 Naoki Suzuki
Naoki Suzuki Tetsuya Akiyama
Tetsuya Akiyama Hitoshi Warita
Hitoshi Warita Masashi Aoki1*
Masashi Aoki1*- 1Department of Neurology, Tohoku University School of Medicine, Sendai, Japan
- 2Department of Neurology, Shodo-kai Southern Tohoku General Hospital, Miyagi, Japan
Amyotrophic lateral sclerosis (ALS) is an intractable adult-onset neurodegenerative disease that leads to the loss of upper and lower motor neurons (MNs). The long axons of MNs become damaged during the early stages of ALS. Genetic and pathological analyses of ALS patients have revealed dysfunction in the MN axon homeostasis. However, the molecular pathomechanism for the degeneration of axons in ALS has not been fully elucidated. This review provides an overview of the proposed axonal pathomechanisms in ALS, including those involving the neuronal cytoskeleton, cargo transport within axons, axonal energy supply, clearance of junk protein, neuromuscular junctions (NMJs), and aberrant axonal branching. To improve understanding of the global changes in axons, the review summarizes omics analyses of the axonal compartments of neurons in vitro and in vivo, including a motor nerve organoid approach that utilizes microfluidic devices developed by this research group. The review also discusses the relevance of intra-axonal transcription factors frequently identified in these omics analyses. Local axonal translation and the relationship among these pathomechanisms should be pursued further. The development of novel strategies to analyze axon fractions provides a new approach to establishing a detailed understanding of resilience of long MN and MN pathology in ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating adult-onset neurodegenerative disorder (Brown and Al-Chalabi, 2017). Both the upper and lower motor neurons (MNs) are affected, such that the disorder is characterized by muscle weakness with spasticity and atrophy. Approximately 10% of ALS occurrence is familial (Ghasemi and Brown, 2018). Since the identification in 1993 (Rosen et al., 1993) of copper/zinc superoxide dismutase 1 (SOD1) in ALS patients with an autosomal dominant trait in 1993 (Aoki et al., 1993), more than 25 genes have been reported as causative genes of familial ALS (Maday et al., 2014; De Vos and Hafezparast, 2017; Ghasemi and Brown, 2018; Cook and Petrucelli, 2019).
The pathomechanisms of ALS have been examined using familial ALS models. Intracellular generation of reactive oxygen species production (Borchelt et al., 1994; Wiedau-Pazos et al., 1996; Howland et al., 2002) and unfolded protein response/endoplasmic reticulum (ER) stress (Kikuchi et al., 2006; Kieran et al., 2007; Urushitani et al., 2008) have been inferred from the discovery of SOD1 as a factor. A cell non-autonomous effect (Boillee et al., 2006; Di Giorgio et al., 2007; Nagai et al., 2007; Yamanaka et al., 2008; de Boer et al., 2014) has also been examined in mutant SOD1-transgenic mouse and cellular models. Mutations in the RNA-binding protein (RBP) TAR DNA-binding protein (TARDBP encoding TDP-43) can result in ALS (Kabashi et al., 2008; Rutherford et al., 2008), and cytoplasmic TDP-43 inclusions have been reported in over 90% of cases of sporadic ALS (Mackenzie et al., 2010). In 2009, fused in sarcoma (FUS) was determined in 2009 to be the causative gene of ALS (Kwiatkowski et al., 2009; Vance et al., 2009). FUS and TDP-43 have similar structural characteristics, including an RNA recognition motif (RRM), a nuclear export signal (NES), a nuclear localization signal (NLS), and prion-like domains (PrLDs) (Kapeli et al., 2017). The C-terminal NLS site regulates the nucleocytoplasmic localization of FUS and is a hotspot for mutations in familial ALS (Kwiatkowski et al., 2009; Vance et al., 2009; Suzuki et al., 2010; Nishiyama et al., 2017). Abnormal phase separation of FUS is involved in this pathomechanism (Guo et al., 2018; Hofweber et al., 2018; Qamar et al., 2018). NLS mutations impair the nuclear import of FUS, and the level of mislocalized cytoplasmic FUS is correlated to the severity of the clinical ALS phenotypes (Dormann et al., 2010). In addition, recent reports have demonstrated that the abnormal NLS function results in the aberrant accumulation of mutant FUS in the cytoplasm (Ichiyanagi et al., 2016; Guo et al., 2018; Hofweber et al., 2018; Qamar et al., 2018; Yoshizawa et al., 2018). Previous studies have found that the toxic gain of function occurring with mutant FUS is crucial for neurodegeneration (Scekic-Zahirovic et al., 2016; Sharma et al., 2016; Shiihashi et al., 2016).
A hexanucleotide repeat expansion in chromosome 9 open reading frame 72 (C9orf72) (DeJesus-Hernandez et al., 2011; Renton et al., 2011) is the most common cause of ALS when examined in Western countries (Balendra and Isaacs, 2018). Loss of function of C9ORF72 (Burberry et al., 2016; O’Rourke et al., 2016), toxic gain of function of C9ORF72 due to repeat RNA (Peters et al., 2015; Jiang et al., 2016), and toxic gain of function due to proteins with dipeptide repeats resulting from repeat-associated non-ATG translation (Mori et al., 2013; Kwon et al., 2014; Mizielinska et al., 2014; Wen et al., 2014; Chew et al., 2015) have been suggested as disease mechanisms.
These findings are mainly focused on the event in the cytoplasm of MNs. Actually, long axons, which have lengths of up to 100 cm in humans, are characteristic of MN morphology, and connect the soma of MNs to the skeletal muscles. In ALS, MNs are dysfunctional due to axonal degeneration (Ferraiuolo et al., 2011), that occurs prior to the motor phenotype in ALS (Fischer et al., 2004; Roy et al., 2005). Consistent with this observation, transgenic models of ALS also demonstrate abnormal axons and other degenerative processes, followed by the death of MNs (Armstrong and Drapeau, 2013; Tian et al., 2016; Fujimori, 2018). Other studies have revealed that axonal damage occurs earlier than the death of cell bodies and subsequent symptoms in patients; such symptoms become apparent only after the loss of many MNs (Dadon-Nachum et al., 2011).
Various reviews have described the physiological and pathological features of neuronal axons, including cargo transport within axons, local translation, and the axonal transcriptome (Jung et al., 2012; Maday et al., 2014; Batista and Hengst, 2016; Neto et al., 2016; Brady and Morfini, 2017; De Vos and Hafezparast, 2017). However, because primary neurons from patients cannot be easily obtained and because axons produce low sample yields and are difficult to culture, the details of the pathological mechanisms of ALS remain unclear. To further elucidate the resilience and pathomechanisms in MN axons, this review summarizes omics analyses of the axon compartment using microfluidic devices and ex vivo samples. Intra-axonal transcription and local axonal translation are the mechanisms of ALS emerging in the field, as discussed in the following.
Accumulating Evidence of Axonal Dysfunction in ALS
The global pathomechanisms of axons in ALS are considered next, in an overview of the current knowledge of axonal events in MNs. This section classifies the pathomechanisms of axonal dysfunction into six subsections, including neuronal cytoskeleton, cargo transport within axons, axonal energy supply, clearance of junk protein, neuromuscular junctions (NMJs), and aberrant axonal branching (Figure 1). As mentioned in the introduction, an increasing number of genes have been found as causative or associated genes for ALS. Evidence of axon pathomechanisms from the genetics of ALS is also accumulating (Table 1). These mechanisms are explained in each subsection.
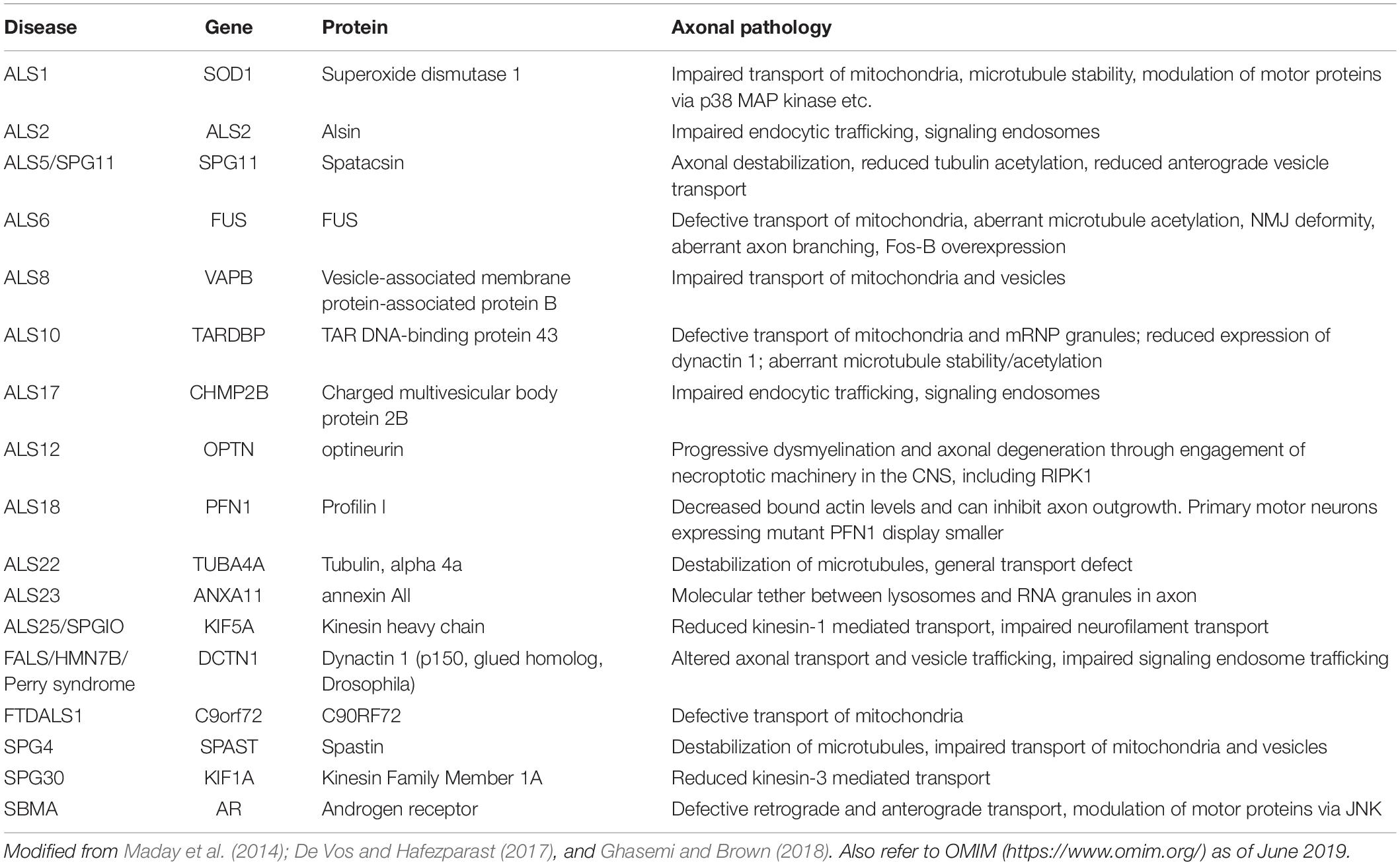
Table 1. Motor neuron disease-associated mutations and axonal pathology.
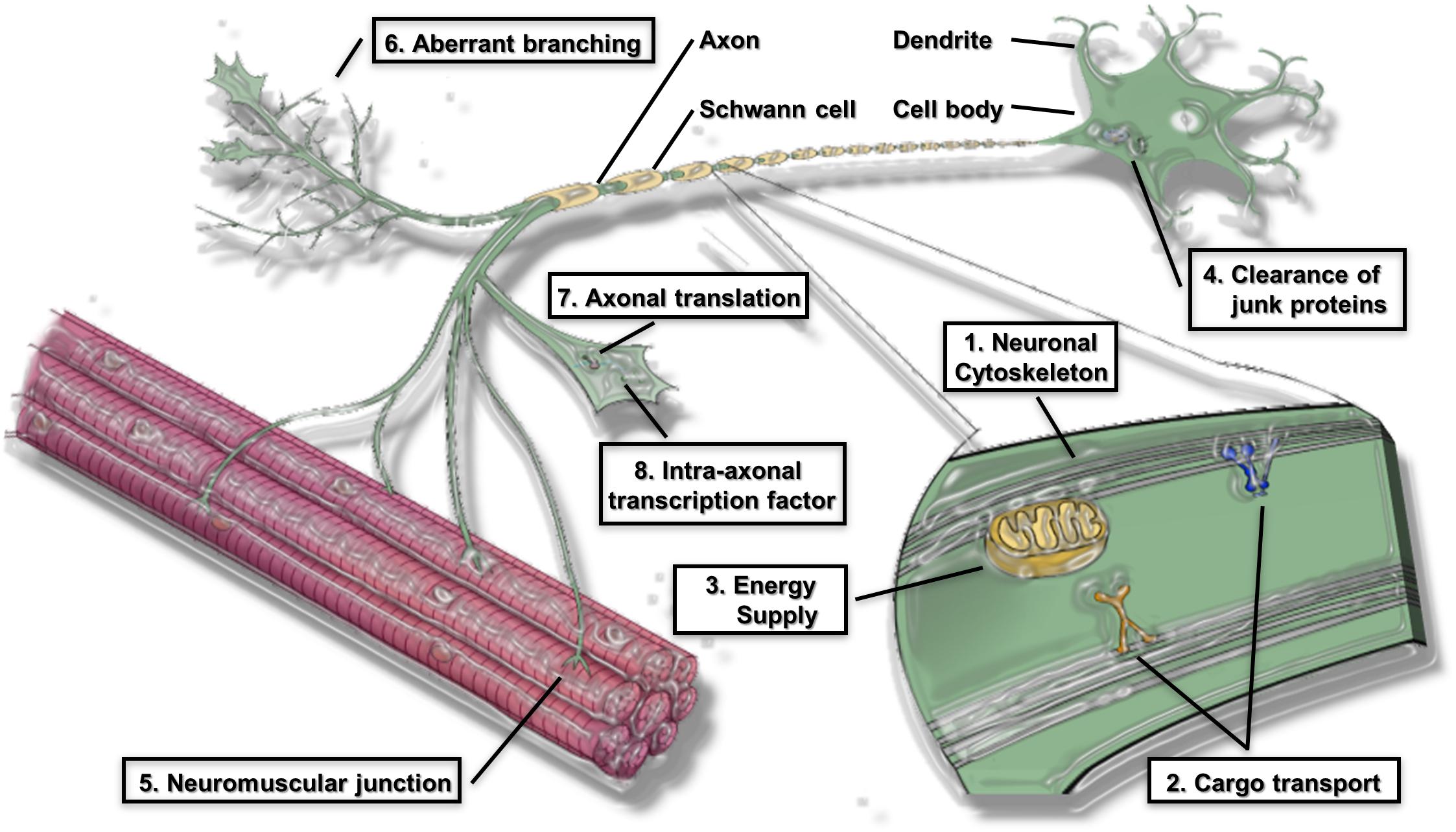
Figure 1. Proposed pathomechanism of ALS in axon compartments. (1) Neuronal cytoskeleton; (2) cargo transport within axons; (3) axonal energy supply; (4) clearance of junk protein; (5) Neuromuscular junction (NMJs); (6) aberrant axonal branching; (7) Axonal translation; (8) Intra-axonal transcription factors are prominent features of the proposed pathomechanism.
