 Qianjie Yang
Qianjie Yang Yunxiang Zhou2†
Yunxiang Zhou2† Yuting Sun
Yuting Sun Anwen Shao
Anwen Shao- 1Department of Ophthalmology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2Department of Surgical Oncology, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 3The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 4Department of Neurosurgery, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
Traumatic brain injury (TBI), a leading cause of morbidity worldwide, induces mechanical, persistent structural, and metabolic abnormalities in neurons and other brain-resident cells. The key pathological features of TBI include neuroinflammation, oxidative stress, excitotoxicity, and mitochondrial dysfunction. These pathological processes persist for a period of time after TBIs. Sirtuins are evolutionarily conserved nicotinamide-adenine dinucleotide (NAD+)-dependent deacetylases and mono-ADP-ribosyl transferases. The mammalian sirtuin family has seven members, referred to as Sirtuin (SIRT) 1–7. Accumulating evidence suggests that SIRT1 and SIRT3 play a neuroprotective role in TBI. Although the evidence is scant, considering the involvement of SIRT2, 4–7 in other brain injury models, they may also intervene in similar pathophysiology in TBI. Neurodegenerative diseases are generally accepted sequelae of TBI. It was found that TBI and neurodegenerative diseases have many similarities and overlaps in pathological features. Besides, sirtuins play some unique roles in some neurodegenerative diseases. Therefore, we propose that sirtuins might be a promising therapeutic target for both TBI and associated neurodegenerative diseases. In this paper, we review the neuroprotective effects of sirtuins on TBI as well as related neurodegeneration and discuss the therapeutic potential of sirtuin modulators.
Introduction
Traumatic brain injury (TBI) is a leading cause of morbidity worldwide. It is associated with long-term disability and significant healthcare expenditures and has become a priority for public health policy (Quaglio et al., 2017). Although most of the neurological injuries are limited and temporary (Cornelius et al., 2013), TBI still has multiple sequelae including sleep disturbance (Barshikar and Bell, 2017), hypopituitarism (Klose and Feldt-Rasmussen, 2018), seizures, epilepsy (Zimmermann et al., 2017), and neurodegenerative diseases (Gardner and Yaffe, 2015). Indeed, TBI is widely recognized as a risk factor for neurodegenerative diseases (Goldstein et al., 2012; Gardner and Yaffe, 2015; Daglas and Adlard, 2018; Gardner et al., 2018), which are characterized by a slow progressive loss of neurons or myelin sheaths and increasing disability and include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease, and multiple sclerosis. Notably, patients with different types of TBI tend to have an increased risk of different neurodegenerative diseases. For example, moderate or severe TBI contributes to the development of late-onset neurodegenerative diseases, in particular AD and PD (Mendez, 2017). Chronic traumatic encephalopathy (CTE), a condition that shares features with neurodegenerative diseases, is linked to mild TBI (Fesharaki-Zadeh, 2019). Numerous random-effects meta-analysis (Perry et al., 2016), epidemiologic study (Gardner and Yaffe, 2015), and retrospective cohort study (Gardner et al., 2018) have lent support to these associations. Besides raising the risk, TBI lowers the age at onset of TBI-related neurodegenerative diseases (Mendez, 2017). Mechanically, it has previously been observed that pathological processes, including oxidative stress, neuroinflammation, excitotoxicity, proteinopathies, and mitochondrial dysfunction, may persist for months or years post-TBI, and these pathophysiological substrates serve as triggers yielding the progression of neurodegenerative diseases (Gentleman et al., 2004; Johnson et al., 2013a; Loane et al., 2014; Zhou et al., 2020a). Accordingly, targeting these post-TBI pathological processes holds the promise for the intervention of TBI and late-onset neurodegenerative diseases.
Homologs of Silent information regulator 2 (Sir2) are collectively known as sirtuins (Gomes et al., 2020). The mammalian sirtuin family has seven members, which are classified into four groups: class I (SIRT1–3), class II (SIRT4), class III (SIRT5), and class IV (SIRT6 and 7) (Figure 1). All sirtuins have an evolutionarily conserved nicotinamide-adenine dinucleotide (NAD+)-dependent catalytic core domain (Beauharnois et al., 2013; Khoury et al., 2018), and each sirtuin has unique N-terminal and C-terminal sequences (Michan and Sinclair, 2007; Li and Kazgan, 2011). SIRT1 and SIRT5 tend to elicit NAD+-dependent deacetylase activity, whereas SIRT4 and SIRT6 are likely to express mono-ADP-ribosyl transferase activity (Lin et al., 2012). Sirtuins occupy different cellular locations including mitochondria (SIRT3–5), the cell nucleus (SIRT6), the nucleolus (SIRT7), and between the cytoplasm and nucleus (SIRT1, 2) (Gomes et al., 2020; Zhou et al., 2020c). In addition, sirtuins are widespread in brain cells, such as neurons (Zakhary et al., 2010; Han et al., 2014; Satoh et al., 2017; Satoh and Shimokawa, 2019), astrocyte (Li et al., 2017), and microglia (Chen et al., 2015).
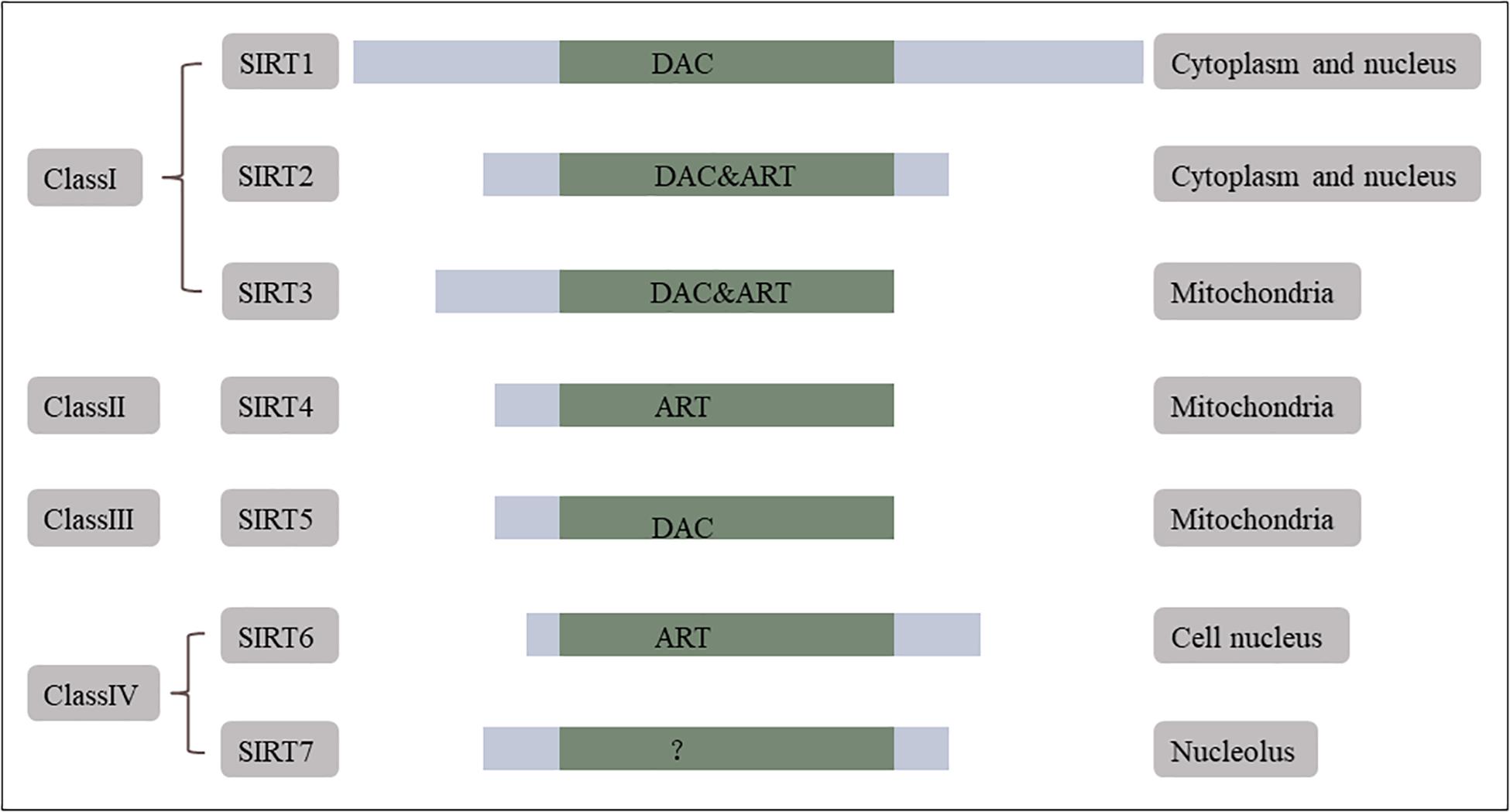
Figure 1. Mammalian sirtuins. Mammals have seven sirtuins. They are classified into four groups. SIRT1, 2, 3 are class I, SIRT4 is class II, SIRT5 is class III, and SIRT6, 7 are class IV. All of them have a conserved NAD+-dependent catalytic core domain. Each of the sirtuins has unique additional N-terminal and C-terminal sequences. SIRT1, 5 tend to show NAD+-dependent deacetylase (DAC) activity first whereas SIRT4, 6 are likely to express its mono-ADP-ribosyl transferase (ART) activity. Sirtuins have distributed cellular localization, mitochondria (SIRT3, 4, 5), cell nucleus (SIRT6), nucleolus (SIRT7), and both in cytoplasm and nucleus (SIRT1, 2). Reprinted and modified from International Journal of Biological Sciences, 7 (5), Xiaoling Li, Mammalian Sirtuins and Energy Metabolism, 575–587, 2011 (Li and Kazgan, 2011).
Sirtuins are of vital interest in oxidative stress, neuroinflammation, blood–brain barrier (BBB) permeability, astrocyte activation, and neural apoptosis in brain injuries. To date, studies regarding SIRT1 and SIRT3 have provided direct evidence about the functions of sirtuins in TBI. Despite the lack of evidence, other sirtuins may also intervene in similar pathophysiology in TBI, as have been shown in other models such as subarachnoid hemorrhage (SAH), ischemic stroke, and ischemia/reperfusion (I/R) injury. In addition, sirtuins also have some considerable impact on neurodegenerative diseases, such as AD and PD. Based on the epidemiological association and pathological similarities of TBI and neurodegenerative diseases, we propose that sirtuins may be a promising therapeutic target for TBI and related neurodegenerative diseases. In this review, we will dissect the similarity on the pathophysiological processes between TBI and neurodegenerative diseases and summarize the neuroprotective effects of sirtuins in these pathophysiological processes. We will also outline sirtuin modulators for pharmacotherapeutics and the probable difficulties in the development of pharmacological agents.
Pathophysiology of TBI and Related Neurodegenerative Diseases
TBI Pathophysiology
TBI is classified into mild, moderate, and severe type, which can lead to different degrees of primary and secondary brain injuries (Cornelius et al., 2013; Khatri et al., 2018). Primary injuries are attributed to the direct result of physical injuries, including hemorrhage and tissue damage (Greve and Zink, 2009). Secondary injuries include cellular hyperexcitability, vasogenic and cytotoxic edema, hypoxia–ischemia (Karve et al., 2016), microglia polarization (Chen et al., 2018a), and astrocyte activation (Li et al., 2017; Zhou et al., 2020b). Reducing primary injuries depends on preventive measures whereas second injuries are sensitive to therapeutic measures (Cornelius et al., 2013). Evidence suggests that second injuries considerably determine the outcomes of TBI patients (Cornelius et al., 2013). Therefore, in this review, we mainly focus on the second injuries and corresponding therapeutic measures.
