 Yunxiang Zhou1
Yunxiang Zhou1 Qiang Chen1Yali Wang1
Qiang Chen1Yali Wang1 Haijian Wu2
Haijian Wu2 Weilin Xu2
Weilin Xu2 Yuanbo Pan2
Yuanbo Pan2 Shiqi Gao2Xiao Dong2
Shiqi Gao2Xiao Dong2 John H. Zhang3,4
John H. Zhang3,4 Anwen Shao2*
Anwen Shao2*- 1Department of Surgical Oncology, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 2Department of Neurosurgery, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 3Department of Physiology and Pharmacology, Basic Sciences, School of Medicine, Loma Linda University, Loma Linda, CA, United States
- 4Department of Anesthesiology, Neurosurgery and Neurology, School of Medicine, Loma Linda University, Loma Linda, CA, United States
Traumatic brain injury (TBI) represents one of the major causes of death worldwide and leads to persisting neurological deficits in many of the survivors. One of the most significant long-term sequelae deriving from TBI is neurodegenerative disease, which is a group of incurable diseases that impose a heavy socio-economic burden. However, mechanisms underlying the increased susceptibility of TBI to neurodegenerative disease remain elusive. The neurovascular unit (NVU) is a functional unit composed of neurons, neuroglia, vascular cells, and the basal lamina matrix. The key role of NVU dysfunction in many central nervous system diseases has been revealed. Studies have proved the presence of prolonged structural and functional abnormalities of the NVU after TBI. Moreover, growing evidence suggests impaired NVU function is also implicated in neurodegenerative diseases. Therefore, we propose the Neurovascular Unit Dysfunction (NVUD) Hypothesis, in which the persistent NVU dysfunction is thought to underlie the development of post-TBI neurodegeneration. We deduce NVUD Hypothesis through relational inference and supporting evidence, and suggest continued NVU abnormalities following TBI serve as the pathophysiological substrate and trigger yielding chronic neuroinflammation, proteinopathies and oxidative stress, consequently leading to the progression of neurodegenerative diseases. The NVUD Hypothesis may provide potential treatment and prevention strategies for TBI and late-onset neurodegenerative diseases.
Introduction
Traumatic brain injury (TBI) represents one of the major causes of mortality and disability worldwide, with a global burden of approximately US$ 400 billion annually (GBD 2016; Maas et al., 2017; de la Tremblaye et al., 2018; GBD 2016 Traumatic Brain Injury and Spinal Cord Injury Collaborators, 2019). Patients surviving the TBI usually suffer from various long-term neurological and neuropsychiatric sequelae, which include (but not limited to) neurodegenerative diseases (Cruz-Haces et al., 2017) and sleep disturbances (Castriotta et al., 2007; Barshikar and Bell, 2017). Actually, TBI-induced neurodegenerative disease was first introduced in the 19th century in professional boxers who repeatedly suffered head trauma (Martland, 1928). From then on, accumulating evidence has suggested that TBI is a significant risk factor for a variety of neurodegenerative diseases such as Alzheimer’s disease (AD) (Mortimer et al., 1991; Fleminger et al., 2003; Hayes et al., 2017; LoBue et al., 2019), Parkinson’s disease (PD) (Goldman et al., 2006; Jafari et al., 2013), and amyotrophic lateral sclerosis (ALS) (Chen et al., 2007; Franz et al., 2019). Notably, sleep deprivation itself is an important predisposing factor for the development of neurodegenerative diseases (Boespflug and Iliff, 2018; Shokri-Kojori et al., 2018; Cordone et al., 2019).
Neurodegenerative diseases are declared a group of chronic diseases burdening the global aging society (de Lau and Breteler, 2006; Li et al., 2013). Alzheimer’s disease is considered the most common neurodegenerative disorders, followed by PD. In 2015, the former incurred worldwide losses totaling US$ 818 billion, an increase of 35% over the previous five years (Fotuhi et al., 2009; Masters et al., 2015; Ascherio and Schwarzschild, 2016; Wimo et al., 2017). Although symptomatic and etiological treatments targeting patients with AD or PD may help alleviate some of the physical or mental symptoms of these incurable, age-related diseases, no treatment strategies have been proved sufficiently effective thus far (Ascherio and Schwarzschild, 2016; Kumar et al., 2016; Mendiola-Precoma et al., 2016; Orimo, 2017). Another neurodegenerative disease, ALS, although relatively less frequent, has a high fatality rate and a median survival of only 14 months from the time of diagnosis (Luna et al., 2019). Therefore, considering the huge medical and social burden of neurodegenerative diseases and the increased incidence in patients with prior TBI, exploring the underlying mechanisms of causality between TBI and post-TBI neurodegenerative diseases is of great significance.
The neurovascular unit (NVU; Figure 1) was originally introduced as a conceptual framework for cerebrovascular diseases, especially ischemic stroke, and has recently been identified as a key player in many other central nervous system (CNS) diseases, including TBI, whose primary and secondary pathologic processes can lead to persistent structural or metabolic abnormalities of the NVU (Zhang et al., 2005; Lok et al., 2015; Perez et al., 2017). Recently, more and more lines of evidence have proved that NVU dysfunction is also related to neurodegenerative diseases (Cai et al., 2017b). Thus, NVU dysfunction participates in both TBI and neurodegenerative diseases, and it may be a candidate mechanism underlying the TBI-induced neurodegenerative diseases. To understand the underlying mechanisms further, we propose the Neurovascular Unit Dysfunction (NVUD) Hypothesis, in which persistent NVU dysfunction accounts for the pathophysiological substrate and trigger condition for post-TBI neurodegeneration. In the present study, we elaborate on the functions of the components of the NVU and their significant roles in the pathophysiology of TBI. We also present scientific evidence supporting the involvement of NVU dysfunction in neurodegenerative disorders and attempt to summarize precise mechanisms linking acute TBI to chronic effects on neurodegeneration. Consequently, we suggest the reasonability of the NVUD Hypothesis and illuminate its value and limitations.
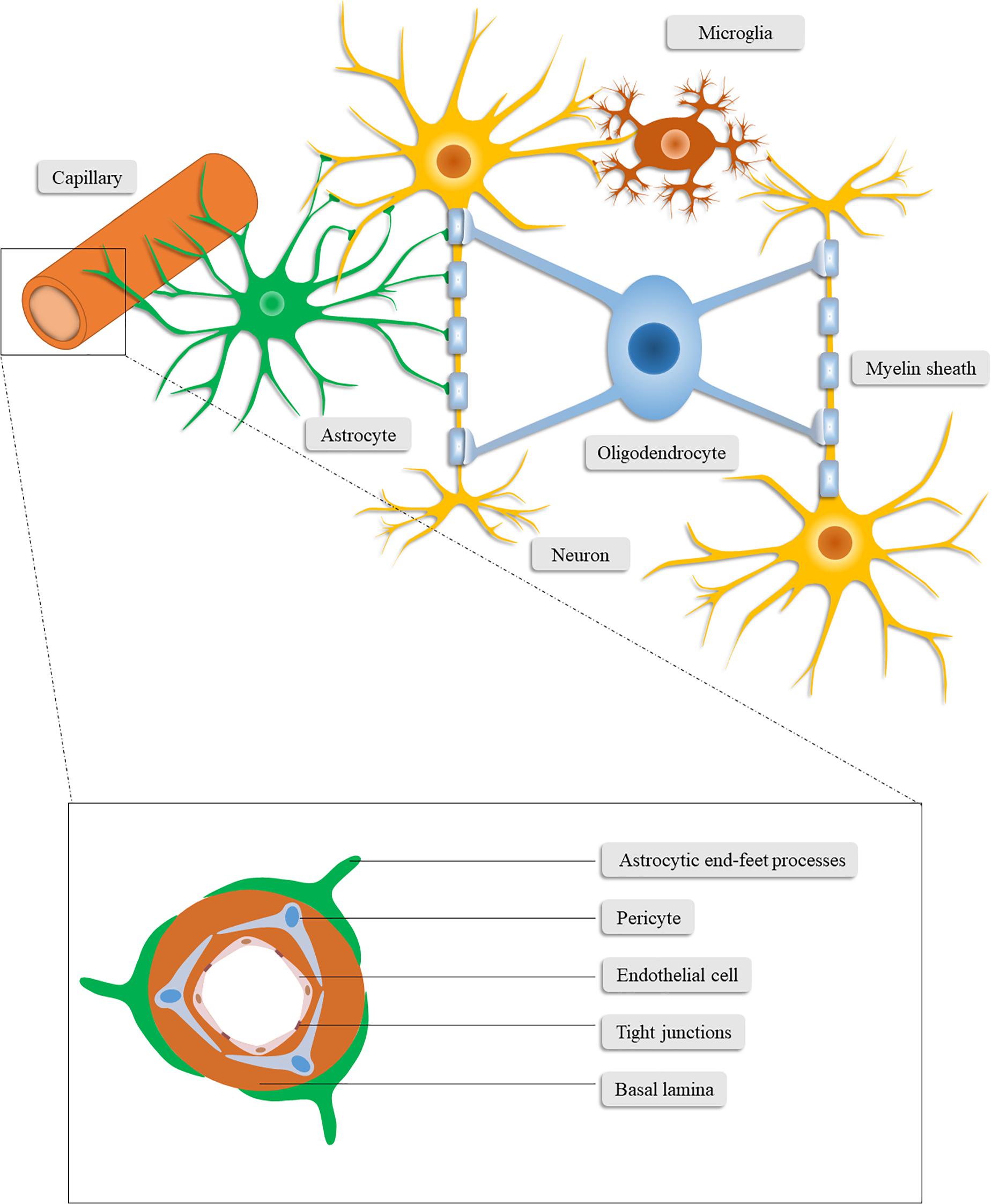
Figure 1. Schematic illustration of the neurovascular unit. The neurovascular unit consists of neurons, neuroglia (astrocytes, microglia, and oligodendrocytes), vascular cells (pericytes, endothelial cells, and vascular smooth muscle cells) and the basal lamina matrix of brain vasculature.
The Components of the NVU Play Critical Roles in the Homeostasis of the CNS
The paradigm of the NVU nowadays encompasses neurons, neuroglia (astrocytes, microglia, and oligodendrocytes), vascular cells (pericytes, endothelial cells, and vascular smooth muscle cells), and the basal lamina matrix of brain vasculature (Lo and Rosenberg, 2009; Cai et al., 2017a). Among these components, neurons may constitute the most pivotal cells for neurological function and have been studied by a great deal of previous work (Cai et al., 2017b). In this section, we mainly focus on the components other than neurons, which also play critical roles in the homeostasis of the CNS and closely interact with neurons.
Glial Components in the NVU
Brain-resident glial cells, which are comprised of astrocytes (astroglia), oligodendrocytes and microglia, exert various functions to maintain CNS homeostasis (Sofroniew and Vinters, 2010; Yang and Wang, 2015). Specifically, astrocytes, the most abundant brain-resident glial cells (Yang and Wang, 2015), stretch out their endfeet to the microvessels to regulate cerebral blood flow and form a functional barrier named glia limitans (Abbott et al., 2006). Through the glia limitans, astrocytes separate neurons from the blood vessels, meninges and perivascular spaces (Sofroniew, 2015). In addition to structural support for neurons, astrocytes also functionally favor neurons in a number of ways (Zhou et al., 2020), including regulating the neurotransmitter glutamate (Rothstein et al., 1996; Anderson and Swanson, 2000; Zou et al., 2010), releasing neurotrophic factors and gliotransmitters (Ye et al., 2003; Kardos et al., 2017; Perez et al., 2017), synthesizing glutamine, cholesterol, glutathione, and thrombospondin (Dringen et al., 2000; Slemmer et al., 2008; Colangelo et al., 2012), converting glucose into lactate (Magistretti and Pellerin, 1999; Danbolt, 2001; Magistretti, 2006), and controlling water homeostasis and neuronal activation (Lang et al., 1998; Walz, 2000; Kofuji and Newman, 2004; Jayakumar and Norenberg, 2010). Furthermore, the concentration of extracellular ions (Colangelo et al., 2012) and the glymphatic system (Jessen et al., 2015) are regulated by astrocytes.
Microglia are immunocompetent cells in the CNS that are easily activated and have the function of recognizing and phagocytizing pathogens, debris and dead cells. They form the first line of defense system of the CNS and conduct phagocytosis-mediated cleanup during senescence or pathological processes (Hu et al., 2014). Conversely, microglia are also involved in the secretion of neurotoxins, such as nitric oxide and pro-inflammatory cytokines, which further deteriorate the cerebral microenvironment in pathological conditions (Hailer, 2008).
