 Yanyan Zhang1
Yanyan Zhang1 Jie Li1Rong Bai1
Jie Li1Rong Bai1 Jianping Wang2Tao Peng1Lijie Chen1Jingtao Wang1Yanru Liu1Tian Tian1*Hong Lu1*
Jianping Wang2Tao Peng1Lijie Chen1Jingtao Wang1Yanru Liu1Tian Tian1*Hong Lu1*- 1Department of Neurology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Department of Neurology, The Fifth Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Adult-onset autosomal dominant leukodystrophy (ADLD) is a lately described rare form of leukodystrophy with only one family report from China. As the only disease associated with increased lamina B1 encoded by LMNB1, ADLDs have different clinical presentations, ranging from autonomic to pyramidal tract and cerebellar ataxia. Here, we report a case of ADLD that presented with positional tremor as the initial symptom. T2-weighted brain MRI showed brain atrophy and diffuse high signal intensity of the cerebral white matter and the brain stem. The precise diagnosis was made by identification of the mutated gene. To the best of our knowledge, this is perhaps the first case report of ADLD presenting as tremor in China.
Introduction
Leukodystrophies refer to a series of rare genetic, progressive disorders primarily characterized by demyelination or hypomyelination of the central nervous system (CNS), representing approximately the epidemiological frequency of 1/50,000-1/7500 (Kohler et al., 2018). Most leukodystrophies display early onset during infancy and childhood with commonly autosomal recessive or X-linked recessive inheritance. In contrast, only few are currently defined as adulthood leukodystrophies, belonging to dominantly inherited diseases. Adult-onset autosomal dominant leukodystrophy (ADLD) is an autosomal dominant inherited demyelinating disorder, with progressive loss of white matter (WM) in the CNS. Autosomal dominant leukodystrophy is an ultra-rare neurodegenerative disease. Eldridge described it for the first time in Eldridge et al. (1984). Until now, very few number of cases from the United States, Ireland, Sweden, Italy, etc. have been reported (Mezaki et al., 1984; Schuster et al., 1984; Brussino et al., 2009; Dos Santos et al., 2012; Molloy et al., 2012). Evidences to date have pointed that ADLD has been identified as the result of overexpression of lamina B1, and it is the first and only neurological disease related to LMNB1, located on chromosome 5q23-31 (Coffeen et al., 2000; Marklund et al., 2006; Padiath et al., 2006; Meijer et al., 2008; Brussino et al., 2010). Under pathological conditions, duplication and deletion upstream of LMNB1 are two different mechanisms of ADLD to result in the increasing expression and accumulation of laminB1 (Mezaki et al., 1984; Giorgio et al., 2015; Padiath, 2015; Nmezi et al., 2019). LaminB1 is a form of nuclear protein and involved in maintaining nuclear integrity and cellular metabolism processes. Its overexpression can lead to a variety of potent effects, including abnormal development of myelin and alterations in nuclear membrane proteins, nucleus integrity, DNA expression, and localization of nuclear envelope proteins (Ferrera et al., 2014; Bartoletti-Stella et al., 2015; Padiath, 2015, 2016; Giacomini et al., 2016; Liu et al., 2018). Previous studies have further proved that the increased laminB1 appears in different cell types of brain, fibroblasts, or peripheral blood (Brussino et al., 2010), especially oligodendrocytes (Padiath, 2015; Rolyan et al., 2015; Lo Martire et al., 2018). In addition, histology studies revealed loss myelin of cerebral and cerebellar, modest reactive gliosis, preservation of oligodendrocytes, and sparing of inflammation (Coffeen et al., 2000; Melberg et al., 2006).
The onset of ADLD often commences at the age of the fourth or fifth decade with autonomic dysfunction, occurring simultaneously with or followed by pyramidal abnormalities and cerebellar signs (Padiath and Fu, 2010; Lin et al., 2011; Brunetti et al., 2014; Finnsson et al., 2015; Terlizzi et al., 2016; Dai et al., 2017). In several atypical patients, however, autonomic dysfunction could occur after somatic motor dysfunction, or fail to be detected (Brussino et al., 2010; Giorgio et al., 2013; Potic et al., 2013). Some updated reports pointed out that cognitive impairment, auditory or visual abnormalities, cardiovascular and skin noradrenergic failure, and REM sleep behavior disorder (RBD) may be clinical features of ADLD (Guaraldi et al., 2011; Flanagan et al., 2013; Laforce et al., 2013; Sandoval-Rodriguez et al., 2017; Table 1). Compared with the onset of clinical symptoms, MRI findings have been observed about a decade earlier (Finnsson et al., 2015). On conventional MRI, ADLDs are characterized by diffuse and symmetrical lesions in WM and cerebellar peduncles, accompanied by the less-effected periventricular region, optic radiations, and U-fibers (Melberg et al., 2006; Brunetti et al., 2014; Corlobe et al., 2015; Finnsson et al., 2015; Zanigni et al., 2015). Several reports have indicated previously that bilateral abnormal signals in corticospinal tracts, internal capsule, corpus callosum, lemniscus medialis, corticonuclear tracts, and cerebellar peduncles have been observed (Melberg et al., 2006; Finnsson et al., 2013; Potic et al., 2013; Corlobe et al., 2015). Recent studies evidenced decreased brain WM metabolism and pathological sediments of lactate in lateral ventricle CSF in using single-voxel proton-MR Spectroscopy (1H-MRS) (Finnsson et al., 2013, 2019; Zanigni et al., 2015). Furthermore, ADLD is a progressive and fatal disease, and affected people usually survive for 10–20 years after the onset of symptoms (Giorgio et al., 2013; Finnsson et al., 2015). A research demonstrated that the patient with duplication and deletion upstream of LMNB1 exhibited earlier onset and more severe clinical symptoms (Mezaki et al., 1984).
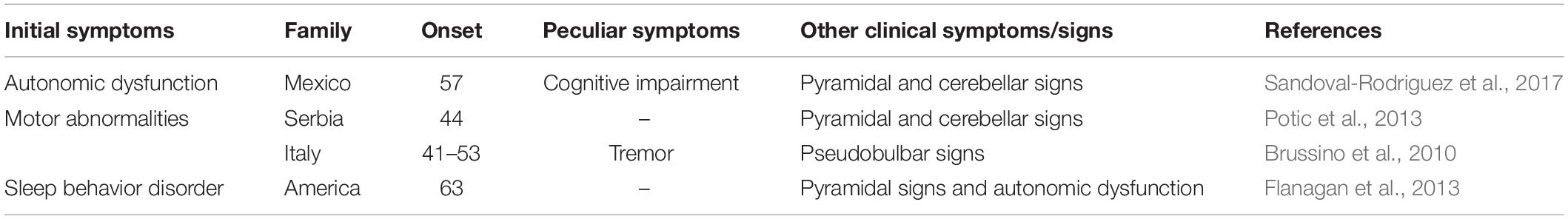
Table 1. The clinical manifestations of variant ADLD patients.
