 Kaitlyn R. Calabro1
Kaitlyn R. Calabro1 Sanford L. Boye2
Sanford L. Boye2 Shreyasi Choudhury1
Shreyasi Choudhury1 Diego Fajardo1James J. Peterson1Wei Li1
Diego Fajardo1James J. Peterson1Wei Li1 Sean M. Crosson1Mi-Jung Kim3
Sean M. Crosson1Mi-Jung Kim3 Dalian Ding4
Dalian Ding4 Richard Salvi4
Richard Salvi4 Shinichi Someya3
Shinichi Someya3 Shannon E. Boye1*
Shannon E. Boye1*- 1Department of Ophthalmology, University of Florida, Gainesville, FL, United States
- 2Department of Pediatrics, University of Florida, Gainesville, FL, United States
- 3Department of Aging and Geriatric Research, University of Florida, Gainesville, FL, United States
- 4Department of Communicative Disorders and Sciences, The State University of New York at Buffalo, Buffalo NY, United States
Usher’s syndrome is the most common combined blindness–deafness disorder with USH1B, caused by mutations in MYO7A, resulting in the most severe phenotype. The existence of numerous, naturally occurring shaker1 mice harboring variable MYO7A mutations on different genetic backgrounds has complicated the characterization of MYO7A knockout (KO) and heterozygote mice. We generated a novel MYO7A KO mouse (Myo7a–/–) that is easily genotyped, maintained, and confirmed to be null for MYO7A in both the eye and inner ear. Like USH1B patients, Myo7a–/– mice are profoundly deaf, and display near complete loss of inner and outer cochlear hair cells (HCs). No gross structural changes were observed in vestibular HCs. Myo7a–/– mice exhibited modest declines in retinal function but, unlike patients, no loss of retinal structure. We attribute the latter to differential expression of MYO7A in mouse vs. primate retina. Interestingly, heterozygous Myo7a+/– mice had reduced numbers of cochlear HCs and concomitant reductions in auditory function relative to Myo7a+/+ controls. Notably, this is the first report that loss of a single Myo7a allele significantly alters auditory structure and function and suggests that audiological characterization of USH1B carriers is warranted. Maintenance of vestibular HCs in Myo7a–/– mice suggests that gene replacement could be used to correct the vestibular dysfunction in USH1B patients. While Myo7a–/– mice do not exhibit sufficiently robust retinal phenotypes to be used as a therapeutic outcome measure, they can be used to assess expression of vectored MYO7A on a null background and generate valuable pre-clinical data toward the treatment of USH1B.
Introduction
Usher syndrome is the most frequent cause of combined deafness–blindness (Smith et al., 1994). Recessive mutations in MyosinVIIa (MYO7A) are associated with the most common form, USH1B (Weil et al., 1995; Astuto et al., 2000; Bharadwaj et al., 2000; Ouyang et al., 2005). Patients are born profoundly deaf, have vestibular dysfunction, and exhibit progressive loss of vision in their first decade (Petit, 2001). MYO7A encodes an unconventional myosin expressed in sensory hair cells (HCs) of the inner ear and the retina. It is expressed early in auditory and vestibular HC development and is required for proper differentiation and development of stereocilia bundles (Roccio et al., 2018). It is therefore unsurprising that mutations rendering MYO7A dysfunctional or absent lead to congenital deafness. The absence/malformation of cochlear HCs suggests that gene supplementation will not be an effective strategy for restoring hearing to USH1B patients. Fortunately, their hearing loss can be addressed with cochlear implantation. In contrast to the structural malformations observed in the inner ear from birth, clinical studies show that patients display progressive retinal degeneration, but can retain both central and peripheral islands of structurally and functionally normal retina into the third decade of life (Jacobson et al., 2008, 2009, 2011; Sumaroka et al., 2016). It is therefore possible that gene supplementation targeted to these preserved regions will be an effective strategy for restoring/preserving vision in these patients.
Development of a retinal gene therapy for USH1B has been hampered by the lack of animal models that faithfully replicate human retinal disease. Although currently available mouse models mimic the hearing loss and vestibular dysfunction seen in patients, they do not exhibit progressive retinal degeneration/dysfunction (el-Amraoui et al., 1996; Self et al., 1998; Libby and Steel, 2001; Lillo et al., 2003). Only subtle retinal phenotypes have been reported. These include (1) modest reductions in retinal function (if backcrossed onto an albino background), defective melanosome transport in the apical RPE, and impaired rhodopsin (RHO) transport through photoreceptor connecting cilia (Williams, 2008; Colella et al., 2013; Trouillet et al., 2018). We, and others, have corrected these subtle phenotypes following treatment with either fragmented or dual AAV-MYO7A vectors (Liu et al., 1997, 1998, 1999; Colella et al., 2013; Lopes et al., 2013). Differences in retinal disease presentation across species are likely related to differential expression of MYO7A in mouse vs. primate retina. In macaques and humans, MYO7A is predominantly localized to photoreceptor inner segments (el-Amraoui et al., 1996; Sahly et al., 2012). In contrast, it is expressed predominantly in mouse retinal pigment epithelium (RPE) (el-Amraoui et al., 1996; Sahly et al., 2012). One explanation for this difference, albeit one that is still debated, relates to structural differences between mouse and primate photoreceptors (Sahly et al., 2012; Volland et al., 2015). Analysis of the apical region of macaque and human photoreceptor inner segments revealed the presence of calyceal processes, within which MYO7A was localized (Sahly et al., 2012). These structures were absent in mouse retina (Sahly et al., 2012), an observation that may explain why mouse models fail to recapitulate the robust retinal degeneration/dysfunction seen in USH1B patients.
The purpose of our study was to generate and characterize a novel, traditional MYO7A knockout (KO) mouse (Myo7a–/–) that could be more easily genotyped, maintained, and studied relative to existing strains (Gibson et al., 1995; Libby and Steel, 2001; Peng et al., 2011). We completed a full characterization of retinal and cochlear HC structure/function, and vestibular HC structure in Myo7a–/–, Myo7a+/–, and Myo7a+/+ littermates. We further investigated the role of MYO7A in RHO trafficking and the differential expression of MYO7A in mouse vs. primate retinas. Results of this study further inform the USH field regarding the impact of complete vs. partial loss of MYO7A on multiple sensory systems, and will aid in the development of therapies aimed to preserve the function of these systems in USH1B patients.
Materials and Methods
Animal Ethics Statement
All mice were bred and maintained at the University of Florida Health Science Center Animal Care Services Facility under a 12 h/12 h light/dark cycle. Food and water were available ad libitum. All experiments were approved by the University of Florida’s Institutional Animal Care and Use Committee and were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and within National Institute of Health regulations. Mice were anesthetized for all in-life experiments with ketamine (100 mg/kg) and xylazine (10 mg/kg) by intraperitoneal injection.
Generation of Myo7a–/– Mice
Mouse ES cells, created as part of the International Knockout Mouse Consortium (IKMC), were acquired from the Sanger Institute, and mice were generated at Jackson Laboratories (Ryder et al., 2013). The targeted trap “tm1a” cassette was inserted between exons 9 and 10 of the Myo7a gene (Skarnes et al., 2011). This cassette contains a flippase recognition target (FRT) followed by lacZ sequence and a loxP site (Figure 1A). The loxP site is followed by kanamycin/neomycin sequence under the control of the human beta-actin promoter, an SV40 polyA, a second FRT site, and a second loxP site. A third loxP site was inserted between Exons 11 and 12 of Myo7a. The critical exons were thus flanked by loxP sites. ES cells were implanted in C57BL/6N mice to create the Myo7atm1a(EUCOMM)Wtsi mouse. C57BL/6N mice carry the retinal disease-causing rd8/Crb1 genes (Mattapallil et al., 2012). The Myo7aTM1a(EUCOMM)Wtsi mice were backcrossed on C57BL/6J mice and screened to ensure removal of the rd8/Crb1 alleles. Resultant weanlings were then crossed with a Sox2-Cre deleter line to create Myo7aTM1b(EUCOMM)/Wtsi, which will from here on be referred to as Myo7a–/– mice. Colony founders were screened and confirmed to have the RPE65-L450M mutation as previously described (Wu et al., 2009). Founders were then backcrossed on a C57BL/6J background for at least five generations prior to use in experiments. The Myo7a–/– colony was maintained by breeding heterozygous females with homozygous males. Myo7a+/+ (wild-type) controls were generated by breeding heterozygous females with heterozygous males. All mice were genotyped prior to use with the following two primer sets: (1) Myo7a WT Allele: Myo7a Forward (GGG AGA GAA AC AGG GTG TG) + Myo7a WT Reverse (AAG CTG GAC TCT CTG GTG GC) and (2) Myo7a KO Allele: Myo7a Forward + Myo7a KO Reverse (TCG TGG TAT CGT TA GCG CC). Myo7a WT primers produce an amplicon of 360 bp, and the Myo7a KO primers produce an amplicon of 178 bp. Myo7a WT was amplified using the following conditions: (1) initial denaturation at 95°C for 2 min; (2) 34 cycles of 95°C denaturation for 30 s, 66°C annealing for 30 s, and 72°C elongation for 45 s; and (3) final elongation at 72°C for 5 min. The Myo7a KO primer set was run using identical PCR settings, with the exception of a 63°C annealing temperature. LacZ also served as a genotyping target for the KO allele using the following primers: LacZ Forward (ACA TCG GGC AAA TAA TAT CG) + LacZ Reverse (ATC ACG ACG CGC TGT ATC). LacZ sequence was amplified using the PCR settings described above with an annealing temperature of 56°C.