Oligodendrocytes, another type of brain-resident glial cell, produce lipid-enriched myelin that wraps axons and accelerates nerve impulses (Plemel et al., 2014). Taken together, glial cells can affect the outcome of patients with CNS disorders given the multifunctional roles of glial cells in the CNS.
Microvascular Components in the NVU
Vascular cells involved in the NVU include pericytes, endothelial cells, and vascular smooth muscle cells. As vascular wall cells embedded in the microvascular basement membrane, pericytes are centrally positioned between endothelial cells, astrocytes and neurons (Sweeney et al., 2016). Pericytes are involved in signal transduction and communication with neighboring cells, through which they regulate blood–brain barrier (BBB) permeability, angiogenesis, neurotoxic metabolism and clearance, neuroinflammation, capillary hemodynamic response, and stem cell activity, demonstrating their vital role in the CNS during health and disease (Sweeney et al., 2016).
Similar to pericytes, endothelial cells are physiologically versatile cells that are involved in the maintenance of vascular homeostasis, regulation of cerebral vasoconstriction and vasodilatation, angiogenesis, and secretion of anti-coagulation factors. Expressing multiple substrate-specific transport systems, endothelial cells also play a vital role in controlling the exchange of CNS with peripheral substances and regulating the transport of ions and nutrients (Cai et al., 2017b). Endothelial cells are highly connected through tight junctions (TJs) which restricts the paracellular diffusion of substances from blood to the CNS (Tso and Macdonald, 2014). These continuously interconnected endothelial cells, together with pericytes, the basal lamina matrix and the astrocytic end-foot processes, constitute the BBB (Najjar et al., 2017). The integrity of the BBB is critical to the homeostasis and immunoprotection of the CNS (Najjar et al., 2017). Of the microvascular components in the NVU, endothelial cells are the main component of the BBB (Cai et al., 2017b) while pericytes primarily maintain the integrity of the BBB (Armulik et al., 2010; Bell et al., 2010; Daneman et al., 2010).
The Extracellular Matrix
The extracellular matrix (ECM) of the basal lamina functions as an anchor for the endothelium through cell-matrix interactions between laminin (or other matrix proteins) and the endothelial integrin receptors. Through these interactions, numerous intracellular signaling pathways can be stimulated (Hynes, 1992; Tilling et al., 2002). The TJs are thought to constitute the primary impediment of paracellular diffusion, therefore, given the involvement of matrix proteins in the regulation of expression of endothelial TJs, the basal lamina proteins are likely to maintain the diffusion as well (Tilling et al., 1998). The ECM and TJs are fundamental to the permeability of the BBB and the degradation of these proteins under pathological circumstances can potently exacerbate BBB dysfunction (Rascher et al., 2002). Additionally, matrix metalloproteinases (MMPs), particularly MMP-9, are capable of degrading the ECM and TJs, which enhances the permeability of the BBB and leads to vasogenic edema (Fujimoto et al., 2008; Halliday et al., 2013).
NVU Dysfunction in the Pathophysiology of TBI
While numerous causes of trauma exist, TBI is consistently related to both primary and secondary damage mechanisms (Logsdon et al., 2015). During TBI, mechanical force directly leads to instant damage, which includes neuronal damage and vascular disruption, followed by the secondary injury mediated by succeeding pathophysiological processes such as oxidative stress, neuroinflammation, BBB dysfunction, and apoptosis (Abdul-Muneer et al., 2015; Chen X. et al., 2018; Khaksari et al., 2018; Main et al., 2018; Sevenich, 2018). All these primary and secondary pathologic processes contribute to the structural and/or metabolic abnormalities of the NVU, resulting in long-term neurological deficits in the patients (Zhang et al., 2005; Perez et al., 2017).
Oxidative Stress
Oxidative stress gives rise to progressive neuropathology during TBI and contributes primarily to secondary injury (Abdul-Muneer et al., 2015; Anthonymuthu et al., 2018). Oxidative stress is considered physiological and biochemical stress or insult deriving mainly from reactive oxygen species (ROS) and reactive nitrogen species (RNS) (Abdul-Muneer et al., 2015). The enhanced production of ROS and RNS following TBI is due to excessive excitotoxicity and insufficient endogenous antioxidant systems. These reactive species result in increased oxidative stress and parallel the production of super-oxidized cellular and vascular structures, oxidized protein, cleaved DNA, and impaired mitochondrial electron transport chain, which can induce more ROS and RNS, thus triggering a vicious circle (Cornelius et al., 2013).
Oxidative stress and elevated toxic proteins can act on astrocytes to induce astrocyte-secreted pro-inflammatory factors, such as interleukin (IL)-6, monocyte chemoattractant protein (MCP)-1, and MMP-9, leading to the BBB compromise and neuroinflammation (Salminen et al., 2011). Oxidative stress is a major contributor to endothelial impairment. Enriched with mitochondria for the normal performance of ATP-dependent active transporters, endothelial cells can thus malfunction under increased oxidative stress and impaired mitochondrial functions (Cai et al., 2017b). Furthermore, oxidative stress and energy depletion induce the dysfunction of cellular ion channels that leads to the depolarization of neurons and the aggregation of excitatory neurotransmitters (e.g., glutamate), which further aggravate neuronal depolarization and the increase of toxic calcium levels (Weilinger et al., 2013). Moreover, enhanced oxidative stress and the subsequent biochemical cascade are adequate to induce early or late apoptosis, immediate cell necrosis and delayed neurodegeneration post TBI (Abdul-Muneer et al., 2015).
Neuroinflammation
Neuroinflammation is a hallmark of different CNS pathologies (Sevenich, 2018). Following the initial injury, the changed microenvironment and released intracellular components from damaged cells trigger the activation and recruitment of local glial cells (Corps et al., 2015; Gyoneva and Ransohoff, 2015; Sharma et al., 2015). Both microglia and astrocytes react within 24 h and peak around day 3–7 post TBI (Fujita et al., 1998; Di Giovanni et al., 2005; Susarla et al., 2014). The activation and proliferation of glia release signaling factors and induce a robust, sterile immune reaction that consists of brain-resident and peripherally recruited inflammatory cells (Corps et al., 2015; Gyoneva and Ransohoff, 2015; Sharma et al., 2015). In addition to danger signals released by damaged cells, mitochondrial stress, glutamate excitotoxicity, and vascular injury have also been identified as inflammatory triggers of neuroinflammation (Simon et al., 2017). This immune reaction is supposed to exert a neuroprotective role and promote wound healing, but it can become neurodestructive and aggravate neuronal damage under certain circumstances (Murakami et al., 2011; Gyoneva and Ransohoff, 2015; Kumar et al., 2017). For instance, resident glia and infiltrating immune cells up-regulate the expression of tumor necrosis factor (TNF)-α, IL-6, and IL-1β rapidly to cope with injury, among which, however, TNF-α is correlated with BBB disruption and brain edema, and can act together with IL-1β to drive astrocyte dysfunction and resultant glutamate excitotoxicity (Ziebell and Morganti-Kossmann, 2010; Viviani et al., 2014; Simon et al., 2017). Notably, many mediators activated after TBI may exhibit pleiotropic effects in a context-dependent manner. In this regard, the conceptual framework of NVU may also help understand the shifting profiles of TBI pathophysiology over time (Lok et al., 2015).
As the inflammatory response progresses, the glial progenitor cells around the damaged tissue form a glial scar, which isolates the injured areas and inhibits the spread of inflammatory cells (Sofroniew, 2009; Karimi-Abdolrezaee and Billakanti, 2012; Burda and Sofroniew, 2014; Peng et al., 2014; Sofroniew, 2015). Nonetheless, the glial scar highly expresses inhibitory components for axonal regeneration and acts as both a physical and chemical barrier to axon elongation. The glial scar is therefore considered the main obstacle for axonal regeneration and the restoration of neuronal connectivity, which makes it a likely cause of the long-term sequelae in TBI patients (Voskuhl et al., 2009; Jeong et al., 2012; Cregg et al., 2014; Sharma et al., 2015).
Blood–Brain Barrier Dysfunction
The BBB functions as a key homeostatic site between the CNS and other major systems in the body (Zhao et al., 2015). As the core feature of TBI, BBB dysfunction may indicate the severity of the injury as well as the length of recovery from TBI (Neuwelt et al., 2008; Main et al., 2018). The disruption of BBB integrity can result from the initial injury or arise secondarily from the succeeding pathological processes, including extensive inflammation, metabolic disturbances, and astrocytic dysfunction (Shlosberg et al., 2010; Badaut and Bix, 2014; Corps et al., 2015; Johnson et al., 2018). Following the brain injury, several pro-inflammatory pathways such as the IL-1β/nuclear factor-κ-gene binding (NF-κB) signaling pathway (Petty and Lo, 2002; Suzuki et al., 2010) and the Janus kinase/signal transducers and activators of transcription 1 (JAK/STAT1) signaling pathway (Gong et al., 2018) have been demonstrated to induce the upregulation of MMPs and the consequential degradation of the ECM and TJs. Moreover, upregulated NF-κB, as a transcription factor, also induces the transcription of pro-inflammatory genes and enzymes, including intracellular adhesion molecule-1 (ICAM-1) and inducible nitric oxide synthase (iNOS; Kumar et al., 2004). The former has been shown to increase BBB permeability through facilitating transendothelial leukocyte migration (Najjar et al., 2017) while the latter has been identified to be excitotoxic and neurotoxic as well as activate MMPs via nitric oxide (Ji et al., 2017; Chen H. et al., 2018). In addition, neuroinflammation and energy depletion result in the impairments of ionic transport, transporters, and mitochondrial oxidative metabolism of endothelial cells, which all exacerbate the breakdown of the BBB (Ott et al., 2007). Moreover, the BBB compromise can, in turn, aggravate the inflammatory response due to the enhanced influx of serum elements, proinflammatory molecules, and infiltrating leukocytes. It can also lead to cerebral hemorrhage, brain edema, and hypoxia (Shlosberg et al., 2010; Badaut and Bix, 2014; Corps et al., 2015; Johnson et al., 2018).
As described above, TJ-interconnected endothelial cells and pericytes are the main constituents that primarily contribute to the maintenance of the BBB (Armulik et al., 2010; Bell et al., 2010; Daneman et al., 2010; Cai et al., 2017b). The impairment of crosstalk between them ultimately leads to BBB dysfunction after TBI onset (Bhowmick et al., 2019). Astrocytes also hold a critical role in BBB function and cerebral water homeostasis (Burda et al., 2016) as astrocytic end-foot processes ensheathe the BBB and densely express perivascular aquaporin-4 (AQP4) channels (Louveau et al., 2017; Sweeney et al., 2018). In addition, BBB permeability can be altered by astrocyte-derived factors (Michinaga and Koyama, 2019). Moreover, other cell types involved in the NVU have also been shown to affect BBB homeostasis after TBI. For example, studies have indicated that oligodendrocyte precursor cells protect BBB against disruption via the transforming growth factor-β (TGF-β) signaling pathway (Seo et al., 2014). The activation, proliferation and phenotypic transformation of microglia indirectly have an impact on BBB function considering their involvement in the inflammatory response, especially the pro-inflammatory role (such as iNOS-expressing) of M1-like phenotype (Ladwig et al., 2017). These glial cells and microvascular cells have a functional interaction with each other in a paracrine manner (Abdul-Muneer et al., 2015). Therefore, the coordinated response of all components within the NVU is the determinant of BBB integrity (Lok et al., 2015).
