 Yukti Vyas
Yukti Vyas Johanna M. Montgomery
Johanna M. Montgomery Juliette E. Cheyne
Juliette E. Cheyne- Department of Physiology, Centre for Brain Research, University of Auckland, Auckland, New Zealand
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that is the most common cause of dementia. Symptoms of AD include memory loss, disorientation, mood and behavior changes, confusion, unfounded suspicions, and eventually, difficulty speaking, swallowing, and walking. These symptoms are caused by neuronal degeneration and cell loss that begins in the hippocampus, and later in disease progression spreading to the rest of the brain. While there are some medications that alleviate initial symptoms, there are currently no treatments that stop disease progression. Hippocampal deficits in amyloid-β-related rodent models of AD have revealed synaptic, behavioral and circuit-level defects. These changes in synaptic function, plasticity, neuronal excitability, brain connectivity, and excitation/inhibition imbalance all have profound effects on circuit function, which in turn could exacerbate disease progression. Despite, the wealth of studies on AD pathology we don’t yet have a complete understanding of hippocampal deficits in AD. With the increasing development of in vivo recording techniques in awake and freely moving animals, future studies will extend our current knowledge of the mechanisms underpinning how hippocampal function is altered in AD, and aid in progression of treatment strategies that prevent and/or delay AD symptoms.
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disease affecting more than 40 million people worldwide (Alzheimer’s Disease International, 2018). AD is clinically characterized as a progressive impairment of memory and other cognitive functions, eventually leading to dementia and death (Förstl and Kurz, 1999; Holtzman et al., 2011). There are three stages of AD: (1) “preclinical” asymptomatic phase, (2) mild cognitive impairment where the first symptoms including changes in mood and behavior, confusion, and some memory loss become evident, and (3) dementia in which patients demonstrate deficits in multiple cognitive domains that are severe enough to produce loss of function (Förstl and Kurz, 1999; Jack et al., 2010; Sperling et al., 2011). Post-mortem AD brain tissue is characterized by pathological markers including amyloid plaques, tau neurofibrillary tangles, vascular damage from the plaque deposition, and profound neuronal cell loss (Blessed et al., 1968; Katzman and Saitoh, 1991; Selkoe, 1991; for a review see Uylings and de Brabander, 2002). There are currently no cures for AD or dementia, and any treatments available are only palliative, therefore, many groups internationally are working to further understand the pathophysiology of AD in order to develop potential treatment strategies.
The hippocampus is widely studied in AD as this brain region is essential for forming new memories, and the progressive degeneration of neurons in the hippocampus responsible for short-term memory loss is a hallmark effect of AD (West et al., 1994, 2004; Fox et al., 1996). Microscopic changes in the hippocampus also precede behavioral symptomology in AD patients (for a review see Mufson et al., 2015) and mouse models; therefore, this review will focus on hippocampal deficits observed in AD.
There are two categories of AD, the early-onset familial AD generally occurring before 65 years of age, and the late-onset sporadic AD occurring after the age of 65. Data from extensive human genetic, histopathological, biomarker and animal model studies indicates that the 39–42 amino-acid (aa) peptide amyloid-β plays a prime role in the pathogenesis of familial and sporadic AD (Oddo et al., 2003; Haass and Selkoe, 2007; Bertram et al., 2010; Jack et al., 2010, 2013; Bateman et al., 2012). Here we will focus on the pathological effects of amyloid-β in AD. The 42aa amyloid-β protein is a hydrophobic peptide with an ominous tendency to assemble into long-lived polymers, and this excessive accumulation and deposition of amyloid-β is hypothesized to underlie the cascade of events that ultimately lead to cell death (Selkoe, 1991; Hardy and Higgins, 1992; Hardy and Selkoe, 2002). Early-onset AD is associated with mutations in the amyloid precursor protein (APP) gene, the presenilin 1 (PS1), and the presenilin 2 gene, which increases the production of the 42aa isoform of amyloid-β, an isoform more closely associated with the development of amyloid plaques than the shorter isoforms (Goate et al., 1991; Borchelt et al., 1996; Duff et al., 1996; Citron et al., 1997; Steiner et al., 1999). The strongest risk factor of late-onset sporadic AD is the ε4 allele of apolipoprotein E (a protein involved in the fat metabolism), which also significantly increases the burden of amyloid plaques in the brain (Verghese et al., 2011). Although familial AD cases represent only approximately 5% of AD cases, they have been critical for understanding the molecular mechanisms of AD, and importantly, similar mechanisms occur in sporadic AD.
Rodent models of AD have been extensively studied to examine neurological changes and to test therapeutic strategies that cannot directly be tested in humans. A frequently used single mutation AD model is the PDAPP model, expressing the AβPPV717F mutation more commonly known as the AβPPInd mutation (Games et al., 1995). To more accurately replicate the human AD pathology, many mouse AD models have multiple mutations. Double mutant models include the Tg2567 and APP23 mouse models which both express the AβPPSwe (AβPPK670N/M671L) mutation but with different promoters (Hsiao et al., 1996; Sturchler-Pierrat et al., 1997). Other multi-mutation models are the TgCRND8 and J20 mouse models expressing the AβPPSwe,Ind mutations, the APP/PS1 mice with the AβPPSwe/PS1M146L, AβPPSwe/PS1P264L, AβPPSwe/PS1L166Por the APPSwe/PS1ΔE9 mutation, the 5xFAD model with AβPPSwe,Lnd,Flo/PS1M146L,L286V mutation, and the 3xTg-AD triple transgenic mouse model with the AβPPSwe/TauP301L/PS1M146V mutations (Holcomb et al., 1998; Mucke et al., 2000; Chishti et al., 2001; Flood et al., 2002; Oddo et al., 2003; Oakley et al., 2006; Radde et al., 2006; Lindström, 2007; Hall and Roberson, 2012; Graybeal et al., 2015). Additional models include transgenic mice expressing the human APP (hAPP), as well as the SAMP8 mouse model that spontaneously develops AD (Mucke et al., 1996; Morley et al., 2000). Although each of these models displays some deficits associated with AD, the difficulty has been generating models where disease progression reaches stage 3 within the shorter lifespan of rodents. For this reason, double and triple knockout models that show faster rates of disease progression are often favored in the field.
While late-stage AD is characterized by profound neuronal loss, more subtle neuronal changes occur early in AD progression including synapse loss and circuit changes, which have been well correlated with cognitive impairments (Davies et al., 1987; Masliah et al., 1991; Terry et al., 1991; for a reviews see Selkoe, 2002; Palop et al., 2006; Palop and Mucke, 2010). Changes in synaptic function, plasticity, neuronal excitability, brain connectivity, and excitation/inhibition imbalance all have profound effects on circuit function, and are thought to exacerbate disease progression in AD. These circuit changes may be crucial targets for slowing or even preventing disease progression before widespread cellular loss has occurred. Changes in circuit function in AD lead to a high co-morbidity with epilepsy, even in the early stages of disease (Vossel et al., 2013; Subota et al., 2017; for reviews see Noebels, 2011; Vossel et al., 2017). Moreover, seizures worsen disease progression (Volicer et al., 1995) whereas anti-epileptic drugs improve memory impairments in individuals with mild cognitive impairment (Bakker et al., 2015).
Hippocampal Deficits in Amyloid-β-Related Alzheimer’s Disease
Behavioral Deficits
Memory impairments are a major feature of AD that are crucial to replicate in rodents to accurately model the disease. Spatial memory encodes information about ones environment and orientation, and is commonly impaired in AD patients (Henderson et al., 1989; Cherrier et al., 2001). A common behavioral paradigm examining spatial memory in mice is the Morris water maze (MWM). Using the MWM it has been demonstrated that mouse models of AD show impaired spatial memory performance (Chen et al., 2000; Cayzac et al., 2015; Rorabaugh et al., 2017; Bergin et al., 2018). In addition, APP mice require considerably more training to reach the same level of competency as the control mice (Zhao et al., 2014). Furthermore AD-associated behavioral deficits have been observed in other hippocampal-dependent tests such as the radial maze, T-maze, olfactory tubing maze, and in the novel object recognition test (Klevanski et al., 2015; Baranger et al., 2017; Biasibetti et al., 2017). Female 3xTg-AD mice perform worse in working memory tests than males which is correlated with a higher amyloid-β load, and is reflective of the higher prevalence of AD in women (Carroll et al., 2007, 2010). Similarly female APP/PS1/tau triple-transgenic mice perform worse than males in the MWM, which is correlated with increased amyloid-β and tau load (Yang et al., 2018). These studies and others demonstrate that mouse models of AD ubiquitously present hippocampal-dependent behavioral deficits (Figure 1), and also reflect the gender differences in AD prevalence observed in human patients.
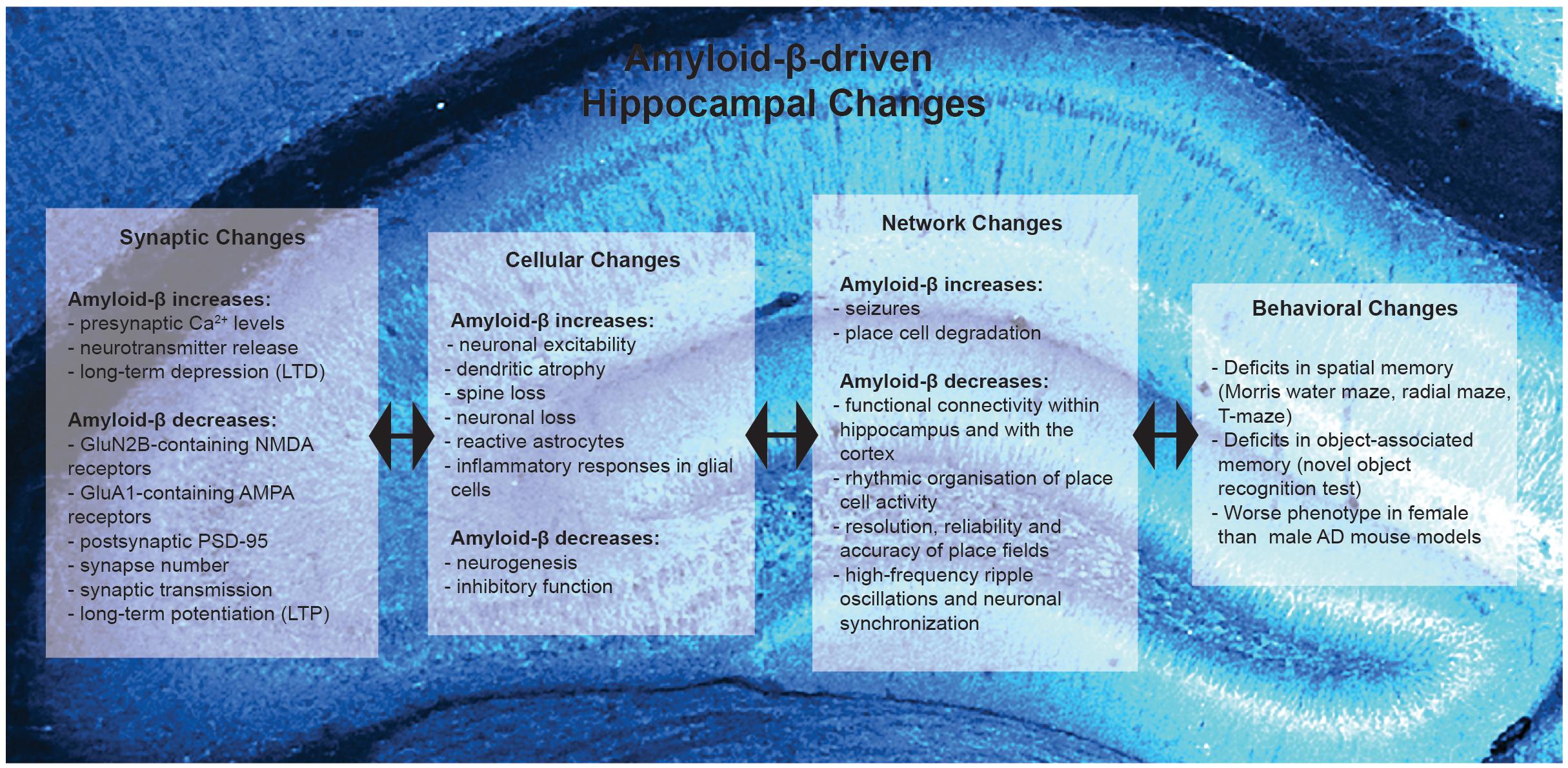
Figure 1. Summary of Amyloid-β-driven hippocampal changes in rodent models of Alzheimer’s disease. Amyloid-β drives changes at the synaptic, cellular, network and behavioral level.
