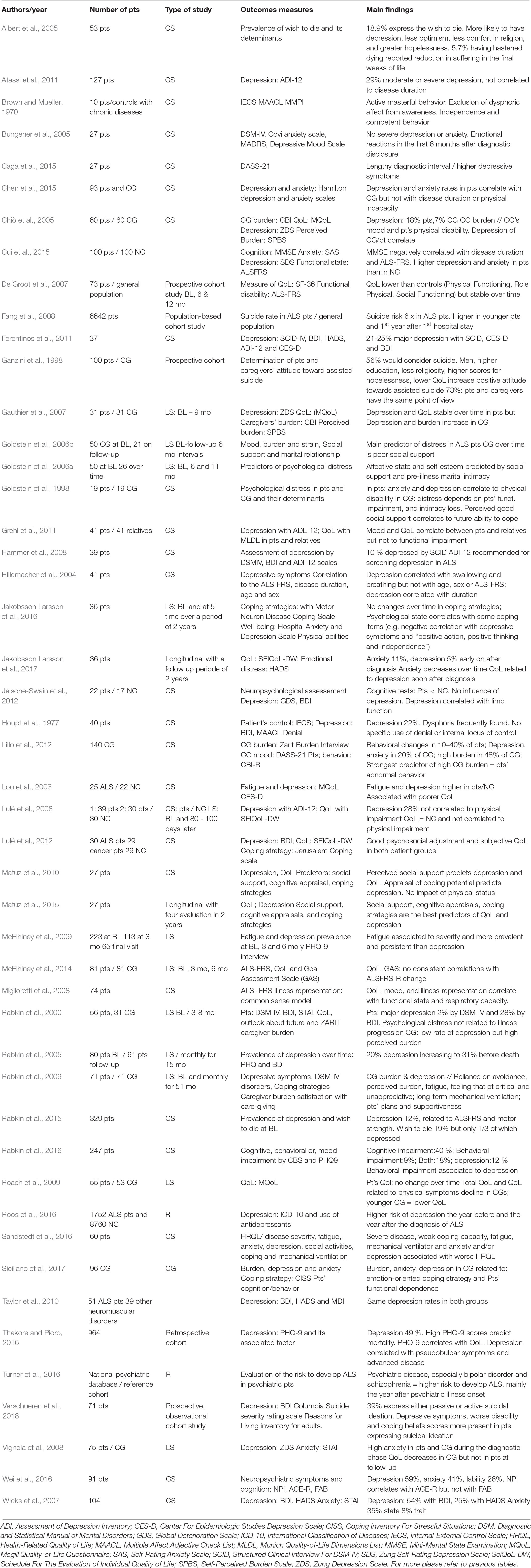
Table 4. Psychosocial adjustment and coping in ALS.
Patients with ALS were initially described as abnormally positive (Brown and Mueller, 1970) and, in contrast to what is usually described in other serious chronic somatic diseases, had a relatively low prevalence of depressive disorders ranging from 0% (Rabkin et al., 2000; Bungener et al., 2005) to 10% (Hammer et al., 2008; McElhiney et al., 2009; Rabkin et al., 2016; Wei et al., 2016), according to their responses to semi-structured questionnaires based on international criteria for depression.
Nevertheless, these results have to be considered in the light of several additional factors. First, when patients are assessed at the extreme stages of the disease (either soon after diagnosis or at a very advanced stage), validated scales indicate that the prevalence of depressive symptoms is around 20% (Rabkin et al., 2005), and this figure rises to above 50% when self-report questionnaires are used (Wicks et al., 2007). Second, a history of depression before the onset of ALS increases the prevalence rate from 10 to 21% when patients are assessed with a semi-structured questionnaire (McElhiney et al., 2009; Ferentinos et al., 2011). An interesting study conducted by Roos et al. (2016) found that the risk of receiving a diagnosis of depression was increased during the year before and the year after the diagnosis of ALS. This is also true for other major psychiatric disorders, namely schizophrenia, bipolar disorder, and anxiety, some of which may predate the diagnosis of ALS by as much as 5 years (Turner et al., 2016). Self-report questionnaires designed to probe depressive symptoms may have their limits, insofar as they are not able to diagnose a depressive state with certainty, leading to potential overestimation of the rate of depression, but they do have the advantage of detecting the potential presence of depressive symptoms that reflect a degree of distress. Studies using this type of instrument report higher rates of depression of above 30% (Lulé et al., 2008; Atassi et al., 2011; Grehl et al., 2011; Carvalho et al., 2016). Thus, even when the official diagnostic criteria are not met, it does not mean that patients do not feel they are affected by their disease. One must thus be aware that depression rates in ALS vary greatly, depending on the tools used to assess it. Finally, studies comparing the rate of depression in patients with ALS versus other chronically and seriously ill patients (e.g., with neuromuscular disease or receiving palliative care for cancer) have failed to find any significant differences, with 8–10% of patients in each group having a diagnosis of major depression according to DSM-IV criteria, and 50% mild-to-moderate depressive symptoms, as measured with the Beck Depressive Inventory (Taylor et al., 2010; Lulé et al., 2012).
Furthermore, more than one third of patients with ALS are on antidepressants (Pisa et al., 2015). Not all studies assessing the prevalence of depression in ALS take this fact into account, which could induce a bias and result in underestimation of the prevalence of depression in this population.
Intuitively, one might assume that the worsening of physical disability increases signs of depression. However, many studies have found either no such link or an inverse link between the severity of the motor disability and scores on mood scales (Lulé et al., 2008; Jelsone-Swain et al., 2012; Chen et al., 2015; Thakore and Pioro, 2016; Wei et al., 2016). This is only true, however, for the consequences of spinal involvement, as the presence of bulbar symptoms, or of breathing difficulty, does increase depressive symptoms (Hillemacher et al., 2004; Goldstein et al., 2006a; Miglioretti et al., 2008; Jelsone-Swain et al., 2012).
Studies looking for a link between disease duration and the severity of depressive symptoms have reported contradictory results, with some finding a positive correlation, some a negative one (Cui et al., 2015; Hillemacher et al., 2004, and others no link at all (Atassi et al., 2011; Caga et al., 2015).
Severe somatic diseases may induce a number of other psychological reactions, such as anxiety, hopelessness, or suicidal thoughts. Kurt et al. (2007) found that the prevalence of anxiety in ALS ranged from 0 to 30%. Vignola et al. (2008) reported that almost 75% of patients experienced moderate-to-severe state anxiety at baseline, which was correlated with trait anxiety. However, Pagnini et al. (2012) found that only 20% of patients had scores above the anxiety cut off. Suicidal thoughts are not rare in patients, ranging from 19 to 39% across studies (Albert et al., 2005; Rabkin et al., 2015; Verschueren et al., 2018). This rate rises to more than 50% when assisted suicide is considered (Ganzini et al., 1998). Patients have an almost 6-fold higher risk of suicide, especially in the first year after symptom onset and when they are younger (Fang et al., 2008). A wish to die is not always associated with a depressive state (Albert et al., 2005) but it is linked to less optimism, less comfort in religion, and greater hopelessness (Albert et al., 2005; Verschueren et al., 2018).
In addition to psychological signs of distress such as depression or anxiety, the estimation of wellbeing is based on the person’s satisfaction with his or her QoL. In patients with ALS, QoL is found to be high, especially when the measurement scales are adapted to the disease (Norris et al., 2010; Jakobsson Larsson et al., 2017) and avoid lending too much importance to the patient’s physical state (Norris et al., 2010; Jakobsson Larsson et al., 2017). The Amyotrophic Lateral Sclerosis Quality of Life (ALSSQOL) was specifically designed for ALS, and has a revised and a short form (Felgoise et al., 2018) that give a balanced appraisal. Qol depends not only on patients’ physical disabilities, but also on their religiosity/spirituality and sociability (Norris et al., 2010; Simmons, 2015). Other factors influencing QoL are anxiety and depression, the ability to cope with physical disabilities, fatigue, and hopelessness (Lou et al., 2003; Abe, 2004; Pagnini et al., 2012; Sandstedt et al., 2016; Jakobsson Larsson et al., 2017).