Neuronal Cytoskeleton
The axon can be visualized as a railway, and the electric signal should be transferred from one train terminal station (the cell body) to another terminal station (the skeletal muscle). Mutations in genes associated with microtubules have been identified as the causative events in ALS.
Several variants of the gene encoding α-tubulin, TUBA4A, destabilize the microtubule network and reduce the repolymerization capability of this network (Smith et al., 2014). A missense mutation in the tubulin-specific chaperone E gene, causing MN degeneration in the progressive motor neuronopathy model mouse, ends in microtubule and axonal defects similar to those induced by the ALS-linked TUBA4A variation in patients (Bommel et al., 2002; Martin et al., 2002).
Mutations in profilin 1 (PFN1) can also lead to familial ALS. PFN1 converts monomeric actin to filamentous actin. Ubiquitinated aggregates are present in cells that express mutant PFN1, and many of these aggregates include TDP-43, which is associated with ALS (Wu et al., 2012). Such cells also have lower levels of bound actin and block axon growth. Primary MNs that harbor mutant PFN1 have a lower ratio of monomeric to filamentous actin and smaller growth cones. The PFN1 transgenic mouse has also been observed to recapitulate the phenotype of MN disease (Fil et al., 2017).
NIMA (never in mitosis gene A)-related kinase 1 (NEK1) has been linked to cilia formation, microtubule stability, and neuronal morphology (Thiel et al., 2011). The NEK1 gene was identified as a susceptibility factor for ALS (Brenner et al., 2016; Kenna et al., 2016). Using in vivo imaging, axonal degeneration was identified as an early event in the SOD1 and C9ORF72 repeat expansion mouse models of ALS (Tian et al., 2016). Neurofilament L transcripts are reduced in ALS (Bergeron et al., 1994). Neurofilaments are also found in a spheroid structure (large axonal swelling) (Corbo and Hays, 1992). Neurofilament light (NFL) and phosphorylated neurofilament heavy (pNFH) are also known as biomarkers for ALS (Brettschneider et al., 2006; Steinacker et al., 2016).
Thus, a dysfunctional cytoskeleton plays a role in ALS pathogenesis.
Cargo Transport Within Axons
Maintenance of the function and structure of all types of cells in mammals requires the intracellular transport of cargo. This transport is especially important in neurons because of their axonal and cell body polarization (De Vos and Hafezparast, 2017). Proteins and mRNA, as well as organelles, are generally synthesized in the soma and transported along the axon. Proper transport is required for the distribution of this cargo at the right time and place in the axon. Using electron microscopy of autopsy samples from ALS cases, defects in the cargo transport within axon transport in ALS have been observed. The studies of this transport defect revealed that the proximal axons of large MNs harbor abnormal accumulation of mitochondria, phosphorylated neurofilaments, and lysosomes (Hirano et al., 1984a, b; Okada et al., 1995; Rouleau et al., 1996). In addition, spheroids present in the axons contain different types of vesicles, lysosomes, mitochondria, neurofilaments, and microtubules (De Vos and Hafezparast, 2017). The accumulation of phosphorylated neurofilaments at the initial segment of MN axons is a major pathological characteristic of ALS (Ackerley et al., 2004; Brady and Morfini, 2017).
Aberrant cargo transport within axons occurs early in ALS disease progression (Williamson and Cleveland, 1999; De Vos et al., 2007). For example, an altered transport of mitochondria in axons has been demonstrated in two different mutant SOD1-G93A transgenic mouse models of ALS (Magrane et al., 2014). The slow anterograde transport of cytoskeletal components is decreased during the months prior to the initial neurodegeneration in mutant SOD1-G37R transgenic mice, a change that has been exhibited using metabolic labeling studies (Williamson and Cleveland, 1999). In mutant SOD1-G93A transgenic mouse models, inhibiting p38 MAPK α rescues retrograde cargo transport defects within axons (Gibbs et al., 2018).
TDP-43 functions as an mRNA transporter across the axonal cytoskeleton, and ALS-related mutations in TDP-43 alter this transport function (Alami et al., 2014). Mitotracker and Lysotracker experiments in FUS-mutant iPSC-derived MNs have demonstrated that defects in the cargo transport within axons can be rescued by histone deacetylase 6 inhibition (Guo et al., 2017).
Mutations in the genes that code for the motor protein dynactin (DCTN1) (Puls et al., 2003) have been identified in the genetic analyses of familial ALS. Mutant dynactin binds weakly to microtubules, compared with the binding of wild-type proteins. ALS and slowly progressing, autosomal dominant, distal hereditary motor neuropathy in vocal paresis (HMN7B) are due to loss-of-function mutations in DCTN1 (Puls et al., 2003; Munch et al., 2004; Yan et al., 2015). DCTN1 expression is also found to be downregulated in ALS-derived autopsy samples (Jiang et al., 2005).
Kinesin family member 5A (KIF5A) is a newly identified gene that plays a role in ALS (Brenner et al., 2018; Nicolas et al., 2018). Mutations that occur in the N-terminal motor domain of KIF5A cause an autosomal dominant type of hereditary spastic paraplegia known as spastic paraplegia (SPG)10, as well as Charcot–Marie–Tooth disease type 2 (Fichera et al., 2004). In contrast, mutations associated with ALS are mainly found in the C-terminal domain, which is important for binding cargo. Patients with loss-of-function KIF5A mutations have longer survival times than those with typical ALS (Brenner et al., 2018; Nicolas et al., 2018). Mutations in KIF5A, as well as KIF1A, which are loss-of-function mutations, are present in the motor or neck domains (Ebbing et al., 2008; Citterio et al., 2015).
In addition to MNs, mature sensory axons also possess a complex series of mRNA. A microtubule-stabilizing agent, paclitaxel, which impairs cargo transport within axons, results in sensory neuropathy (Gumy et al., 2011). Defects in the cargo transport within axons are common to various neurodegenerative diseases. Impaired cargo transport in axons can cause neurodegeneration (Millecamps and Julien, 2013).
Axonal Energy Supply
The mitochondria play an important role in meeting the axonal energy demand as they generate ATP through oxidative phosphorylation (Chamberlain and Sheng, 2019). Following their synthesis in the cell body, the mitochondria enter the axon where they undergo robust trafficking and accumulate at the nodes of Ranvier to meet metabolic needs (Zhang et al., 2012). Disruption of the mitochondrial activity, transport proteins, and microtubule association likely leads to dysfunctional mitochondrial transport in neurodegenerative diseases. Energy deficits in injured axons are caused by damage to the mitochondria following damage to axons, a decrease in mitochondrial transport in axons of mature neurons, and an increased energy consumption (Zhou et al., 2016). During regeneration, the axons adapt to this increased energy demand by changing the dynamics of the mitochondria (Kiryu-Seo and Kiyama, 2019). Mutations in RAPGEF2 mutations impair microtubule stability and the mitochondria distribution in axons (Heo et al., 2018). Reduction in mitochondrial Rho GTPase 1 (Miro 1), the outer mitochondrial membrane protein, leads to anterograde axonal transport defects (Moller et al., 2017). The imbalance between mitochondrial fission and fusion leads to abnormal mitochondrial morphology, underlies axonal damage, and is a potential therapeutic target for treating SPG15 and SPG48 (Denton et al., 2018). The ER and mitochondria form complex sites of interactions known as mitochondria-associated membranes (Gentile et al., 2019). Decreased ER-mitochondria association can occur as a result of loss-of-function mutations in SIGMAR1, leading to impaired retrograde transport and, ultimately, to axonal degeneration and MN death (Bernard-Marissal et al., 2015; Watanabe et al., 2016).
Astrocytes and oligodendrocytes may meet the axonal energy demand (Kang et al., 2010; Lee et al., 2012; Morrison et al., 2013). A deficiency in monocarboxylate transporter 1 (MCT1) was observed in oligodendroglia in the ventral cord of SOD1 transgenic mice and in the motor cortex of ALS patients (Kang et al., 2013; Philips et al., 2013). The removal of the SOD1 mutation from oligodendroglial precursor cells was observed to result in marked attenuation of the progression of the disease (Kang et al., 2013). Reducing the expression of MCT1 in oligodendroglia is the pathomechanism involving the energy supply that contributes to MN degeneration in ALS.
Clearance of Junk Protein
The ubiquitin proteasome and autophagy clearance systems are significant homeostatic processes engaged in eliminating defective organelles and aggregated proteins throughout the life span of the neuron. Impairment of the ubiquitin proteasome degradation system in MNs has been reported to replicate the ALS phenotype in mice (Tashiro et al., 2012). Mice with MN-specific, conditional knockout of the proteasome subunit Rpt3 exhibit locomotor dysfunction, progressive MN loss, and gliosis (Tashiro et al., 2012). Constitutive autophagy in neurons also maintains cellular homeostasis by balancing the synthesis and degradation of proteins, particularly within the distal axonal processes (Maday and Holzbaur, 2016). Several genes, such as valosin-containing protein, are involved in the protein degradation process (Johnson et al., 2010).
What about in the axon fraction? Disruption of the endosomal-lysosomal system due to ALS2/Alsin loss exacerbates the phenotype of SOD1-H46R transgenic mice by accelerating the accumulation of misfolded proteins and immature vesicles in the spinal cord (Hadano et al., 2010). In the early symptomatic and even presymptomatic SOD1-H46R transgenic mice, degenerating and swollen spinal axons with the accumulation of autophagosome-like vesicles have been observed (Hadano et al., 2010). A recent study also reported impairment of the degradation of autophagic vacuoles that engulf damaged mitochondria from distal axons in the SOD1-G93A transgenic mouse model (Xie et al., 2015). The clearance of dysfunctional mitochondria from axons may be mediated by syntaphilin, a mitochondria-anchoring protein, which is expressed at high levels in the early disease stages of ALS in MNs (Lin et al., 2017). FUS mutation causes axonal retention of the FUS protein prior to its aggregation, which is caused by poly(ADP-ribose) polymerase-dependent DNA response signaling (Naumann et al., 2018). The authors of this review also observed the accumulation of mutant FUS protein in the neurites of FUS-mutant induced pluripotent stem cell (iPSC)-derived MNs (Akiyama et al., 2019).
Optineurin (OPTN) mutations are implicated in both familial and sporadic ALS (Maruyama et al., 2010). OPTN binds to ubiquitin and regulates NFκB activation and apoptosis (Nakazawa et al., 2016). OPTN is also involved in several selective autophagy processes regulated by TBK1 (Li et al., 2016). Receptor-interacting kinase (RIPK) 1-dependent signaling is suppressed by OPTN through the regulation of its turnover (Ito et al., 2016). OPTN loss leads to progressive demyelination and axonal degeneration through the activation of necroptotic machinery in the central nervous system (CNS) (Ito et al., 2016). These observations suggest that RIPK1 and RIPK3 are significant in the process of progressive axonal degeneration.
A novel variant in UBQLN4 compromises motor axon morphogenesis in zebrafish, impairing the proteasomal function (Edens et al., 2017; Morrice et al., 2018). Based on these reports, the clearance of junk protein is important in the compartment of the MN axon.
NMJs
Amyotrophic lateral sclerosis can be redefined as a distal axonopathy disease, because many molecular changes influencing MN degeneration occur at the NMJ (Moloney et al., 2014). The NMJ is a highly specialized synapse, that controls signals between muscles and nerves for skeletal muscle function. Neuromuscular remodeling precedes loss of the motor unit in the mutant SOD1-G37R transgenic mouse model (Martineau et al., 2018).
Certain molecules, including galectin-1 (Ferraiuolo et al., 2007; Plachta et al., 2007), CD44 (Schmidt et al., 2011), and amyloid precursor protein (Bryson et al., 2012), affect the function of NMJ. Axon guidance molecules affecting the stability of the cytoskeleton, such as Semaphorin 3A (Venkova et al., 2014), Ephrin A4 (Takata et al., 2013), and Nogo-A (Pradat et al., 2007), have been reported to alter the function of the NMJ in the early stage of ALS. The loss of mitofusin 2 in neurons causes NMJ dysfunction, whereas the upregulation of mitofusin 2 ameliorates the phenotype of mutant SOD1-G93A transgenic mice (Wang et al., 2018).
The expression of mutant FUS or FUS knockdown in zebrafish results in the impairment of motor activity and reduces quantal transmission at NMJs, indicating loss and gain of function of FUS (Armstrong and Drapeau, 2013). These changes in FUS culminate in presynaptic dysfunction at the NMJ (Armstrong and Drapeau, 2013). There is evidence that FUS plays multiple roles in the nucleus and axonal compartments involved in NMJ maintenance and axonal transport (Schoen et al., 2015; So et al., 2018). FUS mediates the regulation of acetylcholine receptor transcription at NMJ and is dysregulated in ALS (Picchiarelli et al., 2019).
C9ORF72 was identified on the presynaptic side where the protein interacts with Rab3 protein family members, suggesting that it has a role in the regulation of synaptic vesicle functions as a guanine nucleotide exchange factor (Frick et al., 2018).
In Drosophila, the protein Arc1 is a component of the capsid-like structures that bind DARC1 mRNA in neurons. These capsids are included in the extracellular vesicles that are transferred across the NMJ from MNs to the muscle cells (Ashley et al., 2018). The transport of mRNA across the NMJ via these retrovirus-like capsids and extracellular vesicles is required for synaptic plasticity (Ashley et al., 2018). Dipeptide repeat proteins related to C9ORF72 spread between cells in vitro and in vivo (Westergard et al., 2016). Tau is another protein that is transported from donor cells to recipient cells through the cell culture medium (Wu et al., 2016b). Evidence suggests that the mechanism of pathogenic molecular transfer, termed the prion hypothesis, may be activated in the extracellular space and across the NMJ synapses during degeneration of the motor cortex with centrifugal spreading (Furukawa et al., 2011; Nonaka et al., 2013; Porta et al., 2018).