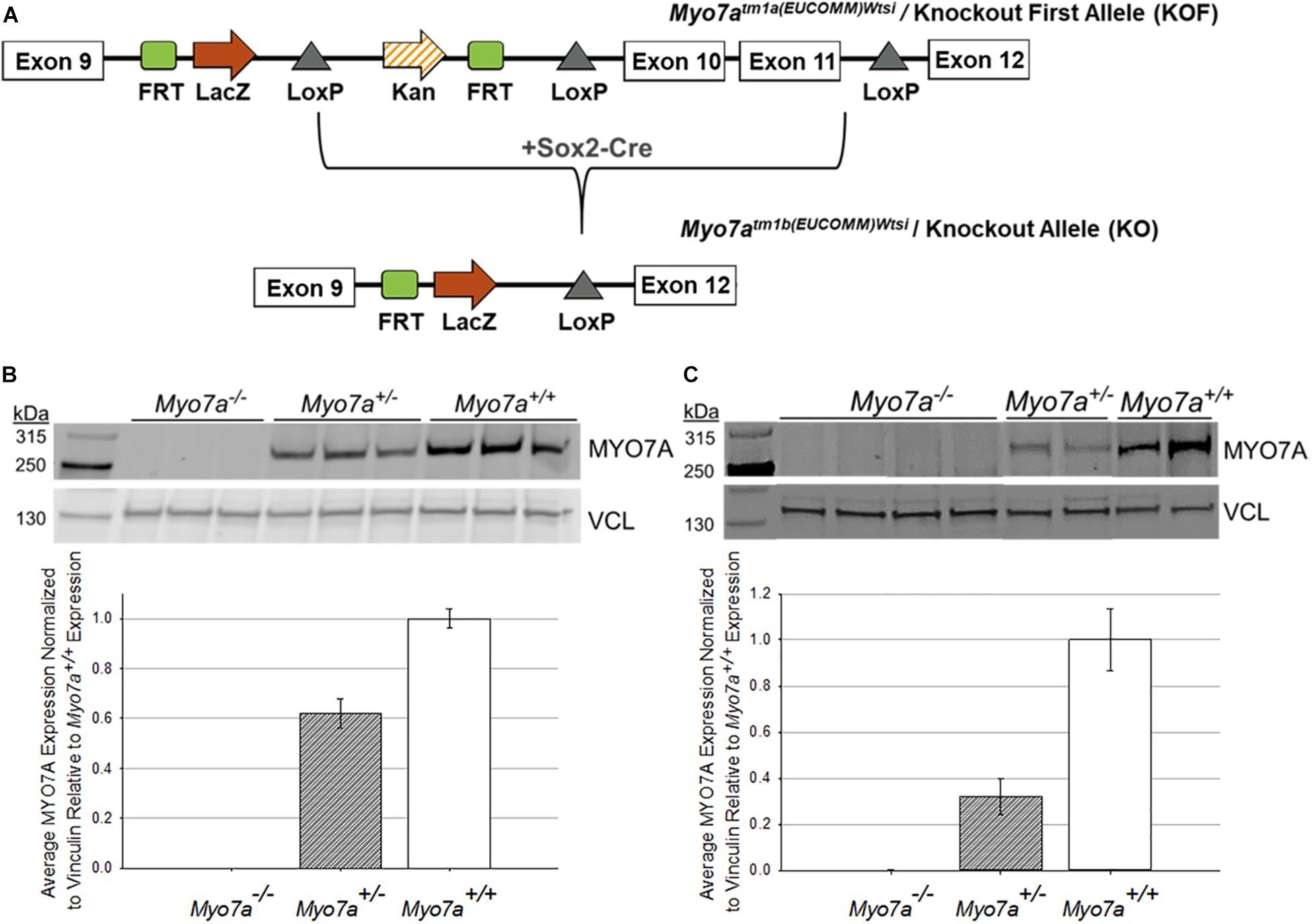
Figure 1. Generation and confirmation of MYO7A knockout in Myo7a–/– mice. (A) Schematic of how Myo7a–/– mice were generated using Cre-lox recombination. Exons 10 and 11 of Myo7a were deleted by breeding Myo7aTM1a(EUCOMM)Wtsi mice with a Sox2-Cre deleter line, creating the Myo7aTM1b(EUCOMM)Wtsi or knockout allele. Immunoblotting was performed to compare differences in MYO7A expression in 4-month-old Myo7a+/+, Myo7a+/–, and Myo7a–/– (B) eyes (n = 3 per genotype) and (C) cochlea (n = 2–4 per genotype). Protein levels were quantified and compared using Fiji. MYO7A expression was normalized to vinculin and analyzed relative to MYO7A expression in Myo7a+/+ mice. FRT, flippase recognition target; Kan, kanamycin; kDa, kilodaltons; VCL, vinculin.
Generation of Myo7a:RHOGFP Mice
Male Myo7a–/– mice were crossed with females homozygous for the hrhoG allele, a knock-in human rhodopsin–GFP fusion (RHOGFP) that were maintained on an albino C57BL/6J background (Chan et al., 2004). All mice were genotyped prior to use for both the Myo7a and RHOGFP genes. RHOGFP alleles were amplified as previously described, producing a 194 bp amplicon for the WT RHO allele or a 922 bp amplicon for the RHOGFP allele (Chan et al., 2004). RHOGFP mice were also crossed to transgenic P23H mice containing the entire human RHO gene to serve as controls (Mao et al., 2011). These mice will hereon be referred to as P23H+:RHOGFP+/–. P23H was amplified using the following primers: P23H Forward (ACC TCT TTC CAC TAC AGG CC) + P23H Reverse (CCT GGT GAT TAT GCC CAA CG). PCR settings were the same as those described above, with an annealing temperature of 60°C. P23H primers produce a 290 bp amplicon when the P23H allele is present.
Western Blots
Eyes from Myo7a+/+, Myo7a+/–, and Myo7a–/– were enucleated, and cochleas were extracted from the temporal bones. Both male and female mice were used for western blot analysis (n = 3–4/genotype). Whole eyes and cochleas were homogenized in RIPA lysis buffer (Santa Cruz, sc-364162). Protein concentration within each sample was determined using BCA assay (Thermo Scientific, 23225). Equal amounts of protein from each sample were loaded on 8–12% Tris-acetate gels (Invitrogen, EA0375) and run for approximately 1 h at 120 V (Invitrogen, A25977). Gels were transferred to a PVDF membrane using a semi-dry system (BioRad, 1704150). The membrane was blocked for 1 h at room temperature in Licor blocking buffer (927-40000) and incubated at 4°C overnight with primary antibodies [MYO7A: Santa Cruz, sc74516, 1:500; Vinculin (VCL): Santa Cruz, sc73614, 1:1000]. Secondary antibody (Licor, 926-68072) incubation took place the following day at room temperature for 1 h. Membranes were then visualized on a Licor Odyssey CLx Imager. Protein quantification was performed by measuring the mean gray for each sample using the open source Fiji software (Schindelin et al., 2012). Protein quantification was normalized to VCL in each sample and written as values relative to MYO7A expression in C57BL/6J mice.
Neural retina and RPE were manually separated from the eyes of 5-month-old male C57BL/6J mice (n = 4) and a 5-year-old male macaque (Macaca fascicularis). Tissues were collected and flash-frozen. MYO7A expression in mouse and non-human primate (NHP) tissues was determined with Western Blot, as described above. The membrane was incubated at 4°C overnight with primary antibodies (MYO7A: Santa Cruz, sc74516, 1:500; β-actin: Santa Cruz, sc47778, 1:1000).
Auditory Brainstem Response Testing
Auditory brainstem response (ABR) thresholds were measured in 5-month-old mice with a tone burst stimulus at 8, 16, and 32 kHz using an ABR recording system (Tucker-Davis Technologies) as previously described (Han et al., 2016; White et al., 2017). Mice were anesthetized and subdermal needle electrodes were placed at the vertex (active), ipsilateral ear (reference), and contralateral ear (ground). At each frequency, the sound level was reduced in 5–10 dB increments from 90 to 10 dB sound pressure level (SPL). A hearing threshold was defined as the lowest level that produced a noticeable ABR as previously described (Han et al., 2016; White et al., 2017). Both male and female mice were used for ABR threshold assessments (n = 5–6/genotype). Two-way analysis of variance (ANOVA) with Bonferroni’s multiple comparisons tests (GraphPad Prism 7) were used to analyze ABR thresholds in Myo7a+/+, Myo7a+/–, and Myo7a–/– mice.
Structural Assessment of Cochlear Hair Cells
Following ABR testing, mice were sacrificed by cervical dislocation. Temporal bones were excised from the head and divided into cochlear and vestibular parts as previously described (Han et al., 2016; White et al., 2017). The excised cochlea was immersed in 10% formalin for 1 day and shipped to the University at Buffalo for analysis. The fixed cochlea was stained with hematoxylin and the cochlear surface preparation was performed as previously described (Ding et al., 2016). HCs were observed in the middle turn of cochlear basilar membrane using a 200× objective as previously described (Ding et al., 2016). For cochleograms, the numbers of inner hair cells (IHCs), first-row outer hair cells (OHC1), second-row outer hair cells (OHC2), and third-row outer hair cells (OHC3) were counted over 0.24 mm intervals from the apex to the base of the cochlea under the microscope at 40× magnification as previously described (Ding et al., 2016). The counting results were then calculated to compute a cochleogram and frequency-place map for mouse cochlea that shows the percentage of missing IHC and OHC as a function of percentage distance from the apex of the cochlea as previously described (Muller et al., 2005; Ding et al., 2016).
Structural Assessment of Spiral Ganglion Neurons and Stria Vasicularis
Excised cochlea was immersed in 4% paraformaldehyde in PBS for 1 day, decalcified in 10% EDTA, pH 7.4, in PBS for 5–7 days, and embedded in paraffin. The paraffin-embedded cochlear specimens were sliced into 5 μm sections, mounted on silane-coated slides, stained with hematoxylin and eosin (H&E), and observed under a light microscope (Leica Microsystems). Spiral ganglion neurons (SGNs) and stria vascularis (SV) were observed in the apical, middle, and basal cochlear tissues using a 40× objective as previously described (Han et al., 2016; White et al., 2017).
Structural Assessment of Vestibular Hair Cells
The excised vestibular apparatus was immersed in 10% formalin for several days. The vestibular end organs were placed in PBS and carefully dissected out as previously described (Zhang et al., 2003). The superior, horizontal, and posterior portions of the vestibular apparatus were opened. The saccule and utricle were separated from surrounding tissue and the overlying otoconia on the saccule and utricle were removed to expose the macula of the saccule and utricle. The ampulla in each of the semicircular canals was separated from the surrounding connective tissue. The maculae and ampullae were stained with hematoxylin and mounted as surface preparation in glycerin on glass slides. Surface preparations were viewed with an Axioskop light microscope (Carl Zeiss) and photographed with a digital camera (SPOT Insight, Diagnostic Instruments Inc.) and processed with software (PowerPoint 2010).
Optical Coherence Tomography
Prior to Optical Coherence Tomography (OCT) analysis, pupils of mice were dilated (atropine 1%, phenylephrine 2.5%). Mice were anesthetized, and retinal cross-section images were obtain with a spectral domain (SD) OCT system (Bioptigen, Inc., Durham, NC, United States). Post-imaging ONL measurements were performed with commercial software (InVivoVue, Bioptigen, Inc.), as previously described (Pang et al., 2011). Equal numbers of males and females were used for each genotype (n = 10–11/genotype). Two-way ANOVA with Bonferroni’s multiple comparisons tests were used to analyze OCT measurements in Myo7a+/+, Myo7a+/–, and Myo7a–/– mice. Significance was defined as a P-value <0.05.