Over Time
Surprisingly, longitudinal studies of changes in patients’ mood as the disease progresses report either stability (Rabkin et al., 2005; Gauthier et al., 2007; McElhiney et al., 2009; Matuz et al., 2015) or a decrease in the depression rate (McElhiney et al., 2014). When Goldstein et al. (2006a) assessed psychosocial factors influencing patients’ psychological state (level of depression and anxiety) at baseline and 6 and 11 months later, they found that the quality of premarital intimacy and social support at disease onset influenced the psychological wellbeing of patients in the more advanced stages of the disease. Anxiety is particularly high in the diagnostic phase, but tends to decrease thereafter in patients, though not in caregivers (Vignola et al., 2008). Importantly, these authors also found that in caregivers, state anxiety was linked to trait anxiety, whereas in patients, it was correlated with clinical features such as a shorter disease course and the presence of depression. Together with other factors, anxiety negatively impacts QoL (Jakobsson Larsson et al., 2017).
Caregivers’ psychological distress and perceived burden is often reported to increase over time (Goldstein et al., 2006b; Gauthier et al., 2007). The burden is determined by a combination of factors, including both the caregiver’s own personality and the patient’s characteristics. Regarding the latter, Lillo et al. (2012) found that behavioral changes are a greater determinant of caregivers’ wellbeing than physical disability, although motor impairment also plays a part, according to Chiò et al. (2005). Behavioral changes may include apathy, loss of empathy, and a lack of appreciation of the efforts made to satisfy their needs (Rabkin et al., 2009). As the disease progresses, patients’ plans for future, particularly regarding life support, may also play a part and influence their caregivers’ QoL. A longitudinal study by Rabkin et al. (2009) found that caregivers tended to be less depressed as the disease advanced, regardless of the outcome (death or tracheostomy), whereas the depression scores of patients remained stable.
Quality of life as reported by patients remains stable even in the advanced stages of the disease (Rabkin et al., 2000; De Groot et al., 2007; Roach et al., 2009). Religiosity, social support, level of anxiety and depression remain the mean determinants of wellbeing, despite the increase in motor impairment (Jakobsson Larsson et al., 2017).
Adaptive Mechanisms in the Face of the Disease
To explain the fact that patients with ALS have a relatively good adaptive reaction to their illness, some authors have suggested that they are in denial of the disease, thus protecting them from depression (Brown and Mueller, 1970; Miglioretti et al., 2008), but this hypothesis has not been confirmed (Houpt et al., 1977).
Psychological adjustment refers to the psychological processes that take place in response to a stressful situation like chronic illness and associated treatment. Various models have been developed, including the stress coping model (Lazarus and Folkman, 1984), illness representation model, adaptive tasks and coping model (Moos and Holahan, 2007), and adjustment model (Moss-Morris, 2013). Based on Lazarus and Folkman’s theoretical framework, Matuz et al. (2010) developed an interesting integrative model of patients’ adaptation in the face of the disease, whereby patients’ mood state and QoL are influenced by social support, cognitive appraisal and coping strategies. The latter include problem management, problem appraisal, emotion regulation, and emotional avoidance. Social support encompasses perceived social support, received social support, and need for social support. Matuz et al. (2010) found that patients used emotion regulation and looked for social support, and the more they used emotion-focused strategies, the lower they scored on a depression scale. When Lulé et al. (2012) compared patients with ALS and patients with cancer on psychological adjustment, they failed to find any significant differences in the use of coping strategies, even if, on average, the ALS group scored lower than the cancer group on a scale measuring active coping strategies (thinking about the situation and trying to solve it, taking an adequate step to deal with their condition). By contrast, depressive symptoms and a high level of burden are more often present in caregivers if they use emotion-focused coping strategies (Siciliano et al., 2017). Factors that have been shown to help caregivers cope with the impact of the disease include social support, as well as anticipatory coping with foreseeable difficulties (Goldstein et al., 1998).
Studies addressing changes in psychological adjustment strategies in ALS over time have yielded conflicting results. Jakobsson Larsson et al. (2016) found that the patients used the same coping strategies throughout the disease course, namely support seeking, positive action, independence, and positive thinking. By contrast, Matuz et al. (2015) reported that while patients used both problem- and emotion-focused coping strategies at disease onset, they used less emotion regulation later on. On the other hand, they continued to have higher perceived social support and an accurate assessment of their own coping potential. These two factors were correlated with depressive scores at follow up. Perceived social support, which reflects patients’ view about the amount and quality of support they receive, can help patients use effective coping strategies, encourage positive health behaviors, and reduce physiological reactivity to stress. A positive subjective appraisal of one’s coping potential indicates that patients feel they are keeping control over their state, which probably decreases their anxiety over physical loss.
At a time when some people advocate legalizing euthanasia for intractable disease, accurate knowledge of the mechanisms by which many patients with ALS succeed in coping with such a dreadful disease is crucial, and could greatly enhance the assistance given to both patients and caregivers in their daily struggle.
Conclusion
The objective of this review was to provide readers with a clear picture of all the manifestations of ALS both at baseline and throughout the course of the disease. Unlike the relentless physical deterioration, cognitive, behavioral and psychological changes are extremely variable. The heterogeneity of clinical situations, and in particular the variety of symptoms present at baseline, seems to strongly influence patients’ clinical course. Regarding their psychological reactions, even if they rarely develop a severe psychiatric illness, they can experience considerable distress, especially at the time of diagnosis and in the advanced stage of the disease. Caution must be taken not to minimize these psychological reactions and to give patients the best possible personalized support at these times. A careful examination of the psychological trajectory shows that the increase in motor disability is not the only determinant of psychological wellbeing. The quality of social support and the use of appropriate adaptive strategies enable patients to cope with their condition and to maintain good psychosocial functioning as the disease progresses.
This review also illustrates the theoretical and practical difficulties that may arise when investigating cognitive, behavioral and psychological aspects of ALS. The first concerns the tools that are to be used. First, these must be adapted to the physical disability of patients with ALS. Potentially confounding variables, such as respiratory insufficiency, have to be controlled. As we have seen, a large number of tests have been used by the different authors. Such a diversity makes comparisons difficult and may at least partially account for the discrepancies that are often observed across studies. Investigators will have eventually to try to agree about which tests should be used, either as routine tests (such as the ECAS for the cognitive assessment, or ALS-FRS or the bulbar Norris scale for physical parameters) or for more specific purposes. It will also be interesting to further study less extensively explored cognitive domains such as memory or social cognition.
Another difficulty is that of longitudinal studies. These are particularly challenging, because of the major physical changes that occur over time. It is not clearly known, for example, whether a cognitive impairment, if any, worsens in line with the physical disability or not. Also, the short life expectancy of many patients is responsible for a substantial drop-out rate. These difficulties can be addressed by recruiting as many patients as possible, preferably in the setting of ALS centers. Bigger samples may also help to tackle the problem of physical and neuropsychological heterogeneity. For example, the seemingly simple question as to whether bulbar-onset patients are more cognitively affected than spinal-onset ones or not is still debated. Likewise, there have been very few studies about the relationships between the cognitive (or psychological, or behavioral, for that matter) and physical profiles of patients. Finally, morphological and functional cerebral imaging studies should be increasingly undertaken in order to learn more about the mechanisms of neuropsychological impairment, and to improve as much as possible the care of patients with ALS.
Author Contributions
BD, SB, and FV contributed to conceptualization and methodology. SB wrote the first draft of the manuscript. BD, FV, and FE reviewed the manuscript. SB, FV, and FE edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Mrs. Elizabeth Portier for assisting in language editing.
References
Abdulla, S., Machts, J., Kaufmann, J., Patrick, K., Kollewe, K., Dengler, R., et al. (2014). Hippocampal degeneration in patients with amyotrophic lateral sclerosis. Neurobiol. Aging 35, 2639–2645. doi: 10.1016/j.neurobiolaging.2014.05.035
Abe, K. (2004). Fatigue and depression are associated with poor quality of life in ALS. Neurology 60, 122–123. doi: 10.1212/01.wnl.0000042781.22278.0a
Abe, K., Fujimura, H., Toyooka, K., Sakoda, S., Yorifuji, S., and Yanagihara, T. (1997). Cognitive function in amyotrophic lateral sclerosis. J. Neurol. Sci. 148, 95–100.