Aberrant Axonal Branching
Axonal branching is a fundamental mechanism of nervous system neuroplasticity (Menon and Gupton, 2018). Accumulating evidence suggests that aberrant axonal branching is involved in the pathomechanisms of ALS.
Overexpression of mutant human TARDBP in zebrafish embryos induces a phenotype that includes shorter MN axons, premature and increased branching, and abnormal swimming (Kabashi et al., 2010). On the other hand, overexpression of progranulin rescues mutant TARDBP-induced aberrant axonal branching and short axonal outgrowth (Laird et al., 2010).
Injection of morpholino antisense oligonucleotides to inhibit the translation of target mRNA and to knock down SMN in zebrafish embryos significantly increases MN branching (McWhorter et al., 2003). C9ORF72 modulates the activity of the small GTPases, resulting in increased activity of LIM kinases 1 and 2 and regulation of axonal actin dynamics (Sivadasan et al., 2016). Various actin isoforms are expressed in primary mouse MNs, and their transcripts have been observed to be translocated into axons (Moradi et al., 2017). It is proposed that short hairpin RNA-mediated depletion of α-actin reduces axonal filopodia dynamics and disrupts collateral branch formation in developing MNs (Moradi et al., 2017).
Temporary overexpression of human cyclin-F (CCNF) in zebrafish embryos increases the levels of cleaved caspase-3 and cell death in the spinal cord. The mutant CCNF zebrafish also developed an MN axonopathy, which consists of shortened primary MN axons and an increased frequency of aberrant axonal branching (Hogan et al., 2017).
A recent study reported that MNs cultured from mutant SOD1-G93A transgenic mouse models exhibit enhanced axonal outgrowth and dendritic branching (Osking et al., 2019). As the level of branching does not correlate with the severity of the disease, in this study, the authors concluded that axonal branching does not affect the disease process. Increased synaptic activity or branching is considered desirable in the field of psychiatric disease (Shao et al., 2019). The authors of the present review identified aberrant axonal branching in FUS-mutant iPSC-derived MNs (Akiyama et al., 2019). The sensory axons branching in the presence of nerve growth factor (NGF) can be observed at sites marked by stalled mitochondria. NGF promotes branching through the generation of ATP and active axonal translation of mRNA (Spillane et al., 2013). The mechanism underlying mitochondrial stalling and growth factor distribution in MNs requires examination.
The meaning of axonal branching might be different in each stage of the development (Jung et al., 2012). In the embryonic stage, axon pathfinding and synaptic formation are important. However, in the developed stage, aberrant axon branching might have a disadvantage in terms of normal function of signal transmission. The significance of aberrant axonal branching in the neurodegenerative model in vivo has not yet been elucidated.
Omics Profiling of the Axonal Compartment
The previous section provided an overview of the important pathomechanisms of MN axons. These pathomechanisms have been found to influence each other and cannot be entirely separated. This section reviews the omics analysis of the axon compartment in order to obtain an overall understanding of this complex process occurring in an important region of the neuron.
Lessons From Different Nervous Systems
The rationale for conducting omics analysis of the axon compartment is as follows (Table 2). Surprisingly complex, constantly changing transcriptomes are present in mature axons. Thus, axonal mRNA localization is likely to be tightly regulated and to play multiple roles. The ribosomal protein S6 has been observed with immunoelectron microscopy in the axons of embryonic sympathetic and hippocampal neurons grown in vitro (Tcherkezian et al., 2010), indicating that local mRNA translation also occurs in growing axons. Further, the local translation of proteins from mRNAs selectively transported from the soma to the synaptic terminal appears to be involved in the regulation of axon outgrowth and regeneration (Zheng et al., 2001; Taylor et al., 2009).
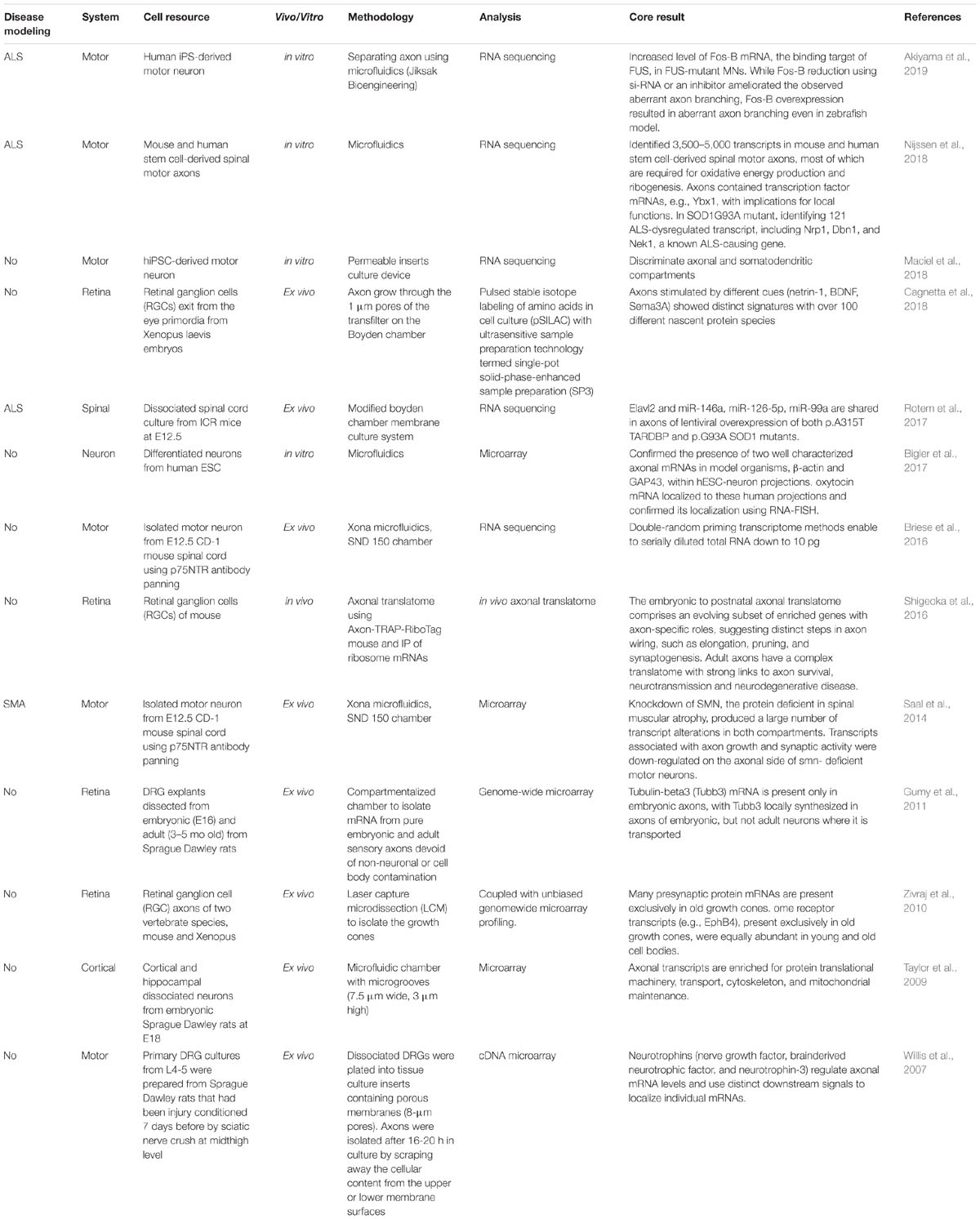
Table 2. Omics analyses of the axon compartment in in vivo, ex vivo, and in vitro models of several types of nervous systems.
Elucidation of what features of axonal function require local translation and determination of the mRNAs that mediate these functions have induced intriguing challenges in the field of axonal biology (Deglincerti and Jaffrey, 2012; Jung et al., 2012). Assessment of the axonal transcriptome using microarray studies has identified important axonal mRNAs and has demonstrated the complexity and dynamic nature of the axonal transcriptome (Zivraj et al., 2010; Gumy et al., 2011).
In a pioneering study involving omics analysis in axons, more than 200 different mRNAs were identified with cDNA microarray analysis in axons derived from rat with injured sensory neurons (Willis et al., 2007). Proteins involved in the transcription, synthesis of proteins, intracellular transport, calcium metabolism, mitochondrial functions, and cytoskeletal functions were identified in the study (Willis et al., 2007). The report raised several important questions regarding axonal translation (Deglincerti and Jaffrey, 2012), including the question of why transcripts for nuclear proteins are localized to distal axons.
Using a microfluidic chamber enabling the isolation of axons without contamination with non-axonal material, mRNA has been purified from mature CNS axons (Taylor et al., 2009). The same study also described the localization of catenin-β1 and neurexin-3 mRNA with fluorescence in situ hybridization in the axonal compartment (Taylor et al., 2009). The somatodendritic compartments are enriched in transcripts with postsynaptic functions and in nuclear non-coding RNAs such as 7SK, whereas transcripts related to translation such as7SL, the cytoplasmic non-coding RNA, are upregulated in the compartment of the axon fraction (Briese et al., 2016).
Transcriptome-wide analyses have revealed numerous transcripts encoding transmembrane or secreted proteins, which comprise about 13% of the total mRNAs found in growth cones (Zivraj et al., 2010). Transcripts present in axons encode many transmembrane proteins, such as integrins and protocadherins, which are cell adhesion molecules, and EphB4 and Nrp2, which are guidance receptors (Zivraj et al., 2010). Thus, local translation may change the cell adhesion capacity of axons and allow axons to respond to extracellular signaling molecules (Gumy et al., 2011). Axons also contain transcripts that code for secreted proteins, including semaphorin and ephrin, which are guidance molecules; BMP1, CTGF, and FGF, which are growth factors; and collagen and TIMP3, which compose and regulate the extracellular matrix. Thus, it is expected that local translation probably plays a role in the regulation of extracellular components by affecting proteins that are secreted from growth cones (Deglincerti and Jaffrey, 2012). Axons also contain structures that resemble the ER and Golgi. Specific labeling of the ER and Golgi exhibits irregular, punctate staining along the axon, suggesting that axon-specific versions of these organelles may be present in nerve terminals (Merianda et al., 2009).
The development of compartmentalization has enabled the examination of axon pathology in MN diseases. The knockdown of SMN, which encodes the protein that is deficient in spinal muscular atrophy (SMA), was shown to produce numerous transcript alterations in both axon and somatic compartments of the microarray (Saal et al., 2014). Transcripts associated with axon growth and synaptic activity are downregulated on the axonal side of SMN-deficient MNs. Improvements in the handling of small quantities of RNA have led to further progress in this field (Briese et al., 2016).
Evaluation of cultured spinal cord neurons grown with a compartmented platform and subjected to next-generation sequencing technology revealed that mRNAs and miRNAs are differentially expressed in the somatic compared with the axonal neuronal compartments (Rotem et al., 2017). In axons with lentiviral overexpression of p.A315T TARDBP or p.G93A SOD1 mutants, Elavl2, miR-146a, miR-126-5p, and miR-99a are commonly expressed. Examination of the local transcriptome revealed that the most abundant mRNAs within human embryonic stem cell-derived neuronal projections are functionally similar to the rat axonal transcriptome of cortical neurons (Bigler et al., 2017).
The use of microfluidic technology has been particularly useful in neuroscience research. Microfluidic platforms have allowed researchers to address specific questions related to axonal guidance, synapse formation, and cargo transport within axons, and led to the development of three-dimensional (3D) CNS models for pharmacological testing and drug screening (Neto et al., 2016). Human iPSC-derived MNs grown in a culture device with permeable inserts were observed to produce large amounts of enriched axonal material that can be harvested for RNA isolation and sequencing (Maciel et al., 2018). Transcriptome profiling has revealed axonal and somatodendritic compartment-specific expression.
Recently, Nijissen and colleagues developed a refined method named Axon-seq, combining microfluidics, RNA sequencing, and bioinformatics analysis (Nijssen et al., 2018). These results demonstrated that the transcriptome of the axon compartment is quite different from that of the soma and includes a smaller number of mRNAs. They identified up to 5,000 mRNAs in mouse and human stem cell-derived MN axons; the functions of the majority of these are oxidative energy and ribosome production. Axons contain transcription factor mRNAs, implicating local functions. Investigation into the response of degenerated ALS motor axons to the SOD1-G93A mutation identified 121 ALS-dysregulated transcripts. Among these, Nrp1 and Dbn1 are involved in axonal function, and Nek1 is a known ALS-causative gene (Brenner et al., 2016; Kenna et al., 2016; Nijssen et al., 2018). Axon-seq is an advanced technique for sequencing the RNA in axons, and thus can provide enhanced knowledge about peripheral nerve biology to explain the vulnerability/resilience of MN (Nijssen et al., 2017; Allodi et al., 2019) and to identify the treatment of MN diseases.
Development of a Microfluidic Device for Larger-Scale Omics Analysis
Despite the improvement offered by the microfluidic device, harvesting a sufficient volume of lysate from the axon compartment remains challenging. In the process of improving the dimensions of the well and materials, a novel microfluidic device was developed by the authors of this study (Table 3). The device enabled comparison of two sets of isogenic FUS-mutant iPSC-derived MNs generated using genome editing technology (Joung and Sander, 2013; Okano and Yamanaka, 2014), and provided observations of increased branching in FUS-mutant MN axons compared with those in isogenic controls (Akiyama et al., 2019). This phenotype was confirmed using other ALS-causative mutations, including SOD1 and TARDBP. Combining this innovative microfluidic device (Kawada et al., 2017) with hiPSC-derived MN organoids further revealed the entire in vitro profile of the human MN axons. This technique identified increased Fos-B mRNA as a binding partner of FUS and as a causative event for aberrant axon morphology both in vitro and in vivo.