Persistent NVU Abnormalities and Imaging/Serum/CSF Biomarkers
Growing evidence has demonstrated these post-TBI NVU abnormalities may persist for months or years, especially in patients with a history of moderate-to-severe TBI (Hayes et al., 2017; LoBue et al., 2019). Reports regarding long-term brain changes after milder injuries have been mixed, and the results were often complicated by factors concerned with the number and severity/complications of injuries, genetic risk, and mental states and behavior (Hayes et al., 2017; LoBue et al., 2019). Diffuse axonal injury has been found in TBI of any severity and post-mortem studies reported widespread axonal pathology could continue for a sufficient length of time (weeks to months, even years) after mild-to-severe TBI in humans (Blumbergs et al., 1989; Johnson et al., 2013a, b; Shetty et al., 2014; Logsdon et al., 2015). A continuum of microglia activation across a wide range of TBI conditions was also observed in animal models (Nagamoto-Combs et al., 2007; Nagamoto-Combs et al., 2010) and human autopsy (form different cause) (Gentleman et al., 2004; Johnson et al., 2013a), even up to 18 years after TBI, and this was related to the consistently ongoing white matter degeneration (Johnson et al., 2013a). However, the functional impact of these microglia is still an enigma, as controversy remains regarding whether they exhibit a pro-inflammatory or pro-regenerative phenotype (Ramlackhansingh et al., 2011; Johnson et al., 2013a; Russo and McGavern, 2016; Scott et al., 2018; Tagge et al., 2018). Evidence of prolonged reactive astrogliosis after even mild injuries was revealed by brain tissue from animal models (Smith et al., 1997) and humans (Goldstein et al., 2012; Shively et al., 2017; Tagge et al., 2018) as well. Likewise, microvascular injury could be detected in the autopsy of long-term survivors from different types of TBI (Hay et al., 2015; Tagge et al., 2018). These continued NVU dysfunctions could bring about chronic pathological processes such as oxidative stress, chronic neuroinflammation, and proteinopathy, which were also reported to be identified years post TBI and could, in turn, aggravate the NVU dysfunction (Johnson et al., 2012; Simon et al., 2017; LoBue et al., 2019). Although chronic oxidative stress has not yet been observed following a single mild human TBI, it appears that oxidative stress after concussion and its associated pathophysiological processes may worsen or prolong symptoms (Hoffer et al., 2013; Cruz-Haces et al., 2017).
Currently, technological advances in clinical TBI neuroimaging make it possible to study NVU dysfunction and ensuing pathological processes in vivo, and findings by neuroimaging studies support the persistent abnormalities as well (LoBue et al., 2019). For example, microglia activation and related chronic neuroinflammation in moderate to severe TBI survivors can be assessed up to 17 years after trauma by positron emission tomography (PET) imaging technology binding to translocator protein, which is expressed by mitochondria of activated microglia (Folkersma et al., 2011; Ramlackhansingh et al., 2011). Moreover, significant increased binding to translocator protein can be identified by PET technology decades after the history of repetitive mild TBI in retired athletes compared to healthy controls (Coughlin et al., 2015). Similarly, selective PET techniques with ligands for neurodegenerative proteins [e.g., amyloid-β peptide (Aβ) and tau protein] can image the deposition of proteinopathy, showing increased binding in moderate-severe TBI victims/retired athletes versus control cases (Hong et al., 2014; Barrio et al., 2015; Scott et al., 2016). Advanced magnetic resonance imaging (MRI) modalities, such as diffusion tensor imaging (DTI, particularly in white matter pathology visualization), magnetic resonance spectroscopy (MRS, particularly in metabolic change assessment), susceptibility weight imaging (SWI, particularly in microscopic bleeding detection), and functional magnetic resonance imaging (fMRI, particularly in neurocognitive function examination) have emerged to allow the discovery of injury biomarkers and the detection of brain changes in patients with even mild TBI over time (Baugh et al., 2012; Xiong et al., 2014; Pavlovic et al., 2019). Additionally, Single Photon Emission Computed Tomography (SPECT) scan also has significance, as being a promising tool for detecting regional functional changes in the brain of patients sustaining TBI (Baugh et al., 2012).
Continued presence of some serum/plasma and CSF biomarkers is also able to persist many years post TBI, which depends on the type and extent of injury, thereby holding great promise for assessing the duration and degree of pathological processes (Williams et al., 2018; LoBue et al., 2019). Proteins including Aβ, tau, α-synuclein, and nuclear transactive response DNA-binding protein 43 (TDP-43) are critically implicated in the pathogenesis of several different neurodegenerative diseases, and have been utilized as neurobiological markers to monitor neuronal damage following TBI, moreover, different marker binding fingerprints may function as predictors for a particular late-onset neurodegenerative disease (Williams et al., 2018). A growing body of literature data has suggested that neurofilament light in the blood and/or CSF also serves as a biomarker indicating neuronal injury after TBI (Shahim et al., 2017; Bernick et al., 2018). Besides, glial fibrillary acidic protein, ubiquitin C-terminal hydrolase L1, S100β, and neuron-specific enolase have shown immense value as reliable blood/CSF markers in predicting poor outcome and evaluating microstructural injuries undetected by computed tomography (Bohmer et al., 2011; Mondello et al., 2016; Papa et al., 2016; Welch et al., 2016). Additionally, chronically elevated expression of serum cytokines may reflect chronic immune activation after TBI (Simon et al., 2017).
Patients With TBI Have Increased Susceptibility to Neurodegenerative Diseases
Accumulating evidence provides support for an association between the risk of developing neurodegenerative disease and a prior TBI event. Previous studies have reported an increased risk of AD among TBI victims (Roberts et al., 1991; Fleminger et al., 2003). Similarly, 1.5–3.8 times higher incidence of PD has been reported in post-TBI cases (Goldman et al., 2006; Jafari et al., 2013). Traumatic brain injury, particularly of repeated TBI, is also considered a predisposing factor for ALS (Chen et al., 2007; Pupillo et al., 2018; Franz et al., 2019). Additionally, age at trauma has been suggested to be a determinant of the TBI victims’ susceptibility to ALS (Seals et al., 2016; Pupillo et al., 2018). Notably, although vulnerability to neurodegenerative diseases has been indicated in patients with TBI of any severity, this increased susceptibility is more common in survivors sustaining moderate-to-severe TBI (Hayes et al., 2017; Graham and Sharp, 2019; LoBue et al., 2019), and these patients have a 1.8-fold increase in neurodegenerative disease risk compared with those with mild TBI (Raj et al., 2017).
Unfortunately, mechanisms underlying this risk remain elusive thus far (LoBue et al., 2019). Early after the initial injury, TBI can induce the production of pathological proteins, whose neurotoxicity contributes directly to persistent abnormalities of brain structure and function, with these proteinopathies lasting for months or years (Goldstein et al., 2012; Johnson et al., 2012, 2013a; Hay et al., 2015; Williams et al., 2018). Intriguingly, post-TBI proteinopathies have similarities to multiple neurodegenerative diseases (Smith et al., 2013; Graham and Sharp, 2019). For instance, pathological accumulation of Aβ and neurofibrillary tangles comprising hyperphosphorylated neuronal tau brings about the development of AD (Zenaro et al., 2017), similarly, α-synuclein is linked to PD (Kalia and Lang, 2016). Furthermore, TDP-43 is considered the primary disease-related protein in ALS, and TDP-43 proteinopathy also features in other neurodegenerative disorders such as AD, PD, and Huntington’s disease (HD; Johnson et al., 2011). However, TDP-43 proteinopathy after TBI is more obscure and several controversial areas remain (Graham and Sharp, 2019). The study by Wiesner et al. (2018) addressed that brain trauma could boost ALS-related TDP-43 pathology, whereas the extent was modulated by ALS-related gene mutations, and the process was indicated to reversible and incapable of triggering ALS progression and neuronal vulnerability. Moreover, a single TBI does not appear to induce TDP-43 proteinopathy in humans (Johnson et al., 2011). In addition to proteinopathies, chronic neuroinflammation and persistent oxidative stress may represent other candidates for the increased susceptibility. Neuroinflammation is a hallmark of CNS disorders, including TBI and neurodegenerative diseases (Heneka et al., 2015; Russo and McGavern, 2016; Sevenich, 2018). Oxidative stress gives rise to progressive neuropathology and contributes primarily to the secondary injury post TBI (Abdul-Muneer et al., 2015; Anthonymuthu et al., 2018), concurrently being involved in the pathogenesis of neurodegenerative diseases (Cruz-Haces et al., 2017).
Taken collectively, proteinopathies, persistent oxidative stress, and chronic neuroinflammation may be the pivotal intermediary pathological processes between early and delayed post-TBI changes (LoBue et al., 2019). Herein we present that there is a pathological correlation between TBI and post-TBI neurodegenerative diseases, and we speculate it is the aforementioned persistent NVU dysfunction following TBI that accounts for the pathophysiological substrate and trigger condition for this pathological correlation. Readers seeking an in-depth discussion regarding the supporting evidence and internal mechanisms for this hypothesis should consult the section “Supporting Evidence and Precise Mechanisms.” But before that, as we have elucidated the bond between NVU dysfunction and TBI pathophysiology, however, is NVU dysfunction involved in the pathophysiology of neurodegenerative diseases?
Is NVU Dysfunction Involved in the Pathophysiology of Neurodegenerative Diseases?
More and more lines of evidence favor the perspective that not only the neuronal degeneration and loss, but also aberrant neuroglia and vascular cells contribute to the pathogenesis of neurodegenerative diseases (Cai et al., 2017b). In this section, we describe several roles of NVU components involved in the pathophysiology of AD, PD, and ALS.
Alzheimer’s Disease
A growing number of studies from post-mortem brain tissue reveal the existence of neurovascular dysfunction in patients with AD, including brain capillary leakage, degeneration of BBB-related cells (e.g., endothelial cells and pericytes), brain infiltration of peripheral cells, abnormal angiogenesis and molecular changes (Sweeney et al., 2018).
Blood–Brain Barrier disruption is considered the key pathway of AD onset (Cai et al., 2017b). This is firstly reflected in its impact on the aggregation of pathological protein. The dysfunction of NVU leads to aberrant cerebral blood flow regulation and impaired BBB transport, resulting in faulty Aβ clearance and increased Aβ production in soluble and fibrillary forms, which are the most neurotoxic and vasculotoxic forms (Benarroch, 2007). Accumulation of Aβ can subsequently induce oxidative stress, neuroinflammation and cell apoptosis, aggravating BBB impairment (Benarroch, 2007). Tau protein is associated with BBB compromise as well, as BBB dysfunction is in concert with the phenomenon that the major hippocampal blood vessels are surrounded by tau protein (Blair et al., 2015). In addition to pathological protein aggregation, AD is also pathologically characterized by chronic inflammation with the involvement of resident microglia and infiltrating peripheral immune cells (Heneka et al., 2015). A dysfunctional BBB is convenient for migrating immune cells to infiltrate the CNS. In this context, when the requisite adhesion molecules are expressed, circulating leukocytes migrate into the brain parenchyma through activated brain endothelial cells and then interact with NVU components to further affect their structural integrity and function (Zenaro et al., 2017).
Parkinson’s Disease
In PD pathogenesis, BBB dysfunction is not as important as it is in AD, but the effects of neuronal and glial cell damage are more crucial. Dopaminergic neuronal loss and Lewy bodies and Lowy neurites containing α-synuclein are the hallmarks of PD (Kalia and Lang, 2016). Dopaminergic neurons are characterized as highly metabolic and thus have abundant amounts of mitochondria, the DNA of which is easily damaged by ROS and DNA repair can meanwhile be reduced in the absence of energy support (Liang et al., 2007; Gredilla et al., 2010). In addition, the metabolic process of dopamine itself will produce a large amount of accumulated ROS under pathological conditions (Cai et al., 2017b). Therefore, oxidative stress and energy depletion can easily give rise to dopaminergic neuronal loss as well as α-synuclein aggregation, which further forms Lewy bodies and Lowy neurites.
Besides neuronal loss, glial components in the NVU also play a pivotal role in PD onset. Equipped with unmyelinated axons, dopaminergic neurons interact most closely with astrocytes (Rodriguez et al., 2014; Rodriguez et al., 2015). In normal physiological conditions, astrocytes provide both structural and functional support for dopaminergic neurons in multifactorial ways (see section “Glial Components in the NVU” for the detailed mechanisms) while dysfunctional astrocytes secrete various cytokines, chemokines, and excitotoxins, which contribute to a series of pathological processes (Salminen et al., 2011; Zhou et al., 2020). Furthermore, microglia–astrocyte interactions can further propel disease progression (Chen et al., 2019).
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is resulting from a complex combination of genetic predispositions and environmental insults. ALS-linked genes include FUS, TARDBP (the one encoding TDP-43), SOD1, C9orf72, among others (Sleigh et al., 2020). Based on the function of these genes, impaired cytoskeletal dynamics and axonal transport have emerged as a key role in ALS (Sleigh et al., 2020). Besides, dysfunctions of other NVU components are also involved in the pathogenesis of ALS. Both astrocytes and microglia have been argued to serve as central players in the pathogenesis. In the early stage of disease preceding the emergence of neuronal death and clinical symptoms, incompetent astrocytes due to astrodegeneration and astrocytic atrophy break the glutamate homeostasis and elicit glutamate excitotoxicity. Microglia are also induced to secrete neurotoxic factors, while in the later stage of disease, reactive astrogliosis and activated microglia may further promote neuronal damage and death (Verkhratsky et al., 2014). Additionally, the involvement of microvascular components in ALS pathology has also been suggested and microvascular compromise appears to precede the neuronal lesions and even the neuroinflammation (Guo and Lo, 2009).
