 Pilar Rojas1,2†
Pilar Rojas1,2† Ana I. Ramírez1,3,4†
Ana I. Ramírez1,3,4† José A. Fernández-Albarral1
José A. Fernández-Albarral1 Inés López-Cuenca1
Inés López-Cuenca1 Elena Salobrar-García1,3,4
Elena Salobrar-García1,3,4 Manuel Cadena2Lorena Elvira-Hurtado1
Manuel Cadena2Lorena Elvira-Hurtado1 Juan J. Salazar1,3,4
Juan J. Salazar1,3,4 Rosa de Hoz1,3,4*
Rosa de Hoz1,3,4* José M. Ramírez1,3,5*
José M. Ramírez1,3,5*- 1Instituto de Investigaciones Oftalmológicas Ramón Castroviejo, Universidad Complutense de Madrid, Madrid, Spain
- 2Hospital General Universitario Gregorio Marañón, Instituto Oftálmico de Madrid, Madrid, Spain
- 3OFTARED, ISCIII, Madrid, Spain
- 4Departamento de Inmunología Oftalmología y ORL, Facultad de Óptica y Optometría, Universidad Complutense de Madrid, Madrid, Spain
- 5Departamento de Inmunología Oftalmología y ORL, Facultad de Medicina, Universidad Complutense de Madrid, Madrid, Spain
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that causes degeneration of the lower and upper motor neurons and is the most prevalent motor neuron disease. This disease is characterized by muscle weakness, stiffness, and hyperreflexia. Patients survive for a short period from the onset of the disease. Most cases are sporadic, with only 10% of the cases being genetic. Many genes are now known to be involved in familial ALS cases, including some of the sporadic cases. It has also been observed that, in addition to genetic factors, there are numerous molecular mechanisms involved in these pathologies, such as excitotoxicity, mitochondrial disorders, alterations in axonal transport, oxidative stress, accumulation of misfolded proteins, and neuroinflammation. This pathology affects the motor neurons, the spinal cord, the cerebellum, and the brain, but recently, it has been shown that it also affects the visual system. This impact occurs not only at the level of the oculomotor system but also at the retinal level, which is why the retina is being proposed as a possible biomarker of this pathology. The current review discusses the main aspects mentioned above related to ALS, such as the main genes involved, the most important molecular mechanisms that affect this pathology, its ocular involvement, and the possible usefulness of the retina as a biomarker.
Introduction: Overview of Amyotrophic Lateral Sclerosis Disease
Motor neuron diseases (MNDs) have a high morbidity and mortality and cause a gradual deterioration of voluntary muscle function due to progressive neuronal damage (Rowland and Shneider, 2001; Turner et al., 2009; Yedavalli et al., 2018; Pinto et al., 2019). MND has a global incidence of one to three cases per 100,000, while its prevalence ranges from one to nine cases per 100,000. Within the group of these diseases, amyotrophic lateral sclerosis (ALS) is the most common, accounting for 80–90% of all MND cases (Román, 1996) and having an incidence per 100,000 people of 0.3–2.5 cases per year (Rowland and Shneider, 2001; Sathasivam, 2010; Kiernan et al., 2011; Pratt et al., 2012). Of all ALS cases, 10% are familial (FALS) (Byrne et al., 2011) ranging from 2% to 15% depending on the population (Conwit, 2006). Overall, both the incidence (Haberlandt, 1959) and the prevalence (Williams et al., 1988) of this pathology can be variable depending on the region and ethnicity.
In ALS, a combination of the symptoms of both upper motor neurons (UMNs) and lower motor neurons (LMNs) occurs (Rowland and Shneider, 2001; Kiernan et al., 2011; Yedavalli et al., 2018). This condition involves muscle weakness and stiffness, overactive reflexes, and sometimes changes in emotions (Carra et al., 2012; Zarei et al., 2015; Yedavalli et al., 2018). ALS involves the swallowing, speech, and respiratory muscles (Carra et al., 2012; Zarei et al., 2015; Yedavalli et al., 2018). The disease usually begins in the extremities (spinal onset), although 25% of patients have a bulbar onset, which has a worse prognosis (Kiernan et al., 2011). ALS is a disease that is asymmetrical with respect to the onset and spread of UMN and LMN dysfunction and constitutes a heterogeneous disease, because not all forms behave in the same way, which makes its classification complex (Zhang et al., 2014).
Although ALS has only been considered a motor disease, neuroimaging tests have recently shown the involvement of non-motor areas such as cerebral global atrophy, decrease in gray matter density, and regional white matter alterations (Ellis et al., 2001; Abrahams et al., 2005; Kassubek et al., 2005; Ringholz et al., 2005; Mezzapesa et al., 2007; Zhang et al., 2014). The alteration of these areas leads to cognitive and behavioral changes (Abrahams et al., 2005; Mezzapesa et al., 2007). Cognitive impairment, mainly featuring executive dysfunction and mild memory loss during the course of the disease, has been found in 50% of ALS patients (Ringholz et al., 2005; Meier et al., 2010).
Mechanisms and Risk Factors Involved in the Pathogenesis of Motor Neuron Diseases
There are different risk factors involved in ALS (Yu et al., 2014; Zarei et al., 2015), which include smoking (Weisskopf et al., 2009; Wang et al., 2014), agricultural chemicals (Ward et al., 2014; Seals et al., 2017), heavy metals (Simons, 1986; Mcguire et al., 1997; Savolainen et al., 1998; Kamel et al., 2002), and low-frequency electromagnetic waves (Zhou et al., 2012). In addition, athletes (Chiò et al., 2004; Turner et al., 2012; Zarei et al., 2015) and hypermetabolic phenotype (Desport et al., 2001; Bouteloup et al., 2009; Vaisman et al., 2009; Dupuis et al., 2011; Ferri and Coccurello, 2017; Steyn et al., 2018; Pape and Grose, 2020) have a higher risk for ALS. A low body mass index can induce disease progression and reduced survival time (Jawaid et al., 2010; Park et al., 2015). Other factors that may affect ALS patients are hyperlipidemia (Palamiuc et al., 2015) and glutamate-rich and fat-rich diets (Iwasaki et al., 2005; Veldink et al., 2007; Morozova et al., 2008; Fitzgerald et al., 2014). However, progesterone and estrogen provide protection against ALS (Gargiulo Monachelli et al., 2011; De Jong et al., 2013; Pape and Grose, 2020).
Several molecular mechanisms can cause neurodegeneration in ALS (De Vos et al., 2000; Magrané and Manfredi, 2009; Shi et al., 2010; Forsberg et al., 2011; Zhu and Sheng, 2011; Donnelly et al., 2013; Jaiswal, 2014; Mitsumoto et al., 2014; Wang et al., 2014), and others can be considered secondary in the development of ALS (Vucic and Kiernan, 2007).
Glutamate Excitotoxicity
Glutamate has a neurotoxic effect when it accumulates at the synapses (Gazulla and Cavero-Nagore, 2006). In patients with ALS (both spinal cord and motor cortex involvement), and in the superoxide dismutase 1 (SOD1) transgenic mouse model, a decrease in glutamate receptors (GluRs) was found in astrocytes. This decrease induces extracellular glutamate accumulation, which causes overstimulation of GluRs and neuronal death via excitotoxicity (Lin et al., 1998; Trotti et al., 1999; Barbeito et al., 2004; Pratt et al., 2012). In ALS patients, a decrease in glutamate transporters is mainly due to an alteration in messenger RNA (Lin et al., 1998; Honig et al., 2000).
Structural and Functional Abnormalities of the Mitochondria
Mitochondrial function disturbances, such as fragmentation and aggregation, are frequently found in ALS patients (Vielhaber et al., 2000; Chung and Suh, 2002; Krasnianski et al., 2005; Echaniz-Laguna et al., 2006; Crugnola et al., 2010; Cozzolino and Carrì, 2012). Increased crests, swelling, and fragmentation have been observed in the mitochondria of the spinal motor neurons and proximal axons of skeletal muscle in ALS-related tissues (Chung and Suh, 2002; Echaniz-Laguna et al., 2002; Boillée et al., 2006a). The increase in the misfolded SOD1 enzyme in the mitochondria of the spinal cord of mice is considered to be the main cause of mitochondrial dysfunction. In addition, aggregates of the enzyme SOD1 may also interact with the apoptosis regulator protein Bcl-2, inducing an apoptotic cascade and contributing to the deterioration of neurons and neuro-muscular degeneration (Boillée et al., 2006a).
Impaired Axonal Structure and Transport Defects
Axonal transport (retrograde and anterograde) is impaired in ALS patients and in mutant SOD1 mice, as evidenced by the accumulation of altered structures, such as mitochondria, neurofilaments, and autophagosomes, in the spinal motor neuron axons (Ikenaka et al., 2012; Zarei et al., 2015). Mutations in the dynein genes have been seen in models of ALS mice. Dyneins are responsible for the transport of mitochondria and autophagosomes, causing both to accumulate in the axon (Ikenaka et al., 2012). Autophagosomes are necessary for the elimination of altered mitochondria and dilated endoplasmic reticules, which accumulate in the axons of motor neurons and cause them to malfunction (Zarei et al., 2015). All of the above suggest that an alteration in axonal transport could be fundamental for the development of ALS.
Free Radical-Mediated Oxidative Stress
Increased free radical and oxidative damage has been found in biopsies from ALS patients, as well as in cerebrospinal fluid, serum, and urine samples (Simpson et al., 2004; Shin Hee and Lee Keun, 2013). This oxidative damage also affects RNA, which has been shown in both human central nervous system (CNS) biopsies and in mouse ALS models of SOD1 (Chang et al., 2008). The enzyme SOD1 is an important anti-oxidant. The alterations in the redox reactions are one of the first theories of how mutations in SOD1 can cause cytotoxicity (Carrì et al., 2015). In addition, it has been observed in the motor cortex, using positron emission tomography (PET), that increased oxidative stress is related to the severity of the disease in ALS patients (Tsujikawa et al., 2015).
Protein Aggregates
It has been shown that in ALS, abnormal protein accumulations are produced that aid in the pathogenesis of the disease (Blokhuis et al., 2013). The ubiquitin–proteasome (UP) system, which repairs and removes proteins, plays an important role in ALS, with ubiquitin-reactive inclusions being characteristic of this pathology (Han-Xiang et al., 2011; Pratt et al., 2012). Among these, the inclusions of proteins TDP-43 and p62 are indicative of this pathology (Al-Sarraj et al., 2011; Williams et al., 2016). Some ALS patients present numerous inclusions positive for p62 but negative for TDP-43 in the hippocampus and cerebellum (Al-Sarraj et al., 2011). These p62-positive and TDP-43-negative inclusions have also been observed in other CNS areas including the retina (Brettchneider et al., 2013; Fawzi et al., 2014).
Neuroinflammation
Neuroinflammation occurs in many neurodegenerative diseases, such as Parkinson’s, Alzheimer’s, and ALS, resulting in the activation of astroglial and microglial cells (Ramírez et al., 2017). Specifically, in ALS, it has been observed that reactive glia (microglia and astrocytes) can influence the damage and subsequent death of motor neurons (Vargas and Johnson, 2010; Philips and Robberecht, 2011; Liao et al., 2012). In the ALS, the presence of mutant proteins (SOD1 and TDP-43), oxidative stress, mitochondrial damage, etc., produces continuous damage, which can trigger a chronic activation of glial cells and, therefore, a sustained inflammatory process that could exacerbate neuronal damage (Philips and Robberecht, 2011). It has been observed that the mutant SOD1 protein can have a toxic function on motor neurons only when the activated microglia is present through a mechanism involving a toll receptor (CD14-TLR) that causes an increase in free radicals (Zhao et al., 2010). Astrocyte activation (Vargas and Johnson, 2010), microglial activation (Alexianu et al., 2001), and lymphocyte appearance (Engelhardt et al., 1993) have been found in animal models of ALS (mutants of SOD1) (Alexianu et al., 2001; Boillée et al., 2006b) and in ALS patients (Kushner et al., 1991; Kawamata et al., 1992; Nagy et al., 1994; Schiffer et al., 1996; Turner et al., 2004). In both cases, the mutant SOD1 causes the microglia to increase the expression of pro-inflammatory cytokines, such as IL-1β and TNF-α (Weydt et al., 2004), and inflammatory mediators, such as cyclooxygenase 2 (COX-2) (Almer et al., 2001) and nitric oxide (NO). The NO released by the microglia could induce apoptosis in motor neurons through the activation of Fas via (Raoul et al., 2002).