Second injuries can further induce metabolic disorders (Hiebert et al., 2015), vascular abnormalities (Cornelius et al., 2013), extensive neuroinflammation (Toklu and Tümer, 2015; Simon et al., 2017), oxidative stress, microglial activation, and excitotoxicity (Zhou et al., 2020a). TBI-induced mitochondrial dysfunction causes cellular metabolic alterations, excitotoxicity, and endogenous antioxidant system exhaustion (Cornelius et al., 2013). The excessive production of reactive oxygen species (ROS) leads to lipid peroxidation, cytotoxicity, necrotic cell apoptosis, and oxidative stress, which in turn, exacerbate mitochondrial dysfunction (Cornelius et al., 2013; Zhou et al., 2020a). Besides, mitochondrial dysfunction can affect cell membrane permeability, resulting in the extensive release of apoptotic proteins (Chen et al., 2018b). Neuroinflammation, the physiological response to the injury, triggers the release of cytokines and chemokines and activates microglia and astrocytes (Cornelius et al., 2013). A recent study of brain injury identified a significant number of rod-shaped microglia arranged like the carriages of a train (Holloway et al., 2019). Microglia are the primary immune sentinels of the central nervous system responding to inflammatory events (Russo and McGavern, 2016) and the main source of ROS (Rangarajan et al., 2015). Studies provided evidence that microglial activity persists for a long-term period in the brain of TBI survivors (Gentleman et al., 2004; Johnson et al., 2013a; Zhou et al., 2020a). Moreover, vascular abnormalities may disrupt the BBB and cause encephaledema (Cornelius et al., 2013). Furthermore, TBI is a common cause of olfactory dysfunction, which is mediated by excitotoxicity, an intercellular cascade initiated by excessive release of glutamate and hyperactivation of the glutamatergic receptors (Marin et al., 2019; Figure 2).
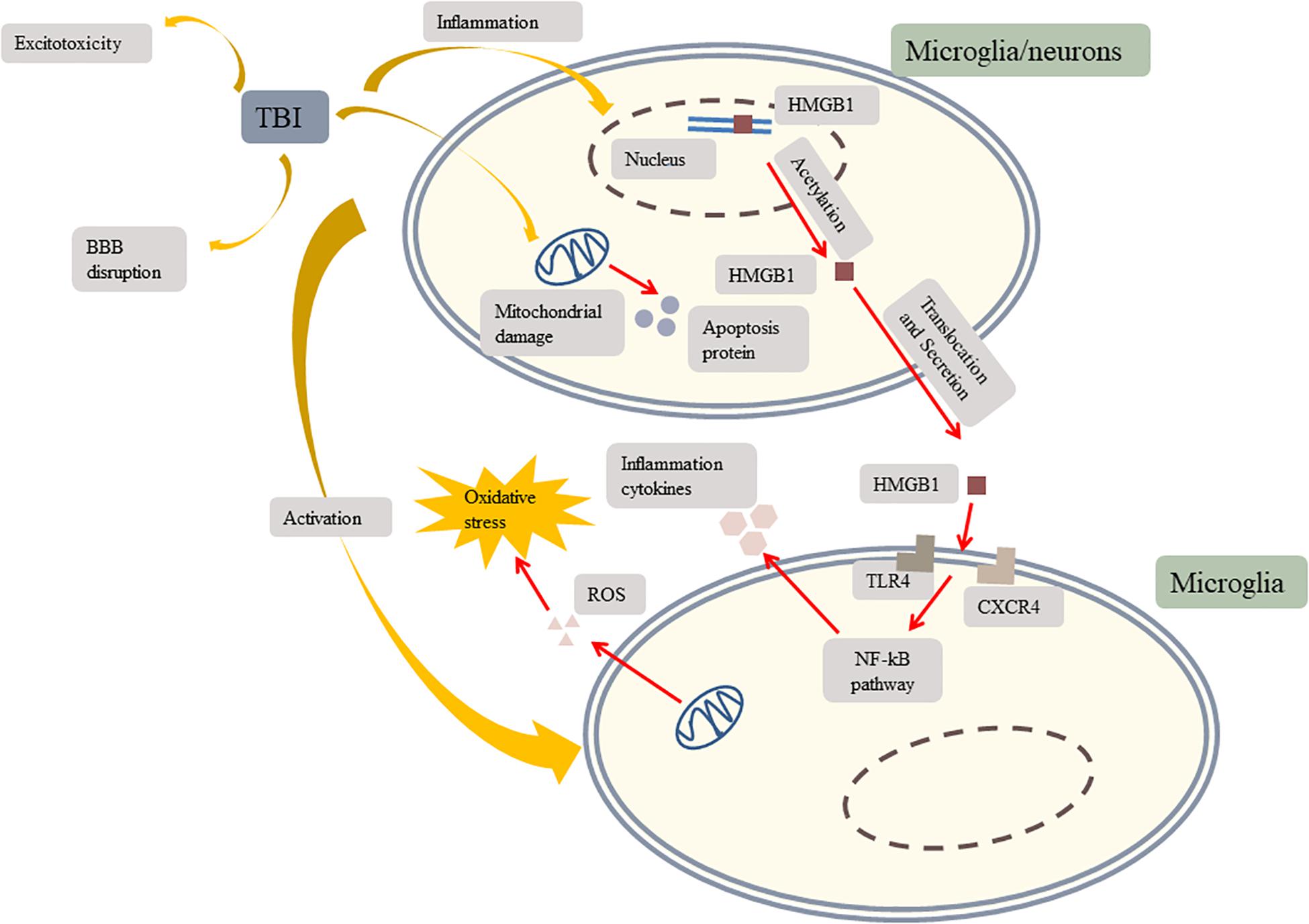
Figure 2. TBI induces inflammation, mitochondrial damage, microglial activation, excitotoxicity, and BBB disruption. TBI results in HMFB1 translocation and secretion. Extracellular HMGB1activates TLR4 and CXCR4 to trigger the NF-kB pathway which induces microglia secreting inflammation cytokines to further activate other microglia. TBI induces mitochondrial damage. Damaged mitochondria produce apoptosis protein and ROS to aggravate oxidative stress. Besides, TBI leads to excitotoxicity and BBB disruption which makes inflammation cytokines leakage more severe. TBI, traumatic brain injury; TLR4, toll-like receptor 4; CXCR4, chemokine (C-X-C motif) receptor 4; BBB, blood–brain barrier; HMGB1, high-mobility group box 1; NF-κB, nuclear factor-κB; ROS, reactive oxygen species.
Mechanisms Underlying TBI-Induced Neurodegenerative Diseases
TBI is one of the well-established contributing factors for late-onset neurodegenerative diseases. TBI may alter the normal trajectory of the aging processes of the affected individual. Multiple injuries produce synergistic effects and accelerate neurodegeneration (DeKosky and Asken, 2017). The common causes of neurodegenerative diseases are neuroinflammation, excitotoxicity, oxidative stress, mitochondrial dysfunction, and proteinopathies, all of which are involved in the pathophysiology of TBI (Zhou et al., 2020a). Inflammation is a normal immune response coping with injuries or harmful stimuli, yet overactive inflammation damages the brain. Post-mortem and animal model data indicate that oxidative stress and neuroinflammation can persist for years after the acute phase of injury as a result of chronic microglial activation (Gentleman et al., 2004; Loane et al., 2014; Zhou et al., 2020a). According to Johnson et al.’s (2013a) research, reactive microglia were still present in 28% brains over a year after a single TBI. Excitotoxicity refers to neuronal cell death caused by the toxic action of excitatory amino acids (Dong et al., 2009). Glutamate is the primary excitatory amino acid neurotransmitter in the central nervous system and is associated with neuronal degeneration, aging, and death (Hernández et al., 2018). Oxidative stress triggers excessive free radical production, leading to neuronal death and stimulates inflammation (Khatri et al., 2018). TBI-induced mitochondrial dysfunction decreases the metabolic energy supply and interferes with synaptic function (Goldstein et al., 2012). The resulting depletion of energy stores stimulates the release of ROS, which in turn, aggravates oxidative stress. Besides, Daglas and Adlard (2018) indicated that the detrimental role of iron deposits in the TBI pathogenesis may constitute one of the reasons for boosting late-onset neurodegenerative diseases.
AD, one of the most common neurodegenerative diseases, is characterized by the aggregation of β-amyloid protein and tau protein. PD, which is caused by the selective degeneration of dopamine and is characterized by bradykinesia and rigidity, is the second most common neurodegenerative disease (Shi et al., 2017). Studies showed that TBI is one of the strong environmental contributing factors for AD and PD (Hayes et al., 2017; Al-Dahhak et al., 2018; Gardner et al., 2018). One of the possible reasons is that TBI causes diffuse axonal injury and leads to proteinopathies. TBI causes mechanical trauma to axons and induces the excessive release of glutamate, which may substantially alter neuronal metabolism (Giza and Hovda, 2014). Dysregulated axonal transport systems lead to axonal swelling, which may disrupt the delivery of necessary proteins and ultimately lead to focal nerve disconnection (Johnson et al., 2012, 2013b). In damaged axons, accumulated amyloid precursor proteins and amyloid β-peptide are markers of axon injury (Smith et al., 2003). These markers are frequently found existing in the brains of TBI survivors (Ikonomovic et al., 2004) and post-mortem brains (Smith et al., 2003). Although the mechanism underlying TBI-induced PD is not clear, it may involve mitochondrial dysfunction and oxidative stress (Shi et al., 2017). Amyloid β-peptide accelerates tau protein pathology, but the underlying mechanism remains unknown (Yin et al., 2018). TBI causes hyperphosphorylation, misfolding, and aggregation of tau proteins, which then form neurofibrillary tangles (NFTs) (Johnson et al., 2012; Du et al., 2014). Hyperphosphorylated tau proteins are released into the extracellular space and stimulate microglia and astrocytes to produce pro-inflammatory cytokines (e.g., IL-1β and TNFα), which further activate tau kinases and aggravate tau hyperphosphorylation (Fesharaki-Zadeh, 2019). Besides phosphorylation, tau acetylation at Lys174 is a crucial change during the post-translational modification of tau proteins, determining the toxicity in mice, tau homeostasis, and the initial step of AD (Min et al., 2015).
CTE, a neurodegenerative disease caused by repeated mild or concussive head injuries, is characterized by an abnormal accumulation of hyperphosphorylated tau protein (Johnson et al., 2012). Clinically, patients with CTE present with two distinct phenotypes: affective changes and cognitive impairment (Fesharaki-Zadeh, 2019). Several large epidemiological studies suggest that a single severe TBI is a significant risk factor for the later development of AD (Johnson et al., 2017). The effects of TBI are long-term and extend beyond the immediate acute phase. Previous findings suggest that neuronal densities in the hippocampus and thalamus continue to decrease 1 year or more after a severe TBI (Maxwell et al., 2003, 2006).
Taken collectively, it appears likely that targeting the aforementioned pathological processes post-TBI holds the promise for the treatment of TBI as well as the treatment and even prevention of TBI-induced neurodegenerative diseases.
Potential Roles of Sirtuins in TBI and Related Neurodegeneration
After TBI, the expressions of sirtuins increase (Yang et al., 2017). Previous research has established that sirtuins exert a neuroprotective effect after TBI (Zhao et al., 2012; Yuan et al., 2016; Chen et al., 2017). Nevertheless, related research concerning the functions of sirtuins, particularly SIRT2, 4, 5, 6, 7, in TBI remains insufficient. Notably, sirtuins are also reported to be involved in the pathophysiology of other acute brain injury models, such as SAH, ischemic stroke, and I/R injury, whose pathophysiologies have some similarities with that of TBI (Table 1). Altogether, sirtuins regulate inflammation, oxidative stress, astrocyte activation, BBB permeability, axon elongation, and synaptic plasticity during acute brain injuries, all of which are characteristic pathological features of TBI (Fujita and Yamashita, 2018). Furthermore, some researchers indicated that sirtuins could reduce the accumulation of amyloid β peptide (Ramesh et al., 2018) and tau protein (Du et al., 2014), suggesting their preventive potential for TBI-induced neurodegenerative diseases. In this section, we outline the potential underlying mechanisms of sirtuins in TBI and related neurodegeneration, particularly with regards to AD. Of note, some of these candidate mechanisms are deduced from the models of other acute brain injuries, which are highlighted in corresponding sentences.