The Neurovascular Unit Dysfunction Hypothesis and its Limitations
Definition and Relational Reasoning
The NVUD Hypothesis underscores the core role of NVU dysfunction in the development of post-TBI neurodegenerative diseases, which is concluded with relational reasoning from the following: (1) The NVU comprises neurons, neuroglial cells, vascular cells, and the basal lamina matrix of brain vasculature, playing critical roles in the homeostasis of the CNS, (2) all of the primary and secondary pathologic processes during TBI contribute to the persisting NVU dysfunction, including neuronal death, neuroglial dysfunction, and BBB compromise, (3) patients with TBI have an increased risk for developing neurodegenerative diseases, based on epidemiology and mutual pathological processes, and (4) NVU dysfunction is also involved in the pathophysiology of neurodegenerative diseases.
Additionally, we have mentioned in the previous sections that the persistent NVU abnormalities after TBI are related to the number and severity of injuries, and there is a dose–response relationship between TBI and later neurodegenerative disease. This similarity may further strengthen the reasonability of our assumption that continued NVU dysfunction serves as the pathophysiological substrate to trigger the development of post-TBI neurodegeneration.
Consistent with our relational inference, increasing supporting evidence obtained by animal models and human postmortem studies has suggested NVU dysfunction an important causative factor for post-TBI neurodegeneration (Figure 2; Graham and Sharp, 2019; LoBue et al., 2019). These evidences also explain the induction of chronic neuroinflammation, persistent oxidative stress, and neurodegenerative protein aggregation after the initial injury.
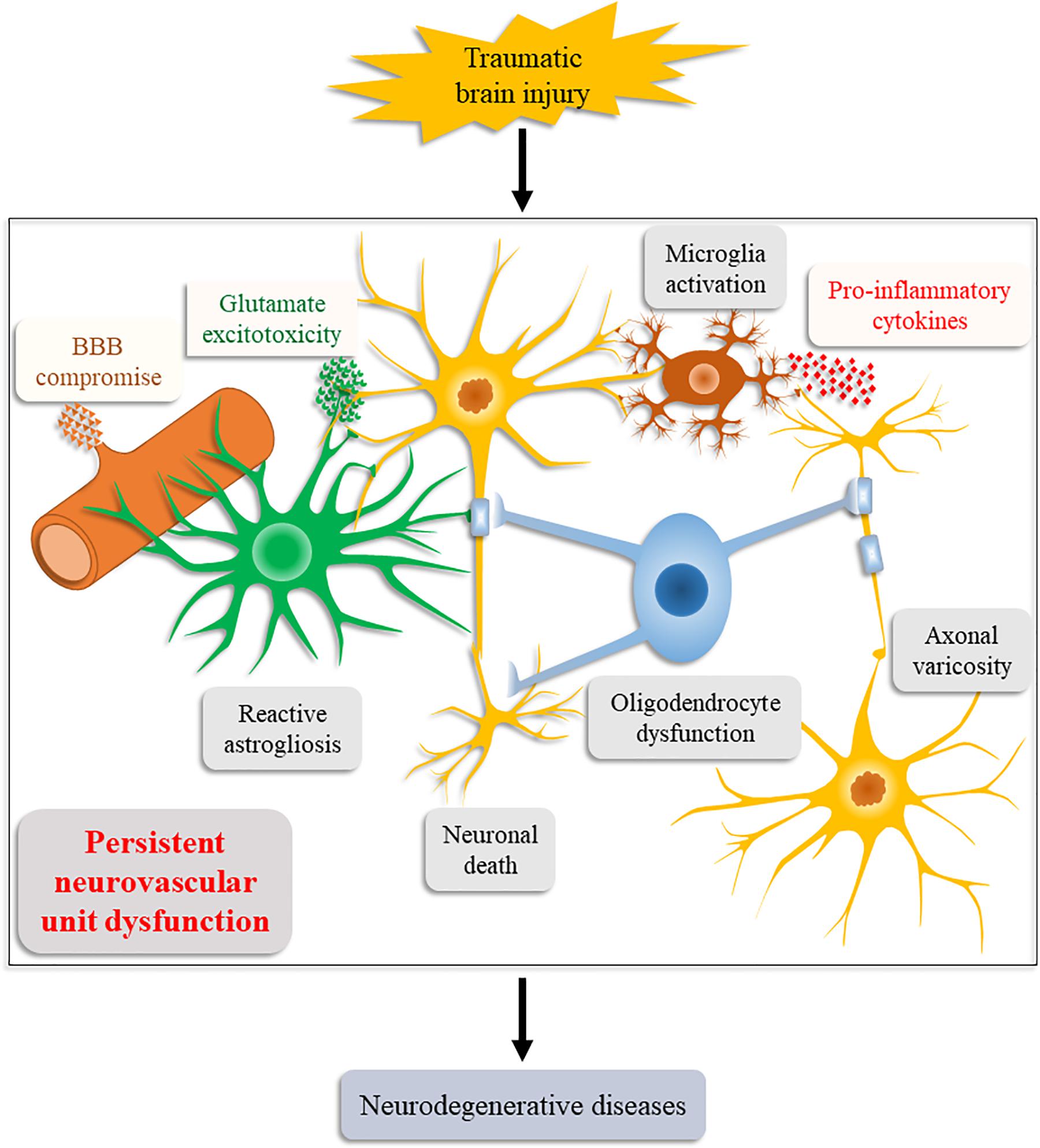
Figure 2. Schematic illustration of the Neurovascular Unit Dysfunction (NVUD) Hypothesis. During TBI, primary and secondary pathologic processes lead to persistent structural and/or functional abnormalities of the neurovascular unit, such as neuronal death, neuroglial dysfunction, and BBB compromise, which propel the progression of neurodegenerative diseases post TBI.
Supporting Evidence and Precise Mechanisms
Prolonged injured axons are considered a source of pathological proteins (Johnson et al., 2013b). Following TBI, the shear forces applied to the cytoskeleton result in damaged microstructure and impaired axonal transport (Maxwell et al., 2003; Tagge et al., 2018), which is also involved in AD (Johnson et al., 2013b) and ALS (Sleigh et al., 2020) pathogenesis. Impaired axonal transport can lead to the co-accumulation of amyloid precursor protein (APP) and AAP-cleaving enzymes in axonal varicosities within just hours of trauma and thus, forming abundant Aβ in the varicosities (Chen et al., 2004; Uryu et al., 2007; Chen et al., 2009; Tran et al., 2011). In the case where damaged axons eventually break down, these intraneuronal Aβ can aggregate in the parenchyma to form plaques (Chen et al., 2004; Johnson et al., 2013b). Shear forces also induce the dissociation of tau from microtubules, which is subsequently processed into a highly pathogenic tau form contributing to mitochondrial damage, neuronal apoptosis, and abnormal long-term potentiation (Kondo et al., 2015; Tagge et al., 2018). Besides, injured axons yield an extensive release of glutamate as well, coupled with cytokine stimulation, distorted glutamate receptor trafficking, and impaired glutamate clearance due to loss of astrocytic functional glutamate transporters, which together contribute to the dysregulation of glutamate homeostasis (Baker et al., 1993; Piao et al., 2019; Zhang et al., 2019). This dysregulation is adequate to incur neuroexcitotoxicity as well as resultant oxidative stress and mitochondrial dysfunction post TBI (Cornelius et al., 2013; Abdul-Muneer et al., 2015), which are strictly implicated in the PD pathogenesis including dopaminergic neuronal loss and α-synuclein aggregation (Liang et al., 2007; Gredilla et al., 2010; Cai et al., 2017b). Indeed, oxidative stress potentially plays a key role in protein carbonylation and pathological protein accumulation (Cruz-Haces et al., 2017). Notably, TBI is suggested to form a transmissible self-propagating proteinopathy (Zanier et al., 2018; Graham and Sharp, 2019). Altogether, the possibility of persistent axonopathy and transmissible proteinopathy provide a potential mechanism for long-term and extensive neurodegeneration in the brain, thereby propelling progression of neurodegenerative diseases (Johnson et al., 2012, 2013b; Zanier et al., 2018; Graham and Sharp, 2019).
Continued BBB dysfunction and neuroglial dysfunction following TBI are also implicated as triggers of post-TBI neurodegeneration, and the underlying mechanisms include, certainly, astrocyte dysfunction-induced impaired glutamate clearance as mentioned above. Perennial BBB alterations recruit brain-resident neuroglia and infiltrating leukocytes to form chronic neuroinflammation (Shetty et al., 2014; Hay et al., 2015), which is also a pathological characteristic of neurodegenerative diseases (Heneka et al., 2015; Sevenich, 2018). Similar to oxidative stress, neuroinflammation is indicated to elicit the abnormal accumulation of pathological proteins (LoBue et al., 2019), which may further exacerbate vascular lesion as the perivascular location of proteinopathies has been observed in autopsies of TBI victims (McKee et al., 2012; Tagge et al., 2018). The glymphatic system is a recently discovered brain-wide waste clearance pathway that utilizes astrocytic AQP4 channels to promote efficient elimination of interstitial solutes, including pathological proteins, from the CNS (Iliff et al., 2012; Jessen et al., 2015). In the acute phase following TBI, reactive astrogliosis is associated with alterations in AQP4 channel distribution and polarization, which are in line with the injury severity and persist for 14–28 days and 28 days in mild TBI and moderate TBI, respectively, resulting in decreased glymphatic influx and increased metabolic waste accumulation (Ren et al., 2013; Rasmussen et al., 2018; Sullan et al., 2018). Besides, decreased glymphatic function is also critically attributed to reactive astrogliosis-induced glial scars (Jessen et al., 2015). Furthermore, the glymphatic system functions primarily during sleep and is largely suppressed during waking state, whereas sleep deprivation constitutes one of the most frequently reported chronic complications following TBI (Jessen et al., 2015; Rasmussen et al., 2018; Sullan et al., 2018). As a result, the function of glymphatic pathway can be down to about 40% and maintained for at least 1 month after TBI (Iliff et al., 2014; Jessen et al., 2015). Therefore, both chronic impairment of BBB transport (Benarroch, 2007; Hay et al., 2015) and glymphatic pathway function (Iliff et al., 2014; Sullan et al., 2018) can induce impaired protein clearance, contributing to the succedent accumulation of neurotoxic and vasculotoxic proteins as well as the onset of post-TBI neurodegeneration.
Neurovascular Unit Dysfunction Hypothesis as a Promising Theoretical Basis for Treatment
Neurovascular Unit Dysfunction abnormalities and ensuing pathological processes evolving over time have cast a spotlight on positive TBI interventions, which may be beneficial over an extended time frame (Ramlackhansingh et al., 2011). In the past, neuronal damage had been considered the main cause of functional impairment in brain injury or neurodegenerative diseases, and almost all the therapeutic strategies were targeted at repairing neuronal injury and rescuing neurons (Cai et al., 2017b). However, in most cases, saving neurons alone seemed to be insufficient for treating brain injuries or diseases (Lok et al., 2015). We have previously reviewed the advance of a variety of stem cells in TBI treatment. Although some preclinical studies and small trials showed a good therapeutic effect, practical value of clinic application has not reached a general success (Zhou et al., 2019). Analogously, stem cell therapies in other CNS diseases, including neurodegenerative diseases and stroke, have not generated substantial clinical improvements (Wang et al., 2017). The NVUD Hypothesis may provide new insights into the treatment strategies for TBI and potential prevention for later neurodegeneration, namely integrated restoration of all neural, neuroglial and vascular connectivity. Indeed, it is increasingly recognized that cell-cell crosstalk within the NVU components is absolutely required for remodeling and repair in acute brain injury and neurodegeneration (Lok et al., 2015). To date, several candidate agents have been found to promote the restoration of NVU integrity in TBI models, such as sonic hedgehog, and troxerutin cerebroprotein hydrolysate (Zhao et al., 2018; Wu et al., 2020), more preclinical and clinical studies are warranted.