The aim of our study was to report a Chinese ADLD patient presenting with postural tremor of the arms as an initial symptom. Brain MRI showed characteristic WM lesion and CNS atrophy.
Clinical Data
The subject came from a family in Northern China. Written informed consent was obtained from the parents or guardians of the participant for the publication of this case report, and the study was conducted in accordance with the principles of the Declaration of Helsinki and relevant policies in China.
The proband (II-4) had been in good health until she presented mild tremor of hands when she was 58 years old. Firstly, she had the difficulty in using her hands with involuntary movement, soon after gait imbalance, urinary incontinence, and constipation. She was diagnosed as Parkinsonism in another hospital, but symptoms still slowly deteriorated. At the age of 61 years old, she was observed with speech slowly and mild inarticulate speech. Moreover, family history revealed that her father and elder sister had similar gait impairment at least 10 years before death. Her 60-year-old brother had several years’ history of autonomic symptoms and gait disturbance (Table 2), and her nephew was reported to have difficulty walking, but further details were not available.
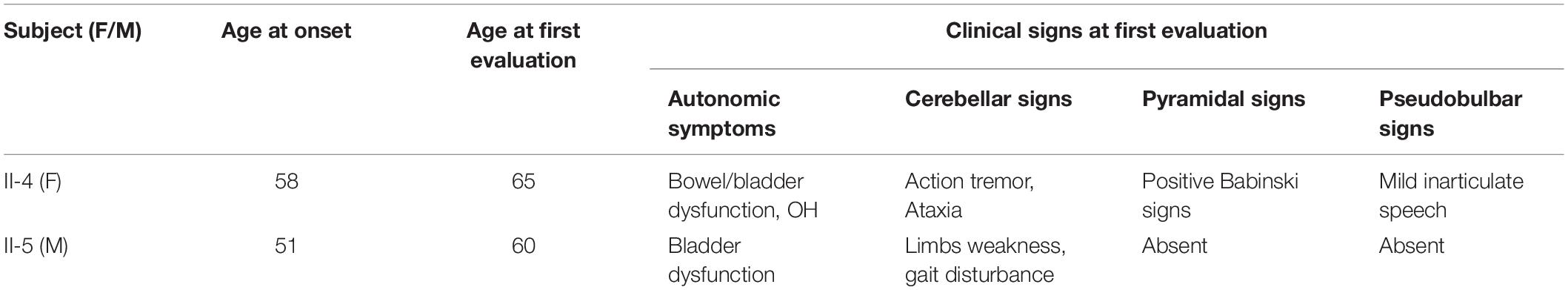
Table 2. ADLD age and clinical features at onset.
Comprehensive clinical evaluations were performed when the proband presented on our hospital for diagnosis and treatment. The physical examination on admission revealed that blood pressure was 153/93 and 143/79 mmHg at rest and after standing for 3 min, respectively. On neurological examination, she had postural tremor of all extremities, and increasing frequency and intensity of involuntary movement when keeping arms flat or when in a standing posture. The proband had slightly increased muscle tone and mild spasticity in the lower limbs, but the muscle strength of all extremities was normal. Bilateral-finger-nose and rapid rotation tests, Babinski signs, and Romberg signs were positive. Tilt test result was also positive. Cognitive function measured by Mini-mental state examination (MMSE) was normal.
The brain MRI scan showed diffuse and symmetrical T2-hyperintense lesions in WM with less affected periventricular rims (Figures 1, 2). Similarly, the cortical spinal tract, cortical nuclear tract, medial thalamus system, and cerebellar foot presented pathological signals on T2-weighted fluid-attenuated inversion-recovery (FLAIR) images (Figure 3). On cross-sectional MRI, the result revealed mild withering of the cerebellum, brain stem, and cerebrum, and diffuse spinal cord atrophy (Figures 4, 5). On the other hand, the genetic tests were in accordance with a diagnosis of ADLD, showing a duplication spanning the entire LMNB1 gene on chromosome 5q in the proband and her younger brother (Figure 6, II-4, II-5).
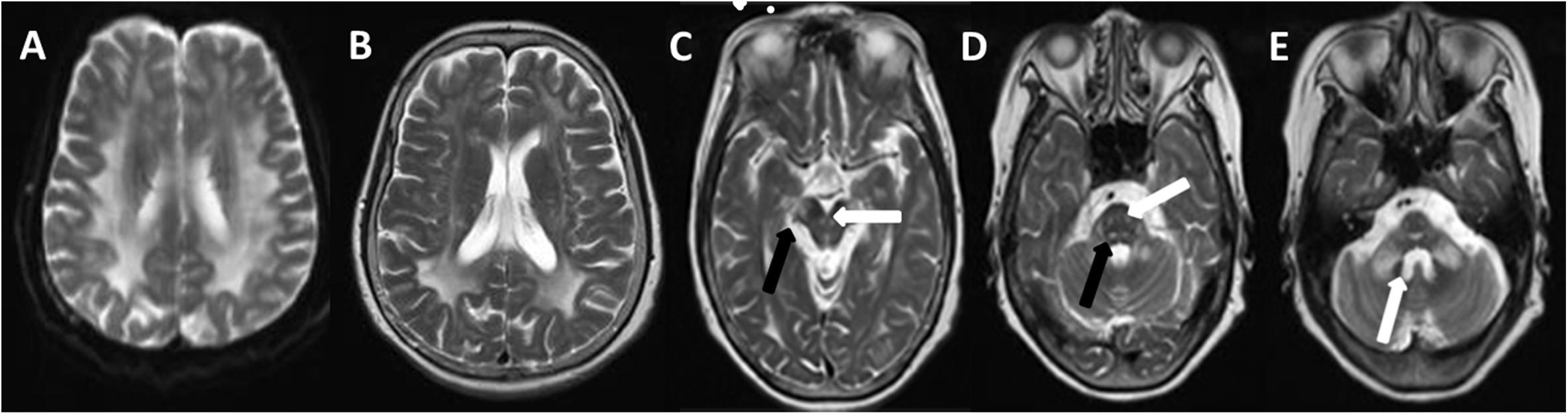
Figure 1. T2W (T2-weighted) changes in the proband. Diffuse hyperintensity of T2W in the white matter of frontals, parietal lobes, basal ganglia, corpus callosum, occipital lobe (A,B), mesencephalon (C, arrow), pons, and pons- and cerebellopontine-binding arms (D, arrow). The medial lemniscuses, the degustation of the superior cerebellar peduncles (C, solid arrow), pyramidal tracts, corticonuclear tracts (D, solid arrow), med peduncles, and cerebellar peduncles (E, arrow) were also affected.