Electroretinogram
Following overnight dark adaptation, mice were dilated (phenylephrine HCL-tropicamide 1%) and anesthetized. Full-field electroretinograms (ERGs) were recorded using a Celeris unit (Diagnosys LLC) according to methods previously described, with minor modifications (McCullough et al., 2019). Briefly, scotopic ERGs were elicited at intensities ranging from 0.025 to 2.5 cds/m2, with interstimulus intervals of 30 s and averaged from five measurements at each intensity. Mice were then light-adapted to a 30 cds/m2 white background light for 5 min. Photopic responses were elicited, with intensities ranging from 1.25 to 25 cds/m2. Fifty responses with interstimulus intervals of 0.4 s were recorded in the presence of a 20 cds/m2 white background and averaged at each intensity. Equal numbers of males and females were used for each genotype (n = 10–11/genotype). Two-way ANOVA with Bonferroni’s multiple comparisons were used to analyze ERG amplitudes in Myo7a+/+, Myo7a+/–, and Myo7a–/– mice. Significance was defined as a P-value <0.05.
Retinal Tissue Processing and Microscopy
Myo7a–/–:RHO-GFP+/– mice were sacrificed between 1 and 6 months of age. Eyes were enucleated and fixed in formalin overnight, then dehydrated using a Tissue-Tek VIP6 automatic processor. Eyes were then embedded in paraffin using a Histocentre embedding station. Paraffin-embedded eyes were sectioned (7 μm) using a Leica microtome (Reichert-Jung BioCut 2030). Paraffin sections were then re-hydrated, and counter-stained with 4′,6-diamidino-2-phenylindole (DAPI) (1:10,000) for 5 min at room temperature. Both DAPI and native GFP expression from RHOGFP were visualized using a Leica TCS SP8 confocal microscope. Myo7a+/+:RHO-GFP+/–, Myo7a+/–:RHO-GFP+/–, and P23H+/–:RHO-GFP+/– mice served as controls. Equal numbers of males and females were used for each genotype (n = 4 mice/genotype per time point). Exposure and gain settings remained constant for all images.
Results
Design and Confirmation of Myo7a–/– Mice
Traditional Myo7a KO mice (Myo7a–/–) were generated using previously described methods (Ryder et al., 2013). Critical exons 10–11, which correspond to the motor and actin-binding domain of MYO7A, were deleted using Cre-lox recombination (Figure 1A). This large deletion allows for clear and simplified genotyping between the wild-type and KO Myo7a alleles. Like shaker1 mice, the Myo7a–/– mice exhibit spinning, hyperactivity, and head tilting behaviors that are identifiable around postnatal day 14 when mouse pups become active and begin walking (data not shown). It was also observed that Myo7a–/– eyes did not dilate as easily/quickly as heterozygous and wild-type controls (data not shown). Complete KO of MYO7A expression in the eye (n = 3 eyes per genotype) and inner ear (n = 2–4 per genotype) of Myo7a–/– mice was confirmed with Western blot (Figures 1B,C). Heterozygous Myo7a+/– mice expressed MYO7A at approximately 60% of wild-type (C57BL/6J) in the eye, and approximately 40% of wild-type in the cochlea. Due to the presence of the lacZ gene in the KO allele, we also probed retinal extracts with α-lacZ. We did not detect lacZ expression in either Myo7a–/– or Myo7a+/– mice (data not shown).
MYO7A Deficiency Leads to Sensorineural Hearing Loss and Degeneration of Cochlear Hair Cells
To confirm that MYO7A deficiency affects hearing in this novel mouse model, ABR thresholds in Myo7a+/+, Myo7a+/–, and Myo7a–/– mice were measured at 5 months of age. Both Myo7a–/– males and females displayed profound hearing loss across all frequencies (Figures 2A,B). To our surprise, heterozygous Myo7a+/– mice also exhibited severe hearing loss at the mid and high frequencies. Myo7a+/– males displayed a 16–22-dB increase in ABR threshold at 16 and 32 kHz relative to age-matched Myo7a+/+ mice (Figure 2A), while Myo7a+/– females displayed a 23–25-dB increase in ABR threshold at 16–32 kHz compared to age-matched Myo7a+/+ mice (Figure 2B). Next, we investigated whether this loss of auditory function corresponded to a loss of cochlear HC structure. Cochleograms were prepared from Myo7a+/+, Myo7a+/–, and Myo7a–/– mice (Figures 2C–E). As expected, Myo7a–/– mice displayed near complete loss of both inner and outer HCs in the middle and basal cochlear regions (Figures 2E,H). While only minimal loss of IHC was observed in the apical and middle cochlear regions, Myo7a+/– mice displayed profound loss of OHC in the basal cochlea (Figures 2D,G) compared to wild-type littermates (Figures 2C,F).
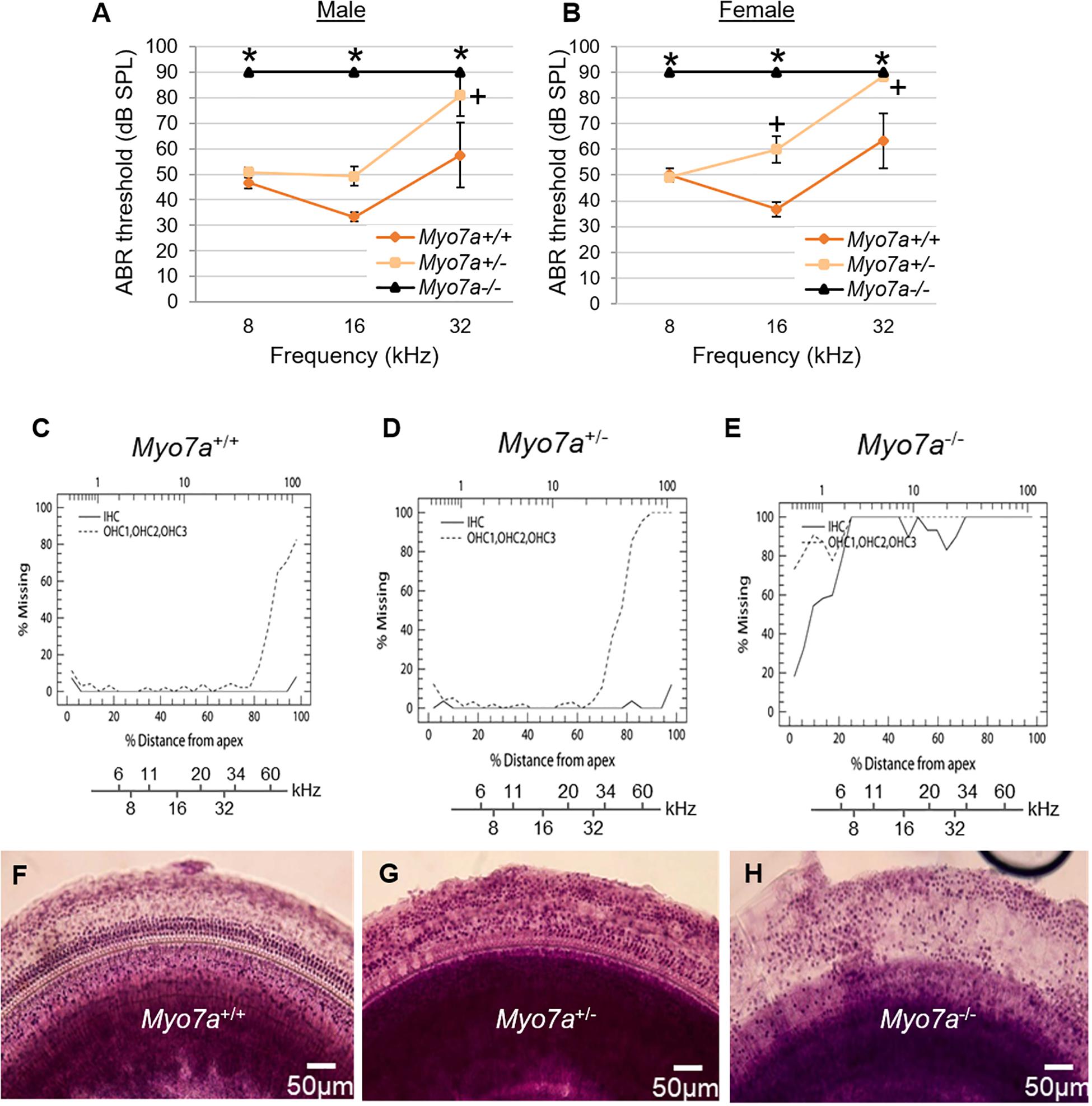
Figure 2. Characterization of cochlear hair cell structure and function in Myo7a+/+, Myo7a+/–, and Myo7a–/– mice at 5 months of age. (A,B) Assessment of auditory brainstem response (ABR) thresholds in male (A) and female (B) mice were measured with a tone burst stimulus at 8, 16, and 32 kHz in (n = 5–6). Two-way ANOVA with Bonferroni’s multiple comparisons tests were used. Data are shown as means ± SEM. ∗p < 0.05 between Myo7a–/– vs. Myo7a+/+; +p < 0.05 between Myo7a+/– vs. Myo7a+/+. (C–E) Cochleograms were recorded in Myo7a+/+ (C), Myo7a+/– (D), and Myo7a–/– (E) mice at 5 months of age. Graphs show percent loss of inner hair cells (IHC, solid line) and outer hair cells (OHC, dotted line) as a function of percent distance from the apex of the cochlea. Lower x-axes show the frequency-place map for mouse cochlea. (F–H) HC morphology in the middle turn of cochlear basilar membranes of Myo7a+/+ (F), Myo7a+/– (G), and Myo7a–/– (H) mice at 5 months of age.
Cochlear pathology also manifests in SGNs and/or SV (Ding et al., 2016; White et al., 2017). To further investigate the impact of MYO7A deficiency on these tissues, histological examination was performed on H&E-stained cochlear sections. Myo7a–/– mice displayed near complete loss of HCs in the organ of Corti, SGNs, and reduced SV thickness or atrophy in the basal cochlea (Figures 3A–C). Myo7a+/– mice also displayed a reduction of SGNs in these regions, though not nearly to the extent seen in Myo7a–/– mice. Degeneration of cochlear structure in Myo7a–/– and Myo7a+/– mice was consistent with the reduced function observed in both genotypes. Taken together, we show that the absence or reduction of MYO7A results in the loss of cochlear HCs, SGNs, and SV, as well as a concomitant loss of hearing in young mice. This is the first report, to our knowledge, that a single functional Myo7a allele is insufficient to support normal hearing in mice. Instead, Myo7a+/– mice display characteristics of early onset age-related hearing loss which, in its early stages, typically manifests as a loss of threshold sensitivity at higher frequencies and a loss of outer HCs in the basal cochlea (Gates and Mills, 2005; Someya and Prolla, 2010).