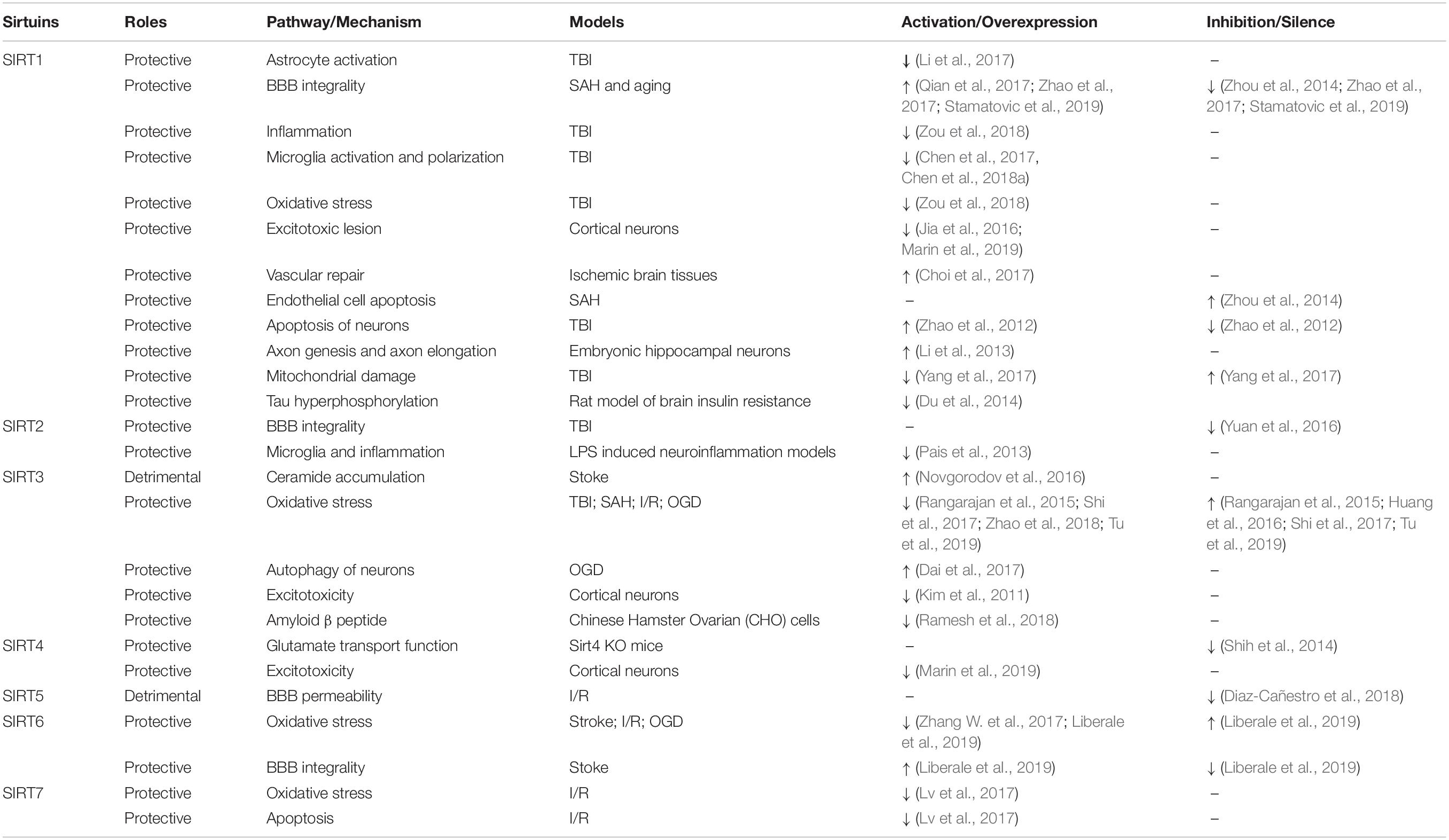
Table 1. Function of sirtuins in brain injury.
Sirtuins Reduce Inflammation
In TBI models, existing research has shown the critical role played by SIRT1 to reduce neuroinflammation (Chen et al., 2017). Moreover, SIRT2 was observed to alleviate inflammation in LPS-induced neuroinflammation models (Pais et al., 2013).
SIRT1 reduces inflammation by modulating high mobility group box 1 (HMGB1). SIRT1 maintains HMGB1 in a deacetylated state to control neuroinflammation resulting from microglial activation after TBI. Acetylated HMGB1 is required for HMGB1 transcription and extracellular secretion. HMGB1 is a non-histone-binding protein. Under normal conditions, HMGB1 binds to DNA and regulates transcription and translation in the nucleus. After TBI damage, HMGB1 translocates from the nucleus to the extracellular space. Extracellular HMGB1 induces microglial activation, which leads to further HMGB1 translocation (Chen et al., 2017). Extracellular HMGB1 binds to receptors on the cell surface, including toll-like receptor 4 and chemokine receptor 4 (Wang et al., 2017). HMGB1 activates receptors via medullary differentiation factor (MyD88) and non-MyD88-dependent pathways, triggering a signaling cascade in the microglia via the nuclear factor-κB (NF-κB) pathway. Nuclear translocation of NF-κB induces microglial polarization and cascade amplification of inflammatory cytokines (Chen et al., 2018a). Furthermore, SIRT1 inhibits activation of the NOD-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasome to attenuate inflammation after TBI. NLRP3 serves as a platform to produce interleukin-1β (IL-1β), an inflammatory initiating cytokine in a caspase reaction (Zou et al., 2018). Inflammation flooding further activates microglia, which in turn, aggravates inflammation (Chen et al., 2017). Previous studies have shown that microglial activation up to 1 year after TBI may cause chronic inflammation (Gentleman et al., 2004; Loane et al., 2014). Activated microglia have been identified around lesions in various neurodegenerative diseases (Xu et al., 2016). Excessive activation of microglia further damages neurons by releasing cytotoxic factors and accelerating the progression of neurodegenerative diseases (Glass et al., 2010). Moreover, SIRT1 was shown to mitigate astrocyte activation by inhibiting the p38 mitogen-activated protein kinase (MAPK) signaling pathway in an experimental model of TBI (Li et al., 2017). Astrocyte activation results in the overproduction of glial scar and inflammatory cytokines, which further activates the astrocytes, creating a positive inflammatory feedback loop that may injure the remaining neurons (Gayen et al., 2020).
Sirtuins Reduce Excitotoxicity
Excitotoxicity is implicated in the pathogenesis of several neurodegenerative diseases. Excessive release of glutamate results in glutamate receptor hyperactivation, calcium buffering impairment, free radical overproduction, and mitochondrial hyperpermeability, consequently leading to further neuronal degeneration and programmed cell death (Dong et al., 2009). Studies have shown that SIRT1, SIRT3, and SIRT4 are key players in reducing excitotoxic lesion in cortical neurons models (Kim et al., 2011; Jia et al., 2016; Marin et al., 2019).
An in vitro study in cortical neurons found that SIRT1 deacetylated and decreased peroxisome proliferator-activated receptor-coactivator 1α (PGC1α) to reduce glutamate excitotoxicity (Jia et al., 2016). PGC1α is a key factor in the maintenance of mitochondrial biogenesis and respiratory function (Scarpulla, 2011). Previous studies showed that SIRT4 promotes glutamate transport capacity by inhibiting excitotoxicity (Shih et al., 2014) and interacts with glutamine catabolism by hampering glutamate dehydrogenase (GDH) (Marin et al., 2019). Glutamine catabolism is crucial during the DNA repair response (Jia et al., 2016). Excessive glutamine catabolism leads to neuronal dysfunction and cell death (Jia et al., 2016). Furthermore, mitochondrial SIRT3 expression increases after poly(ADP-ribose) polymerase-1-mediated NAD depletion in cortical neurons. Kim et al. (2011) concluded that SIRT3 is essential for neuroprotection against excitotoxicity. The authors showed that NAD depletion further increased ROS production, aggravated oxidative stress, and upregulated the expression of SIRT3 in mitochondria.
Sirtuins Improve Mitochondrial Function and Reduce Oxidative Stress
Mitochondrial dysfunction is a fundamental pathogenic event in most neurodegenerative diseases. Findings from rat models of TBI have revealed that ceramide is the first lipid to accumulate in the mitochondria after TBI (Roux et al., 2016), suggesting it may serve as a therapeutic indicator (Barbacci et al., 2017). Ceramide accumulation causes mitochondrial dysfunction (Novgorodov et al., 2016), which is associated with increased mitochondrial fission and excessive ROS production (Zhao et al., 2018).
According to existing research, SIRT1 and SIRT3 have been detected to alleviate oxidative stress in TBI models (Rangarajan et al., 2015; Zou et al., 2018). The study by Yang et al. (2017) showed that SIRT1 reduces the mitochondrial damage to play its neuroprotective roles in TBI. SIRT3, SIRT6, and SIRT7 showed their functions of reducing oxidative stress in other brain models (Lv et al., 2017; Shi et al., 2017; Zhang W. et al., 2017; Zhao et al., 2018; Liberale et al., 2019; Tu et al., 2019).
SIRT1 has been shown to improve mitochondrial function by inhibiting the p38 MAPK pathway in animal models of TBI. Given that MAPK protein is associated with multiple phosphorylation/dephosphorylation signaling cascades, SIRT1 may inhibit this pathway by dephosphorylating p38 MAPK (Yang et al., 2017). A previous study found that SIRT1 activity was downregulated in the brains of patients with neurodegenerative diseases including PD. Furthermore, the authors reported that SIRT1 reduced oxidative stress-induced neural cell death and concluded that SIRT1 is a pro-survival protein that is downregulated under cellular stress (Singh et al., 2017). These findings suggest that SIRT1 has the potential to reduce oxidative stress in TBI and associated neurodegenerative diseases.
Investigations of the effects of SIRT3 on mitochondrial dysfunction and oxidative stress have yielded contradictory findings. Results from animal models provide evidence supporting a promoting role for SIRT3 in the accumulation of ceramide within the mitochondria (Novgorodov et al., 2016), which serves as a causal factor of mitochondrial dysfunction by inhibiting the activity of respiratory chain complex III (Gudz et al., 1997; Anderson and Sims, 1999; Yu et al., 2007). Indeed, SIRT3 deacetylates ceramide synthase 1, 2, and 6 to keep them in hyperacetylated state (primed state), propelling the ceramide accumulation (Novgorodov et al., 2016). However, some study showed that the inhibition of SIRT3 had no beneficial effects on brain injury in a model of stroke (Verma et al., 2019). Inversely, inhibition of SIRT3 in microglia leads to excessive ROS production. SIRT3 deacetylates Forkhead box O 3a (Foxo3a), a transcription factor that transactivates antioxidant genes, to enable it to translocate into the nucleus. Thus, SIRT3 increases the production of antioxidants, including catalase and manganese superoxide dismutase (mnSOD), to protect against oxidative stress (Rangarajan et al., 2015). Another study pointed out that it is a compensatory increase in SIRT1 rather than the inhibition of SIRT3 that exerts neuroprotective effect (Verma et al., 2019). Not only that, recent work by researchers proved that SIRT3 exerted neuroprotective effect against oxidative stress not only in models of TBI (Rangarajan et al., 2015) but also in SAH (Huang et al., 2016), I/R (Zhao et al., 2018; Tu et al., 2019), and oxygen-glucose deprivation (OGD) (Wang et al., 2015) models. Moreover, SIRT3 protects dopaminergic neurons against oxidative stress (Shi et al., 2017). The degeneration of dopaminergic neurons is the primary cause of PD. SIRT3 regulation of mnSOD activity via deacetylation at the lysine 68 site attenuates oxidative stress and restores mitochondrial membrane potential in dopaminergic neurons (Shi et al., 2017). Collectively, these findings support SIRT3-induced attenuation of oxidative stress after TBI and related neurodegeneration.
SIRT6 locates in the nucleus, where it promotes resistance to DNA damage and oxidative stress by supporting DNA double-strand break repair (Beauharnois et al., 2013). SIRT6 levels decrease after I/R injury. Nuclear factor erythroid-derived 2-like 2 (NRF2) is activated via the deacetylation by SIRT6. NRF2 is a basic leucine zipper transcription factor that regulates the expression of antioxidant proteins, such as heme oxygenase-1 and SOD, that resist oxidative stress and have a neuroprotective function (Zhang W. et al., 2017). Although SIRT7 is expressed ubiquitously in the cortex, striatum, hippocampus, and thalamus (Islam et al., 2018), its function in the brain is unclear. A recent study in an I/R model found that SIRT7 regulated apoptosis and resisted oxidative stress via deacetylation of the p53 protein (Lv et al., 2017). This finding suggests that SIRT6 and 7 may have similar effects on oxidative stress after TBI.