Changes in Glutamatergic Synapse Function
Changes in synapse function are a vital aspect during the early stages of AD progression with amyloid-β playing a complex role as, in addition to its effects on synapses, amyloid-β levels are regulated by synaptic activity. Synapse loss is highly correlated with cognitive impairments in AD (Terry et al., 1991). Synapse loss is also correlated with amyloid-β burden (Terry et al., 1991), and many studies have focused on how amyloid-β influences presynaptic function, postsynaptic receptors and proteins, and consequently synapse function. However, neuronal and synaptic activity also influence the metabolism of amyloid-β (Kamenetz et al., 2003). Furthermore, the extracellular concentration of amyloid-β is critical in determining whether it will aggregate into toxic species (Kamenetz et al., 2003). Consequently, the areas of the brain with the highest basal rates of metabolic and neuronal activity develop the most amyloid-β plaques (Buckner et al., 2005). Synaptic activity rapidly regulates interstitial fluid amyloid-β levels in vivo and correlates with local amyloid-β burden (Cirrito et al., 2005). Extracellular amyloid-β levels have been linked to synaptic vesicle release, suggesting that the synaptic amyloid-β levels are regulated presynaptically (Cirrito et al., 2005). Amyloid-β evokes sustained increases in presynaptic Ca2+, and acts as a positive endogenous regulator of neurotransmitter vesicle release probability at hippocampal synapses (Abramov et al., 2009). These studies indicate that amyloid-β increases neurotransmitter release and the consequent hyperactivity further leads to more amyloid-β and its subsequent aggregation, resulting in a positive feedback loop (which has been proposed to be a major feature of AD; for a review see Doig, 2018). However, β-amyloid can also lead to depletion of presynaptic proteins involved in neurotransmitter release such as dynamin (Kelly et al., 2005; for a review see Honer, 2003).
Amyloid-β effects multiple postsynaptic proteins and there is evidence that correcting postsynaptic changes can improve impairments in mouse models of AD. More than 90% of synaptic oligomeric amyloid-β is colocalized in the postsynaptic density (Lacor et al., 2004). Amyloid pathology appears to progress in a neurotransmitter-specific manner with the glutamatergic and cholinergic terminals being the most vulnerable, whereas GABAergic terminals appear to be more resilient (for a review see Bell and Claudio Cuello, 2006). In early stages of AD, amyloid-β disrupts neuronal signaling via glutamatergic and acetylcholine receptors (Dougherty et al., 2003; Abramov et al., 2009). Amyloid-β regulates N-methyl-D-aspartate receptor (NMDAR) trafficking (Snyder et al., 2005) and oligomeric amyloid-β leads to a selective loss of GluN2B-containing NMDAR function (Kessels et al., 2013). Increases in the intracellular domain of APP (AICD), which occur in AD, affect NMDAR composition by increasing the prevalence of GluN2B containing receptors (Pousinha et al., 2017). Furthermore, increased ACID reduces excitability of CA1 neurons and impairs spatial memory (Pousinha et al., 2019). Amyloid-β induces NMDAR-dependent degradation of postsynaptic density 95 (PSD-95) at glutamatergic synapses (Roselli et al., 2005). In addition, accumulation of amyloid-β in APP mutant neurons reduces synaptic PSD-95 and GluA1 (Almeida et al., 2005). Interestingly, restoration of PSD-95 levels can rescue memory deficits in AbPPSwe/PS1 mice (Bustos et al., 2017). Therefore, amyloid-β also acts postsynaptically to reduce the expression of glutamatergic receptors and proteins, which is directly linked to cognitive impairments in AD.
These amyloid-β-induced pre and post-synaptic alterations consequently impair glutamatergic synaptic transmission in several mouse models of AD. Amyloid-β depresses synaptic transmission, and this was initially noted in APPInd mice which displayed severe impairments in synaptic transmission between hippocampal CA3 and CA1 cells (Hsia et al., 1999; Kamenetz et al., 2003). Additionally, the APP/PS1 model of AD, which overexpresses mutant human genes for APP and PS1, display deficits in synaptic transmission at a younger age than Tg4510 mice, which overexpress the mutant human Tau gene (Gelman et al., 2018). Therefore, amyloid-β plays a dominant role in causing synaptic deficits in the hippocampus, from the structural to the functional level (Figure 1).
In summary, amyloid-β increases presynaptic transmitter release but its postsynaptic negative effects override these leading to impaired synaptic function in AD. However, many of these studies examined the influence of amyloid-β using via external application in vitro. Thus, more in vivo studies are required to decipher the influence of intrinsically-released amyloid-β on synapse function, and to understand the temporal relation between AD-associated presynaptic and postsynaptic changes in the hippocampus. Exactly how these complex synaptic changes affect circuit function also remains somewhat a mystery. Nevertheless, disrupted synapse function could directly impact the ability of synapses to undergo synaptic plasticity, which in turn could underlie the memory deficits characteristic of AD.
Changes in Synaptic Plasticity
Synaptic plasticity has been well studied in AD as deficits in the ability of synapses to undergo changes in strength could be responsible for memory deficits. There is good consensus in the field that impaired synaptic strengthening is a key feature of AD as deficits in long-term potentiation (LTP) occur in many mouse models of AD (Nalbantoglu et al., 1997; Chapman et al., 1999; Gruart et al., 2008; Gengler et al., 2010; Klevanski et al., 2015; Gelman et al., 2018; Viana da Silva et al., 2019). Even transgenic mice expressing only the carboxy-terminal 104 amino acids of APP display deficits in the maintenance of LTP (Nalbantoglu et al., 1997). On the contrary, amyloid-β facilitates synapse weakening in the form of long-term depression (LTD) and depotentiation in vivo (Kim et al., 2001). Amyloid-β drives the loss of surface α-amino-3-hydroxy-5-methylisoxazole-4-proprionic acid receptors (AMPAR) by employing signaling pathways of LTD and can also lead to reduced synaptic NMDAR currents (Hsieh et al., 2006; Kessels et al., 2013). The synaptic depression and memory deficits induced by amyloid-β require the AMPAR subunit GluA3, as they are absent in GluA3 knockout mice (Reinders et al., 2016). This suggests that amyloid-β initiates removal of GluA3-containing AMPARs from synapses leading to synaptic and memory deficits (Reinders et al., 2016). Taken together, synaptic strengthening is impaired and synaptic depression is enhanced in AD (Figure 1) (for a review see Mucke and Selkoe, 2012).
Upon investigation of the mechanisms affecting synaptic plasticity in AD, several genes and pathways have been implicated. APP/PS1 mice showed reduced expression of synaptic plasticity genes such as Arc, Zif268, NR2B, GluR1, and Homer-1a (Dickey et al., 2003). In transgenic mice producing hAPP, dentate granule cells in particular were vulnerable to disruption of Arc expression as well as reductions in actin-binding protein α-actinin-2, which was tightly correlated with reductions in Fos and calbindin, shown previously to reflect learning deficits in these hAPP mice (Palop et al., 2005). Amyloid-β precursor protein (β-APP) fragments and amyloid-β oligomers impair LTP in vivo, and in hippocampal slices, this process is mediated via activation of several different kinases, such as c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5 (Cullen et al., 1997; Walsh et al., 2002; Wang et al., 2004; Klyubin et al., 2012). LTP deficits in APP/PS1 mice are also linked to disruption of the hippocampal pro-opiomelanocortin (POMC)/melanocortin 4 receptor (MC4R) circuit as the suppression of hippocampal MC4R activity exacerbated LTP impairments in these mice, and is alleviated by activation of the hippocampal MC4R-coupled Gs signaling and POMC/MC4R activity (Shen et al., 2016). MC4R activation also rescues amyloid-β-induced synaptic dysfunction thereby implicating the POMC/MC4R as a potential therapeutic target to rescue synaptic dysfunction in AD. Contextual fear conditioning deficits in aged 5XFAD mice is associated with different expression of hippocampal proteins than normal aging (Neuner et al., 2017). Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to AD-related cognitive deficits (Palop et al., 2003). Overall, these reflect amyloid-β’s multi-faceted disruption of processes involved in synaptic plasticity in AD.
Several studies have examined whether rescuing synaptic plasticity deficits in mouse models of AD can improve behavioral symptoms. Neuron-specific postnatal deficiency of PS1 prevented amyloid pathology and rescued LTP in AβPPInd mice but failed to prevent cognitive deficits observed in the object recognition test in these mice (Dewachter et al., 2002), suggesting that LTP deficits do not underlie all behavioral deficits in this model. Another approach of preventing LTP deficits in AD mice included activation of Wnt signaling as several studies have shown Wnt signaling activation to facilitate LTP in wildtype mice (Chen et al., 2006; Beaumont et al., 2007; Cerpa et al., 2011). Vargas et al. (2014) found that chronic activation of Wnt signaling enhanced basal excitatory synaptic transmission, facilitated LTP and improved episodic memory in APP/PS1 mice (Vargas et al., 2014). In attempts to rescue synaptic plasticity deficits in AD mice, Cissé et al. (2011) regulated NMDAR function using receptor tyrosine kinase EphB2 which phosphorylates NMDARs via Src-mediated tyrosine phosphorylation (Dalva et al., 2000; Henderson et al., 2001; Takasu et al., 2002; Chen et al., 2008; Cissé et al., 2011). The phosphorylation status of NMDAR subunits is correlated with cognitive performance, and levels of EphB2 and tyrosine-phosphorylated NMDARs are depleted in hAPP mice (Sze et al., 2001; Palop et al., 2005, 2007; Simón et al., 2009). Reversing EphB2 depletion in the dentate gyrus reversed LTP and memory impairments in hAPP mice (Cissé et al., 2011). Furthermore, neutralization of adenosine A2A receptors could restore associative CA3 LTP and revert memory deficits in APP/PS1 mice (Viana da Silva et al., 2016). Chronic intranasal administration of Colivelin (a novel and strong humanin derivative) reduced amyloid-β deposition in the hippocampus, rescued suppression of hippocampal LTP, and prevented AD-associated behavioral impairments in APP/PS1 mice (Wu et al., 2017). In addition, increasing levels of the secreted APPα (sAPPα, an alternative cleavage product of APP that has neuroprotective and neurotrophic properties) completely reversed deficits in LTP and spatial memory tasks in APPSwe/PS1ΔE9 mice (Tan et al., 2018). Therefore, it is important to keep in mind the protective roles of some biproducts of APP (for a review see Montagna et al., 2017), although the pathological biproducts of APP are often the focus in AD literature. Together these studies demonstrate potential for therapeutics that target LTP and its downstream pathways using a range of different methods to provide behavioral improvements. It is currently unknown whether these treatment strategies are applicable to idiopathic AD.