Mutant protein TDP-43 can produce microglial activation through the surface receptor CD14, which stimulates the NF-κB and AP-1 pathways and the inflammasome. This can cause a neurotoxic cascade leading to motor neuron death (Zhao et al., 2015). In the ALS SOD1 model, the microglia isolated at the onset of the disease has an M2 or anti-inflammatory phenotype; however, the microglia isolated at the end of the disease has a neurotoxic M1 phenotype. This demonstrates a dual role of microglial cells during the disease process in this ALS model (Liao et al., 2012).
Astrogliosis has been observed in ALS models with mutations in C9orf72, FUS, SOD1, and TARDBP genes (Wong et al., 1995; Liu et al., 2016; Sharma et al., 2016). In the last model, the death of the motor neurons could be due, in part, to a loss of the function of the TDP-43 protein in the astrocytes (Yang et al., 2014). In ALS SOD1 models, astrocytes can release ATP, causing the activation of microglial cells via purinergic receptors (P2X7) (Gandelman et al., 2010). They can also release transforming growth factor-β1 (TGF-β1), which can induce microglial inactivation, thereby eliminating the possible beneficial effects of the microglia and, thus, accelerating the progression of the pathology (Endo et al., 2015). Moreover, the microglia in ALS can affect astrocytes by favoring the appearance of a neurotoxic subtype (Liddelow et al., 2017).
Considering all of the above, both the microglia and reactive astrocytes can affect neural function in ALS by playing an important role in the progression of the disease.
Genetics
Although most cases (90–95%) of ALS are sporadic (SALS) and not inherited, 10% of cases are of genetic origin (Byrne et al., 2011). Specific genetic locus mutations have been found to constitute the cases of FALS (Deivasigamani et al., 2014). The pattern of inheritance depends on the genes involved. Most cases are inherited in an autosomal dominant pattern (He et al., 2015). Men show more intense symptoms of the disease than women. There are many genes involved in the development of this disease. In FALS, mutations in the C9orf72 gene represent 30–40% of cases, those in the SOD1 gene make up 15–20%, those in the FUS and TARDBP genes each represent approximately 5% of cases, and the remaining genes that have been associated with FALS each represent a small proportion of cases (Pratt et al., 2012; Blokhuis et al., 2013). We next describe some of these genes.
C9orf72 Gene
This gene is located at the locus 9p21 of chromosome 9. It has been proposed that the C9orf72 mutation can decrease the C9orf72 protein, thus causing less endocytosis, which is necessary for autophagy (Mathis et al., 2019). Moreover, the massive accumulation of the expanded hexanucleotide GGGGCC could be neurotoxic and sequester proteins that bind to RNA, thereby causing the disruption of the machinery that processes RNA (Mathis et al., 2019). This mutation could also lead to increased vulnerability to excitotoxicity due to increased calcium permeability mediated by AMPA type receptors (Selvaraj et al., 2018). The form of inheritance is autosomal dominant, although some carriers do not develop the disease, so it has an incomplete penetrance (Renton et al., 2011). This variant represents the most frequent cause of SALS (7%) and FALS (30–40%) (Majounie et al., 2012). The manifestations at the onset of the disease are typical of ALS and have a bulbar onset. This variant is often associated with an earlier age of onset, a faster clinical course, and shorter survival (Schymick and Traynor, 2010).
FUS Gene
The FUS gene, located on the short arm of chromosome 16 (16p11.2), encodes a protein called sarcoma fusion protein (FUS) (Hübers et al., 2015; Scekic-Zahirovic et al., 2017). In ALS, mutations in the FUS gene have been observed, most of which can produce changes in amino acids in the region of the protein related to DNA binding and mRNA processing. These mutations may interfere with the importation of FUS into the cell nucleus, which will cause an accumulation of FUS in the cytoplasm. This has been observed particularly in the nerve cells that control muscle movement (Scekic-Zahirovic et al., 2017). This mutation occurs in 3–5% of FALS and in 1% of SALS cases (Deng et al., 2010; Mackenzie et al., 2010). In addition, patients with FUS mutations tend to develop the disease earlier and have a shorter life expectancy than those observed with mutations in other genes. Patients with ALS and FUS mutations may also develop frontotemporal dementia (FTD) (Kwiatkowski et al., 2009; Vance et al., 2009).
OPTN Gene
The optineurin gene (OPTN) is located on chromosome 10 and encodes a protein called optineurin, which is a multifunctional ubiquitin-binding phosphoprotein found in the cytoplasm. Changes of this protein can cause alterations in intracellular traffic and lead to inclusions in ALS. In addition, this protein is also involved in the signaling of the tumor necrosis factor α/NF-κB pathway (Zhu et al., 2007) and the mGluR (Ying et al., 2010). In 2010, Maruyama et al. (2010) and Schymick and Traynor (2010) found mutations in the OPTN gene in ALS patients. These mutations are present in 1.2% of patients with FALS (Del Bo et al., 2011). ALS patients with OPTN mutations presented typical spinal-onset disease (Weishaupt et al., 2013). Mutations in this gene were previously shown to be involved in primary open-angle glaucoma (Maruyama et al., 2010). This gene has also been linked to normotensional glaucoma in ALS patients (Weishaupt et al., 2013). In cells from patients with SALS and FALS, optineurin can be placed in inclusion bodies with TDP-43 (Maruyama et al., 2010), FUS (Ito et al., 2011), and SOD1 (Maruyama et al., 2010). Both retinal ganglion cells (RGCs) and motor neurons share common susceptibility factors (Weishaupt et al., 2013).
SOD1 Gene
The SOD1 gene is located on the long arm of chromosome 21 (21q22.11). This gene encodes a cytosolic enzyme called copper zinc (Cu/Zn) SOD1 (Gatchel and Zoghbi, 2005; Valentine et al., 2005) that plays a very important role in the elimination of superoxide radicals, thereby protecting against free radicals (Azadmanesh and Borgstahl, 2018). Around 2.5–23% of patients with FALS and 0.44–7% of those with SALS have mutations in SOD1 (Andersen, 2006; Van Es et al., 2010). Most of the inherited forms of SOD1 gene mutations are dominant (Hayward et al., 1998; Marucci et al., 2007). Mutations in the SOD1 enzyme can induce configurational changes in the SOD1 protein leading to motor neuron toxicity (Mathis et al., 2019). ALS patients who present mutations in SOD1 show an earlier time of onset and a longer duration of disease. In addition, they usually do not present cognitive disorders, and their motor symptoms usually begin in the lower extremities (Mathis et al., 2019).
TARDBP Gene
The Tar DNA binding protein (TARDBP) gene is located on the short arm of chromosome 1 (1p36.22) and encodes the TDP-43 protein. More than 40 mutations have been identified in the TARDBP gene and result in 6.5% of FALS cases. The form of inheritance is dominant (Kühnlein et al., 2008; Van Deerlin et al., 2008) and represents 0–5% of SALS cases (Kühnlein et al., 2008; Rutherford et al., 2008; Sreedharan et al., 2008). Most of the mutations cause changes in the amino acids in the TDP-43 protein and affect the region of the protein involved in the processing of RNA (Neumann et al., 2006; Da Cruz and Cleveland, 2011). Cytoplasmic inclusions of ubiquitin-reactive hyperphosphorylated TDP-43 proteins have been found in tissues of patients with FTD (Arai et al., 2006; Neumann et al., 2006). These inclusions have also been seen in the glial tissue and neurons of patients with SALS (Maekawa et al., 2009). These inclusions can often be placed with p62 and ubiquitin but are not found in FALS with FUS mutation (Vance et al., 2009) or SOD1 mutation (Mackenzie et al., 2007). Some patients with FALS and TARDBP mutation may also develop FTD (Arai et al., 2006; Neumann et al., 2006).
Materials and Methods
A literature search was performed up to May 2020 using the “MESH” terms in PubMed with the following keywords and word combinations: “Amyotrophic Lateral Sclerosis,” AND “Environmental Exposure,” “Nerve Degeneration,” “mitochondria,” “oxidative stress,” “Protein Aggregates,” “Microglia,” “Genetics,” “Oculomotor Muscles,” “Visual Pathways,” “Evoked Potentials, Visual,” “Contrast Sensitivity,” “Visual Fields,” “Visual Field Tests,” “Visual Acuity,” “Retina,” OR “Optical Coherence Tomography.” After filtering by author criteria (articles published in the last 10 years), English or Spanish language, and the condition that all address the relationship between ALS and visual pathway as the main subject, 196 articles were considered, and 304 articles did not satisfy the selection criteria (Figure 1).
Figure 1. Flow chart materials and methods.
Inclusion criteria: Articles were selected according to the following criteria: (i) articles focused on general features of ALS pathology, (ii) research that related the ALS with visual system alterations, (iii) retinal research developed in both ALS patients and animal experimental models of ALS, and (iv) articles based on human clinical trials with optical coherence tomography (OCT) analysis.
Exclusion criteria: Articles were excluded with the following characteristics: (i) research not carried out in mammalian experimental models of ALS and (ii) articles that did not have the sufficient outcomes associated with the objective of this review or did not meet the selection criteria of the authors.
Amyotrophic Lateral Sclerosis and the Eye
As discussed above, ALS affects not only motor neurons, the spinal cord, the cerebellum, and large areas of the brain but also the visual system, including the oculomotor and visual pathways. However, patients usually do not have visual complaints. For this reason, studies focusing on the visual pathway are not common. Most previous studies were related to the oculomotor function and the study of visual evoked potentials (VEPs) that analyze the visual pathway. However, some studies of ALS have subsequently emerged that are more related to visual function, using tests such as visual acuity, contrast sensitivity, and visual field (VF). In addition, the retina has now been described as a “window to the brain,” and the changes that the brain suffers in neurodegenerative diseases can also appear in the retinal tissue (MacCormick et al., 2015). Changes in retinal tissue can be detected using OCT, which is a diagnostic technique widely used in ophthalmology and has recently been used for the analysis of retinal and optic nerve changes in neurodegenerative diseases such as ALS disease. This technique can help in the diagnosis and follow-up of these pathologies (Salobrar-García et al., 2015a,b, 2016).
The following is a description of the main alterations found in this visual system using the aforementioned techniques and tests (Figure 2).
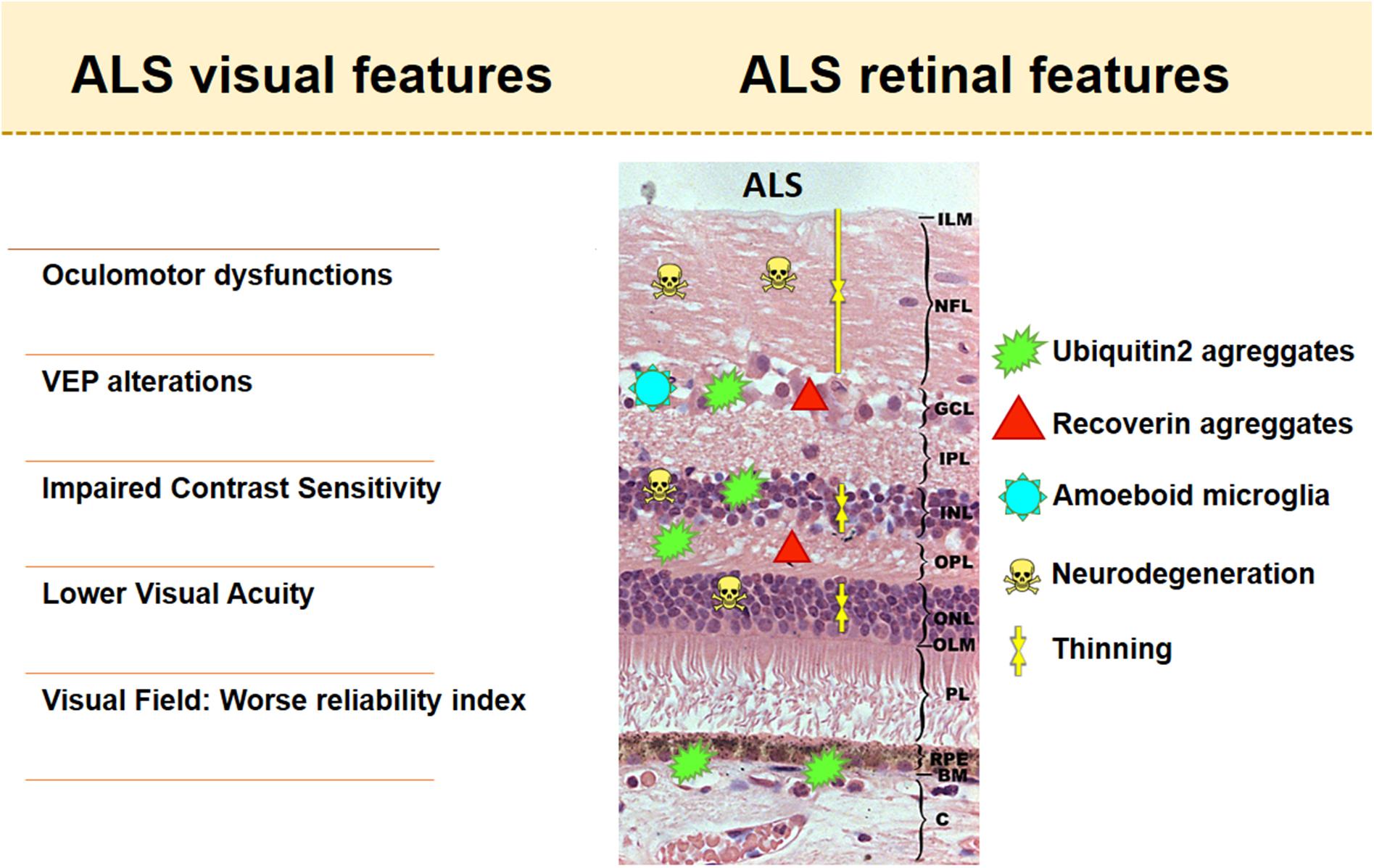
Figure 2. Visual and retinal features in amyotrophic lateral sclerosis (ALS). The retinal scheme represents the main histological findings found in retinas with ALS.