Sirtuins Mitigate Proteinopathies
Sirtuins are a kind of longevity assurance factor and involved in the promotion of healthy aging mechanisms (Braidy et al., 2012). Sirtuins have some special functions on lowering the risk of neurodegenerative diseases such as reducing the accumulation of tau proteins and amyloid β-peptide (Braidy et al., 2012; Du et al., 2014).
Tau proteins and amyloid β-peptide accumulations serve as indispensable parts in the development of AD (Braidy et al., 2012). Cytological studies have shown that SIRT1 in growth cones promotes axon genesis and elongation in embryonic hippocampal neurons (Li et al., 2013). The accumulation of amyloid precursor protein and amyloid β-peptide in injured axons after TBI has been demonstrated in vivo (Smith et al., 2003) and in vitro (Tang et al., 2017) studies. These pathologies are associated with AD (Smith et al., 2003). SIRT1 locates in the distal regions of the axon, the axonal growth cone in particular. SIRT1 regulates the deacetylation and activation of protein kinase B (Akt). Akt is the upstream inhibitory kinase of glycogen synthase kinase3 (GSK3). Akt activation inhibits GSK3 activity (Li et al., 2013). GSK3 is a multifunctional serine/threonine kinase and a key regulator of neurogenesis, polarization, neurite outgrowth, and plasticity in the nervous system (Hur and Zhou, 2010). Intracellular mechanisms involved in axon genesis include actin filaments and the reorganization of microtubules (Li et al., 2013). In addition to reducing amyloid β-peptide accumulation, SIRT1 upregulates the amount of lysosome in primary astrocytes to promote the degradation of amyloid β-peptide by its deacetylase activity (Li M.Z. et al., 2018). Recent studies showed that SIRT1 may exert its anti-amyloidogenic effects by direct activating gene ADAM10, which encodes disintegrin protein (Braidy et al., 2012). Furthermore, an in vitro study in Chinese hamster ovarian cells found that SIRT3 reduced ROS production and lipid peroxidation and improved mitochondrial function, resulting in the reduction of amyloid β-peptide production (Ramesh et al., 2018).
Hyperphosphorylated tau protein accumulation is widespread after a single TBI (Johnson et al., 2012). Hyperphosphorylated tau proteins misfold and aggregate to form NFTs, consequently resulting in impaired tau function (Johnson et al., 2012). Hyperphosphorylated tau proteins are liberated into the extracellular space, stimulating microglia and astrocytes to release pro-inflammatory cytokines (e.g., IL-1β and TNFα), which further activate tau kinases and aggravate tau hyperphosphorylation (Fesharaki-Zadeh, 2019). In addition to phosphorylation, acetylation is a crucial step in the post-translational modification of tau proteins. The acetylation of tau protein has been demonstrated in all stages of AD. An in vitro study in cortical neurons argued that SIRT3 deacetylated tau proteins, which attenuated the pathological accumulation of tau proteins (Yin et al., 2018). Du et al.’s (2014) research has shown that SIRT1 suppressed tau protein accumulation in hippocampus neurons in a rat model of brain insulin resistance.
Sirtuins and BBB Integrity
The BBB is an interface between the brain parenchyma and cerebral circulation. BBB permeability is regulated by pericytes, astrocytes, and cerebral microvascular endothelial cells through the expression of tight and adherent junctions (Liberale et al., 2019). BBB disruption serves as one of the critical parts in the development of AD and other neurodegenerative disorders. Disruption of the BBB allows neurotoxic cells, pathogens, and inflammatory cytokines to leak from the cerebral circulation to the brain parenchyma, where they activate inflammatory and immune responses associated with severe TBI and the development of neurodegenerative diseases (Qian et al., 2017; Sweeney et al., 2018; Zhou et al., 2020a).
SIRT2 serves as a pivotal factor in BBB integrity. SIRT2 inhibition has been shown to increase BBB permeability and exacerbate inflammation and brain edema after TBI (Yuan et al., 2016). Matrix metalloproteinase (MMP)-9 degrades tight junction proteins of BBB. SIRT2 inhibition stimulates MMP-9 expression and disturbs the integrity of BBB, allowing plasma components (e.g., macrophage and inflammatory factors) crossing the BBB more easily, consequently resulting in the exacerbation of neuroinflammation. Moreover, SIRT2 inhibition promotes microglia and macrophage activation, which intensifies the inflammatory processes. SIRT2 inhibition promotes nuclear translocation of NF-kB p65 via acetylation of the p65 subunit to increase NF-kB activity and upregulate its targets including aquaporin 4, MMP-9, and proinflammatory cytokines (Yuan et al., 2016). By contrast, SIRT5 adversely affects the maintenance of BBB integrity. SIRT5 has previously been observed to exacerbate inflammation in an experimental model of I/R injury, while SIRT5 inhibition decreased endothelial permeability of the BBB and increased the expression of occludin and claudin-5 via the phosphatidylinositol 3-kinase/Akt pathway (Diaz-Cañestro et al., 2018).
In experimental models of SAH and aging, SIRT1 prevents BBB hyperpermeability (Qian et al., 2017; Stamatovic et al., 2019). This effect can be reversed by inhibiting SIRT1 (Zhou et al., 2014). Tight junction proteins, such as claudin-5 and occludin, are the main components of the BBB structure (Kniesel and Wolburg, 2000). SIRT1 upregulates tight junction protein expression in response to brain edema to protect BBB integrity. In addition, SIRT1 attenuates damage caused by brain edema via the deacetylation of p53. P53 is a protein upregulating endothelial MMP-9 via the NF-kB pathway to reduce occlusion in animal models of SAH (Yan et al., 2008; Qian et al., 2017). Furthermore, a study of ischemic brain tissue from mice found that SIRT1 was involved in the repair of BBB vascular destruction by upregulating vascular endothelial growth factor expression in astrocytes (Choi et al., 2017). SIRT1 was proved to accelerate endothelial cell apoptosis in an animal model of SAH (Zhou et al., 2014). Furthermore, a recent study suggested that endothelial SIRT6 protected BBB integrity after an I/R injury in a stroke model; however, the mechanism is not fully understood (Liberale et al., 2019).
Sirtuins and Neuronal Apoptosis
Sirtuins reduce the apoptosis of normal neurons and promote apoptosis in damaged neurons. TBI induces SIRT1 expression and activates the MAPK/extracellular signal-related kinase (ERK) pathway. Inhibition of SIRT1 and the MAPK/ERK pathway decreases neuronal apoptosis. Moreover, inhibition of the MAPK/ERK pathway further decreases SIRT1 levels. The dynamic relationship between SIRT1 and apoptosis has been demonstrated in in vivo and in vitro models of TBI (Zhao et al., 2012). Moreover, sirtuins play an essential role in reducing apoptosis in other models of brain injury. SIRT1 is a nicotinamide-adenine dinucleotide-dependent p53 deacetylase. P53 induces apoptosis in the acetylated state (Sykes et al., 2006). SIRT1 promotes p53 proteolysis via deacetylation (Qian et al., 2017) to inhibit p53 transactivation activity and suppress apoptosis in response to oxidative stress in an experimental model of SAH. SIRT3 was found to promote autophagy of damaged neurons to protect ischemic neurons in a model of OGD (Dai et al., 2017). Cortical neurons overexpress SIRT3 under ischemic and hypoxia conditions, further increasing the phosphorylation of adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK), which inhibits the phosphorylation of mTOR and induces autophagy of ischemia neurons (Dai et al., 2017).
Sirtuins as Promising Therapeutic Targets
Given that primary TBIs are irreversible, interventional strategies to prevent secondary injuries and promote plasticity and the recovery process are of primary concern. As mentioned previously, the sirtuin family are vividly involved in several TBI-induced pathophysiological processes including microglial and astrocyte activation, inflammation, proteinopathies, oxidative stress, and mitochondrial damage, all of which may facilitate the onset of neurodegenerative diseases. Moreover, sirtuins have a wide range of effects on BBB integrity, apoptosis, and excitotoxicity in other in vivo and in vitro models of brain injury. As a result of these pleiotropic effects, sirtuins have been acknowledged to be emerging targets for TBI and TBI-related neurodegeneration (Donmez and Outeiro, 2013; Foolad et al., 2019).
There is a growing body of literature that recognizes the therapeutic potential of the modulators of sirtuins (Table 2). Although these modulators have not been tested clinically, there is an abundance of preclinical data in cortical neuron cultures and animal models that describe specific mechanisms of modulator actions. Identification of a modulator that allows precise control of sirtuin expression will enable clinicians to treat TBI and prevent pathological processes that trigger late-onset neurodegenerative diseases.
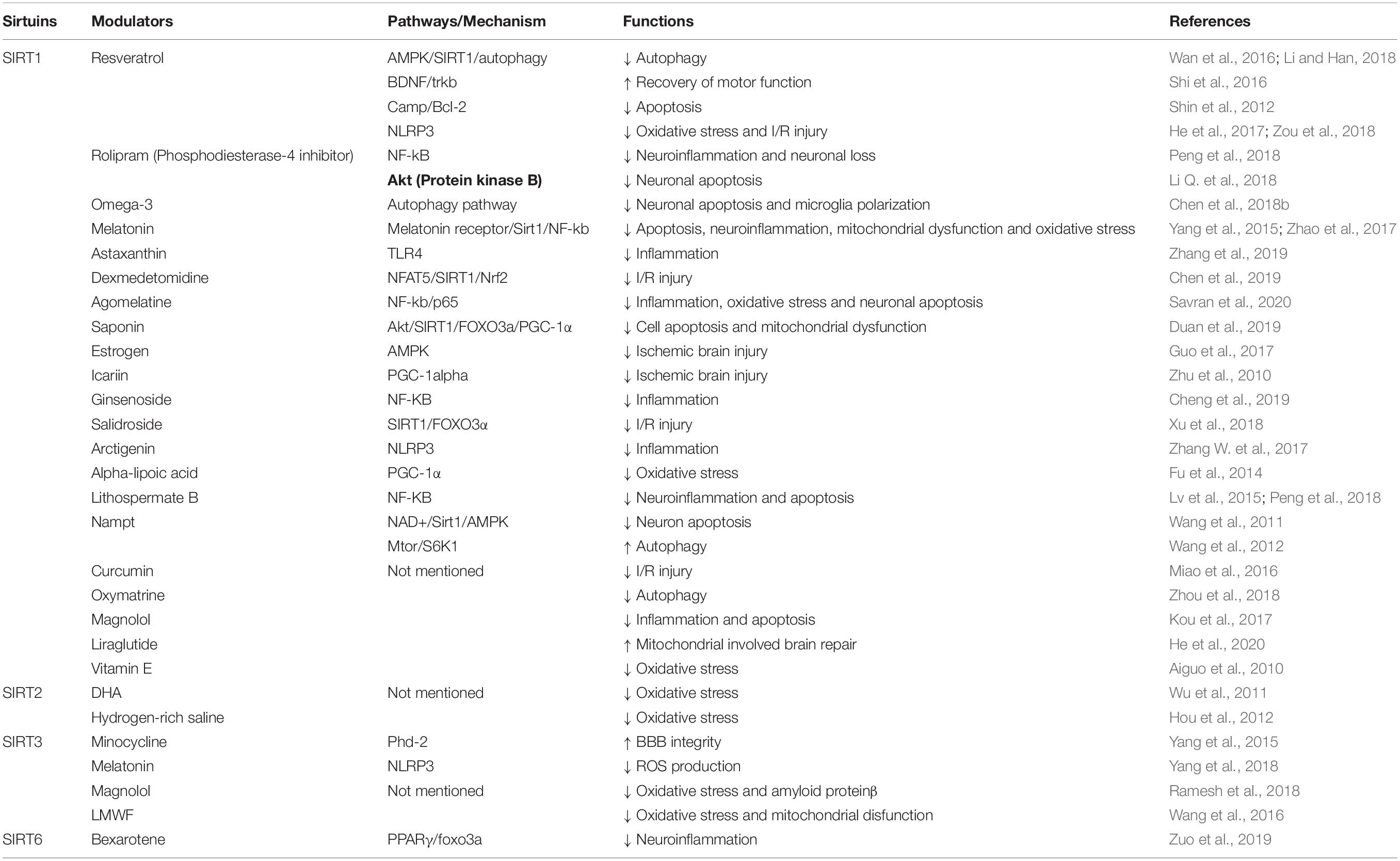
Table 2. Modulators of sirtuins.
