 Dylan J. Dues
Dylan J. Dues Darren J. Moore
Darren J. Moore- Center for Neurodegenerative Science, Van Andel Institute, Grand Rapids, MI, United States
Mutations in leucine-rich repeat kinase 2 (LRRK2) instigate an autosomal dominant form of Parkinson’s disease (PD). Despite the neuropathological heterogeneity observed in LRRK2-PD, accumulating evidence suggests that alpha-synuclein and tau pathology are observed in a vast majority of cases. Intriguingly, the presence of protein aggregates spans both LRRK2-PD and idiopathic disease, supportive of a common pathologic mechanism. Thus, it is important to consider how LRRK2 mutations give rise to such pathology, and whether targeting LRRK2 might modify the accumulation, transmission, or toxicity of protein aggregates. Likewise, it is not clear how LRRK2 mutations drive PD pathogenesis, and whether protein aggregates are implicated in LRRK2-dependent neurodegeneration. While animal models have been instrumental in furthering our understanding of a potential interaction between LRRK2 and protein aggregation, the biology is far from clear. We aim to provide a thoughtful overview of the evidence linking LRRK2 to protein aggregation in animal models.
Introduction
Leucine-rich repeat kinase 2 (LRRK2) is a large multidomain and dual enzymatic protein that exists intracellularly and predominantly as a dimer in its active form (Islam and Moore, 2017). LRRK2 has a number of purported functions, the most well studied of which include roles in the endolysosomal pathway and autophagy (Berwick et al., 2019; Cunningham and Moore, 2020). Most significant, however, is the role that LRRK2 plays in human health. Mutations in LRRK2 are a common genetic cause of Parkinson’s disease (PD) (Biskup and West, 2008). To date, at least seven missense mutations have been identified as causative in disease. These pathogenic mutations appear to cluster within the Roc-COR tandem (R1441C/G/H, N1437H, and Y1699C) and kinase (G2019S and I2020T) domains, implicating both GTPase and kinase activity in disease pathogenesis (Healy et al., 2008; Islam and Moore, 2017). Additional findings from GWAS have repeatedly linked variation at the LRRK2 locus as an important risk factor in sporadic PD susceptibility (Nalls et al., 2014, 2019). Thus, LRRK2 represents a pleomorphic risk locus for PD pathogenesis as both a causative and risk-modifying factor.
Familial PD caused by LRRK2 mutations (LRRK2-PD) generally develops as a late-onset autosomal dominant disorder and is thought to be driven by a toxic gain-of-function mechanism (Paisán-Ruıíz et al., 2004; Zimprich et al., 2004). A consensus view holds that elevated kinase activity underlies the pathogenic nature of LRRK2, as all causative mutations appear to enhance kinase activity, though GTPase activity seems to be required as well (Sheng et al., 2012; Steger et al., 2016; Nguyen et al., 2019). Despite LRRK2 mutations being one of the most common identifiable causes of PD, the vast majority of PD occurs via an unknown etiology. In spite of this, recent evidence suggests that LRRK2 activation (in the absence of mutation) may be present in sporadic PD (Maio et al., 2018). While being clinically indistinguishable from sporadic disease, LRRK2-PD frequently harbors neuropathological features synonymous with sporadic PD (Biskup and West, 2008). These features include dopaminergic neurodegeneration in the midbrain along with the appearance of intraneuronal proteinaceous aggregates. These inclusions are generally associated with neurotoxicity, and inclusion burden in cortical and limbic regions is associated with the manifestation of cognitive and neuropsychiatric symptoms, a feature that spans both sporadic disease and LRRK2-PD (Aarsland et al., 2005; Braak et al., 2005; Goldwurm et al., 2006; Smith et al., 2019). Thus, persuasive clinical findings infer that mutations in LRRK2 might influence PD-associated pathology and provoke PD pathogenesis through mechanisms similar to those present in sporadic disease. While the application of LRRK2 pathobiology to idiopathic PD is alluring, it is critical that conserved pathologic features are reconciled with experimental evidence. Accordingly, the contribution of LRRK2 toward protein aggregation is of great interest and likely to be of therapeutic relevance.
A looming question in LRRK2 pathophysiology is whether protein aggregation is a primary or secondary effect of LRRK2 mutations. Given the variety of neuropathology in LRRK2-PD, in many cases being diverse (i.e., co-pathology), it remains to be elucidated whether aggregation is simply resultant to LRRK2-mediated cellular dysfunction or whether pathogenic LRRK2 activity plays a more direct role in driving inclusion formation. Likewise, the requirement of protein aggregate substrates, such as α-synuclein or tau (reviewed in Moussaud et al., 2014), in mediating LRRK2-dependent neurodegeneration is far from clear. Notably, “pure” nigral degeneration in the absence of protein aggregate pathology, though rare, has been observed in LRRK2-PD (Gaig et al., 2007; Takanashi et al., 2018). While it is possible that soluble oligomeric protein or other toxic species are still present in these atypical cases, the non-appearance of typical Lewy pathology composed of insoluble fibrillar α-synuclein, or neurofibrillary tangles composed of tau, suggest that inclusion pathology may be a by-product of LRRK2-PD pathogenesis rather than a required element. Even when present, it may be the case that the composition of protein aggregates in LRRK2-PD is wholly distinct from that of sporadic disease. Toward this concept, examination of α-synuclein derived from the LRRK2-PD brain was found to possess a divergent biochemical profile despite a comparable histopathological appearance to sporadic PD (Mamais et al., 2013). Further, the range of pathological findings in the LRRK2-PD have been far from consistent. Though accruing post-mortem studies suggest that the common G2019S mutation appears to more faithfully produce typical Lewy pathology, examination of LRRK2-PD caused by other mutations has found substantial variation. Divergent pathology has even been observed in LRRK2-PD cases between affected members of the same family harboring an identical missense mutation (Zimprich et al., 2004). Thus, while the specific LRRK2 mutation may have an impact on neuropathological variation, it appears likely that stochastic events may also play a role in the formation of protein aggregates. Though the apparent heterogeneity of LRRK2-PD neuropathology might at first suggest that inclusion formation is merely non-specific, accumulating evidence suggest a more selective and subtle impact of LRRK2 in facilitating protein aggregation. Toward addressing some of these questions, past and present genetic studies have granted some insight into the interplay of LRRK2 and the aggregate-prone proteins, α-synuclein and tau.
Common variation at the SNCA (encoding α-synuclein) and MAPT (encoding tau) loci have been associated with PD risk (Davis et al., 2015). Additionally, variation at these same loci has been implicated in affecting aspects of LRRK2-PD, such as the age of disease-onset (Kobayashi et al., 2006; Golub et al., 2009; Gan-Or et al., 2011; Botta-Orfila et al., 2012). Thus, LRRK2, tau, and α-synuclein appear to converge in mysterious and seemingly complex ways in PD pathogenesis. Accordingly, elucidation of the interplay between LRRK2 and PD-associated proteins α-synuclein and tau is highly relevant to the application of LRRK2-targeted therapeutics in suppressing PD-associated neuropathology. Here, we will provide a concise review of current advances in understanding the contribution of LRRK2 toward protein aggregation in human studies and experimental models.
Protein Aggregation in Human LRRK2-PD
Substantial evidence supports a role for protein aggregation in the pathophysiology of PD (Moore et al., 2005). In sporadic disease, the presence of Lewy pathology composed of misfolded α-synuclein protein is a neuropathological hallmark. While predominantly restricted to the brainstem, Lewy pathology is believed to seemingly progress in a caudal to rostral fashion throughout the brain (Braak et al., 2003; Brundin and Melki, 2017). The observation of Lewy pathology in surviving dopaminergic neurons of the substantia nigra has led to the hypothesis that inclusions are ultimately toxic, leading to the progressive loss of dopaminergic innervation of the striatum and associated hypokinetic motor phenotype (Poewe et al., 2017). Further, an abundance of Lewy pathology in cortical and limbic brain regions has been associated with the clinical correlates of dementia and neuropsychiatric disease, respectively (Aarsland et al., 2005; Braak et al., 2005; Smith et al., 2019). Thus, the presence of α-synuclein aggregation appears to drive a heterogenous clinical phenotype by disrupting functional circuits in addition to driving progressive neurodegeneration.
Importantly, protein aggregation encompasses both sporadic and LRRK2-PD, suggesting a common pathologic basis of disease. However, whether neurodegeneration in sporadic and LRRK2-PD are equally dependent on protein aggregation, and whether all LRRK2 mutations act through a common pathway is unknown. While progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta remains the primary pathology, a feature that is universal across PD etiology, the nature, extent, and contribution of protein aggregation is far from clear. The majority of post-mortem neuropathological assessments have demonstrated that typical Lewy pathology is a predominant finding in LRRK2-PD (Biskup and West, 2008). This is likely due to the overrepresentation of the G2019S mutation in post-mortem studies, as the G2019S mutation is by far the most common LRRK2 mutation and appears to present with typical Lewy pathology. Pathological examination of LRRK2-PD brains harboring the G2019S mutation has identified typical, transitional, and diffuse Lewy pathology, tau and neurofibrillary tangle pathology, TDP-43 aggregates, ubiquitin inclusions, and even “pure” neurodegeneration without overt pathology (Paisán-Ruıíz et al., 2004; Zimprich et al., 2004; Biskup and West, 2008; Wider et al., 2010; Ujiie et al., 2012; Ling et al., 2013). Thus, while α-synuclein aggregation appears to be the most prevalent finding, neuropathology in LRRK2-PD is pleomorphic. Aside from the G2019S mutation, a number of other post-mortem studies incorporating less common LRRK2 mutations have discovered equally diverse pathological findings. Notably, several members of an affected family harboring the R1441C mutation were revealed to exhibit either Lewy pathology, neurofibrillary tangle pathology, or ubiquitin pathology (Zimprich et al., 2004). Ubiquitin inclusions in the absence of Lewy pathology was also observed in two patients with the Y1699C mutation (Zimprich et al., 2004). Intriguingly, no inclusions were detected in a patient with the R1441G mutation (Martí-Massó et al., 2009). Similarly, the I2020T mutation was demonstrated to induce nigral degeneration without significant Lewy pathology, though some cases were later found to harbor extensive brainstem tau pathology (Ujiie et al., 2012; Takanashi et al., 2018).