Neurogenesis
Neurogenesis in the adult hippocampus is a dynamic process that continuously changes the dentate gyrus, and is important for hippocampal plasticity, learning, and memory (Altman and Das, 1965; Eriksson et al., 1998; Aimone et al., 2011; Gu et al., 2012). Adult hippocampal neurogenesis consists of three main stages: proliferation, differentiation, and survival (Dard et al., 2019). Controversy exists in the literature as to whether hippocampal neurogenesis is increased or decreased in mouse models of AD (Rodríguez et al., 2008; Demars et al., 2010; Hamilton et al., 2010; Krezymon et al., 2013; for reviews see Wirths, 2017; Dard et al., 2019). However, altered neurogenesis must still provide some cognitive benefit as conditional ablation of adult neurogenesis in APPSwe/PS1ΔE9 mice worsened behavioral performance in contextual conditioning and pattern separation tasks (Hollands et al., 2017). Together these studies suggest that deficits in adult neurogenesis may contribute to the pathology of AD, and points toward the possibility of increasing neurogenesis or using neural stem cells transplantation as an approach for preventing AD-associated changes in neuronal circuitry. In support of this idea Richetin et al. (2017) demonstrated that amplification of mitochondrial function rescued adult neurogenesis in APP/PS1 mice, and overexpression of the pro-neuronal marker Neurod1 increased dendritic growth and spine formation, and consequently rescued spatial memory in these mice (Richetin et al., 2017). Furthermore, neural stem cell engrafts into APP/PS1 mice were able to restore memory and promote endogenous neurogenesis and synaptic remodeling in these mice (Zhang et al., 2017).
Changes in Neuronal Excitability, and Excitation/Inhibition Imbalance
AD-associated alterations have also been observed beyond the synapse, with hyperexcitability of hippocampal neurons observed both in vitro and in vivo. Hippocampal neurons show increased excitability in the 3xTg-AD model due to altered Kv2.1 potassium channel function (Frazzini et al., 2016). Similarly, neurons of aged SAMP8 mice are hyperexcitable and show altered voltage-dependent Ca2+ currents (Wang et al., 2017). In APP/PS1 mice hyperexcitability has been linked to structural degeneration of dendrites (Šišková et al., 2014). The dendritic structure is known to determine the electrical properties of neurons as it defines the input-to-output conversion, therefore, when dendritic integrity is impaired neuronal function is aberrant (Šišková et al., 2014). These in vitro demonstrations of hyperexcitability in AD were later confirmed in vivo in APP/PS1 mice using two-photon imaging in the hippocampus (Busche et al., 2012). Neuronal hyperactivity in the hippocampus in vivo was correlated with soluble amyloid-β levels (Busche et al., 2012). Together, these studies demonstrate that hippocampal hyperexcitability is a common feature of different mouse models of AD (for a review see Busche and Konnerth, 2015). In addition to alterations in neuronal excitability, mouse models of AD demonstrate deficits in γ-aminobutyric acid (GABA) pathways and altered excitation/inhibition balance leading to seizures. In APP/PS1 mice deficits in the GABAergic pathway and feed forward inhibition are age-dependent (Oyelami et al., 2016; Viana da Silva et al., 2019). In hAPP mice parvalbumin interneuron dysfunction and reduced levels of voltage-gated sodium channel subunit Nav1.1 have also been linked to abnormal oscillatory rhythms, network synchrony and cognitive function (Verret et al., 2012). Furthermore, APP/PS1 mice show somatostatin-positive interneuron axon loss, enhanced spine turnover, and impaired learning–dependent spine gain in association with memory deficits in these mice (Schmid et al., 2016). Similarly, soluble amyloid-β oligomers increase neuronal excitability by disrupting glutamatergic/GABAergic balance in the hippocampus, and this could be prevented by increasing GABA tone or partially blocking NMDAR activity (Lei et al., 2016). Moreover, APP/PS1 mice are also susceptible to seizures, the frequency of which is correlated with the load of amyloid-β plaques (Minkeviciene et al., 2009; Busche et al., 2012; Reyes-Marin and Nuñez, 2017). Seizure activity appears to trigger compensatory mechanisms in the dentate gyrus of hAPP mice as enhanced synaptic inhibition and GABAergic sprouting have been observed (Palop et al., 2007). Furthermore, synaptic and cognitive deficits in hAPP and APP23 mice are reversed by antiepileptic drugs which suppress neuronal network dysfunction (Bromley-Brits et al., 2011; Sanchez et al., 2012). Together these data show that deficits in inhibition leading to overexcitation and seizures is commonly seen in mouse models of AD and contributes to our understanding of epilepsy co-morbidity in AD patients.
Overall these studies show that inhibition is reduced in AD, which combined with hyperactive excitatory neurons massively shifts the ratio toward excess excitation leading to seizures (Figure 1), which negatively impact cognition. Increased excitability may eventually promote the excitotoxic damage observed in the AD brain. Therefore, restoration of excitation/inhibition balance may hold therapeutic potential in AD. The relation between the synaptic and plasticity changes to hyperexcitable networks seems counterintuitive as weaker synapses, impaired strengthening, and enhanced depression should lead to reduced network activity. However, changes in dendritic structure and activity levels can increase excitability of neurons leading to action potentials being triggered by fewer inputs. Other homeostatic changes, such as inhibitory alterations, that aim to restore activity levels may overcompensate and fail to restore balance.
Astrocytic Changes
Alterations in glial function have also been observed in AD, and growing evidence shows that glial changes may precede neuronal changes and behavioral impairment noted in the progression of AD (Heneka et al., 2010; for a review see De Strooper and Karran, 2016). Astrogliosis is a universal feature of AD brains (Nagele et al., 2004; Rodríguez et al., 2009; Heneka et al., 2010, 2013). Inflammatory responses in glial cells contribute to the pathogenesis of AD, and several studies have highlighted specific therapeutic targets for the treatment of AD, such as targeting the inflammasome NLRP3 or RIPK1, an enzyme abundantly expressed in microglia (Heneka et al., 2013; Ofengeim et al., 2017). Pathological astroglial changes have been shown to be prevented by environmental enrichment in PDAPP-J20 transgenic mice (Beauquis et al., 2013). Additionally, it was identified that gamma frequency entrainment could recruit both glial and neuronal responses to attenuate AD-associated pathology (Iaccarino et al., 2016). Reactive astrocytes likely play a role in clearing damaged synapses and dendrites, however, they are limited in their ability to fully clear away debris (Gomez-Arboledas et al., 2018). The role of astrocytes in synaptic plasticity is also affected in AD (for a review see Singh and Abraham, 2017). Therefore, preventing glial pathology may represent a new therapeutic intervention for AD, and preventing abhorrent glial changes can be achieved by altering network activity, either naturally by changing the environment or artificially by stimulation.
Changes in Brain Connectivity and Circuit Function
Brain connectivity and circuit function are disrupted in AD, in part due to synaptic and neuronal loss (Figure 1). At the synaptic level, amyloid-β induced LTD results in loss of dendritic spines (Hsieh et al., 2006; Wei et al., 2010). Amyloid-β-induced synapse loss and dendritic spine abnormalities have been noted by other studies in several mouse models of AD, such as the APP mice, APP/PS1 mice, PDAPP, and Tg2576 mice (Lanz et al., 2003; Spires et al., 2005; Shankar et al., 2007; Knafo et al., 2009). In hippocampal slice cultures from APPSDL mice, spine loss was accompanied by changes in spine shape from mushroom to stubby spines (Tackenberg and Brandt, 2009; for a review see Tackenberg et al., 2009). Use of adeno-associated virus to express oligomeric amyloid-β in the hippocampus also resulted in spine loss (Forner et al., 2019). Interestingly, extracellular amyloid-β lead to a greater reduction in stubby spines than intracellular overexpression, while other spine types were equally affected (Forner et al., 2019). Amyloid-β pathology also results in dendritic abnormalities and atrophy. High-resolution confocal microscopy has revealed that, in the PSAPP mouse model of AD, dendrites passing within 40 μm of amyloid deposits displayed loss of dendritic spines, shaft atrophy, varicosity formation, and sprouting (Tsai et al., 2004; Grutzendler et al., 2007). Similarly, post-mortem human brains from AD patients also display similar dendritic alterations (Merino-Serrais et al., 2013), further emphasizing that amyloid deposits and their surroundings microenvironments are toxic to dendrites.
The hippocampal CA1 subregion is particularly more susceptible to AD-associated atrophy in comparison to CA2 or CA3 subregions (West et al., 2000; Frisoni et al., 2008; Apostolova et al., 2010). Selective neuronal death in brain regions most affected by AD has also been demonstrated in APP mice, and this was directly correlated with amyloid plaque formation (Calhoun et al., 1998). As a result, mouse models of AD demonstrate decreased functional connectivity within the hippocampus as well as the cortex, as examined by resting state fMRI and optical intrinsic signal imaging technique, respectively (Bero et al., 2012; Shah et al., 2013). Furthermore, functional coupling between the hippocampus CA1 region and medial frontal cortex is also impaired in mouse models of AD (Zhurakovskaya et al., 2019).
Spatial memory deficits in AD mice are attributed to changes in circuit function due to altered cellular responses in the hippocampus. At a cellular level, place cells play a critical role in spatial memory and these have been shown to be affected in AD. Place fields from control mice become spatially restricted and stable after repeated exposures of a new environment; however, APP mice produce a spatial code of lower resolution, reliability and accuracy (Zhao et al., 2014). Furthermore, hippocampal place cell degradation and MWM training deficits correlate with amyloid-β plaque burden, respectively in Tg2576 and PDAPP mouse models of AD (Chen et al., 2000; Cacucci et al., 2008). A lack of learning dependent changes in place cells in APP-PS1 mice has been correlated with impaired action-reward association tasks in a spatially defined environment (Cayzac et al., 2015). Impairments in rhythmic organization of place cell activity have also been observed in the 3xTg mouse model of AD, and may contribute to the unstable spatial representation and spatial memory deficits (Mably et al., 2017). Furthermore, in young rTg4510 mice high-frequency ripple oscillations and neuronal synchronization are reduced even though place fields of hippocampal CA1 cells are largely normal (Ciupek et al., 2015). Impaired cellular and network activity in the hippocampus therefore appear to contribute to spatial memory deficits in mouse models of AD.