Abrahams, S., Leigh, P. N., and Goldstein, L. H. (2005). Cognitive change in ALS: a prospective study. Neurology 64, 1222–1226. doi: 10.1212/01.WNL.0000156519.41681.27
Abrahams, S., Leigh, P. N., Harvey, A., Vythelingum, G. N., Grisé, D., and Goldstein, L. H. (2000). Verbal fluency and executive dysfunction in amyotrophic lateral sclerosis (ALS). Neuropsychologia 38, 734–747. doi: 10.1016/s0028-3932(99)00146-3
Agosta, F., Ferraro, P. M., Riva, N., Spinelli, E. G., Chiò, A., Canu, E., et al. (2016). Structural brain correlates of cognitive and behavioral impairment in MND. Hum. Brain Mapp. 37, 1614–1626. doi: 10.1002/hbm.23124
Aho-Özhan, H. E. A., Keller, J., Heimrath, J., Uttner, I., Kassubek, J., Birbaumer, N., et al. (2016). Perception of emotional facial expressions in amyotrophic lateral sclerosis (ALS) at behavioural and brain metabolic level. PLoS One 11:e0164655. doi: 10.1371/journal.pone.0164655
Albert, S. M., Rabkin, J. G., Del Bene, M. L., Tider, T., O’Sullivan, I., Rowland, L. P., et al. (2005). Wish to die in end-stage ALS. Neurology 65, 68–74. doi: 10.1212/01.wnl.0000168161.54833.bb
Andrews, S. C., Pavlis, A., Staios, M., and Fisher, F. (2017a). Which behaviours? Identifying the most common and burdensome behaviour changes in amyotrophic lateral sclerosis. Psychol. Health Med. 22, 483–492. doi: 10.1080/13548506.2016.1164871
Andrews, S. C., Staios, M., Howe, J., Reardon, K., and Fisher, F. (2017b). Multimodal emotion processing deficits are present in amyotrophic lateral sclerosis. Neuropsychology 31, 304–310. doi: 10.1037/neu0000323
Atassi, N., Cook, A., Pineda, C. M. E., Yerramilli-Rao, P., Pulley, D., and Cudkowicz, M. (2011). Depression in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 12, 109–112. doi: 10.3109/17482968.2010.536839
Beeldman, E., Raaphorst, J., Klein Twennaar, M., de Visser, M., Schmand, B. A., and de Haan, R. J. (2016). The cognitive profile of ALS: a systematic review and meta-analysis update. J. Neurol. Neurosurg. Psychiatr. 87, 611–619. doi: 10.1136/jnnp-2015-310734
Benbrika, S., Doidy, F., Carluer, L., Mondou, A., Buhour, M.-S., Eustache, F., et al. (2018). Alexithymia in amyotrophic lateral sclerosis and its neural correlates. Front. Neurol. 9:566. doi: 10.3389/fneur.2018.00566
Bock, M., Duong, Y.-N., Kim, A., Allen, I., Murphy, J., and Lomen-Hoerth, C. (2016). Cognitive-behavioral changes in amyotrophic lateral sclerosis: screening prevalence and impact on patients and caregivers. Amyotroph. Lateral Scler. Front. Degener. 17, 366–373. doi: 10.3109/21678421.2016.1165257
Bock, M., Duong, Y.-N., Kim, A., Allen, I., Murphy, J., and Lomen-Hoerth, C. (2017). Progression and effect of cognitive-behavioral changes in patients with amyotrophic lateral sclerosis. Neurol. Clin. Pract. 7, 488–498. doi: 10.1212/CPJ.0000000000000397
Bora, E. (2017). Meta-analysis of social cognition in amyotrophic lateral sclerosis. Cortex J. Devoted Study Nerv. Syst. Behav. 88, 1–7. doi: 10.1016/j.cortex.2016.11.012
Bortolato, B., Miskowiak, K. W., Köhler, C. A., Maes, M., Fernandes, B. S., Berk, M., et al. (2016). Cognitive remission: a novel objective for the treatment of major depression? BMC Med. 14:9. doi: 10.1186/s12916-016-0560-3
Braber, W. (2016). Measurements of Cognitive Functioning in ALS Patients: a Longitudinal Study [WWW Document]. Available at: http://dspace.library.uu.nl/handle/1874/340869 (accessed February.19, 2019)Google Scholar
Branco, L. M. T., de Rezende, T. J. R., Roversi, C. O., Zanao, T., Casseb, R. F., de Campos, B. M., et al. (2018). Brain signature of mild stages of cognitive and behavioral impairment in amyotrophic lateral sclerosis. Psychiatr. Res. Neuroimaging 272, 58–64. doi: 10.1016/j.pscychresns.2017.11.010
Brown, W. A., and Mueller, P. S. (1970). Psychological function in individuals with amyotrophic lateral sclerosis (ALS). Psychosom. Med. 32, 141–152. doi: 10.1097/00006842-197003000-00002
Buhour, M.-S., Doidy, F., Mondou, A., Pélerin, A., Carluer, L., Eustache, F., et al. (2017). Voxel-based mapping of grey matter volume and glucose metabolism profiles in amyotrophic lateral sclerosis. EJNMMI Res. 7, 21. doi: 10.1186/s13550-017-0267-2
Bungener, C., Piquard, A., Pradat, P.-F., Salachas, F., Meininger, V., and Lacomblez, L. (2005). Psychopathology in amyotrophic lateral sclerosis: a preliminary study with 27 ALS patients. Amyotroph. Lateral Scler. Mot. Neuron Disord. 6, 221–225. doi: 10.1080/14660820510037863
Burke, T., Elamin, M., Galvin, M., Hardiman, O., and Pender, N. (2015). Caregiver burden in amyotrophic lateral sclerosis: a cross-sectional investigation of predictors. J. Neurol. 262, 1526–1532. doi: 10.1007/s00415-015-7746-z
Burke, T., Pinto-Grau, M., Lonergan, K., Bede, P., O’Sullivan, M., Heverin, M., et al. (2017). A Cross-sectional population-based investigation into behavioral change in amyotrophic lateral sclerosis: subphenotypes, staging, cognitive predictors, and survival. Ann. Clin. Transl. Neurol. 4, 305–317. doi: 10.1002/acn3.407
Burke, T., Pinto-Grau, M., Lonergan, K., Elamin, M., Bede, P., Costello, E., et al. (2016). Measurement of social cognition in amyotrophic lateral sclerosis: a population based study. PLoS One 11:e0160850. doi: 10.1371/journal.pone.0160850
Burkhardt, C., Neuwirth, C., and Weber, M. (2017). Longitudinal assessment of the edinburgh cognitive and behavioural amyotrophic lateral sclerosis screen (ECAS): lack of practice effect in ALS patients? Amyotroph. Lateral Scler. Front. Degener. 18, 202–209. doi: 10.1080/21678421.2017.1283418
Byrne, S., Elamin, M., Bede, P., Shatunov, A., Walsh, C., Corr, B., et al. (2012). Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 11, 232–240. doi: 10.1016/S1474-4422(12)70014-5
Caga, J., Hsieh, S., Highton-Williamson, E., Zoing, M. C., Ramsey, E., Devenney, E., et al. (2018). Apathy and its impact on patient outcome in amyotrophic lateral sclerosis. J. Neurol. 265, 187–193. doi: 10.1007/s00415-017-8688-4
Caga, J., Ramsey, E., Hogden, A., Mioshi, E., and Kiernan, M. C. (2015). A longer diagnostic interval is a risk for depression in amyotrophic lateral sclerosis. Palliat. Support. Care 13, 1019–1024. doi: 10.1017/S1478951514000881
Canosa, A., Pagani, M., Cistaro, A., Montuschi, A., Iazzolino, B., Fania, P., et al. (2016). 18F-FDG-PET correlates of cognitive impairment in ALS. Neurology 86, 44–49. doi: 10.1212/WNL.0000000000002242
Carelli, L., Solca, F., Faini, A., Madotto, F., Lafronza, A., Monti, A., et al. (2018). The complex interplay between depression/anxiety and executive functioning: insights from the ECAS in a LARGE ALS population. Front. Psychol. 9:450. doi: 10.3389/fpsyg.2018.00450
Carluer, L., Mondou, A., Buhour, M.-S., Laisney, M., Pélerin, A., Eustache, F., et al. (2015). Neural substrate of cognitive theory of mind impairment in amyotrophic lateral sclerosis. Cortex J. Devoted Study Nerv. Syst. Behav. 65, 19–30. doi: 10.1016/j.cortex.2014.12.010
Carvalho, T. L., de Almeida, L. M. S., Lorega, C. M. A., Barata, M. F. O., Ferreira, M. L. B., de Brito-Marques, P. R., et al. (2016). Depression and anxiety in individuals with amyotrophic lateral sclerosis: a systematic review. Trends Psychiatry Psychother. 38, 1–5. doi: 10.1590/2237-6089-2015-0030
Cavallo, M., Adenzato, M., Macpherson, S. E., Karwig, G., Enrici, I., and Abrahams, S. (2011). Evidence of social understanding impairment in patients with amyotrophic lateral sclerosis. PLoS One 6:e25948. doi: 10.1371/journal.pone.0025948
Cerami, C., Dodich, A., Canessa, N., Crespi, C., Iannaccone, S., Corbo, M., et al. (2014). Emotional empathy in amyotrophic lateral sclerosis: a behavioural and voxel-based morphometry study. Amyotroph. Lateral Scler. Front. Degener. 15, 21–29. doi: 10.3109/21678421.2013.785568
Chen, D., Guo, X., Zheng, Z., Wei, Q., Song, W., Cao, B., et al. (2015). Depression and anxiety in amyotrophic lateral sclerosis: correlations between the distress of patients and caregivers. Muscle Nerve 51, 353–357. doi: 10.1002/mus.24325
Chiò, A., Gauthier, A., Calvo, A., Ghiglione, P., and Mutani, R. (2005). Caregiver burden and patients’ perception of being a burden in ALS. Neurology 64, 1780–1782. doi: 10.1212/01.WNL.0000162034.06268.37
Christidi, F., Karavasilis, E., Riederer, F., Zalonis, I., Ferentinos, P., Velonakis, G., et al. (2018). Gray matter and white matter changes in non-demented amyotrophic lateral sclerosis patients with or without cognitive impairment: a combined voxel-based morphometry and tract-based spatial statistics whole-brain analysis. Brain Imag. Behav. 12, 547–563. doi: 10.1007/s11682-017-9722-y
Cistaro, A., Valentini, M. C., Chiò, A., Nobili, F., Calvo, A., Moglia, C., et al. (2012). Brain hypermetabolism in amyotrophic lateral sclerosis: a FDG PET study in ALS of spinal and bulbar onset. Eur. J. Nucl. Med. Mol. Imag. 39, 251–259. doi: 10.1007/s00259-011-1979-6
Consonni, M., Cappa, S. F., Dalla Bella, E., Contarino, V. E., and Lauria, G. (2018). Cortical correlates of behavioural change in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatr. [Epub ahead of print].