More recent observations have amplified the significance of tau pathology in LRRK2-PD. Indeed, a recent examination of post-mortem LRRK2-PD brains further illuminated the prominence of tau aggregation as a common pathologic substrate in LRRK2-PD (Henderson et al., 2019c). Though the sample size was limited, a larger proportion of LRRK2-PD brains were found to harbor tau pathology relative to α-synuclein pathology (100% vs. ∼64%). While phosphorylated α-synuclein (pSer129+) pathology was abundant in the substantia nigra, amygdala, hippocampus, and cingulate cortex, phosphorylated tau (AT8+) was more prominent in the amygdala, hippocampus, and entorhinal cortex. The regional mapping of each particular protein to a pathologic “territory” is plausibly in reference to a cell or tissue-dependent aspect of LRRK2 pathobiology. Given the preponderance of LRRK2-PD cases harboring mixed pathological features, future studies should make note of the regional distribution of pathology in dissecting mechanisms related to LRRK2 and protein aggregation. Returning to the findings of the above study, the midbrain was determined to be largely devoid of tau pathology, suggesting that while tau aggregation may be a typical pathologic feature in LRRK2-PD, the application of tau pathology to dopaminergic neuron loss remains ambiguous. The burden of tau pathology was, however, strongly associated with the level of α-synuclein pathology, reminiscent of evidence supporting the potential of tau and α-synuclein cross seeding (Guo et al., 2013). Neither tau nor α-synuclein burden was found to be associated with the level of amyloid-β pathology, further supporting a synergistic relationship between tau and α-synuclein rather than a general disruption in neuronal proteostasis. While a direct interaction between LRRK2 and tau or α-synuclein seems unlikely, it is notable that most regions harboring mixed pathology only rarely exhibited co-pathology within the same cell. It is interesting to hypothesize why some neuronal populations appear to foster one type of protein aggregate, while other populations of neurons another. As well, from an in vivo perspective, the requirement of tau or α-synuclein cross seeding in fostering LRRK2-dependent neurodegeneration is fascinating. Toward this concept, examining region-autonomous variation in LRRK2 biology may be a promising endeavor. While human studies have proven invaluable to our collective understanding of protein aggregation in LRRK2-PD, much of what is known regarding the mechanistic role of LRRK2 in driving protein aggregation and neuropathology has been garnered from animal models.
LRRK2 and Protein Aggregation in Animal Models
The identification of pathogenic mutations in the LRRK2 gene has allowed for the generation of LRRK2-PD animal models. These models have provided valuable insight into the biology and pathobiology of LRRK2, though many key questions remain unanswered. While extensive work has been conducted in the development and characterization of these models, here, we will focus specifically on observations of protein aggregation and neuropathology. Toward this aim, experimental findings from traditional genetic, viral vector, and protein-based animal models will be discussed.
LRRK2 in Transgenic and Knockin Models
Multiple pathogenic substitutions in the R1441 residue of the GTPase domain have been identified in LRRK2-PD and are associated with variable neuropathology (Zimprich et al., 2004). Some of the first LRRK2 animal models sought to examine the impact of mutations at this residue in the rodent brain (Table 1), and the development of R1441C knockin mice led to a primary identification of altered dopaminergic neurotransmission. However, the integrity of the nigrostriatal tract in R1441C knockin mice remained relatively unaffected (Tong et al., 2009). Furthermore, biochemical and histological assessment of these mice failed to detect any indications of tau, α-synuclein, or ubiquitin aggregation (Tong et al., 2009). In contrast, a BAC transgenic mouse line expressing human R1441G LRRK2 was found to harbor modest signs of tau pathology. Specifically, elevated protein levels of phosphorylated tau were identified in R1441G BAC mice relative to controls. In addition, neuritic inclusion pathology (AT8+) was observed in the cortex and striatum (Li et al., 2009). While neurodegeneration was not observed in either of these genetic models, subtle alterations in the dopaminergic system were detected, along with a promising implication for involvement of tau.
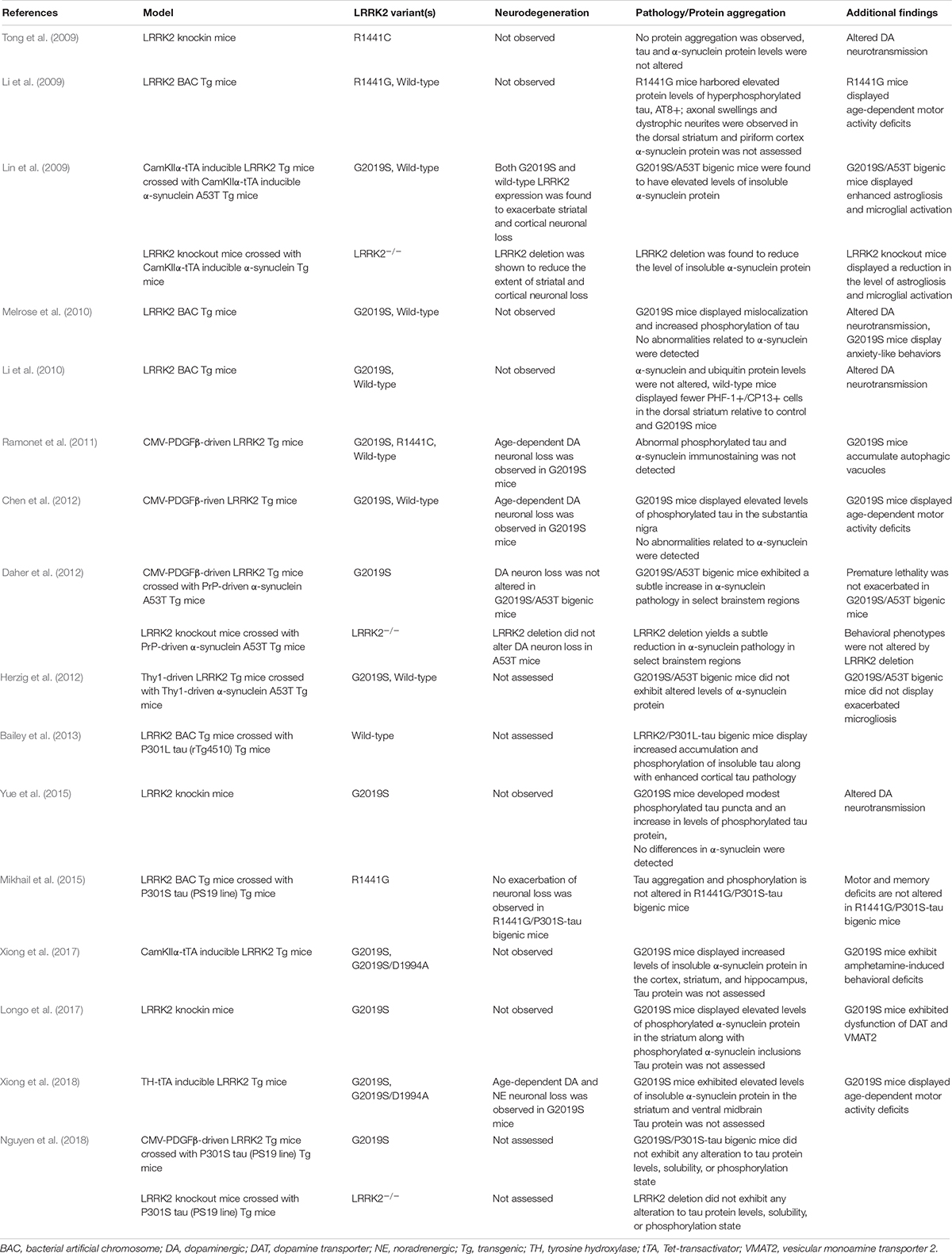
Table 1. LRRK2 in transgenic and knockin models.