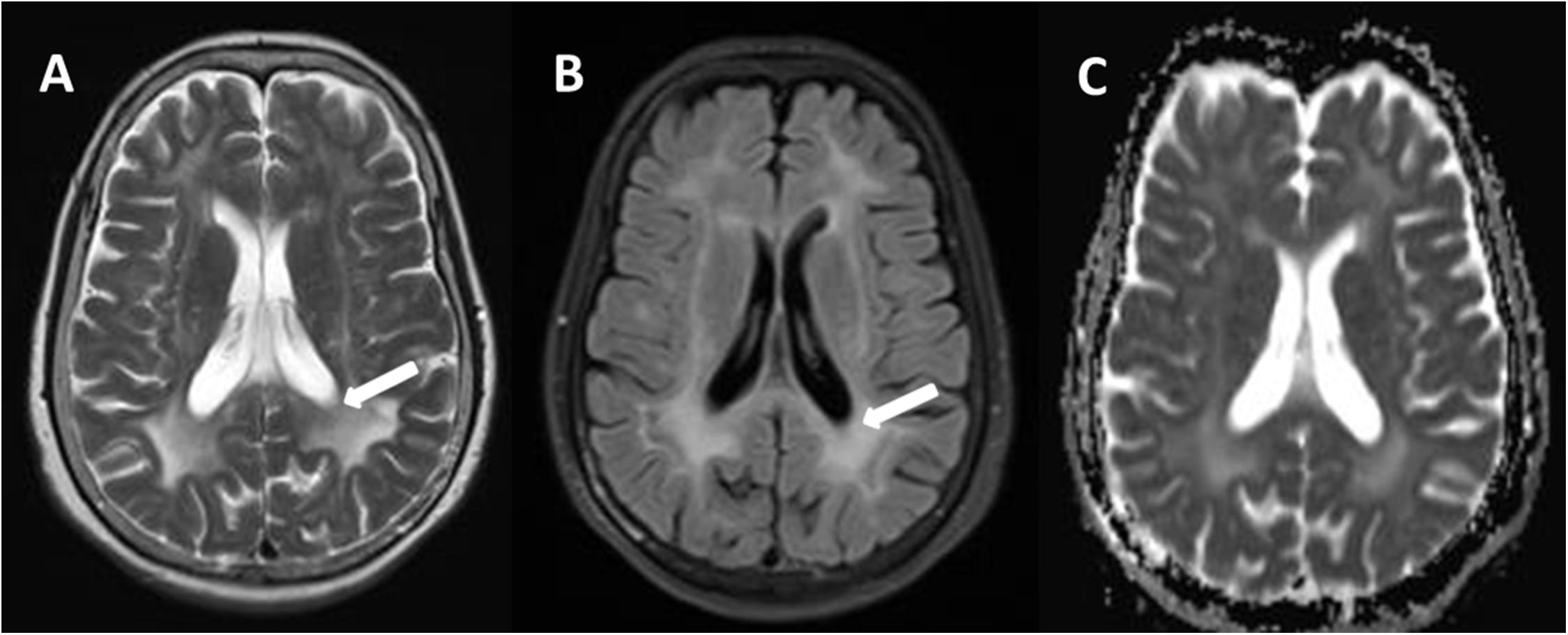
Figure 2. Comparison of a T2W (A) and fluid-attenuated inversion-recovery (FLAIR) (B), and apparent diffusion coefficient (ADC) (C) in transverse (axial) cross section through hemispheres at lateral ventricles. There are relative preservation of the periventricular region (A, arrows) and sparing of the subcortical U fibers on T2W. Compared to T2W, intensity of periventricular rims is suppressed in the most severely affected areas (B, arrows). The hyperintense lesion areas in ADC were smaller on T2W (C).
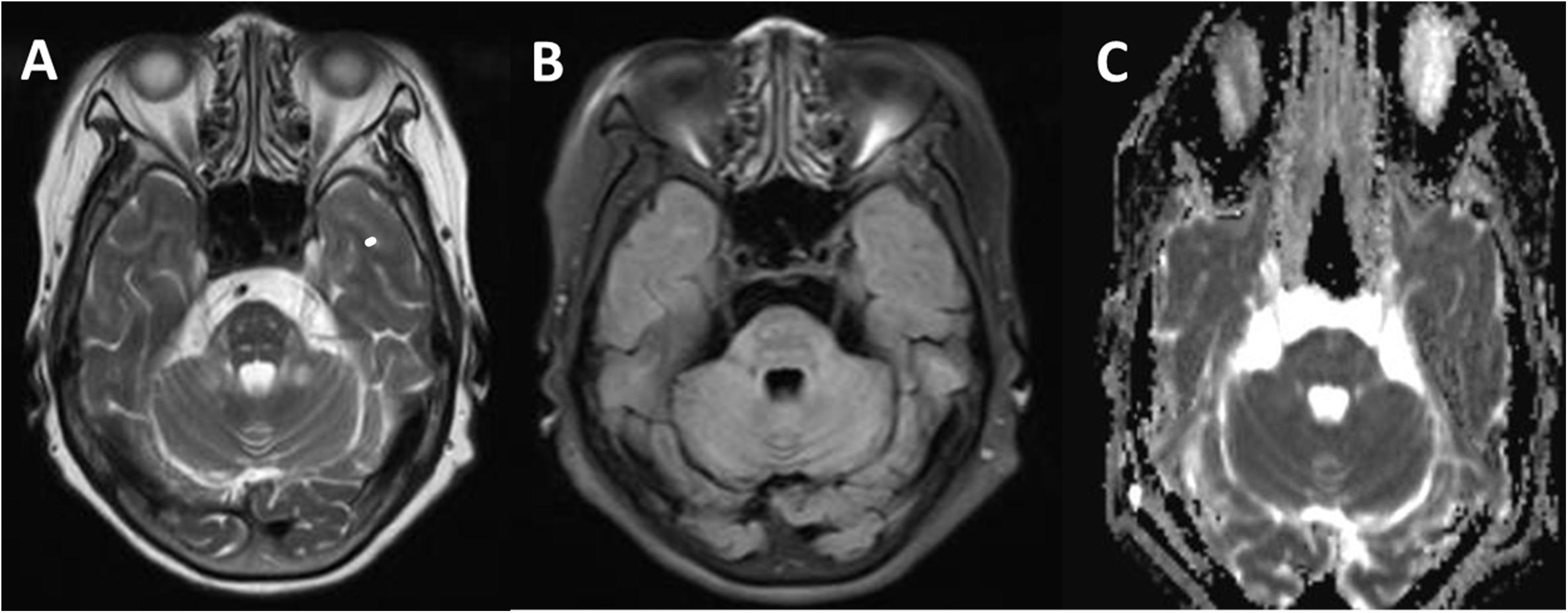
Figure 3. Comparison of a T2W (A), fluid-attenuated inversion-recovery (FLAIR) (B), and apparent diffusion coefficient (ADC) (C) in transverse section of the pituitary gland. Signal alterations were extended to pyramidal tracts, corticonuclear tracts, medial lemniscuses, and the degustation of the superior cerebellar peduncles (A). These signals above lesion areas were suppressed on FLAIR and not affected on the ADC map (C).