Alterations in networks in other brain regions which are connected to the hippocampus are also observed in AD. For example cortical principle cells become hyperexcitable at the early stages of amyloid pathology, and via the thalamo-cortical pathway, drive thalamic cells too (Gurevicius et al., 2013; Busche et al., 2015). This precedes hippocampal electrophysiological abnormalities, and is hypothesized to underlie the network reorganization which leads to epileptic seizures (Palop et al., 2007; Minkeviciene et al., 2009).
Treatment Strategies in Amyloid-β-Related AD
The prevention of behavioral deficits in AD mice has been studied extensively with a variety of different approaches: For example: Learning and age-related memory deficits can be prevented in APP/PS1, TgCRND8, Tg2576, and PDAPP mice with immunization against the amyloid-β peptide (Janus et al., 2000; Morgan et al., 2000; Dodart et al., 2002; Kotilinek et al., 2002). Such immunizations reduce pathological changes including plaque formation in PDAPP mice (Schenk et al., 1999). In 3xTg-AD mice, immunizations against amyloid-β have also been shown to act at the synaptic level by reducing synaptic impairments (Baglietto-Vargas et al., 2018). In this same AD mouse model, accumulation of intraneuronal amyloid-β precedes plaque and tangle pathology. Using immunotherapy to clear intraneuronal amyloid-β pathology rescued the early cognitive deficits seen in the MWM (Billings et al., 2005). Re-emergence of the amyloid-β pathology could again lead to cognitive deficits, implicating intraneuronal amyloid-β in the onset of cognitive dysfunction (Billings et al., 2005).
Despite billions of dollars being invested into drug development for AD, over 100 compounds have failed in clinical trials (Mehta et al., 2017). These potential disease-modifying drugs fall into four categories: monoclonal antibodies, gamma secretase inhibitors, tau aggregation inhibitors, and symptomatic treatments. Some examples of previously failed clinical trials include (i) bapineuzumab, one of the first monoclonal amyloid-β antibodies to reach phase 3 clinical trials, but unfortunately was found to have no significant clinical benefit (Salloway et al., 2014), (ii) solanezumab, which despite demonstrating an excellent safety profile and low incidence of vasogenic edema, failed to meet primary and secondary endpoints in the phase2B-3A study (Doody et al., 2014; Siemers et al., 2016), (iii) crenezumab did not show a significant benefit in treatment in comparison to placebo in a phase 2 trial (Miller, 2012), and (iv) gantenerumab did not meet a significant clinical efficacy endpoint in phase 3 trials at its administered dosage (Ostrowitzki et al., 2017). However, more recently, Aducanumab, a human monoclonal antibody that is selective for aggregated forms of amyloid-β has been examined as a potential treatment for amyloid-β-associated pathologies. In vivo multiphoton imaging of calcium homeostasis in aged Tg2576 mice demonstrated that acute topical application of aducanumab to the brain resulted in clearance of amyloid plaques, and chronic systemic administration ameliorated calcium overload and restoring intracellular calcium to control levels (Kastanenka et al., 2016). Aducanumab also restored NMDAR GluN1 and GluN2A subunit-expressing cell numbers to wildtype levels, thus indicating a potential restoration of neuronal network function and cognitive function in these mice. Phase I clinical trials using Aducanumab demonstrated an acceptable safety and tolerability profile of the drug, and it was shown to reduce amyloid deposition in the brain in a dose- and time-dependent manner (Ferrero et al., 2016; Sevigny et al., 2016). Phase III clinical trials were performed in 3200 individuals across 20 countries, but early analyses showed no promising effects of Aducanumab in decreasing amyloid burden or improving symptomology in patients and thus the study has been halted (Selkoe, 2019). However, longitudinal studies are required to investigate any potential long-term benefits of antibodies against amyloid-β.
Genetic, social, environmental, and pharmacological approaches have also been used to prevent behavioral deficits in AD models. For example, development of memory deficits was prevented in APPSwe/PS1ΔE9 mice by constitutive deletion of the amyloid-β-binding cellular prion protein (Gimbel et al., 2010). Conditional deletion of PrPc at 12 or 16 months of age completely rescued MWM deficits, novel object recognition, and passive avoidance test in APPSwe/PS1ΔE9 mice, together with reversal of hippocampal synapse loss (Salazar et al., 2017). Memory deficits in APP/PS1 mice could be rescued by social interaction, and this effect was linked to increased levels of BDNF in the hippocampus (Hsiao et al., 2014). In addition, environmental enrichment led to reduced amyloid-β levels and amyloid deposition in APPSwe/PS1ΔE9 (Lazarov et al., 2005). Environmental enrichment also changes the function of microglia in a way that prevents their inflammatory response to human soluble amyloid-β oligomers (Xu et al., 2016). Recently it was demonstrated that environmental enrichment and voluntary exercise revives adult neurogenesis, reverses AD-associated memory deficits, and prevents amyloid-β seeding (representing early stages of plaque formation) via activated phagocytic microglia cells (Ziegler-Waldkirch et al., 2018). Therefore, prolonged environmental enrichment could protect against AD by regulating the brain’s innate immune system. 5xFAD mice displayed improved cognitive abilities, decreased amyloid plaque and neuroinflammation in the entorhinal cortex after treatment with RS67333, a partial 5-HT4R agonist, for 4 months (Baranger et al., 2017). Genetic reduction of tau expression has also been shown to prevent behavioral impairments and neuronal deficits (Roberson et al., 2007; Vossel et al., 2010). Similarly, expression of truncated versions of tau that lack dendritic localization has beneficial effects in APPswe transgenic mice (Ittner et al., 2010), fitting with evidence that shows the amyloid-β-induced mis-localization of endogeneous tau into dendrites is detrimental (Zempel et al., 2010). These are just a few of the treatment strategies that have shown promise in animal trials, however, there are not yet many that have translated well in human trials. However manipulating circuit function still holds promise for future treatments (for a review see Canter et al., 2016).
Future Directions in Understanding Circuit Function in Amyloid-β-Related Alzheimer’s Disease
Given the complex multifaceted nature of the identified issues in AD it is becoming increasingly important to understand changes in brain networks in vivo. Examining circuit dynamics during behavior will give the next breakthroughs in our understanding. To date there have been several studies that have taken advantage of in vivo two-photon imaging to better understand circuit changes in the hippocampus and other brain regions. For example in vivo studies that have examined hyperactivity near plaques in both hippocampus and cortex (Busche et al., 2008, 2012) have identified significant heterogeneity in cell responses within the same brain region. In the visual cortex neuronal hyperactivity has been shown to affect function (Grienberger et al., 2012). Hyperactive neurons exhibited poor orientation tuning, which was correlated with impaired performance in visual-pattern discrimination (Grienberger et al., 2012). Furthermore, visual experience driven-expression of Arc is impaired in AD mice, providing further in vivo evidence of altered memory processes (Rudinskiy et al., 2012). Astrocytes in the cortex have also shown synchronous hyperactivity and intercellular calcium waves in APP/PS1 mice (Kuchibhotla et al., 2009). In future studies it will be crucial to understand the link between aberrant neuronal and glial activity in vivo in AD mice.
Examining in vivo dynamics of axons and dendrites longitudinally with disease progression and/or treatment also offer promise for understanding complex changes in AD models. For example, long-term imaging revealed how axon pathology proceeds around amyloid-β plaques in APP-PS1 mice (Blazquez-Llorca et al., 2017). Axons near plaques appeared swollen before becoming disconnected, over a time course of several months (Blazquez-Llorca et al., 2017). In addition, instability of dendritic spines and axonal boutons near plaques was revealed in this way and could be prevented by γ-secretase inhibitor treatment (Liebscher et al., 2014). Further studies are needed to reveal not just structural but also functional changes in dendrites and axons using calcium and voltage imaging in vivo. Furthermore in vivo imaging in freely moving animals using miniaturized microscopes is another exciting possibility for future studies (for a review see Werner et al., 2019).
Activation of specific subsets of neurons using channel rhodopsin is also an important approach to understand circuit changes in AD. By utilizing learning-dependent expression of channel rhodopsin it is possible to label memory engram cells (Ryan et al., 2015). It is then possible to re-activate these cells ontogenetically and trigger memory retrieval (Ryan et al., 2015). This approach has been used to restore fear memory in young AD mice (Roy et al., 2016). More studies are required to increase our understanding of the specific pathways involved in memory deficits in AD so that treatments can be targeted to the right networks at the right time in disease progression.
In conclusion, hippocampal deficits in synapse and neuronal function manifest into behavioral abnormalities in mouse models of AD. However, more research and consensus in the field are required to completely understand hippocampal deficits in AD. With the increasing development of in vivo recording techniques in awake and freely moving animals, future studies will extend our current knowledge about how hippocampal function is altered in AD by combining network imaging with behavior. It will be crucial to identify network changes early and treat them before pathology becomes widespread. However, because network changes likely contribute to disease progression this could lead to future treatments that prevent AD symptoms from worsening.
Author Contributions
YV, JM, and JC wrote the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abramov, E., Dolev, I., Fogel, H., Ciccotosto, G. D., Ruff, E., and Slutsky, I. (2009). Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 12, 1567–1576. doi: 10.1038/nn.2433
Aimone, J. B., Deng, W., and Gage, F. H. (2011). Resolving new memories: a critical look at the dentate gyrus, adult neurogenesis, and pattern separation. Neuron 70, 589–596. doi: 10.1016/j.neuron.2011.05.010
Almeida, C. G., Tampellini, D., Takahashi, R. H., Greengard, P., Lin, M. T., Snyder, E. M., et al. (2005). Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 20, 187–198. doi: 10.1016/j.nbd.2005.02.008
Altman, J., and Das, G. D. (1965). Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 124, 319–335. doi: 10.1002/cne.901240303
Alzheimer’s Disease International, (2018). World Alzheimer Reports | Alzheimer’s Disease International. Available at: https://www.alz.co.uk/research/world-report (accessed October 30, 2019).
Apostolova, L. G., Mosconi, L., Thompson, P. M., Green, A. E., Hwang, K. S., Ramirez, A., et al. (2010). Subregional hippocampal atrophy predicts Alzheimer’s dementia in the cognitively normal. Neurobiol. Aging 31, 1077–1088. doi: 10.1016/j.neurobiolaging.2008.08.008
Baglietto-Vargas, D., Prieto, G. A., Limon, A., Forner, S., Rodriguez-Ortiz, C. J., Ikemura, K., et al. (2018). Impaired AMPA signaling and cytoskeletal alterations induce early synaptic dysfunction in a mouse model of Alzheimer’s disease. Aging Cell 17:e12791. doi: 10.1111/acel.12791
Bakker, A., Albert, M. S., Krauss, G., Speck, C. L., and Gallagher, M. (2015). Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. Neuroimage Clin. 7, 688–698. doi: 10.1016/j.nicl.2015.02.009
Baranger, K., Giannoni, P., Girard, S. D., Girot, S., Gaven, F., Stephan, D., et al. (2017). Chronic treatments with a 5-HT4 receptor agonist decrease amyloid pathology in the entorhinal cortex and learning and memory deficits in the 5xFAD mouse model of Alzheimer’s disease. Neuropharmacology 126, 128–141. doi: 10.1016/j.neuropharm.2017.08.031
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804.