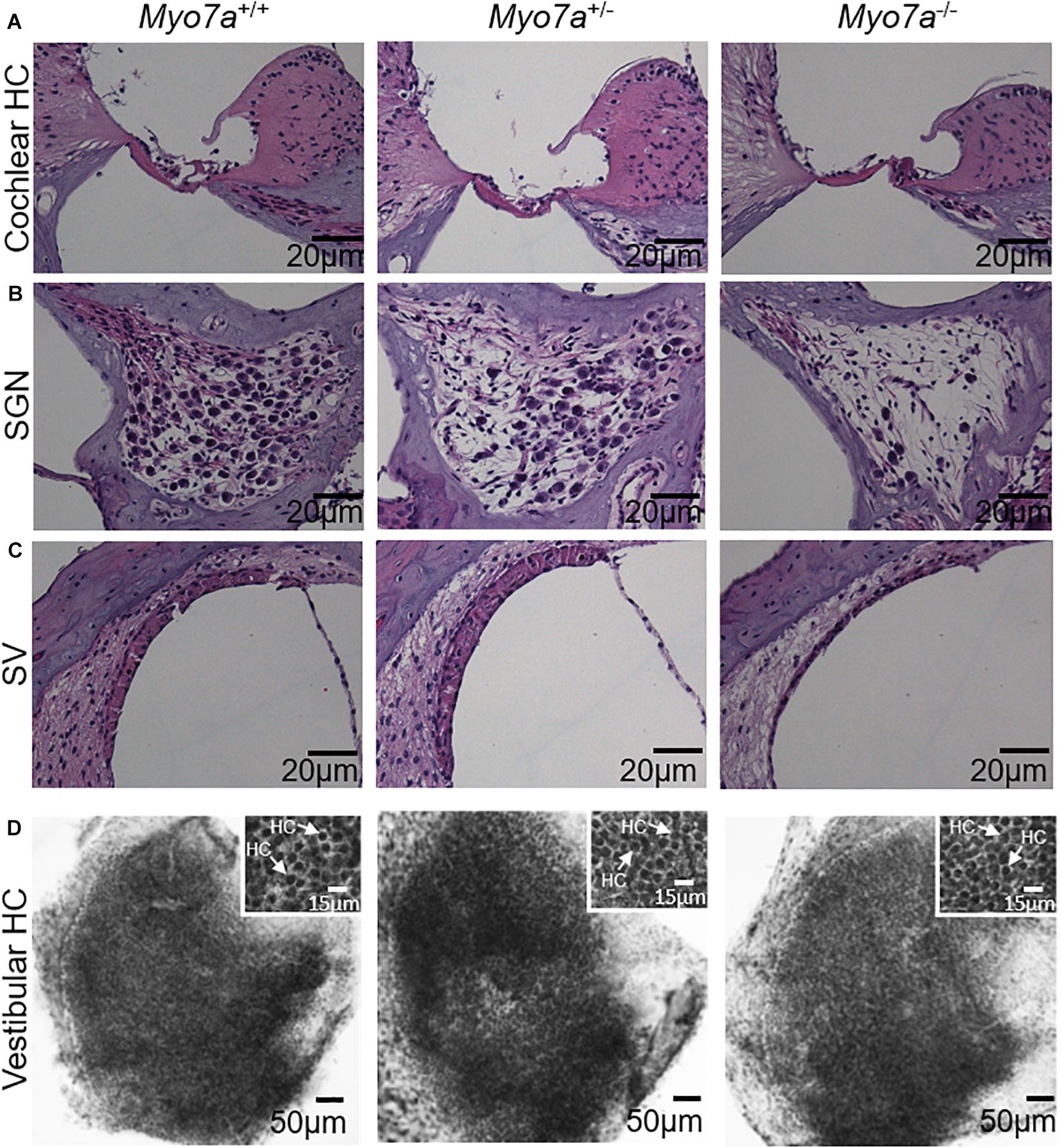
Figure 3. Assessment of basal–cochlear and utricle structure. Organ of Corti (A), spiral ganglion neurons (B), stria vascularis (C), and vestibular hair cells (D) in 5-month-old Myo7a+/+, Myo7a+/–, and Myo7a–/– mice. (A–C) Representative H&E-stained sections from the basal-cochlear region, imaged at 40× magnification. (D) Low magnification photomicrograph of representative hematoxylin-stained surface preparations of the macular utricle showing a dense array of vestibular hair cells covering the sensory epithelium (scale bar: 50 μm). Inset panels show a higher magnification view of densely packed vestibular hair cells with darkly stained nuclei (arrows, scale bar: 15 μm). SGN, spiral ganglion neuron; SV, stria vascularis. Scale bars for organ of Corti, SGN, and SV = 20 μm.
Vestibular HCs Are Preserved in Myo7a–/– Mice
Next, we investigated the impact of MYO7A on the structural integrity of vestibular HCs. Histological examination was performed on hematoxylin-stained HCs of the utricle macula of the vestibular tissue (Figure 3D). We did not observe any gross structural changes in the vestibular HCs of 5-month-old Myo7a–/– mice. Similarly, no obvious changes were observed in Myo7a+/– and Myo7a+/+ mice.
MYO7A-Deficient Mice Do Not Recapitulate the Retinal Degeneration/Dysfunction Seen in USH1B Patients
Retinal function and structure in age matched Myo7a–/–, Myo7a+/–, and Myo7a+/+ mice were assessed with ERG and OCT, respectively, every 30 days from 1 to 6 months of age. Scotopic and photopic amplitudes were only modestly reduced in both Myo7a+/– and Myo7a–/– mice relative to Myo7a+/+ controls after the 1 month time point (Figures 4A–E, photopic a-wave data not shown). By 6 months, the ERG amplitudes of Myo7a+/– and Myo7a–/– mice were reduced by approximately 20% that seen in Myo7a+/+ control mice. The marginal functional decreases observed in Myo7a+/– and Myo7a–/– mice were not associated with gross structural changes in the retina of these animals. OCT analysis confirmed that outer nuclear layer thickness (a measure of photoreceptor preservation) was maintained in all cohorts (Figure 4F).
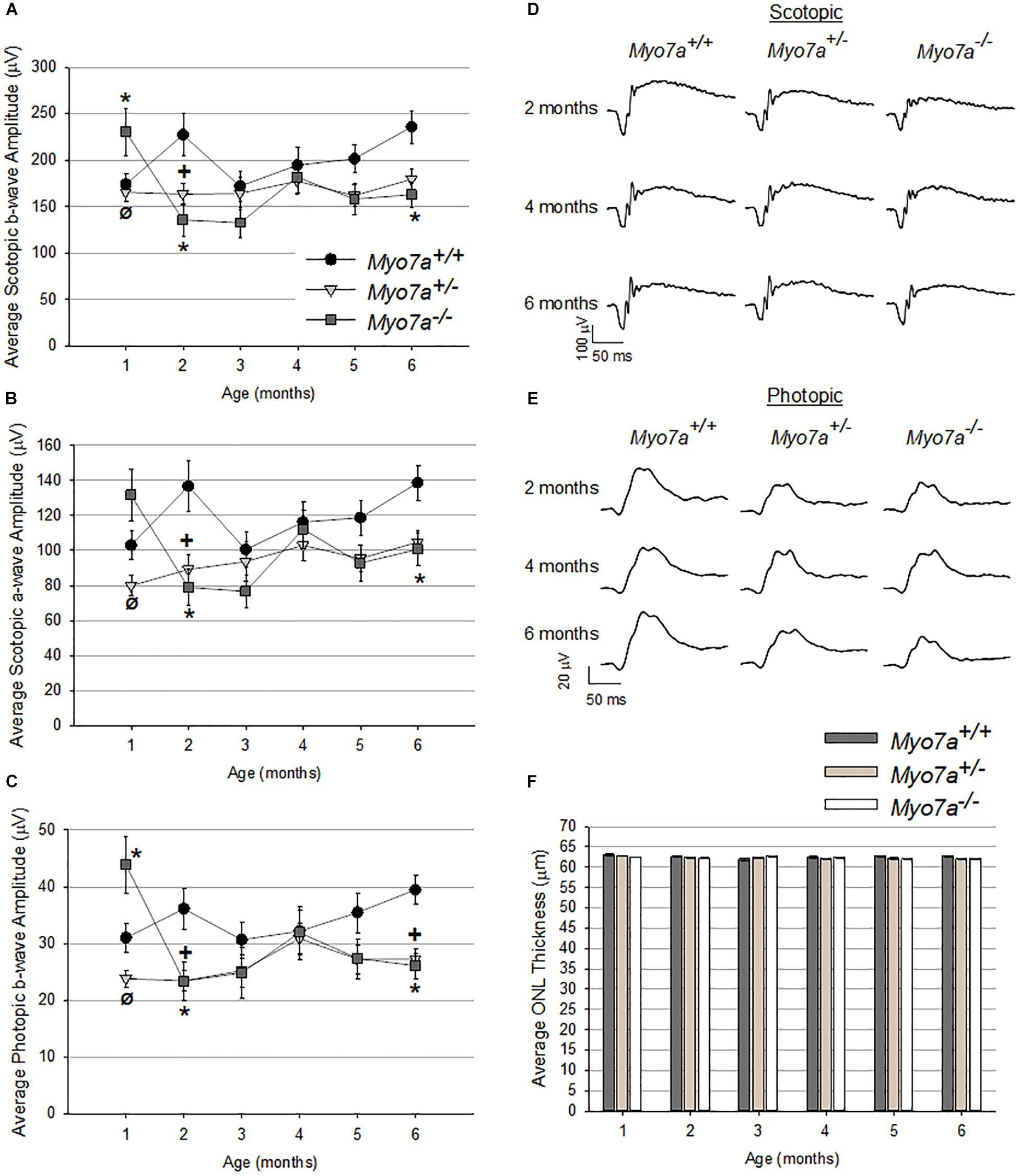
Figure 4. Characterization of retinal function and structure in Myo7a+/+, Myo7a+/–, and Myo7a–/– mice (n = 11–12 mice/genotype) over 6 months. Average scotopic b-wave (A), scotopic a-wave (B), and photopic b-wave (C) amplitudes. Equal numbers of males and females were used. Representative scotopic (D) and photopic (E) ERG waveforms at 2-, 4-, and 6-months of age. Scotopic and photopic values in panels (A–D) were recorded at 1.0 and 25.0 cd.s/m2, respectively. Average outer nuclear layer (ONL) thickness (F) in Myo7a+/+, Myo7a+/–, and Myo7a–/– mice over 6 months (n = 11–12 mice/genotype). Equal numbers of males and females were used. No gross changes in ONL thickness were observed. ∗p < 0.5 between Myo7a–/– vs. Myo7a+/+; +p < 0.5 between Myo7a+/– vs. Myo7a+/+.