Limitations of NVUD Hypothesis
There are some aspects that NVUD Hypothesis cannot explain. For instance, studies have revealed that cell-cell signaling within the NVU is crucial in the pathogenesis of HD and that TDP-43 pathology may feature in HD (Gu et al., 2005; Shin et al., 2005; Gu et al., 2007; Faideau et al., 2010; Johnson et al., 2011; Jansen et al., 2017). However, the increased incidence of HD in TBI victims has been rarely reported. Moreover, despite universal occurred NVU dysfunction, only a subgroup of TBI victims develop late-onset neurodegenerative diseases, indicating other factors should be taken into consideration, such as genetic susceptibility (e.g., APOE genotyping) (Baugh et al., 2012; Goldstein et al., 2012; Shively et al., 2017; LoBue et al., 2019). Furthermore, aging not only increases the NVU’s vulnerability to neurodegeneration, but also attenuates its self-repair capabilities (Cai et al., 2017b; Wang et al., 2017). Thus, the crosstalk between NVUD Hypothesis and aging merits further explanation. The NVUD Hypothesis also needs further refinement. Although the NVU paradigm in this study focuses on the components surrounding the cerebral capillaries, critical roles of vascular components and perivascular nerve fibers connecting with larger blood vessels in CNS diseases have also been proposed (Zhang et al., 2012). These groups of structures may need to be incorporated into an expanded NVUD paradigm, which will revise the NVUD Hypothesis.
Conclusion
In the present study, we propose the NVUD Hypothesis and discuss the reasonability. We also present its value as a promising theoretical basis for treatment and illustrate the limitations of this theory. The NVUD Hypothesis emphasizes that persistent NVU dysfunction functions as the pathophysiological substrate and trigger for late-onset neurodegeneration after TBI. Specifically, continued NVU abnormalities following TBI incur the chronic neuroinflammation, persistent oxidative stress, and neurodegenerative proteins aggregation, which in turn exacerbate NVU dysfunction and thus, forming a vicious circle and consequently leading to the progression of neurodegenerative diseases.
Author Contributions
YZ and AS conceptualized the research project. YZ, YW, HW, SG, and YP wrote the manuscript and made the original figures. QC, AS, WX, JZ, and XD critically revised the texts and figures. AS supervised the research and led the discussion. All authors read and approved the final manuscript.
Funding
This work was funded by China Postdoctoral Science Foundation (2017M612010), National Natural Science Foundation of China (81701144), and Zhejiang Province Natural Science Foundation (LQ17H090003).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
A β, amyloid β peptide; AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; AQP4, aquaporin-4; BBB, blood–brain barrier; CNS, central nervous system; DTI, diffusion tensor imaging; ECM, extracellular matrix; fMRI, functional magnetic resonance imaging; HD, Huntington disease; HTT, Huntingtin gene; ICAM-1, intracellular adhesion molecule-1; IL, interleukin; iNOS, inducible nitric oxide synthase; JAK, Janus kinase; MCP, monocyte chemoattractant protein; MMP, matrix metalloprotein; MRS, magnetic resonance spectroscopy; MSNs, medium spiny neurons; NF κ B, nuclear factor- κ B; NVU, neurovascular unit; NVUD, Neurovascular Unit Dysfunction; PD, Parkinson’s disease; PET, positron emission tomography; RNS, reactive nitrogen species; ROS, reactive oxygen species; SPECT, Single Photon Emission Computed Tomography; STAT1, signal transducers and activators of transcription 1 SWI susceptibility weight imaging; TBI, traumatic brain injury; TGF-β, transforming growth factor- β,TJ, tight junction; TNF, tumor necrosis factor.
References
Abbott, N. J., Ronnback, L., and Hansson, E. (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53. doi: 10.1038/nrn1824
Abdul-Muneer, P. M., Chandra, N., and Haorah, J. (2015). Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 51, 966–979. doi: 10.1007/s12035-014-8752-3
Anderson, C. M., and Swanson, R. A. (2000). Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32, 1–14. doi: 10.1002/1098-1136(200010)32:1<1::aid-glia10>3.0.co;2-w
Anthonymuthu, T. S., Kenny, E. M., Lamade, A. M., Kagan, V. E., and Bayir, H. (2018). Oxidized phospholipid signaling in traumatic brain injury. Free Rad. Biol. Med. 124, 493–503. doi: 10.1016/j.freeradbiomed.2018.06.031
Armulik, A., Genove, G., Mae, M., Nisancioglu, M. H., Wallgard, E., Niaudet, C., et al. (2010). Pericytes regulate the blood-brain barrier. Nature 468, 557–561.
Ascherio, A., and Schwarzschild, M. A. (2016). The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 15, 1257–1272. doi: 10.1016/s1474-4422(16)30230-7
Badaut, J., and Bix, G. J. (2014). Vascular neural network phenotypic transformation after traumatic injury: potential role in long-term sequelae. Transl. Stroke Res. 5, 394–406. doi: 10.1007/s12975-013-0304-z
Baker, A. J., Moulton, R. J., MacMillan, V. H., and Shedden, P. M. (1993). Excitatory amino acids in cerebrospinal fluid following traumatic brain injury in humans. J. Neurosurg. 79, 369–372. doi: 10.3171/jns.1993.79.3.0369
Barrio, J. R., Small, G. W., Wong, K. P., Huang, S. C., Liu, J., Merrill, D. A., et al. (2015). In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proc. Natl. Acad. Sci. U.S.A. 112, E2039–E2047.
Barshikar, S., and Bell, K. R. (2017). Sleep disturbance after TBI. Curr. Neurol. Neurosci. Rep. 17:87.
Baugh, C. M., Stamm, J. M., Riley, D. O., Gavett, B. E., Shenton, M. E., Lin, A., et al. (2012). Chronic traumatic encephalopathy: neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imag. Behav. 6, 244–254. doi: 10.1007/s11682-012-9164-5
Bell, R. D., Winkler, E. A., Sagare, A. P., Singh, I., LaRue, B., Deane, R., et al. (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. doi: 10.1016/j.neuron.2010.09.043
Benarroch, E. E. (2007). Neurovascular unit dysfunction: a vascular component of Alzheimer disease? Neurology 68, 1730–1732. doi: 10.1212/01.wnl.0000264502.92649.ab
Bernick, C., Zetterberg, H., Shan, G., Banks, S., and Blennow, K. (2018). Longitudinal performance of plasma neurofilament light and tau in professional fighters: the professional fighters brain health study. J. Neurotr. 35, 2351–2356. doi: 10.1089/neu.2017.5553
Bhowmick, S., D’Mello, V., Caruso, D., Wallerstein, A., and Abdul-Muneer, P. M. (2019). Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol. 317, 260–270. doi: 10.1016/j.expneurol.2019.03.014
Blair, L. J., Frauen, H. D., Zhang, B., Nordhues, B. A., Bijan, S., Lin, Y. C., et al. (2015). Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol. Commun. 3:8.
Blumbergs, P. C., Jones, N. R., and North, J. B. (1989). Diffuse axonal injury in head trauma. J. Neurol. Neurosurg. Psychiatry 52, 838–841.
Boespflug, E. L., and Iliff, J. J. (2018). The emerging relationship between interstitial fluid-cerebrospinal fluid exchange, amyloid-beta, and sleep. Biol. Psychiatry 83, 328–336. doi: 10.1016/j.biopsych.2017.11.031
Bohmer, A. E., Oses, J. P., Schmidt, A. P., Peron, C. S., Krebs, C. L., Oppitz, P. P., et al. (2011). Neuron-specific enolase, S100B, and glial fibrillary acidic protein levels as outcome predictors in patients with severe traumatic brain injury. Neurosurgery 68, 1624–1630.
Burda, J. E., Bernstein, A. M., and Sofroniew, M. V. (2016). Astrocyte roles in traumatic brain injury. Exp. Neurol. 275(Pt 3), 305–315. doi: 10.1016/j.expneurol.2015.03.020
Burda, J. E., and Sofroniew, M. V. (2014). Reactive gliosis and the multicellular response to CNS damage and Disease. Neuron 81, 229–248. doi: 10.1016/j.neuron.2013.12.034
Cai, W., Liu, H., Zhao, J., Chen, L. Y., Chen, J., Lu, Z., et al. (2017a). Pericytes in brain injury and repair after ischemic stroke. Transl. Stroke Res. 8, 107–121. doi: 10.1007/s12975-016-0504-4
Cai, W., Zhang, K., Li, P., Zhu, L., Xu, J., Yang, B., et al. (2017b). Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: an aging effect. Ageing Res. Rev. 34, 77–87. doi: 10.1016/j.arr.2016.09.006
Chen, H., Guan, B., Chen, X., Chen, X., Li, C., Qiu, J., et al. (2018). Baicalin attenuates blood-brain barrier disruption and hemorrhagic transformation and improves neurological outcome in ischemic stroke rats with delayed t-PA treatment: involvement of ONOO(-)-MMP-9 pathway. Transl. Stroke Res. 9, 515–529. doi: 10.1007/s12975-017-0598-3
Chen, X., Pan, Z., Fang, Z., Lin, W., Wu, S., Yang, F., et al. (2018). Omega-3 polyunsaturated fatty acid attenuates traumatic brain injury-induced neuronal apoptosis by inducing autophagy through the upregulation of SIRT1-mediated deacetylation of Beclin-1. J. Neuroinflamm. 15:310.
Castriotta, R. J., Wilde, M. C., Lai, J. M., Atanasov, S., Masel, B. E., and Kuna, S. T. (2007). Prevalence and consequences of sleep disorders in traumatic brain injury. J. Clin. Sleep Med. 3, 349–356. doi: 10.5664/jcsm.26855
Chen, H., Richard, M., Sandler, D. P., Umbach, D. M., and Kamel, F. (2007). Head injury and amyotrophic lateral sclerosis. Am. J. Epidemiol. 166, 810–816.
Chen, X. H., Johnson, V. E., Uryu, K., Trojanowski, J. Q., and Smith, D. H. (2009). A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 19, 214–223. doi: 10.1111/j.1750-3639.2008.00176.x
Chen, X. H., Siman, R., Iwata, A., Meaney, D. F., Trojanowski, J. Q., and Smith, D. H. (2004). Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 165, 357–371. doi: 10.1016/s0002-9440(10)63303-2
Chen, Z., Zhong, D., and Li, G. (2019). The role of microglia in viral encephalitis: a review. J. Neuroinflamm. 16:76.
Colangelo, A. M., Cirillo, G., Lavitrano, M. L., Alberghina, L., and Papa, M. (2012). Targeting reactive astrogliosis by novel biotechnological strategies. Biotechnol. Adv. 30, 261–271. doi: 10.1016/j.biotechadv.2011.06.016
Cordone, S., Annarumma, L., Rossini, P. M., and De Gennaro, L. (2019). Sleep and beta-Amyloid deposition in Alzheimer Disease: insights on mechanisms and possible innovative treatments. Front. Pharmacol. 10:695. doi: 10.3389/fphar.2019.00695
Cornelius, C., Crupi, R., Calabrese, V., Graziano, A., Milone, P., Pennisi, G., et al. (2013). Traumatic brain injury: oxidative stress and neuroprotection. Antioxid. Redox Signal. 19, 836–853.
Corps, K. N., Roth, T. L., and McGavern, D. B. (2015). Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 72, 355–362.
Coughlin, J. M., Wang, Y., Munro, C. A., Ma, S., Yue, C., Chen, S., et al. (2015). Neuroinflammation and brain atrophy in former NFL players: an in vivo multimodal imaging pilot study. Neurobiol. Dis. 74, 58–65. doi: 10.1016/j.nbd.2014.10.019
Cregg, J. M., DePaul, M. A., Filous, A. R., Lang, B. T., Tran, A., and Silver, J. (2014). Functional regeneration beyond the glial scar. Exp. Neurol. 253, 197–207. doi: 10.1016/j.expneurol.2013.12.024
Cruz-Haces, M., Tang, J., Acosta, G., Fernandez, J., and Shi, R. (2017). Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Transl. Neurodegen. 6:20.
Daneman, R., Zhou, L., Kebede, A. A., and Barres, B. A. (2010). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. doi: 10.1038/nature09513
de la Tremblaye, P. B., O’Neil, D. A., LaPorte, M. J., Cheng, J. P., Beitchman, J. A., Thomas, T. C., et al. (2018). Elucidating opportunities and pitfalls in the treatment of experimental traumatic brain injury to optimize and facilitate clinical translation. Neurosci. Biobehav. Rev. 85, 160–175. doi: 10.1016/j.neubiorev.2017.05.022
de Lau, L. M. L., and Breteler, M. M. B. (2006). Epidemiology of Parkinson’s disease. Lancet Neurol. 5, 525–535.