Subsequent genetic models sought to examine the impact of the G2019S mutation (Table 1). To date, a number of G2019S LRRK2 transgenic models have been developed, though findings have been mixed. Melrose et al. found that human G2019S LRRK2 BAC transgenic mice displayed phosphorylated tau inclusions (CP13+, 12E8+) in cortical and limbic brain regions at advanced ages (18–24 month) (Melrose et al., 2010). Human wild-type LRRK2 BAC mice were also examined in this study and found to harbor tau pathology. However, in the wild-type LRRK2 line, tau pathology was primarily restricted to the hippocampus and to a much lesser burden than in G2019S mice. Notably, neither background was found to exhibit any apparent α-synuclein pathology. Similar to the R1441C knockin model, these mice also displayed abnormal dopaminergic neurotransmission, furthering support for a common pathogenic role in modulating synaptic function (Melrose et al., 2010). While Melrose et al. found an additive effect of the G2019S mutation on tau phosphorylation, a separate study assessing a different mouse G2019S LRRK2 BAC transgenic model failed to detect any alterations (Li et al., 2010; Melrose et al., 2010). In contrast, accumulation of phosphorylated tau inclusions (PHF1+, CP13+) was not significantly elevated relative to control mice (Li et al., 2010). While a number of factors might explain this discrepancy, including variation in strain background, or transgene copy number and/or genomic integration site, it is notable that neither model exhibited neurodegeneration of midbrain dopaminergic neurons (Li et al., 2010; Melrose et al., 2010).
Despite the lack of overt neurodegeneration in these early LRRK2 mouse models, although exhibiting modest neuropathologic phenotypes, parallel efforts developed a number of additional models focused on selectively boosting transgene expression within dopaminergic neurons of the substantia nigra (Table 1). While some of these models were successful in provoking dopaminergic neurodegeneration, the impact of protein aggregation in many of these models was generally not assessed or simply not detected (Ramonet et al., 2011; Chen et al., 2012; Tsika et al., 2014; Liu et al., 2015). In one of these studies, transgenic mice expressing human G2019S LRRK2 under control of a CMV-enhanced human PDGFβ promoter were found to have an age-dependent (12–16 months) loss of dopaminergic neurons in the substantia nigra relative to controls expressing human wild-type LRRK2 from the same promoter (Chen et al., 2012). While no changes in α-synuclein or ubiquitin protein levels were observed, a nearly two-fold increase in phosphorylated tau (AT8+) was detected in the substantia nigra of G2019S LRRK2 mice (Chen et al., 2012). Unfortunately, the requirement of tau as a necessary pathologic substrate for neurodegeneration in this model was not determined. In a separate study, the G2019S mutation was expressed throughout the mouse forebrain under an inducible CamKIIα-tTA (Tet-transactivator) conditional expression system (Xiong et al., 2017). This study also examined a kinase-inactive variant, G2019S/D1994A LRRK2, under the same expression system (Xiong et al., 2017). While dopaminergic neurodegeneration was not observed with this approach, a substantial increase in the level of insoluble α-synuclein was detected in select forebrain regions of G2019S LRRK2 mice but not in G2019S/D1994A LRRK2 mice (Xiong et al., 2017). However, a similar approach examining G2019S and G2019S/D1994A LRRK2 mutations under a TH-tTA conditional expression system discovered robust age- and kinase-dependent neurodegeneration coupled with an increase in the levels of insoluble α-synuclein (Xiong et al., 2018).
In order to examine the G2019S mutation under more physiologic conditions, more recent studies have utilized G2019S LRRK2 knockin mice. While G2019S LRRK2 BAC transgenic mice previously displayed alterations in tau phosphorylation, Yue et al. (2015) found that G2019S knockin mice failed to exhibit, similarly, significant changes. However, these mice did harbor modest pathology upon examination at advanced age (18 months). Cytoplasmic accumulation of phosphorylated tau (pSer202+) was observed, including some puncta formation and neuritic pathology (Yue et al., 2015). More readily apparent were deficits in striatal dopamine release and altered mitochondrial dynamics, again supporting synaptic alterations as an early sign of LRRK2 pathogenicity (Yue et al., 2015). In a separate study, G2019S LRRK2 knockin mice were found to exhibit a two-fold increase in the level of phosphorylated α-synuclein (pSer129) in the striatum (Longo et al., 2017). This finding was coupled with histological evidence of striatal α-synuclein inclusions. Importantly, both of these findings were age-dependent (12 months) (Longo et al., 2017). In a follow-up study, primary neuronal cultures generated from G2019S knockin mice were found to exhibit elevated levels of both phosphorylated tau (AT8+) and insoluble α-synuclein (Schapansky et al., 2018). Remarkably, administration of a LRRK2 kinase inhibitor was demonstrated to reverse the effect of the G2019S mutation on altered α-synuclein solubility, though the effect of kinase inhibition on tau phosphorylation was not determined (Schapansky et al., 2018). Certain species of α-synuclein and tau are thought to synergistically interact, and experimental evidence suggest that α-synuclein and tau are able to cross seed (Lim, 2019). While the finding of alterations to both α-synuclein and tau in a LRRK2 model are intriguing, the process by which this occurs may not necessarily be linear, and subsequent studies should address this concern.
While the collective findings from LRRK2 genetic animal models have not fully clarified the impact of LRRK2 on protein aggregation, the use of LRRK2 mice in a cross breeding approach has allowed for an alternative strategy in dissecting this relationship (Table 1). One of the first in vivo studies examining a potential interaction between LRRK2 and α-synuclein found that crossing inducible transgenic mice lines expressing human LRRK2 (either wild-type or G2019S) and A53T α-synuclein led to an increase in the burden of α-synuclein pathology (Lin et al., 2009). The mouse lines used in this study were generated using the CamKIIα-tTA conditional expression system, with transgene expression being restricted to the forebrain. While both LRRK2 lines independently were without phenotype, crossing either line with A53T mice had an additive effect on the level of cortical α-synuclein pathology. Intriguingly, A53T mice were also crossed with LRRK2 knockout mice and a dramatic reduction in the burden of α-synuclein pathology was observed. Taken together, these findings first suggested that LRRK2 may be involved in the progression of α-synuclein neuropathology (Lin et al., 2009). Other studies examining the interaction of LRRK2 and α-synuclein have suggested that the impact of LRRK2 on protein aggregation may be more complex. In two related studies, distinct human mutant α-synuclein transgenic mouse models with predominant hindbrain pathology were found to have no pathological interaction with LRRK2 transgenic or knockout mice (Daher et al., 2012; Herzig et al., 2012). It is possible that the discrepancy between these two studies and the former result from a regionally-restricted interaction between LRRK2 and α-synuclein, with greater potential for interaction in the forebrain where LRRK2 expression is enriched (Biskup et al., 2006; Higashi et al., 2007; West et al., 2014).
Similar to the potential interaction between LRRK2 and α-synuclein, a pathologic interplay between LRRK2 and tau has also been pursued using a cross breeding approach (Table 1). A human wild-type LRRK2 BAC transgenic mouse line crossed with inducible transgenic tau mice (expressing human P301L mutant tau under a CamKIIα-tTA conditional expression system) was shown to enhance cortical tau aggregation and increase tau phosphorylation (AT8+, CP13+, MC1+) (Bailey et al., 2013). In contrast, a human R1441G LRRK2 BAC transgenic mouse line crossed with human P301S tau mice (driven by the mouse prion promoter, PrP) found no significant interaction (Mikhail et al., 2015). While the P301S tau mice used in this study developed widespread tau pathology, no additional alterations in tau aggregation or phosphorylation were identified in these bigenic mice (Mikhail et al., 2015). Similarly, modest hippocampal neuron loss specific to the P301S tau mouse background was not exacerbated by mutant LRRK2 (Mikhail et al., 2015). As this study did not generate P301S tau/LRRK2 knockout mice, it was not clear whether tau pathology was dependent on endogenous LRRK2 activity. However, a subsequent study was able to address this question. Nguyen et al. crossed the same PrP-P301S tau mice with either LRRK2 knockout or human G2019S transgenic mice (under the CMVe-PDGFβ promoter) (Nguyen et al., 2018). Neither genetic deletion of LRRK2 nor pathogenic mutation altered tau protein levels, solubility, or phosphorylation state. In addition, histopathological assessment of these mice found no interaction between tau pathology and LRRK2 genotype (Nguyen et al., 2018).
LRRK2 in Viral Vector-Based Models
Additional evidence supporting the involvement of LRRK2 in protein aggregation, propagation, and toxicity has come from the implementation of viral vectors in rodent models (Table 2). Importantly, the use of viral vectors offers several advantages over traditional genetic models. First, viral vector delivery offers spatiotemporal control of gene expression and avoids the potential confounding effects of developmental compensation. As well, higher levels of transgene expression are likely attainable when using this approach. In a practical sense, viral vectors may be applied across multiple genetic lines, amplifying the utility of viral vectors toward understanding disease-relevant interactions. Large-capacity viral vectors carrying full-length human LRRK2 have been used to uncover the impact of LRRK2 overexpression and pathogenic mutations on nigrostriatal pathway integrity (Table 2).