Oculomotor Function Alterations
Amyotrophic lateral sclerosis is a condition that affects motor neurons and large areas of the brain, so ocular movements may be affected. Thus, numerous studies on oculomotor function have appeared since the 1980s and continue to appear today (Table 1 and Figure 2). Although oculomotor function is generally retained in ALS patients, they may manifest various oculomotor dysfunctions both at a relative early stage and in advanced stages of the disease. These include (i) a worsening of saccadic and pursuit eye movements (Leveille et al., 1982; Cohen and Caroscio, 1983; Saito and Yamamoto, 1989; Gizzi et al., 1992; Marti-Fàbregas and Roig, 1993; Ohki et al., 1994; Abel et al., 1995; Shaunak et al., 1995; Okuda et al., 2009; Donaghy et al., 2010; Moss et al., 2012; Kang et al., 2018); (ii) a lack of suppression of the vestibulo-ocular reflex (Ohki et al., 1994); (iii) a significant increase in error rates (distraction) and latency in anti-saccadic movements (Shaunak et al., 1995; Donaghy et al., 2010) that may evoke saccadic paradigms, head shaking (Kang et al., 2018), and positional nystagmus of central origin (Saito and Yamamoto, 1989; Marti-Fàbregas and Roig, 1993; Ohki et al., 1994; Kang et al., 2018); (iv) gaze fixation instability (Saito and Yamamoto, 1989; Palmowski et al., 1995; Shaunak et al., 1995; Donaghy et al., 2009; Moss et al., 2012; Kang et al., 2018); (v) eyelid opening apraxia (Averbuch-Heller et al., 1998; Moss et al., 2012); and (vi) square wave jerks (Gizzi et al., 1992; Shaunak et al., 1995; Kang et al., 2018), which may reflect the incidence of secondary abnormalities, such as parkinsonism (Gizzi et al., 1992), and facilitate prefrontal dysfunction in these patients (Shaunak et al., 1995). In bulbar onset compared with spinal onset, saccadic dysmetria and abnormal cogwheeling smooth pursuits are increased, which suggests neurodegeneration in ALS involving more than motor neurons (dysfunction of vestibule cerebellar connections), especially in bulbar-onset disease (Kang et al., 2018). In a longitudinal study (Proudfoot et al., 2016), ALS patients, even those with normal saccadic function, were shown to have problems with executive and visual search tasks. These impairments were often more severe than expected in ALS patients. However, no significant progression was observed in the longitudinal study, nor were changes found in the connectivity of the R-FMRI network (Proudfoot et al., 2016). In the postmortem histopathological examination of ALS patients, cell loss was found in the substantia nigra and the rostral interstitial nucleus of the medial longitudinal fasciculus, suggesting that the early involvement of vertical saccades might correspond to a different clinical–pathological type (Averbuch-Heller et al., 1998). Therefore, as suggested by Sharma et al. (2011), changes in the oculomotor function of ALS patients could be promising biomarkers for the mechanical diagnosis, prognosis, and follow-up of ALS.
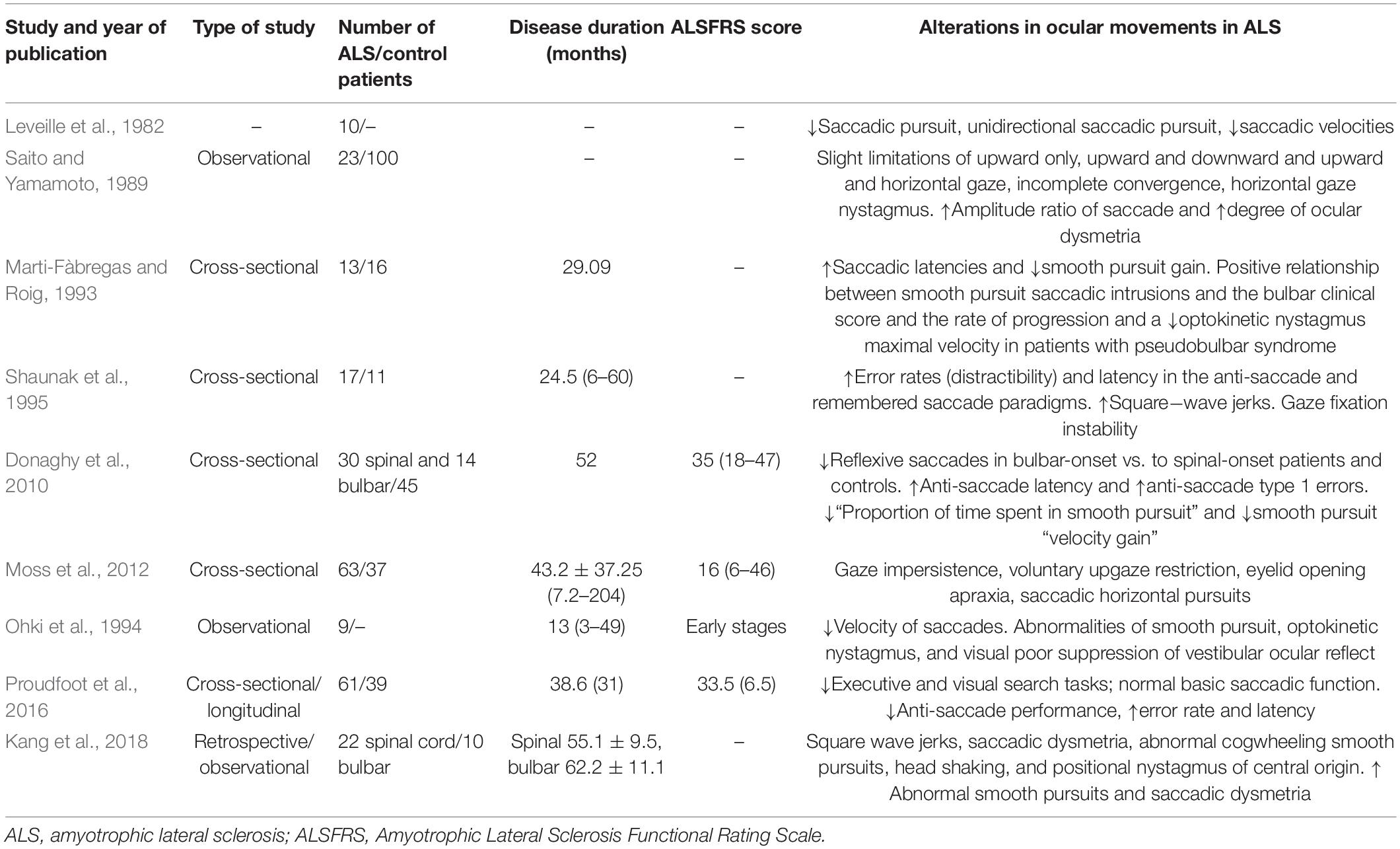
Table 1. Previous studies of extraocular movements in ALS patients.
The extraocular muscles (EOMs) and their motor neurons are retained in ALS. However, from the onset of this disease, the muscles of the limbs show axon retraction at the neuromuscular junctions. Wnt is a preserved family of secreted signaling molecules that are primarily involved in the formation of neuromuscular junctions. This signaling pathway was analyzed both in ALS patients and in a SOD1G93A mouse model for its possible implications in the preservation of normal morphology and the function in EOMs in this disease (McLoon et al., 2014). The authors found differential patterns of expression for Wnt1 and Wnt3a isoforms in the EOMs for limb muscles, especially at the neuromuscular junction level. This suggests that in ALS patients, this signaling pathway is preserved in the EOMs and dysregulated in the muscles of the limbs that subsequently develop a pathology (McLoon et al., 2014).
Visual Pathway Alterations
The alteration of the visual pathway can be analyzed using VEPs, contrast sensitivity, and VF. Next, we describe the different alterations found in the visual pathway of each.
Visual Evoked Potentials
Few studies have analyzed the VEPs in ALS patients (Table 2 and Figure 2). It was not until 1986 that alterations in VEPs were found in patients with ALS, with abnormal relative difference in latencies between each eye. In this study of 32 patients analyzed, only four had VEP abnormalities (increased latency time); however, these alterations were mild (Matheson et al., 1986). In other studies, while wave latency and amplitudes were within normal limits in all ALS patients (Ghezzi et al., 1989; Palma et al., 1993), somatosensory evoked potentials were abnormally delayed (N9–N13 and N13–N19 latencies), but no correlation was found among these abnormalities and the duration and severity of the disease (Ghezzi et al., 1989).
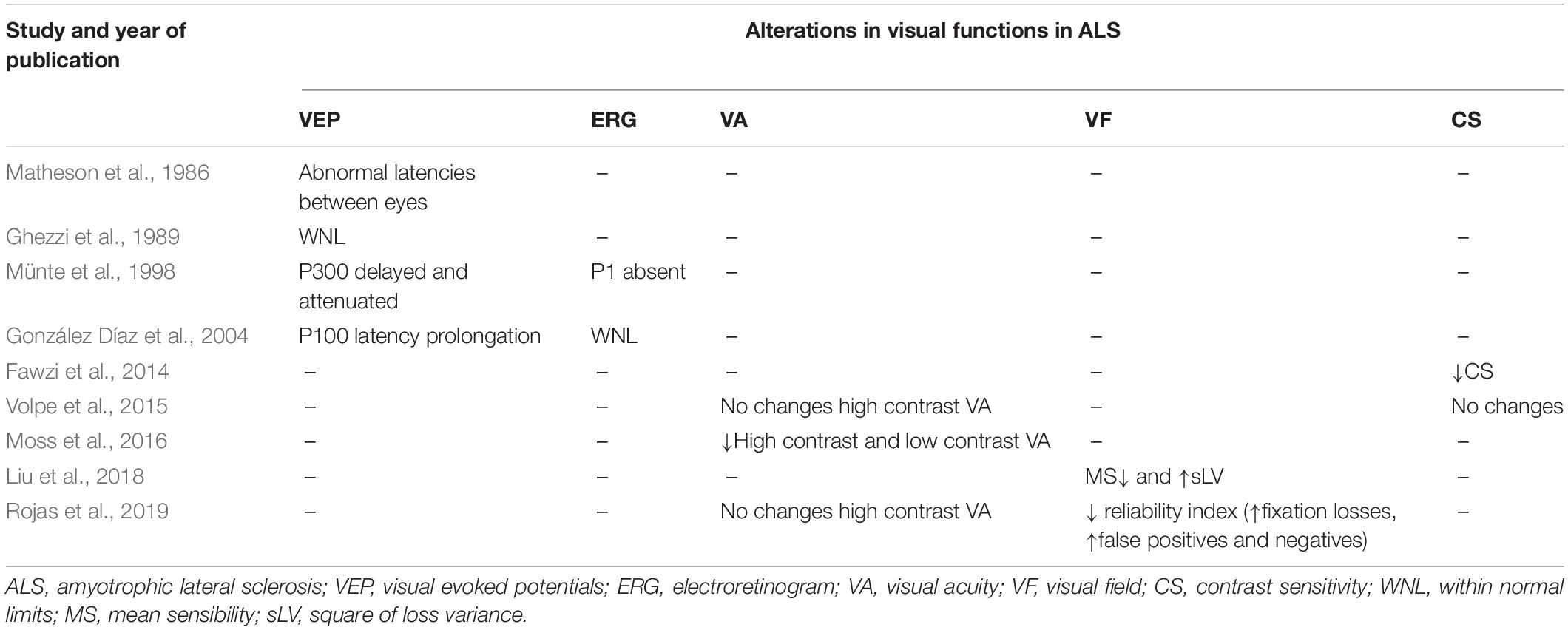
Table 2. Psychophysics visual test studies in ALS patients.