Beaumont, V., Thompson, S.-A., Choudhry, F., Nuthall, H., Glantschnig, H., Lipfert, L., et al. (2007). Evidence for an enhancement of excitatory transmission in adult CNS by Wnt signaling pathway modulation. Mol. Cell. Neurosci. 35, 513–524. doi: 10.1016/j.mcn.2007.03.004
Beauquis, J., Pavía, P., Pomilio, C., Vinuesa, A., Podlutskaya, N., Galvan, V., et al. (2013). Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer’s disease. Exp. Neurol. 239, 28–37. doi: 10.1016/j.expneurol.2012.09.009
Bell, K. F. S., and Claudio Cuello, A. (2006). Altered synaptic function in Alzheimer’s disease. Eur. J. Pharmacol. 545, 11–21. doi: 10.1016/j.ejphar.2006.06.045
Bergin, D. H., Jing, Y., Mockett, B. G., Zhang, H., Abraham, W. C., and Liu, P. (2018). Altered plasma arginine metabolome precedes behavioural and brain arginine metabolomic profile changes in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease. Transl. Psychiatry 8:108.
Bero, A. W., Bauer, A. Q., Stewart, F. R., White, B. R., Cirrito, J. R., Raichle, M. E., et al. (2012). Bidirectional relationship between functional connectivity and amyloid-β deposition in mouse brain. J. Neurosci. 32, 4334–4340. doi: 10.1523/jneurosci.5845-11.2012
Bertram, L., Lill, C. M., and Tanzi, R. E. (2010). The genetics of Alzheimer disease: back to the future. Neuron 68, 270–281. doi: 10.1016/j.neuron.2010.10.013
Biasibetti, R., Almeida dos Santos, J. P., Rodrigues, L., Wartchow, K. M., Suardi, L. Z., Nardin, P., et al. (2017). Hippocampal changes in STZ-model of Alzheimer’s disease are dependent on sex. Behav. Brain Res. 316, 205–214. doi: 10.1016/j.bbr.2016.08.057
Billings, L. M., Oddo, S., Green, K. N., McGaugh, J. L., and LaFerla, F. M. (2005). Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688. doi: 10.1016/j.neuron.2005.01.040
Blazquez-Llorca, L., Valero-Freitag, S., Rodrigues, E. F., Merchán-Pérez, Á., Rodríguez, J. R., Dorostkar, M. M., et al. (2017). High plasticity of axonal pathology in Alzheimer’s disease mouse models. Acta Neuropathol. Commun. 5:14.
Blessed, G., Tomlinson, B. E., and Roth, M. (1968). The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br. J. Psychiatry 114, 797–811. doi: 10.1192/bjp.114.512.797
Borchelt, D. R., Eckman, C. B., Thinakaran, G., Lee, M. K., Kim, G., Yager, D., et al. (1996). Familial Alzheimer’s disease–linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 17, 1005–1013. doi: 10.1016/s0896-6273(00)80230-5
Bromley-Brits, K., Deng, Y., and Song, W. (2011). Morris water maze test for learning and memory deficits in Alzheimer’s disease model mice. J. Vis. Exp. 53:2920. doi: 10.3791/2920
Buckner, R. L., Snyder, A. Z., Shannon, B. J., LaRossa, G., Sachs, R., Fotenos, A. F., et al. (2005). Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J. Neurosci. 25, 7709–7717. doi: 10.1523/jneurosci.2177-05.2005
Busche, M. A., Chen, X., Henning, H. A., Reichwald, J., Staufenbiel, M., Sakmann, B., et al. (2012). Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 109, 8740–8745. doi: 10.1073/pnas.1206171109
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K.-H., Haass, C., et al. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689. doi: 10.1126/science.1162844
Busche, M. A., Kekuš, M., Adelsberger, H., Noda, T., Förstl, H., Nelken, I., et al. (2015). Rescue of long-range circuit dysfunction in Alzheimer’s disease models. Nat. Neurosci. 18, 1623–1630. doi: 10.1038/nn.4137
Busche, M. A., and Konnerth, A. (2015). Neuronal hyperactivity–A key defect in Alzheimer’s disease? Bioessays 37, 624–632. doi: 10.1002/bies.201500004
Bustos, F. J., Ampuero, E., Jury, N., Aguilar, R., Falahi, F., Toledo, J., et al. (2017). Epigenetic editing of the Dlg4/PSD95 gene improves cognition in aged and Alzheimer’s disease mice. Brain J. Neurol. 140, 3252–3268. doi: 10.1093/brain/awx272
Cacucci, F., Yi, M., Wills, T. J., Chapman, P., and O’Keefe, J. (2008). Place cell firing correlates with memory deficits and amyloid plaque burden in Tg2576 Alzheimer mouse model. Proc. Natl. Acad. Sci. U.S.A. 105, 7863–7868. doi: 10.1073/pnas.0802908105
Calhoun, M. E., Wiederhold, K.-H., Abramowski, D., Phinney, A. L., Probst, A., Sturchler-Pierrat, C., et al. (1998). Neuron loss in APP transgenic mice. Nature 395, 755–756. doi: 10.1038/27351
Canter, R. G., Penney, J., and Tsai, L.-H. (2016). The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 539, 187–196. doi: 10.1038/nature20412
Carroll, J. C., Rosario, E. R., Chang, L., Stanczyk, F. Z., Oddo, S., LaFerla, F. M., et al. (2007). Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J. Neurosci. 27, 13357–13365. doi: 10.1523/jneurosci.2718-07.2007
Carroll, J. C., Rosario, E. R., Kreimer, S., Villamagna, A., Gentzschein, E., Stanczyk, F. Z., et al. (2010). Sex differences in β-amyloid accumulation in 3xTg-AD mice: role of neonatal sex steroid hormone exposure. Brain Res. 1366, 233–245. doi: 10.1016/j.brainres.2010.10.009
Cayzac, S., Mons, N., Ginguay, A., Allinquant, B., Jeantet, Y., and Cho, Y. H. (2015). Altered hippocampal information coding and network synchrony in APP-PS1 mice. Neurobiol. Aging 36, 3200–3213. doi: 10.1016/j.neurobiolaging.2015.08.023
Cerpa, W., Gambrill, A., Inestrosa, N. C., and Barria, A. (2011). Regulation of NMDA-Receptor Synaptic Transmission by Wnt Signaling. J. Neurosci. 31, 9466–9471. doi: 10.1523/JNEUROSCI.6311-10.2011
Chapman, P. F., White, G. L., Jones, M. W., Cooper-Blacketer, D., Marshall, V. J., Irizarry, M., et al. (1999). Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 2, 271–276. doi: 10.1038/6374
Chen, G., Chen, K. S., Knox, J., Inglis, J., Bernard, A., Martin, S. J., et al. (2000). A learning deficit related to age and β-amyloid plaques in a mouse model of Alzheimer’s disease. Nature 408, 975–979. doi: 10.1038/35050103
Chen, J., Park, C. S., and Tang, S.-J. (2006). Activity-dependent synaptic wnt release regulates hippocampal long term potentiation. J. Biol. Chem. 281, 11910–11916. doi: 10.1074/jbc.m511920200
Chen, Y., Fu, A. K. Y., and Ip, N. Y. (2008). Bidirectional signaling of ErbB and Eph receptors at synapses. Neuron Glia Biol. 4, 211–221. doi: 10.1017/S1740925X09990287
Cherrier, M. M., Mendez, M., and Perryman, K. (2001). Route learning performance in Alzheimer disease patients. Cogn. Behav. Neurol. 14, 159–168.
Chishti, M. A., Yang, D. S., Janus, C., Phinney, A. L., Horne, P., Pearson, J., et al. (2001). Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570. doi: 10.1074/jbc.m100710200
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Cissé, M., Halabisky, B., Harris, J., Devidze, N., Dubal, D. B., Sun, B., et al. (2011). Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52. doi: 10.1038/nature09635
Citron, M., Westaway, D., Xia, W., Carlson, G., Diehl, T., Levesque, G., et al. (1997). Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat. Med. 3, 67–72. doi: 10.1038/nm0197-67
Ciupek, S. M., Cheng, J., Ali, Y. O., Lu, H.-C., and Ji, D. (2015). Progressive functional impairments of hippocampal neurons in a tauopathy mouse model. J. Neurosci. 35, 8118–8131. doi: 10.1523/JNEUROSCI.3130-14.2015
Cullen, W. K., Suh, Y. H., Anwyl, R., and Rowan, M. J. (1997). Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport 8, 3213–3217. doi: 10.1097/00001756-199710200-00006
Dalva, M. B., Takasu, M. A., Lin, M. Z., Shamah, S. M., Hu, L., Gale, N. W., et al. (2000). EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 103, 945–956. doi: 10.1016/s0092-8674(00)00197-5
Dard, R. F., Dahan, L., and Rampon, C. (2019). Targeting hippocampal adult neurogenesis using transcription factors to reduce Alzheimer’s disease-associated memory impairments. Hippocampus 29, 579–586. doi: 10.1002/hipo.23052
Davies, C. A., Mann, D. M. A., Sumpter, P. Q., and Yates, P. O. (1987). A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J. Neurol. Sci. 78, 151–164. doi: 10.1016/0022-510x(87)90057-8
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615.
Demars, M., Hu, Y.-S., Gadadhar, A., and Lazarov, O. (2010). Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J. Neurosci. Res. 88, 2103–2117. doi: 10.1002/jnr.22387
Dewachter, I., Reversé, D., Caluwaerts, N., Ris, L., Kuipéri, C., Van den Haute, C., et al. (2002). Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J. Neurosci. 22, 3445–3453. doi: 10.1523/jneurosci.22-09-03445.2002
Dickey, C. A., Loring, J. F., Montgomery, J., Gordon, M. N., Eastman, P. S., and Morgan, D. (2003). Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein + presenilin-1 transgenic mice. J. Neurosci. 23, 5219–5226. doi: 10.1523/jneurosci.23-12-05219.2003
Dodart, J.-C., Bales, K. R., Gannon, K. S., Greene, S. J., DeMattos, R. B., Mathis, C., et al. (2002). Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat. Neurosci. 5, 452–457. doi: 10.1038/nn842
Doig, A. J. (2018). Positive feedback loops in Alzheimer’s disease: the Alzheimer’s feedback hypothesis. J. Alzheimers. Dis. 66, 25–36. doi: 10.3233/jad-180583
Doody, R. S., Thomas, R. G., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2014). Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 311–321. doi: 10.1056/NEJMoa1312889
Dougherty, J. J., Wu, J., and Nichols, R. A. (2003). Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J. Neurosci. 23, 6740–6747. doi: 10.1523/jneurosci.23-17-06740.2003
Duff, K., Eckman, C., Zehr, C., Yu, X., Prada, C.-M., Perez-tur, J., et al. (1996). Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713. doi: 10.1038/383710a0
Eriksson, P. S., Perfilieva, E., Björk-Eriksson, T., Alborn, A.-M., Nordborg, C., Peterson, D. A., et al. (1998). Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317.
Ferrero, J., Williams, L., Stella, H., Leitermann, K., Mikulskis, A., O’Gorman, J., et al. (2016). First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement. 2, 169–176. doi: 10.1016/j.trci.2016.06.002
Flood, D. G., Reaume, A. G., Dorfman, K. S., Lin, Y.-G., Lang, D. M., Trusko, S. P., et al. (2002). FAD mutant PS-1 gene-targeted mice: increased Aβ42 and Aβ deposition without APP overproduction. Neurobiol. Aging 23, 335–348. doi: 10.1016/s0197-4580(01)00330-x
Forner, S., Martini, A. C., Aleph Prieto, A. C., Dang, C. T., Rodriguez-Ortiz, C. J., Reyes-Ruiz, J. M., et al. (2019). Intra- and extracellular β-amyloid overexpression via adeno-associated virus-mediated gene transfer impairs memory and synaptic plasticity in the hippocampus. Sci. Rep. 9:15936.