Di Giovanni, S., Movsesyan, V., Ahmed, F., Cernak, I., Schinelli, S., Stoica, B., et al. (2005). Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. U.S.A. 102, 8333–8338. doi: 10.1073/pnas.0500989102
Dringen, R., Gutterer, J. M., and Hirrlinger, J. (2000). Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 267, 4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x
Faideau, M., Kim, J., Cormier, K., Gilmore, R., Welch, M., Auregan, G., et al. (2010). In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum. Mol. Genet. 19, 3053–3067. doi: 10.1093/hmg/ddq212
Fleminger, S., Oliver, D. L., Lovestone, S., Rabe-Hesketh, S., and Giora, A. (2003). Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on, a partial replication. J. Neurol. Neurosurg. Psychiatry 74, 857–862. doi: 10.1136/jnnp.74.7.857
Folkersma, H., Boellaard, R., Yaqub, M., Kloet, R. W., Windhorst, A. D., Lammertsma, A. A., et al. (2011). Widespread and prolonged increase in (R)-(11)C-PK11195 binding after traumatic brain injury. J. Nuclear Med. 52, 1235–1239. doi: 10.2967/jnumed.110.084061
Fotuhi, M., Hachinski, V., and Whitehouse, P. J. (2009). Changing perspectives regarding late-life dementia. Nat. Rev. Neurol. 5, 649–658. doi: 10.1038/nrneurol.2009.175
Franz, C. K., Joshi, D., Daley, E. L., Grant, R. A., Dalamagkas, K., Leung, A., et al. (2019). Impact of traumatic brain injury on amyotrophic lateral sclerosis: from bedside to bench. J. Neurophysiol. 122, 1174–1185. doi: 10.1152/jn.00572.2018
Fujimoto, M., Takagi, Y., Aoki, T., Hayase, M., Marumo, T., Gomi, M., et al. (2008). Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 28, 1674–1685. doi: 10.1038/jcbfm.2008.59
Fujita, T., Yoshimine, T., Maruno, M., and Hayakawa, T. (1998). Cellular dynamics of macrophages and microglial cells in reaction to stab wounds in rat cerebral cortex. Acta Neurochir. 140, 275–279. doi: 10.1007/s007010050095
GBD 2016 Traumatic Brain Injury and Spinal Cord Injury Collaborators (2019). Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990-2016: a systematic analysis for the Global burden of disease study 2016. Lancet Neurol. 18, 56–87.
Gentleman, S. M., Leclercq, P. D., Moyes, L., Graham, D. I., Smith, C., Griffin, W. S., et al. (2004). Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci. Int. 146, 97–104. doi: 10.1016/j.forsciint.2004.06.027
Goldman, S. M., Tanner, C. M., Oakes, D., Bhudhikanok, G. S., Gupta, A., and Langston, J. W. (2006). Head injury and Parkinson’s disease risk in twins. Ann. Neurol. 60, 65–72. doi: 10.1002/ana.20882
Goldstein, L. E., Fisher, A. M., Tagge, C. A., Zhang, X. L., Velisek, L., Sullivan, J. A., et al. (2012). Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4:134ra60.
Gong, L., Manaenko, A., Fan, R., Huang, L., Enkhjargal, B., McBride, D., et al. (2018). Osteopontin attenuates inflammation via JAK2/STAT1 pathway in hyperglycemic rats after intracerebral hemorrhage. Neuropharmacology 138, 160–169. doi: 10.1016/j.neuropharm.2018.06.009
Graham, N. S., and Sharp, D. J. (2019). Understanding neurodegeneration after traumatic brain injury: from mechanisms to clinical trials in dementia. J. Neurol. Neurosurg. Psychiatry 90, 1221–1233. doi: 10.1136/jnnp-2017-317557
Gredilla, R., Bohr, V. A., and Stevnsner, T. (2010). Mitochondrial DNA repair and association with aging–an update. Exp. Gerontol. 45, 478–488. doi: 10.1016/j.exger.2010.01.017
Gu, X., Andre, V. M., Cepeda, C., Li, S. H., Li, X. J., Levine, M. S., et al. (2007). Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington’s disease. Mol. Neurodegen. 2:8. doi: 10.1186/1750-1326-2-8
Gu, X., Li, C., Wei, W., Lo, V., Gong, S., Li, S. H., et al. (2005). Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron 46, 433–444. doi: 10.1016/j.neuron.2005.03.025
Guo, S., and Lo, E. H. (2009). Dysfunctional cell-cell signaling in the neurovascular unit as a paradigm for central nervous system disease. Stroke 40, S4–S7.
Gyoneva, S., and Ransohoff, R. M. (2015). Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol. Sci. 36, 471–480. doi: 10.1016/j.tips.2015.04.003
Hailer, N. P. (2008). Immunosuppression after traumatic or ischemic CNS damage: it is neuroprotective and illuminates the role of microglial cells. Prog. Neurobiol. 84, 211–233. doi: 10.1016/j.pneurobio.2007.12.001
Halliday, M. R., Pomara, N., Sagare, A. P., Mack, W. J., Frangione, B., and Zlokovic, B. V. (2013). Relationship between cyclophilin a levels and matrix metalloproteinase 9 activity in cerebrospinal fluid of cognitively normal apolipoprotein e4 carriers and blood-brain barrier breakdown. JAMA Neurol. 70, 1198–1200.
Hay, J. R., Johnson, V. E., Young, A. M., Smith, D. H., and Stewart, W. (2015). Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J. Neuropathol. Exp. Neurol. 74, 1147–1157. doi: 10.1093/jnen/74.12.1147
Hayes, J. P., Logue, M. W., Sadeh, N., Spielberg, J. M., Verfaellie, M., Hayes, S. M., et al. (2017). Mild traumatic brain injury is associated with reduced cortical thickness in those at risk for Alzheimer’s disease. Brain 140, 813–825.
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405.
Hoffer, M. E., Balaban, C., Slade, M. D., Tsao, J. W., and Hoffer, B. (2013). Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One 8:e54163. doi: 10.1371/journal.pone.054163
Hong, Y. T., Veenith, T., Dewar, D., Outtrim, J. G., Mani, V., Williams, C., et al. (2014). Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 71, 23–31.
Hu, X., Liou, A. K., Leak, R. K., Xu, M., An, C., Suenaga, J., et al. (2014). Neurobiology of microglial action in CNS injuries: receptor-mediated signaling mechanisms and functional roles. Prog. Neurobiol. 119-120, 60–84. doi: 10.1016/j.pneurobio.2014.06.002
Hynes, R. O. (1992). Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69, 11–25. doi: 10.1016/0092-8674(92)90115-s
Iliff, J. J., Chen, M. J., Plog, B. A., Zeppenfeld, D. M., Soltero, M., Yang, L., et al. (2014). Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 34, 16180–16193. doi: 10.1523/jneurosci.3020-14.2014
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 4:147ra111. doi: 10.1126/scitranslmed.3003748
Jafari, S., Etminan, M., Aminzadeh, F., and Samii, A. (2013). Head injury and risk of Parkinson disease: a systematic review and meta-analysis. Mov. Disord. 28, 1222–1229. doi: 10.1002/mds.25458
Jansen, A. H., van Hal, M., Op den Kelder, I. C., Meier, R. T., de Ruiter, A. A., Schut, M. H., et al. (2017). Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia 65, 50–61. doi: 10.1002/glia.23050
Jayakumar, A. R., and Norenberg, M. D. (2010). The Na–K–Cl Co-transporter in astrocyte swelling. Metab. Brain Dis. 25, 31–38. doi: 10.1007/s11011-010-9180-3
Jeong, S. R., Kwon, M. J., Lee, H. G., Joe, E. H., Lee, J. H., Kim, S. S., et al. (2012). Hepatocyte growth factor reduces astrocytic scar formation and promotes axonal growth beyond glial scars after spinal cord injury. Exp. Neurol. 233, 312–322. doi: 10.1016/j.expneurol.2011.10.021
Jessen, N. A., Munk, A. S., Lundgaard, I., and Nedergaard, M. (2015). The glymphatic system: a beginner’s guide. Neurochem. Res. 40, 2583–2599.
Ji, B., Zhou, F., Han, L., Yang, J., Fan, H., Li, S., et al. (2017). Sodium tanshinone IIA sulfonate enhances effectiveness Rt-PA treatment in acute ischemic stroke patients associated with ameliorating blood-brain barrier damage. Transl. Stroke Res. 8, 334–340. doi: 10.1007/s12975-017-0526-6
Johnson, V. E., Stewart, J. E., Begbie, F. D., Trojanowski, J. Q., Smith, D. H., and Stewart, W. (2013a). Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136, 28–42. doi: 10.1093/brain/aws322
Johnson, V. E., Stewart, W., and Smith, D. H. (2013b). Axonal pathology in traumatic brain injury. Exp. Neurol. 246, 35–43. doi: 10.1016/j.expneurol.2012.01.013
Johnson, V. E., Stewart, W., and Smith, D. H. (2012). Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149. doi: 10.1111/j.1750-3639.2011.00513.x
Johnson, V. E., Stewart, W., Trojanowski, J. Q., and Smith, D. H. (2011). Acute and chronically increased immunoreactivity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans. Acta Neuropathol. 122, 715–726. doi: 10.1007/s00401-011-0909-9
Johnson, V. E., Weber, M. T., Xiao, R., Cullen, D. K., Meaney, D. F., Stewart, W., et al. (2018). Mechanical disruption of the blood-brain barrier following experimental concussion. Acta Neuropathol. 135, 711–726. doi: 10.1007/s00401-018-1824-0
Kalia, L. V., and Lang, A. E. (2016). Parkinson disease in 2015: evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 12, 65–66. doi: 10.1038/nrneurol.2015.249
Kardos, J., Heja, L., Jemnitz, K., Kovacs, R., and Palkovits, M. (2017). The nature of early astroglial protection-Fast activation and signaling. Prog. Neurobiol. 153, 86–99. doi: 10.1016/j.pneurobio.2017.03.005
Karimi-Abdolrezaee, S., and Billakanti, R. (2012). Reactive astrogliosis after spinal cord injury-beneficial and detrimental effects. Mol. Neurobiol. 46, 251–264. doi: 10.1007/s12035-012-8287-4
Khaksari, M., Soltani, Z., and Shahrokhi, N. (2018). Effects of female sex steroids administration on pathophysiologic mechanisms in traumatic brain injury. Transl. Stroke Res. 9, 393–416. doi: 10.1007/s12975-017-0588-5
Kofuji, P., and Newman, E. A. (2004). Potassium buffering in the central nervous system. Neuroscience 129, 1045–1056.
Kondo, A., Shahpasand, K., Mannix, R., Qiu, J., Moncaster, J., Chen, C. H., et al. (2015). Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523, 431–436. doi: 10.1038/nature14658
Kumar, A., Nisha, C. M., Silakari, C., Sharma, I., Anusha, K., Gupta, N., et al. (2016). Current and novel therapeutic molecules and targets in Alzheimer’s disease. J. Form. Med. Assoc. 115, 3–10.
Kumar, A., Stoica, B. A., Loane, D. J., Yang, M., Abulwerdi, G., Khan, N., et al. (2017). Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J. Neuroinflamm. 14:47.
Kumar, A., Takada, Y., Boriek, A. M., and Aggarwal, B. B. (2004). Nuclear factor-kappaB: its role in health and disease. J. Mol. Med. 82, 434–448.
Ladwig, A., Walter, H. L., Hucklenbroich, J., Willuweit, A., Langen, K. J., Fink, G. R., et al. (2017). Osteopontin augments M2 microglia response and separates M1- and M2-polarized microglial activation in permanent focal cerebral ischemia. Med. Inflamm. 2017:7189421.
Lang, F., Busch, G. L., Ritter, M., Völkl, H., Waldegger, S., Gulbins, E., et al. (1998). Functional significance of cell volume regulatory mechanisms. Physiol. Rev. 78, 247–306. doi: 10.1152/physrev.1998.78.1.247
Li, C., Ebrahimi, A., and Schluesener, H. (2013). Drug pipeline in neurodegeneration based on transgenic mice models of Alzheimer’s disease. Ageing Res. Rev. 12, 116–140. doi: 10.1016/j.arr.2012.09.002
Liang, C. L., Wang, T. T., Luby-Phelps, K., and German, D. C. (2007). Mitochondria mass is low in mouse substantia nigra dopamine neurons: implications for Parkinson’s disease. Exp. Neurol. 203, 370–380. doi: 10.1016/j.expneurol.2006.08.015
Lo, E. H., and Rosenberg, G. A. (2009). The neurovascular unit in health and disease: introduction. Stroke 40, S2–S3.