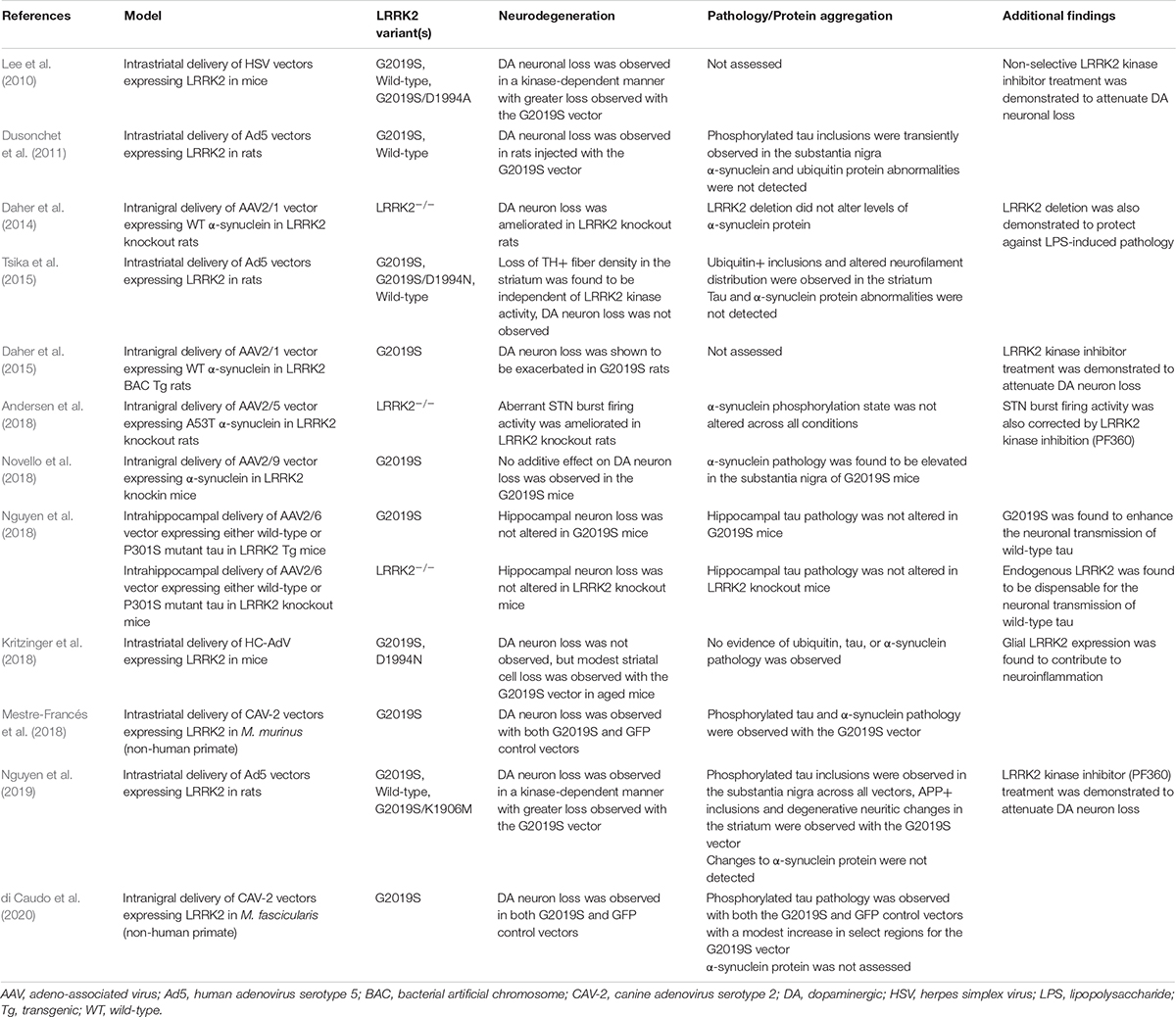
Table 2. LRRK2 in viral vector-based models.
In a herpes simplex virus (HSV) vector mouse model, either human wild-type, G2019S, or G2019S/D1994A LRRK2 variants (kinase-inactive) were delivered via intrastriatal injection (Lee et al., 2010). Using this approach, modest dopaminergic neuronal loss was observed with the wild-type variant with significantly greater loss with the G2019S variant. Concurrently, the kinase-inactive variant did not induce any observable loss and concurrent administration of early yet non-selective LRRK2 kinase inhibitors was shown to attenuate neurodegeneration in this model. Accordingly, these findings support the importance of functional kinase activity in mediating dopaminergic neurodegeneration. Unfortunately, protein aggregation was not assessed in this study so it is not clear how tau or α-synuclein might be involved with LRRK2 delivered via HSV vectors.
Another study examining human LRRK2 utilized human adenoviral vectors (Ad5) driven by a neuronal-specific synapsin-1 promoter and found similar findings to the HSV model (Dusonchet et al., 2011). In this approach, intrastriatal delivery of Ad5 carrying G2019S LRRK2 induced robust neurodegeneration in the substantia nigra of adult rats, while no effect was observed with the wild-type LRRK2 or GFP vectors. Intriguingly, transient induction of neuritic inclusions was observed in the substantia nigra with both wild-type and G2019S vectors. These inclusions exhibited positive immunostaining for phosphorylated tau (AT8+) but not for α-synuclein (Dusonchet et al., 2011). Following these initial findings, a second study examined the impact of the Ad5-LRRK2 on striatal pathology (Tsika et al., 2015). Intraneuronal ubiquitin-positive inclusions were observed in the striatum of rats injected with the G2019S LRRK2 vector, but not with a G2019S/D1994N vector. As well, degenerative neuritic changes and altered phosphorylated neurofilament distribution were observed in the striatum in a kinase-dependent fashion (Tsika et al., 2015). Unfortunately, the G2019S/D1994N mutation was shown to alter LRRK2 protein stability, partly confounding the validity of this synthetic inactive mutation in determining kinase-dependency. However, an additional study performing intrastriatal injection of Ad5-LRRK2 vectors at a higher viral titer in rats utilized an alternative stable kinase-inactivating mutation, G2019S/K1906M (Nguyen et al., 2019). In this study, phosphorylated tau inclusions were detected in the substantia nigra across all LRRK2 variants, suggesting that the effect is not specific to the G2019S mutation or kinase activity (Nguyen et al., 2019). Concurrently, biochemical analysis determined that tau protein levels, phosphorylation state, and solubility were not altered in response to Ad5-LRRK2 vectors (Nguyen et al., 2019). Additionally, ubiquitin-positive inclusions in the striatum did not appear to be dependent on kinase activity and none of the LRRK2 vectors induced detectable α-synuclein pathology. However, in this study, the G2019S LRRK2 vector alone induced APP-positive axonal inclusions and degenerative neuritic changes in the striatum (Nguyen et al., 2019). In a separate study, high-capacity adenoviral vectors (HC-AdV) expressing G2019S or G2019S/D1994N LRRK2 variants from a ubiquitous CAG promoter were delivered to the striatum of mice (Kritzinger et al., 2018). In this instance, modest age-dependent neuroinflammation and vacuolization of striatal white fiber tracts was induced by G2019S LRRK2 only in old animals, but protein aggregation was not detected (Kritzinger et al., 2018). Outside of rodents, a helper-dependent canine adenovirus type 2 (CAV-2) vector expressing human wild-type or G2019S LRRK2 was utilized in a non-human primate model, Microcebus murinus (Mestre-Francés et al., 2018). In this model, nigral neurodegeneration was equivalently observed with both LRRK2 variants and with a GFP control, making interpretation of this LRRK2 model challenging, although an increase in α-synuclein and tau phosphorylation (pSer129-αSyn+ and pSer396-tau+, respectively) was identified due to the G2019S variant (Mestre-Francés et al., 2018). An additional study assessing CAV-2-LRRK2 vectors in macaques, similarly, found an elevation of phosphorylated tau (PHF+) immunostaining in the substantia nigra of G2019S LRRK2-injected animals (di Caudo et al., 2020).
In contrast to viral vectors delivering LRRK2, viral vectors have also been used to introduce α-synuclein overexpression in neuronal populations of interest. Moreover, these vectors have proven to reliably induce neurodegeneration indicative of α-synuclein-dependent toxicity (Ulusoy et al., 2010). In order to investigate the contribution of LRRK2 toward protein aggregation and neuropathology, α-synuclein viral vectors have been introduced into LRRK2 rodent models in an approach that mimics the generation of bigenic models discussed previously. Toward this, an AAV2/1 vector delivering human wild-type α-synuclein to the substantia nigra of adult rats was shown to induce ∼30% loss of dopaminergic neurons (Daher et al., 2014). Remarkably, when the same vector was introduced in LRRK2 knockout rats, the neurodegenerative phenotype was ameliorated. While it was difficult to assess the extent of α-synuclein pathology in this model due to neuronal loss, the level of α-synuclein pathology was shown to inversely correlate with cell loss (Daher et al., 2014). In a subsequent study, AAV2/1-α-synuclein was delivered to the substantia nigra of G2019S LRRK2 BAC transgenic rats (Daher et al., 2015). Here, dopaminergic cell loss was determined to be significantly increased in the G2019S LRRK2 rats relative to control animals, and importantly, this effect was found to be kinase-dependent as pharmacologic kinase inhibition reversed the effect (Daher et al., 2015).
Use of an AAV2/5-α-synuclein vector delivered to the substantia nigra of rats was shown to induce aberrant neuronal firing activity in the subthalamic nucleus (Andersen et al., 2018). Intriguingly, this pathologic effect was not present in LRRK2 knockout rats and was reversed in wild-type rats following administration of a LRRK2 kinase inhibitor (Andersen et al., 2018). However, a later study found that long-term administration of a LRRK2 kinase inhibitor failed to protect against this abnormal α-synuclein-mediated neuronal dysfunction in rats (Andersen et al., 2019). Another study examined the impact of the G2019S mutation on virally mediated α-synuclein pathology using a more physiologically relevant model. Using G2019S LRRK2 knockin mice, along with wild-type controls, Novello et al. (2018) found that stereotactic injection of AAV2/9 vectors expressing human A53T α-synuclein was sufficient to induce ∼50% loss of dopaminergic neurons in the substantia nigra. However, no additive neurodegenerative effect was conferred by the G2019S mutation in the knockin model. Rather, G2019S knockin mice were found to exhibit a nearly two-fold increase in the level of nigral α-synuclein pathology (as measured by pSer129+ immunostaining) (Novello et al., 2018). This study also sufficiently characterized the observed α-synuclein pathology as resistant to proteinase-K treatment, indicative of fibrillar aggregates (Novello et al., 2018). While these findings suggest that LRRK2 may, in part, mediate the neurodegenerative phenotype of α-synuclein, a separate study identified LRRK2 activation via proximity ligation assays in the substantia nigra of rats following intranigral injection of AAV2/1 vector expressing human wild-type α-synuclein (Maio et al., 2018). Thus, while overexpression of LRRK2 via viral vector delivery is not generally sufficient to induce α-synuclein pathology in the face of neurodegeneration, the reverse seems to be true in that virally-mediated α-synuclein pathology may be potentiated by LRRK2 activity.