Förstl, H., and Kurz, A. (1999). Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 249, 288–290.
Fox, N. C., Warrington, E. K., Freeborough, P. A., Hartikainen, P., Kennedy, A. M., Stevens, J. M., et al. (1996). Presymptomatic hippocampal atrophy in Alzheimer’s diseaseA longitudinal MRI study. Brain 119, 2001–2007. doi: 10.1093/brain/119.6.2001
Frazzini, V., Guarnieri, S., Bomba, M., Navarra, R., Morabito, C., Mariggiò, M. A., et al. (2016). Altered Kv2.1 functioning promotes increased excitability in hippocampal neurons of an Alzheimer’s disease mouse model. Cell Death Dis. 7:e2100. doi: 10.1038/cddis.2016.18
Frisoni, G. B., Ganzola, R., Canu, E., Rüb, U., Pizzini, F. B., Alessandrini, F., et al. (2008). Mapping local hippocampal changes in Alzheimer’s disease and normal ageing with MRI at 3 Tesla. Brain 131, 3266–3276. doi: 10.1093/brain/awn280
Games, D., Adams, D., Alessandrini, R., Barbour, R., Berthelette, P., Blackwell, C., et al. (1995). Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373, 523–527. doi: 10.1038/373523a0
Gelman, S., Palma, J., Tombaugh, G., and Ghavami, A. (2018). Differences in synaptic dysfunction between rTg4510 and APP/PS1 mouse models of Alzheimer’s disease. J. Alzheimers Dis. 61, 195–208. doi: 10.3233/jad-170457
Gengler, S., Hamilton, A., and Hölscher, C. (2010). Synaptic plasticity in the hippocampus of a APP/PS1 mouse model of Alzheimer’s disease is impaired in old but not young mice. PLoS One 5:e9764. doi: 10.1371/journal.pone.0009764
Gimbel, D. A., Nygaard, H. B., Coffey, E. E., Gunther, E. C., Laurén, J., Gimbel, Z. A., et al. (2010). Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010
Goate, A., Chartier-Harlin, M. C., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706.
Gomez-Arboledas, A., Davila, J. C., Sanchez-Mejias, E., Navarro, V., Nuñez-Diaz, C., Sanchez-Varo, R., et al. (2018). Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 66, 637–653. doi: 10.1002/glia.23270
Graybeal, J. J., Bozzelli, P. L., Graybeal, L. L., Groeber, C. M., McKnight, P. E., Cox, D. N., et al. (2015). Human ApoE ε4 alters circadian rhythm activity, IL-1β, and GFAP in CRND8 Mice. J. Alzheimers Dis. 43, 823–834. doi: 10.3233/JAD-132009
Grienberger, C., Rochefort, N. L., Adelsberger, H., Henning, H. A., Hill, D. N., Reichwald, J., et al. (2012). Staged decline of neuronal function in vivo in an animal model of Alzheimer’s disease. Nat. Commun. 3:774. doi: 10.1038/ncomms1783
Gruart, A., López-Ramos, J. C., Muñoz, M. D., and Delgado-García, J. M. (2008). Aged wild-type and APP, PS1, and APP + PS1 mice present similar deficits in associative learning and synaptic plasticity independent of amyloid load. Neurobiol. Dis. 30, 439–450. doi: 10.1016/j.nbd.2008.03.001
Grutzendler, J., Helmin, K., Tsai, J., and Gan, W.-B. (2007). Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1097, 30–39. doi: 10.1196/annals.1379.003
Gu, Y., Arruda-Carvalho, M., Wang, J., Janoschka, S. R., Josselyn, S. A., Frankland, P. W., et al. (2012). Optical controlling reveals time-dependent roles for adult-born dentate granule cells. Nat. Neurosci. 15, 1700–1706. doi: 10.1038/nn.3260
Gurevicius, K., Lipponen, A., and Tanila, H. (2013). Increased cortical and thalamic excitability in freely moving APPswe/PS1dE9 mice modeling epileptic activity associated with Alzheimer’s disease. Cereb. Cortex 1991, 1148–1158. doi: 10.1093/cercor/bhs105
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hall, A. M., and Roberson, E. D. (2012). Mouse models of Alzheimer’s disease. Brain Res. Bull. 88, 3–12.
Hamilton, L. K., Aumont, A., Julien, C., Vadnais, A., Calon, F., and Fernandes, K. J. L. (2010). Widespread deficits in adult neurogenesis precede plaque and tangle formation in the 3xTg mouse model of Alzheimer’s disease. Eur. J. Neurosci. 32, 905–920. doi: 10.1111/j.1460-9568.2010.07379.x
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185.
Henderson, J. T., Georgiou, J., Jia, Z., Robertson, J., Elowe, S., Roder, J. C., et al. (2001). The receptor tyrosine kinase EphB2 Regulates NMDA-dependent synaptic function. Neuron 32, 1041–1056. doi: 10.1016/s0896-6273(01)00553-0
Henderson, V. W., Mack, W., and Williams, B. W. (1989). Spatial disorientation in Alzheimer’s disease. Arch. Neurol. 46, 391–394.
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Heneka, M. T., Rodríguez, J. J., and Verkhratsky, A. (2010). Neuroglia in neurodegeneration. Brain Res. Rev. 63, 189–211. doi: 10.1016/j.brainresrev.2009.11.004
Holcomb, L., Gordon, M. N., McGowan, E., Yu, X., Benkovic, S., Jantzen, P., et al. (1998). Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4, 97–100. doi: 10.1038/nm0198-097
Hollands, C., Tobin, M. K., Hsu, M., Musaraca, K., Yu, T.-S., Mishra, R., et al. (2017). Depletion of adult neurogenesis exacerbates cognitive deficits in Alzheimer’s disease by compromising hippocampal inhibition. Mol. Neurodegener. 12:64.
Holtzman, D. M., Morris, J. C., and Goate, A. M. (2011). Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3:77sr1. doi: 10.1126/scitranslmed.3002369
Honer, W. G. (2003). Pathology of presynaptic proteins in Alzheimer’s disease: more than simple loss of terminals. Neurobiol. Aging 24, 1047–1062. doi: 10.1016/j.neurobiolaging.2003.04.005
Hsia, A. Y., Masliah, E., McConlogue, L., Yu, G. Q., Tatsuno, G., Hu, K., et al. (1999). Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. U.S.A. 96, 3228–3233. doi: 10.1073/pnas.96.6.3228
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., et al. (1996). Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–103. doi: 10.1126/science.274.5284.99
Hsiao, Y.-H., Hung, H.-C., Chen, S.-H., and Gean, P.-W. (2014). Social interaction rescues memory deficit in an animal model of Alzheimer’s disease by increasing BDNF-dependent hippocampal neurogenesis. J. Neurosci. 34, 16207–16219. doi: 10.1523/jneurosci.0747-14.2014
Hsieh, H., Boehm, J., Sato, C., Iwatsubo, T., Tomita, T., Sisodia, S., et al. (2006). AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. doi: 10.1016/j.neuron.2006.10.035
Iaccarino, H. F., Singer, A. C., Martorell, A. J., Rudenko, A., Gao, F., Gillingham, T. Z., et al. (2016). Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235. doi: 10.1038/nature20587
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jack, C. R., Knopman, D. S., Jagust, W. J., Petersen, R. C., Weiner, M. W., Aisen, P. S., et al. (2013). Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. doi: 10.1016/s1474-4422(12)70291-0
Jack, C. R., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128. doi: 10.1016/s1474-4422(09)70299-6
Janus, C., Pearson, J., McLaurin, J., Mathews, P. M., Jiang, Y., Schmidt, S. D., et al. (2000). Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 408, 979–982. doi: 10.1038/35050110
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., et al. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/s0896-6273(03)00124-7
Kastanenka, K. V., Bussiere, T., Shakerdge, N., Qian, F., Weinreb, P. H., Rhodes, K., et al. (2016). Immunotherapy with aducanumab restores calcium homeostasis in Tg2576 mice. J. Neurosci. 36, 12549–12558. doi: 10.1523/jneurosci.2080-16.2016
Kelly, B. L., Vassar, R., and Ferreira, A. (2005). Beta-amyloid-induced dynamin 1 depletion in hippocampal neurons. A potential mechanism for early cognitive decline in Alzheimer disease. J. Biol. Chem. 280, 31746–31753. doi: 10.1074/jbc.m503259200
Kessels, H. W., Nabavi, S., and Malinow, R. (2013). Metabotropic NMDA receptor function is required for β-amyloid-induced synaptic depression. Proc. Natl. Acad. Sci. U.S.A. 110, 4033–4038. doi: 10.1038/cddis.2015.160
Kim, J. H., Anwyl, R., Suh, Y. H., Djamgoz, M. B., and Rowan, M. J. (2001). Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J. Neurosci. 21, 1327–1333. doi: 10.1523/jneurosci.21-04-01327.2001
Klevanski, M., Herrmann, U., Weyer, S. W., Fol, R., Cartier, N., Wolfer, D. P., et al. (2015). The APP intracellular domain is required for normal synaptic morphology, synaptic plasticity, and hippocampus-dependent behavior. J. Neurosci. 35, 16018–16033. doi: 10.1523/JNEUROSCI.2009-15.2015
Klyubin, I., Cullen, W. K., Hu, N.-W., and Rowan, M. J. (2012). Alzheimer’s disease Aβ assemblies mediating rapid disruption of synaptic plasticity and memory. Mol. Brain 5:25. doi: 10.1186/1756-6606-5-25
Knafo, S., Alonso-Nanclares, L., Gonzalez-Soriano, J., Merino-Serrais, P., Fernaud-Espinosa, I., Ferrer, I., et al. (2009). Widespread changes in dendritic spines in a model of Alzheimer’s disease. Cereb. Cortex 19, 586–592. doi: 10.1093/cercor/bhn111
Kotilinek, L. A., Bacskai, B., Westerman, M., Kawarabayashi, T., Younkin, L., Hyman, B. T., et al. (2002). Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J. Neurosci. 22, 6331–6335. doi: 10.1523/jneurosci.22-15-06331.2002
Krezymon, A., Richetin, K., Halley, H., Roybon, L., Lassalle, J.-M., Francès, B., et al. (2013). Modifications of hippocampal circuits and early disruption of adult neurogenesis in the tg2576 mouse model of Alzheimer’s disease. PLoS One 8:e76497. doi: 10.1371/journal.pone.0076497
Kuchibhotla, K. V., Lattarulo, C. R., Hyman, B. T., and Bacskai, B. J. (2009). Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323, 1211–1215. doi: 10.1126/science.1169096
Lacor, P. N., Buniel, M. C., Chang, L., Fernandez, S. J., Gong, Y., Viola, K. L., et al. (2004). Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. 24, 10191–10200. doi: 10.1523/jneurosci.3432-04.2004
Lanz, T. A., Carter, D. B., and Merchant, K. M. (2003). Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol. Dis. 13, 246–253. doi: 10.1016/s0969-9961(03)00079-2
Lazarov, O., Robinson, J., Tang, Y.-P., Hairston, I. S., Korade-Mirnics, Z., Lee, V. M.-Y., et al. (2005). Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell 120, 701–713. doi: 10.1016/j.cell.2005.01.015
Lei, M., Xu, H., Li, Z., Wang, Z., O’Malley, T. T., Zhang, D., et al. (2016). Soluble Aβ oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol. Dis. 85, 111–121. doi: 10.1016/j.nbd.2015.10.019
Liebscher, S., Page, R. M., Käfer, K., Winkler, E., Quinn, K., Goldbach, E., et al. (2014). Chronic γ-secretase inhibition reduces amyloid plaque-associated instability of pre- and postsynaptic structures. Mol. Psychiatry 19, 937–946. doi: 10.1038/mp.2013.122
Lindström, P. (2007). The physiology of obese-hyperglycemic mice [ob/ob Mice]. Sci. World J. 7, 666–685. doi: 10.1100/tsw.2007.117
Mably, A. J., Gereke, B. J., Jones, D. T., and Colgin, L. L. (2017). Impairments in spatial representations and rhythmic coordination of place cells in the 3xTg mouse model of Alzheimer’s disease. Hippocampus 27, 378–392. doi: 10.1002/hipo.22697
Masliah, E., Terry, R. D., Alford, M., DeTeresa, R., and Hansen, L. A. (1991). Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer’s disease. Am. J. Pathol. 138, 235–246.