Resveratrol (3, 5, 4′-trihydroxystilbene) is one of the most effective natural SIRT1-activating compounds. Resveratrol is a natural polyphenolic substance extracted from grapes that has been found to increase SIRT1 activity about 10-fold. By modifying at the B ring 4′ position, scientists developed a variety of synthetic resveratrol derivatives to lower toxicity to cells or improve potency (Dai et al., 2018). Resveratrol and its derivatives play various roles in apoptosis (Shin et al., 2012), autophagy (Wan et al., 2016; Li and Han, 2018), oxidative stress (He et al., 2017), and recovery of motor functions (Shi et al., 2016). Moreover, resveratrol modulates the activity of SIRT3 and SIRT5 (Dai et al., 2018). Several other natural extracts also activate SIRT1, including magnolol (Kou et al., 2017), oxymatrine (Zhou et al., 2018), curcumin (Miao et al., 2016), salvianolic acid B (Lv et al., 2015), arctigenin (Zhang S. et al., 2017), astaxanthin (Zhang et al., 2019), salidroside (Xu et al., 2018), ginsenoside (Cheng et al., 2019), icariin (Zhu et al., 2010), low molecular weight fucoidan (Wang et al., 2016), and saponin from Aralia taibaiensis (Duan et al., 2019). These findings suggest that Chinese traditional medicine may be a source of sirtuin modulators.
Moreover, some hormones modulate SIRT1. Melatonin, a kind of hormone mainly secreted by the pineal gland, regulates circadian rhythms. Melatonin is a powerful antioxidant that attenuates early brain injury–induced oxidative stress and neuronal apoptosis by activating the SIRT1/NF-kB pathway (Zhao et al., 2017). Furthermore, melatonin upregulates the expression of SIRT3 to reduce the production of ROS and resist oxidative stress (Yang et al., 2018). Estrogen also regulates SIRT1. Estrogen restores resistance to cell apoptosis and attenuates ischemic brain injury via the SIRT1/AMPK pathway (Guo et al., 2017). Activation of the SIRT1/AMPK pathway may underlie the neuroprotective effect of estrogen.
Some drugs that are not originally developed to regulate sirtuins have currently been found to have neuroprotective properties via a SIRT-involved pathway. For instance, the long-acting glucagon-like peptide-1 analogs, liraglutide (He et al., 2020), agomelatine (Savran et al., 2020), and dexmedetomidine (Chen et al., 2019), exert a SIRT1-dependent neuroprotective effect and critically regulate the metabolism. Furthermore, the antibiotic, minocycline, modulates SIRT3 to protect BBB integrity (Yang et al., 2015), and bexarotene attenuates neuroinflammation by modulating SIRT6 (Zuo et al., 2019).
Intermittent fasting, and daily caloric (Ran et al., 2015) or protein restrictions (de Carvalho et al., 2019) have been shown to reduce inflammation and oxidative stress by upregulating SIRT1. Specifically, reduced energy intake without affecting nutritional requirements has a neuroprotective effect after brain injury (Ran et al., 2015). Besides, docosahexaenoic acid (Wu et al., 2011), vitamin E (Aiguo et al., 2010), and hydrogen-rich saline (Hou et al., 2012) reduce oxidative stress. The antioxidant, alpha-lipoic acid, reduces oxidative stress by upregulating SIRT1 (Fu et al., 2014).
Besides, NAD+ nicotinamide mononucleotide (NMN; precursor of NAD+) provides neuroprotection by directly activating sirtuins (Klimova et al., 2019). Dysregulation of NAD+ metabolism may cause neurodegenerative diseases and acute brain injury. NMN is a multi-targeted modulator: it activates sirtuins, inhibits mitochondrial fission, and is an NAD+ supplement. Therefore, NMN is a promising therapeutic agent for TBI and associated neurodegenerative diseases (Klimova and Kristian, 2019). NMN is an endogenous cellular metabolic compound that may have lower toxicity and fewer side effects at high doses (Park et al., 2016).
Furthermore, some enzymes modulate sirtuin. Nicotinamide phosphoribosyltransferase (Nampt) is the rate-limiting enzyme in mammalian NAD+ biosynthesis (Wang et al., 2011). Nampt, which is located primarily in neurons, regulates autophagy and apoptosis (Wang et al., 2012). Rolipram, a phosphodiesterase-4 inhibitor, modulates inflammation and neuronal apoptosis via different SIRT1-dependent pathways (Li Q. et al., 2018; Peng et al., 2018).
However, a better understanding of sirtuins and their molecular structures is still needed to facilitate the discovery of additional modulators. Referring to Table 1, we can find that sirtuins seem to play contradictory roles under certain circumstances, which may bring difficulties to the development of TBI pharmacological agents (Gomes et al., 2019).
Conclusion and Limitations
TBI is an accepted risk factor for neurodegenerative diseases. The key pathological features of TBI and neurodegenerative diseases include neuroinflammation, oxidative stress, excitotoxicity, proteinopathies, and mitochondrial dysfunction, with many similarities and overlaps. Therefore, targeting these processes has the therapeutic potential for TBI and associated neurodegenerative diseases. The sirtuin family have seven members, namely SIRT1–7 and are strictly implicated in the pathological mechanisms of acute brain injuries. Among these sirtuins, SIRT1 and SIRT3 have been demonstrated to reduce the post-TBI damage by alleviating inflammation, oxidative stress and proteinopathies, neuronal apoptosis, and BBB compromise. The rest sirtuins intervene in similar pathophysiology in other brain injury models and merit further exploration in TBI models. Recent progress on sirtuin modulators has made sirtuins promising therapeutic targets. However, sirtuins may show contradictory effects under certain conditions, and certain off-target effects should be taken into consideration.
This review has some limitations. First, studies about sirtuins’ effects on TBI are not enough yet, especially SIRT2, 4, 5, 6, and 7, which lack support from direct evidence in TBI models. We speculate that functions of SIRT2, 4, 5, 6, and 7 in other kinds of brain injury models may be also valid in TBI models. Second, the demonstration of the functions of sirtuins in this article is all based on animal test results. Hence, further exploration of human and clinical evidence is warranted. Third, the hypothetical linking between TBI and neurodegenerative diseases is supported by similarities of pathological processes. We should explore more supporting evidence to offer this association a more solid foundation.
Author Contributions
YZ and AS conceptualized the research project. QY, YZ, YuS, and YL wrote the manuscript and made the original figures. AS, YeS, and QY critically revised the texts and figures. AS, YeS, and YZ supervised the research and led the discussion. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Natural Science Foundation of China (81701144).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AD, Alzheimer’s disease; Akt, protein kinase B; AMPK, adenosine monophosphate activated protein kinase; AQP4, aquaporin 4; BBB, blood–brain barrier; Cat, catalase; COX-1, cyclooxygenase-1; CTE, chronic traumatic encephalopathy; Foxo3a, forkhead box O 3a; GDH, glutamate dehydrogenase; GSK3, glycogen synthase kinase3; HMGB1, high-mobility group box 1; HO-1, heme oxygenase-1; I/R, ischemia/reperfusion; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinases; MARK, mitogen-activated protein kinase; MMP-9, matrix metalloproteinase 9; mnSod, manganese superoxide dismutase; MyD88, medullary differentiation factor; Nampt, nicotinamide phosphoribosyl-transferase; NF- κ B, nuclear factor- κ B; NFTs, neurofibrillary tangles; NLRP3, NLR family pyrin domain containing 3; NMN, NAD+ nicotinamide mononucleotide; NRF2, nuclear factor (erythroid-derived 2)-like 2; OGD, oxygen-glucose deprivation; PARP-1, poly (ADP-ribose) polymerase-1; PD, Parkinson’s disease; PGC1 α, peroxisome proliferator-activated receptor-coactivator 1 α; PI3K, phosphatidylinositol 3-kinase; ROS, reactive oxygen species; Sir2, Silence information regulator 2; SIRT, sirtuin; SOD, superoxide dismutase; STACs, sirtuin activating compounds; TBI, traumatic brain injury; TLR4, toll-like receptor 4.
References
Aiguo, W., Zhe, Y., and Gomez-Pinilla, F. (2010). Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabil. Neural Repair 24, 290–298. doi: 10.1177/1545968309348318
Al-Dahhak, R., Khoury, R., Qazi, E., and Grossberg, G. T. (2018). Traumatic brain injury, chronic traumatic encephalopathy, and Alzheimer disease. Clin. Geriatr. Med. 34, 617–635. doi: 10.1016/j.cger.2018.06.008
Anderson, M. F., and Sims, N. R. (1999). Mitochondrial respiratory function and cell death in focal cerebral ischemia. J. Neurochem. 73, 1189–1199. doi: 10.1046/j.1471-4159.1999.0731189.x
Barbacci, D. C., Roux, A., Muller, L., Jackson, S. N., Post, J., Baldwin, K., et al. (2017). Mass spectrometric imaging of ceramide biomarkers tracks therapeutic response in traumatic brain injury. ACS Chem. Neurosci. 8, 2266–2274. doi: 10.1021/acschemneuro.7b00189
Barshikar, S., and Bell, K. R. (2017). Sleep disturbance after TBI. Curr. Neurol. Neurosci. Rep. 17:87. doi: 10.1007/s11910-017-0792-4
Beauharnois, J. M., Bolívar, B. E., and Welch, J. T. (2013). Sirtuin 6: a review of biological effects and potential therapeutic properties. Mol. Biosyst. 9, 1789–1806. doi: 10.1039/c3mb00001j
Braidy, N., Jayasena, T., Poljak, A., and Sachdev, P. S. (2012). Sirtuins in cognitive ageing and Alzheimer’s disease. Curr. Opin. Psychiatry 25, 226–230. doi: 10.1097/YCO.0b013e32835112c1
Chen, H., Wu, D., Ding, X., and Ying, W. (2015). SIRT2 is required for lipopolysaccharide-induced activation of BV2 microglia. Neuroreport 26, 88–93. doi: 10.1097/WNR.0000000000000305
Chen, L., Cao, J., Cao, D., Wang, M., Xiang, H., Yang, Y., et al. (2019). Protective effect of dexmedetomidine against diabetic hyperglycemia-exacerbated cerebral ischemia/reperfusion injury: an in vivo and in vitro study. Life Sci. 235:116553. doi: 10.1016/j.lfs.2019.116553
Chen, X., Chen, C., Fan, S., Wu, S., Yang, F., Fang, Z., et al. (2018a). Omega-3 polyunsaturated fatty acid attenuates the inflammatory response by modulating microglia polarization through SIRT1-mediated deacetylation of the HMGB1/NF-kappaB pathway following experimental traumatic brain injury. J. Neuroinflammation 15:116. doi: 10.1186/s12974-018-1151-3
Chen, X., Pan, Z., Fang, Z., Lin, W., Wu, S., Yang, F., et al. (2018b). Omega-3 polyunsaturated fatty acid attenuates traumatic brain injury-induced neuronal apoptosis by inducing autophagy through the upregulation of SIRT1-mediated deacetylation of Beclin-1. J. Neuroinflammation 15:310. doi: 10.1186/s12974-018-1345-8
Chen, X., Wu, S., Chen, C., Xie, B., Fang, Z., Hu, W., et al. (2017). Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-kappaB pathway following experimental traumatic brain injury. J. Neuroinflammation 14:143. doi: 10.1186/s12974-017-0917-3