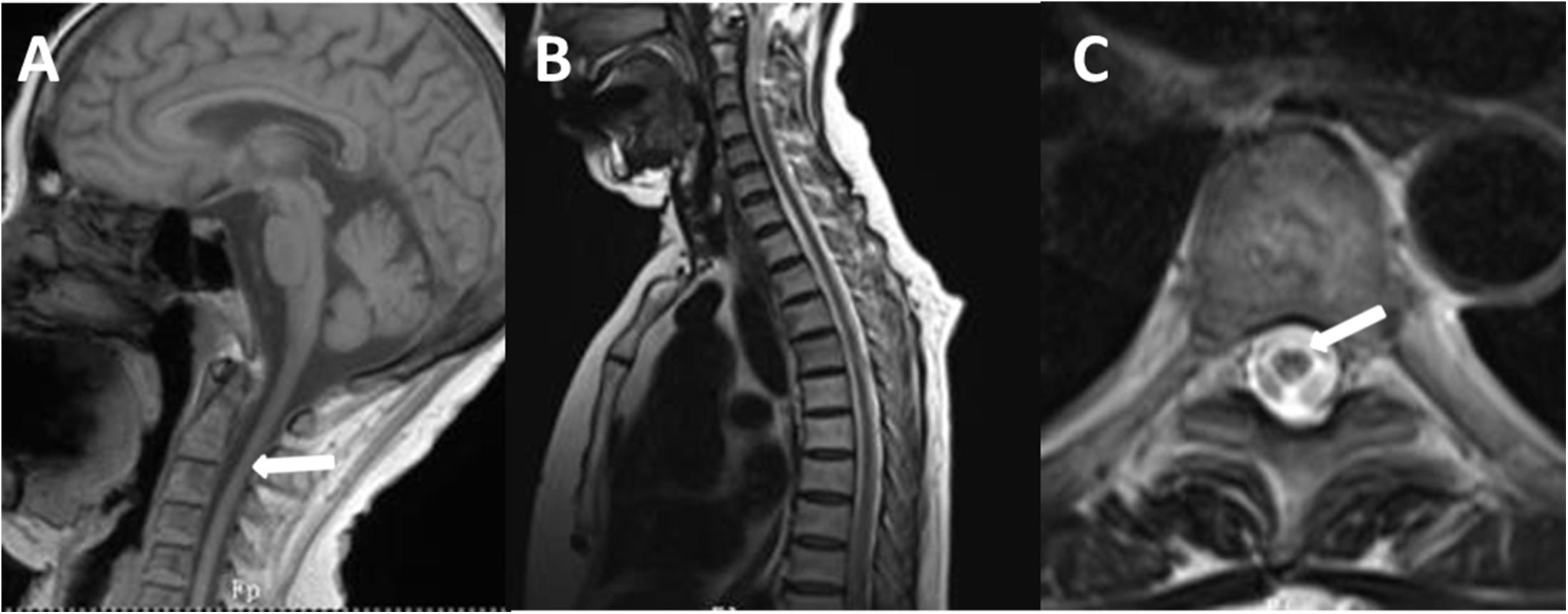
Figure 4. T1-weighted spin-echo image (T1W) (A), T2W (B), and cross-sectional (C) MR image of the spinal cord of the proband. T1W on sagittal surface and cross-sectional MRI showed diffuse withering, with accompanying abnormal signal in white matter of thoracic one to four level spinal cord on T2WI.
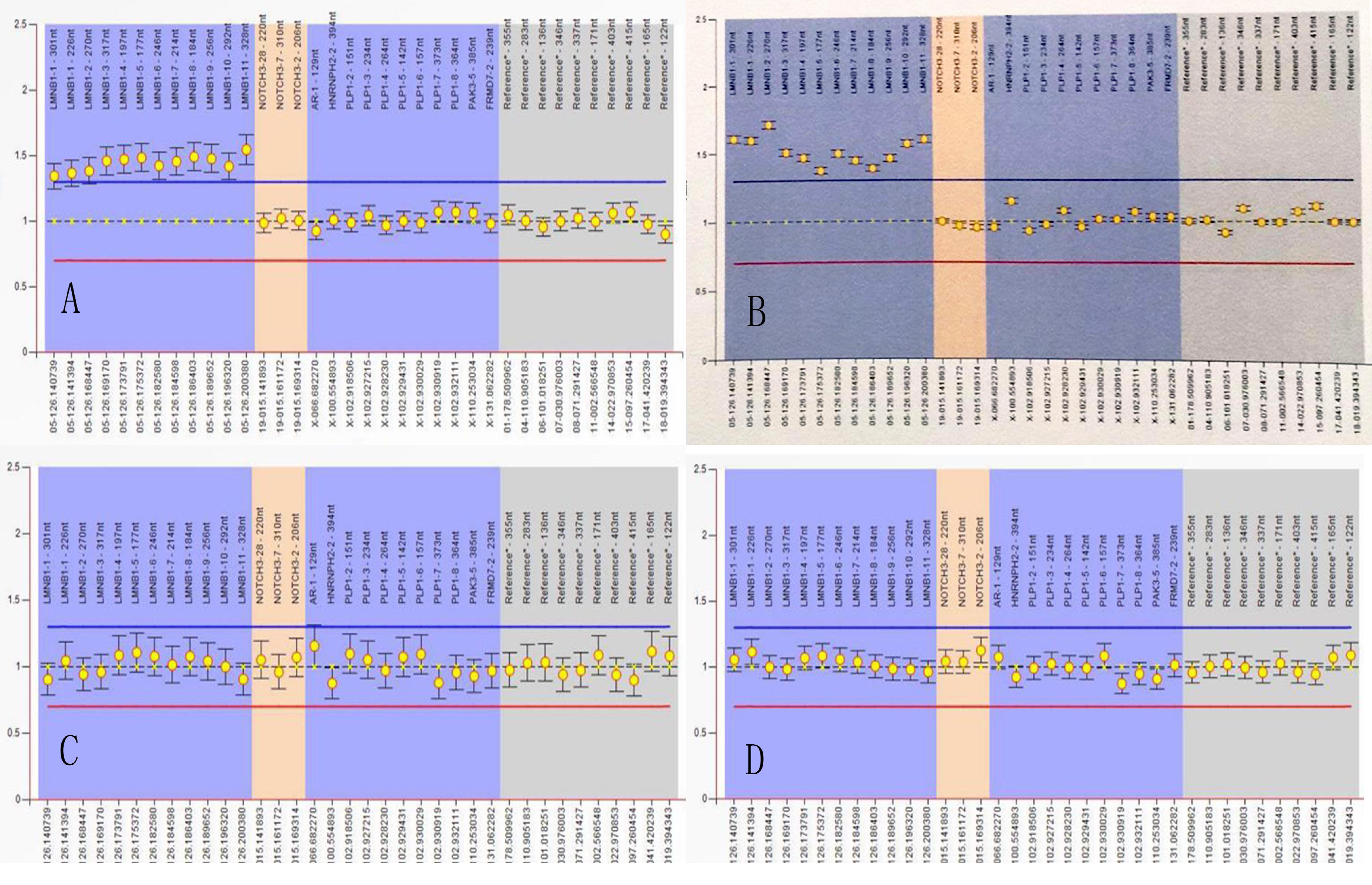
Figure 5. Multiplex ligand-dependent probe amplification (MLPA) experiment. MLPA of II-4 (A) and II-5 (B) showed genomic duplications of LMNB1, extending between nucleotide positions 126.140.739 and 126.200.380, with an estimated size of 59,651 bp (A,B), III-9 (C), and III-11 (D) were normal.
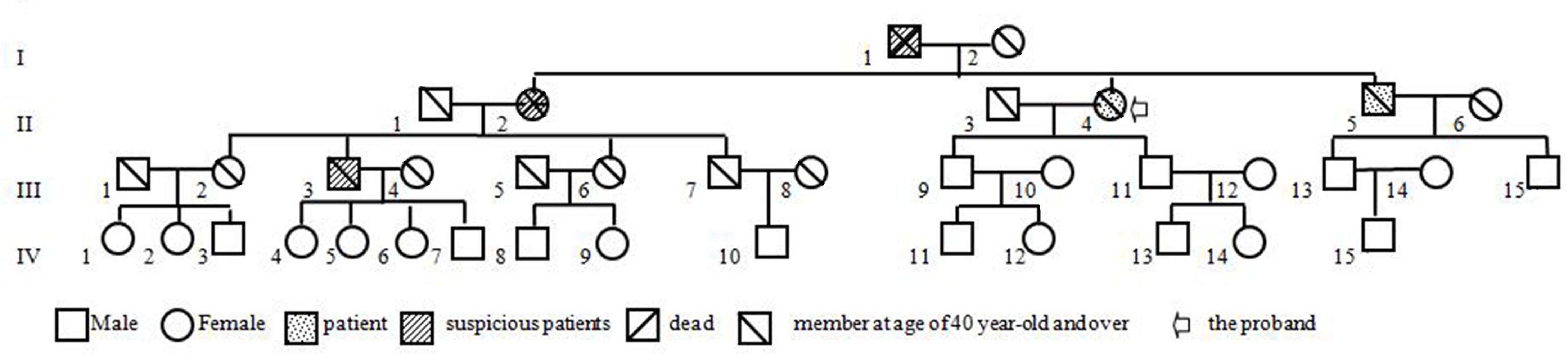
Figure 6. The pedigree of the laminB1-duplicated ADLD family.
Discussion
Based on clinical feature and neuroimaging data above, the diagnosis of our patient was confirmed by genetic testing. Subsequent to a comprehensive analysis of previous reports, we believe that the female with tremor of limbs as the initial manifestation will be the first Chinese patient.
Tremor is characterized by a rhythmic and involuntary movement of any body part. It commonly has been reported with diseases such as essential tremor, Parkinson disease (PD), multiple sclerosis (MS), and psychogenic and drug-induced tremor (Elias and Shah, 2014). Unlike these diseases, LMNB1-related ADLD patients with tremor are extremely rare and ADLDs with tremor as the initial symptom have never been reported previously in China (Dai et al., 2017). There are four studies with patients presenting tremor of limbs or trunk from different countries, including Sweden and Italy (Schuster et al., 1984; Sundblom et al., 2009; Brussino et al., 2010; Terlizzi et al., 2016; Table 3). Analysis result showed that patients from Italy-2 (Schuster et al., 1984), Sweden (Sundblom et al., 2009), and Italy-3 (Terlizzi et al., 2016) were consistent with characteristics of this type of leukodystrophy. They had an onset in about the fifth decade with autonomic dysfunction, characteristic MRI signs, and positive genetic test. By contrast, the Italy-1 family was distinguished from others and defined as ADLD-1-TO because of variant features of the absence of the autonomic dysfunction, relative sparing of cerebellar WM, and the increased laminB1 mRNA without LMNB1 duplication (Brussino et al., 2010; Giorgio et al., 2015). Although these reports referred to tremor of limbs or trunk, the symptom was thought to be manifestations in the course of disease progression.