Mehta, D., Jackson, R., Paul, G., Shi, J., and Sabbagh, M. (2017). Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Investig. Drugs 26, 735–739. doi: 10.1080/13543784.2017.1323868
Merino-Serrais, P., Benavides-Piccione, R., Blazquez-Llorca, L., Kastanauskaite, A., Rábano, A., Avila, J., et al. (2013). The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer’s disease. Brain 136, 1913–1928. doi: 10.1093/brain/awt088
Miller, G. (2012). Alzheimer’s research. Stopping Alzheimer’s before it starts. Science 337, 790–792.
Minkeviciene, R., Rheims, S., Dobszay, M. B., Zilberter, M., Hartikainen, J., Fülöp, L., et al. (2009). Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 29, 3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009
Montagna, E., Dorostkar, M. M., and Herms, J. (2017). The role of APP in structural spine plasticity. Front. Mol. Neurosci. 10:136. doi: 10.3389/fnmol.2017.00136
Morgan, D., Diamond, D. M., Gottschall, P. E., Ugen, K. E., Dickey, C., Hardy, J., et al. (2000). Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408, 982–985. doi: 10.1038/35050116
Morley, J. E., Kumar, V. B., Bernardo, A. E., Farr, S. A., Uezu, K., Tumosa, N., et al. (2000). β-Amyloid precursor polypeptide in SAMP8 mice affects learning and memory. Peptides 21, 1761–1767. doi: 10.1016/s0196-9781(00)00342-9
Mucke, L., Abraham, C. R., and Masliah, E. (1996). Neurotrophic and neuroprotective effects of hAPP in transgenic micea. Ann. N. Y. Acad. Sci. 777, 82–88. doi: 10.1111/j.1749-6632.1996.tb34405.x
Mucke, L., Masliah, E., Yu, G.-Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. doi: 10.1523/jneurosci.20-11-04050.2000
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2:a006338. doi: 10.1101/cshperspect.a006338
Mufson, E. J., Mahady, L., Waters, D., Counts, S. E., Perez, S. E., DeKosky, S. T., et al. (2015). Hippocampal plasticity during the progression of Alzheimer’s disease. Neuroscience 309, 51–67. doi: 10.1016/j.neuroscience.2015.03.006
Nagele, R. G., Wegiel, J., Venkataraman, V. K., Imaki, H., and Wegiel, J. (2004). Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 25, 663–674. doi: 10.1016/j.neurobiolaging.2004.01.007
Nalbantoglu, J., Tiradosantiago, G., Lahsaini, A., Poirier, J., Goncalves, O., Verge, A., et al. (1997). Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature 387, 500–505. doi: 10.1038/387500a0
Neuner, S. M., Wilmott, L. A., Hoffmann, B. R., Mozhui, K., and Kaczorowski, C. C. (2017). Hippocampal proteomics defines pathways associated with memory decline and resilience in normal aging and Alzheimer’s disease mouse models. Behav. Brain Res. 322, 288–298. doi: 10.1016/j.bbr.2016.06.002
Noebels, J. (2011). A perfect storm: Converging paths of epilepsy and Alzheimer’s dementia intersect in the hippocampal formation. Epilepsia 52(Suppl. 1), 39–46. doi: 10.1111/j.1528-1167.2010.02909.x
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/jneurosci.1202-06.2006
Oddo, S., Caccamo, A., Kitazawa, M., Tseng, B. P., and LaFerla, F. M. (2003). Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 24, 1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012
Ofengeim, D., Mazzitelli, S., Ito, Y., DeWitt, J. P., Mifflin, L., Zou, C., et al. (2017). RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, E8788–E8797.
Ostrowitzki, S., Lasser, R. A., Dorflinger, E., Scheltens, P., Barkhof, F., Nikolcheva, T., et al. (2017). A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 9:95. doi: 10.1186/s13195-017-0318-y
Oyelami, T., Bondt, A. D., den Wyngaert, I. V., Hoorde, K. V., Hoskens, L., Shaban, H., et al. (2016). Age-dependent concomitant changes in synaptic dysfunction and GABAergic pathway in the APP/PS1 mouse model. Acta Neurobiol. Exp. 76, 282–293. doi: 10.21307/ane-2017-027
Palop, J. J., Chin, J., Bien-Ly, N., Massaro, C., Yeung, B. Z., Yu, G.-Q., et al. (2005). Vulnerability of dentate granule cells to disruption of arc expression in human amyloid precursor protein transgenic mice. J. Neurosci. 25, 9686–9693. doi: 10.1523/jneurosci.2829-05.2005
Palop, J. J., Chin, J., and Mucke, L. (2006). A network dysfunction perspective on neurodegenerative diseases. Nature 443, 768–773. doi: 10.1038/nature05289
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Palop, J. J., Jones, B., Kekonius, L., Chin, J., Yu, G.-Q., Raber, J., et al. (2003). Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc. Natl. Acad. Sci. U.S.A. 100, 9572–9577. doi: 10.1073/pnas.1133381100
Palop, J. J., and Mucke, L. (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. doi: 10.1038/nn.2583
Pousinha, P. A., Mouska, X., Bianchi, D., Temido-Ferreira, M., Rajão-Saraiva, J., Gomes, R., et al. (2019). The amyloid precursor protein C-terminal domain alters CA1 neuron firing, modifying hippocampus oscillations and impairing spatial memory encoding. Cell Rep. 29, 317–331.e5. doi: 10.1016/j.celrep.2019.08.103
Pousinha, P. A., Mouska, X., Raymond, E. F., Gwizdek, C., Dhib, G., Poupon, G., et al. (2017). Physiological and pathophysiological control of synaptic GluN2B-NMDA receptors by the C-terminal domain of amyloid precursor protein. eLife 6:e25659. doi: 10.7554/eLife.25659
Radde, R., Bolmont, T., Kaeser, S. A., Coomaraswamy, J., Lindau, D., Stoltze, L., et al. (2006). Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 7, 940–946. doi: 10.1038/sj.embor.7400784
Reinders, N. R., Pao, Y., Renner, M. C., da Silva-Matos, C. M., Lodder, T. R., Malinow, R., et al. (2016). Amyloid-β effects on synapses and memory require AMPA receptor subunit GluA3. Proc. Natl. Acad. Sci. U.S.A. 113, E6526–E6534.
Reyes-Marin, K. E., and Nuñez, A. (2017). Seizure susceptibility in the APP/PS1 mouse model of Alzheimer’s disease and relationship with amyloid β plaques. Brain Res. 1677, 93–100. doi: 10.1016/j.brainres.2017.09.026
Richetin, K., Moulis, M., Millet, A., Arràzola, M. S., Andraini, T., Hua, J., et al. (2017). Amplifying mitochondrial function rescues adult neurogenesis in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 102, 113–124. doi: 10.1016/j.nbd.2017.03.002
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
Rodríguez, J. J., Jones, V. C., Tabuchi, M., Allan, S. M., Knight, E. M., LaFerla, F. M., et al. (2008). Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS One 3:e2935. doi: 10.1371/journal.pone.0002935
Rodríguez, J. J., Olabarria, M., Chvatal, A., and Verkhratsky, A. (2009). Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 16, 378–385.
Rorabaugh, J. M., Chalermpalanupap, T., Botz-Zapp, C. A., Fu, V. M., Lembeck, N. A., Cohen, R. M., et al. (2017). Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer’s disease. Brain J. Neurol. 140, 3023–3038. doi: 10.1093/brain/awx232
Roselli, F., Tirard, M., Lu, J., Hutzler, P., Lamberti, P., Livrea, P., et al. (2005). Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J. Neurosci. 25, 11061–11070. doi: 10.1523/jneurosci.3034-05.2005
Roy, D. S., Arons, A., Mitchell, T. I., Pignatelli, M., Ryan, T. J., and Tonegawa, S. (2016). Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature 531, 508–512. doi: 10.1038/nature17172
Rudinskiy, N., Hawkes, J. M., Betensky, R. A., Eguchi, M., Yamaguchi, S., Spires-Jones, T. L., et al. (2012). Orchestrated experience-driven Arc responses are disrupted in a mouse model of Alzheimer’s disease. Nat. Neurosci. 15, 1422–1429. doi: 10.1038/nn.3199
Ryan, T. J., Roy, D. S., Pignatelli, M., Arons, A., and Tonegawa, S. (2015). Engram cells retain memory under retrograde amnesia. Science 348, 1007–1013. doi: 10.1126/science.aaa5542
Salazar, S. V., Gallardo, C., Kaufman, A. C., Herber, C. S., Haas, L. T., Robinson, S., et al. (2017). Conditional deletion of Prnp rescues behavioral and synaptic deficits after disease onset in transgenic Alzheimer’s disease. J. Neurosci. 37, 9207–9221. doi: 10.1523/jneurosci.0722-17.2017
Salloway, S., Sperling, R., Fox, N. C., Blennow, K., Klunk, W., Raskind, M., et al. (2014). Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 322–333. doi: 10.1056/NEJMoa1304839
Sanchez, P. E., Zhu, L., Verret, L., Vossel, K. A., Orr, A. G., Cirrito, J. R., et al. (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. U.S.A. 109, E2895–E2903.
Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., et al. (1999). Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. doi: 10.1038/22124
Schmid, L. C., Mittag, M., Poll, S., Steffen, J., Wagner, J., Geis, H.-R., et al. (2016). Dysfunction of somatostatin-positive interneurons associated with memory deficits in an Alzheimer’s disease model. Neuron 92, 114–125. doi: 10.1016/j.neuron.2016.08.034
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1007/978-981-10-7757-9_11
Selkoe, D. J. (2019). Alzheimer disease and aducanumab: adjusting our approach. Nat. Rev. Neurol. 15, 365–366. doi: 10.1038/s41582-019-0205-1
Sevigny, J., Chiao, P., Bussière, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56.