While less is known about the function of LRRK2 activity on virally-induced tau pathology, alterations to tau biology have been a common observation in LRRK2 animal models. The generation of bigenic mice examining the impact of LRRK2 genotype in mutant tau transgenic mice model previously failed to detect substantial changes to tau (Nguyen et al., 2018). However, additional experimentation suggests that LRRK2 genotype may alter other aspects of virally-mediated tau pathology. Utilizing AAV2/6 vectors to deliver human P301S tau via intrahippocampal injection, Nguyen et al. compared LRRK2 knockout and G2019S LRRK2 transgenic mice (Nguyen et al., 2018). While P301S tau induced ample tau pathology in the hippocampus, LRRK2 genotype was not determined to alter the burden of tau pathology or hippocampal neuronal loss (Nguyen et al., 2018). In contrast, use of an AAV2/6 vector co-expressing human wild-type tau and a GFP reporter revealed an impact of LRRK2 genotype on the neuronal transmission of tau within defined hippocampal circuits (Nguyen et al., 2018). While endogenous LRRK2 was found to be dispensable for tau transmission, a two-fold increase in transmission to both ipsilateral and contralateral CA3 regions was observed in G2019S LRRK2 transgenic mice (Nguyen et al., 2018). Notably, this experiment found no evidence of tau aggregation or neurodegeneration following viral vector delivery of wild-type tau, suggesting that LRRK2 activity may instead serve to accelerate the spread of tau pathology from cell-to-cell within defined circuits.
LRRK2 in Protein-Based Models
Accumulating evidence from human and experimental animal studies supports the pathological transmission of α-synuclein and tau species in PD and other neurodegenerative diseases (Frost and Diamond, 2009). Notably, both α-synuclein and tau pathology have been observed in fetal neural grafts of PD patients at autopsy (Kordower et al., 2008; Li et al., 2008, 2016; Cisbani et al., 2017). While the mechanistic explanations surrounding the stereotypical spatiotemporal spread of tau and α-synuclein pathology remains uncertain, it is clear that protein aggregation is inducible across interconnected neuronal populations and in a sequential pattern. Experimental models have demonstrated the cell-to-cell release and uptake of toxic protein species (Volpicelli-Daley et al., 2011, 2016; Mao et al., 2016). Whether such direct transmission models are relevant to human disease still remains to be determined but understanding the progression of inclusion formation within the context of in vivo models is certain to be invaluable. The development of α-synuclein pre-formed fibrils (PFF) as an experimental tool has allowed for the examination of cell-to-cell transmission of α-synuclein and resulting induction of pathology (Luk et al., 2009; Volpicelli-Daley et al., 2011; Duffy et al., 2018; Polinski et al., 2018; Patterson et al., 2019). Importantly, PFF-induced α-synuclein inclusions recapitulate key features of human Lewy pathology. Direct delivery of α-synuclein PFFs into the rodent brain has provided a robust model in which to dissect mechanisms of protein misfolding, aggregation, and spread in vivo. While the advancement and implementation of tau PFFs in vivo have been more limited, both α-synuclein and tau have been used to further a mechanistic understanding of pathological aggregation in the mammalian brain.
The introduction of α-synuclein PFFs into LRRK2 rodent models has provided a robust system in which to determine how LRRK2 activity relates to the progression and propagation of α-synuclein aggregation as well as the impact of pathogenic LRRK2 activity on α-synuclein mediated neurodegeneration (Table 3). Importantly, inoculation with α-synuclein PFFs and subsequent induction of Lewy-like pathology has been demonstrated to be dependent on the presence of endogenous α-synuclein (Volpicelli-Daley et al., 2011; Luk et al., 2012). Moreover, the level of pathologic burden across brain regions appears related to the regional variation in α-synuclein expression, such that brain regions associated with elevated levels of α-synuclein are associated with a greater severity of α-synuclein pathology while regions with lower levels of expression are associated with relative resistance to pathology (Henderson et al., 2019a). While the PFF model is less useful in determining the contribution of LRRK2 mutations toward the direct initiation of α-synuclein or tau pathology, other aspects of this model have proven instrumental. Notably, the PFF model has robustly demonstrated the importance of neural connectivity rather than area locality for the transmission of pathology, further supporting a cell-to-cell transmission mechanism in the progression of pathology (Henderson et al., 2019a). This feature is readily observed in the established rodent intrastriatal injection paradigm, in which the induction of α-synuclein pathology progressively accumulates in neuronal populations innervating the striatum. This observation is readily apparent in the substantia nigra, where progressive α-synuclein pathology peaks at 2–3 months following injection followed by modest neurodegeneration by 6 months (Luk et al., 2012; Paumier et al., 2015). The PFF model thus allows for examination of the spatiotemporal spread of protein substrates coupled with insight into selective vulnerability of affected regions.
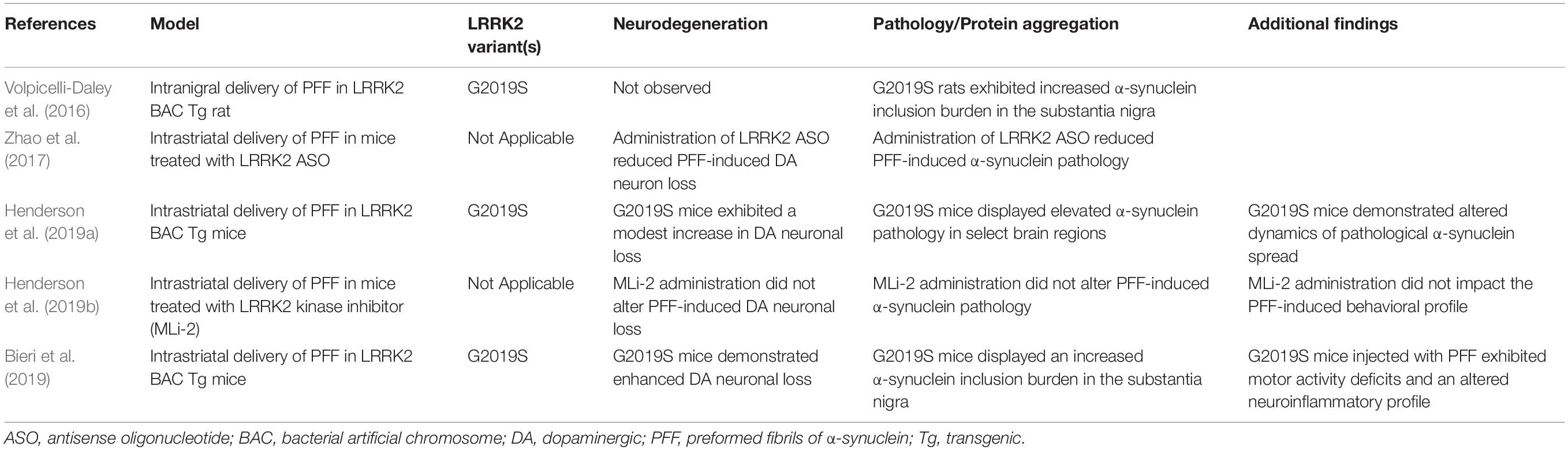
Table 3. LRRK2 in protein-based models.
The implementation of the PFF model in comparative genetic mouse models has facilitated a greater understanding of how LRRK2 activity might alter the progression of pathology at the connectome level (Table 3). In a recent study by Henderson et al., regional variation in the burden of α-synuclein pathology was observed in G2019S LRRK2 BAC transgenic mice compared to non-transgenic controls (following intrastriatal PFF injection), such that G2019S mice displayed both higher and lower levels of α-synuclein pathology across distinct brain regions (Henderson et al., 2019a). Intriguingly, a significant increase in the burden of α-synuclein pathology was observed in the cortex of G2019S mice. Robust LRRK2 transgene expression is present in the cortex of G2019S mice, suggesting that heightened LRRK2 levels may potentiate α-synuclein pathology in this region. Indeed, G2019S mice were shown to harbor a greater burden of pathology in regions that were resilient in non-transgenic mice. In terms of dopaminergic neurodegeneration, G2019S mice displayed a modest increase in nigral neuronal loss which was reflective of elevated α-synuclein pathology in the substantia nigra. Additional studies have sought to identify whether LRRK2 is required for α-synuclein transmission and neurodegeneration in the PFF model. Administration of MLi-2, a selective LRRK2 kinase inhibitor, in mice inoculated with α-synuclein PFFs demonstrated no significant impact on the induction of α-synuclein pathology in several regions of interest or alterations in dopaminergic cell loss in the substantia nigra (Henderson et al., 2019b). While these findings suggest an additive, but not critical, role for mutant LRRK2 in promoting α-synuclein pathology in vivo, other studies have produced contrasting results. Toward this, administration of antisense oligonucleotides directed against LRRK2 were found to reduce α-synuclein inclusion burden and neurodegeneration in an α-synuclein PFF model (Zhao et al., 2017). As well, primary neuronal cultures derived from either LRRK2 knockin or knockout mice were found to exhibit differential levels of α-synuclein aggregate pathology following PFF administration, with LRRK2 knockout playing a protective role (MacIsaac et al., 2020).