Consonni, M., Iannaccone, S., Cerami, C., Frasson, P., Lacerenza, M., Lunetta, C., et al. (2013). The cognitive and behavioural profile of amyotrophic lateral sclerosis: application of the consensus criteria. Behav. Neurol. 27, 143–153. doi: 10.3233/BEN-2012-110202
Couratier, P., Corcia, P., Lautrette, G., Nicol, M., Preux, P.-M., and Marin, B. (2016). Epidemiology of amyotrophic lateral sclerosis: a review of literature. Rev. Neurol. 172, 37–45. doi: 10.1016/j.neurol.2015.11.002
Crespi, C., Cerami, C., Dodich, A., Canessa, N., Arpone, M., Iannaccone, S., et al. (2014). Microstructural white matter correlates of emotion recognition impairment in amyotrophic lateral sclerosis. Cortex J. Devoted Study Nerv. Syst. Behav. 53, 1–8. doi: 10.1016/j.cortex.2014.01.002
Crockford, C., Newton, J., Lonergan, K., Chiwera, T., Booth, T., Chandran, S., et al. (2018). ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology 91, e1370–e1380. doi: 10.1212/WNL.0000000000006317
Cui, F., Zhu, W., Zhou, Z., Ren, Y., Li, Y., Li, M., et al. (2015). Frequency and risk factor analysis of cognitive and anxiety-depressive disorders in patients with amyotrophic lateral sclerosis/motor neuron disease. Neuropsychiatr. Dis. Treat. 11, 2847–2854. doi: 10.2147/NDT.S90520
Cukor, D., Peterson, R. A., Cohen, S. D., and Kimmel, P. L. (2006). Depression in end-stage renal disease hemodialysis patients. Nat. Clin. Pract. Nephrol. 2, 678–687. doi: 10.1038/ncpneph0359
Dary-Auriol, M., Ingrand, P., Bonnaud, V., Dumas, P., Neau, J. P., and Gil, R. (1997). [Amyotrophic lateral sclerosis and cognition disorders. neuropsychological study of a population of 26 patients]. Rev. Neurol. 153, 244–250.
De Groot, I. J. M., Post, M. W. M., van Heuveln, T., Van den Berg, L. H., and Lindeman, E. (2007). Cross-sectional and longitudinal correlations between disease progression and different health-related quality of life domains in persons with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 8, 356–361. doi: 10.1080/17482960701553949
Elamin, M., Bede, P., Byrne, S., Jordan, N., Gallagher, L., Wynne, B., et al. (2013). Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology 80, 1590–1597. doi: 10.1212/WNL.0b013e31828f18ac
Elamin, M., Phukan, J., Bede, P., Jordan, N., Byrne, S., Pender, N., et al. (2011). Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology 76, 1263–1269. doi: 10.1212/WNL.0b013e318214359f
Fang, F., Valdimarsdóttir, U., Fürst, C. J., Hultman, C., Fall, K., Sparén, P., et al. (2008). Suicide among patients with amyotrophic lateral sclerosis. Brain J. Neurol. 131, 2729–2733. doi: 10.1093/brain/awn161
Felgoise, S. H., Feinberg, R., Stephens, H. E., Barkhaus, P., Boylan, K., Caress, J., et al. (2018). Amyotrophic lateral sclerosis-specific quality of life-short form (ALSSQOL-SF): a brief, reliable, and valid version of the ALSSQOL-R. Muscle Nerve 58, 646–654. doi: 10.1002/mus.26203
Femiano, C., Trojsi, F., Caiazzo, G., Siciliano, M., Passaniti, C., Russo, A., et al. (2018). apathy is correlated with widespread diffusion tensor imaging (DTI) impairment in amyotrophic lateral sclerosis. Behav. Neurol. 2018:2635202. doi: 10.1155/2018/2635202
Ferentinos, P., Paparrigopoulos, T., Rentzos, M., Zouvelou, V., Alexakis, T., and Evdokimidis, I. (2011). Prevalence of major depression in ALS: comparison of a semi-structured interview and four self-report measures. Amyotroph. Lateral Scler. 12, 297–302. doi: 10.3109/17482968.2011.556744
Flaherty, C., Kraft, J., Brothers, A., Harrison, M., Legro, R. S., Manni, A., et al. (2017). The relationship between oestrogen and executive functioning in ALS females with emerging frontotemporal lobar degeneration (FTLD) supports a neuroendocrine model of FTLD attenuation. Amyotroph. Lateral Scler. Front. Degener. 18, 74–85. doi: 10.1080/21678421.2016.1249487
Floeter, M. K., Bageac, D., Danielian, L. E., Braun, L. E., Traynor, B. J., and Kwan, J. Y. (2016). Longitudinal imaging in C9orf72 mutation carriers: relationship to phenotype. NeuroImage Clin. 12, 1035–1043. doi: 10.1016/j.nicl.2016.10.014
Floeter, M. K., Traynor, B. J., Farren, J., Braun, L. E., Tierney, M., Wiggs, E. A., et al. (2017). Disease progression in C9orf72 mutation carriers. Neurology 89, 234–241. doi: 10.1212/WNL.0000000000004115
Ganzini, L., Johnston, W. S., McFarland, B. H., Tolle, S. W., and Lee, M. A. (1998). Attitudes of patients with amyotrophic lateral sclerosis and their care givers toward assisted suicide. N. Engl. J. Med. 339, 967–973. doi: 10.1056/NEJM199810013391406
Gauthier, A., Vignola, A., Calvo, A., Cavallo, E., Moglia, C., Sellitti, L., et al. (2007). A longitudinal study on quality of life and depression in ALS patient-caregiver couples. Neurology 68, 923–926. doi: 10.1212/01.wnl.0000257093.53430.a8
Gibbons, Z. C., Richardson, A., Neary, D., and Snowden, J. S. (2008). Behaviour in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 9, 67–74. doi: 10.1080/17482960701642437
Gibbons, Z. C., Snowden, J. S., Thompson, J. C., Happé, F., Richardson, A., and Neary, D. (2007). Inferring thought and action in motor neurone disease. Neuropsychologia 45, 1196–1207. doi: 10.1016/j.neuropsychologia.2006.10.008
Gillingham, S. M., Yunusova, Y., Ganda, A., Rogaeva, E., Black, S. E., Stuss, D. T., et al. (2017). Assessing cognitive functioning in ALS: a focus on frontal lobe processes. Amyotroph. Lateral Scler. Front. Degener. 18, 182–192. doi: 10.1080/21678421.2016.1248977
Giordana, M. T., Ferrero, P., Grifoni, S., Pellerino, A., Naldi, A., and Montuschi, A. (2011). Dementia and cognitive impairment in amyotrophic lateral sclerosis: a review. Neurol. Sci. 32, 9–16. doi: 10.1007/s10072-010-0439-6
Girardi, A., MacPherson, S. E., and Abrahams, S. (2011). Deficits in emotional and social cognition in amyotrophic lateral sclerosis. Neuropsychology 25, 53–65. doi: 10.1037/a0020357
Goldstein, L. H., Adamson, M., Jeffrey, L., Down, K., Barby, T., Wilson, C., et al. (1998). The psychological impact of MND on patients and carers. J. Neurol. Sci. 160(Suppl. 1), S114–S121.