In addition, in ALS patients the P1 component was also found to be absent, and the P300 component was delayed and attenuated (Münte et al., 1998). In a study of six ALS patients, only one patient with a 9-month disease evolution had abnormal P100 bilateral extension VEPs and a significant interocular difference of P100. In addition, none of the patients presented alterations in their electroretinogram (González Díaz et al., 2004). Subsequently, electrophysiological studies of ALS patients showed new evidence of cortical participation involving visual areas, with alterations in the early sensory components of VEPs (Münte et al., 1998).
Contrast Sensitivity
While one study revealed impaired contrast sensitivity during an eye examination in two ALS patients with C9orf72 mutation (Fawzi et al., 2014), another study concluded that this function is not affected in ALS patients (Volpe et al., 2015; Table 2 and Figure 2).
Visual Acuity
Visual acuity in ALS patients is controversial. While a study found a lower visual acuity in ALS patients in both high-contrast and low-contrast (2.5% and 1.5%) visual acuity with Sloan charts (Moss et al., 2012), in others, the visual acuity exam revealed no differences in monocular high-contrast visual acuity (Volpe et al., 2015; Moss et al., 2016; Rojas et al., 2019) or low-contrast visual acuity (Moss et al., 2016).
Visual Field
Only two studies describe VF in early ALS spinal onset patients. With a decrease of mean sensitivity and an increase of the square of loss variance (Liu et al., 2018), ALS patients presented a worse reliability index (fixation losses, false positives, and negatives) due to motor difficulties. For this reason, the authors suggested that VF is not a suitable test to assess ALS patients (Rojas et al., 2019; Table 2 and Figure 2).
Retinal Abnormalities in Amyotrophic Lateral Sclerosis
Retinal tissue can be analyzed using histological techniques on postmortem tissues or using the OCT technique mentioned above. This technique is an optical analog of ultrasonic imaging using low-coherence interferometry to produce cross-sectional images of the retina and allows in vivo observation of retinal tissue alterations (Bhende et al., 2018).
Retinal Histopathological Studies in Amyotrophic Lateral Sclerosis Patients and Amyotrophic Lateral Sclerosis Experimental Models of Mammals
Few studies have focused on the histopathology of retinal tissue in both ALS patients and animal models of mammals with this condition. Histopathological studies in ALS patients demonstrated intraretinal protein inclusions. The first histopathological analysis of the retinas of patients with ALS was performed in 2014 on a patient with the C9orf72 mutation. In this study, the authors found p62-positive and pTDP43-negative intracytoplasmic perinuclear inclusions in the inner nuclear layer (INL). These deposits were similar to those observed in the dentate gyrus of ALS patients with the C9orf72 mutation. The p62-positive staining was colocalized for both the poly-(GA)n dipeptide repeat and ubiquitin in the retina, which is similar to the perinuclear inclusions located in the brains of patients with this mutation. The p62-positive inclusions found were likely located in specific cone bipolar cells and within the amacrine and horizontal cells, as they were also marked with GLT-1 and recoverin. The authors suggested that these deposits may be related to the affectation of contrast sensitivity (Fawzi et al., 2014). In addition, in other ALS patients with C9orf72 mutations, specific p62 inclusions were observed in the retinal ganglion cell layer (GCL) in a far smaller proportion than in INL (94.9% in INL vs. 5.1% in GCL). Numerous positive ubiquitin2 + aggregates were also observed in a mutant UBQLN2 transgenic mice experimental model, mainly in the INL, with fewer in the outer plexiform layer (OPL) and some in the GCL (Volpe et al., 2015). The accumulation of ubiquitin2 aggregates in the layers of the retina with more synapses is related to the accumulation of these aggregates in the dendritic spines of the hippocampus. The location of the aggregates at the synapses, and their spines, may be associated with the dementia observed in this experimental model of ALS. In addition, few ubiquitin2 positive aggregates were detected in the subretinal space of the retinal pigment epithelium, which sometimes elevated these levels in the same way as druses (Volpe et al., 2015). Furthermore, lipofuscin deposits sometimes related to subretinal drusen-like aggregates were found in progranulin-deficient FTD patients (Ward et al., 2017). Retinal thinning in these patients was detected by OCT before symptoms, suggesting that the eye is affected in progranulin-deficient FTD disease (Ward et al., 2014).
As noted previously, microglial and astroglial cell activation occurs in ALS. However, to our knowledge, there are only two works that analyzed the glial cells of the retina in relation to ALS. hSOD1 + vacuoles located in the dendrites of excitatory retinal neurons were observed in a mouse model of ALS SOD1 (SOD1G93A), mainly in the inner plexiform layer (IPL) and rarely in the GCL and INL. However, there were no signs of activation of either the astroglia or the microglia of the retina compared with those of wild-type mice (Ringer et al., 2017). In contrast, microglial activation was demonstrated in a mouse model of ALS [devoid of ran-binding protein2 (Ranbp2)]. Ranbp2 is a protein involved in nucleo-cytoplasmic transport whose regulation is impaired in both SALS and FALS (Ferreira, 2019). In ALS mice without this protein (with respect to wild-type controls), there was an increase in the number of CD45 +, CD11b +, and F4/80 + microglial cells surrounding the RGCs, in addition to a notable increase in amoeboid forms. An increase of metalloproteinase 28, which is an immune regulator, was also observed in the RGCs, suggesting that Ranbp2 may be involved in the signaling between the microglia and the RGCs in the immune response of ALS disease (Cho et al., 2019; Figure 2).
Optical Coherence Tomography
Although the first study performed in 75 ALS patients with Cirrus OCT showed no changes in the peripapillary and macular areas (Roth et al., 2013), further studies have shown that there are changes in the retina of ALS patients (Ringelstein et al., 2014; Volpe et al., 2015; Hübers et al., 2016; Simonett et al., 2016; Mukherjee et al., 2017; Abdelhak et al., 2018; Liu et al., 2018; Rohani et al., 2018; Rojas et al., 2019; Table 3).
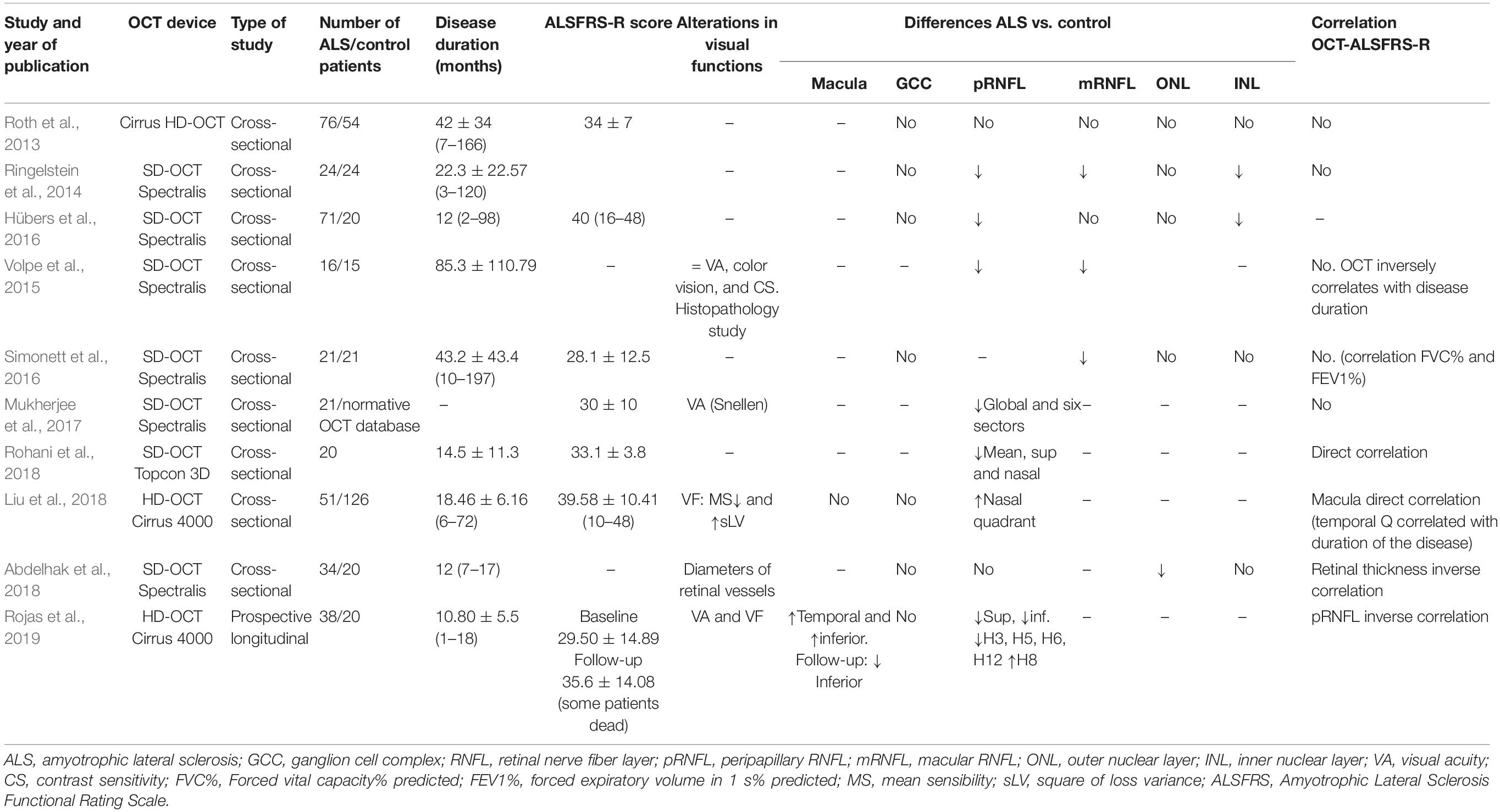
Table 3. OCT studies in ALS patients.
In the retina of ALS patients, high-resolution SD-OCT revealed reduction in the mean total macular thickness, in the peripapillary retinal nerve fiber layer (pRNFL), the INL (Ringelstein et al., 2014; Hübers et al., 2016), and the outer nuclear layer (ONL) (Abdelhak et al., 2018), suggesting neurodegeneration of the retina in ALS patients.
Volpe et al. studied whether clinical and histopathological findings were present in the eyes of ALS patients and explored their correlation with an animal model (ALS/dementia transgenic mice with dysfunctional ubiquilin2, UBQLN2P497H) (Volpe et al., 2015). Using SD-OCT, the authors observed that (i) in ALS patients compared with the controls, there was a decrease in total macular volume; (ii) 37.5% of ALS patients showed an average pRNFL below the first percentile, and temporal and papillomacular bundle were more affected; and (iii) in ALS patients, the total macular thickness and pRNFL thickness correlated inversely with the time of evolution of ALS (Volpe et al., 2015).
Recently, in a study performed in early ALS patients with spinal onset and without ocular diseases, a significant macular thickness increase was found in the temporal and inferior areas of the inner macular ring in comparison with that in a healthy control, suggesting that this retinal thickening in early ALS patients could be due a microglial activation in the neuroinflammatory process (Ringelstein et al., 2014; Rojas et al., 2019). In contrast, in another study in ALS patients, only the macular retinal nerve fiber layer (mRNFL) showed a significant thickness decrease by OCT, which correlated positively with pulmonary function tests (Simonett et al., 2016). However, neither total macular thickness nor macular thickness showed changes (Simonett et al., 2016).