Next, we evaluated the impact of MYO7A on RHO trafficking in Myo7a–/–, Myo7a+/–, and Myo7a+/+ mice. Previous studies employed immunogold labeling to demonstrate that MYO7A deficiency impaired/slowed transport of RHO through the photoreceptor connecting cilium (Liu et al., 1997, 1999). We used alternative methods to further investigate this phenomenon. Myo7a–/– mice were crossed with RHOGFP+/+ mice. The addition of GFP to the C-terminus of the RHO protein allows direct visualization of the protein and its expression is known to cause retinal degeneration (Chan et al., 2004). We hypothesized that, if RHO trafficking is impaired/slowed in the absence of MYO7A, then this phenotype may be compounded by the addition of a single RHOGFP allele and would be observable under fluorescent microscopy. We therefore generated Myo7a–/–:RHOGFP+/– mice by crossing Myo7a–/– and RHOGFP+/+ mice. To confirm that RHOGFP exacerbates degeneration in the presence of RHO mislocalization we also generated P23H+:RHOGFP+/– by crossing RHOGFP+/+ mice to transgenic RHO(P23H) mice as the P23H model exhibits mislocalized RHO as well as rapid degeneration (Illing et al., 2002; Mao et al., 2011). In all cases, mouse strains maintained one, wild-type Rho allele.
Rhodopsin–GFP fusion was imaged in retinal cross sections from all mouse strains (Figure 5A). RHOGFP mislocalization was not observed in Myo7a–/–:RHOGFP+/–, Myo7a+/–:RHOGFP+/–, and Myo7a+/+:RHOGFP+/– mice. In P23H+:RHOGFP+/– mice, however, mislocalization of RHOGFP in the photoreceptor inner segments was seen, especially at the 1- and 2-month time points before the onset of advanced degeneration.
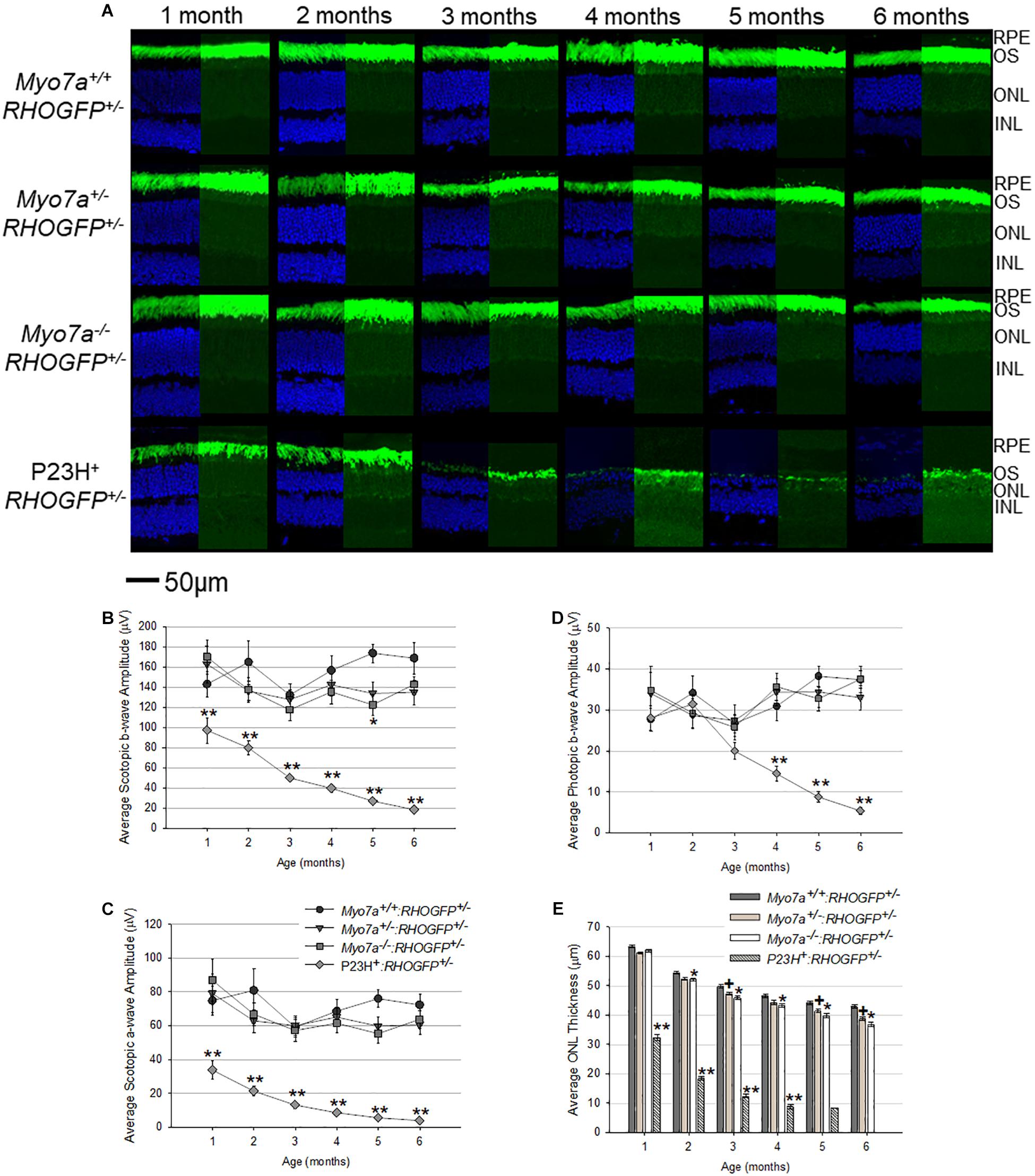
Figure 5. Characterization of retinal structure and function in Myo7a+/+:RHOGFP+/–, Myo7a+/–:RHOGFP+/–, Myo7a–/–:RHOGFP+/–, and P23H+:RHOGFP+/– mice (n = 11–12 mice/genotype) over 6 months. (A) Retinal cross sections (n = 4 mice/genotype) were co-stained with DAPI (left). Endogenous GFP expression from RHOGFP in the same section (without DAPI) is shown (right). Endogenous GFP expression on the right is overexposed for optimal visualization of RHOGFP localization in photoreceptor inner segments and ONL. All images taken at 60× magnification with identical exposure settings. Scale = 50 μm. Average (B) scotopic b-wave, (C) scotopic a-wave, and (D) photopic b-wave amplitudes in Myo7a+/+:RHOGFP+/–, Myo7a+/–:RHOGFP+/–, Myo7a–/–:RHOGFP+/–, and P23H+:RHOGFP+/– mice (n = 10–12 mice/genotype) over 6 months. Equal numbers of males and females were used. Scotopic and photopic averages were recorded at 1.0 and 25.0 cd.s/m2, respectively. (E) Average outer nuclear layer (ONL) thickness in Myo7a+/+:RHOGFP+/–, Myo7a+/–:RHOGFP+/–, Myo7a–/–:RHOGFP+/–, and P23H+:RHOGFP+/– mice (n = 10–12 mice/genotype) over 6 months. Equal numbers of males and females were used. No P23H+:RHOGFP+/– ONL data are included at 6 months because retinas were too thin for accurate measurement. ∗p < 0.5 between Myo7a–/–:RHOGFP+/– vs. Myo7a+/+:RHOGFP+/–; +p < 0.5 between Myo7a+/–:RHOGFP+/– vs. Myo7a+/+:RHOGFP+/–; ∗∗p < 0.5 between P23H+:RHOGFP+/– vs. Myo7a+/+:RHOGFP+/–.
Electroretinograms and OCT were performed on age-matched mice every 30 days from 1 to 6 months of age. Both Myo7a–/–:RHOGFP+/– and Myo7a+/–:RHOGFP+/– mice had slightly lower scotopic and photopic a- and b-wave amplitudes compared to Myo7a+/+:RHOGFP+/– controls, but this difference did not achieve statistical significance at most time points (Figures 5B–D). In contrast, the P23H+:RHOGFP+/– mice displayed significantly reduced ERG amplitudes at all time points and more rapid loss of retinal function relative to previous P23H + mouse models, a result presumably due to the addition of the RHOGFP allele (Price et al., 2011; Sakami et al., 2011; Mao et al., 2012). OCT analysis revealed slow, yet progressive, thinning in all Myo7a:RHOGFP mice (Figure 5E). This was expected as the fusion of GFP to the C-terminus of RHO was previously shown to cause degeneration (Chan et al., 2004). The ONLs of Myo7a+/–:RHOGFP+/– and Myo7a–/–:RHOGFP+/– mice were significantly thinner than Myo7a+/+:RHOGFP+/– controls by 2 months of age. MYO7A deficiency exacerbates the thinning caused by RHOGFP expression, as can be seen in the decreased ONL thickness in Myo7a–/–:RHOGFP+/– mice compared to Myo7a+/–:RHOGFP+/– mice (complete loss of MYO7A had a greater impact than partial loss). In contrast, photoreceptor degeneration in P23H+:RHOGFP+/– mice was rapid, with ONL thickness approximately 50% thinner relative to other strains at 1 month of age.
MYO7A Is Differentially Expressed in Mouse vs. NHP
MYO7A is expressed in the calyceal processes of primate photoreceptors, and these cells are the initial site of disease in USH1B patients (Jacobson et al., 2008, 2009, 2011; Sahly et al., 2012; Sumaroka et al., 2016; Subira et al., 2019). Mice potentially lack calyceal processes (Sahly et al., 2012) and do not exhibit retinal degeneration in the absence of MYO7A. One possible explanation for this discrepancy is that, unlike primates, mouse photoreceptors do not express MYO7A or rely on this protein to function. To investigate this further, we measured the relative expression of MYO7A in mouse vs. NHP photoreceptors and RPE via Western blot. The majority of MYO7A expression in adult (5-month-old) C57BL/6J mice is located in the RPE (Figure 6). In contrast, the majority of MYO7A expression is located in the neural retina of adult (5 years old) macaque (Figure 6). Because manual separation of the neural retina from the RPE is never 100% effective (some RPE cells remain attached to neural retina and vice versa), we cannot exclude the possibility that the minor signals observed in primate RPE or mouse neural retina were experimental artifact. This finding is consistent with previous reports that MYO7A is localized exclusively in mouse RPE, and not neural retina (el-Amraoui et al., 1996).