Despite the mixed findings, several additional studies have noted either an acceleration or exacerbation of PFF-induced α-synuclein pathology in the context of the pathogenic G2019S mutation. While enhanced LRRK2 activity does not appear to enhance neuronal uptake of fibrils in vitro, it is possible that mutant LRRK2 impacts diverse biological processes that result in the potentiation of aggregation. Volpicelli-Daley et al. found that overexpression of G2019S LRRK2 in a BAC transgenic rat model increased the number of α-synuclein and ubiquitin-positive inclusions in the substantia nigra at 1 month following unilateral intranigral PFF injection (Volpicelli-Daley et al., 2016). Likewise, Bieri et al. (2019) found that bilateral intrastriatal injection of α-synuclein PFF into G2019S LRRK2 BAC mice led to an exacerbation of phosphorylated α-synuclein aggregates across interconnected brain regions including the substantia nigra. While the effect of the G2019S mutation on protein aggregation was significant at 1, 3, and 6 months following PFF injection, the effect size was diminished over time. This observation supports that PFF-induced pathology may be subject to a thresholding effect, and, additionally, that LRRK2 may have both an immediate impact on the induction of pathology as well as longitudinal control of aggregate formation. Of interest, dopaminergic cell loss was more severe in the G2019S background, which could be resultant from the increased burden of aggregate pathology or from an intrinsic impact of G2019S LRRK2 on neuronal survival (Bieri et al., 2019). In total, LRRK2 appears to subtlety modulate the burden of protein aggregate pathology (Table 3), though the mechanism by which this occurs remains to be fully uncovered.
Conclusion
Given the heterogenous pathological landscape of LRRK2-PD, it remains uncertain whether and how LRRK2 activity might impact the development of protein aggregate pathology. Although there is evidence for a potential interaction between LRRK2 and α-synuclein, or LRRK2 and tau, many studies in animal models have had conflicting results. This likely arises from variations in experimental approach and models, and further lack of understanding of LRRK2 biology. Even when an interaction is identified, the mechanisms by which LRRK2 induces, promotes, or accelerates aggregation or neurodegeneration are not well-defined. While some studies suggest that LRRK2 is not critical for the development of α-synuclein or tau pathology, the opposite paradigm still remains to be fully explored. Whether α-synuclein or tau are essential for mutant LRRK2-induced neuropathology has not been formally evaluated in vivo. Subsequent studies will clarify this interaction and seek to place the significance and interaction of LRRK2, α-synuclein, and tau within PD-associated neurodegenerative processes. Toward this aim, LRRK2 animal models have been critical in developing our current understanding of LRRK2 pathobiology and, along with technological refinements and advances in this area, they are poised to play a significant role in future mechanistic studies of LRRK2, including protein aggregation pathways.
Author Contributions
DD: writing – original draft. DM: writing – reviewing and editing. Both authors contributed to the article and approved the submitted version.
Funding
Work in the authors’ laboratory was supported by funding from the National Institutes of Health (R01 NS091719 and R01 NS105432), Michael J. Fox Foundation for Parkinson’s Research, and the Van Andel Institute Graduate School.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aarsland, D., Perry, R., Brown, A., Larsen, J. P., and Ballard, C. (2005). Neuropathology of dementia in Parkinson’s disease: a prospective, community-based study. Ann. Neurol. 58, 773–776. doi: 10.1002/ana.20635
Andersen, M. A., Christensen, K. V., Badolo, L., Smith, G. P., Jeggo, R., Jensen, P. H., et al. (2018). Parkinson’s disease-like burst firing activity in subthalamic nucleus induced by AAV-α-synuclein is normalized by LRRK2 modulation. Neurobiol. Dis. 116, 13–27. doi: 10.1016/j.nbd.2018.04.011
Andersen, M. A., Sotty, F., Jensen, P. H., Badolo, L., Jeggo, R., Smith, G. P., et al. (2019). Long-term exposure to PFE-360 in the AAV-α-synuclein rat model: findings and implications. eNeuro 6: ENEURO.0453-18.2019. doi: 10.1523/eneuro.0453-18.2019
Bailey, R. M., Covy, J. P., Melrose, H. L., Rousseau, L., Watkinson, R., Knight, J., et al. (2013). LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol. 126, 809–827. doi: 10.1007/s00401-013-1188-4
Berwick, D. C., Heaton, G. R., Azeggagh, S., and Harvey, K. (2019). LRRK2 Biology from structure to dysfunction: research progresses, but the themes remain the same. Mol. Neurodegener. 14: 49. doi: 10.1186/s13024-019-0344-2
Bieri, G., Brahic, M., Bousset, L., Couthouis, J., Kramer, N. J., Ma, R., et al. (2019). LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 137, 961–980. doi: 10.1007/s00401-019-01995-0
Biskup, S., Moore, D. J., Celsi, F., Higashi, S., West, A. B., Andrabi, S. A., et al. (2006). Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 60, 557–569. doi: 10.1002/ana.21019
Biskup, S., and West, A. B. (2008). Zeroing in on LRRK2-linked pathogenic mechanisms in Parkinson’s disease. Biochim. Biophys. Acta 1792, 625–633. doi: 10.1016/j.bbadis.2008.09.015
Botta-Orfila, T., Ezquerra, M., Pastor, P., Fernández-Santiago, R., Pont-Sunyer, C., Compta, Y., et al. (2012). Age at onset in LRRK2-associated PD is modified by SNCA variants. J. Mol. Neurosci. 48, 245–247. doi: 10.1007/s12031-012-9820-7
Braak, H., Rüb, U., Steur, E. N. H. J., Tredici, K. D., and de Vos, R. A. I. (2005). Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64, 1404–1410. doi: 10.1212/01.wnl.0000158422.41380.82
Braak, H., Tredici, K. D., Rüb, U., de Vos, R. A. I., Steur, E. N. H. J., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Brundin, P., and Melki, R. (2017). Prying into the prion hypothesis for Parkinson’s disease. J. Neurosci. 37, 9808–9818. doi: 10.1523/jneurosci.1788-16.2017
Chen, C.-Y., Weng, Y.-H., Chien, K.-Y., Lin, K.-J., Yeh, T.-H., Cheng, Y.-P., et al. (2012). (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 19, 1623–1633. doi: 10.1038/cdd.2012.42
Cisbani, G., Maxan, A., Kordower, J. H., Planel, E., Freeman, T. B., and Cicchetti, F. (2017). Presence of tau pathology within foetal neural allografts in patients with Huntington’s and Parkinson’s disease. Brain J. Neurol. 140, 2982–2992. doi: 10.1093/brain/awx255
Cunningham, L. A., and Moore, D. J. (2020). Endosomal sorting pathways in the pathogenesis of Parkinson’s disease. Prog. Brain Res. 252, 271–306. doi: 10.1016/bs.pbr.2020.02.001
Daher, J. P. L., Abdelmotilib, H. A., Hu, X., Volpicelli-Daley, L. A., Moehle, M. S., Fraser, K. B., et al. (2015). Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates α-synuclein gene-induced neurodegeneration. J. Biol. Chem. 290, 19433–19444. doi: 10.1074/jbc.m115.660001
Daher, J. P. L., Pletnikova, O., Biskup, S., Musso, A., Gellhaar, S., Galter, D., et al. (2012). Neurodegenerative phenotypes in an A53T α-synuclein transgenic mouse model are independent of LRRK2. Hum. Mol. Genet. 21, 2420–2431. doi: 10.1093/hmg/dds057
Daher, J. P. L., Volpicelli-Daley, L. A., Blackburn, J. P., Moehle, M. S., and West, A. B. (2014). Abrogation of α-synuclein–mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc. Natl. Acad. Sci. U.S.A. 111, 9289–9294. doi: 10.1073/pnas.1403215111
Davis, A. A., Andruska, K. M., Benitez, B. A., Racette, B. A., Perlmutter, J. S., and Cruchaga, C. (2015). Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol. Aging 37, 209.e1–209.e7. doi: 10.1016/j.neurobiolaging.2015.09.014
di Caudo, C., Martínez-Valbuena, I., Mundiñano, I.-C., Gennetier, A., Hernandez, M., Carmona-Abellan, M., et al. (2020). CAV-2-mediated GFP and LRRK2G2019S expression in the Macaca fascicularis Brain. Front. Mol. Neurosci. 13:49. doi: 10.3389/fnmol.2020.00049