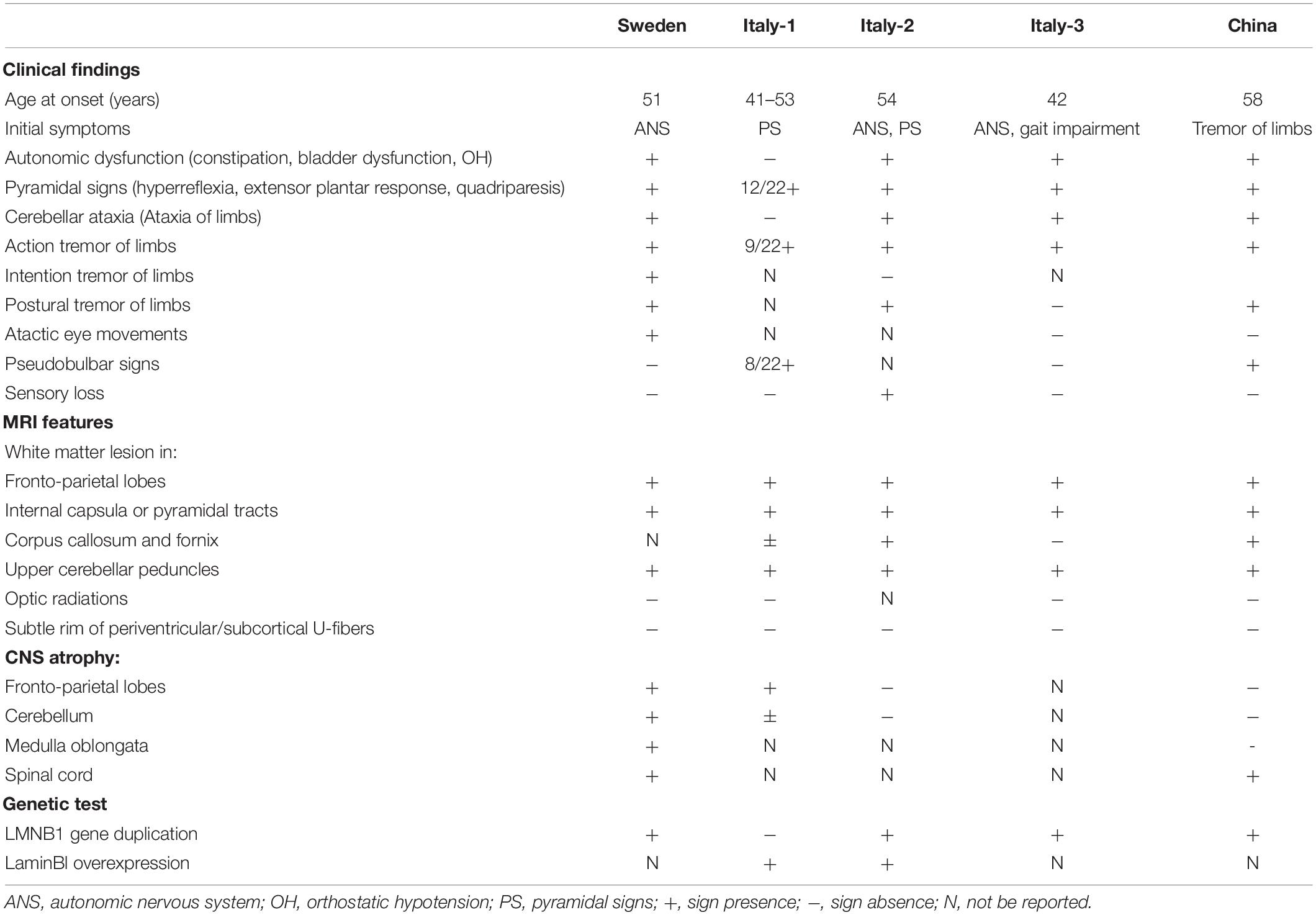
Table 3. Clinical features of individuals affected by ADLD with tremor.
Our patient is a peculiar ADLD with the clinical presenting feature being a tremor of limbs. We attribute the movement disorder to ADLD on the basis of the following three pieces of evidence. Firstly, the patient was in good health before first symptoms, excluding the possibility of secondary manifestations of the disease. Secondly, neuroradiological findings display typical clinical signs. T2 high intensities under the cerebral cortex extended down through midbrain, brain stem, to the cerebellum adjacent to the fourth ventricle. Thirdly, and most importantly, gait dysfunction was noted in members over three generations, and five out of nine family members at the age of 40–60 years had similar symptoms, mainly with gait disturbance as the early symptom (Figure 6, I-1, II-1, II-4, II-5, III-3). This study also gets support from gene test, keeping a leading place in the pathogenesis of ADLD.
Unfortunately, present mechanism theories failed to explain the tremor of ADLD in more detail. To the best of our knowledge, the tremor could be associated with neurodegenerative disorders involving the cerebellum and brain structures (Fratkin and Vig, 2012). Moreover, pathological studies suggested that the cerebellum, thalamus, pons, premotor cortical regions, or the basal ganglia may be involved in tremor, especially since the cerebellothalamocortical pathway is essentially involved in all pathologic tremors (Elble, 2013; Muthuraman et al., 2018; Saifee, 2019). Given the above data, we understand that ADLD cases with such heterogeneous presentations had varying degrees of cerebellar lesion. These seem to support the speculation that the demyelination in cerebellothalamocortical network would be responsible for the tremor of ADLD patients. Furthermore, the non-recurrent mutation of LMNB1 and potential compensatory mechanisms with individual differences may be interpreted as part of the reason why only a few patients present movement disorders in this family (Giorgio et al., 2013).
Conclusion
To summarize, our study revealed that cerebellar dysfunction presentation may appear as initial onset, and this could be the first case report of ADLD disguising as movement disorder in China. Given the small number of cases and various clinical manifestations, ADLD was often misdiagnosed as other neurodegenerative diseases leading to delay therapy. Neurologists should take comprehensive assessment and take into account the differential diagnosis of patients presenting with movement disorders.
Data Availability Statement
All datasets generated for this study are included in the manuscript/supplementary files.
Ethics Statement
The subjects were from a family in Northern China. Written informed consent was obtained from the parents or guardians of the participant for the publication of this case report and the study was conducted in accordance with the principles of the Declaration of Helsinki and relevant policies in China.
Author Contributions
YZ drafted the manuscript. TT and HL designed the study and TT repeatedly modified the text and details. JL, RB, JPW, TP, LC, and YL participated in the literature review and discussion about article writing and revision. All authors read and approved the final manuscript.
Funding
This study was supported by funds from the National Natural Science Foundation of China (Grant number: U1804171).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patient and the families for their participation in this study.