Cheng, Z., Zhang, M., Ling, C., Zhu, Y., Ren, H., Hong, C., et al. (2019). Neuroprotective effects of ginsenosides against cerebral ischemia. Molecules 24:1102. doi: 10.3390/molecules24061102
Choi, Y. K., Kim, J. H., Lee, D. K., Lee, K. S., Won, M. H., Jeoung, D., et al. (2017). Carbon monoxide potentiation of L-type Ca(2+) channel activity increases HIF-1α-independent VEGF expression via an AMPKα/SIRT1-mediated PGC-1α/ERRα axis. Antioxid. Redox Signal. 27, 21–36. doi: 10.1089/ars.2016.6684
Cornelius, C., Crupi, R., Calabrese, V., Graziano, A., Milone, P., Pennisi, G., et al. (2013). Traumatic brain injury: oxidative stress and neuroprotection. Antioxid. Redox Signal. 19, 836–853. doi: 10.1089/ars.2012.4981
Daglas, M., and Adlard, P. A. (2018). The involvement of iron in traumatic brain injury and neurodegenerative disease. Front. Neurosci. 12:981. doi: 10.3389/fnins.2018.00981
Dai, H., Sinclair, D. A., Ellis, J. L., and Steegborn, C. (2018). Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharmacol. Ther. 188, 140–154. doi: 10.1016/j.pharmthera.2018.03.004
Dai, S. H., Chen, T., Li, X., Yue, K. Y., Luo, P., Yang, L. K., et al. (2017). Sirt3 confers protection against neuronal ischemia by inducing autophagy: involvement of the AMPK-mTOR pathway. Free Radic. Biol. Med. 108, 345–353. doi: 10.1016/j.freeradbiomed.2017.04.005
de Carvalho, T. S., Sanchez-Mendoza, E. H., Nascentes, L. M., Schultz Moreira, A. R., Sardari, M., Dzyubenko, E., et al. (2019). Moderate protein restriction protects against focal cerebral ischemia in mice by mechanisms involving anti-inflammatory and anti-oxidant responses. Mol. Neurobiol. 56, 8477–8488. doi: 10.1007/s12035-019-01679-6
DeKosky, S. T., and Asken, B. M. (2017). Injury cascades in TBI-related neurodegeneration. Brain Inj. 31, 1177–1182. doi: 10.1080/02699052.2017.1312528
Diaz-Cañestro, C., Merlini, M., Bonetti, N. R., Liberale, L., Wüst, P., Briand-Schumacher, S., et al. (2018). Sirtuin 5 as a novel target to blunt blood-brain barrier damage induced by cerebral ischemia/reperfusion injury. Int. J. Cardiol. 260, 148–155. doi: 10.1016/j.ijcard.2017.12.060
Dong, X. X., Wang, Y., and Qin, Z. H. (2009). Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 30, 379–387. doi: 10.1038/aps.2009.24
Donmez, G., and Outeiro, T. F. (2013). SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol. Med. 5, 344–352. doi: 10.1002/emmm.201302451
Du, L. L., Xie, J. Z., Cheng, X. S., Li, X. H., Kong, F. L., Jiang, X., et al. (2014). Activation of sirtuin 1 attenuates cerebral ventricular streptozotocin-induced tau hyperphosphorylation and cognitive injuries in rat hippocampi. Age 36, 613–623. doi: 10.1007/s11357-013-9592-1
Duan, J., Cui, J., Zheng, H., Xi, M., Guo, C., Weng, Y., et al. (2019). Aralia taibaiensis protects against I/R-induced brain cell injury through the Akt/SIRT1/FOXO3a pathway. Oxid. Med. Cell. Longev. 2019:7609765. doi: 10.1155/2019/7609765
Fesharaki-Zadeh, A. (2019). Chronic traumatic encephalopathy: a brief overview. Front. Neurol. 10:713. doi: 10.3389/fneur.2019.00713
Foolad, F., Khodagholi, F., and Javan, M. (2019). Sirtuins in multiple sclerosis: the crossroad of neurodegeneration, autoimmunity and metabolism. Mult. Scler. Relat. Disord. 34, 47–58. doi: 10.1016/j.msard.2019.06.004
Fu, B., Zhang, J., Zhang, X., Zhang, C., Li, Y., Zhang, Y., et al. (2014). Alpha-lipoic acid upregulates SIRT1-dependent PGC-1alpha expression and protects mouse brain against focal ischemia. Neuroscience 281, 251–257. doi: 10.1016/j.neuroscience.2014.09.058
Fujita, Y., and Yamashita, T. (2018). Sirtuins in neuroendocrine regulation and neurological diseases. Front. Neurosci. 12:778. doi: 10.3389/fnins.2018.00778
Gardner, R. C., Byers, A. L., Barnes, D. E., Li, Y., Boscardin, J., and Yaffe, K. (2018). Mild TBI and risk of Parkinson disease: a chronic effects of neurotrauma consortium study. Neurology 90, e1771–e1779. doi: 10.1212/wnl.0000000000005522
Gardner, R. C., and Yaffe, K. (2015). Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol. Cell. Neurosci. 66(Pt B), 75–80. doi: 10.1016/j.mcn.2015.03.001
Gayen, M., Bhomia, M., Balakathiresan, N., and Knollmann-Ritschel, B. (2020). Exosomal MicroRNAs released by activated astrocytes as potential neuroinflammatory biomarkers. Int. J. Mol. Sci. 21:2312. doi: 10.3390/ijms21072312
Gentleman, S. M., Leclercq, P. D., Moyes, L., Graham, D. I., Smith, C., Griffin, W. S., et al. (2004). Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci. Int. 146, 97–104. doi: 10.1016/j.forsciint.2004.06.027
Giza, C. C., and Hovda, D. A. (2014). The new neurometabolic cascade of concussion. Neurosurgery 75(Suppl. 4), S24–S33. doi: 10.1227/neu.0000000000000505
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Goldstein, L. E., Fisher, A. M., Tagge, C. A., Zhang, X. L., Velisek, L., Sullivan, J. A., et al. (2012). Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4:134ra60. doi: 10.1126/scitranslmed.3003716
Gomes, P., Leal, H., Mendes, A. F., Reis, F., and Cavadas, C. (2019). Dichotomous sirtuins: implications for drug discovery in neurodegenerative and cardiometabolic diseases. Trends Pharmacol. Sci. 40, 1021–1039. doi: 10.1016/j.tips.2019.09.003
Gomes, P., Viana, S. D., Nunes, S., Rolo, A. P., Palmeira, C. M., and Reis, F. (2020). The yin and yang faces of the mitochondrial deacetylase sirtuin 3 in age-related disorders. Ageing Res. Rev. 57:100983. doi: 10.1016/j.arr.2019.100983
Greve, M. W., and Zink, B. J. (2009). Pathophysiology of traumatic brain injury. Mt. Sinai J. Med. 76, 97–104. doi: 10.1002/msj.20104
Gudz, T. I., Tserng, K. Y., and Hoppel, C. L. (1997). Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J. Biol. Chem. 272, 24154–24158. doi: 10.1074/jbc.272.39.24154
Guo, J. M., Shu, H., Wang, L., Xu, J. J., Niu, X. C., and Zhang, L. (2017). SIRT1-dependent AMPK pathway in the protection of estrogen against ischemic brain injury. CNS Neurosci. Ther. 23, 360–369. doi: 10.1111/cns.12686
Han, P., Tang, Z., Yin, J., Maalouf, M., Beach, T. G., Reiman, E. M., et al. (2014). Pituitary adenylate cyclase-activating polypeptide protects against β-amyloid toxicity. Neurobiol. Aging 35, 2064–2071. doi: 10.1016/j.neurobiolaging.2014.03.022
Hayes, J. P., Logue, M. W., Sadeh, N., Spielberg, J. M., Verfaellie, M., Hayes, S. M., et al. (2017). Mild traumatic brain injury is associated with reduced cortical thickness in those at risk for Alzheimer’s disease. Brain 140, 813–825. doi: 10.1093/brain/aww344
He, Q., Li, Z., Wang, Y., Hou, Y., Li, L., and Zhao, J. (2017). Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int. Immunopharmacol. 50, 208–215. doi: 10.1016/j.intimp.2017.06.029
He, W., Wang, H., Zhao, C., Tian, X., Li, L., and Wang, H. (2020). Role of liraglutide in brain repair promotion through Sirt1-mediated mitochondrial improvement in stroke. J. Cell. Physiol. 235, 2986–3001. doi: 10.1002/jcp.29204
Hernández, D. E., Salvadores, N. A., Moya-Alvarado, G., Catalán, R. J., Bronfman, F. C., and Court, F. A. (2018). Axonal degeneration induced by glutamate excitotoxicity is mediated by necroptosis. J. Cell Sci. 131:jcs214684. doi: 10.1242/jcs.214684
Hiebert, J. B., Shen, Q., Thimmesch, A. R., and Pierce, J. D. (2015). Traumatic brain injury and mitochondrial dysfunction. Am. J. Med. Sci. 350, 132–138. doi: 10.1097/maj.0000000000000506
Holloway, O. G., Canty, A. J., King, A. E., and Ziebell, J. M. (2019). Rod microglia and their role in neurological diseases. Semin. Cell Dev. Biol. 94, 96–103. doi: 10.1016/j.semcdb.2019.02.005
Hou, Z., Luo, W., Sun, X., Hao, S., Zhang, Y., Xu, F., et al. (2012). Hydrogen-rich saline protects against oxidative damage and cognitive deficits after mild traumatic brain injury. Brain Res. Bull. 88, 560–565. doi: 10.1016/j.brainresbull.2012.06.006
Huang, W., Huang, Y., Huang, R. Q., Huang, C. G., Wang, W. H., Gu, J. M., et al. (2016). SIRT3 expression decreases with reactive oxygen species generation in rat cortical neurons during early brain injury induced by experimental subarachnoid hemorrhage. Biomed Res. Int. 2016:8263926. doi: 10.1155/2016/8263926
Hur, E. M., and Zhou, F. Q. (2010). GSK3 signalling in neural development. Nat. Rev. Neurosci. 11, 539–551. doi: 10.1038/nrn2870
Ikonomovic, M. D., Uryu, K., Abrahamson, E. E., Ciallella, J. R., Trojanowski, J. Q., Lee, V. M. Y., et al. (2004). Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol. 190, 192–203. doi: 10.1016/j.expneurol.2004.06.011
Islam, M. S., Wei, F. Y., Ohta, K., Shigematsu, N., Fukuda, T., Tomizawa, K., et al. (2018). Sirtuin 7 is involved in the consolidation of fear memory in mice. Biochem. Biophys. Res. Commun. 495, 261–266. doi: 10.1016/j.bbrc.2017.10.159
Jia, N., Sun, Q., Su, Q., and Chen, G. (2016). SIRT1-mediated deacetylation of PGC1α attributes to the protection of curcumin against glutamate excitotoxicity in cortical neurons. Biochem. Biophys. Res. Commun. 478, 1376–1381. doi: 10.1016/j.bbrc.2016.08.132
Johnson, V. E., Stewart, J. E., Begbie, F. D., Trojanowski, J. Q., Smith, D. H., and Stewart, W. (2013a). Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136(Pt 1), 28–42. doi: 10.1093/brain/aws322
Johnson, V. E., Stewart, W., and Smith, D. H. (2013b). Axonal pathology in traumatic brain injury. Exp. Neurol. 246, 35–43. doi: 10.1016/j.expneurol.2012.01.013
Johnson, V. E., Stewart, W., Arena, J. D., and Smith, D. H. (2017). Traumatic brain injury as a trigger of neurodegeneration. Adv. Neurobiol. 15, 383–400. doi: 10.1007/978-3-319-57193-5_15
Johnson, V. E., Stewart, W., and Smith, D. H. (2012). Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149. doi: 10.1111/j.1750-3639.2011.00513.x
Karve, I. P., Taylor, J. M., and Crack, P. J. (2016). The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 173, 692–702. doi: 10.1111/bph.13125
Khatri, N., Thakur, M., Pareek, V., Kumar, S., Sharma, S., and Datusalia, A. K. (2018). Oxidative stress: major threat in traumatic brain injury. CNS Neurol. Disord. Drug Targets 17, 689–695. doi: 10.2174/1871527317666180627120501
Khoury, N., Koronowski, K. B., Young, J. I., and Perez-Pinzon, M. A. (2018). The NAD(+)-dependent family of sirtuins in cerebral ischemia and preconditioning. Antioxid. Redox Signal. 28, 691–710. doi: 10.1089/ars.2017.7258
Kim, S. H., Lu, H. F., and Alano, C. C. (2011). Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PLoS One 6:e14731. doi: 10.1371/journal.pone.0014731
Klimova, N., and Kristian, T. (2019). Multi-targeted effect of nicotinamide mononucleotide on brain bioenergetic metabolism. Neurochem. Res. 44, 2280–2287. doi: 10.1007/s11064-019-02729-0