Duffy, M. F., Collier, T. J., Patterson, J. R., Kemp, C. J., Fischer, D. L., Stoll, A. C., et al. (2018). Quality over quantity: advantages of using alpha-synuclein preformed fibril triggered synucleinopathy to model idiopathic Parkinson’s disease. Front. Neurosci. Switz 12:621. doi: 10.3389/fnins.2018.00621
Dusonchet, J., Kochubey, O., Stafa, K., Young, S. M., Zufferey, R., Moore, D. J., et al. (2011). A rat model of progressive nigral neurodegeneration induced by the Parkinson’s disease-associated G2019S mutation in LRRK2. J. Neurosci. 31, 907–912. doi: 10.1523/jneurosci.5092-10.2011
Frost, B., and Diamond, M. I. (2009). Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11, 155–159. doi: 10.1038/nrn2786
Gaig, C., Marti, M. J., Ezquerra, M., Rey, M. J., Cardozo, A., and Tolosa, E. (2007). G2019S LRRK2 mutation causing Parkinson’s disease without Lewy bodies. J. Neurol. Neurosurg. Psychiatry 78, 626–628. doi: 10.1136/jnnp.2006.107904
Gan-Or, Z., Bar-Shira, A., Mirelman, A., Gurevich, T., Giladi, N., and Orr-Urtreger, A. (2011). The age at motor symptoms onset in LRRK2-associated Parkinson’s disease is affected by a variation in the MAPT locus: a possible interaction. J. Mol. Neurosci. 46, 541–544. doi: 10.1007/s12031-011-9641-0
Goldwurm, S., Zini, M., Fonzo, A. D., Gaspari, D. D., Siri, C., Simons, E. J., et al. (2006). LRRK2 G2019S mutation and Parkinson’s disease: a clinical, neuropsychological and neuropsychiatric study in a large Italian sample. Parkins. Relat. D 12, 410–419. doi: 10.1016/j.parkreldis.2006.04.001
Golub, Y., Berg, D., Calne, D. B., Pfeiffer, R. F., Uitti, R. J., Stoessl, A. J., et al. (2009). Genetic factors influencing age at onset in LRRK2-linked Parkinson disease. Parkins. Relat. D 15, 539–541. doi: 10.1016/j.parkreldis.2008.10.008
Guo, J. L., Covell, D. J., Daniels, J. P., Iba, M., Stieber, A., Zhang, B., et al. (2013). Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117. doi: 10.1016/j.cell.2013.05.057
Healy, D. G., Falchi, M., O’Sullivan, S. S., Bonifati, V., Durr, A., Bressman, S., et al. (2008). Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 7, 583–590. doi: 10.1016/s1474-4422(08)70117-0
Henderson, M. X., Cornblath, E. J., Darwich, A., Zhang, B., Brown, H., Gathagan, R. J., et al. (2019a). Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat. Neurosci. 22, 1248–1257. doi: 10.1038/s41593-019-0457-5
Henderson, M. X., Sengupta, M., McGeary, I., Zhang, B., Olufemi, M. F., Brown, H., et al. (2019b). LRRK2 inhibition does not impart protection from α-synuclein pathology and neuron death in non-transgenic mice. Acta Neuropathol. Commun. 7:28. doi: 10.1186/s40478-019-0679-5
Henderson, M. X., Sengupta, M., Trojanowski, J. Q., and Lee, V. M. Y. (2019c). Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 7:183. doi: 10.1186/s40478-019-0836-x
Herzig, M. C., Bidinosti, M., Schweizer, T., Hafner, T., Stemmelen, C., Weiss, A., et al. (2012). High LRRK2 levels fail to induce or exacerbate neuronal alpha-synucleinopathy in mouse brain. PLoS One 7:e36581. doi: 10.1371/journal.pone.0036581
Higashi, S., Moore, D. J., Colebrooke, R. E., Biskup, S., Dawson, V. L., Arai, H., et al. (2007). Expression and localization of Parkinson’s disease-associated leucine-rich repeat kinase 2 in the mouse brain. J. Neurochem. 100, 368–381. doi: 10.1111/j.1471-4159.2006.04246.x
Islam, M. S., and Moore, D. J. (2017). Mechanisms of LRRK2-dependent neurodegeneration: role of enzymatic activity and protein aggregation. Biochem. Soc. T 45, 163–172. doi: 10.1042/bst20160264
Kobayashi, H., Ujike, H., Hasegawa, J., Yamamoto, M., Kanzaki, A., and Sora, I. (2006). Correlation of tau gene polymorphism with age at onset of Parkinson’s disease. Neurosci. Lett. 405, 202–206. doi: 10.1016/j.neulet.2006.06.057
Kordower, J. H., Chu, Y., Hauser, R. A., Freeman, T. B., and Olanow, C. W. (2008). Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 14, 504–506. doi: 10.1038/nm1747
Kritzinger, A., Ferger, B., Gillardon, F., Stierstorfer, B., Birk, G., Kochanek, S., et al. (2018). Age-related pathology after adenoviral overexpression of the leucine-rich repeat kinase 2 in the mouse striatum. Neurobiol. Aging 66, 97–111. doi: 10.1016/j.neurobiolaging.2018.02.008
Lee, B. D., Shin, J.-H., VanKampen, J., Petrucelli, L., West, A. B., Ko, H. S., et al. (2010). Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 16:998. doi: 10.1038/nm.2199
Li, J.-Y., Englund, E., Holton, J. L., Soulet, D., Hagell, P., Lees, A. J., et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503. doi: 10.1038/nm1746
Li, W., Englund, E., Widner, H., Mattsson, B., Westen, D., van Lätt, J., et al. (2016). Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc. Natl. Acad. Sci. 113, 6544–6549. doi: 10.1073/pnas.1605245113
Li, X., Patel, J. C., Wang, J., Avshalumov, M. V., Nicholson, C., Buxbaum, J. D., et al. (2010). Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J. Neurosci. 30, 1788–1797. doi: 10.1523/jneurosci.5604-09.2010
Li, Y., Liu, W., Oo, T. F., Wang, L., Tang, Y., Jackson-Lewis, V., et al. (2009). Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 12, 826–828. doi: 10.1038/nn.2349
Lim, K. H. (2019). Diverse misfolded conformational strains and cross-seeding of misfolded proteins implicated in neurodegenerative diseases. Front. Mol. Neurosci. 12:158. doi: 10.3389/fnmol.2019.00158
Lin, X., Parisiadou, L., Gu, X.-L., Wang, L., Shim, H., Sun, L., et al. (2009). Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant α-synuclein. Neuron 64, 807–827. doi: 10.1016/j.neuron.2009.11.006
Ling, H., Kara, E., Bandopadhyay, R., Hardy, J., Holton, J., Xiromerisiou, G., et al. (2013). TDP-43 pathology in a patient carrying G2019S LRRK2 mutation and a novel p.Q124E MAPT. Neurobiol. Aging 34, 2889.e5–2889.e9. doi: 10.1016/j.neurobiolaging.2013.04.011
Liu, G., Sgobio, C., Gu, X., Sun, L., Lin, X., Yu, J., et al. (2015). Selective expression of Parkinson’s disease-related Leucine-rich repeat kinase 2 G2019S missense mutation in midbrain dopaminergic neurons impairs dopamine release and dopaminergic gene expression. Hum. Mol. Genet. 24, 5299–5312. doi: 10.1093/hmg/ddv249
Longo, F., Mercatelli, D., Novello, S., Arcuri, L., Brugnoli, A., Vincenzi, F., et al. (2017). Age-dependent dopamine transporter dysfunction and Serine129 phospho-α-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol. Commun 5:22. doi: 10.1186/s40478-017-0426-8
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J. Q., et al. (2012). Pathological α-synuclein transmission initiates parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Luk, K. C., Song, C., O’Brien, P., Stieber, A., Branch, J. R., Brunden, K. R., et al. (2009). Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. 106, 20051–20056. doi: 10.1073/pnas.0908005106
MacIsaac, S., Quevedo-Melo, T., Zhang, Y., Volta, M., Farrer, M. J., and Milnerwood, A. J. (2020). Neuron-autonomous susceptibility to induced synuclein aggregation is exacerbated by endogenous Lrrk2 mutations and ameliorated by Lrrk2 genetic knock-out. Brain Commun. 2:fcz052. doi: 10.1093/braincomms/fcz052
Maio, R. D., Hoffman, E. K., Rocha, E. M., Keeney, M. T., Sanders, L. H., Miranda, B. R. D., et al. (2018). LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 10:eaar5429. doi: 10.1126/scitranslmed.aar5429
Mamais, A., Raja, M., Manzoni, C., Dihanich, S., Lees, A., Moore, D., et al. (2013). Divergent α-synuclein solubility and aggregation properties in G2019S LRRK2 Parkinson’s disease brains with Lewy Body pathology compared to idiopathic cases. Neurobiol. Dis. 58, 183–190. doi: 10.1016/j.nbd.2013.05.017
Mao, X., Ou, M. T., Karuppagounder, S. S., Kam, T.-I., Yin, X., Xiong, Y., et al. (2016). Pathological -synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353:aah3374. doi: 10.1126/science.aah3374
Martí-Massó, J.-F., Ruiz-Martínez, J., Bolaño, M. J., Ruiz, I., Gorostidi, A., Moreno, F., et al. (2009). Neuropathology of Parkinson’s disease with the R1441G mutation in LRRK2. Mov. Disord. 24, 1998–2001. doi: 10.1002/mds.22677