Only one study exists that detected a thinning in the ONL (Abdelhak et al., 2018), suggesting a possible impact on the photoreceptors and linking this finding with this subclinical visual acuity impairment (Moss et al., 2012; Abdelhak et al., 2018). Using SD-OCT, this work analyzed the retinal vessels, finding that in ALS patients compared with the control, the outer wall thickness of the retinal vessels was higher than in the control group (Abdelhak et al., 2018). Similar microvascular alterations in the brain and spinal cord of ALS model mice were found to precede the degeneration of motor neurons (Zhong et al., 2008).
In ALS patients, an analysis of the relationship of clinical features and retinal changes using SD-OCT and diffusion tensor imaging (DTI) found no significant correlation between clinical features and retinal thickness; however, there was a direct correlation between retinal thickness and fractional anisotropy of the corticospinal tract (Hübers et al., 2016). On the basis of these observations, it was suggested that retinal changes could be related to damage of white matter in the corticospinal tract and may be a possible biomarker in ALS (Hübers et al., 2016).
Although several papers have demonstrated the pRNFL decrease in ALS patients without ocular pathology (Mukherjee et al., 2017; Rohani et al., 2018; Rojas et al., 2019), there is controversy about its correlation with ALSFRS-R values. While one study did not demonstrate a correlation between RNFL thickness and the ALSFRS-R score and their progression rates (Mukherjee et al., 2017), other authors found a correlation with some OCT parameters (Abdelhak et al., 2018; Liu et al., 2018; Rohani et al., 2018; Rojas et al., 2019; Salobrar-García et al., 2019). In a study of the pRNFL thickness in four quadrants, the average pRNFL thickness showed a significant positive correlation with the ALSFRS-R score (Rohani et al., 2018), and the pRNFL thickness in the inferior sector was negative (Rojas et al., 2019). When the analysis was more detailed, dividing the papilla into 12 hourly sectors, sectors H5 and H6 had a positive direct significant correlation, and H8 had an inverse significant correlation of pRNFL with the ALSFRS-R values (Rojas et al., 2019). In addition, the entire retinal thickness correlated negatively with the ALSFRS-R score (Abdelhak et al., 2018).
Follow-up OCT studies of ALS patients are very rare. In the only one performed of early ALS patients with spinal onset (basal patients) who were examined 6 months after the basal scan (follow-up patients), the SD-OCT follow-up analysis of ALS patients showed a significant macular thickness decrease in the inferior areas of the inner and outer macular ring and a significant pRNFL thickness decrease in the superior and inferior quadrants, compared with the baseline (Rojas et al., 2019).
Asymmetry is a typical hallmark of ALS, as mentioned above. In the left eye (LE) of ALS patients, after adjustment for multiplicity, there was a significant decrease in pRNFL thickness in the nasal quadrant compared with the corresponding quadrant thickness in the right eye (RE) (Rohani et al., 2018). In the follow-up study mentioned above (Rojas et al., 2019), there were significant interocular asymmetries in some areas between the LE, which were always thinner, and the RE were observed. In the baseline ALS group, in the LE, the inferior-nasal quadrant of the macular ganglion cell complex (GCC), and the H7 and H9 hourly-sectors of the pRNFL were significantly thinner than those in the RE, while in the follow-up ALS group, both the supero-nasal quadrant of macular GCC and the temporal quadrant, and the H8 and H9 hourly sectors of the pRNFL, were significantly thinner in the LE than those in the RE. Therefore, the asymmetric participation of the CNS in this disease is not exclusive to the motor system (Rohani et al., 2018).
Thinning of the inner retina was also observed by SD-OCT in other neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease, in which both RNFL and GCL thinning were detected (Moreno-Ramos et al., 2013; Coppola et al., 2015; Salobrar-García et al., 2015b; Garcia-Martin et al., 2016). However, in FTD, OCT identified ONL thinning, specifically in the ellipsoid zone, and this thinning of the outer retina correlated with cognitive changes (Kim et al., 2017). Moreover, the relationship between ONL thinning and FTD was especially apparent in the subgroup of patients considered to have “likely tauopathy” based on their symptoms or their genetics (Kim et al., 2017).
The differences observed in retinal thickness measurements using OCT may be due to the following: (i) the different disease stages of ALS patients included in the studies; (ii) the heterogeneous nature of this pathology (Hübers et al., 2016); and (iii) the small number of participants in these studies, since most ALS patients do not manifest visual problems.
Many authors observed changes in retinal thickness, as shown in this section. Therefore, retinal changes constitute a biomarker of neurodegeneration and progression of ALS disease (Hübers et al., 2016; Rohani et al., 2018), and OCT analysis is a useful tool for the study of this pathology (Rojas et al., 2019).
Conclusion
In view of the above, changes in visual function are moderate in ALS. The main changes occur at the oculomotor level, but with no great affect. Functional tests such as VA, contrast sensitivity, VF, and VEPs show mild alterations in ALS. However, the low rate of functional impairment does not mean that there are no structural changes in the visual pathway in ALS patients. New techniques such as OCT have made it possible to detect structural changes in the retina (due to retrograde or anterograde involvement), but which ultimately reflect changes in the visual pathway. These changes monitored with OCT could help to follow up this pathology. However, these studies are recent and are not homogeneous due to differences in the patients’ degree of disease, time of evolution, age group, or ALS scores. In addition, differences in the OCT technology make it difficult to compare between the different studies. Thus, it is necessary to perform more studies to analyze the retinal changes that occur in ALS disease. In addition, there are also many unanswered questions, including when and where the changes in the retina first occur, which subgroups of patients exhibit retinal phenotypes, and whether these phenotypes change over time. Longer studies are needed to determine whether retinal thickness might be a useful way to measure disease progression and whether patients with different genetic mutations that cause ALS experience more severe retinal involvement.
We conclude that, in addition to CNS disorders, there are also peripheral neurological diseases that can affect the retina. We have shown that the retina has great sensitivity as a biomarker for susceptibility/risk in MND, despite the fact that the retinal changes do not produce clinical visual symptoms. Therefore, the use of OCT in ALS patients could be a recommended test within the diagnostic techniques of this pathology.
Author Contributions
PR, AR, IL-C, ES-G, RH, and JR designed the concept and drafted the manuscript. JF-A, IL-C, ES-G, MC, LE-H, and JS collected the literature, analyzed the data, and edited the language in the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Ophthalmological Network OFTARED (Enfermedades oculares: Prevención, detección precoz, tratamiento y rehabilitación de las patologías oculares) (RD16/0008/0005), Institute of Health of Carlos III, Spanish Ministry of Economy and the European program FEDER and Network RETiBRAIN (La retina un modelo para investigar Neuroprotección en patologías del Sistema Nervioso Central) (RED2018-102499-T), Spanish Ministry of Science, Innovation and Universities. JF-A was currently supported by a Predoctoral Fellowship (FPU17/01023) from the Spanish Ministry of Science, Innovation and Universities. IL-C was currently supported by a Predoctoral Fellowship (CT42/18-CT43/18) from the Complutense University of Madrid.
References
Abdelhak, A., Hübers, A., Böhm, K., Ludolph, A. C., Kassubek, J., and Pinkhardt, E. H. (2018). In vivo assessment of retinal vessel pathology in amyotrophic lateral sclerosis. J. Neurol. 265, 949–953. doi: 10.1007/s00415-018-8787-x
Abel, L. A., Williams, I. M., Gibson, K. L., and Levi, L. (1995). Effects of stimulus velocity and acceleration on smooth pursuit in motor neuron disease. J. Neurol. 242, 419–424. doi: 10.1007/BF00873543
Abrahams, S., Goldstein, L. H., Suckling, J., Ng, V., Simmons, A., Chitnis, X., et al. (2005). Frontotemporal white matter changes in amyotrophic lateral sclerosis. J. Neurol. 252, 321–331. doi: 10.1007/s00415-005-0646-x
Alexianu, M. E., Kozovska, M., and Appel, S. H. (2001). Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 57, 1282–1289. doi: 10.1212/WNL.57.7.1282
Almer, G., Guégan, C., Teismann, P., Naini, A., Rosoklija, G., Hays, A. P., et al. (2001). Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann. Neurol. 49, 176–185. doi: 10.1002/1531-8249(20010201)49:2<176::aid-ana37>3.0.co;2-x
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. doi: 10.1007/s00401-011-0911-2
Andersen, P. M. (2006). Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 6, 37–46. doi: 10.1007/s11910-996-0008-9
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Averbuch-Heller, L., Helmchen, C., Horn, A. K., Leigh, R. J., and Büttner-Ennerver, J. A. (1998). Slow vertical saccades in motor neuron disease: correlation of structure and function. Ann. Neurol. 44, 641–648. doi: 10.1002/ana.410440410
Azadmanesh, J., and Borgstahl, G. E. O. (2018). A review of the catalytic mechanism of human manganese superoxide dismutase. Antioxidants 7:25. doi: 10.3390/antiox7020025
Barbeito, L. H., Pehar, M., Cassina, P., Vargas, M. R., Peluffo, H., Viera, L., et al. (2004). A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res. Rev. 47, 263–274. doi: 10.1016/j.brainresrev.2004.05.003
Bhende, M., Shetty, S., Parthasarathy, M., and Ramya, S. (2018). Optical coherence tomography: a guide to interpretation of common macular diseases. Indian J. Ophthalmol. 66, 20–35. doi: 10.4103/ijo.ijo_902_17
Blokhuis, A. M., Groen, E. J., Koppers, M., van den Berg, L. H., and Pasterkamp, R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794. doi: 10.1007/s00401-013-1125-6
Boillée, S., Vande Velde, C., and Cleveland, D. W. (2006a). ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. doi: 10.1016/j.neuron.2006.09.018
Boillée, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006b). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Bouteloup, C., Desport, J.-C., Clavelou, P., Guy, N., Derumeaux-Burel, H., Ferrier, A., et al. (2009). Hypermetabolism in ALS patients: an early and persistent phenomenon. J. Neurol. 256, 1236–1242. doi: 10.1007/s00415-009-5100-z
Brettchneider, J., Del Tredici, K., Toledo, J. B., Robinson, J. L., Irwin, D. J., Grossman, M., et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. doi: 10.1002/ana.23937.Stages
Byrne, S., Walsh, C., Lynch, C., Bede, P., Elamin, M., Kenna, K., et al. (2011). Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 82, 623–627. doi: 10.1136/jnnp.2010.224501
Carra, S., Crippa, V., Rusmini, P., Boncoraglio, A., Minoia, M., Giorgetti, E., et al. (2012). Alteration of protein folding and degradation in motor neuron diseases: implications and protective functions of small heat shock proteins. Prog. Neurobiol. 97, 83–100. doi: 10.1016/j.pneurobio.2011.09.009
Carrì, M. T., Valle, C., Bozzo, F., and Cozzolino, M. (2015). Oxidative stress and mitochondrial damage: importance in non-SOD1 ALS. Front. Cell. Neurosci. 9:41. doi: 10.3389/fncel.2015.00041
Chang, Y., Kong, Q., Shan, X., Tian, G., Ilieva, H., Cleveland, D. W., et al. (2008). Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS One 3:e2849. doi: 10.1371/journal.pone.0002849
Chiò, A., Gauthier, A., Montuschi, A., Calvo, A., Vito, D. N., Ghiglione, P., et al. (2004). A cross sectional study on determinants of quality of life in ALS. J. Neurol. Neurosurg. Psychiatry 75, 1597–1601. doi: 10.1136/jnnp.2003.033100
Cho, K., Yoon, D., Yu, M., Peachey, N. S., and Ferreira, P. A. (2019). Microglial activation in an amyotrophic lateral sclerosis-like model caused by Ranbp2 loss and nucleocytoplasmic transport impairment in retinal ganglion neurons. Cell. Mol. Life Sci. 76, 3407–3432. doi: 10.1007/s00018-019-03078-5
Chung, M. J., and Suh, Y.-L. (2002). Ultrastructural changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct. Pathol. 26, 3–7. doi: 10.1080/01913120252934260
Cohen, B., and Caroscio, J. (1983). Eye movements in amyotrophic lateral sclerosis. J. Neural Transm. Suppl. 19, 305–315.