Klimova, N., Long, A., and Kristian, T. (2019). Nicotinamide mononucleotide alters mitochondrial dynamics by SIRT3-dependent mechanism in male mice. J. Neurosci. Res. 97, 975–990. doi: 10.1002/jnr.24397
Klose, M., and Feldt-Rasmussen, U. (2018). Chronic endocrine consequences of traumatic brain injury - what is the evidence? Nat. Rev. Endocrinol. 14, 57–62. doi: 10.1038/nrendo.2017.103
Kniesel, U., and Wolburg, H. (2000). Tight junctions of the blood-brain barrier. Cell. Mol. Neurobiol. 20, 57–76. doi: 10.1023/a:1006995910836
Kou, D. Q., Jiang, Y. L., Qin, J. H., and Huang, Y. H. (2017). Magnolol attenuates the inflammation and apoptosis through the activation of SIRT1 in experimental stroke rats. Pharmacol. Rep. 69, 642–647. doi: 10.1016/j.pharep.2016.12.012
Li, D., Liu, N., Zhao, H. H., Zhang, X., Kawano, H., Liu, L., et al. (2017). Interactions between Sirt1 and MAPKs regulate astrocyte activation induced by brain injury in vitro and in vivo. J. Neuroinflammation 14:67. doi: 10.1186/s12974-017-0841-6
Li, M. Z., Zheng, L. J., Shen, J., Li, X. Y., Zhang, Q., Bai, X., et al. (2018). SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes. Neural Regen. Res. 13, 2005–2013. doi: 10.4103/1673-5374.239449
Li, Q., Peng, Y., Fan, L., Xu, H., He, P., Cao, S., et al. (2018). Phosphodiesterase-4 inhibition confers a neuroprotective efficacy against early brain injury following experimental subarachnoid hemorrhage in rats by attenuating neuronal apoptosis through the SIRT1/Akt pathway. Biomed. Pharmacother. 99, 947–955. doi: 10.1016/j.biopha.2018.01.093
Li, X., and Kazgan, N. (2011). Mammalian sirtuins and energy metabolism. Int. J. Biol. Sci. 7, 575–587. doi: 10.7150/ijbs.7.575
Li, X. H., Chen, C., Tu, Y., Sun, H. T., Zhao, M. L., Cheng, S. X., et al. (2013). Sirt1 promotes axonogenesis by deacetylation of Akt and inactivation of GSK3. Mol. Neurobiol. 48, 490–499. doi: 10.1007/s12035-013-8437-3
Li, Z., and Han, X. (2018). Resveratrol alleviates early brain injury following subarachnoid hemorrhage: possible involvement of the AMPK/SIRT1/autophagy signaling pathway. Biol. Chem. 399, 1339–1350. doi: 10.1515/hsz-2018-0269
Liberale, L., Gaul, D. S., Akhmedov, A., Bonetti, N. R., Nageswaran, V., Costantino, S., et al. (2019). Endothelial SIRT6 blunts stroke size and neurological deficit by preserving blood-brain barrier integrity: a translational study. Eur. Heart J. 41, 1575–1587. doi: 10.1093/eurheartj/ehz712
Lin, H., Su, X., and He, B. (2012). Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem. Biol. 7, 947–960. doi: 10.1021/cb3001793
Loane, D. J., Kumar, A., Stoica, B. A., Cabatbat, R., and Faden, A. I. (2014). Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J. Neuropathol. Exp. Neurol. 73, 14–29. doi: 10.1097/NEN.0000000000000021
Lv, H., Wang, L., Shen, J., Hao, S., Ming, A., Wang, X., et al. (2015). Salvianolic acid B attenuates apoptosis and inflammation via SIRT1 activation in experimental stroke rats. Brain Res. Bull. 115, 30–36. doi: 10.1016/j.brainresbull.2015.05.002
Lv, J., Tian, J., Zheng, G., and Zhao, J. (2017). Sirtuin7 is involved in protecting neurons against oxygen-glucose deprivation and reoxygenation-induced injury through regulation of the p53 signaling pathway. J. Biochem. Mol. Toxicol. 31:e21955. doi: 10.1002/jbt.21955
Marin, C., Langdon, C., Alobid, I., Fuentes, M., Bonastre, M., and Mullol, J. (2019). Recovery of olfactory function after excitotoxic lesion of the olfactory bulbs is associated with increases in bulbar SIRT1 and SIRT4 expressions. Mol. Neurobiol. 56, 5643–5653. doi: 10.1007/s12035-019-1472-y
Maxwell, W. L., Dhillon, K., Harper, L., Espin, J., MacIntosh, T. K., Smith, D. H., et al. (2003). There is differential loss of pyramidal cells from the human hippocampus with survival after blunt head injury. J. Neuropathol. Exp. Neurol. 62, 272–279. doi: 10.1093/jnen/62.3.272
Maxwell, W. L., MacKinnon, M. A., Smith, D. H., McIntosh, T. K., and Graham, D. I. (2006). Thalamic nuclei after human blunt head injury. J. Neuropathol. Exp. Neurol. 65, 478–488. doi: 10.1097/01.jnen.0000229241.28619.75
Mendez, M. F. (2017). What is the relationship of traumatic brain injury to dementia? J. Alzheimers Dis. 57, 667–681. doi: 10.3233/jad-161002
Miao, Y., Zhao, S., Gao, Y., Wang, R., Wu, Q., Wu, H., et al. (2016). Curcumin pretreatment attenuates inflammation and mitochondrial dysfunction in experimental stroke: the possible role of Sirt1 signaling. Brain Res. Bull. 121, 9–15. doi: 10.1016/j.brainresbull.2015.11.019
Michan, S., and Sinclair, D. (2007). Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13. doi: 10.1042/BJ20070140
Min, S. W., Chen, X., Tracy, T. E., Li, Y., Zhou, Y., Wang, C., et al. (2015). Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 21, 1154–1162. doi: 10.1038/nm.3951
Novgorodov, S. A., Riley, C. L., Keffler, J. A., Yu, J., Kindy, M. S., Macklin, W. B., et al. (2016). SIRT3 deacetylates ceramide synthases: IMPLICATIONS FOR MITOCHONDRIAL DYSFUNCTION AND BRAIN INJURY. J. Biol. Chem. 291, 1957–1973. doi: 10.1074/jbc.M115.668228
Pais, T. F., Szegö, É. M., Marques, O., Miller-Fleming, L., Antas, P., Guerreiro, P., et al. (2013). The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J. 32, 2603–2616. doi: 10.1038/emboj.2013.200
Park, J. H., Long, A., Owens, K., and Kristian, T. (2016). Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol. Dis. 95, 102–110. doi: 10.1016/j.nbd.2016.07.018
Peng, Y., Jin, J., Fan, L., Xu, H., He, P., Li, J., et al. (2018). Rolipram attenuates early brain injury following experimental subarachnoid hemorrhage in rats: possibly via regulating the SIRT1/NF-kappaB pathway. Neurochem. Res. 43, 785–795. doi: 10.1007/s11064-018-2480-4
Perry, D. C., Sturm, V. E., Peterson, M. J., Pieper, C. F., Bullock, T., Boeve, B. F., et al. (2016). Association of traumatic brain injury with subsequent neurological and psychiatric disease: a meta-analysis. J. Neurosurg. 124, 511–526. doi: 10.3171/2015.2.Jns14503
Qian, C., Jin, J., Chen, J., Li, J., Yu, X., Mo, H., et al. (2017). SIRT1 activation by resveratrol reduces brain edema and neuronal apoptosis in an experimental rat subarachnoid hemorrhage model. Mol. Med. Rep. 16, 9627–9635. doi: 10.3892/mmr.2017.7773
Quaglio, G., Gallucci, M., Brand, H., Dawood, A., and Cobello, F. (2017). Traumatic brain injury: a priority for public health policy. Lancet Neurol. 16, 951–952. doi: 10.1016/s1474-4422(17)30370-8
Ramesh, S., Govindarajulu, M., Lynd, T., Briggs, G., Adamek, D., Jones, E., et al. (2018). SIRT3 activator Honokiol attenuates β-Amyloid by modulating amyloidogenic pathway. PLoS One 13:e0190350. doi: 10.1371/journal.pone.0190350
Ran, M., Li, Z., Yang, L., Tong, L., Zhang, L., and Dong, H. (2015). Calorie restriction attenuates cerebral ischemic injury via increasing SIRT1 synthesis in the rat. Brain Res. 1610, 61–68. doi: 10.1016/j.brainres.2015.03.043
Rangarajan, P., Karthikeyan, A., Lu, J., Ling, E. A., and Dheen, S. T. (2015). Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia. Neuroscience 311, 398–414. doi: 10.1016/j.neuroscience.2015.10.048
Roux, A., Muller, L., Jackson, S. N., Post, J., Baldwin, K., Hoffer, B., et al. (2016). Mass spectrometry imaging of rat brain lipid profile changes over time following traumatic brain injury. J. Neurosci. Methods 272, 19–32. doi: 10.1016/j.jneumeth.2016.02.004
Russo, M. V., and McGavern, D. B. (2016). Inflammatory neuroprotection following traumatic brain injury. Science 353, 783–785. doi: 10.1126/science.aaf6260
Satoh, A., Imai, S. I., and Guarente, L. (2017). The brain, sirtuins, and ageing. Nat. Rev. Neurosci. 18, 362–374. doi: 10.1038/nrn.2017.42
Satoh, K., and Shimokawa, H. (2019). Sirtuin 5 promotes ischemia/reperfusion-induced blood-brain barrier damage after stroke. Int. J. Cardiol. 284, 77–78. doi: 10.1016/j.ijcard.2018.11.019
Savran, M., Aslankoc, R., Ozmen, O., Erzurumlu, Y., Savas, H. B., Temel, E. N., et al. (2020). Agomelatine could prevent brain and cerebellum injury against LPS-induced neuroinflammation in rats. Cytokine 127:154957. doi: 10.1016/j.cyto.2019.154957
Scarpulla, R. C. (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278. doi: 10.1016/j.bbamcr.2010.09.019
Shi, H., Deng, H.-X., Gius, D., Schumacker, P. T., Surmeier, D. J., and Ma, Y.-C. (2017). Sirt3 protects dopaminergic neurons from mitochondrial oxidative stress. Hum. Mol. Genet. 26, 1915–1926. doi: 10.1093/hmg/ddx100
Shi, N., Zhu, C., and Li, L. (2016). Rehabilitation training and resveratrol improve the recovery of neurological and motor function in rats after cerebral ischemic injury through the Sirt1 signaling pathway. Biomed Res. Int. 2016:1732163. doi: 10.1155/2016/1732163
Shih, J., Liu, L., Mason, A., Higashimori, H., and Donmez, G. (2014). Loss of SIRT4 decreases GLT-1-dependent glutamate uptake and increases sensitivity to kainic acid. J. Neurochem. 131, 573–581. doi: 10.1111/jnc.12942
Shin, J. A., Lee, K.-E., Kim, H.-S., and Park, E.-M. (2012). Acute resveratrol treatment modulates multiple signaling pathways in the ischemic brain. Neurochem. Res. 37, 2686–2696. doi: 10.1007/s11064-012-0858-2
Simon, D. W., McGeachy, M. J., Bayir, H., Clark, R. S., Loane, D. J., and Kochanek, P. M. (2017). The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 13, 171–191. doi: 10.1038/nrneurol.2017.13
Singh, P., Hanson, P. S., and Morris, C. M. (2017). SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson’s disease. BMC Neurosci. 18:46. doi: 10.1186/s12868-017-0364-1
Smith, D. H., Chen, X.-H., Iwata, A., and Graham, D. I. (2003). Amyloid beta accumulation in axons after traumatic brain injury in humans. J. Neurosurg. 98, 1072–1077. doi: 10.3171/jns.2003.98.5.1072
Stamatovic, S. M., Martinez-Revollar, G., Hu, A., Choi, J., Keep, R. F., and Andjelkovic, A. V. (2019). Decline in Sirtuin-1 expression and activity plays a critical role in blood-brain barrier permeability in aging. Neurobiol. Dis. 126, 105–116. doi: 10.1016/j.nbd.2018.09.006
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. doi: 10.1038/nrneurol.2017.188
Sykes, S. M., Mellert, H. S., Holbert, M. A., Li, K., Marmorstein, R., Lane, W. S., et al. (2006). Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 24, 841–851. doi: 10.1016/j.molcel.2006.11.026
Tang, F., Guo, S., Liao, H., Yu, P., Wang, L., Song, X., et al. (2017). Resveratrol enhances neurite outgrowth and synaptogenesis via sonic hedgehog signaling following oxygen-glucose deprivation/reoxygenation injury. Cell. Physiol. Biochem. 43, 852–869. doi: 10.1159/000481611
Toklu, H. Z., and Tümer, N. (2015). “Oxidative stress, brain edema, blood–brain barrier permeability, and autonomic dysfunction from traumatic brain injury,” in Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects, ed. F. H. Kobeissy (Boca Raton, FL: CRC Press).