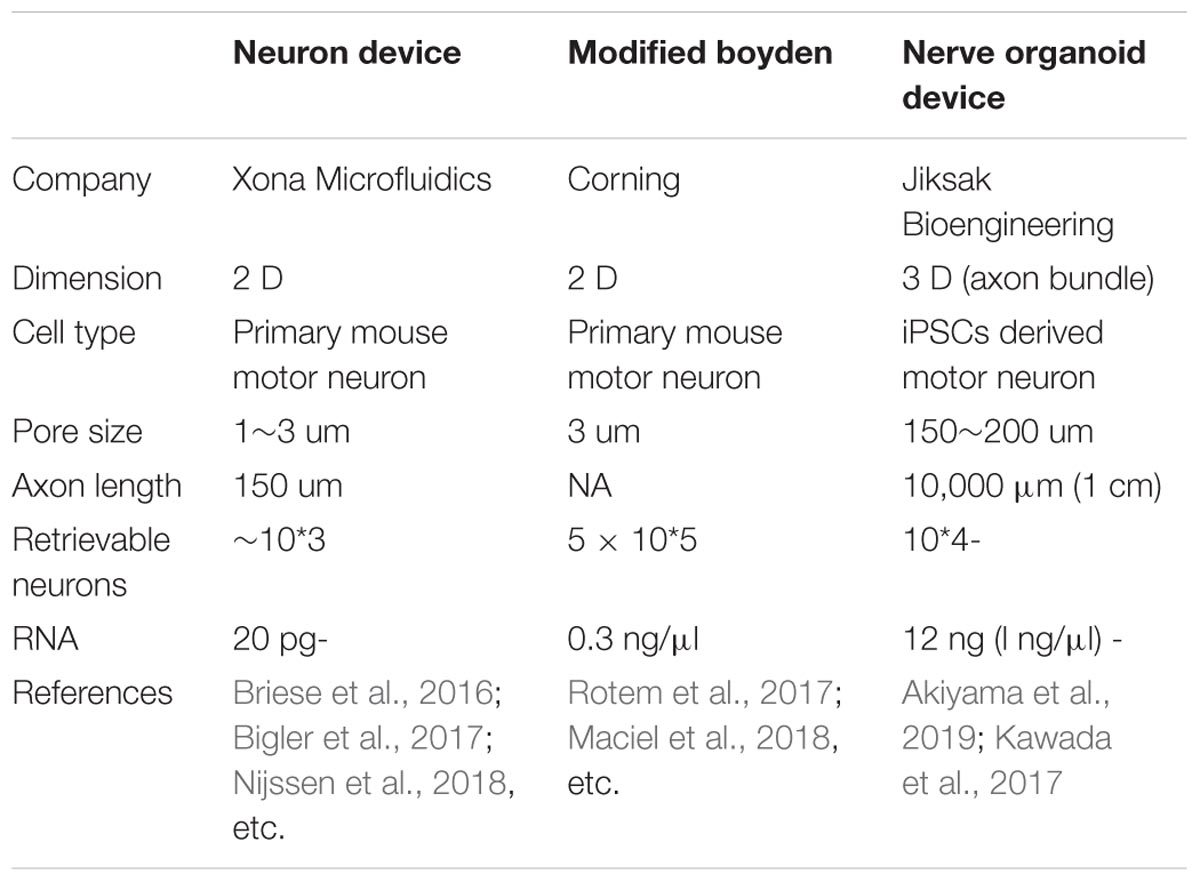
Table 3. Comparison of our microfluidic devices with those of previous studies.
Morphological changes in MN axon branching have been found to precede MN death in the mutant SOD1-G93A transgenic mouse model (Tian et al., 2016), and abnormal neural branching has been detected in zebrafish that overexpress mutant FUS (Armstrong and Drapeau, 2013). Improvements in axon morphology following suppression of abnormally upregulated Fos-B in FUS mutants suggested a novel therapeutic candidate for FUS-mutant ALS.
Previous studies have also reported that upregulation of Fos-B mRNA is associated with increases in spines (Lafragette et al., 2017; Cahill et al., 2018) and growth cones (Anastasiadou and Knoll, 2016). δFos-B modulates immature spines of the nucleus accumbens in a model of drug addiction (Grueter et al., 2013). Certain chemical stimulators such as kainic acid lead to neurodegeneration via upregulated expression of immediate early genes, including that of Fos-B (Pereno et al., 2011). The hyperexcitability hypothesis is a major theme in proposing the pathomechanism of ALS (Wainger et al., 2014). A recent report of activator protein-1 (AP-1) and MN degeneration in the mutant SOD1-G93A transgenic mouse model has attracted attention (Bhinge et al., 2017). Additionally, the suppression of dual leucine zipper kinase, the upstream signal protein for c-Jun (AP-1 family member), may become a therapeutic target for ALS (Bhinge et al., 2017). Although substantial differences have been reported in SOD1-ALS compared with FUS-ALS and TARDBP-ALS (Fujimori, 2018), SOD1-, TARDBP-, and FUS-mutant MNs have common features, suggesting a role for AP-1 in the neurodegeneration observed in ALS. The Fos-B protein accumulates abnormally in the MNs of ALS patients, including in sporadic cases. Thus, Fos-B appears to be a potential therapeutic target molecule.
The novel microfluidic device described in the preceding paragraph comprises a large canal that enables the collection of sufficient samples of isolated MN axons for RNA sequencing (Kawada et al., 2017). This device has proven useful in visualizing the global profile of the axon compartment. Although other types of microfluidic devices, some of which are specific to cell fraction analysis, are available on the market (Briese et al., 2016; Rotem et al., 2017), they are typically restricted by the limited amount of specimen obtained (Table 3). As only a very small amount of specimen can be analyzed, variation in conditions, such as cell purity and culture procedures, may influence the results. Kawada’s microfluidic device enables analysis with fewer technical biases because it involves the collection of large amounts of macroscopically observable axon bundles. RNA profiles from the axon samples have reproduced the previously reported profiles of the MN axon (Briese et al., 2016; Rotem et al., 2017), justifying the methodology of this novel device. Furthermore, the data obtained may provide important resources for the subcellular fractional analysis of stem cell-derived MN axons.
Are These mRNAs Translated in Axons?
An important question is whether these mRNAs are translated in axons or transported to the nucleus/cell body. The importance of axonal translation for CNS maintenance is under debate (Spaulding and Burgess, 2017). Several types of mature polarized cells utilize asymmetrical mRNA localization as a means of synaptic communication with other types of cells (Xing and Bassell, 2013). In vivo, the longest axons, such as those of mature sensory and motor peripheral neurons, rely most strongly on mRNA transport and local translation to maintain homeostasis.
Upregulation of ribosome synthesis in axons has been found to occur early in the pathogenesis of both mutant SOD1-G93A transgenic mouse models and human ALS autopsy samples, which suggests the involvement of Schwann cells in ALS pathology and in aberrant axonal RNA metabolism (Verheijen et al., 2014). Gene expression analyses of the anterior branch of human obturator MNs biopsied from patients with ALS demonstrated upregulation of a cluster of genes that play important roles in biological processes involving RNA processing and protein metabolism (Riva et al., 2016).
Direct evidence for neurodegeneration has been obtained from the observation of mRNA transport dysregulation due to mutations in the RBP SMN1, which causes SMA (Wang et al., 2016b). SMN is present ubiquitously, and its deletion is lethal. However, MNs are more sensitive to SMN reduction than other cell types, possibly because reduced SMN decreases the axonal localization of several mRNAs (Rage et al., 2013) and inhibits the activity of the mammalian target of rapamycin in axons (Kye et al., 2014). An additional role for SMN is in the regulation of axonal localization and local translation of growth-associated protein 43 (GAP43) mRNA in growth cones (Fallini et al., 2016). The overexpression of two mRNA-binding proteins, HuD and IGF2 mRNA-binding protein 1, restores the mRNA and protein levels of GAP43 and has been shown to rescue the axon outgrowth defects in the neurons of an SMA patient (Fallini et al., 2016).
In previous studies, protein interaction screening intended to elucidate FUS-mutant phenotypes also identified several molecules that interact with FUS, including SMN (Yamazaki et al., 2012; Groen et al., 2013). Aberrant distribution of SMN in cytosolic FUS accumulations induces SMN reduction in axons. Accumulation of mutant human FUS induces an integrated stress response and reduces protein synthesis in nearby axons (Lopez-Erauskin et al., 2018).
Non-nuclear pools of splicing factor, proline-glutamine rich (SFPQ) are essential for normal motor development via local mRNA maintenance or processing, and the coiled-coil domain of SFPQ is required for axonal localization (Thomas-Jinu et al., 2017). The RBPs modulate nuclear processing, intracellular transport, and local translation of target mRNAs for an accurate spatial and temporal gene expression. SFPQ functions as an RBP because it binds to and modulates numerous neuronal mRNAs, including in cells, such as dorsal root ganglion neurons. SFPQ, which has been identified by subcellular compartmentalization analysis (Cosker et al., 2016; Takeuchi et al., 2018), has been found to orchestrate spatial gene expression, which is essential for axonal viability.
Local translation is also involved in several neurodevelopmental disorders (Batista and Hengst, 2016). Local translation defects are associated with fragile X mental retardation and autism spectrum disorders (Kelleher and Bear, 2008). Fragile X mental retardation protein, which is present in dendritic spines, growth cones, and axons, modulates plasticity (Bassell and Warren, 2008) and the presynaptic proteome (Christie et al., 2009; Akins et al., 2012). In mouse brain slices, loss of the fragile X mental retardation protein was found to perturb the development of presynaptic nerve terminals (Hanson and Madison, 2007).
Degeneration of motor axons results from mutations in various tRNA synthetases, which is consistent with the notion that local translation of transported mRNA is necessary for axonal homeostasis (He et al., 2015; Storkebaum, 2016). Recently, cytoplasmic polyadenylation element-binding protein 4 was found to orchestrate the dysregulation of mRNA expression in autism (Parras et al., 2018). Identification of a master regulator of RNA metabolism would be beneficial in understanding and treating for both diseases that affect MNs and psychiatric diseases.
Nascent chain tracking is a novel technique for visualizing local translation. This method uses multi-epitope tags and antibody-based fluorescent probes to quantify the dynamics of protein synthesis at the level of individual mRNAs (Morisaki et al., 2016). Due to its sensitivity and versatility, nascent chain tracking is a useful tool for quantifying mRNA translation kinetics. Synaptic activity can induce mRNA localization and the local translation of β-actin, which stabilizes expanding synapses at dendritic spines (Wu et al., 2016a; Yoon et al., 2016). Real-time visualization of mRNA translation in the axonal compartment is an innovative method enabling analysis of axonal pathology in vivo (Wang et al., 2016a; Yan et al., 2016). The inducible fluorescent probe can be regulated in time and space in neurons and is used to examine the maturation of miRNA. The local maturation of miRNA by synaptic stimulation results in a spatially restricted protein synthesis reduction from the mRNA (Sambandan et al., 2017). The proteomics approach described in a later section adds to the understanding of the global change of nascent proteins produced in the axon fraction.
Role of Intra-Axonal Transcription Factors
Why transcripts for nuclear proteins are localized to the distal axons is a big question raised by the omics analysis. Fos-B, a mediator of abnormal axonal branching in FUS-mutated MNs, is a transcription factor. Another research group also reported dysregulated transcription factors in ALS MNs (Nijssen et al., 2018). In determining the role of transcription factors in the axon compartment, a comprehensive transcription of the action fraction has identified mRNAs encoding a larger amount of transcription factors and co-factors (Ji and Jaffrey, 2014). One example is that of axonal STAT3, which is translated locally, activated upon nerve injury, and is transported retrogradely with dynein and importin α5, modulating the survival of peripheral sensory neurons (Ben-Yaakov et al., 2012). Recently, Tp53inp2 was reported to be an atypical mRNA regulating axon growth by enhancing the NGF-TrkA pathway independently with translation (Crerar et al., 2019). Importantly, data have indicated that axonal degeneration shared early molecular change in the neurodegenerative process of neurological disorders in aged populations (Dadon-Nachum et al., 2011; Tagliaferro and Burke, 2016; Salvadores et al., 2017).
In the brain of a person with Alzheimer’s disease, inhibition of local translation of Atf4 mRNA overproduction eliminates amyloid β-induced cell loss (Baleriola et al., 2014; Peng et al., 2016). Atf4 mRNA translation is controlled by phosphorylation of elongation initiation factor 2a, pivotal for an integrated stress response (Batista and Hengst, 2016). The role of axonal transcription factors in relation to translated proteins and non-translated RNA requires further elucidation.
Interaction Among the Mechanisms Already Described
The hallmark feature in the majority of autopsy cases of ALS is nuclear depletion and cytoplasmic accumulation of TDP-43 in degenerated neurons (Kim and Taylor, 2017). Thus, dysfunctional trafficking between the nucleus and cytoplasm likely plays a role in the pathology of ALS (Nedelsky and Taylor, 2019) and may also be important in normal physiological aging, Huntington’s disease, and Alzheimer’s disease (Nedelsky and Taylor, 2019). RBPs with prion-like domains (PrLDs) undergo liquid-liquid phase separation to form functional liquids, which can be converted into abnormal hydrogels that contain pathological fibrils that are often seen in neurodegenerative diseases. TDP-43, FUS, heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), and hnRNPA2 are nuclear RBPs with PrLDs that are incorrectly sent to cytoplasmic inclusions in neurodegenerative diseases. Mutations in PrLDs increase the rate of fibril formation and initiate disease (Guo et al., 2018). Karyopherin-β2, also known as transportin-1, binds the proline-tyrosine NLS and then blocks and reverses FUS, TATA-box-binding protein associated factor (TAF) 15, Ewing sarcoma RBP1 (EWSR1), hnRNPA1, and hnRNPA2 fibril formation. Importin-α and karyopherin-β1 also block and reverse TDP-43 fibril formation. Phase separation, like stress granule formation, is an emerging property of proteins containing PrLD such as FUS (Guo et al., 2018; Hofweber et al., 2018; Qamar et al., 2018; Yoshizawa et al., 2018). T-cell-restricted intracellular antigen-1 (TIA1) mutations were found to delay stress granule disassembly and to promote the accumulation of granules harboring TDP-43 (Mackenzie et al., 2017). C. elegans TIAR-2/TIA protein functions cell autonomously to inhibit axon regeneration (Andrusiak et al., 2019). One of the important roles of phase separation is transcription enhancement (Sabari et al., 2018), which might be related to local translation/transcription. The association between axonal dysfunction and these cytoplasmic events, including phase separation, has not yet been elucidated.
There is emerging evidence of interactions among different processes of axonal pathology in ALS. Annexin A11 (ANXA11), a phosphoinositide-binding protein associated with the RNA granule, has the role of a molecular tether between lysosomes and RNA granules. Such tethering is impaired by the ALS-associated ANXA11 mutation (Smith et al., 2017; Liao et al., 2019). Late endosome bearing mRNA encoding mitochondrial functional molecules stops at mitochondria and these mRNAs are translated on Rab7a endosomes locally in the axon (Cioni et al., 2019).