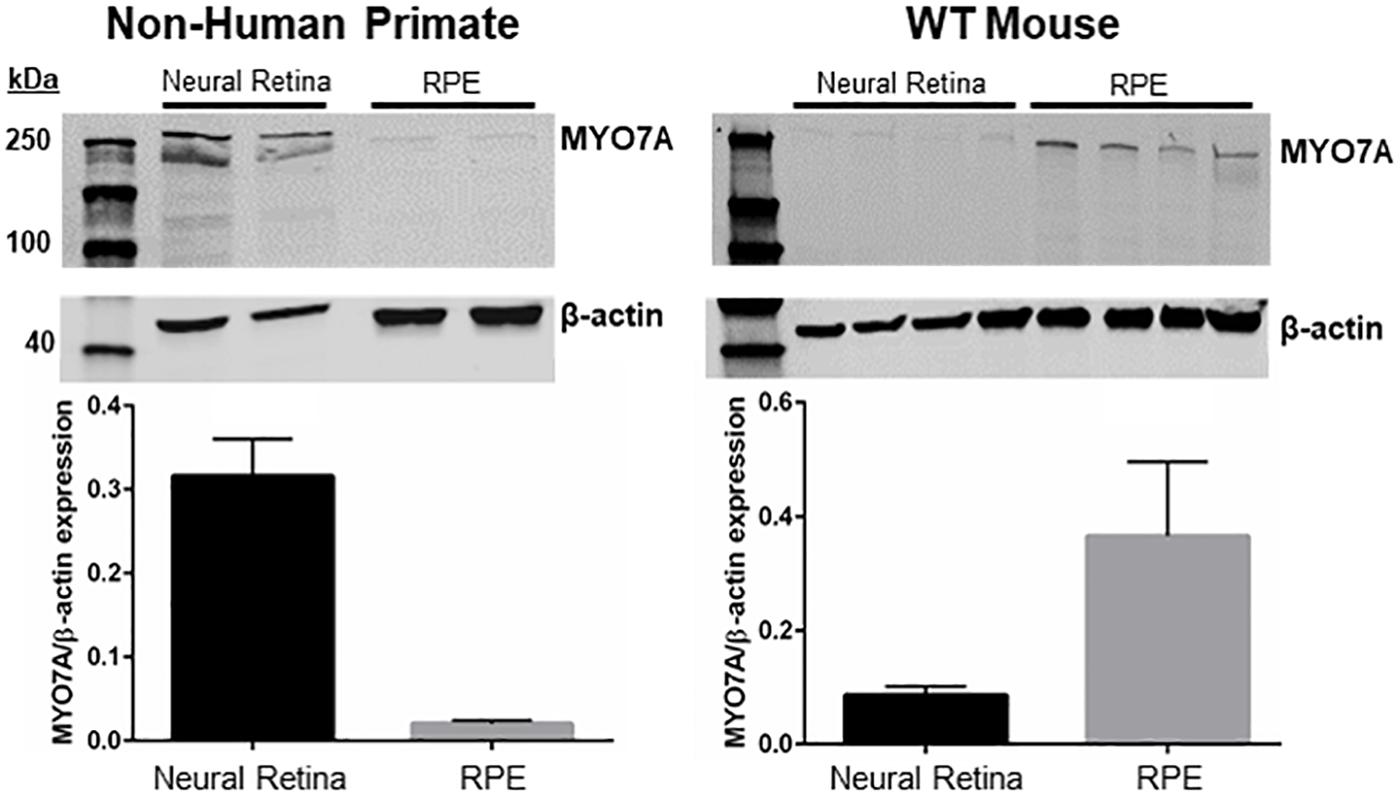
Figure 6. Expression of MYO7A in non-human primate vs. mouse. Neural retina and RPE from a 5-year-old male macaque and 5-month-old WT mice were manually separated. Protein from each compartment was immunoblotted with an antibody raised against MYO7A. The majority of MYO7A expression in macaque was found in neural retina (left). The majority of MYO7A expression in mouse was found in RPE (right). Protein levels were quantified and compared using Fiji. MYO7A expression was normalized to beta-actin. WT mouse = C57BL/6J.
Discussion
Our results demonstrate that Myo7a–/– mice, a newly established traditional KO model of USH1B, faithfully model the sensorineural hearing loss and degeneration of cochlear HCs, but not the robust retinal dysfunction/degeneration seen in patients. These findings are consistent with those previously described in other MYO7A-deficient mouse lines (Self et al., 1998; Libby and Steel, 2001; Lillo et al., 2003). A novel finding of this study is that Myo7a+/– mice also exhibited sensorineural hearing loss, degeneration of basal HCs in the cochlea, and marginal reductions in retinal function. This finding prompted us to look more deeply at previous reports of different Myo7a mutant mice for any evidence of haploinsufficiency.
Earlier studies that identified a role for MYO7A in cochlear development/function compared homozygous Myo7ash1, Myo7a6J, and Myo7a816SB mice to heterozygous controls (no wild-type littermates were included) and thus haploinsufficiency was not interrogated (Self et al., 1998). The finding that MYO7A was necessary for migration of melanosomes into apical RPE was based on comparisons between Myo7ash1–/– and Myo7ash1+/– mice (Liu et al., 1998). Melanosome localization was reported to be normal in heterozygous mice, but no comparison to wild-type controls was performed (i.e., haploinsufficiency appears unlikely but was not fully interrogated). The conclusion that MYO7A was necessary for efficient RHO transport through photoreceptor connecting cilia was based on comparisons between homozygous and heterozygous Myo7ash1, Myo7a4626SB, and Myo7a4494SB mice (Liu et al., 1999). Liu and colleagues concluded that opsin localization was normal in heterozygous mice, but no comparison to wild-type littermate controls was shown (i.e., haploinsufficiency appears unlikely but was not fully interrogated). The finding that MYO7A was required for normal phagocytosis of photoreceptor outer segments by RPE was based on comparisons between homozygous Myo7a4626SB/4626SB and heterozygous Myo7a+/4626SB mice (no wild-type littermate controls were used) (Gibbs et al., 2003). That study concluded that phagocytosis proceeded normally in heterozygous mice (i.e., haploinsufficiency appears unlikely but was not fully interrogated). Finally, the finding that MYO7A deficiency results in reduced retinal activity (∼20% reductions in ERG amplitudes) was based on comparisons between homozygous Myo7a4626SB/4626SB and heterozygous Myo7a+/4626SB mice (no wild-type littermate controls were used) and thus haploinsufficiency was not investigated (Libby and Steel, 2001; Colella et al., 2013). Taken together, previous studies did not explore whether heterozygous, carrier mice displayed an altered phenotype compared to “normal,” Myo7a+/+ mice.
To our knowledge, this is the first report of haploinsufficiency in mice carrying a single mutant Myo7a allele. This prompted us to search for evidence of cochlear/retinal dysfunction in USH1B carriers. Work from the 1970s showed that some Usher’s syndrome carriers exhibited (1) a tendency for hearing loss when compared to the normal population, (2) dark adaptation abnormalities, and (3) fundus abnormalities similar to retinitis pigmentosa (Dehaas et al., 1970; Holland et al., 1972; Sondheimer et al., 1979). These findings were associated with Usher’s syndrome as a whole, however, because genetic linkage had not yet been established. By the 1990s, USH1B had been linked to chromosome region 11p13-15 (Kimberling et al., 1992; Smith et al., 1992; Bonnetamir et al., 1994; Weil et al., 1995) and studies found that a number of obligate carriers exhibited significant audiological and ophthalmological abnormalities (Wagenaar et al., 1995). These clinical findings are intriguing, and align with our findings in heterozygous Myo7a+/– mice, but a more comprehensive assessment of genetically confirmed USH1B carriers is warranted to determine their significance.
Age-related hearing loss, or presbycusis, typically begins with a loss of sensitivity to high-frequency sounds and a loss of outer HCs in the basal cochlea (Gates and Mills, 2005). Over time, these deficits progress toward lower-frequencies and more apical regions of the cochlea. The loss of high-frequency ABR thresholds and basal cochlear HCs we observed in 5-month-old Myo7a+/– mice appear to mimic the early stages of presbycusis. Longer-term analysis is needed to determine whether sensorineural hearing loss and degeneration of cochlear structure in heterozygous mice worsens over time. Recently, it was reported that high frequency hearing loss occurred in rats with reduced estrogen levels (as a result of ovariectomy) and that this loss was associated with decreases in MYO7A expression (Diao et al., 2019). Investigators hypothesize that reduced levels of estrogen and MYO7A are responsible for the hearing loss observed in post-menopausal women (Svedbrant et al., 2015; Zhang et al., 2018). Notably, the hearing loss we observed in heterozygous Myo7a+/– mice was more pronounced in females at high frequencies.
While MYO7A localization in cochlear tissues and its role in auditory function are well-established (Hasson et al., 1995; Hosoya et al., 2016), its impact on the vestibular system has been less studied. In the vestibular system, MYO7A is expressed in the stereocilia, cuticular plates, and cell bodies in macular and semicircular canal HCs of mice and guinea pigs (Hasson et al., 1997) and clearly has a functional role in the vestibular system because MYO7A-deficient mice manifest behaviors (circling, head tilting) associated with its dysfunction. Our results show that, unlike cochlear HCs which are almost completely degenerated, vestibular HCs are present in Myo7a–/– mice at 5 months of age. Similar results were seen in Myo7aI487N/I487N ewaso mice, the only other MYO7A-deficient mouse line in which vestibular HC structure has been described (Miller et al., 2012). Rows of stereocilia are missing from the vestibular HCs and disrupted staircase morphology were observed in this mouse (Miller et al., 2012). However, the vestibular HCs were still present and contained stereocilia of various lengths. These findings shed light on results obtained in gene replacement studies recently conducted in other USH mouse models wherein vestibular HCs proved more amenable to therapy than cochlear HCs (Emptoz et al., 2017; Isgrig et al., 2017). Although our results do not rule out ultrastructural defects that may exist in MYO7A-deficient vestibular HCs, they suggest that these cells either do not degenerate, or are lost at a slower rate than their cochlear counterparts in the absence of MYO7A and support further analysis of vestibular HC structure in USH1B patients. If vestibular HCs are maintained, there exists the potential to treat USH1B-associated vestibular dysfunction via gene supplementation. This is an attractive prospect as currently available treatments for hearing loss (cochlear implants) and planned treatments for vision loss (gene supplementation) will not alleviate the loss of equilibrium caused by dysfunction of the vestibular system.