Tu, L.-F., Cao, L.-F., Zhang, Y.-H., Guo, Y.-L., Zhou, Y.-F., Lu, W.-Q., et al. (2019). Sirt3-dependent deacetylation of COX-1 counteracts oxidative stress-induced cell apoptosis. FASEB J. 33, 14118–14128. doi: 10.1096/fj.201900708R
Verma, R., Ritzel, R. M., Crapser, J., Friedler, B. D., and McCullough, L. D. (2019). Evaluation of the neuroprotective effect of Sirt3 in experimental stroke. Transl. Stroke Res. 10, 57–66. doi: 10.1007/s12975-017-0603-x
Wan, D., Zhou, Y., Wang, K., Hou, Y., Hou, R., and Ye, X. (2016). Resveratrol provides neuroprotection by inhibiting phosphodiesterases and regulating the cAMP/AMPK/SIRT1 pathway after stroke in rats. Brain Res. Bull. 121, 255–262. doi: 10.1016/j.brainresbull.2016.02.011
Wang, P., Guan, Y.-F., Du, H., Zhai, Q.-W., Su, D.-F., and Miao, C.-Y. (2012). Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 8, 77–87. doi: 10.4161/auto.8.1.18274
Wang, P., Xu, T. Y., Guan, Y. F., Tian, W. W., Viollet, B., Rui, Y. C., et al. (2011). Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann. Neurol. 69, 360–374. doi: 10.1002/ana.22236
Wang, Q., Li, L., Li, C. Y., Pei, Z., Zhou, M., and Li, N. (2015). SIRT3 protects cells from hypoxia via PGC-1α- and MnSOD-dependent pathways. Neuroscience 286, 109–121. doi: 10.1016/j.neuroscience.2014.11.045
Wang, T., Zhu, M., and He, Z.-Z. (2016). Low-molecular-weight fucoidan attenuates mitochondrial dysfunction and improves neurological outcome after traumatic brain injury in aged mice: involvement of Sirt3. Cell. Mol. Neurobiol. 36, 1257–1268. doi: 10.1007/s10571-015-0323-2
Wang, T.-H., Xiong, L.-L., Yang, S.-F., You, C., Xia, Q.-J., Xu, Y., et al. (2017). LPS pretreatment provides neuroprotective roles in rats with subarachnoid hemorrhage by downregulating MMP9 and Caspase3 associated with TLR4 signaling activation. Mol. Neurobiol. 54, 7746–7760. doi: 10.1007/s12035-016-0259-7
Wu, A., Ying, Z., and Gomez-Pinilla, F. (2011). The salutary effects of DHA dietary supplementation on cognition, neuroplasticity, and membrane homeostasis after brain trauma. J. Neurotrauma 28, 2113–2122. doi: 10.1089/neu.2011.1872
Xu, L., He, D., and Bai, Y. (2016). Microglia-mediated inflammation and neurodegenerative disease. Mol. Neurobiol. 53, 6709–6715. doi: 10.1007/s12035-015-9593-4
Xu, L., Jia, L., Wang, Q., Hou, J., Li, S., and Teng, J. (2018). Salidroside attenuates hypoxia/reoxygenation-induced human brain vascular smooth muscle cell injury by activating the SIRT1/FOXO3alpha pathway. Exp. Ther. Med. 15, 822–830. doi: 10.3892/etm.2017.5446
Yan, J., Chen, C., Hu, Q., Yang, X., Lei, J., Yang, L., et al. (2008). The role of p53 in brain edema after 24 h of experimental subarachnoid hemorrhage in a rat model. Exp. Neurol. 214, 37–46. doi: 10.1016/j.expneurol.2008.07.006
Yang, F., Zhou, L., Wang, D., Wang, Z., and Huang, Q. Y. (2015). Minocycline ameliorates hypoxia-induced blood-brain barrier damage by inhibition of HIF-1α through SIRT-3/PHD-2 degradation pathway. Neuroscience 304, 250–259. doi: 10.1016/j.neuroscience.2015.07.051
Yang, H., Gu, Z.-T., Li, L., Maegele, M., Zhou, B.-Y., Li, F., et al. (2017). SIRT1 plays a neuroprotective role in traumatic brain injury in rats via inhibiting the p38 MAPK pathway. Acta Pharmacol. Sin. 38, 168–181. doi: 10.1038/aps.2016.130
Yang, S., Chen, X., Li, S., Sun, B., and Hang, C. (2018). Melatonin treatment regulates SIRT3 expression in early brain injury (EBI) due to reactive oxygen species (ROS) in a mouse model of subarachnoid hemorrhage (SAH). Med. Sci. Monit. 24, 3804–3814. doi: 10.12659/msm.907734
Yin, J., Han, P., Song, M., Nielsen, M., Beach, T. G., Serrano, G. E., et al. (2018). Amyloid-β increases tau by mediating Sirtuin 3 in Alzheimer’s disease. Mol. Neurobiol. 55, 8592–8601. doi: 10.1007/s12035-018-0977-0
Yu, J., Novgorodov, S. A., Chudakova, D., Zhu, H., Bielawska, A., Bielawski, J., et al. (2007). JNK3 signaling pathway activates ceramide synthase leading to mitochondrial dysfunction. J. Biol. Chem. 282, 25940–25949. doi: 10.1074/jbc.M701812200
Yuan, F., Xu, Z.-M., Lu, L.-Y., Nie, H., Ding, J., Ying, W.-H., et al. (2016). SIRT2 inhibition exacerbates neuroinflammation and blood-brain barrier disruption in experimental traumatic brain injury by enhancing NF-κB p65 acetylation and activation. J. Neurochem. 136, 581–593. doi: 10.1111/jnc.13423
Zakhary, S. M., Ayubcha, D., Dileo, J. N., Jose, R., Leheste, J. R., Horowitz, J. M., et al. (2010). Distribution analysis of deacetylase SIRT1 in rodent and human nervous systems. Anat. Rec. 293, 1024–1032. doi: 10.1002/ar.21116
Zhang, S., Jiang, L., Che, F., Lu, Y., Xie, Z., and Wang, H. (2017). Arctigenin attenuates ischemic stroke via SIRT1-dependent inhibition of NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 493, 821–826. doi: 10.1016/j.bbrc.2017.08.062
Zhang, W., Wei, R., Zhang, L., Tan, Y., and Qian, C. (2017). Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 366, 95–104. doi: 10.1016/j.neuroscience.2017.09.035
Zhang, X., Lu, Y., Wu, Q., Dai, H., Li, W., Lv, S., et al. (2019). Astaxanthin mitigates subarachnoid hemorrhage injury primarily by increasing sirtuin 1 and inhibiting the Toll-like receptor 4 signaling pathway. FASEB J. 33, 722–737. doi: 10.1096/fj.201800642RR
Zhao, H., Luo, Y., Chen, L., Zhang, Z., Shen, C., Li, Y., et al. (2018). Sirt3 inhibits cerebral ischemia-reperfusion injury through normalizing Wnt/beta-catenin pathway and blocking mitochondrial fission. Cell Stress Chaperones 23, 1079–1092. doi: 10.1007/s12192-018-0917-y
Zhao, L., Liu, H., Yue, L., Zhang, J., Li, X., Wang, B., et al. (2017). Melatonin attenuates early brain injury via the melatonin receptor/Sirt1/NF-κB signaling pathway following subarachnoid hemorrhage in mice. Mol. Neurobiol. 54, 1612–1621. doi: 10.1007/s12035-016-9776-7
Zhao, Y., Luo, P., Guo, Q., Li, S., Zhang, L., Zhao, M., et al. (2012). Interactions between SIRT1 and MAPK/ERK regulate neuronal apoptosis induced by traumatic brain injury in vitro and in vivo. Exp. Neurol. 237, 489–498. doi: 10.1016/j.expneurol.2012.07.004
Zhou, S., Qiao, B., Chu, X., and Kong, Q. (2018). Oxymatrine attenuates cognitive deficits through SIRT1-mediated autophagy in ischemic stroke. J. Neuroimmunol. 323, 136–142. doi: 10.1016/j.jneuroim.2018.06.018
Zhou, X. M., Zhang, X., Zhang, X. S., Zhuang, Z., Li, W., Sun, Q., et al. (2014). SIRT1 inhibition by sirtinol aggravates brain edema after experimental subarachnoid hemorrhage. J. Neurosci. Res. 92, 714–722. doi: 10.1002/jnr.23359
Zhou, Y., Chen, Q., Wang, Y., Wu, H., Xu, W., Pan, Y., et al. (2020a). Persistent neurovascular unit dysfunction: pathophysiological substrate and trigger for late-onset neurodegeneration after traumatic brain injury. Front. Neurosci. 14:581. doi: 10.3389/fnins.2020.00581
Zhou, Y., Shao, A., Yao, Y., Tu, S., Deng, Y., and Zhang, J. (2020b). Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Cell Commun. Signal. 18:62. doi: 10.1186/s12964-020-00549-2
Zhou, Y., Xu, Y., and Shao, A. (2020c). Letter by Zhou et al regarding article, “mitochondrial deacetylase Sirt3 reduces vascular dysfunction and hypertension while Sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress”. Circ. Res. 126, e31–e32. doi: 10.1161/CIRCRESAHA.120.316755
Zhu, H. R., Wang, Z. Y., Zhu, X. L., Wu, X. X., Li, E. G., and Xu, Y. (2010). Icariin protects against brain injury by enhancing SIRT1-dependent PGC-1alpha expression in experimental stroke. Neuropharmacology 59, 70–76. doi: 10.1016/j.neuropharm.2010.03.017
Zimmermann, L. L., Martin, R. M., and Girgis, F. (2017). Treatment options for posttraumatic epilepsy. Curr. Opin. Neurol. 30, 580–586. doi: 10.1097/WCO.0000000000000505
Zou, P., Liu, X., Li, G., and Wang, Y. (2018). Resveratrol pretreatment attenuates traumatic brain injury in rats by suppressing NLRP3 inflammasome activation via SIRT1. Mol. Med. Rep. 17, 3212–3217. doi: 10.3892/mmr.2017.8241
Keywords: sirtuin, traumatic brain injury, neurodegenerative disease, neuroinflammation, excitotoxicity, oxidative stress
Citation: Yang Q, Zhou Y, Sun Y, Luo Y, Shen Y and Shao A (2020) Will Sirtuins Be Promising Therapeutic Targets for TBI and Associated Neurodegenerative Diseases? Front. Neurosci. 14:791. doi: 10.3389/fnins.2020.00791
Received: 21 April 2020; Accepted: 06 July 2020;
Published: 31 July 2020.
Edited by:
Andreas Martin Grabrucker, University of Limerick, IrelandReviewed by:
Pedro Gomes, University of Coimbra, PortugalPengcheng Han, Medical University of South Carolina, United States
Copyright © 2020 Yang, Zhou, Sun, Luo, Shen and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ye Shen, aWRyc2hlbkB6anUuZWR1LmNu; Anwen Shao, MjExMTgxMTZAemp1LmVkdS5jbg==; YW53ZW5zaGFvQHNpbmEuY29t
†These authors have contributed equally to this work