In summary, exactly how these complex mechanisms are influenced by each other is still unknown. There is a need for understanding how cytoskeletons are maintained, and how molecules are transported/metabolized/synthesized, or abolished when unnecessary. Elucidating the interaction of these mechanisms might answer the vital question of why MNs are vulnerable in ALS.
Advanced Omics Analysis and Further Consideration
Conducting a comprehensive analysis of the newly produced proteome from limited samples of subcellular compartments that are uncontaminated by the somatodendrite remains a major technical problem (Eichelbaum and Krijgsveld, 2014). Stable isotope labeling of amino acids in cell culture (SILAC) has been combined with single-well solid phase-enhanced sample prep. Using this method, the newly produced proteome of isolated retinal axons was obtained rapidly (in approximately 5 min) (Cagnetta et al., 2018). Treating axons treated with stimuli such as netrin-1, brain-derived neurotropic factor, and Sema3A, has demonstrated distinct proteomes with more than 100 different nascent proteins. Compartment analysis using pulsed SILAC may be applied to ALS cells with a sophisticated culture device.
Using an axon-TRAP-RiboTag approach in mice, the dynamic translatome of axons in the retina in vivo matches the subcellular function (Shigeoka et al., 2016). The translatome of the embryonic and postnatal axons includes a changing, enriched set of genes with axon-specific roles. Thus, specific steps in axon wiring, such as axon growth, elimination of unnecessary axons, and synaptogenesis, may be present. Adult axons harbor a complicated translatome that plays a role in axon survival, neurotransmission, and neurodegenerative diseases. Mating of several ALS mouse models can help in precisely understanding mRNA dysregulation. Further transcriptome and proteome analyses using labeled growth cones of single projections in the mouse cerebral cortex in vivo may also be of use (Poulopoulos et al., 2019). Spatial transcriptomics is another method for elucidating gene expression in the mouse spinal cord over the disease course, and in postmortem tissue from patients with ALS (Maniatis et al., 2019). Another important approach is the single-cell transcriptomics of nerve organoids in vitro (Quadrato et al., 2017); pseudo-time analysis or single-cell trajectory analysis can help establish the relationship between the cause and effect of the transcriptome of the organoids (Xiang et al., 2017; Klaus et al., 2019). Sophisticated neuromuscular co-culture organoids would be beneficial for these studies.
Stimulated Raman scattering microscopy is a new technique for chemical imaging that can be used to map the distribution of various molecules–including lipids, proteins, and nucleic acids–in live cells and tissues, as determined by their intrinsic molecular vibration (Freudiger et al., 2008). The authors of this review used this type of imaging to visualize peripheral degeneration in several ALS mouse models and human postmortem tissue (Tian et al., 2016). Non-labeled live imaging of motor axons may assist in monitoring the time course of axonal pathology in vivo.
In clinical settings, the strength-duration time constant, which represents the hyperexcitability of an MN axon, is significantly increased in patients (Kanai et al., 2006, 2012; Geevasinga et al., 2015). Hyperexcitability is thought to be the target of MN death in ALS (Wainger et al., 2014). In cell culture settings, the shortened isoform of TDP-43 is upregulated by neuronal hyperactivation (Weskamp et al., 2019). The role of these short isoform of TDP-43, which might be the product of dysregulation of RNA metabolism, should be considered in the axon fraction. Recent studies have revealed the importance of stathmin-2 (STMN2), a regulator of microtubule stability, in the pathomechanism of TARDBP mutation (Klim et al., 2019; Melamed et al., 2019). The expression of a microtubule regulator, STMN2, is decreased following TARDBP knockdown, when TDP-43 is mis-localized, and in MNs from patients and the spinal cord of postmortem samples. The reduced function of TDP-43 results in the loss of STMN2 due to altered splicing. This is functionally important, as STMN2 is necessary for the outgrowth and regeneration of MN axons. Post-translational STMN2 stabilization rescues neurite outgrowth and axon regeneration deficits by TDP-43 depletion (Klim et al., 2019). A reduction in TDP-43 inhibits axonal regeneration of iPSC-derived MNs, whereas rescue of the expression of STMN2 restores the axonal regeneration capacity (Melamed et al., 2019). The effect of the short form of TDP-43 or cryptic exons under the control of TDP-43 (Ling et al., 2015) should be examined in the axon fraction.
Concluding Remarks
As described in the preceding section, advanced omics approaches, in vivo analysis, and axon–cytoplasmic interactions should be examined as the next steps in investigating axonal pathology in neurodegenerative disease research. The novel concept of microfluidic devices, including the nerve organoid device presented by the authors of this review, should be applied to other neuron types, co-culture systems, or proteomics analyses using human pluripotent cells, because this technique may help elucidate the resilience of long MN and the pathomechanism of ALS.
Author Contributions
NS, TA, and MA prepared the manuscript. All authors read, revised, and approved the final version of the manuscript.
Funding
This work was supported by funding from Grant-in-Aid for Young Scientists (A) (15H05667), Grant-in-Aid for Scientific Research (C) (18K07519), and Grant-in-Aid for Scientific Research (B) (16H05318) from Japanese Ministry of Education, Culture, Sports, Science and Technology; the Research Project for Practical Applications of Regenerative Medicine from the Japan Agency for Medical Research and Development (AMED) (Grant number 19bm0804003h0003); Japan Intractable Diseases (Nanbyo) Research Foundation, the Kanae Foundation for the Promotion of Medical Science, and “Inochi-no-Iro” ALS research grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MK declared a past co-authorship with the authors NS and MA to the handling Editor.
Acknowledgments
The authors thank H. Okano, T. Fujii, J. Kawada, S. Mitsuzawa, and K. Eggan for the excellent comments, Y. Akiyama for the illustrations. The authors also would like to thank Enago (www.enago.jp) for the English language review.
References
Ackerley, S., Grierson, A. J., Banner, S., Perkinton, M. S., Brownlees, J., Byers, H. L., et al. (2004). p38alpha stress-activated protein kinase phosphorylates neurofilaments and is associated with neurofilament pathology in amyotrophic lateral sclerosis. Mol. Cell. Neurosci. 26, 354–364. doi: 10.1016/j.mcn.2004.02.009
Akins, M. R., Leblanc, H. F., Stackpole, E. E., Chyung, E., and Fallon, J. R. (2012). Systematic mapping of fragile X granules in the mouse brain reveals a potential role for presynaptic FMRP in sensorimotor functions. J. Comp. Neurol. 520, 3687–3706. doi: 10.1002/cne.23123
Akiyama, T., Suzuki, N., Ishikawa, M., Fujimori, K., Sone, T., Kawada, J., et al. (2019). Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons. EBiomedicine 45, 362–378. doi: 10.1016/j.ebiom.2019.06.013
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S. W., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Allodi, I., Nijssen, J., Benitez, J. A., Schweingruber, C., Fuchs, A., Bonvicini, G., et al. (2019). Modeling motor neuron resilience in ALS using stem cells. Stem Cell Rep. 12, 1329–1341. doi: 10.1016/j.stemcr.2019.04.009
Anastasiadou, S., and Knoll, B. (2016). The multiple sclerosis drug fingolimod (FTY720) stimulates neuronal gene expression, axonal growth and regeneration. Exp. Neurol. 279, 243–260. doi: 10.1016/j.expneurol.2016.03.012
Andrusiak, M. G., Sharifnia, P., Lyu, X., Wang, Z., Dickey, A. M., Wu, Z., et al. (2019). Inhibition of axon regeneration by liquid-like TIAR-2 granules. Neuron 104, 290–304.e8. doi: 10.1016/j.neuron.2019.07.004
Aoki, M., Ogasawara, M., Matsubara, Y., Narisawa, K., Nakamura, S., Itoyama, Y., et al. (1993). Mild ALS in Japan associated with novel SOD mutation. Nat. Genet. 5, 323–324. doi: 10.1038/ng1293-323
Armstrong, G. A., and Drapeau, P. (2013). Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum. Mol. Genet. 22, 4282–4292. doi: 10.1093/hmg/ddt278
Ashley, J., Cordy, B., Lucia, D., Fradkin, L. G., Budnik, V., and Thomson, T. (2018). Retrovirus-like gag protein Arc1 binds RNA and traffics across synaptic boutons. Cell 172, 262–274.e11. doi: 10.1016/j.cell.2017.12.022
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi: 10.1038/s41582-018-0047-2
Baleriola, J., Walker, C. A., Jean, Y. Y., Crary, J. F., Troy, C. M., Nagy, P. L., et al. (2014). Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172. doi: 10.1016/j.cell.2014.07.001
Bassell, G. J., and Warren, S. T. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. doi: 10.1016/j.neuron.2008.10.004
Batista, A. F., and Hengst, U. (2016). Intra-axonal protein synthesis in development and beyond. Int. J. Dev. Neurosci. 55, 140–149. doi: 10.1016/j.ijdevneu.2016.03.004
Ben-Yaakov, K., Dagan, S. Y., Segal-Ruder, Y., Shalem, O., Vuppalanchi, D., Willis, D. E., et al. (2012). Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J. 31, 1350–1363. doi: 10.1038/emboj.2011.494
Bergeron, C., Beric-Maskarel, K., Muntasser, S., Weyer, L., Somerville, M. J., and Percy, M. E. (1994). Neurofilament light and polyadenylated mRNA levels are decreased in amyotrophic lateral sclerosis motor neurons. J. Neuropathol. Exp. Neurol. 53, 221–230. doi: 10.1097/00005072-199405000-00002
Bernard-Marissal, N., Medard, J. J., Azzedine, H., and Chrast, R. (2015). Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138, 875–890. doi: 10.1093/brain/awv008
Bhinge, A., Namboori, S. C., Zhang, X., Vandongen, A. M. J., and Stanton, L. W. (2017). Genetic correction of SOD1 mutant iPSCs reveals ERK and JNK activated AP1 as a driver of neurodegeneration in amyotrophic lateral sclerosis. Stem Cell Rep. 8, 856–869. doi: 10.1016/j.stemcr.2017.02.019
Bigler, R. L., Kamande, J. W., Dumitru, R., Niedringhaus, M., and Taylor, A. M. (2017). Messenger RNAs localized to distal projections of human stem cell derived neurons. Sci. Rep. 7:611. doi: 10.1038/s41598-017-00676-w
Boillee, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Bommel, H., Xie, G., Rossoll, W., Wiese, S., Jablonka, S., Boehm, T., et al. (2002). Missense mutation in the tubulin-specific chaperone E (Tbce) gene in the mouse mutant progressive motor neuronopathy, a model of human motoneuron disease. J. Cell Biol. 159, 563–569. doi: 10.1083/jcb.200208001
Borchelt, D. R., Lee, M. K., Slunt, H. S., Guarnieri, M., Xu, Z. S., Wong, P. C., et al. (1994). Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. U.S.A. 91, 8292–8296. doi: 10.1073/pnas.91.17.8292
Brady, S. T., and Morfini, G. A. (2017). Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases. Neurobiol. Dis. 105, 273–282. doi: 10.1016/j.nbd.2017.04.010
Brenner, D., Muller, K., Wieland, T., Weydt, P., Bohm, S., Lule, D., et al. (2016). NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139:e28.
Brenner, D., Yilmaz, R., Muller, K., Grehl, T., Petri, S., Meyer, T., et al. (2018). Hot-spot KIF5A mutations cause familial ALS. Brain 141, 688–697. doi: 10.1093/brain/awx370
Brettschneider, J., Petzold, A., Sussmuth, S. D., Ludolph, A. C., and Tumani, H. (2006). Axonal damage markers in cerebrospinal fluid are increased in ALS. Neurology 66, 852–856. doi: 10.1212/01.wnl.0000203120.85850.54
Briese, M., Saal, L., Appenzeller, S., Moradi, M., Baluapuri, A., and Sendtner, M. (2016). Whole transcriptome profiling reveals the RNA content of motor axons. Nucleic Acids Res. 44:e33. doi: 10.1093/nar/gkv1027
Brown, R. H. Jr., and Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. N. Engl. J. Med. 377:1602.