Goldstein, L. H., Atkins, L., Landau, S., Brown, R., and Leigh, P. N. (2006b). Predictors of psychological distress in carers of people with amyotrophic lateral sclerosis: a longitudinal study. Psychol. Med. 36, 865–875. doi: 10.1017/S0033291706007124
Goldstein, L. H., Atkins, L., Landau, S., Brown, R. G., and Leigh, P. N. (2006a). Longitudinal predictors of psychological distress and self-esteem in people with ALS. Neurology 67, 1652–1658. doi: 10.1212/01.wnl.0000242886.91786.47
Gordon, P. H., Delgadillo, D., Piquard, A., Bruneteau, G., Pradat, P.-F., Salachas, F., et al. (2011). The range and clinical impact of cognitive impairment in French patients with ALS: a cross-sectional study of neuropsychological test performance. Amyotroph. Lateral Scler. 12, 372–378. doi: 10.3109/17482968.2011.580847
Govaarts, R., Beeldman, E., Kampelmacher, M. J., van Tol, M.-J., van den Berg, L. H., van der Kooi, A. J., et al. (2016). The frontotemporal syndrome of ALS is associated with poor survival. J. Neurol. 263, 2476–2483. doi: 10.1007/s00415-016-8290-1
Grehl, T., Rupp, M., Budde, P., Tegenthoff, M., and Fangerau, H. (2011). Depression and QOL in patients with ALS: how do self-ratings and ratings by relatives differ? Qual. Life Res. Int. J. Qual. Life Asp. Treat. Care Rehabil. 20, 569–574. doi: 10.1007/s11136-010-9781-7
Grossman, A. B., Woolley-Levine, S., Bradley, W. G., and Miller, R. G. (2007). Detecting neurobehavioral changes in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 8, 56–61. doi: 10.1080/17482960601044106
Hammer, E. M., Häcker, S., Hautzinger, M., Meyer, T. D., and Kübler, A. (2008). Validity of the ALS-Depression-Inventory (ADI-12)–a new screening instrument for depressive disorders in patients with amyotrophic lateral sclerosis. J. Affect. Disord. 109, 213–219. doi: 10.1016/j.jad.2007.11.012
Hervieu-Bègue, M., Rouaud, O., Graule Petot, A., Catteau, A., and Giroud, M. (2016). Semantic memory assessment in 15 patients with amyotrophic lateral sclerosis. Rev. Neurol. 172, 307–312. doi: 10.1016/j.neurol.2015.10.009
Hillemacher, T., Grässel, E., Tigges, S., Bleich, S., Neundörfer, B., Kornhuber, J., et al. (2004). Depression and bulbar involvement in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Mot. Neuron Disord. 5, 245–249. doi: 10.1080/14660820410021294
Houpt, J. L., Gould, B. S., and Norris, F. H. (1977). Psychological characteristics of patients with amyotrophic lateral sclerosis (ALS). Psychosom. Med. 39, 299–303.
Hu, W. T., Shelnutt, M., Wilson, A., Yarab, N., Kelly, C., Grossman, M., et al. (2013). Behavior matters–cognitive predictors of survival in amyotrophic lateral sclerosis. PLoS One 8:e57584. doi: 10.1371/journal.pone.0057584
Irwin, D., Lippa, C. F., and Swearer, J. M. (2007). Cognition and amyotrophic lateral sclerosis (ALS). Am. J. Alzheimers Dis. Other Demen. 22, 300–312. doi: 10.1177/1533317507301613
Iwasaki, Y., Kinoshita, M., Ikeda, K., Takamiya, K., and Shiojima, T. (1990). Neuropsychological dysfunctions in amyotrophic lateral sclerosis: relation to motor disabilities. Int. J. Neurosci. 54, 191–195. doi: 10.3109/00207459008986635
Jakobsson Larsson, B., Nordin, K., and Nygren, I. (2016). Coping with amyotrophic lateral sclerosis; from diagnosis and during disease progression. J. Neurol. Sci. 361, 235–242. doi: 10.1016/j.jns.2015.12.042
Jakobsson Larsson, B., Ozanne, A. G., Nordin, K., and Nygren, I. (2017). A prospective study of quality of life in amyotrophic lateral sclerosis patients. Acta Neurol. Scand. 136, 631–638. doi: 10.1111/ane.12774
Jelsone-Swain, L., Persad, C., Votruba, K. L., Weisenbach, S. L., Johnson, T., Gruis, K. L., et al. (2012). The relationship between depressive symptoms, disease state, and cognition in amyotrophic lateral sclerosis. Front. Psychol. 3:542. doi: 10.3389/fpsyg.2012.00542
Kamminga, J., Leslie, F. V. C., Hsieh, S., Caga, J., Mioshi, E., Hornberger, M., et al. (2016). Syntactic comprehension deficits across the FTD-ALS continuum. Neurobiol. Aging 41, 11–18. doi: 10.1016/j.neurobiolaging.2016.02.002
Kasper, E., Schuster, C., Machts, J., Bittner, D., Vielhaber, S., Benecke, R., et al. (2015). Dysexecutive functioning in ALS patients and its clinical implications. Amyotroph. Lateral Scler. Front. Degener. 16, 160–171. doi: 10.3109/21678421.2015.1026267
Kasper, E., Schuster, C., Machts, J., Kaufmann, J., Bittner, D., Vielhaber, S., et al. (2014). Microstructural white matter changes underlying cognitive and behavioural impairment in ALS–an in vivo study using DTI. PLoS One 9:e114543. doi: 10.1371/journal.pone.0114543
Kasper, E., Zydatiss, K., Schuster, C., Machts, J., Bittner, D., Kaufmann, J., et al. (2016). No change in executive performance in ALS patients: a longitudinal neuropsychological study. Neurodegener. Dis. 16, 184–191. doi: 10.1159/000440957
Katon, W., Lin, E. H. B., and Kroenke, K. (2007). The association of depression and anxiety with medical symptom burden in patients with chronic medical illness. Gen. Hosp. Psychiatr. 29, 147–155. doi: 10.1016/j.genhosppsych.2006.11.005
Kilani, M., Micallef, J., Soubrouillard, C., Rey-Lardiller, D., Dematteï, C., Dib, M., et al. (2004). A longitudinal study of the evolution of cognitive function and affective state in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Mot. Neuron Disord. 5, 46–54. doi: 10.1080/14660820310017560
Kurt, A., Nijboer, F., Matuz, T., and Kübler, A. (2007). Depression and anxiety in individuals with amyotrophic lateral sclerosis: epidemiology and management. CNS Drugs 21, 279–291. doi: 10.2165/00023210-200721040-00003
Lazarus, R. S., and Folkman, S. (1984). Stress, Appraisal, and Coping. New York, NY: Springer Publishing Company.