USH1B patients can retain both central and peripheral islands of structurally and functionally intact photoreceptors, suggesting that they may be good candidates for retinal gene therapy (Jacobson et al., 2008, 2009, 2011; Sumaroka et al., 2016). Toward that end, various studies, including ours, have been conducted to determine whether MYO7A-deficient mice have ocular phenotypes that are sufficiently robust to serve as outcome measures in preclinical gene therapy studies. Like previously examined mouse models, we show that Myo7a–/– do not exhibit retinal degeneration and have only marginal decrements in retinal function. Contrary to previous reports that MYO7A deficiency in homozygous Myo7ash1, Myo7a4494SB, and Myo7a4626B mice led to impaired/slowed transport of RHO through the photoreceptor connecting cilium, we did not observe accumulation of RHOGFP in the inner segments of photoreceptors in Myo7a–/–:RHOGFP+/– mice (Liu et al., 1999). OCT scans of USH1B patients suggest that the physiopathologic basis of this disease occurs at the level of the photoreceptors (Jacobson et al., 2008, 2011; Sumaroka et al., 2016; Subira et al., 2019). Specific findings include disruption/loss of the external limiting membrane, disruption of the myoid zone and ellipsoid zone, and loss of the outer segments. None of these disruptions were observed in OCT scans of Myo7a–/– or Myo7a+/– mice (data not shown). Our results, and others (Sahly et al., 2012), suggest this discrepancy is due to differential expression of MYO7A/photoreceptor structure across species. The majority of primate MYO7A is expressed in neural retina (not RPE), whereas it is marginally detected in mouse neural retina but expresses strongly in mouse RPE. These results argue that MYO7A-deficient mice are of limited value for elucidating the mechanisms underlying the retinal degeneration/dysfunction seen in USH1B patients. However, they do faithfully mimic the auditory and vestibular phenotypes and thus could reasonably be used to study these aspects of the disease and potential therapeutic interventions targeted to the inner ear. In addition, the observation that MYO7A haploinsufficiency impacts auditory function in mice warrants further investigation in USH1B carriers.
Due to the large size of the MYO7A cDNA, dual AAV vector-based strategies are currently being developed to address USH1B (Colella et al., 2013; Lopes et al., 2013; Dyka et al., 2014). While the novel Myo7a–/– mice described herein are unlikely be useful for measuring therapeutic efficacy of these therapies in retina (no sufficient outcome measures are available), they will be instrumental in evaluating the efficiency of dual vector platforms because they are definitively null, and can be easily genotyped/maintained. The best performing platforms will be defined in part by their propensity to drive full-length MYO7A expression in Myo7a–/– retina. Ultimately, these dual vectors will be best evaluated for their ability to recapitulate endogenous MYO7A expression in primate retina.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the University of Florida’s Institutional Animal Care and Use Committee.
Author Contributions
KC performed the experiments, analyzed the data, and wrote the initial draft of the manuscript. DF, JP, WL, and SC performed the experiments. SMC analyzed the data. M-JK, DD, RS, and SS designed and performed the experiments, and analyzed the data. SEB and SLB designed and supervised the project and edited the manuscript.
Funding
This work was supported the National Institutes of Health (NIH) Grant T32 EY00713224, NIH Grant F31 EY029139, NIH/NIDCD Grant R01 DC012552, NIH/NIDCD R01 DC014437, and the Foundation Fighting Blindness.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Paul Gamlin for providing non-human primate tissue, Dr. Alfred Lewin for providing us with the transgenic P23H mouse model, and Drs. Jim Resnick and Scott Wilson for providing advice on mouse construction.
References
Astuto, L. M., Weston, M. D., Carney, C. A., Hoover, D. M., Cremers, C. W., Wagenaar, M., et al. (2000). Genetic heterogeneity of Usher syndrome: analysis of 151 families with Usher type I. Am. J. Hum. Genet. 67, 1569–1574. doi: 10.1086/316889
Bharadwaj, A. K., Kasztejna, J. P., Huq, S., Berson, E. L., and Dryja, T. P. (2000). Evaluation of the myosin VIIA gene and visual function in patients with usher syndrome type I. Exp. Eye Res. 71, 173–181. doi: 10.1006/exer.2000.0863
Bonnetamir, B., Korostishevsky, M., Kalinsky, H., Seroussi, E., Beker, R., Weiss, S., et al. (1994). Genetic-mapping of the gene for usher syndrome - linkage analysis in a large samaritan kindred. Genomics 20, 36–42. doi: 10.1006/geno.1994.1124
Chan, F., Bradley, A., Wensel, T. G., and Wilson, J. H. (2004). Knock-in human rhodopsin-GFP fusions as mouse models for human disease and targets for gene therapy. Proc. Natl. Acad. Sci. U.S.A. 101, 9109–9114. doi: 10.1073/pnas.0403149101
Colella, P., Sommella, A., Marrocco, E., Di Vicino, U., Polishchuk, E., Garrido, M. G., et al. (2013). Myosin7a deficiency results in reduced retinal activity which is improved by gene therapy. PLoS One 8:e72027. doi: 10.1371/journal.pone.0072027
Dehaas, E. B. H., Vanlith, G. H. M., Rijnders, J., Rumke, A. M. L., and Volmer, C. (1970). Ushers syndrome - with special reference to heterozygous manifestations. Doc. Ophthalmol. 28, 166–190. doi: 10.1007/bf00153876
Diao, H., Zhao, L., Qin, L., Bai, W., Wang, K., Zhang, J., et al. (2019). Lower expression of prestin and MYO7A correlates with menopause-associated hearing loss. Climacteric 22, 361–369. doi: 10.1080/13697137.2018.1547698
Ding, D. L., Jiang, H. Y., Chen, G. D., Longo-Guess, C., Muthaiah, V. P. K., Tian, C., et al. (2016). N-acetyl-cysteine prevents age-related hearing loss and the progressive loss of inner hair cells in gamma-glutamyl transferase 1 deficient mice. Aging 8, 730–750. doi: 10.18632/aging.100927
Dyka, F. M., Boye, S. L., Chiodo, V. A., Hauswirth, W. W., and Boye, S. E. (2014). Dual adeno-associated virus vectors result in efficient in vitro and in vivo expression of an oversized gene, MYO7A. Hum. Gene Ther. Methods 25, 166–177. doi: 10.1089/hgtb.2013.212
el-Amraoui, A., Sahly, I., Picaud, S., Sahel, J., Abitbol, M., and Petit, C. (1996). Human usher 1B/mouse shaker-1: the retinal phenotype discrepancy explained by the presence/absence of myosin VIIA in the photoreceptor cells. Hum. Mol. Genet. 5, 1171–1178. doi: 10.1093/hmg/5.8.1171
Emptoz, A., Michel, V., Lelli, A., Akil, O., de Monvel, J. B., Lahlou, G., et al. (2017). Local gene therapy durably restores vestibular function in a mouse model of usher syndrome type 1G. Proc. Natl. Acad. Sci. U.S.A. 114, 9695–9700. doi: 10.1073/pnas.1708894114
Gibbs, D., Kitamoto, J., and Williams, D. S. (2003). Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin Vila, the usher syndrome 1B protein. Proc. Natl. Acad. Sci. U.S.A. 100, 6481–6486. doi: 10.1073/pnas.1130432100
Gibson, F., Walsh, J., Mburu, P., Varela, A., Brown, K. A., Antonio, M., et al. (1995). A type-vii myosin encoded by the mouse deafness gene shaker-1. Nature 374, 62–64. doi: 10.1038/374062a0
Han, C., Ding, D., Lopez, M.-C., Manohar, S., Zhang, Y., Kim, M.-J., et al. (2016). Effects of long-term exercise on age-related hearing loss in mice. J. Neurosci. 36, 11308–11319. doi: 10.1523/jneurosci.2493-16.2016
Hasson, T., Gillespie, P. G., Garcia, J. A., MacDonald, R. B., Zhao, Y. D., Yee, A. G., et al. (1997). Unconventional myosins in inner-ear sensory epithelia. J. Cell Biol. 137, 1287–1307. doi: 10.1083/jcb.137.6.1287
Hasson, T., Heintzelman, M. B., Santos-Sacchi, J., Corey, D. P., and Mooseker, M. S. (1995). expression in cochlea and retina of myosin VIIa, the gene-product defective in usher syndrome type 1b. Proc. Natl. Acad. Sci. U.S.A. 92, 9815–9819. doi: 10.1073/pnas.92.21.9815
Holland, M. G., Cambie, E., and Kloepfer, W. (1972). An evaluation of genetic carriers of ushers syndrome. Am. J. Ophthalmol. 74, 940–947. doi: 10.1016/0002-9394(72)91215-9
Hosoya, M., Fujioka, M., Ogawa, K., and Okano, H. (2016). Distinct expression patterns of causative genes responsible for hereditary progressive hearing loss in non-human primate cochlea. Sci. Rep. 6:22250. doi: 10.1038/srep22250
Illing, M. E., Rajan, R. S., Bence, N. F., and Kopito, R. R. (2002). A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J. Biol. Chem. 277, 34150–34160. doi: 10.1074/jbc.m204955200
Isgrig, K., Shteamer, J. W., Belyantseva, I. A., Drummond, M. C., Fitzgerald, T. S., Vijayakumar, S., et al. (2017). Gene therapy restores balance and auditory functions in a mouse model of usher syndrome. Mol. Ther. 25, 780–791. doi: 10.1016/j.ymthe.2017.01.007
Jacobson, S. G., Aleman, T. S., Sumaroka, A., Cideciyan, A. V., Roman, A. J., Windsor, E. A. M., et al. (2009). Disease boundaries in the retina of patients with usher syndrome caused by MYO7A gene mutations. Investig. Ophthalmol. Vis. Sci. 50, 1886–1894. doi: 10.1167/iovs.08-3122
Jacobson, S. G., Cideciyan, A. V., Aleman, T. S., Sumaroka, A., Roman, A. J., Gardner, L. M., et al. (2008). Usher syndromes due to MYO7A, PCDH15, USH2A or GPR98 mutations share retinal disease mechanism. Hum. Mol. Genet. 17, 2405–2415. doi: 10.1093/hmg/ddn140
Jacobson, S. G., Cideciyan, A. V., Gibbs, D., Sumaroka, A., Roman, A. J., Aleman, T. S., et al. (2011). Retinal disease course in usher syndrome 1B Due to MYO7A mutations. Investig. Ophthalmol. Vis. Sci. 52, 7924–7936. doi: 10.1167/iovs.11-8313
Kimberling, W. J., Moller, C. G., Davenport, S., Priluck, I. A., Beighton, P. H., Greenberg, J., et al. (1992). linkage of usher syndrome type-i gene (ush1b) to the long arm of chromosome-11. Genomics 14, 988–994. doi: 10.1016/s0888-7543(05)80121-1
Libby, R. T., and Steel, K. P. (2001). Electroretinographic anomalies in mice with mutations in Myo7a, the gene involved. in human Usher syndrome type 1B. Investig. Ophthalmol. Vis. Sci. 42, 770–778.