References
Bartoletti-Stella, A., Gasparini, L., Giacomini, C., Corrado, P., Terlizzi, R., Giorgio, E., et al. (2015). Messenger RNA processing is altered in autosomal dominant leukodystrophy. Hum. Mol. Genet. 24, 2746–2756. doi: 10.1093/hmg/ddv034
Brunetti, V., Ferilli, M. A., Nociti, V., and Silvestri, G. (2014). Teaching NeuroImages: Autosomal dominant leukodystrophy in a sporadic case. Neurology 83, e121. doi: 10.1212/wnl.0000000000000803
Brussino, A., Vaula, G., Cagnoli, C., Mauro, A., Pradotto, L., Daniele, D., et al. (2009). A novel family with Lamin B1 duplication associated with adult-onset leucoencephalopathy. J. Neurol. Neurosurg. Psychiatry 80, 237–240. doi: 10.1136/jnnp.2008.147330
Brussino, A., Vaula, G., Cagnoli, C., Panza, E., Seri, M., Di Gregorio, E., et al. (2010). A family with autosomal dominant leukodystrophy linked to 5q23.2-q23.3 without lamin B1 mutations. European journal of neurology. 17, 541–549. doi: 10.1111/j.1468-1331.2009.02844.x
Coffeen, C. M., McKenna, C. E., Koeppen, A. H., Plaster, N. M., Maragakis, N., Mihalopoulos, J., et al. (2000). Genetic localization of an autosomal dominant leukodystrophy mimicking chronic progressive multiple sclerosis to chromosome 5q31. Hum. Mol. Genet. 9, 787–793. doi: 10.1093/hmg/9.5.787
Corlobe, A., Taithe, F., Clavelou, P., Pierre, E., Carra-Dallière, C., Ayrignac, X., et al. (2015). A novel autosomal dominant leukodystrophy with specific MRI pattern. J. Neurol. 262, 988–991. doi: 10.1007/s00415-015-7660-4
Dai, Y., Ma, Y., Li, S., Banerjee, S., Liang, S., Liu, Q., et al. (2017). An LMNB1 Duplication Caused Adult-Onset Autosomal Dominant Leukodystrophy in Chinese Family: Clinical Manifestations, Neuroradiology and Genetic Diagnosis. Frontiers in molecular neuroscience. 10:215. doi: 10.3389/fnmol.2017.00215
Dos Santos, M. M., Grond-Ginsbach, C., Aksay, S. S., Chen, B., Tchatchou, S., Wolf, N. I., et al. (2012). Adult-onset autosomal dominant leukodystrophy due to LMNB1 gene duplication. J. Neurol. 259, 579–581. doi: 10.1007/s00415-011-6225-4
Elble, R. J. (2013). Tremor disorders. Current opinion in neurology 26, 413–419. doi: 10.1097/WCO.0b013e3283632f46
Eldridge, R., Anayiotos, C. P., Schlesinger, S., Cowen, D., Bever, C., Patronas, N., et al. (1984). Hereditary adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. N. Engl. J. Med. 311, 948–953. doi: 10.1056/NEJM198410113111504
Ferrera, D., Canale, C., Marotta, R., Mazzaro, N., Gritti, M., Mazzanti, M., et al. (2014). Lamin B1 overexpression increases nuclear rigidity in autosomal dominant leukodystrophy fibroblasts. FASEB journal 28, 3906–3918. doi: 10.1096/fj.13-247635
Finnsson, J., Lubberink, M., Savitcheva, I., Fällmar, D., Melberg, A., Kumlien, E., et al. (2019). Glucose metabolism in the brain in LMNB1-related autosomal dominant leukodystrophy. Acta Neurol. Scand. 139, 135–142. doi: 10.1111/ane.13024
Finnsson, J., Melberg, A., and Raininko, R. (2013). 1H-MR spectroscopy of adult-onset autosomal dominant leukodystrophy with autonomic symptoms. Neuroradiology 55, 933–939. doi: 10.1007/s00234-013-1174-5
Finnsson, J., Sundblom, J., Dahl, N., Melberg, A., and Raininko, R. (2015). LMNB1-related autosomal-dominant leukodystrophy: Clinical and radiological course. Ann. Neurol. 78, 412–425. doi: 10.1002/ana.24452
Flanagan, E. P., Gavrilova, R. H., Boeve, B. F., Kumar, N., Jelsing, E. J., and Silber, M. H. (2013). Adult-onset autosomal dominant leukodystrophy presenting with REM sleep behavior disorder. Neurology. 80, 118–120. doi: 10.1212/WNL.0b013e31827b1b2a
Fratkin, J. D., and Vig, P. J. (2012). Neuropathology of degenerative ataxias. Handbook of clinical neurology. 103, 111–125. doi: 10.1016/B978-0-444-51892-7.00005-X
Giacomini, C., Mahajani, S., Ruffilli, R., Marotta, R., and Gasparini, L. (2016). Lamin B1 protein is required for dendrite development in primary mouse cortical neurons. Mol. Biol. Cell 27, 35–47. doi: 10.1091/mbc.E15-05-0307
Giorgio, E., Robyr, D., Spielmann, M., Ferrero, E., Di Gregorio, E., Imperiale, D., et al. (2015). A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD). Hum. Mol. Genet 24, 3143–3154. doi: 10.1093/hmg/ddv065
Giorgio, E., Rolyan, H., Kropp, L., Chakka, A. B., Yatsenko, S., Di Gregorio, E., et al. (2013). Analysis of LMNB1 duplications in autosomal dominant leukodystrophy provides insights into duplication mechanisms and allele-specific expression. Hum. Mutat. 34, 1160–1171. doi: 10.1002/humu.22348
Guaraldi, P., Donadio, V., Capellari, S., Contin, M., Casadio, M. C., Montagna, P., et al. (2011). Isolated noradrenergic failure in adult-onset autosomal dominant leukodystrophy. Autonomic neuroscience : basic & clinical. 159, 123–126. doi: 10.1016/j.autneu.2010.07.011
Kohler, W., Curiel, J., and Vanderver, A. (2018). Adulthood leukodystrophies. Nature reviews Neurology. 14, 94–105. doi: 10.1038/nrneurol.2017.175
Laforce, R., Roy, M., Descoteaux, M., Berthelot, C., and Bouchard, J.-P. (2013). Neurocognitive deficits and diffusion MR imaging abnormalities in a case of adult-onset autosomal dominant leukodystrophy. Translational Neuroscience. 4, 513–515.