LoBue, C., Munro, C., Schaffert, J., Didehbani, N., Hart, J., Batjer, H., et al. (2019). Traumatic brain injury and risk of long-term brain changes, accumulation of pathological markers, and developing dementia: a review. J. Alzheimer Dis. 70, 629–654. doi: 10.3233/jad-190028
Logsdon, A. F., Lucke-Wold, B. P., Turner, R. C., Huber, J. D., Rosen, C. L., and Simpkins, J. W. (2015). Role of microvascular disruption in brain damage from traumatic brain injury. Comprehens. Physiol. 5, 1147–1160. doi: 10.1002/cphy.c140057
Lok, J., Wang, X. S., Xing, C. H., Maki, T. K., Wu, L. M., Guo, S. Z., et al. (2015). Targeting the neurovascular unit in brain trauma. CNS Neurosci. Therap. 21, 304–308. doi: 10.1111/cns.12359
Louveau, A., Plog, B. A., Antila, S., Alitalo, K., Nedergaard, M., and Kipnis, J. (2017). Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Invest. 127, 3210–3219. doi: 10.1172/jci90603
Luna, J., Diagana, M., Ait Aissa, L., Tazir, M., Ali Pacha, L., Kacem, I., et al. (2019). Clinical features and prognosis of amyotrophic lateral sclerosis in Africa: the TROPALS study. J. Neurol. Neurosurg. Psychiatry 90, 20–29.
Maas, A. I. R., Menon, D. K., Adelson, P. D., Andelic, N., Bell, M. J., Belli, A., et al. (2017). Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 16, 987–1048.
Magistretti, P. J. (2006). Neuron-glia metabolic coupling and plasticity. J. Exp. Biol. 209, 2304–2311. doi: 10.1242/jeb.02208
Magistretti, P. J., and Pellerin, L. (1999). Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 1155–1163. doi: 10.1098/rstb.1999.0471
Main, B. S., Villapol, S., Sloley, S. S., Barton, D. J., Parsadanian, M., Agbaegbu, C., et al. (2018). Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol. Neurodegen. 13:17.
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer’s disease. Nat. Rev. Dis. Prim. 1:15056.
Maxwell, W. L., Domleo, A., McColl, G., Jafari, S. S., and Graham, D. I. (2003). Post-acute alterations in the axonal cytoskeleton after traumatic axonal injury. J. Neurotrauma 20, 151–168. doi: 10.1089/08977150360547071
McKee, A. C., Stein, T. D., Nowinski, C. J., Stern, R. A., Daneshvar, D. H., Alvarez, V. E., et al. (2012). The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64.
Mendiola-Precoma, J., Berumen, L. C., Padilla, K., and Garcia-Alcocer, G. (2016). Therapies for prevention and treatment of Alzheimer’s Disease. Biomed. Res. Intern. 2016:2589276.
Michinaga, S., and Koyama, Y. (2019). Dual roles of astrocyte-derived factors in regulation of blood-brain barrier function after brain damage. Intern. J. Mol. Sci. 20:E571.
Mondello, S., Kobeissy, F., Vestri, A., Hayes, R. L., Kochanek, P. M., and Berger, R. P. (2016). Serum concentrations of ubiquitin C-terminal hydrolase-L1 and glial fibrillary acidic protein after pediatric traumatic brain injury. Sci. Rep. 6:28203.
Mortimer, J. A., Van Duijn, C. M., Chandra, V., Fratiglioni, L., Graves, A. B., Heyman, A., et al. (1991). Head trauma as a risk factor for alzheimer’s disease: A collaborativere-analysis of case-control studies. Int. J. Epidemiol. 20, S28–S35.
Murakami, K., Koide, M., Dumont, T. M., Russell, S. R., Tranmer, B. I., and Wellman, G. C. (2011). Subarachnoid hemorrhage induces gliosis and increased expression of the pro-inflammatory cytokine high mobility group box 1 protein. Transl. Stroke Res. 2, 72–79. doi: 10.1007/s12975-010-0052-2
Nagamoto-Combs, K., McNeal, D. W., Morecraft, R. J., and Combs, C. K. (2007). Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. J. Neurotrauma 24, 1719–1742. doi: 10.1089/neu.2007.0377
Nagamoto-Combs, K., Morecraft, R. J., Darling, W. G., and Combs, C. K. (2010). Long-term gliosis and molecular changes in the cervical spinal cord of the rhesus monkey after traumatic brain injury. J. Neurotrauma 27, 565–585. doi: 10.1089/neu.2009.0966
Najjar, S., Pahlajani, S., De Sanctis, V., Stern, J. N. H., Najjar, A., and Chong, D. (2017). Neurovascular unit dysfunction and blood-brain barrier hyperpermeability contribute to schizophrenia neurobiology: a theoretical integration of clinical and experimental evidence. Front. Psychiatry 8:83. doi: 10.3389/fpsyt.2017.00083
Neuwelt, E., Abbott, N. J., Abrey, L., Banks, W. A., Blakley, B., Davis, T., et al. (2008). Strategies to advance translational research into brain barriers. Lancet Neurol. 7, 84–96. doi: 10.1016/s1474-4422(07)70326-5
Orimo, S. (2017). New development of diagnosis and treatment for Parkinson’s disease. Clin. Neurol. 57, 259–273.
Ott, M., Gogvadze, V., Orrenius, S., and Zhivotovsky, B. (2007). Mitochondria, oxidative stress and cell death. Apoptosis 12, 913–922. doi: 10.1007/s10495-007-0756-2
Papa, L., Mittal, M. K., Ramirez, J., Ramia, M., Kirby, S., Silvestri, S., et al. (2016). In children and youth with mild and moderate traumatic brain injury, glial fibrillary acidic protein out-performs s100beta in detecting traumatic intracranial lesions on computed tomography. J. Neurotrauma 33, 58–64. doi: 10.1089/neu.2015.3869
Pavlovic, D., Pekic, S., Stojanovic, M., and Popovic, V. (2019). Traumatic brain injury: neuropathological, neurocognitive and neurobehavioral sequelae. Pituitary 22, 270–282. doi: 10.1007/s11102-019-00957-9
Peng, L., Parpura, V., and Verkhratsky, A. (2014). Editorial neuroglia as a central element of neurological diseases: an underappreciated target for therapeutic intervention. Curr. Neuropharmacol. 12, 303–307. doi: 10.2174/1570159x12999140829152550
Perez, E. J., Tapanes, S. A., Loris, Z. B., Balu, D. T., Sick, T. J., Coyle, J. T., et al. (2017). Enhanced astrocytic d-serine underlies synaptic damage after traumatic brain injury. J. Clin. Invest. 127, 3114–3125. doi: 10.1172/jci92300
Petty, M. A., and Lo, E. H. (2002). Junctional complexes of the blood-brain barrier: permeability changes in neuroinflammation. Prog. Neurobiol. 68, 311–323. doi: 10.1016/s0301-0082(02)00128-4
Piao, C. S., Holloway, A. L., Hong-Routson, S., and Wainwright, M. S. (2019). Depression following traumatic brain injury in mice is associated with down-regulation of hippocampal astrocyte glutamate transporters by thrombin. J. Cereb. Blood Flow Metab. 39, 58–73. doi: 10.1177/0271678x17742792
Plemel, J. R., Keough, M. B., Duncan, G. J., Sparling, J. S., Yong, V. W., Stys, P. K., et al. (2014). Remyelination after spinal cord injury: is it a target for repair? Prog. Neurobiol. 117, 54–72.
Pupillo, E., Poloni, M., Bianchi, E., Giussani, G., Logroscino, G., Zoccolella, S., et al. (2018). Trauma and amyotrophic lateral sclerosis: a european population-based case-control study from the EURALS consortium. Amyotrop. Later. Scler. Frontotemp. Degen. 19, 118–125. doi: 10.1080/21678421.2017.1386687
Raj, R., Kaprio, J., Korja, M., Mikkonen, E. D., Jousilahti, P., and Siironen, J. (2017). Risk of hospitalization with neurodegenerative disease after moderate-to-severe traumatic brain injury in the working-age population: a retrospective cohort study using the Finnish national health registries. PLoS Med. 14:e1002316. doi: 10.1371/journal.pone.01002316
Ramlackhansingh, A. F., Brooks, D. J., Greenwood, R. J., Bose, S. K., Turkheimer, F. E., Kinnunen, K. M., et al. (2011). Inflammation after trauma: microglial activation and traumatic brain injury. Ann. Neurol. 70, 374–383. doi: 10.1002/ana.22455
Rascher, G., Fischmann, A., Kroger, S., Duffner, F., Grote, E. H., and Wolburg, H. (2002). Extracellular matrix and the blood-brain barrier in glioblastoma multiforme: spatial segregation of tenascin and agrin. Acta Neuropathol. 104, 85–91. doi: 10.1007/s00401-002-0524-x
Rasmussen, M. K., Mestre, H., and Nedergaard, M. (2018). The glymphatic pathway in neurological disorders. Lancet Neurol. 17, 1016–1024. doi: 10.1016/s1474-4422(18)30318-1
Ren, Z., Iliff, J. J., Yang, L., Yang, J., Chen, X., Chen, M. J., et al. (2013). ‘Hit & Run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J. Cereb. Blood Flow Metab. 33, 834–845.
Roberts, G. W., Gentleman, S. M., Lynch, A., and Graham, D. I. (1991). beta A4 amyloid protein deposition in brain after head trauma. Lancet 338, 1422–1423. doi: 10.1016/0140-6736(91)92724-g
Rodriguez, M., Morales, I., Rodriguez-Sabate, C., Sanchez, A., Castro, R., Brito, J. M., et al. (2014). The degeneration and replacement of dopamine cells in Parkinson’s disease: the role of aging. Front. Neuroanat. 8:80. doi: 10.3389/fphar.2019.0080
Rodriguez, M., Rodriguez-Sabate, C., Morales, I., Sanchez, A., and Sabate, M. (2015). Parkinson’s disease as a result of aging. Aging Cell 14, 293–308.
Rothstein, J. D., Dykes-Hoberg, M., Pardo, C. A., Bristol, L. A., Jin, L., Kuncl, R. W., et al. (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686. doi: 10.1016/s0896-6273(00)80086-0
Russo, M. V., and McGavern, D. B. (2016). Inflammatory neuroprotection following traumatic brain injury. Science 353, 783–785. doi: 10.1126/science.aaf6260
Salminen, A., Ojala, J., Kaarniranta, K., Haapasalo, A., Hiltunen, M., and Soininen, H. (2011). Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 34, 3–11. doi: 10.1111/j.1460-9568.2011.07738.x
Scott, G., Ramlackhansingh, A. F., Edison, P., Hellyer, P., Cole, J., Veronese, M., et al. (2016). Amyloid pathology and axonal injury after brain trauma. Neurology 86, 821–828. doi: 10.1212/wnl.0000000000002413
Scott, G., Zetterberg, H., Jolly, A., Cole, J. H., De Simoni, S., Jenkins, P. O., et al. (2018). Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain 141, 459–471. doi: 10.1093/brain/awx339
Seals, R. M., Hansen, J., Gredal, O., and Weisskopf, M. G. (2016). Physical trauma and amyotrophic lateral sclerosis: a population-based study using danish national registries. Am. J. Epidemiol. 183, 294–301. doi: 10.1093/aje/kwv169
Seo, J. H., Maki, T., Maeda, M., Miyamoto, N., Liang, A. C., Hayakawa, K., et al. (2014). Oligodendrocyte precursor cells support blood-brain barrier integrity via TGF-beta signaling. PLoS One 9:e103174. doi: 10.1371/journal.pone.0103174
Sevenich, L. (2018). Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front. Immunol. 9:697.
Shahim, P., Zetterberg, H., Tegner, Y., and Blennow, K. (2017). Serum neurofilament light as a biomarker for mild traumatic brain injury in contact sports. Neurology 88, 1788–1794. doi: 10.1212/wnl.0000000000003912
Sharma, K., Zhang, G., and Li, S. (2015). Chapter 11 - Astrogliosis and Axonal Regeneration. Oxford: Academic Press.