Leslie, F. V. C., Hsieh, S., Caga, J., Savage, S. A., Mioshi, E., Hornberger, M., et al. (2015). Semantic deficits in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 16, 46–53. doi: 10.3109/21678421.2014.987301
Lillo, P., Mioshi, E., and Hodges, J. R. (2012). Caregiver burden in amyotrophic lateral sclerosis is more dependent on patients’ behavioral changes than physical disability: a comparative study. BMC Neurol. 12:156. doi: 10.1186/1471-2377-12-156
Lillo, P., Mioshi, E., Zoing, M. C., Kiernan, M. C., and Hodges, J. R. (2011). How common are behavioural changes in amyotrophic lateral sclerosis? Amyotroph. Lateral Scler. 12, 45–51. doi: 10.3109/17482968.2010.520718
Lou, J.-S., Reeves, A., Benice, T., and Sexton, G. (2003). Fatigue and depression are associated with poor quality of life in ALS. Neurology 60, 122–123. doi: 10.1212/01.wnl.0000042781.22278.0a
Lulé, D., Diekmann, V., Anders, S., Kassubek, J., Kübler, A., Ludolph, A. C., et al. (2007). Brain responses to emotional stimuli in patients with amyotrophic lateral sclerosis (ALS). J. Neurol. 254, 519–527. doi: 10.1007/s00415-006-0409-3
Lulé, D., Häcker, S., Ludolph, A., Birbaumer, N., and Kübler, A. (2008). Depression and quality of life in patients with amyotrophic lateral sclerosis. Dtsch. Arzteblatt Int. 105, 397–403. doi: 10.3238/arztebl.2008.0397
Lulé, D., Kurt, A., Jürgens, R., Kassubek, J., Diekmann, V., Kraft, E., et al. (2005). Emotional responding in amyotrophic lateral sclerosis. J. Neurol. 252, 1517–1524. doi: 10.1007/s00415-005-0907-8
Lulé, D., Pauli, S., Altintas, E., Singer, U., Merk, T., Uttner, I., et al. (2012). Emotional adjustment in amyotrophic lateral sclerosis (ALS). J. Neurol. 259, 334–341. doi: 10.1007/s00415-011-6191-x
Lulé, D. E., Aho-Özhan, H. E. A., Vázquez, C., Weiland, U., Weishaupt, J. H., Otto, M., et al. (2018). Story of the ALS-FTD continuum retold: rather two distinct entities. J. Neurol. Neurosurg. Psychiatr. 90, 586–589. doi: 10.1136/jnnp-2018-318800
Machts, J., Vielhaber, S., Kollewe, K., Petri, S., Kaufmann, J., and Schoenfeld, M. A. (2018). global hippocampal volume reductions and local CA1 shape deformations in amyotrophic lateral sclerosis. Front. Neurol. 9:565. doi: 10.3389/fneur.2018.00565
Marconi, A., Meloni, G., Fossati, F., Lunetta, C., Bastianello, S., Melazzini, M., et al. (2012). Aggressiveness, sexuality, and obsessiveness in late stages of ALS patients and their effects on caregivers. Amyotroph. Lateral Scler. 13, 452–458. doi: 10.3109/17482968.2012.696658
Matuz, T., Birbaumer, N., Hautzinger, M., and Kübler, A. (2010). Coping with amyotrophic lateral sclerosis: an integrative view. J. Neurol. Neurosurg. Psychiatr. 81, 893–898. doi: 10.1136/jnnp.2009.201285
Matuz, T., Birbaumer, N., Hautzinger, M., and Kübler, A. (2015). Psychosocial adjustment to ALS: a longitudinal study. Front. Psychol. 6:1197. doi: 10.3389/fpsyg.2015.01197
McElhiney, M., Rabkin, J. G., Goetz, R., Katz, J., Miller, R. G., Forshew, D. A., et al. (2014). Seeking a measure of clinically meaningful change in ALS. Amyotroph. Lateral Scler. Front. Degener. 15, 398–405. doi: 10.3109/21678421.2014.942668
McElhiney, M. C., Rabkin, J. G., Gordon, P. H., Goetz, R., and Mitsumoto, H. (2009). Prevalence of fatigue and depression in ALS patients and change over time. J. Neurol. Neurosurg. Psychiatr. 80, 1146–1149. doi: 10.1136/jnnp.2008.163246
Meier, S. L., Charleston, A. J., and Tippett, L. J. (2010). Cognitive and behavioural deficits associated with the orbitomedial prefrontal cortex in amyotrophic lateral sclerosis. Brain J. Neurol. 133, 3444–3457. doi: 10.1093/brain/awq254
Menke, R. A. L., Proudfoot, M., Talbot, K., and Turner, M. R. (2018). The two-year progression of structural and functional cerebral MRI in amyotrophic lateral sclerosis. NeuroImage Clin. 17, 953–961. doi: 10.1016/j.nicl.2017.12.025
Miglioretti, M., Mazzini, L., Oggioni, G. D., Testa, L., and Monaco, F. (2008). Illness perceptions, mood and health-related quality of life in patients with amyotrophic lateral sclerosis. J. Psychosom. Res. 65, 603–609. doi: 10.1016/j.jpsychores.2008.05.012
Mioshi, E., Caga, J., Lillo, P., Hsieh, S., Ramsey, E., Devenney, E., et al. (2014). Neuropsychiatric changes precede classic motor symptoms in ALS and do not affect survival. Neurology 82, 149–155. doi: 10.1212/WNL.0000000000000023
Mioshi, E., Lillo, P., Yew, B., Hsieh, S., Savage, S., Hodges, J. R., et al. (2013). Cortical atrophy in ALS is critically associated with neuropsychiatric and cognitive changes. Neurology 80, 1117–1123. doi: 10.1212/WNL.0b013e31828869da
Montuschi, A., Iazzolino, B., Calvo, A., Moglia, C., Lopiano, L., Restagno, G., et al. (2015). Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J. Neurol. Neurosurg. Psychiatr. 86, 168–173. doi: 10.1136/jnnp-2013-307223
Moos, R. H., and Holahan, C. J. (2007). “Adaptive tasks and methods of coping with illness and disability,” in Coping with Chronic Illness and Disability – Theoretical, Empirical, and Clinical Aspects. ed E. Martz and H. Livneh, (New York, NY: Springer), 107–126.
Moss-Morris, R. (2013). Adjusting to chronic illness: time for a unified theory. Br. J. Health Psychol. 18, 681–686. doi: 10.1111/bjhp.12072
Murphy, J., Factor-Litvak, P., Goetz, R., Lomen-Hoerth, C., Nagy, P. L., Hupf, J., et al. (2016). Cognitive-behavioral screening reveals prevalent impairment in a large multicenter ALS cohort. Neurology 86, 813–820. doi: 10.1212/WNL.0000000000002305
Murphy, J., Henry, R., and Lomen-Hoerth, C. (2007). Establishing subtypes of the continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch. Neurol. 64, 330–334. doi: 10.1001/archneur.64.3.330
Murphy, J. M., Henry, R. G., Langmore, S., Kramer, J. H., Miller, B. L., and Lomen-Hoerth, C. (2007). Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch. Neurol. 64, 530–534. doi: 10.1001/archneur.64.4.530
Neary, D., Snowden, J. S., and Mann, D. M. (2000). Cognitive change in motor neurone disease/amyotrophic lateral sclerosis (MND/ALS). J. Neurol. Sci. 180, 15–20. doi: 10.1016/s0022-510x(00)00425-1
Norris, L., Que, G., and Bayat, E. (2010). Psychiatric aspects of amyotrophic lateral sclerosis (ALS). Curr. Psychiatry Rep. 12, 239–245. doi: 10.1007/s11920-010-0118-6
Oh, S. I., Oh, K. W., Kim, H. J., Park, J. S., and Kim, S. H. (2016). Impaired perception of emotional expression in amyotrophic lateral sclerosis. J. Clin. Neurol. Seoul Korea 12, 295–300. doi: 10.3988/jcn.2016.12.3.295
Ohta, Y., Sato, K., Takemoto, M., Takahashi, Y., Morihara, R., Nakano, Y., et al. (2017). Behavioral and affective features of amyotrophic lateral sclerosis patients. J. Neurol. Sci. 381, 119–125. doi: 10.1016/j.jns.2017.08.024
Olney, R. K., Murphy, J., Forshew, D., Garwood, E., Miller, B. L., Langmore, S., et al. (2005). The effects of executive and behavioral dysfunction on the course of ALS. Neurology 65, 1774–1777. doi: 10.1212/01.wnl.0000188759.87240.8b
Pagnini, F., Simmons, Z., Corbo, M., and Molinari, E. (2012). Amyotrophic lateral sclerosis: time for research on psychological intervention? Amyotroph. Lateral Scler. 13, 416–417. doi: 10.3109/17482968.2011.653572
Palmieri, A., Mento, G., Calvo, V., Querin, G., D’Ascenzo, C., Volpato, C., et al. (2015). Female gender doubles executive dysfunction risk in ALS: a case-control study in 165 patients. J. Neurol. Neurosurg. Psychiatr. 86, 574–579. doi: 10.1136/jnnp-2014-307654