Lin, S. T., Ptacek, L. J., and Fu, Y. H. (2011). Adult-onset autosomal dominant leukodystrophy: linking nuclear envelope to myelin. The Journal of neuroscience: the official journal of the Society for Neuroscience. 31, 1163–1166. doi: 10.1523/JNEUROSCI.5994-10.2011
Liu, S., Wu, Y., Yang, L., Li, X., Huang, L., and Xing, X. (2018). Functions of lamin B1 and the new progress of its roles in neurological diseases and tumors. Sheng wu gong cheng xue bao = Chinese journal of biotechnology. 34, 1742–1749. doi: 10.13345/j.cjb.180063
Lo Martire, V., Alvente, S., Bastianini, S., Berteotti, C., Bombardi, C., Calandra-Buonaura, G., et al. (2018). Mice overexpressing lamin B1 in oligodendrocytes recapitulate the age-dependent motor signs, but not the early autonomic cardiovascular dysfunction of autosomal-dominant leukodystrophy (ADLD). Exp. Neurol. 301, 1–12. doi: 10.1016/j.expneurol.2017.12.006
Marklund, L., Melin, M., Melberg, A., Giedraitis, V., and Dahl, N. (2006). Adult-onset autosomal dominant leukodystrophy with autonomic symptoms restricted to 1.5 Mbp on chromosome 5q23. Am J Med Genet B Neuropsychiatr Genet. 141B, 608–614. doi: 10.1002/ajmg.b.30342
Meijer, I. A., Simoes-Lopes, A. A., Laurent, S., Katz, T., St-Onge, J., Verlaan, D. J., et al. (2008). A novel duplication confirms the involvement of 5q23.2 in autosomal dominant leukodystrophy. Archives of neurology. 65, 1496–1501. doi: 10.1001/archneur.65.11.1496
Melberg, A., Hallberg, L., Kalimo, H., and Raininko, R. (2006). MR characteristics and neuropathology in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. AJNR American journal of neuroradiology. 27, 904–911.
Mezaki, N., Miura, T., Ogaki, K., Eriguchi, M., Mizuno, Y., Komatsu, K., et al. (1984). Duplication and deletion upstream of LMNB1 in autosomal dominant adult-onset leukodystrophy. Neurology Genetics. x 4, e292. doi: 10.1212/nxg.0000000000000292
Molloy, A., Cotter, O., van Spaendonk, R., Sistermans, E., and Sweeney, B. (2012). A patient with a rare leukodystrophy related to lamin B1 duplication. Ir. Med. J. 105, 186–187.
Muthuraman, M., Schnitzler, A., and Groppa, S. (2018). Pathophysiology of tremor. Der Nervenarzt. 89, 408–415. doi: 10.1007/s00115-018-0490-8
Nmezi, B., Giorgio, E., Raininko, R., Lehman, A., Spielmann, M., Koenig, M. K., et al. (2019). Genomic deletions upstream of lamin B1 lead to atypical autosomal dominant leukodystrophy. Neurology Genetics 5, e305. doi: 10.1212/NXG.0000000000000305
Padiath, Q. S. (2015). Autosomal Dominant Leukodystrophy: A Disease of the Nuclear Lamina. Frontiers in cell and developmental biology. x 7:41. doi: 10.3389/fcell.2019.00041
Padiath, Q. S. (2016). Lamin B1 mediated demyelination: Linking Lamins, Lipids and Leukodystrophies. Nucleus. 7, 547–553. doi: 10.1080/19491034.2016.1260799
Padiath, Q. S., and Fu, Y. H. (2010). Autosomal dominant leukodystrophy caused by lamin B1 duplications a clinical and molecular case study of altered nuclear function and disease. Methods Cell Biol. 98, 337–357. doi: 10.1016/S0091-679X(10)98014-X
Padiath, Q. S., Saigoh, K., Schiffmann, R., Asahara, H., Yamada, T., Koeppen, A., et al. (2006). Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 38, 1114–1123. doi: 10.1038/ng1872
Potic, A., Pavlovic, A. M., Uziel, G., Kozic, D., Ostojic, J., Rovelli, A., et al. (2013). Adult-onset autosomal dominant leukodystrophy without early autonomic dysfunctions linked to lamin B1 duplication: a phenotypic variant. J. Neurol. 260, 2124–2129. doi: 10.1007/s00415-013-6958-3
Rolyan, H., Tyurina, Y. Y., Hernandez, M., Amoscato, A. A., Sparvero, L. J., Nmezi, B. C., et al. (2015). Defects of Lipid Synthesis Are Linked to the Age-Dependent Demyelination Caused by Lamin B1 Overexpression. The Journal of neuroscience 35, 12002–12017. doi: 10.1523/JNEUROSCI.1668-15.2015
Sandoval-Rodriguez, V., Cansino-Torres, M. A., Saenz-Farret, M., Castaneda-Cisneros, G., Moreno, G., and Zuniga-Ramirez, C. (2017). Autosomal dominant leukodystrophy presenting as Alzheimer’s-type dementia. Multiple sclerosis and related disorders 17, 230–233. doi: 10.1016/j.msard.2017.08.014
Schuster, J., Sundblom, J., Thuresson, A. C., Hassin-Baer, S., Klopstock, T., Dichgans, M., et al. (1984). Genomic duplications mediate overexpression of lamin B1 in adult-onset autosomal dominant leukodystrophy (ADLD) with autonomic symptoms. Neurogenetics. x 12, 65–72. doi: 10.1007/s10048-010-0269-y
Sundblom, J., Melberg, A., Kalimo, H., Smits, A., and Raininko, R. (2009). MR imaging characteristics and neuropathology of the spinal cord in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. AJNR American journal of neuroradiology. 30, 328–335. doi: 10.3174/ajnr.A1354
Terlizzi, R., Calandra-Buonaura, G., Zanigni, S., Barletta, G., Capellari, S., Guaraldi, P., et al. (2016). A longitudinal study of a family with adult-onset autosomal dominant leukodystrophy: Clinical, autonomic and neuropsychological findings. Autonomic neuroscience: basic & clinical. 195, 20–26. doi: 10.1016/j.autneu.2016.02.005
Keywords: adult-onset autosomal dominant leukodystrophy, LMNB1 gene, movement disorder, tremor, neurodegenerative disease
Citation: Zhang Y, Li J, Bai R, Wang J, Peng T, Chen L, Wang J, Liu Y, Tian T and Lu H (2019) LMNB1-Related Adult-Onset Autosomal Dominant Leukodystrophy Presenting as Movement Disorder: A Case Report and Review of the Literature. Front. Neurosci. 13:1030. doi: 10.3389/fnins.2019.01030
Received: 18 March 2019; Accepted: 11 September 2019;
Published: 21 October 2019.
Edited by:
Odete A. B. da Cruz e Silva, University of Aveiro, PortugalReviewed by:
Sandra Rebelo, University of Aveiro, PortugalSabina Capellari, University of Bologna, Italy
Pietro Cortelli, University of Bologna, Italy
Copyright © 2019 Zhang, Li, Bai, Wang, Peng, Chen, Wang, Liu, Tian and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tian Tian, dGlhbnRpYW5fMjAxNkAxMjYuY29t; Hong Lu, c25sdWhvbmdAMTYzLmNvbQ==