Shetty, A. K., Mishra, V., Kodali, M., and Hattiangady, B. (2014). Blood brain barrier dysfunction and delayed neurological deficits in mild traumatic brain injury induced by blast shock waves. Front. Cell. Neurosci. 8:232. doi: 10.3389/fphar.2019.00232
Shin, J. Y., Fang, Z. H., Yu, Z. X., Wang, C. E., Li, S. H., and Li, X. J. (2005). Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 171, 1001–1012. doi: 10.1083/jcb.200508072
Shively, S. B., Edgerton, S. L., Iacono, D., Purohit, D. P., Qu, B. X., Haroutunian, V., et al. (2017). Localized cortical chronic traumatic encephalopathy pathology after single, severe axonal injury in human brain. Acta Neuropathol. 133, 353–366. doi: 10.1007/s00401-016-1649-7
Shlosberg, D., Benifla, M., Kaufer, D., and Friedman, A. (2010). Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 6, 393–403. doi: 10.1038/nrneurol.2010.74
Shokri-Kojori, E., Wang, G. J., Wiers, C. E., Demiral, S. B., Guo, M., Kim, S. W., et al. (2018). beta-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. U.S.A. 115, 4483–4488. doi: 10.1073/pnas.1721694115
Simon, D. W., McGeachy, M. J., Bayir, H., Clark, R. S., Loane, D. J., and Kochanek, P. M. (2017). The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 13, 171–191. doi: 10.1038/nrneurol.2017.13
Sleigh, J. N., Tosolini, A. P., Gordon, D., Devoy, A., Fratta, P., Fisher, E. M. C., et al. (2020). Mice carrying ALS mutant TDP-43, but not mutant FUS, display in vivo defects in axonal transport of signaling endosomes. Cell Rep. 30, 3655–3662.
Slemmer, J. E., Shacka, J. J., Sweeney, M. I., and Weber, J. T. (2008). Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr. Med. Chem. 15, 404–414. doi: 10.2174/092986708783497337
Smith, D. H., Chen, X. H., Pierce, J. E., Wolf, J. A., Trojanowski, J. Q., Graham, D. I., et al. (1997). Progressive atrophy and neuron death for one year following brain trauma in the rat. J. Neurotrauma 14, 715–727. doi: 10.1089/neu.1997.14.715
Smith, D. H., Johnson, V. E., and Stewart, W. (2013). Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat. Rev. Neurol. 9, 211–221. doi: 10.1038/nrneurol.2013.29
Sofroniew, M. V. (2009). dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. doi: 10.1016/j.tins.2009.08.002
Sofroniew, M. V. (2015). Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 16, 249–263. doi: 10.1038/nrn3898
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Sullan, M. J., Asken, B. M., Jaffee, M. S., DeKosky, S. T., and Bauer, R. M. (2018). Glymphatic system disruption as a mediator of brain trauma and chronic traumatic encephalopathy. Neurosci. Biobehav. Rev. 84, 316–324. doi: 10.1016/j.neubiorev.2017.08.016
Susarla, B. T., Villapol, S., Yi, J. H., Geller, H. M., and Symes, A. J. (2014). Temporal patterns of cortical proliferation of glial cell populations after traumatic brain injury in mice. ASN Neuro 6, 159–170.
Suzuki, H., Ayer, R., Sugawara, T., Chen, W., Sozen, T., Hasegawa, Y., et al. (2010). Protective effects of recombinant osteopontin on early brain injury after subarachnoid hemorrhage in rats. Crit. Care Med. 38, 612–618. doi: 10.1097/ccm.0b013e3181c027ae
Sweeney, M. D., Ayyadurai, S., and Zlokovic, B. V. (2016). Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci. 19, 771–783. doi: 10.1038/nn.4288
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. doi: 10.1038/nrneurol.2017.188
Tagge, C. A., Fisher, A. M., Minaeva, O. V., Gaudreau-Balderrama, A., Moncaster, J. A., Zhang, X. L., et al. (2018). Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 141, 422–458. doi: 10.1093/brain/awx350
Tilling, T., Engelbertz, C., Decker, S., Korte, D., Huwel, S., and Galla, H. J. (2002). Expression and adhesive properties of basement membrane proteins in cerebral capillary endothelial cell cultures. Cell Tissue Res. 310, 19–29. doi: 10.1007/s00441-002-0604-1
Tilling, T., Korte, D., Hoheisel, D., and Galla, H. J. (1998). Basement membrane proteins influence brain capillary endothelial barrier function in vitro. J. Neurochem. 71, 1151–1157. doi: 10.1046/j.1471-4159.1998.71031151.x
Tran, H. T., LaFerla, F. M., Holtzman, D. M., and Brody, D. L. (2011). Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J. Neurosci. 31, 9513–9525. doi: 10.1523/jneurosci.0858-11.2011
Tso, M. K., and Macdonald, R. L. (2014). Subarachnoid hemorrhage: a review of experimental studies on the microcirculation and the neurovascular unit. Transl. Stroke Res. 5, 174–189. doi: 10.1007/s12975-014-0323-4
Uryu, K., Chen, X. H., Martinez, D., Browne, K. D., Johnson, V. E., Graham, D. I., et al. (2007). Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 208, 185–192. doi: 10.1016/j.expneurol.2007.06.018
Verkhratsky, A., Parpura, V., Pekna, M., Pekny, M., and Sofroniew, M. (2014). Glia in the pathogenesis of neurodegenerative diseases. Biochem. Soc. Trans. 42, 1291–1301. doi: 10.1042/bst20140107
Viviani, B., Boraso, M., Marchetti, N., and Marinovich, M. (2014). Perspectives on neuroinflammation and excitotoxicity: a neurotoxic conspiracy? Neurotoxicology 43, 10–20. doi: 10.1016/j.neuro.2014.03.004
Voskuhl, R. R., Peterson, R. S., Song, B., Ao, Y., Morales, L. B., Tiwari-Woodruff, S., et al. (2009). Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J. Neurosci. 29, 11511–11522. doi: 10.1523/jneurosci.1514-09.2009
Walz, W. (2000). Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 36, 291–300. doi: 10.1016/s0197-0186(99)00137-0
Wang, Y., Ji, X., Leak, R. K., Chen, F., and Cao, G. (2017). Stem cell therapies in age-related neurodegenerative diseases and stroke. Ageing Res. Rev. 34, 39–50. doi: 10.1016/j.arr.2016.11.002
Weilinger, N. L., Maslieieva, V., Bialecki, J., Sridharan, S. S., Tang, P. L., and Thompson, R. J. (2013). Ionotropic receptors and ion channels in ischemic neuronal death and dysfunction. Acta Pharmacol. Sin. 34, 39–48. doi: 10.1038/aps.2012.95
Welch, R. D., Ayaz, S. I., Lewis, L. M., Unden, J., Chen, J. Y., Mika, V. H., et al. (2016). Ability of serum glial fibrillary acidic protein, Ubiquitin C-terminal hydrolase-L1, and S100B to differentiate normal and abnormal head computed tomography findings in patients with suspected mild or moderate traumatic brain injury. J. Neurotrauma 33, 203–214. doi: 10.1089/neu.2015.4149
Wiesner, D., Tar, L., Linkus, B., Chandrasekar, A., Olde Heuvel, F., Dupuis, L., et al. (2018). Reversible induction of TDP-43 granules in cortical neurons after traumatic injury. Exp. Neurol. 299, 15–25. doi: 10.1016/j.expneurol.2017.09.011
Williams, S. M., Peltz, C., Yaffe, K., Schulz, P., and Sierks, M. R. (2018). CNS disease-related protein variants as blood-based biomarkers in traumatic brain injury. Neurology 91, 702–709. doi: 10.1212/wnl.0000000000006322
Wimo, A., Guerchet, M., Ali, G.-C., Wu, Y.-T., Prina, A. M., Winblad, B., et al. (2017). The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimer Dement. 13, 1–7. doi: 10.1016/j.jalz.2016.07.150
Wu, J., He, J., Tian, X., Zhong, J., Li, H., and Sun, X. (2020). Activation of the hedgehog pathway promotes recovery of neurological function after traumatic brain injury by protecting the neurovascular unit. Transl. Stroke Res. doi: 10.1007/s12975-019-00771-2 [Epub ahead of print].
Xiong, K. L., Zhu, Y. S., and Zhang, W. G. (2014). Diffusion tensor imaging and magnetic resonance spectroscopy in traumatic brain injury: a review of recent literature. Brain Imag. Behav. 8, 487–496. doi: 10.1007/s11682-013-9288-2
Yang, Z., and Wang, K. K. (2015). Glial fibrillary acidic protein: from intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 38, 364–374. doi: 10.1016/j.tins.2015.04.003
Ye, Z. C., Wyeth, M. S., Baltan-Tekkok, S., and Ransom, B. R. (2003). hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci. 23, 3588–3596. doi: 10.1523/jneurosci.23-09-03588.2003
Zanier, E. R., Bertani, I., Sammali, E., Pischiutta, F., Chiaravalloti, M. A., Vegliante, G., et al. (2018). Induction of a transmissible tau pathology by traumatic brain injury. Brain 141, 2685–2699.
Zenaro, E., Piacentino, G., and Constantin, G. (2017). The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 107, 41–56.
Zhang, D., Xiao, M., Wang, L., and Jia, W. (2019). Blood-based glutamate scavengers reverse traumatic brain injury-induced synaptic plasticity disruption by decreasing glutamate level in hippocampus interstitial fluid, but not cerebral spinal fluid, in vivo. Neurotox. Res. 35, 360–372. doi: 10.1007/s12640-018-9961-8
Zhang, J. H., Badaut, J., Tang, J., Obenaus, A., Hartman, R., and Pearce, W. J. (2012). The vascular neural network–a new paradigm in stroke pathophysiology. Nat. Rev. Neurol. 8, 711–716. doi: 10.1038/nrneurol.2012.210
Zhang, X., Chen, Y., Jenkins, L. W., Kochanek, P. M., and Clark, R. S. (2005). Bench-to-bedside review: apoptosis/programmed cell death triggered by traumatic brain injury. Crit. Care 9, 66–75.
Zhao, H., Liu, Y., Zeng, J., Li, D., and Huang, Y. (2018). Troxerutin cerebroprotein hydrolysate injection ameliorates neurovascular injury induced by traumatic brain injury - via endothelial nitric oxide synthase pathway regulation. Int. J. Neurosci. 128, 1118–1127. doi: 10.1080/00207454.2018.1486828
Zhao, Z., Nelson, A. R., Betsholtz, C., and Zlokovic, B. V. (2015). Establishment and dysfunction of the blood-brain barrier. Cell 163, 1064–1078. doi: 10.1016/j.cell.2015.10.067
Zhou, Y., Shao, A., Xu, W., Wu, H., and Deng, Y. (2019). Advance of stem cell treatment for traumatic brain injury. Front. Cell. Neurosci. 13:301. doi: 10.3389/fphar.2019.00301
Zhou, Y., Shao, A., Yao, Y., Tu, S., Deng, Y., and Zhang, J. (2020). Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Cell Commun. Signal. 18:62.
Ziebell, J. M., and Morganti-Kossmann, M. C. (2010). Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7, 22–30. doi: 10.1016/j.nurt.2009.10.016
Keywords: traumatic brain injury, neurodegenerative disease, neurovascular unit, hypothesis, neuroinflammation, proteinopathy, oxidative stress, blood–brain barrier
Citation: Zhou Y, Chen Q, Wang Y, Wu H, Xu W, Pan Y, Gao S, Dong X, Zhang JH and Shao A (2020) Persistent Neurovascular Unit Dysfunction: Pathophysiological Substrate and Trigger for Late-Onset Neurodegeneration After Traumatic Brain Injury. Front. Neurosci. 14:581. doi: 10.3389/fnins.2020.00581
Received: 03 February 2020; Accepted: 12 May 2020;
Published: 09 June 2020.
Edited by:
Eng-King Tan, National Neuroscience Institute (NNI), SingaporeReviewed by:
Ye Xiong, Henry Ford Health System, United StatesZezong Gu, University of Missouri, United States
Copyright © 2020 Zhou, Chen, Wang, Wu, Xu, Pan, Gao, Dong, Zhang and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anwen Shao, MjExMTgxMTZAemp1LmVkdS5jbg==; YW53ZW5zaGFvQHNpbmEuY29t