Palmieri, A., Naccarato, M., Abrahams, S., Bonato, M., D’Ascenzo, C., Balestreri, S., et al. (2010). Right hemisphere dysfunction and emotional processing in ALS: an fMRI study. J. Neurol. 257, 1970–1978. doi: 10.1007/s00415-010-5640-2
Papps, B., Abrahams, S., Wicks, P., Leigh, P. N., and Goldstein, L. H. (2005). Changes in memory for emotional material in amyotrophic lateral sclerosis (ALS). Neuropsychologia 43, 1107–1114. doi: 10.1016/j.neuropsychologia.2004.11.027
Phukan, J., Elamin, M., Bede, P., Jordan, N., Gallagher, L., Byrne, S., et al. (2012). The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J. Neurol. Neurosurg. Psychiatr. 83, 102–108. doi: 10.1136/jnnp-2011-300188
Pisa, F. E., Logroscino, G., Casetta, A., Cecotti, L., Verriello, L., Bratina, A., et al. (2015). The use of antidepressant medication before and after the diagnosis of amyotrophic lateral sclerosis: a population-based cohort study. Neuroepidemiology 44, 91–98. doi: 10.1159/000374119
Poletti, B., Solca, F., Carelli, L., Faini, A., Madotto, F., Lafronza, A., et al. (2018). Cognitive-behavioral longitudinal assessment in ALS: the Italian Edinburgh Cognitive and Behavioral ALS screen (ECAS). Amyotroph. Lateral Scler. Front. Degener. 19, 387–395. doi: 10.1080/21678421.2018.1473443
Raaphorst, J., de Visser, M., Linssen, W. H. J. P., de Haan, R. J., and Schmand, B. (2010). The cognitive profile of amyotrophic lateral sclerosis: a meta-analysis. Amyotroph. Lateral Scler. 11, 27–37. doi: 10.3109/17482960802645008
Raaphorst, J., van Tol, M. J., de Visser, M., van der Kooi, A. J., Majoie, C. B., van den Berg, L. H., et al. (2015). Prose memory impairment in amyotrophic lateral sclerosis patients is related to hippocampus volume. Eur. J. Neurol. 22, 547–554. doi: 10.1111/ene.12615
Rabkin, J., Goetz, R., Murphy, J. M., Factor-Litvak, P., Mitsumoto, H., and Als Cosmos Study Group, (2016). Cognitive impairment, behavioral impairment, depression, and wish to die in an ALS cohort. Neurology 87, 1320–1328. doi: 10.1212/WNL.0000000000003035
Rabkin, J. G., Albert, S. M., Del Bene, M. L., O’Sullivan, I., Tider, T., Rowland, L. P., et al. (2005). Prevalence of depressive disorders and change over time in late-stage ALS. Neurology 65, 62–67. doi: 10.1212/01.wnl.0000167187.14501.0c
Rabkin, J. G., Albert, S. M., Rowland, L. P., and Mitsumoto, H. (2009). How common is depression among ALS caregivers? a longitudinal study. Amyotroph. Lateral Scler. 10, 448–455. doi: 10.1080/17482960802459889
Rabkin, J. G., Goetz, R., Factor-Litvak, P., Hupf, J., McElhiney, M., Singleton, J., et al. (2015). Depression and wish to die in a multicenter cohort of ALS patients. Amyotroph. Lateral Scler. Front. Degener. 16, 265–273. doi: 10.3109/21678421.2014.980428
Rabkin, J. G., Wagner, G. J., and Del Bene, M. (2000). Resilience and distress among amyotrophic lateral sclerosis patients and caregivers. Psychosom. Med. 62, 271–279. doi: 10.1097/00006842-200003000-00020
Radakovic, R., Stephenson, L., Newton, J., Crockford, C., Swingler, R., Chandran, S., et al. (2017). Multidimensional apathy and executive dysfunction in amyotrophic lateral sclerosis. Cortex J. Devoted Study Nerv. Syst. Behav. 94, 142–151. doi: 10.1016/j.cortex.2017.06.023
Ringholz, G. M., Appel, S. H., Bradshaw, M., Cooke, N. A., Mosnik, D. M., and Schulz, P. E. (2005). Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65, 586–590. doi: 10.1212/01.wnl.0000172911.39167.b6
Roach, A. R., Averill, A. J., Segerstrom, S. C., and Kasarskis, E. J. (2009). The dynamics of quality of life in ALS patients and caregivers. Ann. Behav. Med. Publ. Soc. Behav. Med. 37, 197–206. doi: 10.1007/s12160-009-9092-9
Roberts-South, A., Findlater, K., Strong, M. J., and Orange, J. B. (2012). Longitudinal changes in discourse production in amyotrophic lateral sclerosis. Semin. Speech Lang. 33, 79–94. doi: 10.1055/s-0031-1301165
Robinson, K. M., Lacey, S. C., Grugan, P., Glosser, G., Grossman, M., and McCluskey, L. F. (2006). Cognitive functioning in sporadic amyotrophic lateral sclerosis: a six month longitudinal study. J. Neurol. Neurosurg. Psychiatr. 77, 668–670. doi: 10.1136/jnnp.2005.073403
Roos, E., Mariosa, D., Ingre, C., Lundholm, C., Wirdefeldt, K., Roos, P. M., et al. (2016). Depression in amyotrophic lateral sclerosis. Neurology 86, 2271–2277. doi: 10.1212/WNL.0000000000002671
Roy-Bellina, S., Brunel, H., Almohsen, C., Gely-Nargeot, M., Carton, S., and Camu, W. (2008). Alexithymia in amyotrophic lateral sclerosis. Neuropsychol Approach. J. Neurol. 255:51.
Saberi, S., Stauffer, J. E., Schulte, D. J., and Ravits, J. (2015). neuropathology of amyotrophic lateral sclerosis and its variants. Neurol. Clin. 33, 855–876. doi: 10.1016/j.ncl.2015.07.012
Sandstedt, P., Johansson, S., Ytterberg, C., Ingre, C., Holmqvist, L. W., and Kierkegaard, M. (2016). Predictors of health-related quality of life in people with amyotrophic lateral sclerosis. J. Neurol. Sci. 370, 269–273. doi: 10.1016/j.jns.2016.09.034
Sarro, L., Agosta, F., Canu, E., Riva, N., Prelle, A., Copetti, M., et al. (2011). Cognitive functions and white matter tract damage in amyotrophic lateral sclerosis: a diffusion tensor tractography study. AJNR Am. J. Neuroradiol. 32, 1866–1872. doi: 10.3174/ajnr.A2658
Schreiber, H., Gaigalat, T., Wiedemuth-Catrinescu, U., Graf, M., Uttner, I., Muche, R., et al. (2005). Cognitive function in bulbar- and spinal-onset amyotrophic lateral sclerosis. a longitudinal study in 52 patients. J. Neurol. 252, 772–781. doi: 10.1007/s00415-005-0739-6
Schuster, C., Kasper, E., Dyrba, M., Machts, J., Bittner, D., Kaufmann, J., et al. (2014a). Cortical thinning and its relation to cognition in amyotrophic lateral sclerosis. Neurobiol. Aging 35, 240–246. doi: 10.1016/j.neurobiolaging.2013.07.020
Schuster, C., Kasper, E., Machts, J., Bittner, D., Kaufmann, J., Benecke, R., et al. (2014b). Longitudinal course of cortical thickness decline in amyotrophic lateral sclerosis. J. Neurol. 261, 1871–1880. doi: 10.1007/s00415-014-7426-4
Sedda, A. (2014). Disorders of emotional processing in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 27, 659–665. doi: 10.1097/WCO.0000000000000147
Siciliano, M., Santangelo, G., Trojsi, F., Di Somma, C., Patrone, M., Femiano, C., et al. (2017). Coping strategies and psychological distress in caregivers of patients with amyotrophic lateral sclerosis (ALS). Amyotroph. Lateral Scler. Front. Degener. 18, 367–377. doi: 10.1080/21678421.2017.1285316
Simmons, Z. (2015). Patient-perceived outcomes and quality of life in ALS. Neurother. J. Am. Soc. Exp. Neurother. 12, 394–402. doi: 10.1007/s13311-014-0322-x
Sterling, L. E., Jawaid, A., Salamone, A. R., Murthy, S. B., Mosnik, D. M., McDowell, E., et al. (2010). Association between dysarthria and cognitive impairment in ALS: a prospective study. Amyotroph. Lateral Scler. 11, 46–51. doi: 10.3109/17482960903207997
Stojkovic, T., Stefanova, E., Pekmezovic, T., Peric, S., and Stevic, Z. (2016). Executive dysfunction and survival in patients with amyotrophic lateral sclerosis: preliminary report from a serbian centre for motor neuron disease. Amyotroph. Lateral Scler. Front. Degener. 17, 543–547. doi: 10.1080/21678421.2016.1211148
Strong, M. J. (2017). Revisiting the concept of amyotrophic lateral sclerosis as a multisystems disorder of limited phenotypic expression. Curr. Opin. Neurol. 30, 599–607. doi: 10.1097/WCO.0000000000000488
Strong, M. J., Grace, G. M., Orange, J. B., Leeper, H. A., Menon, R. S., and Aere, C. (1999). A prospective study of cognitive impairment in ALS. Neurology 53, 1665–1670.