Shah, D., Jonckers, E., Praet, J., Vanhoutte, G., Delgado, Y., Palacios, R., et al. (2013). Resting state FMRI reveals diminished functional connectivity in a mouse model of amyloidosis. PLoS One 8:e84241. doi: 10.1371/journal.pone.0084241
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/jneurosci.4970-06.2007
Shen, Y., Tian, M., Zheng, Y., Gong, F., Fu, A. K. Y., and Ip, N. Y. (2016). Stimulation of the hippocampal POMC/MC4R circuit alleviates synaptic plasticity impairment in an Alzheimer’s disease model. Cell Rep. 17, 1819–1831. doi: 10.1016/j.celrep.2016.10.043
Siemers, E. R., Sundell, K. L., Carlson, C., Case, M., Sethuraman, G., Liu-Seifert, H., et al. (2016). Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. 12, 110–120. doi: 10.1016/j.jalz.2015.06.1893
Simón, A. M., de Maturana, R. L., Ricobaraza, A., Escribano, L., Schiapparelli, L., Cuadrado-Tejedor, M., et al. (2009). Early changes in hippocampal Eph receptors precede the onset of memory decline in mouse models of Alzheimer’s disease. J. Alzheimers Dis. 17, 773–786. doi: 10.3233/jad-2009-1096
Singh, A., and Abraham, W. C. (2017). Astrocytes and synaptic plasticity in health and disease. Exp. Brain Res. 235, 1645–1655. doi: 10.1007/s00221-017-4928-1
Šišková, Z., Justus, D., Kaneko, H., Friedrichs, D., Henneberg, N., Beutel, T., et al. (2014). Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 84, 1023–1033. doi: 10.1016/j.neuron.2014.10.024
Snyder, E. M., Nong, Y., Almeida, C. G., Paul, S., Moran, T., Choi, E. Y., et al. (2005). Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 8, 1051–1058.
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 280–292.
Spires, T. L., Meyer-Luehmann, M., Stern, E. A., McLean, P. J., Skoch, J., Nguyen, P. T., et al. (2005). Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J. Neurosci. 25, 7278–7287. doi: 10.1523/jneurosci.1879-05.2005
Steiner, H., Capell, A., Leimer, U., and Haass, C. (1999). Genes and mechanisms involved in β-amyloid generation and Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 249, 266–270.
Sturchler-Pierrat, C., Abramowski, D., Duke, M., Wiederhold, K.-H., Mistl, C., Rothacher, S., et al. (1997). Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U.S.A. 94, 13287–13292. doi: 10.1073/pnas.94.24.13287
Subota, A., Pham, T., Jetté, N., Sauro, K., Lorenzetti, D., and Holroyd-Leduc, J. (2017). The association between dementia and epilepsy: a systematic review and meta-analysis. Epilepsia 58, 962–972. doi: 10.1111/epi.13744
Sze, C.-I., Bi, H., Kleinschmidt-DeMasters, B. K., Filley, C. M., and Martin, L. J. (2001). N-Methyl-d-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer’s disease. J. Neurol. Sci. 182, 151–159. doi: 10.1016/s0022-510x(00)00467-6
Tackenberg, C., and Brandt, R. (2009). Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J. Neurosci. 29, 14439–14450. doi: 10.1523/JNEUROSCI.3590-09.2009
Tackenberg, C., Ghori, A., and Brandt, R. (2009). Thin, stubby or mushroom: spine pathology in Alzheimer’s disease. Curr. Alzheimer Res. 6, 261–268. doi: 10.2174/156720509788486554
Takasu, M. A., Dalva, M. B., Zigmond, R. E., and Greenberg, M. E. (2002). Modulation of NMDA receptor- dependent calcium influx and gene expression through EphB receptors. Science 295, 491–495. doi: 10.1126/science.1065983
Tan, V. T. Y., Mockett, B. G., Ohline, S. M., Parfitt, K. D., Wicky, H. E., Peppercorn, K., et al. (2018). Lentivirus-mediated expression of human secreted amyloid precursor protein-alpha prevents development of memory and plasticity deficits in a mouse model of Alzheimer’s disease. Mol. Brain 11:7.
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Tsai, J., Grutzendler, J., Duff, K., and Gan, W.-B. (2004). Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 7, 1181–1183. doi: 10.1038/nn1335
Uylings, H. B. M., and de Brabander, J. M. (2002). Neuronal changes in normal human aging and Alzheimer’s disease. Brain Cogn. 49, 268–276. doi: 10.1093/hmg/ddy304
Vargas, J. Y., Fuenzalida, M., and Inestrosa, N. C. (2014). In vivo activation of Wnt signaling pathway enhances cognitive function of adult mice and reverses cognitive deficits in an Alzheimer’s disease model. J. Neurosci. 34, 2191–2202. doi: 10.1523/jneurosci.0862-13.2014
Verghese, P. B., Castellano, J. M., and Holtzman, D. M. (2011). Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 10, 241–252.
Verret, L., Mann, E. O., Hang, G. B., Barth, A. M. I., Cobos, I., Ho, K., et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. doi: 10.1016/j.cell.2012.02.046
Viana da Silva, S., Haberl, M. G., Zhang, P., Bethge, P., Lemos, C., Gonçalves, N., et al. (2016). Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Commun. 7:11915.
Viana da Silva, S., Zhang, P., Haberl, M. G., Labrousse, V., Grosjean, N., Blanchet, C., et al. (2019). Hippocampal mossy fibers synapses in CA3 pyramidal cells are altered at an early stage in a mouse model of Alzheimer’s disease. J. Neurosci. 39, 4193–4205. doi: 10.1523/jneurosci.2868-18.2019
Volicer, L., Smith, S., and Volicer, B. J. (1995). Effect of seizures on progression of dementia of the Alzheimer type. Dementia 6, 258–263. doi: 10.1159/000106956
Vossel, K. A., Beagle, A. J., Rabinovici, G. D., Shu, H., Lee, S. E., Naasan, G., et al. (2013). Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 70, 1158–1166. doi: 10.1001/jamaneurol.2013.136
Vossel, K. A., Tartaglia, M. C., Nygaard, H. B., Zeman, A. Z., and Miller, B. L. (2017). Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol. 16, 311–322. doi: 10.1016/s1474-4422(17)30044-3
Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A. C., Sharma, P., Finkbeiner, S., et al. (2010). Tau reduction prevents Abeta-induced defects in axonal transport. Science 330:198. doi: 10.1126/science.1194653
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi: 10.1038/416535a
Wang, H.-L., Xian, X.-H., Song, Q.-Y., Pang, C., Wang, J.-L., Wang, M.-W., et al. (2017). Age-related alterations of neuronal excitability and voltage-dependent Ca2+ current in a spontaneous mouse model of Alzheimer’s disease. Behav. Brain Res. 321, 209–213. doi: 10.1016/j.bbr.2017.01.009
Wang, Q., Walsh, D. M., Rowan, M. J., Selkoe, D. J., and Anwyl, R. (2004). Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J. Neurosci. 24, 3370–3378. doi: 10.1523/jneurosci.1633-03.2004
Wei, W., Nguyen, L. N., Kessels, H. W., Hagiwara, H., Sisodia, S., and Malinow, R. (2010). Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 13, 190–196. doi: 10.1038/nn.2476
Werner, C. T., Williams, C. J., Fermelia, M. R., Lin, D.-T., and Li, Y. (2019). Circuit mechanisms of neurodegenerative diseases: a new frontier with miniature fluorescence microscopy. Front. Neurosci. 13:1174. doi: 10.3389/fnins.2019.01174
West, M. J., Coleman, P. D., Flood, D. G., and Troncoso, J. C. (1994). Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 344, 769–772. doi: 10.1016/s0140-6736(94)92338-8
West, M. J., Kawas, C. H., Martin, L. J., and Troncoso, J. C. (2000). The CA1 region of the human hippocampus is a hot spot in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 908, 255–259. doi: 10.1111/j.1749-6632.2000.tb06652.x
West, M. J., Kawas, C. H., Stewart, W. F., Rudow, G. L., and Troncoso, J. C. (2004). Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol. Aging 25, 1205–1212. doi: 10.1016/j.neurobiolaging.2003.12.005
Wirths, O. (2017). Altered neurogenesis in mouse models of Alzheimer disease. Neurogenesis 4:e1327002. doi: 10.1080/23262133.2017.1327002
Wu, M., Shi, H., He, Y., Yuan, L., Qu, X., Zhang, J., et al. (2017). Colivelin ameliorates impairments in cognitive behaviors and synaptic plasticity in APP/PS1 transgenic mice. J. Alzheimers Dis. 59, 1067–1078. doi: 10.3233/JAD-170307
Xu, H., Gelyana, E., Rajsombath, M., Yang, T., Li, S., and Selkoe, D. (2016). Environmental enrichment potently prevents microglia-mediated neuroinflammation by human amyloid β-protein oligomers. J. Neurosci. 36, 9041–9056. doi: 10.1523/JNEUROSCI.1023-16.2016
Yang, J.-T., Wang, Z.-J., Cai, H.-Y., Yuan, L., Hu, M.-M., Wu, M.-N., et al. (2018). Sex differences in neuropathology and cognitive behavior in APP/PS1/tau triple-transgenic mouse model of Alzheimer’s disease. Neurosci. Bull. 34, 736–746. doi: 10.1007/s12264-018-0268-9
Zempel, H., Thies, E., Mandelkow, E., and Mandelkow, E.-M. (2010). Abeta oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 30, 11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010
Zhang, W., Gu, G.-J., Zhang, Q., Liu, J.-H., Zhang, B., Guo, Y., et al. (2017). NSCs promote hippocampal neurogenesis, metabolic changes and synaptogenesis in APP/PS1 transgenic mice. Hippocampus 27, 1250–1263. doi: 10.1002/hipo.22794
Zhao, R., Fowler, S. W., Chiang, A. C. A., Ji, D., and Jankowsky, J. L. (2014). Impairments in experience-dependent scaling and stability of hippocampal place fields limit spatial learning in a mouse model of Alzheimer’s disease. Hippocampus 24, 963–978. doi: 10.1002/hipo.22283
Zhurakovskaya, E., Ishchenko, I., Gureviciene, I., Aliev, R., Gröhn, O., and Tanila, H. (2019). Impaired hippocampal-cortical coupling but preserved local synchrony during sleep in APP/PS1 mice modeling Alzheimer’s disease. Sci. Rep. 9:5380.
Keywords: hippocampus, Alzheheimer’s disease, mouse models, synaptic plasticity, circuit changes
Citation: Vyas Y, Montgomery JM and Cheyne JE (2020) Hippocampal Deficits in Amyloid-β-Related Rodent Models of Alzheimer’s Disease. Front. Neurosci. 14:266. doi: 10.3389/fnins.2020.00266
Received: 17 December 2019; Accepted: 09 March 2020;
Published: 07 April 2020.
Edited by:
Sabine Liebscher, Ludwig-Maximilians-Universität München, GermanyReviewed by:
Melanie Meyer-Luehmann, University of Freiburg, GermanyIoannis Sotiropoulos, University of Minho, Portugal
Copyright © 2020 Vyas, Montgomery and Cheyne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johanna M. Montgomery, am0ubW9udGdvbWVyeUBhdWNrbGFuZC5hYy5ueg==; Juliette E. Cheyne, ai5jaGV5bmVAYXVja2xhbmQuYWMubno=