Conwit, R. A. (2006). Preventing familial ALS: A clinical trial may be feasible but is an efficacy trial warranted? J. Neurol. Sci. 251, 1–2. doi: 10.1016/j.jns.2006.07.009
Coppola, G., Di Renzo, A., Ziccardi, L., Martelli, F., Fadda, A., Manni, G., et al. (2015). Optical coherence tomography in Alzheimer’s disease: a meta-analysis. PLoS One 10:e0134750. doi: 10.1371/journal.pone.0134750
Cozzolino, M., and Carrì, M. T. (2012). Mitochondrial dysfunction in ALS. Prog. Neurobiol. 97, 54–66. doi: 10.1016/j.pneurobio.2011.06.003
Crugnola, V., Lamperti, C., Lucchini, V., Ronchi, D., Peverelli, L., Prelle, A., et al. (2010). Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch. Neurol. 67, 849–854. doi: 10.1001/archneurol.2010.128
Da Cruz, S., and Cleveland, D. W. (2011). Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 21, 904–919. doi: 10.1016/j.conb.2011.05.029
De Jong, S., Huisman, M., Sutedja, N., Van Der Kooi, A., De Visser, M., Schelhaas, J., et al. (2013). Endogenous female reproductive hormones and the risk of amyotrophic lateral sclerosis. J. Neurol. 260, 507–512. doi: 10.1007/s00415-012-6665-5
De Vos, K., Severin, F., Van Herreweghe, F., Vancompernolle, K., Goossens, V., Hyman, A., et al. (2000). Tumor necrosis factor induces hyperphosphorylation of kinesin light chain and inhibits kinesin-mediated transport of mitochondria. J. Cell Biol. 149, 1207–1214. doi: 10.1083/jcb.149.6.1207
Deivasigamani, S., Verma, H. K., Ueda, R., Ratnaparkhi, A., and Ratnaparkhi, G. S. (2014). A genetic screen identifies Tor as an interactor of VAPB in a Drosophila model of amyotrophic lateral sclerosis. Biol. Open 3, 1127–1138. doi: 10.1242/bio.201410066
Del, Bo, R., Tiloca, C., Pensato, V., Corrado, L., Ratti, A., et al. (2011). Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 82, 1239–1243. doi: 10.1136/jnnp.2011.242313
Deng, H. X., Zhai, H., Bigio, E. H., Yan, J., Fecto, F., Ajroud, K., et al. (2010). FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 67, 739–748. doi: 10.1002/ana.22051
Desport, J., Preux, P., Magy, L., Boirie, Y., Vallat, J., Beaufrère, B., et al. (2001). Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am. J. Clin. Nutr. 74, 328–334. doi: 10.1093/ajcn/74.3.328
Donaghy, C., Pinnock, R., Abrahams, S., Cardwell, C., Hardiman, O., Patterson, V., et al. (2009). Ocular fixation instabilities in motor neurone disease: AA marker of frontal lobe dysfunction? J. Neurol. 256, 420–426. doi: 10.1007/s00415-009-0109-x
Donaghy, C., Pinnock, R., Abrahams, S., Cardwell, C., Hardiman, O., Patterson, V., et al. (2010). Slow saccades in bulbar-onset motor neurone disease. J. Neurol. 257, 1134–1140. doi: 10.1007/s00415-010-5478-7
Donnelly, C. J., Zhang, P.-W., Pham, J. T., Heusler, A. R., Mistry, N. A., Vidensky, S., et al. (2013). RNA Toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 415–428. doi: 10.1016/j.neuron.2013.10.015
Dupuis, L., Pradat, P. F., Ludolph, A. C., and Loeffler, J. P. (2011). Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 10, 75–82. doi: 10.1016/S1474-4422(10)70224-6
Echaniz-Laguna, A., Zoll, J., Ponsot, E., N’guessan, B., Tranchant, C., Loeffler, J.-P., et al. (2006). Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: a temporal study in man. Exp. Neurol. 198, 25–30. doi: 10.1016/j.expneurol.2005.07.020
Echaniz-Laguna, A., Zoll, J., Ribera, F., Tranchant, C., Warter, J. M., Lonsdorfer, J., et al. (2002). Mitochondrial respiratory chain function in skeletal muscle of ALS patients. Ann. Neurol. 52, 623–627. doi: 10.1002/ana.10357
Ellis, C. M., Suckling, J., Amaro, E., Bullmore, E. T., Simmons, A., Williams, S. C., et al. (2001). Volumetric analysis reveals corticospinal tract degeneration and extramotor involvement in ALS. Neurology 57, 1571–1578. doi: 10.1212/wnl.57.9.1571
Endo, F., Komine, O., Fujimori-Tonou, N., Katsuno, M., Jin, S., Watanabe, S., et al. (2015). Astrocyte-Derived TGF-β1 accelerates disease progression in als mice by interfering with the neuroprotective functions of microglia and T Cells. Cell Rep. 11, 592–604. doi: 10.1016/j.celrep.2015.03.053
Engelhardt, J. I., Tajti, J., and Appel, S. H. (1993). Lymphocytic Infiltrates in the Spinal Cord in Amyotrophic Lateral Sclerosis. Arch. Neurol. 50, 30–36. doi: 10.1001/archneur.1993.00540010026013
Fawzi, A. A., Simonett, J. M., Purta, P., Moss, H. E., Lowry, J. L., Deng, H.-X., et al. (2014). Clinicopathologic report of ocular involvement in ALS patients with C9orf72 mutation. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 569–580. doi: 10.3109/21678421.2014.951941
Ferreira, P. A. (2019). The coming-of-age of nucleocytoplasmic transport in motor neuron disease and neurodegeneration. Cell. Mol. Life Sci. 76, 2247–2273. doi: 10.1007/s00018-019-03029-0
Ferri, A., and Coccurello, R. (2017). What is “Hyper” in the ALS Hypermetabolism? Mediat. Inflamm. 2017:7821672. doi: 10.1155/2017/7821672
Fitzgerald, K. C., O’Reilly, É. J., Falcone, G. J., McCullough, M. L., Park, Y., Kolonel, L. N., et al. (2014). Dietary ω-3 polyunsaturated fatty acid intake and risk for amyotrophic lateral sclerosis. JAMA Neurol. 71, 1102–1110. doi: 10.1001/jamaneurol.2014.1214
Forsberg, K., Andersen, P. M., Marklund, S. L., and Brännström, T. (2011). Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 121, 623–634. doi: 10.1007/s00401-011-0805-3
Gandelman, M., Peluffo, H., Beckman, J. S., Cassina, P., and Barbeito, L. (2010). Extracellular ATP and the P2X7receptor in astrocyte-mediated motor neuron death: implications for amyotrophic lateral sclerosis. J. Neuroinflammation 7:33. doi: 10.1186/1742-2094-7-33
Garcia-Martin, E., Bambo, M. P., Marques, M. L., Satue, M., Otin, S., Larrosa, J. M., et al. (2016). Ganglion cell layer measurements correlate with disease severity in patients with Alzheimer’s disease. Acta Ophthalmol. 94, e454–e459. doi: 10.1111/aos.12977
Gargiulo Monachelli, G., Meyer, M., Rodríguez, G. E., Garay, L. I., Sica, R. E. P., De Nicola, A. F., et al. (2011). Endogenous progesterone is associated to amyotrophic lateral sclerosis prognostic factors. Acta Neurol. Scand. 123, 60–67. doi: 10.1111/j.1600-0404.2010.01385.x
Gatchel, J. R., and Zoghbi, H. Y. (2005). Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 6, 743–755. doi: 10.1038/nrg1691
Gazulla, J., and Cavero-Nagore, M. (2006). [Glutamate and Alzheimer’s disease]. Rev. Neurol. 42, 427–432. doi: 10.33588/rn.4207.2005223
Ghezzi, A., Mazzalovo, E., Locatelli, C., Zibetti, A., Zaffaroni, M., and Montanini, R. (1989). Multimodality evoked potentials in amyotrophic lateral sclerosis. Acta Neurol. Scand. 79, 353–356. doi: 10.1111/j.1600-0404.1989.tb03799.x
Gizzi, M., DiRocco, A., Sivak, M., and Cohen, B. (1992). Ocular motor function in motor neuron disease. Neurology 42, 1037–1046. doi: 10.1212/wnl.42.5.1037
González Díaz, N., Barrios, E. E., Escamilla Chávez, C., and Escobar Rodríguez, D. A. (2004). Multimodal-evoked potentials in patients with amyotrophic lateral sclerosis. Rev. Med. IMSS 42, 477–486.
Haberlandt, W. (1959). Genetic aspects of amyotrophic lateral sclerosis and progressive bulbar paralysis. Acta Genet. Med. Gemellol. 8, 369–374. doi: 10.1017/s1120962300018801
Han-Xiang, D., Chen, W., Seong-Tshool, S., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. doi: 10.1038/nature10353
Hayward, C., Brock, D. J., Minns, R. A., and Swingler, R. J. (1998). Homozygosity for Asn86Ser mutation in the CuZn-superoxide dismutase gene produces a severe clinical phenotype in a juvenile onset case of familial amyotrophic lateral sclerosis. J. Med. Genet. 35:174. doi: 10.1136/jmg.35.2.174
He, J., Mangelsdorf, M., Fan, D., Bartlett, P., and Brown, M. A. (2015). Amyotrophic lateral sclerosis genetic studies: from genome-wide association mapping to genome sequencing. Neuroscientist 21, 599–615. doi: 10.1177/1073858414555404
Honig, L. S., Chambliss, D. D., Bigio, E. H., Carroll, S. L., and Elliott, J. L. (2000). Glutamate transporter EAAT2 splice variants occur not only in ALS, but also in AD and controls. Neurology 55, 1082–1088. doi: 10.1212/WNL.55.8.1082
Hübers, A., Just, W., Rosenbohm, A., Müller, K., Marroquin, N., Goebel, I., et al. (2015). De novo FUS mutations are the most frequent genetic cause in early-onset German ALS patients. Neurobiol. Aging 36, 3117.e1–3117.e6. doi: 10.1016/j.neurobiolaging.2015.08.005
Hübers, A., Müller, H. P., Dreyhaupt, J., Böhm, K., Lauda, F., Tumani, H., et al. (2016). Retinal involvement in amyotrophic lateral sclerosis: a study with optical coherence tomography and diffusion tensor imaging. J. Neural Transm. 123, 281–287. doi: 10.1007/s00702-015-1483-4
Ikenaka, K., Katsuno, M., Kawai, K., Ishigaki, S., Tanaka, F., and Sobue, G. (2012). Disruption of axonal transport in motor neuron diseases. Int. J. Mol. Sci. 13, 1225–1238. doi: 10.3390/ijms13011225
Ito, H., Fujita, K., Nakamura, M., Wate, R., Kaneko, S., Sasaki, S., et al. (2011). Optineurin is co-localized with FUS in basophilic inclusions of ALS with FUS mutation and in basophilic inclusion body disease. Acta Neuropathol. 121, 555–557. doi: 10.1007/s00401-011-0809-z
Iwasaki, Y., Ikeda, K., and Kinoshita, M. (2005). Molecular and cellular mechanism of glutamate receptors in relation to amyotrophic lateral sclerosis. Curr. Drug Targets CNS Neurol. Disord. 1, 511–518. doi: 10.2174/1568007023339021
Jaiswal, M. K. (2014). Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation. Mol. Cell. Ther. 2, 2–15. doi: 10.1186/2052-8426-2-26
Jawaid, A., Murthy, S. B., Wilson, A. M., Qureshi, S. U., Amro, M. J., Wheaton, M., et al. (2010). A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph. Lateral Scler. 11, 542–548. doi: 10.3109/17482968.2010.482592
Kamel, F., Umbach, D. M., Munsat, T. L., Shefner, J. M., Hu, H., and Sandler, D. P. (2002). Lead exposure and amyotrophic lateral sclerosis. Epidemiology 13, 311–319. doi: 10.1097/00001648-200205000-00012
Kang, B.-H., Kim, J.-I., Lim, Y.-M., and Kim, K.-K. (2018). Abnormal oculomotor functions in amyotrophic lateral sclerosis. J. Clin. Neurol. 14:464. doi: 10.3988/jcn.2018.14.4.464
Kassubek, J., Unrath, A., Huppertz, H.-J., Lulé, D., Ethofer, T., Sperfeld, A.-D., et al. (2005). Global brain atrophy and corticospinal tract alterations in ALS, as investigated by voxel-based morphometry of 3-D MRI. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 6, 213–220. doi: 10.1080/14660820510038538
Kawamata, T., Akiyama, H., Yamada, T., and McGeer, P. L. (1992). Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 140, 691–707.