Bryson, J. B., Hobbs, C., Parsons, M. J., Bosch, K. D., Pandraud, A., Walsh, F. S., et al. (2012). Amyloid precursor protein (APP) contributes to pathology in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 21, 3871–3882. doi: 10.1093/hmg/dds215
Burberry, A., Suzuki, N., Wang, J. Y., Moccia, R., Mordes, D. A., Stewart, M. H., et al. (2016). Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 8:347ra93. doi: 10.1126/scitranslmed.aaf6038
Cagnetta, R., Frese, C. K., Shigeoka, T., Krijgsveld, J., and Holt, C. E. (2018). Rapid cue-specific remodeling of the nascent axonal proteome. Neuron 99, 29–46.e4. doi: 10.1016/j.neuron.2018.06.004
Cahill, M. E., Walker, D. M., Gancarz, A. M., Wang, Z. J., Lardner, C. K., Bagot, R. C., et al. (2018). The dendritic spine morphogenic effects of repeated cocaine use occur through the regulation of serum response factor signaling. Mol. Psychiatry 23, 1474–1486. doi: 10.1038/mp.2017.116
Chamberlain, K. A., and Sheng, Z. H. (2019). Mechanisms for the maintenance and regulation of axonal energy supply. J. Neurosci. Res. 97, 897–913. doi: 10.1002/jnr.24411
Chew, J., Gendron, T. F., Prudencio, M., Sasaguri, H., Zhang, Y. J., Castanedes-Casey, M., et al. (2015). Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348, 1151–1154. doi: 10.1126/science.aaa9344
Christie, S. B., Akins, M. R., Schwob, J. E., and Fallon, J. R. (2009). The FXG: a presynaptic fragile X granule expressed in a subset of developing brain circuits. J. Neurosci. 29, 1514–1524. doi: 10.1523/JNEUROSCI.3937-08.2009
Cioni, J. M., Lin, J. Q., Holtermann, A. V., Koppers, M., Jakobs, M. A. H., Azizi, A., et al. (2019). Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176, 56–72.e15. doi: 10.1016/j.cell.2018.11.030
Citterio, A., Arnoldi, A., Panzeri, E., Merlini, L., D’angelo, M. G., Musumeci, O., et al. (2015). Variants in KIF1A gene in dominant and sporadic forms of hereditary spastic paraparesis. J. Neurol. 262, 2684–2690. doi: 10.1007/s00415-015-7899-9
Cook, C., and Petrucelli, L. (2019). Genetic convergence brings clarity to the enigmatic red line in ALS. Neuron 101, 1057–1069. doi: 10.1016/j.neuron.2019.02.032
Corbo, M., and Hays, A. P. (1992). Peripherin and neurofilament protein coexist in spinal spheroids of motor neuron disease. J. Neuropathol. Exp. Neurol. 51, 531–537. doi: 10.1097/00005072-199209000-00008
Cosker, K. E., Fenstermacher, S. J., Pazyra-Murphy, M. F., Elliott, H. L., and Segal, R. A. (2016). The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat. Neurosci. 19, 690–696. doi: 10.1038/nn.4280
Crerar, H., Scott-Solomon, E., Bodkin-Clarke, C., Andreassi, C., Hazbon, M., Logie, E., et al. (2019). Regulation of NGF signaling by an axonal untranslated mRNA. Neuron 102, 553–563.e8. doi: 10.1016/j.neuron.2019.02.011
Dadon-Nachum, M., Melamed, E., and Offen, D. (2011). The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 43, 470–477. doi: 10.1007/s12031-010-9467-1
de Boer, A. S., Koszka, K., Kiskinis, E., Suzuki, N., Davis-Dusenbery, B. N., and Eggan, K. (2014). Genetic validation of a therapeutic target in a mouse model of ALS. Sci. Transl. Med. 6:248ra104. doi: 10.1126/scitranslmed.3009351
De Vos, K. J., Chapman, A. L., Tennant, M. E., Manser, C., Tudor, E. L., Lau, K. F., et al. (2007). Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 16, 2720–2728. doi: 10.1093/hmg/ddm226
De Vos, K. J., and Hafezparast, M. (2017). Neurobiology of axonal transport defects in motor neuron diseases: opportunities for translational research? Neurobiol. Dis. 105, 283–299. doi: 10.1016/j.nbd.2017.02.004
Deglincerti, A., and Jaffrey, S. R. (2012). Insights into the roles of local translation from the axonal transcriptome. Open Biol. 2:120079. doi: 10.1098/rsob.120079
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Denton, K., Mou, Y., Xu, C. C., Shah, D., Chang, J., Blackstone, C., et al. (2018). Impaired mitochondrial dynamics underlie axonal defects in hereditary spastic paraplegias. Hum. Mol. Genet. 27, 2517–2530. doi: 10.1093/hmg/ddy156
Di Giorgio, F. P., Carrasco, M. A., Siao, M. C., Maniatis, T., and Eggan, K. (2007). Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 10, 608–614. doi: 10.1038/nn1885
Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 29, 2841–2857. doi: 10.1038/emboj.2010.143
Ebbing, B., Mann, K., Starosta, A., Jaud, J., Schols, L., Schule, R., et al. (2008). Effect of spastic paraplegia mutations in KIF5A kinesin on transport activity. Hum. Mol. Genet. 17, 1245–1252. doi: 10.1093/hmg/ddn014
Edens, B. M., Yan, J., Miller, N., Deng, H. X., Siddique, T., and Ma, Y. C. (2017). A novel ALS-associated variant in UBQLN4 regulates motor axon morphogenesis. eLife 6:e25453. doi: 10.7554/eLife.25453
Eichelbaum, K., and Krijgsveld, J. (2014). Rapid temporal dynamics of transcription, protein synthesis, and secretion during macrophage activation. Mol. Cell. Proteomics 13, 792–810. doi: 10.1074/mcp.M113.030916
Fallini, C., Donlin-Asp, P. G., Rouanet, J. P., Bassell, G. J., and Rossoll, W. (2016). Deficiency of the survival of motor neuron protein impairs mRNA localization and local translation in the growth cone of motor neurons. J. Neurosci. 36, 3811–3820. doi: 10.1523/JNEUROSCI.2396-15.2016
Ferraiuolo, L., Heath, P. R., Holden, H., Kasher, P., Kirby, J., and Shaw, P. J. (2007). Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci. 27, 9201–9219. doi: 10.1523/jneurosci.1470-07.2007
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630. doi: 10.1038/nrneurol.2011.152
Fichera, M., Lo Giudice, M., Falco, M., Sturnio, M., Amata, S., Calabrese, O., et al. (2004). Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia. Neurology 63, 1108–1110. doi: 10.1212/01.wnl.0000138731.60693.d2
Fil, D., Deloach, A., Yadav, S., Alkam, D., Macnicol, M., Singh, A., et al. (2017). Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum. Mol. Genet. 26, 686–701. doi: 10.1093/hmg/ddw429
Fischer, L. R., Culver, D. G., Tennant, P., Davis, A. A., Wang, M., Castellano-Sanchez, A., et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. doi: 10.1016/j.expneurol.2003.10.004
Freudiger, C. W., Min, W., Saar, B. G., Lu, S., Holtom, G. R., He, C., et al. (2008). Label-free biomedical imaging with high sensitivity by stimulated Raman scattering microscopy. Science 322, 1857–1861. doi: 10.1126/science.1165758
Frick, P., Sellier, C., Mackenzie, I. R. A., Cheng, C. Y., Tahraoui-Bories, J., Martinat, C., et al. (2018). Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol. Commun. 6:72. doi: 10.1186/s40478-018-0579-0
Fujimori, K. (2018). Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 24, 1579–1589. doi: 10.1038/s41591-018-0140-5
Furukawa, Y., Kaneko, K., Watanabe, S., Yamanaka, K., and Nukina, N. (2011). A seeding reaction recapitulates intracellular formation of sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem. 286, 18664–18672. doi: 10.1074/jbc.M111.231209
Geevasinga, N., Menon, P., Howells, J., Nicholson, G. A., Kiernan, M. C., and Vucic, S. (2015). Axonal ion channel dysfunction in C9orf72 familial amyotrophic lateral sclerosis. JAMA Neurol. 72, 49–57. doi: 10.1001/jamaneurol.2014.2940
Gentile, F., Scarlino, S., Falzone, Y. M., Lunetta, C., Tremolizzo, L., Quattrini, A., et al. (2019). The peripheral nervous system in amyotrophic lateral sclerosis: opportunities for translational research. Front. Neurosci. 13:601. doi: 10.3389/fnins.2019.00601
Ghasemi, M., and Brown, R. H. Jr. (2018). Genetics of amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Med. 8:a024125. doi: 10.1101/cshperspect.a024125
Gibbs, K. L., Kalmar, B., Rhymes, E. R., Fellows, A. D., Ahmed, M., Whiting, P., et al. (2018). Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis. 9:596. doi: 10.1038/s41419-018-0624-8
Groen, E. J., Fumoto, K., Blokhuis, A. M., Engelen-Lee, J., Zhou, Y., Van Den Heuvel, D. M., et al. (2013). ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum. Mol. Genet. 22, 3690–3704. doi: 10.1093/hmg/ddt222
Grueter, B. A., Robison, A. J., Neve, R. L., Nestler, E. J., and Malenka, R. C. (2013). FosB differentially modulates nucleus accumbens direct and indirect pathway function. Proc. Natl. Acad. Sci. U.S.A. 110, 1923–1928. doi: 10.1073/pnas.1221742110
Gumy, L. F., Yeo, G. S., Tung, Y. C., Zivraj, K. H., Willis, D., Coppola, G., et al. (2011). Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA 17, 85–98. doi: 10.1261/rna.2386111
Guo, L., Kim, H. J., Wang, H., Monaghan, J., Freyermuth, F., Sung, J. C., et al. (2018). Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173, 677–692.e20. doi: 10.1016/j.cell.2018.03.002
Guo, W., Naujock, M., Fumagalli, L., Vandoorne, T., Baatsen, P., Boon, R., et al. (2017). HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 8:861. doi: 10.1038/s41467-017-00911-y
Hadano, S., Otomo, A., Kunita, R., Suzuki-Utsunomiya, K., Akatsuka, A., Koike, M., et al. (2010). Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS One 5:e9805. doi: 10.1371/journal.pone.0009805
Hanson, J. E., and Madison, D. V. (2007). Presynaptic FMR1 genotype influences the degree of synaptic connectivity in a mosaic mouse model of fragile X syndrome. J. Neurosci. 27, 4014–4018. doi: 10.1523/jneurosci.4717-06.2007
He, W., Bai, G., Zhou, H., Wei, N., White, N. M., Lauer, J., et al. (2015). CMT2D neuropathy is linked to the neomorphic binding activity of glycyl-tRNA synthetase. Nature 526, 710–714. doi: 10.1038/nature15510
Heo, K., Lim, S. M., Nahm, M., Kim, Y. E., Oh, K. W., Park, H. T., et al. (2018). A de novo RAPGEF2 variant identified in a sporadic amyotrophic lateral sclerosis patient impairs microtubule stability and axonal mitochondria distribution. Exp. Neurobiol. 27, 550–563. doi: 10.5607/en.2018.27.6.550
Hirano, A., Donnenfeld, H., Sasaki, S., and Nakano, I. (1984a). Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 43, 461–470. doi: 10.1097/00005072-198409000-00001
Hirano, A., Nakano, I., Kurland, L. T., Mulder, D. W., Holley, P. W., and Saccomanno, G. (1984b). Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 43, 471–480. doi: 10.1097/00005072-198409000-00002
Hofweber, M., Hutten, S., Bourgeois, B., Spreitzer, E., Niedner-Boblenz, A., Schifferer, M., et al. (2018). Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 173, 706–719.e13. doi: 10.1016/j.cell.2018.03.004
Hogan, A. L., Don, E. K., Rayner, S. L., Lee, A., Laird, A. S., Watchon, M., et al. (2017). Expression of ALS/FTD-linked mutant CCNF in zebrafish leads to increased cell death in the spinal cord and an aberrant motor phenotype. Hum. Mol. Genet. 26, 2616–2626. doi: 10.1093/hmg/ddx136
Howland, D. S., Liu, J., She, Y., Goad, B., Maragakis, N. J., Kim, B., et al. (2002). Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. U.S.A. 99, 1604–1609. doi: 10.1073/pnas.032539299
Ichiyanagi, N., Fujimori, K., Yano, M., Ishihara-Fujisaki, C., Sone, T., Akiyama, T., et al. (2016). Establishment of in vitro FUS-associated familial amyotrophic lateral sclerosis model using human induced pluripotent stem cells. Stem Cell Rep. 6, 496–510. doi: 10.1016/j.stemcr.2016.02.011
Ito, Y., Ofengeim, D., Najafov, A., Das, S., Saberi, S., Li, Y., et al. (2016). RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608. doi: 10.1126/science.aaf6803
Ji, S. J., and Jaffrey, S. R. (2014). Axonal transcription factors: novel regulators of growth cone-to-nucleus signaling. Dev. Neurobiol. 74, 245–258. doi: 10.1002/dneu.22112
Jiang, J., Zhu, Q., Gendron, T. F., Saberi, S., Mcalonis-Downes, M., Seelman, A., et al. (2016). Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550. doi: 10.1016/j.neuron.2016.04.006
Jiang, Y. M., Yamamoto, M., Kobayashi, Y., Yoshihara, T., Liang, Y., Terao, S., et al. (2005). Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 57, 236–251. doi: 10.1002/ana.20379
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
Joung, J. K., and Sander, J. D. (2013). TALENs: a widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 14, 49–55. doi: 10.1038/nrm3486
Jung, H., Yoon, B. C., and Holt, C. E. (2012). Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat. Rev. Neurosci. 13, 308–324. doi: 10.1038/nrn3210
Kabashi, E., Lin, L., Tradewell, M. L., Dion, P. A., Bercier, V., Bourgouin, P., et al. (2010). Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 19, 671–683. doi: 10.1093/hmg/ddp534
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., Mcconkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574. doi: 10.1038/ng.132
Kanai, K., Kuwabara, S., Misawa, S., Tamura, N., Ogawara, K., Nakata, M., et al. (2006). Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage. Brain 129, 953–962. doi: 10.1093/brain/awl024
Kanai, K., Shibuya, K., Sato, Y., Misawa, S., Nasu, S., Sekiguchi, Y., et al. (2012). Motor axonal excitability properties are strong predictors for survival in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 83, 734–738. doi: 10.1136/jnnp-2011-301782
Kang, S. H., Fukaya, M., Yang, J. K., Rothstein, J. D., and Bergles, D. E. (2010). NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681. doi: 10.1016/j.neuron.2010.09.009
Kang, S. H., Li, Y., Fukaya, M., Lorenzini, I., Cleveland, D. W., Ostrow, L. W., et al. (2013). Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 16, 571–579. doi: 10.1038/nn.3357
Kapeli, K., Martinez, F. J., and Yeo, G. W. (2017). Genetic mutations in RNA-binding proteins and their roles in ALS. Hum. Genet. 136, 1193–1214. doi: 10.1007/s00439-017-1830-7
Kawada, J., Kaneda, S., Kirihara, T., Maroof, A., Levi, T., Eggan, K., et al. (2017). Generation of a motor nerve organoid with human stem cell-derived neurons. Stem Cell Rep. 9, 1441–1449. doi: 10.1016/j.stemcr.2017.09.021
Kelleher, R. J. III, and Bear, M. F. (2008). The autistic neuron: troubled translation? Cell 135, 401–406. doi: 10.1016/j.cell.2008.10.017
Kenna, K. P., Van Doormaal, P. T., Dekker, A. M., Ticozzi, N., Kenna, B. J., Diekstra, F. P., et al. (2016). NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 48, 1037–1042. doi: 10.1038/ng.3626
Kieran, D., Woods, I., Villunger, A., Strasser, A., and Prehn, J. H. (2007). Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc. Natl. Acad. Sci. U.S.A. 104, 20606–20611. doi: 10.1073/pnas.0707906105
Kikuchi, H., Almer, G., Yamashita, S., Guegan, C., Nagai, M., Xu, Z., et al. (2006). Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. U.S.A. 103, 6025–6030. doi: 10.1073/pnas.0509227103
Kim, H. J., and Taylor, J. P. (2017). Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 96, 285–297. doi: 10.1016/j.neuron.2017.07.029
Kiryu-Seo, S., and Kiyama, H. (2019). Mitochondrial behavior during axon regeneration/degeneration in vivo. Neurosci. Res. 139, 42–47. doi: 10.1016/j.neures.2018.08.014
Klaus, J., Kanton, S., Kyrousi, C., Ayo-Martin, A. C., Di Giaimo, R., Riesenberg, S., et al. (2019). Altered neuronal migratory trajectories in human cerebral organoids derived from individuals with neuronal heterotopia. Nat. Med. 25, 561–568. doi: 10.1038/s41591-019-0371-0
Klim, J. R., Williams, L. A., Limone, F., Guerra San Juan, I., Davis-Dusenbery, B. N., Mordes, D. A., et al. (2019). ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 22, 167–179. doi: 10.1038/s41593-018-0300-4
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Kwon, I., Xiang, S., Kato, M., Wu, L., Theodoropoulos, P., Wang, T., et al. (2014). Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345, 1139–1145. doi: 10.1126/science.1254917
Kye, M. J., Niederst, E. D., Wertz, M. H., Goncalves Ido, C., Akten, B., Dover, K. Z., et al. (2014). SMN regulates axonal local translation via miR-183/mTOR pathway. Hum. Mol. Genet. 23, 6318–6331. doi: 10.1093/hmg/ddu350
Lafragette, A., Bardo, M. T., Lardeux, V., Solinas, M., and Thiriet, N. (2017). Reduction of cocaine-induced locomotor effects by enriched environment is associated with cell-specific accumulation of DeltaFosB in striatal and cortical subregions. Int. J. Neuropsychopharmacol. 20, 237–246. doi: 10.1093/ijnp/pyw097
Laird, A. S., Van Hoecke, A., De Muynck, L., Timmers, M., Van Den Bosch, L., Van Damme, P., et al. (2010). Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS One 5:e13368. doi: 10.1371/journal.pone.0013368
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi: 10.1038/nature11314
Li, F., Xie, X., Wang, Y., Liu, J., Cheng, X., Guo, Y., et al. (2016). Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat. Commun. 7:12708. doi: 10.1038/ncomms12708
Liao, Y. C., Fernandopulle, M. S., Wang, G., Choi, H., Hao, L., Drerup, C. M., et al. (2019). RNA granules hitchhike on lysosomes for long-distance transport, using annexin A11 as a molecular tether. Cell 179, 147–164.e20. doi: 10.1016/j.cell.2019.08.050
Lin, M. Y., Cheng, X. T., Xie, Y., Cai, Q., and Sheng, Z. H. (2017). Removing dysfunctional mitochondria from axons independent of mitophagy under pathophysiological conditions. Autophagy 13, 1792–1794. doi: 10.1080/15548627.2017.1356552
Ling, J. P., Pletnikova, O., Troncoso, J. C., and Wong, P. C. (2015). TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655. doi: 10.1126/science.aab0983
Lopez-Erauskin, J., Tadokoro, T., Baughn, M. W., Myers, B., Mcalonis-Downes, M., Chillon-Marinas, C., et al. (2018). ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 100, 816–830.e7. doi: 10.1016/j.neuron.2018.09.044
Maciel, R., Bis, D. M., Rebelo, A. P., Saghira, C., Zuchner, S., and Saporta, M. A. (2018). The human motor neuron axonal transcriptome is enriched for transcripts related to mitochondrial function and microtubule-based axonal transport. Exp. Neurol. 307, 155–163. doi: 10.1016/j.expneurol.2018.06.008
Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., et al. (2017). TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95, 808–816.e9. doi: 10.1016/j.neuron.2017.07.025
Mackenzie, I. R., Rademakers, R., and Neumann, M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. doi: 10.1016/S1474-4422(10)70195-2
Maday, S., and Holzbaur, E. L. (2016). Compartment-specific regulation of autophagy in primary neurons. J. Neurosci. 36, 5933–5945. doi: 10.1523/JNEUROSCI.4401-15.2016
Maday, S., Twelvetrees, A. E., Moughamian, A. J., and Holzbaur, E. L. (2014). Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292–309. doi: 10.1016/j.neuron.2014.10.019
Magrane, J., Cortez, C., Gan, W. B., and Manfredi, G. (2014). Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 23, 1413–1424. doi: 10.1093/hmg/ddt528
Maniatis, S., Aijo, T., Vickovic, S., Braine, C., Kang, K., Mollbrink, A., et al. (2019). Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 364, 89–93. doi: 10.1126/science.aav9776
Martin, N., Jaubert, J., Gounon, P., Salido, E., Haase, G., Szatanik, M., et al. (2002). A missense mutation in Tbce causes progressive motor neuronopathy in mice. Nat. Genet. 32, 443–447. doi: 10.1038/ng1016
Martineau, E., Di Polo, A., Vande Velde, C., and Robitaille, R. (2018). Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. eLife 7:e41973. doi: 10.7554/eLife.41973
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
McWhorter, M. L., Monani, U. R., Burghes, A. H., and Beattie, C. E. (2003). Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 162, 919–931.