Melrose, H. L., Dächsel, J. C., Behrouz, B., Lincoln, S. J., Yue, M., Hinkle, K. M., et al. (2010). Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 40, 503–517. doi: 10.1016/j.nbd.2010.07.010
Mestre-Francés, N., Serratrice, N., Gennetier, A., Devau, G., Cobo, S., Trouche, S. G., et al. (2018). Exogenous LRRK2G2019S induces parkinsonian-like pathology in a nonhuman primate. Jci Insight 3:e98202. doi: 10.1172/jci.insight.98202
Mikhail, F., Calingasan, N., Parolari, L., Subramanian, A., Yang, L., and Beal, M. F. (2015). Lack of exacerbation of neurodegeneration in a double transgenic mouse model of mutant LRRK2 and tau. Hum. Mol. Genet. 24, 3545–3556. doi: 10.1093/hmg/ddv105
Moore, D. J., West, A. B., Dawson, V. L., and Dawson, T. M. (2005). Molecular pathophysiology of parkinson’s disease. Annu. Rev. Neurosci. 28, 57–87. doi: 10.1146/annurev.neuro.28.061604.135718
Moussaud, S., Jones, D. R., Moussaud-Lamodière, E. L., Delenclos, M., Ross, O. A., and McLean, P. J. (2014). Alpha-synuclein and tau: teammates in neurodegeneration? Mol. Neurodegener. 9:43. doi: 10.1186/1750-1326-9-43
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102. doi: 10.1016/s1474-4422(19)30320-5
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993. doi: 10.1038/ng.3043
Nguyen, A. P. T., Daniel, G., Valdés, P., Islam, M. S., Schneider, B. L., and Moore, D. J. (2018). G2019S LRRK2 enhances the neuronal transmission of tau in the mouse brain. Hum. Mol. Genet. 27, 120–134. doi: 10.1093/hmg/ddx389
Nguyen, A. P. T., Tsika, E., Kelly, K., Levine, N., Chen, X., West, A. B., et al. (2019). Dopaminergic neurodegeneration Induced by Parkinson’s Disease-Linked G2019S LRRK2 is dependent on kinase and GTPase activity. bioRxiv 2019.12.17.879759. doi: 10.1101/2019.12.17.879759
Novello, S., Arcuri, L., Dovero, S., Dutheil, N., Shimshek, D. R., Bezard, E., et al. (2018). G2019S LRRK2 mutation facilitates α-synuclein neuropathology in aged mice. Neurobiol. Dis. 120, 21–33. doi: 10.1016/j.nbd.2018.08.018
Paisán-Ruıíz, C., Jain, S., Evans, E. W., Gilks, W. P., Simón, J., van der Brug, M., et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. doi: 10.1016/j.neuron.2004.10.023
Patterson, J. R., Polinski, N. K., Duffy, M. F., Kemp, C. J., Luk, K. C., Volpicelli-Daley, L. A., et al. (2019). Generation of alpha-synuclein preformed fibrils from monomers and use in vivo. J. Vis. Exp. 148:e59758. doi: 10.3791/59758
Paumier, K. L., Luk, K. C., Manfredsson, F. P., Kanaan, N. M., Lipton, J. W., Collier, T. J., et al. (2015). Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 82, 185–199. doi: 10.1016/j.nbd.2015.06.003
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:nrd201713. doi: 10.1038/nrdp.2017.13
Polinski, N. K., Volpicelli-Daley, L. A., Sortwell, C. E., Luk, K. C., Cremades, N., Gottler, L. M., et al. (2018). Best practices for generating and using alpha-synuclein pre-formed fibrils to model Parkinson’s disease in rodents. J. Park. Dis. 8, 303–322. doi: 10.3233/jpd-171248
Ramonet, D., Daher, J. P. L., Lin, B. M., Stafa, K., Kim, J., Banerjee, R., et al. (2011). Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One 6:e18568. doi: 10.1371/journal.pone.0018568
Schapansky, J., Khasnavis, S., DeAndrade, M. P., Nardozzi, J. D., Falkson, S. R., Boyd, J. D., et al. (2018). Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble α-synuclein in neurons. Neurobiol. Dis. 111, 26–35. doi: 10.1016/j.nbd.2017.12.005
Sheng, Z., Zhang, S., Bustos, D., Kleinheinz, T., Pichon, C. E. L., Dominguez, S. L., et al. (2012). Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci. Transl. Med. 4:164ra161. doi: 10.1126/scitranslmed.3004485
Smith, C., Malek, N., Grosset, K., Cullen, B., Gentleman, S., and Grosset, D. G. (2019). Neuropathology of dementia in patients with Parkinson’s disease: a systematic review of autopsy studies. J. Neurol. Neurosurg. Psychiatry 90:1243. doi: 10.1136/jnnp-2019-321111
Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. elife 5:e12813. doi: 10.7554/elife.12813
Takanashi, M., Funayama, M., Matsuura, E., Yoshino, H., Li, Y., Tsuyama, S., et al. (2018). Isolated nigral degeneration without pathological protein aggregation in autopsied brains with LRRK2 p.R1441H homozygous and heterozygous mutations. Acta Neuropathol. Commun. 6:105. doi: 10.1186/s40478-018-0617-y
Tong, Y., Pisani, A., Martella, G., Karouani, M., Yamaguchi, H., Pothos, E. N., et al. (2009). R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl. Acad. Sci. U.S.A. 106, 14622–14627. doi: 10.1073/pnas.0906334106
Tsika, E., Kannan, M., Foo, C. S.-Y., Dikeman, D., Glauser, L., Gellhaar, S., et al. (2014). Conditional expression of Parkinson’s disease-related R1441C LRRK2 in midbrain dopaminergic neurons of mice causes nuclear abnormalities without neurodegeneration. Neurobiol. Dis. 71, 345–358. doi: 10.1016/j.nbd.2014.08.027
Tsika, E., Nguyen, A. P. T., Dusonchet, J., Colin, P., Schneider, B. L., and Moore, D. J. (2015). Adenoviral-mediated expression of G2019S LRRK2 induces striatal pathology in a kinase-dependent manner in a rat model of Parkinson’s disease. Neurobiol. Dis. 77, 49–61. doi: 10.1016/j.nbd.2015.02.019
Ujiie, S., Hatano, T., Kubo, S.-I., Imai, S., Sato, S., Uchihara, T., et al. (2012). LRRK2 I2020T mutation is associated with tau pathology. Parkins. Relat. D 18, 819–823. doi: 10.1016/j.parkreldis.2012.03.024
Ulusoy, A., Decressac, M., Kirik, D., and Björklund, A. (2010). Chapter 5 Viral vector-mediated overexpression of α-synuclein as a progressive model of Parkinson’s disease. Prog. Brain Res. 184, 89–111. doi: 10.1016/s0079-6123(10)84005-1
Volpicelli-Daley, L. A., Abdelmotilib, H., Liu, Z., Stoyka, L., Daher, J. P. L., Milnerwood, A. J., et al. (2016). G2019S-LRRK2 expression augments α-synuclein sequestration into inclusions in neurons. J. Neurosci. 36, 7415–7427. doi: 10.1523/jneurosci.3642-15.2016
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
West, A. B., Cowell, R. M., Daher, J. P. L., Moehle, M. S., Hinkle, K. M., Melrose, H. L., et al. (2014). Differential LRRK2 expression in the cortex, striatum, and substantia nigra in transgenic and nontransgenic rodents. J. Comp. Neurol. 522, 2465–2480. doi: 10.1002/cne.23583
Wider, C., Dickson, D. W., and Wszolek, Z. K. (2010). Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neurodegener. Dis. 7, 175–179. doi: 10.1159/000289232
Xiong, Y., Neifert, S., Karuppagounder, S. S., Liu, Q., Stankowski, J. N., Lee, B. D., et al. (2018). Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 115, 201712648. doi: 10.1073/pnas.1712648115
Xiong, Y., Neifert, S., Karuppagounder, S. S., Stankowski, J. N., Lee, B. D., Grima, J. C., et al. (2017). Overexpression of Parkinson’s disease-associated mutation LRRK2 G2019S in mouse forebrain induces behavioral deficits and α-synuclein pathology. Eneuro 4:ENEURO.0004-.17. doi: 10.1523/eneuro.0004-17.2017
Yue, M., Hinkle, K. M., Davies, P., Trushina, E., Fiesel, F. C., Christenson, T. A., et al. (2015). Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 78, 172–195. doi: 10.1016/j.nbd.2015.02.031
Zhao, H. T., John, N., Delic, V., Ikeda-Lee, K., Kim, A., Weihofen, A., et al. (2017). LRRK2 antisense oligonucleotides ameliorate α-synuclein inclusion formation in a Parkinson’s disease mouse model. Mol. Ther. Nucl. Acids 8, 508–519. doi: 10.1016/j.omtn.2017.08.002
Keywords: Parkinson’s disease, LRRK2, tau, alpha-synuclein, protein aggregation, neurodegeneration, animal models
Citation: Dues DJ and Moore DJ (2020) LRRK2 and Protein Aggregation in Parkinson’s Disease: Insights From Animal Models. Front. Neurosci. 14:719. doi: 10.3389/fnins.2020.00719
Received: 01 May 2020; Accepted: 16 June 2020;
Published: 08 July 2020.
Edited by:
Hardy Rideout, Biomedical Research Foundation of the Academy of Athens (BRFAA), GreeceReviewed by:
Maria Shadrina, Institute of Molecular Genetics (RAS), RussiaPetr A. Slominsky, Institute of Molecular Genetics (RAS), Russia
Copyright © 2020 Dues and Moore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Darren J. Moore, ZGFycmVuLm1vb3JlQHZhaS5vcmc=