Kiernan, M. C., Vucic, S., Cheah, B. C., Turner, M. R., Eisen, A., Hardiman, O., et al. (2011). Amyotrophic lateral sclerosis. Lancet 377, 942–955. doi: 10.1016/S0140-6736(10)61156-7
Kim, B. J., Irwin, D. J., Song, D., Daniel, E., Leveque, J. D., Raquib, A. R., et al. (2017). Optical coherence tomography identifies outer retina thinning in frontotemporal degeneration. Neurology 89, 1604–1611. doi: 10.1212/WNL.0000000000004500
Krasnianski, A., Deschauer, M., Neudecker, S., Gellerich, F. N., Müller, T., Schoser, B. G., et al. (2005). Mitochondrial changes in skeletal muscle in amyotrophic lateral sclerosis and other neurogenic atrophies. Brain 128, 1870–1876. doi: 10.1093/brain/awh540
Kühnlein, P., Sperfeld, A.-D., Vanmassenhove, B., Van Deerlin, V., Lee, V. M.-Y., Trojanowski, J. Q., et al. (2008). Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch. Neurol. 65, 1185–1189. doi: 10.1001/archneur.65.9.1185
Kushner, P. D., Stephenson, D. T., and Wright, S. (1991). Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J. Neuropathol. Exp. Neurol. 50, 263–277. doi: 10.1097/00005072-199105000-00008
Kwiatkowski, T. J., Bosco, D. A., LeClerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. doi: 10.1126/science.1166066
Leveille, A., Kiernan, J., Goodwin, J. A., and Antel, J. (1982). Eye movements in amyotrophic lateral sclerosis. Arch. Neurol. 39, 684–686. doi: 10.1001/archneur.1982.00510230010003
Liao, B., Zhao, W., Beers, D. R., Henkel, J. S., and Appel, S. H. (2012). Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 237, 147–152. doi: 10.1016/j.expneurol.2012.06.011
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lin, C.-L. G., Bristol, L. A., Jin, L., Dykes-Hoberg, M., Crawford, T., Clawson, L., et al. (1998). Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in Amyotrophic Lateral Sclerosis. Neuron 20, 589–602. doi: 10.1016/S0896-6273(00)80997-6
Liu, Y., Pattamatta, A., Zu, T., Reid, T., Bardhi, O., Borchelt, D. R., et al. (2016). C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 90, 521–534. doi: 10.1016/j.neuron.2016.04.005
Liu, Z., Wang, H., Fan, D., and Wang, W. (2018). Comparison of optical coherence tomography findings and visual field changes in patients with primary open-angle glaucoma and amyotrophic lateral sclerosis. J. Clin. Neurosci. 48, 233–237. doi: 10.1016/j.jocn.2017.10.080
MacCormick, I. J., Czanner, G., and Faragher, B. (2015). Developing retinal biomarkers of neurological disease: an analytical perspective. Biomark. Med. 9, 691–701. doi: 10.2217/bmm.15.17
Mackenzie, I. R. A., Bigio, E. H., Ince, P. G., Geser, F., Neumann, M., Cairns, N. J., et al. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61, 427–434. doi: 10.1002/ana.21147
Mackenzie, I. R. A., Rademakers, R., and Neumann, M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. doi: 10.1016/S1474-4422(10)70195-2
Maekawa, S., Leigh, P. N., King, A., Jones, E., Steele, J. C., Bodi, I., et al. (2009). TDP-43 is consistently co-localized with ubiquitinated inclusions in sporadic and Guam amyotrophic lateral sclerosis but not in familial amyotrophic lateral sclerosis with and without SOD1 mutations. Neuropathology 29, 672–683. doi: 10.1111/j.1440-1789.2009.01029.x
Magrané, J., and Manfredi, G. (2009). Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid. Redox Signal. 11, 1615–1626. doi: 10.1089/ars.2009.2604
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G. P., Waite, A., Rollinson, S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330.
Marti-Fàbregas, J., and Roig, C. (1993). Oculomotor abnormalities in motor neuron disease. J. Neurol. 240, 475–478. doi: 10.1007/BF00874116
Marucci, G., Morandi, L., Bartolomei, I., Salvi, F., Pession, A., Righi, A., et al. (2007). Amyotrophic lateral sclerosis with mutation of the Cu/Zn superoxide dismutase gene (SOD1) in a patient with Down syndrome. Neuromuscul. Disord. 17, 673–676. doi: 10.1016/j.nmd.2007.06.003
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
Matheson, J. K., Harrington, H. J., and Hallett, M. (1986). Abnormalities of multimodality evoked potentials in amyotrophic lateral sclerosis. Arch. Neurol. 43, 338–340. doi: 10.1001/archneur.1986.00520040026013
Mathis, S., Goizet, C., Soulages, A., Vallat, J. M., Masson, G., and Le. (2019). Genetics of amyotrophic lateral sclerosis: a review. J. Neurol. Sci. 399, 217–226. doi: 10.1016/j.jns.2019.02.030
Mcguire, V., Longstreth, W. T., Nelson, L. M., Koepsell, T. D., Checkoway, H., Morgan, M. S., et al. (1997). Occupational exposures and Amyotrophic Lateral Sclerosis a population-based case-control study. Am. J. Epidemiol. 145, 1076–1088. doi: 10.1093/oxfordjournals.aje.a009070
McLoon, L. K., Harandi, V. M., Brännström, T., Andersen, P. M., and Liu, J. X. (2014). Wnt and extraocular muscle sparing in amyotrophic lateral sclerosis. Invest. Ophthalmol. Vis. Sci. 55, 5482–5496. doi: 10.1167/iovs.14-14886
Meier, S. L., Charleston, A. J., and Tippett, L. J. (2010). Cognitive and behavioural deficits associated with the orbitomedial prefrontal cortex in amyotrophic lateral sclerosis. Brain 133, 3444–3457. doi: 10.1093/brain/awq254
Mezzapesa, D., Ceccarelli, A., Dicuonzo, F., Carella, A., Caro, M. F., and De et al. (2007). Whole-brain and regional brain atrophy in amyotrophic lateral sclerosis. AJNR Am. J. Neuroradiol. 28, 255–259.
Mitsumoto, H., Factor-Litvak, P., Andrews, H., Goetz, R. R., Andrews, L., Rabkin, J. G., et al. (2014). ALS Multicenter Cohort Study of Oxidative Stress (ALS COSMOS): the study methodology, recruitment, and baseline demographic and disease characteristics. Amyotroph. Lateral Scler. Front. Degener. 15, 192–203. doi: 10.3109/21678421.2013.864312
Moreno-Ramos, T., Benito-León, J., Villarejo, A., and Bermejo-Pareja, F. (2013). Retinal nerve fiber layer thinning in dementia associated with parkinson’s disease, dementia with lewy bodies, and alzheimer’s disease. J. Alzheimers Dis. 34, 659–664. doi: 10.3233/JAD-121975
Morozova, N., Weisskopf, M. G., McCullough, M. L., Munger, K. L., Calle, E. E., Thun, M. J., et al. (2008). Diet and amyotrophic lateral sclerosis. Epidemiology 19, 324–337. doi: 10.1097/EDE.0b013e3181632c5d
Moss, H. E., McCluskey, L., Elman, L., Hoskins, K., Talman, L., Grossman, M., et al. (2012). Cross-sectional evaluation of clinical neuro-ophthalmic abnormalities in an amyotrophic lateral sclerosis population1. Moss, H. E. et al. Cross-sectional evaluation of clinical neuro-ophthalmic abnormalities in an amyotrophic lateral sclerosis population. J. Neurol. Sci. 314, 97–101. doi: 10.1016/j.jns.2011.10.016
Moss, H. E., Samelson, M., Mohan, G., and Jiang, Q. L. (2016). High and low contrast visual acuity are not affected in amyotrophic lateral sclerosis. PLoS One 11:e0168714. doi: 10.1371/journal.pone.0168714
Mukherjee, N., McBurney-Lin, S., Kuo, A., Bedlack, R., and Tseng, H. (2017). Retinal thinning in amyotrophic lateral sclerosis patients without ophthalmic disease. PLoS One 12:e0185242. doi: 10.1371/journal.pone.0185242
Münte, T. F., Tröger, M. C., Nusser, I., Wieringa, B. M., Johannes, S., Matzke, M., et al. (1998). Alteration of early components of the visual evoked potential in amyotrophic lateral sclerosis. J. Neurol. 245, 206–210. doi: 10.1007/s004150050206
Nagy, D., Kato, T., and Kushner, P. D. (1994). Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J. Neurosci. Res. 38, 336–347. doi: 10.1002/jnr.490380312
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Ohki, M., Kanayama, R., Nakamura, T., Okuyama, T., Kimura, Y., and Koike, Y. (1994). Ocular abnormalities in amyotrophic lateral sclerosis. Acta Otolaryngol. 114, 138–142. doi: 10.3109/00016489409128318
Okuda, B., Yamamoto, T., Yamasaki, M., Maya, K., and Imai, T. (2009). Motor neuron disease with slow eye movements and vertical gaze palsy. Acta Neurol. Scand. 85, 71–76. doi: 10.1111/j.1600-0404.1992.tb03999.x
Palamiuc, L., Schlagowski, A., Ngo, S. T., Vernay, A., Dirrig-Grosch, S., Henriques, A., et al. (2015). A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med 7, 526–546. doi: 10.15252/emmm.201404433
Palma, V., Guadagnino, M., Brescia Morra, V., and Nolfe, G. (1993). Multimodality evoked potentials in sporadic amyotrophic lateral sclerosis: a statistical approach. Electromyogr. Clin. Neurophysiol. 33, 167–171.
Palmowski, A., Jost, W. H., Osterhage, J., Prudlo, J., Käsmann, B., Schimrigk, K., et al. (1995). [Disorders of eye movement in amyotrophic lateral sclerosis–report of 2 patients]. Klin. Monbl. Augenheilkd. 206, 170–172. doi: 10.1055/s-2008-1035424
Pape, J. A., and Grose, J. H. (2020). The effects of diet and sex in amyotrophic lateral sclerosis. Rev. Neurol. 176, 301–315. doi: 10.1016/j.neurol.2019.09.008
Park, Y., Park, J., Kim, Y., Baek, H., and Kim, S. H. (2015). Association between nutritional status and disease severity using the amyotrophic lateral sclerosis (ALS) functional rating scale in ALS patients. Nutrition 31, 1362–1367. doi: 10.1016/j.nut.2015.05.025
Philips, T., and Robberecht, W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263. doi: 10.1016/S1474-4422(11)70015-1
Pinto, W. B. V. R., Debona, R., Nunes, P. P., Assis, A. C. D., Lopes, C. G., Bortholin, T., et al. (2019). Atypical motor neuron disease variants: still a diagnostic challenge in neurology. Rev. Neurol. 175, 221–232. doi: 10.1016/j.neurol.2018.04.016
Pratt, A. J., Getzoff, E. D., and Perry, J. J. P. (2012). Amyotrophic lateral sclerosis: update and new developments. Degener. Neurol. Neuromuscul. Dis. 2012, 1–14. doi: 10.2147/DNND.S19803
Proudfoot, M., Menke, R. A. L., Sharma, R., Berna, C. M., Hicks, S. L., Kennard, C., et al. (2016). Eye-tracking in amyotrophic lateral sclerosis: a longitudinal study of saccadic and cognitive tasks. Amyotroph. Lateral Scler. Front. Degener. 17, 101–111. doi: 10.3109/21678421.2015.1054292
Ramírez, A. I., de Hoz, R., Salobrar-García, E., Salazar, J. J., Rojas, B., Ajoy, D., et al. (2017). The role of microglia in retinal neurodegeneration: Alzheimer’s disease, Parkinson, and glaucoma. Front. Aging Neurosci. 9:214. doi: 10.3389/fnagi.2017.00214
Raoul, C., Estévez, A. G., Nishimune, H., Cleveland, D. W., DeLapeyrière, O., Henderson, C. E., et al. (2002). Motoneuron death triggered by a specific pathway downstream of fas: potentiation by ALS-linked SOD1 mutations. Neuron 35, 1067–1083. doi: 10.1016/S0896-6273(02)00905-4
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Ringelstein, M., Albrecht, P., Südmeyer, M., Harmel, J., Müller, A. K., Keser, N., et al. (2014). Subtle retinal pathology in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 1, 290–297. doi: 10.1002/acn3.46
Ringer, C., Weihe, E., and Schütz, B. (2017). SOD1 G93A mutant mice develop a neuroinflammation-independent dendropathy in excitatory neuronal subsets of the olfactory bulb and retina. J. Neuropathol. Exp. Neurol. 76, 769–778. doi: 10.1093/jnen/nlx057
Ringholz, G. M., Appel, S. H., Bradshaw, M., Cooke, N. A., Mosnik, D. M., and Schulz, P. E. (2005). Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65, 586–590. doi: 10.1212/01.wnl.0000172911.39167.b6
Rohani, M., Meysamie, A., Zamani, B., Sowlat, M. M., and Akhoundi, F. H. (2018). Reduced retinal nerve fiber layer (RNFL) thickness in ALS patients: a window to disease progression. J. Neurol. 265, 1557–1562. doi: 10.1007/s00415-018-8863-2
Rojas, P., Hoz, R., De, Ramírez, A. I., Ferreras, A., Salobrar-Garcia, E., et al. (2019). Changes in Retinal OCT and their correlations with neurological disability in early ALS patients, a follow-up study. Brain Sci. 9, 1–18. doi: 10.3390/brainsci9120337
Román, G. C. (1996). Neuroepidemiology of amyotrophic lateral sclerosis: clues to aetiology and pathogenesis. J. Neurol. Neurosurg. Psychiatry 61, 131–137. doi: 10.1136/jnnp.61.2.131
Roth, N. M., Saidha, S., Zimmermann, H., Brandt, A. U., Oberwahrenbrock, T., Maragakis, N. J., et al. (2013). Optical coherence tomography does not support optic nerve involvement in amyotrophic lateral sclerosis. Eur. J. Neurol. 20, 1170–1176. doi: 10.1111/ene.12146
Rowland, L. P., and Shneider, N. A. (2001). Amyotrophic lateral sclerosis. N. Engl. J. Med. 344, 1688–1700. doi: 10.1056/NEJM200105313442207
Rutherford, N. J., Zhang, Y. J., Baker, M., Gass, J. M., Finch, N. C. A., Xu, Y. F., et al. (2008). Novel mutations in TARDBP(TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 4:e1000193. doi: 10.1371/journal.pgen.1000193
Saito, S., and Yamamoto, H. (1989). [Analysis of oculomotor disorders in motor neuron disease]. Clin. Neurol. 29, 969–975.