Melamed, Z., Lopez-Erauskin, J., Baughn, M. W., Zhang, O., Drenner, K., Sun, Y., et al. (2019). Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 22, 180–190. doi: 10.1038/s41593-018-0293-z
Menon, S., and Gupton, S. (2018). Recent advances in branching mechanisms underlying neuronal morphogenesis. F1000Res. 7:F1000 Faculty Rev-1779.
Merianda, T. T., Lin, A. C., Lam, J. S., Vuppalanchi, D., Willis, D. E., Karin, N., et al. (2009). A functional equivalent of endoplasmic reticulum and Golgi in axons for secretion of locally synthesized proteins. Mol. Cell. Neurosci. 40, 128–142. doi: 10.1016/j.mcn.2008.09.008
Millecamps, S., and Julien, J. P. (2013). Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 14, 161–176. doi: 10.1038/nrn3380
Mizielinska, S., Gronke, S., Niccoli, T., Ridler, C. E., Clayton, E. L., Devoy, A., et al. (2014). C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194. doi: 10.1126/science.1256800
Moller, A., Bauer, C. S., Cohen, R. N., Webster, C. P., and De Vos, K. J. (2017). Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum. Mol. Genet. 26, 4668–4679. doi: 10.1093/hmg/ddx348
Moloney, E. B., De Winter, F., and Verhaagen, J. (2014). ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 8:252. doi: 10.3389/fnins.2014.00252
Moradi, M., Sivadasan, R., Saal, L., Luningschror, P., Dombert, B., Rathod, R. J., et al. (2017). Differential roles of alpha-, beta-, and gamma-actin in axon growth and collateral branch formation in motoneurons. J. Cell Biol. 216, 793–814. doi: 10.1083/jcb.201604117
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morisaki, T., Lyon, K., Deluca, K. F., Deluca, J. G., English, B. P., Zhang, Z., et al. (2016). Real-time quantification of single RNA translation dynamics in living cells. Science 352, 1425–1429. doi: 10.1126/science.aaf0899
Morrice, J. R., Gregory-Evans, C. Y., and Shaw, C. A. (2018). Modeling environmentally-induced motor neuron degeneration in zebrafish. Sci. Rep. 8:4890. doi: 10.1038/s41598-018-23018-w
Morrison, B. M., Lee, Y., and Rothstein, J. D. (2013). Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 23, 644–651. doi: 10.1016/j.tcb.2013.07.007
Munch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. doi: 10.1212/01.wnl.0000134608.83927.b1
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nakazawa, S., Oikawa, D., Ishii, R., Ayaki, T., Takahashi, H., Takeda, H., et al. (2016). Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat. Commun. 7:12547. doi: 10.1038/ncomms12547
Naumann, M., Pal, A., Goswami, A., Lojewski, X., Japtok, J., Vehlow, A., et al. (2018). Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 9:335. doi: 10.1038/s41467-017-02299-1
Nedelsky, N. B., and Taylor, J. P. (2019). Bridging biophysics and neurology: aberrant phase transitions in neurodegenerative disease. Nat. Rev. Neurol. 15, 272–286. doi: 10.1038/s41582-019-0157-5
Neto, E., Leitao, L., Sousa, D. M., Alves, C. J., Alencastre, I. S., Aguiar, P., et al. (2016). Compartmentalized microfluidic platforms: the unrivaled breakthrough of in vitro tools for neurobiological research. J. Neurosci. 36, 11573–11584. doi: 10.1523/jneurosci.1748-16.2016
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283.e6. doi: 10.1016/j.neuron.2018.02.027
Nijssen, J., Aguila, J., Hoogstraaten, R., Kee, N., and Hedlund, E. (2018). Axon-Seq decodes the motor axon transcriptome and its modulation in response to ALS. Stem Cell Rep. 11, 1565–1578. doi: 10.1016/j.stemcr.2018.11.005
Nijssen, J., Comley, L. H., and Hedlund, E. (2017). Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 133, 863–885. doi: 10.1007/s00401-017-1708-8
Nishiyama, A., Niihori, T., Warita, H., Izumi, R., Akiyama, T., Kato, M., et al. (2017). Comprehensive targeted next-generation sequencing in Japanese familial amyotrophic lateral sclerosis. Neurobiol. Aging 53, e191–e194. doi: 10.1016/j.neurobiolaging.2017.01.004
Nonaka, T., Masuda-Suzukake, M., Arai, T., Hasegawa, Y., Akatsu, H., Obi, T., et al. (2013). Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134. doi: 10.1016/j.celrep.2013.06.007
Okada, Y., Yamazaki, H., Sekine-Aizawa, Y., and Hirokawa, N. (1995). The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81, 769–780. doi: 10.1016/0092-8674(95)90538-3
Okano, H., and Yamanaka, S. (2014). iPS cell technologies: significance and applications to CNS regeneration and disease. Mol. Brain 7:22. doi: 10.1186/1756-6606-7-22
O’Rourke, J. G., Bogdanik, L., Yanez, A., Lall, D., Wolf, A. J., Muhammad, A. K., et al. (2016). C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329. doi: 10.1126/science.aaf1064
Osking, Z., Ayers, J. I., Hildebrandt, R., Skruber, K., Brown, H., Ryu, D., et al. (2019). ALS-linked SOD1 mutants enhance neurite outgrowth and branching in adult motor neurons. iScience 11, 294–304. doi: 10.1016/j.isci.2018.12.026
Parras, A., Anta, H., Santos-Galindo, M., Swarup, V., Elorza, A., Nieto-Gonzalez, J. L., et al. (2018). Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature 560, 441–446. doi: 10.1038/s41586-018-0423-5
Peng, Y., Kim, M. J., Hullinger, R., O’riordan, K. J., Burger, C., Pehar, M., et al. (2016). Improved proteostasis in the secretory pathway rescues Alzheimer’s disease in the mouse. Brain 139, 937–952. doi: 10.1093/brain/awv385
Pereno, G. L., Balaszczuk, V., and Beltramino, C. A. (2011). Kainic acid-induced early genes activation and neuronal death in the medial extended amygdala of rats. Exp. Toxicol. Pathol. 63, 291–299. doi: 10.1016/j.etp.2010.02.001
Peters, O. M., Cabrera, G. T., Tran, H., Gendron, T. F., Mckeon, J. E., Metterville, J., et al. (2015). Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 88, 902–909. doi: 10.1016/j.neuron.2015.11.018
Philips, T., Bento-Abreu, A., Nonneman, A., Haeck, W., Staats, K., Geelen, V., et al. (2013). Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136, 471–482. doi: 10.1093/brain/aws339
Picchiarelli, G., Demestre, M., Zuko, A., Been, M., Higelin, J., Dieterle, S., et al. (2019). FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat. Neurosci. 22, 1793–1805. doi: 10.1038/s41593-019-0498-9
Plachta, N., Annaheim, C., Bissiere, S., Lin, S., Ruegg, M., Hoving, S., et al. (2007). Identification of a lectin causing the degeneration of neuronal processes using engineered embryonic stem cells. Nat. Neurosci. 10, 712–719. doi: 10.1038/nn1897
Porta, S., Xu, Y., Restrepo, C. R., Kwong, L. K., Zhang, B., Brown, H. J., et al. (2018). Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 9:4220. doi: 10.1038/s41467-018-06548-9
Poulopoulos, A., Murphy, A. J., Ozkan, A., Davis, P., Hatch, J., Kirchner, R., et al. (2019). Subcellular transcriptomes and proteomes of developing axon projections in the cerebral cortex. Nature 565, 356–360. doi: 10.1038/s41586-018-0847-y
Pradat, P. F., Bruneteau, G., Gonzalez De Aguilar, J. L., Dupuis, L., Jokic, N., Salachas, F., et al. (2007). Muscle Nogo-A expression is a prognostic marker in lower motor neuron syndromes. Ann. Neurol. 62, 15–20. doi: 10.1002/ana.21122
Puls, I., Jonnakuty, C., Lamonte, B. H., Holzbaur, E. L., Tokito, M., Mann, E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456.
Qamar, S., Wang, G., Randle, S. J., Ruggeri, F. S., Varela, J. A., Lin, J. Q., et al. (2018). FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-pi interactions. Cell 173, 720–734.e15. doi: 10.1016/j.cell.2018.03.056
Quadrato, G., Nguyen, T., Macosko, E. Z., Sherwood, J. L., Min Yang, S., Berger, D. R., et al. (2017). Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545, 48–53. doi: 10.1038/nature22047
Rage, F., Boulisfane, N., Rihan, K., Neel, H., Gostan, T., Bertrand, E., et al. (2013). Genome-wide identification of mRNAs associated with the protein SMN whose depletion decreases their axonal localization. RNA 19, 1755–1766. doi: 10.1261/rna.040204.113
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Riva, N., Clarelli, F., Domi, T., Cerri, F., Gallia, F., Trimarco, A., et al. (2016). Unraveling gene expression profiles in peripheral motor nerve from amyotrophic lateral sclerosis patients: insights into pathogenesis. Sci. Rep. 6:39297. doi: 10.1038/srep39297
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62.
Rotem, N., Magen, I., Ionescu, A., Gershoni-Emek, N., Altman, T., Costa, C. J., et al. (2017). ALS along the axons - expression of coding and noncoding RNA differs in axons of ALS models. Sci. Rep. 7:44500. doi: 10.1038/srep44500
Rouleau, G. A., Clark, A. W., Rooke, K., Pramatarova, A., Krizus, A., Suchowersky, O., et al. (1996). SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann. Neurol. 39, 128–131.