Lillo, C., Kitamoto, J., Liu, X. R., Quint, E., Steel, K. P., and Williams, D. S. (2003). Mouse models for usher syndrome 1B. Adv. Exp. Med. Biol. 533, 143–150. doi: 10.1007/978-1-4615-0067-4_18
Liu, X. R., Ondek, B., and Williams, D. S. (1998). Mutant myosin VIIa causes defective melanosome distribution in the RPE of shaker-1 mice. Nat. Genet. 19, 117–118. doi: 10.1038/470
Liu, X. R., Udovichenko, I. P., Brown, S. D. M., Steel, K. P., and Williams, D. S. (1999). Myosin VIIa participates in opsin transport through the photoreceptor cilium. J. Neurosci. 19, 6267–6274. doi: 10.1523/jneurosci.19-15-06267.1999
Liu, X. R., Vansant, G., Udovichenko, I. P., Wolfrum, U., and Williams, D. S. (1997). Myosin VIIa, the product of the usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motil. Cytoskeleton 37, 240–252. doi: 10.1002/(sici)1097-0169(1997)37:3<240::aid-cm6>3.3.co;2-2
Lopes, V. S., Boye, S. E., Louie, C. M., Boye, S., Dyka, F., Chiodo, V., et al. (2013). Retinal gene therapy with a large MYO7A cDNA using adeno-associated virus. Gene Ther. 20, 824–833. doi: 10.1038/gt.2013.3
Mao, H., James, T. Jr., Schwein, A., Shabashvili, A. E., Hauswirth, W. W., Gorbatyuk, M. S., et al. (2011). AAV delivery of wild-type rhodopsin preserves retinal function in a mouse model of autosomal dominant retinitis pigmentosa. Hum. Gene Ther. 22, 567–575. doi: 10.1089/hum.2010.140
Mao, H. Y., Gorbatyuk, M. S., Rossmiller, B., Hauswirth, W. W., and Lewin, A. S. (2012). Long-Term rescue of retinal structure and function by rhodopsin RNA replacement with a single adeno-associated viral vector in P23H RHO transgenic mice. Hum. Gene Ther. 23, 356–366. doi: 10.1089/hum.2011.213
Mattapallil, M. J., Wawrousek, E. F., Chan, C.-C., Zhao, H., Roychoudhury, J., Ferguson, T. A., et al. (2012). The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Investig. Ophthalmol. Vis. Sci. 53, 2921–2927. doi: 10.1167/iovs.12-9662
McCullough, K. T., Boye, S. L., Fajardo, D., Calabro, K., Peterson, J. J., Strang, C. E., et al. (2019). Somatic gene editing of GUCY2D by AAV-CRISPR/Cas9 alters retinal structure and function in mouse and macaque. Hum. Gene Ther. 30, 571–589. doi: 10.1089/hum.2018.193
Miller, K. A., Williams, L. H., Rose, E., Kuiper, M., Dahl, H. H., and Manji, S. S. (2012). Inner ear morphology is perturbed in two novel mouse models of recessive deafness. PLoS One 7:e51284. doi: 10.1371/journal.pone.0051284
Muller, M., von Hunerbein, K., Holdis, S., and Smolders, J. W. T. (2005). A physiological place-frequency map of the cochlea in the CBA/J mouse. Hear. Res. 202, 63–73. doi: 10.1016/j.heares.2004.08.011
Ouyang, X. M., Yan, D., Du, L. L., Hejtmancik, J. F., Jacobson, S. G., Nance, W. E., et al. (2005). Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population. Hum. Genet. 116, 292–299. doi: 10.1007/s00439-004-1227-2
Pang, J.-J., Dai, X., Boye, S. E., Barone, I., Boye, S. L., Mao, S., et al. (2011). Long-term retinal function and structure rescue using capsid mutant AAV8 vector in the rd10 mouse, a model of recessive retinitis pigmentosa. Mol. Ther. 19, 234–242. doi: 10.1038/mt.2010.273
Peng, Y. W., Zallocchi, M., Wang, W. M., Delimont, D., and Cosgrove, D. (2011). Moderate light-induced degeneration of rod photoreceptors with delayed transducin translocation in shaker1 Mice. Investig. Ophthalmol. Vis. Sci. 52, 6421–6427.
Petit, C. (2001). Usher syndrome: from genetics to pathogenesis. Annu. Rev. Genomics Hum. Genet. 2, 271–297. doi: 10.1146/annurev.genom.2.1.271
Price, B. A., Sandoval, I. M., Chan, F., Simons, D. L., Wu, S. M., Wensel, T. G., et al. (2011). Mislocalization and degradation of human P23H-Rhodopsin-GFP in a knockin mouse model of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 52, 9728–9736. doi: 10.1167/iovs.11-8654
Roccio, M., Perny, M., Ealy, M., Widmer, H. R., Heller, S., and Senn, P. (2018). Molecular characterization and prospective isolation of human fetal cochlear hair cell progenitors. Nat. Commun. 9:4027. doi: 10.1038/s41467-018-06334-7
Ryder, E., Gleeson, D., Sethi, D., Vyas, S., Miklejewska, E., Dalvi, P., et al. (2013). Molecular characterization of mutant mouse strains generated from the EUCOMM/KOMP-CSD ES cell resource. Mamm. Genome 24, 286–294. doi: 10.1007/s00335-013-9467-x
Sahly, I., Dufour, E., Schietroma, C., Michel, V., Bahloul, A., Perfettini, I., et al. (2012). Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J. Cell Biol. 199, 381–399. doi: 10.1083/jcb.201202012
Sakami, S., Maeda, T., Bereta, G., Okano, K., Golczak, M., Sumaroka, A., et al. (2011). Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J. Biol. Chem. 286, 10551–10567. doi: 10.1074/jbc.M110.209759
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Self, T., Mahony, M., Fleming, J., Walsh, J., Brown, S. D. M., and Steel, K. P. (1998). Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development 125, 557–566.
Skarnes, W. C., Rosen, B., West, A. P., Koutsourakis, M., Bushell, W., Iyer, V., et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342. doi: 10.1038/nature10163
Smith, R. J. H., Berlin, C. I., Hejtmancik, J. F., Keats, B. J. B., Kimberling, W. J., Lewis, R. A., et al. (1994). Clinical-Diagnosis of the usher syndromes. Am. J. Med. Genet. 50, 32–38.
Smith, R. J. H., Lee, E. C., Kimberling, W. J., Daiger, S. P., Pelias, M. Z., Keats, B. J. B., et al. (1992). localization of 2 genes for usher syndrome Type-I to chromosome-11. Genomics 14, 995–1002. doi: 10.1016/s0888-7543(05)80122-3
Someya, S., and Prolla, T. A. (2010). Mitochondrial oxidative damage and apoptosis in age-related hearing loss. Mech. Ageing Dev. 131, 480–486. doi: 10.1016/j.mad.2010.04.006
Sondheimer, S., Fishman, G. A., Young, R. S., and Vasquez, V. A. (1979). dark-adaptation testing in heterozygotes of ushers syndrome. Br. J. Ophthalmol. 63, 547–550. doi: 10.1136/bjo.63.8.547
Subira, O., Catala-Mora, J., Diaz-Cascajosa, J., Padron-Perez, N., Claveria, M. A., Coll-Alsina, N., et al. (2019). Retinal findings in pediatric patients with usher syndrome Type 1 due to mutations in MYO7A gene. Eye doi: 10.1038/s41433-019-0536-6 [Epub ahead of print].
Sumaroka, A., Matsui, R., Cideciyan, A. V., McGuigan, D. B. III, Sheplock, R., Schwartz, S. B., et al. (2016). Outer retinal changes including the ellipsoid zone band in usher syndrome 1B due to MYO7A mutations. Investig. Ophthalmol. Vis. Sci. 57, OCT253–OCT261. doi: 10.1167/iovs.15-18860
Svedbrant, J., Bark, R., Hultcrantz, M., and Hederstierna, C. (2015). Hearing decline in menopausal women - a 10-year follow-up. Acta Otolaryngol. 135, 807–813. doi: 10.3109/00016489.2015.1023354
Trouillet, A., Dubus, E., Degardin, J., Estivalet, A., Ivkovic, I., Godefroy, D., et al. (2018). Cone degeneration is triggered by the absence of USH1 proteins but prevented by antioxidant treatments. Sci. Rep. 8:1968. doi: 10.1038/s41598-018-20171-0
Volland, S., Hughes, L. C., Kong, C., Burgess, B. L., Linberg, K. A., Luna, G., et al. (2015). Three-dimensional organization of nascent rod outer segment disk membranes. Proc. Natl. Acad. Sci. U.S.A. 112, 14870–14875. doi: 10.1073/pnas.1516309112
Wagenaar, M., Terrahe, B., Vanaarem, A., Huygen, P., Admiraal, R., and Bleekerwagemakers, E. (1995). Clinical findings in obligate carriers of Type-I usher syndrome. Am. J. Med. Genet. 59, 375–379. doi: 10.1002/ajmg.1320590319
Weil, D., Blanchard, S., Kaplan, J., Guilford, P., Gibson, F., Walsh, J., et al. (1995). Defective myosin VIIA gene responsible for usher syndrome type 1B. Nature 374, 60–61. doi: 10.1038/374060a0
White, K., Kim, M.-J., Ding, D., Han, C., Park, H.-J., Meneses, Z., et al. (2017). G6pd deficiency does not affect the cytosolic glutathione or thioredoxin antioxidant defense in mouse cochlea. J. Neurosci. 37, 5770–5781. doi: 10.1523/jneurosci.0519-17.2017
Williams, D. S. (2008). Usher syndrome: animal models, retinal function of usher proteins, and prospects for gene therapy. Vis. Res. 48, 433–441. doi: 10.1016/j.visres.2007.08.015
Wu, Y., Fishkin, N. E., Pande, A., Pande, J., and Sparrow, J. R. (2009). Novel lipofuscin bisretinoids prominent in human retina and in a model of recessive stargardt disease. J. Biol. Chem. 284, 20155–20166. doi: 10.1074/jbc.M109.021345
Zhang, J. F., Zhang, T. Y., Yu, L. S., Ruan, Q. Y., Yin, L. X., Liu, D., et al. (2018). Effects of ovarian reserve and hormone therapy on hearing in premenopausal and postmenopausal women: a cross-sectional study. Maturitas 111, 77–81. doi: 10.1016/j.maturitas.2018.01.019
Keywords: retina, cochlea, Usher syndrome, myosin-7A, vision, hearing
Citation: Calabro KR, Boye SL, Choudhury S, Fajardo D, Peterson JJ, Li W, Crosson SM, Kim M-J, Ding D, Salvi R, Someya S and Boye SE (2019) A Novel Mouse Model of MYO7A USH1B Reveals Auditory and Visual System Haploinsufficiencies. Front. Neurosci. 13:1255. doi: 10.3389/fnins.2019.01255
Received: 04 September 2019; Accepted: 05 November 2019;
Published: 22 November 2019.
Edited by:
Stylianos Michalakis, Ludwig-Maximilians-Universität München, GermanyReviewed by:
Elvir Becirovic, Ludwig-Maximilians-Universität München, GermanyJan Wijnholds, Leiden University Medical Center, Netherlands
Copyright © 2019 Calabro, Boye, Choudhury, Fajardo, Peterson, Li, Crosson, Kim, Ding, Salvi, Someya and Boye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shannon E. Boye, c2hhaXJlQHVmbC5lZHU=