Strutt, A. M., Palcic, J., Wager, J. G., Titus, C., Macadam, C., Brown, J., et al. (2012). Cognition, behavior, and respiratory function in amyotrophic lateral sclerosis. ISRN Neurol. 2012:912123. doi: 10.5402/2012/912123
Taylor, L., Wicks, P., Leigh, P. N., and Goldstein, L. H. (2010). Prevalence of depression in amyotrophic lateral sclerosis and other motor disorders. Eur. J. Neurol. 17, 1047–1053. doi: 10.1111/j.1468-1331.2010.02960.x
Terada, T., Obi, T., Yoshizumi, M., Murai, T., Miyajima, H., and Mizoguchi, K. (2011). Frontal lobe-mediated behavioral changes in amyotrophic lateral sclerosis: are they independent of physical disabilities? J. Neurol. Sci. 309, 136–140. doi: 10.1016/j.jns.2011.06.049
Thakore, N. J., and Pioro, E. P. (2016). Depression in ALS in a large self-reporting cohort. Neurology 86, 1031–1038. doi: 10.1212/WNL.0000000000002465
Tremolizzo, L., Pellegrini, A., Susani, E., Lunetta, C., Woolley, S. C., Ferrarese, C., et al. (2016). Behavioural but not cognitive impairment is a determinant of caregiver burden in amyotrophic lateral sclerosis. Eur. Neurol. 75, 191–194. doi: 10.1159/000445110
Trojsi, F., Di Nardo, F., Santangelo, G., Siciliano, M., Femiano, C., Passaniti, C., et al. (2017). Resting state fMRI correlates of Theory of Mind impairment in amyotrophic lateral sclerosis. Cortex 97, 1–16. doi: 10.1016/j.cortex.2017.09.016
Trojsi, F., Siciliano, M., Russo, A., Passaniti, C., Femiano, C., Ferrantino, T., et al. (2016). Theory of mind and its neuropsychological and quality of life correlates in the early stages of amyotrophic lateral sclerosis. Front. Psychol. 7:1934. doi: 10.3389/fpsyg.2016.01934
Tsujimoto, M., Senda, J., Ishihara, T., Niimi, Y., Kawai, Y., Atsuta, N., et al. (2011). Behavioral changes in early ALS correlate with voxel-based morphometry and diffusion tensor imaging. J. Neurol. Sci. 307, 34–40. doi: 10.1016/j.jns.2011.05.025
Turner, M. R., Goldacre, R., Talbot, K., and Goldacre, M. J. (2016). Psychiatric disorders prior to amyotrophic lateral sclerosis. Ann. Neurol. 80, 935–938. doi: 10.1002/ana.24801
Unglik, J., Bungener, C., Delgadillo, D., Salachas, F., Pradat, P. F., Bruneteau, G., et al. (2018). Emotional feeling in patients suffering from amyotrophic lateral sclerosis. Geriatr. Psychol. Neuropsychiatr. Vieil. 16, 414–422. doi: 10.1684/pnv.2018.0762
van der Hulst, E.-J., Bak, T. H., and Abrahams, S. (2015). Impaired affective and cognitive theory of mind and behavioural change in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 86, 1208–1215. doi: 10.1136/jnnp-2014-309290
Verschueren, A., Kianimehr, G., Belingher, C., Salort-Campana, E., Loundou, A., Grapperon, A.-M., et al. (2018). Wish to die and reasons for living among patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 20, 68–73. doi: 10.1080/21678421.2018.1530265
Verstraete, E., Veldink, J. H., Hendrikse, J., Schelhaas, H. J., van den Heuvel, M. P., and van den Berg, L. H. (2012). Structural MRI reveals cortical thinning in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 83, 383–388. doi: 10.1136/jnnp-2011-300909
Vignola, A., Guzzo, A., Calvo, A., Moglia, C., Pessia, A., Cavallo, E., et al. (2008). Anxiety undermines quality of life in ALS patients and caregivers. Eur. J. Neurol. 15, 1231–1236. doi: 10.1111/j.1468-1331.2008.02303.x
Watermeyer, T. J., Brown, R. G., Sidle, K. C. L., Oliver, D. J., Allen, C., Karlsson, J., et al. (2015). Executive dysfunction predicts social cognition impairment in amyotrophic lateral sclerosis. J. Neurol. 262, 1681–1690. doi: 10.1007/s00415-015-7761-0
Wei, Q., Chen, X., Cao, B., Ou, R., Zhao, B., Wu, Y., et al. (2016). Associations between neuropsychiatric symptoms and cognition in Chinese patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 17, 358–365. doi: 10.3109/21678421.2016.1154574
Wicks, P., Abrahams, S., Masi, D., Hejda-Forde, S., Leigh, P. N., and Goldstein, L. H. (2007). Prevalence of depression in a 12-month consecutive sample of patients with ALS. Eur. J. Neurol. 14, 993–1001. doi: 10.1111/j.1468-1331.2007.01843.x
Witgert, M., Salamone, A. R., Strutt, A. M., Jawaid, A., Massman, P. J., Bradshaw, M., et al. (2010). Frontal-lobe mediated behavioral dysfunction in amyotrophic lateral sclerosis. Eur. J. Neurol. 17, 103–110. doi: 10.1111/j.1468-1331.2009.02801.x
Woolley, S., Goetz, R., Factor-Litvak, P., Murphy, J., Hupf, J., Lomen-Hoerth, C., et al. (2018). Longitudinal screening detects cognitive stability and behavioral deterioration in ALS patients. Behav. Neurol. 2018:5969137. doi: 10.1155/2018/5969137
Woolley, S. C., and Katz, J. S. (2008). Cognitive and behavioral impairment in amyotrophic lateral sclerosis. Phys. Med. Rehabil. Clin. N. Am. 19, 607–617. doi: 10.1016/j.pmr.2008.04.002
Woolley, S. C., Moore, D. H., and Katz, J. S. (2010). Insight in ALS: awareness of behavioral change in patients with and without FTD. Amyotroph. Lateral Scler. 11, 52–56. doi: 10.3109/17482960903171110
Woolley, S. C., and Rush, B. K. (2017). Considerations for clinical neuropsychological evaluation in amyotrophic lateral sclerosis. Arch. Clin. Neuropsychol. 32, 906–916. doi: 10.1093/arclin/acx089
Woolley, S. C., Zhang, Y., Schuff, N., Weiner, M. W., and Katz, J. S. (2011). Neuroanatomical correlates of apathy in ALS using 4 Tesla diffusion tensor MRI. Amyotroph. Lateral Scler. 12, 52–58. doi: 10.3109/17482968.2010.521842
Xu, Z., Alruwaili, A. R. S., Henderson, R. D., and McCombe, P. A. (2017). Screening for cognitive and behavioural impairment in amyotrophic lateral sclerosis: frequency of abnormality and effect on survival. J. Neurol. Sci. 376, 16–23. doi: 10.1016/j.jns.2017.02.061
Zago, S., Poletti, B., Morelli, C., Doretti, A., and Silani, V. (2011). Amyotrophic lateral sclerosis and frontotemporal dementia (ALS-FTD). Arch. Ital. Biol. 149, 39–56. doi: 10.4449/aib.v149i1.1263
Zalonis, I., Christidi, F., Paraskevas, G., Zabelis, T., Evdokimidis, I., and Kararizou, E. (2012). Can executive cognitive measures differentiate between patients with spinal- and bulbar-onset amyotrophic lateral sclerosis? Arch. Clin. Neuropsychol. 27, 348–354. doi: 10.1093/arclin/acs031
Keywords: amyotrophic lateral sclerosis, extra-motor manifestations, cognition, emotion, psychological adjustment, coping
Citation: Benbrika S, Desgranges B, Eustache F and Viader F (2019) Cognitive, Emotional and Psychological Manifestations in Amyotrophic Lateral Sclerosis at Baseline and Overtime: A Review. Front. Neurosci. 13:951. doi: 10.3389/fnins.2019.00951
Received: 28 March 2019; Accepted: 22 August 2019;
Published: 10 September 2019.
Edited by:
Francesca Trojsi, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Judith Machts, Universitätsklinikum Magdeburg, GermanyJohannes Prudlo, Rostock University Hospital, Germany
Copyright © 2019 Benbrika, Desgranges, Eustache and Viader. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Béatrice Desgranges, YmVhdHJpY2UuZGVzZ3Jhbmdlc0B1bmljYWVuLmZy