Roy, S., Zhang, B., Lee, V. M., and Trojanowski, J. Q. (2005). Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol. 109, 5–13. doi: 10.1007/s00401-004-0952-x
Rutherford, N. J., Zhang, Y. J., Baker, M., Gass, J. M., Finch, N. A., Xu, Y. F., et al. (2008). Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 4:e1000193. doi: 10.1371/journal.pgen.1000193
Saal, L., Briese, M., Kneitz, S., Glinka, M., and Sendtner, M. (2014). Subcellular transcriptome alterations in a cell culture model of spinal muscular atrophy point to widespread defects in axonal growth and presynaptic differentiation. RNA 20, 1789–1802. doi: 10.1261/rna.047373.114
Sabari, B. R., Dall’agnese, A., Boija, A., Klein, I. A., Coffey, E. L., Shrinivas, K., et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361:eaar3958. doi: 10.1126/science.aar3958
Salvadores, N., Sanhueza, M., Manque, P., and Court, F. A. (2017). Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front. Neurosci. 11:451. doi: 10.3389/fnins.2017.00451
Sambandan, S., Akbalik, G., Kochen, L., Rinne, J., Kahlstatt, J., Glock, C., et al. (2017). Activity-dependent spatially localized miRNA maturation in neuronal dendrites. Science 355, 634–637. doi: 10.1126/science.aaf8995
Scekic-Zahirovic, J., Sendscheid, O., El Oussini, H., Jambeau, M., Sun, Y., Mersmann, S., et al. (2016). Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 35, 1077–1097. doi: 10.15252/embj.201592559
Schmidt, N., Akaaboune, M., Gajendran, N., Martinez-Pena, Y., Valenzuela, I., Wakefield, S., et al. (2011). Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of alpha-dystrobrevin. J. Cell Biol. 195, 1171–1184. doi: 10.1083/jcb.201107083
Schoen, M., Reichel, J. M., Demestre, M., Putz, S., Deshpande, D., Proepper, C., et al. (2015). Super-resolution microscopy reveals presynaptic localization of the ALS/FTD related protein FUS in hippocampal neurons. Front. Cell. Neurosci. 9:496. doi: 10.3389/fncel.2015.00496
Shao, Z., Noh, H., Bin Kim, W., Ni, P., Nguyen, C., Cote, S. E., et al. (2019). Dysregulated protocadherin-pathway activity as an intrinsic defect in induced pluripotent stem cell-derived cortical interneurons from subjects with schizophrenia. Nat. Neurosci. 22, 229–242. doi: 10.1038/s41593-018-0313-z
Sharma, A., Lyashchenko, A. K., Lu, L., Nasrabady, S. E., Elmaleh, M., Mendelsohn, M., et al. (2016). ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 7:10465. doi: 10.1038/ncomms10465
Shigeoka, T., Jung, H., Jung, J., Turner-Bridger, B., Ohk, J., Lin, J. Q., et al. (2016). Dynamic axonal translation in developing and mature visual circuits. Cell 166, 181–192. doi: 10.1016/j.cell.2016.05.029
Shiihashi, G., Ito, D., Yagi, T., Nihei, Y., Ebine, T., and Suzuki, N. (2016). Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain 139, 2380–2394. doi: 10.1093/brain/aww161
Sivadasan, R., Hornburg, D., Drepper, C., Frank, N., Jablonka, S., Hansel, A., et al. (2016). C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat. Neurosci. 19, 1610–1618. doi: 10.1038/nn.4407
Smith, B. N., Ticozzi, N., Fallini, C., Gkazi, A. S., Topp, S., Kenna, K. P., et al. (2014). Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. doi: 10.1016/j.neuron.2014.09.027
Smith, B. N., Topp, S. D., Fallini, C., Shibata, H., Chen, H. J., Troakes, C., et al. (2017). Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci. Transl. Med. 9:eaad9157. doi: 10.1126/scitranslmed.aad9157
So, E., Mitchell, J. C., Memmi, C., Chennell, G., Vizcay-Barrena, G., Allison, L., et al. (2018). Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet. 27, 463–474. doi: 10.1093/hmg/ddx415
Spaulding, E. L., and Burgess, R. W. (2017). Accumulating evidence for axonal translation in neuronal homeostasis. Front. Neurosci. 11:312. doi: 10.3389/fnins.2017.00312
Spillane, M., Ketschek, A., Merianda, T. T., Twiss, J. L., and Gallo, G. (2013). Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep. 5, 1564–1575. doi: 10.1016/j.celrep.2013.11.022
Steinacker, P., Feneberg, E., Weishaupt, J., Brettschneider, J., Tumani, H., Andersen, P. M., et al. (2016). Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J. Neurol. Neurosurg. Psychiatry 87, 12–20. doi: 10.1136/jnnp-2015-311387
Storkebaum, E. (2016). Peripheral neuropathy via mutant tRNA synthetases: inhibition of protein translation provides a possible explanation. Bioessays 38, 818–829. doi: 10.1002/bies.201600052
Suzuki, N., Aoki, M., Warita, H., Kato, M., Mizuno, H., Shimakura, N., et al. (2010). FALS with FUS mutation in Japan, with early onset, rapid progress and basophilic inclusion. J. Hum. Genet. 55, 252–254. doi: 10.1038/jhg.2010.16
Tagliaferro, P., and Burke, R. E. (2016). Retrograde axonal degeneration in Parkinson disease. J. Parkinsons Dis. 6, 1–15. doi: 10.3233/JPD-150769
Takata, M., Tanaka, H., Kimura, M., Nagahara, Y., Tanaka, K., Kawasaki, K., et al. (2013). Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental models of amyotrophic lateral sclerosis. Br. J. Pharmacol. 170, 341–351. doi: 10.1111/bph.12277
Takeuchi, A., Iida, K., Tsubota, T., Hosokawa, M., Denawa, M., Brown, J. B., et al. (2018). Loss of Sfpq causes long-gene transcriptopathy in the brain. Cell Rep. 23, 1326–1341. doi: 10.1016/j.celrep.2018.03.141
Tashiro, Y., Urushitani, M., Inoue, H., Koike, M., Uchiyama, Y., Komatsu, M., et al. (2012). Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J. Biol. Chem. 287, 42984–42994. doi: 10.1074/jbc.M112.417600
Taylor, A. M., Berchtold, N. C., Perreau, V. M., Tu, C. H., Li Jeon, N., and Cotman, C. W. (2009). Axonal mRNA in uninjured and regenerating cortical mammalian axons. J. Neurosci. 29, 4697–4707. doi: 10.1523/JNEUROSCI.6130-08.2009
Tcherkezian, J., Brittis, P. A., Thomas, F., Roux, P. P., and Flanagan, J. G. (2010). Transmembrane receptor DCC associates with protein synthesis machinery and regulates translation. Cell 141, 632–644. doi: 10.1016/j.cell.2010.04.008
Thiel, C., Kessler, K., Giessl, A., Dimmler, A., Shalev, S. A., Von Der Haar, S., et al. (2011). NEK1 mutations cause short-rib polydactyly syndrome type Majewski. Am. J. Hum. Genet. 88, 106–114. doi: 10.1016/j.ajhg.2010.12.004
Thomas-Jinu, S., Gordon, P. M., Fielding, T., Taylor, R., Smith, B. N., Snowden, V., et al. (2017). Non-nuclear pool of splicing factor SFPQ regulates axonal transcripts required for normal motor development. Neuron 94, 322–336.e5. doi: 10.1016/j.neuron.2017.03.026
Tian, F., Yang, W., Mordes, D. A., Wang, J. Y., Salameh, J. S., Mok, J., et al. (2016). Monitoring peripheral nerve degeneration in ALS by label-free stimulated Raman scattering imaging. Nat. Commun. 7:13283. doi: 10.1038/ncomms13283
Urushitani, M., Ezzi, S. A., Matsuo, A., Tooyama, I., and Julien, J. P. (2008). The endoplasmic reticulum-Golgi pathway is a target for translocation and aggregation of mutant superoxide dismutase linked to ALS. FASEB J. 22, 2476–2487. doi: 10.1096/fj.07-092783
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Venkova, K., Christov, A., Kamaluddin, Z., Kobalka, P., Siddiqui, S., and Hensley, K. (2014). Semaphorin 3A signaling through neuropilin-1 is an early trigger for distal axonopathy in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 73, 702–713. doi: 10.1097/NEN.0000000000000086
Verheijen, M. H., Peviani, M., Hendricusdottir, R., Bell, E. M., Lammens, M., Smit, A. B., et al. (2014). Increased axonal ribosome numbers is an early event in the pathogenesis of amyotrophic lateral sclerosis. PLoS One 9:e87255. doi: 10.1371/journal.pone.0087255
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019
Wang, C., Han, B., Zhou, R., and Zhuang, X. (2016a). Real-time imaging of translation on single mRNA transcripts in live cells. Cell 165, 990–1001. doi: 10.1016/j.cell.2016.04.040
Wang, E. T., Taliaferro, J. M., Lee, J. A., Sudhakaran, I. P., Rossoll, W., Gross, C., et al. (2016b). Dysregulation of mRNA localization and translation in genetic disease. J. Neurosci. 36, 11418–11426. doi: 10.1523/jneurosci.2352-16.2016
Wang, L., Gao, J., Liu, J., Siedlak, S. L., Torres, S., Fujioka, H., et al. (2018). Mitofusin 2 regulates axonal transport of calpastatin to prevent neuromuscular synaptic elimination in skeletal muscles. Cell Metab. 28, 400–414.e8. doi: 10.1016/j.cmet.2018.06.011
Watanabe, S., Ilieva, H., Tamada, H., Nomura, H., Komine, O., Endo, F., et al. (2016). Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 8, 1421–1437. doi: 10.15252/emmm.201606403
Wen, X., Tan, W., Westergard, T., Krishnamurthy, K., Markandaiah, S. S., Shi, Y., et al. (2014). Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225. doi: 10.1016/j.neuron.2014.12.010
Weskamp, K., Tank, E. M., Miguez, R., Mcbride, J. P., Gomez, N. B., White, M., et al. (2019). Shortened TDP43 isoforms upregulated by neuronal hyperactivity drive TDP43 pathology in ALS. J. Clin. Invest. 130, 1139–1155. doi: 10.1172/JCI130988
Westergard, T., Jensen, B. K., Wen, X., Cai, J., Kropf, E., Iacovitti, L., et al. (2016). Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 17, 645–652. doi: 10.1016/j.celrep.2016.09.032
Wiedau-Pazos, M., Goto, J. J., Rabizadeh, S., Gralla, E. B., Roe, J. A., Lee, M. K., et al. (1996). Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science 271, 515–518.
Williamson, T. L., and Cleveland, D. W. (1999). Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 2, 50–56. doi: 10.1038/4553
Willis, D. E., Van Niekerk, E. A., Sasaki, Y., Mesngon, M., Merianda, T. T., Williams, G. G., et al. (2007). Extracellular stimuli specifically regulate localized levels of individual neuronal mRNAs. J. Cell Biol. 178, 965–980. doi: 10.1083/jcb.200703209
Wu, B., Eliscovich, C., Yoon, Y. J., and Singer, R. H. (2016a). Translation dynamics of single mRNAs in live cells and neurons. Science 352, 1430–1435. doi: 10.1126/science.aaf1084
Wu, C. H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012b). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280
Wu, J. W., Hussaini, S. A., Bastille, I. M., Rodriguez, G. A., Mrejeru, A., Rilett, K., et al. (2016b). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092. doi: 10.1038/nn.4328
Xiang, Y., Tanaka, Y., Patterson, B., Kang, Y. J., Govindaiah, G., Roselaar, N., et al. (2017). Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell 21, 383–398.e7. doi: 10.1016/j.stem.2017.07.007
Xie, Y., Zhou, B., Lin, M. Y., Wang, S., Foust, K. D., and Sheng, Z. H. (2015). Endolysosomal deficits augment mitochondria pathology in spinal motor neurons of asymptomatic fALS mice. Neuron 87, 355–370. doi: 10.1016/j.neuron.2015.06.026
Xing, L., and Bassell, G. J. (2013). mRNA localization: an orchestration of assembly, traffic and synthesis. Traffic 14, 2–14. doi: 10.1111/tra.12004
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253. doi: 10.1038/nn2047
Yamazaki, T., Chen, S., Yu, Y., Yan, B., Haertlein, T. C., Carrasco, M. A., et al. (2012). FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2, 799–806. doi: 10.1016/j.celrep.2012.08.025
Yan, S., Guo, C., Hou, G., Zhang, H., Lu, X., Williams, J. C., et al. (2015). Atomic-resolution structure of the CAP-Gly domain of dynactin on polymeric microtubules determined by magic angle spinning NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 112, 14611–14616. doi: 10.1073/pnas.1509852112
Yan, X., Hoek, T. A., Vale, R. D., and Tanenbaum, M. E. (2016). Dynamics of translation of single mRNA molecules in vivo. Cell 165, 976–989. doi: 10.1016/j.cell.2016.04.034
Yoon, Y. J., Wu, B., Buxbaum, A. R., Das, S., Tsai, A., English, B. P., et al. (2016). Glutamate-induced RNA localization and translation in neurons. Proc. Natl. Acad. Sci. U.S.A. 113, E6877–E6886.
Yoshizawa, T., Ali, R., Jiou, J., Fung, H. Y. J., Burke, K. A., Kim, S. J., et al. (2018). Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell 173, 693–705.e22. doi: 10.1016/j.cell.2018.03.003
Zhang, C. L., Rodenkirch, L., Schultz, J. R., and Chiu, S. Y. (2012). A novel method to study the local mitochondrial fusion in myelinated axons in vivo. J. Neurosci. Methods 207, 51–58. doi: 10.1016/j.jneumeth.2012.03.013
Zheng, J. Q., Kelly, T. K., Chang, B., Ryazantsev, S., Rajasekaran, A. K., Martin, K. C., et al. (2001). A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J. Neurosci. 21, 9291–9303. doi: 10.1523/jneurosci.21-23-09291.2001
Zhou, B., Yu, P., Lin, M. Y., Sun, T., Chen, Y., and Sheng, Z. H. (2016). Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J. Cell Biol. 214, 103–119. doi: 10.1083/jcb.201605101
Keywords: amyotrophic lateral sclerosis (ALS), omics analysis, axonal dysfunction, local translation, axon branching, motor nerve organoid, human induced pluripotent stem cell (hiPSC)-derived motor neuron
Citation: Suzuki N, Akiyama T, Warita H and Aoki M (2020) Omics Approach to Axonal Dysfunction of Motor Neurons in Amyotrophic Lateral Sclerosis (ALS). Front. Neurosci. 14:194. doi: 10.3389/fnins.2020.00194
Received: 28 November 2019; Accepted: 24 February 2020;
Published: 25 March 2020.
Edited by:
Manoj Kumar Jaiswal, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Philip Van Damme, KU Leuven, BelgiumLudo M. Van Den Bosch, KU Leuven, Belgium
Masahisa Katsuno, Nagoya University Hospital, Japan
Copyright © 2020 Suzuki, Akiyama, Warita and Aoki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naoki Suzuki, bmFva2lAbWVkLnRvaG9rdS5hYy5qcA==; Masashi Aoki, YW9raW1AbWVkLnRvaG9rdS5hYy5qcA==