Salobrar-García, E., de Hoz, R., Ramírez, A. I., Salazar, J. J., Rojas, P., López-Cuenca, I., et al. (2019). “Ocular Exploration in the Diagnosis and Follow-Up of the Alzheimer’s Dementia,” in Alzheimer’s Disease [Internet], ed. T. Wisniewski (Brisbane: Codon Publications), 159–178. doi: 10.15586/alzheimersdisease.2019.ch10
Salobrar-García, E., de Hoz, R., Rojas, B., Ramirez, A. I., Salazar, J. J., Yubero, R., et al. (2015a). Ophthalmologic psychophysical tests support OCT findings in mild Alzheimer’s disease. J. Ophthalmol. 2015:736949. doi: 10.1155/2015/736949
Salobrar-García, E., Hoyas, I., Leal, M., de Hoz, R., Rojas, B., Ramirez, A. I., et al. (2015b). Analysis of retinal peripapillary segmentation in early Alzheimer’s disease patients. Biomed Res. Int. 2015:636548. doi: 10.1155/2015/636548
Salobrar-García, E., Ramírez, A. I., de Hoz, R., Rojas, P., Salazar, J. J., Rojas, B., et al. (2016). “The impact of the eye in dementia: the eye and its role in diagnosis and follow-up,” in Update on Dementia, ed. D. Moretti (London: InTech), 379–426. doi: 10.5772/64490
Sathasivam, S. (2010). Motor neurone disease: clinical features, diagnosis, diagnostic pitfalls and prognostic markers. Singapore Med. J. 51, 367–372.
Savolainen, K. M., Loikkanen, J., Eerikäinen, S., and Naarala, J. (1998). Interactions of excitatory neurotransmitters and xenobiotics in excitotoxicity and oxidative stress: glutamate and lead. Toxicol. Lett. 10, 363–367. doi: 10.1016/S0378-4274(98)00233-1
Scekic-Zahirovic, J., Oussini, H., El, Mersmann, S., Drenner, K., Wagner, M., et al. (2017). Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta Neuropathol. 133, 887–906. doi: 10.1007/s00401-017-1687-9
Schiffer, D., Cordera, S., Cavalla, P., and Migheli, A. (1996). Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J. Neurol. Sci. 139, 27–33. doi: 10.1016/0022-510X(96)00073-1
Schymick, J. C., and Traynor, B. J. (2010). Expanding the genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Alzheimer Res. Ther. 4:30. doi: 10.1186/alzrt133
Seals, R. M., Kioumourtzoglou, M. A., Gredal, O., Hansen, J., and Weisskopf, M. G. (2017). Occupational formaldehyde and amyotrophic lateral sclerosis. Eur. J. Epidemiol. 32, 893–899. doi: 10.1007/s10654-017-0249-8
Selvaraj, B. T., Livesey, M. R., Zhao, C., Gregory, J. M., James, O. T., Cleary, E. M., et al. (2018). C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 9:347. doi: 10.1038/s41467-017-02729-0
Sharma, A., Lyashchenko, A. K., Lu, L., Nasrabady, S. E., Elmaleh, M., Mendelsohn, M., et al. (2016). ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 4:10465. doi: 10.1038/ncomms10465
Sharma, R., Hicks, S., Berna, C. M., Kennard, C., Talbot, K., and Turner, M. R. (2011). Oculomotor dysfunction in amyotrophic lateral sclerosis: a comprehensive review. Arch. Neurol. 68, 857–861. doi: 10.1001/archneurol.2011.130
Shaunak, S., Orrell, R. W., O’Sullivan, E., Hawken, M. B., Lane, R. J., Henderson, L., et al. (1995). Oculomotor function in amyotrophic lateral sclerosis: evidence for frontal impairment. Ann. Neurol. 38, 38–44. doi: 10.1002/ana.410380109
Shi, P., Ström, A.-L., Gal, J., and Zhu, H. (2010). Effects of ALS-related SOD1 mutants on dynein-and KIF5-mediated retrograde and anterograde axonal transport. Biochim. Biophys. Acta 1802, 707–716. doi: 10.1016/j.bbadis.2010.05.008
Shin Hee, J., and Lee Keun, J. (2013). “Multiple routes of motor neuron degeneration in ALS,” in Current Advances in Amyotrophic Lateral Sclerosis, ed. A. Éstevez (London: InTech), 35–71. doi: 10.5772/56625
Simonett, J. M., Huang, R., Siddique, N., Farsiu, S., Siddique, T., Volpe, N. J., et al. (2016). Macular sub-layer thinning and association with pulmonary function tests in Amyotrophic Lateral Sclerosis. Sci. Rep. 6:29187. doi: 10.1038/srep29187
Simons, T. J. (1986). Cellular interactions between lead and calcium. Br. Med. Bull. 42, 431–434. doi: 10.1093/oxfordjournals.bmb.a072162
Simpson, E. P., Henry, Y. K., Henkel, J. S., Smith, R. G., and Appel, S. H. (2004). Increased lipid peroxidation in sera of ALS patients: a potential biomarker of disease burden. Neurology 62, 1758–1765. doi: 10.1212/WNL.62.10.1758
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Steyn, F. J., Ioannides, Z., van Eijk, R., Heggie, S., Thorpe, K., Ceslis, A., et al. (2018). Hypermetabolism in ALS is associated with greater functional decline and shorter survival Neurodegeneration. J. Neurol. Neurosurg. Psychiatry 89, 1016–1023. doi: 10.1136/jnnp-2017-317887
Trotti, D., Rolfs, A., Danbolt, N. C., Brown, R. H. Jr., and Hediger, M. A. (1999). SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat. Neurosci. 2, 427–433. doi: 10.1038/8091
Tsujikawa, T., Kiyono, Y., Matsunaga, A., Ikawa, M., Nakamoto, Y., Mori, T., et al. (2015). Increased oxidative stress is related to disease severity in the ALS motor cortex: a PET study. Neurology 84, 2033–2039. doi: 10.1212/wnl.0000000000001588
Turner, M. R., Cagnin, A., Turkheimer, F. E., Miller, C. C. J., Shaw, C. E., Brooks, D. J., et al. (2004). Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 15, 601–609. doi: 10.1016/j.nbd.2003.12.012
Turner, M. R., Kiernan, M. C., Leigh, P. N., and Talbot, K. (2009). Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. 8, 94–109. doi: 10.1016/S1474-4422(08)70293-X
Turner, M. R., Wotton, C., Talbot, K., and Goldacre, M. J. (2012). Cardiovascular fitness as a risk factor for amyotrophic lateral sclerosis: indirect evidence from record linkage study. J. Neurol. Neurosurg. Psychiatry 83, 395–398. doi: 10.1136/jnnp-2011-301161
Vaisman, N., Lusaus, M., Nefussy, B., Niv, E., Comaneshter, D., Hallack, R., et al. (2009). Do patients with amyotrophic lateral sclerosis (ALS) have increased energy needs? J. Neurol. Sci. 279, 26–29. doi: 10.1016/j.jns.2008.12.027
Valentine, J. S., Doucette, P. A., and Zittin Potter, S. (2005). Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593. doi: 10.1146/annurev.biochem.72.121801.161647
Van Deerlin, V. M., Leverenz, J. B., Bekris, L. M., Bird, T. D., Yuan, W., Elman, L. B., et al. (2008). TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 7, 409–416. doi: 10.1016/S1474-4422(08)70071-1
Van Es, M. A., Dahlberg, C., Birve, A., Veldink, J. H., van den Berg, L. H., and Andersen, P. M. (2010). Large-scale SOD1 mutation screening provides evidence for genetic heterogeneity in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 81, 562–566. doi: 10.1136/jnnp.2009.181453
Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vargas, M. R., and Johnson, J. A. (2010). Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurother. J. Am. Soc. Exp. Neurother. 7, 471–481. doi: 10.2174/1381612823666170622095802
Veldink, J. H., Kalmijn, S., Groeneveld, G. J., Wunderink, W., Koster, A., de Vries, J. H., et al. (2007). Intake of Polyunsaturated fatty acids and vitamin E reduce the risk of developing ALS. J. Neurol. Neurosurg. Psychiatry 78, 367–371. doi: 10.1136/jnnp.2005.083378
Vielhaber, S., Kunz, D., Winkler, K., Wiedemann, F. R., Kirches, E., Feistner, H., et al. (2000). Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 123(Pt 7), 1339–1348. doi: 10.1093/brain/123.7.1339
Volpe, N. J., Simonett, J., Fawzi, A. A., and Siddique, T. (2015). Opthalmic manifestations of amyotrophic lateral sclerosis (an American Ophthalmological Society thesis). Trans. Am. Ophthalmol. Soc. 113, 1–15. doi: 10.3109/21678421.2013.838413
Vucic, S., and Kiernan, M. C. (2007). Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 78, 849–852. doi: 10.1136/jnnp.2006.105056
Wang, W., Zhang, F., Li, L., Tang, F., Siedlak, S. L., Fujioka, H., et al. (2014). MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J. Biol. Chem. 290, 168–182. doi: 10.1074/jbc.M114.617167
Ward, M. E., Chen, R., Huang, H. Y., Ludwig, C., Telpoukhovskaia, M., Taubes, A., et al. (2017). Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci. Transl. Med. 9:eaah5642. doi: 10.1126/scitranslmed.aah5642
Ward, M. E., Taubes, A., Chen, R., Miller, B. L., Sephton, C. F., Gelfand, J. M., et al. (2014). Early retinal neurodegeneration and impaired Ran-mediated nuclear import of TDP-43 in progranulin-deficient FTLD. J. Exp. Med. 211, 1937–1945. doi: 10.1084/jem.20140214
Weishaupt, J. H., Waibel, S., Birve, A., Volk, A. E., Mayer, B., Meyer, T., et al. (2013). A novel optineurin truncating mutation and three glaucoma-associated missense variants in patients with familial amyotrophic lateral sclerosis in Germany. Neurobiol. Aging 34, 1516.e9–1516.e15. doi: 10.1016/j.neurobiolaging.2012.09.007
Weisskopf, M. G., Morozova, N., O’Reilly, E. J., McCullough, M. L., Calle, E. E., Thun, M. J., et al. (2009). Prospective study of chemical exposures and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 80, 558–561. doi: 10.1136/jnnp.2008.156976
Weydt, P., Yuen, E. C., Ransom, B. R., and Möller, T. (2004). Increased cytotoxic potential of microglia from ALS-transgenic mice. Glia 48, 179–182. doi: 10.1002/glia.20062
Williams, D. B., Floate, D. A., and Leicester, J. (1988). Familial motor neuron disease: differing penetrance in large pedigrees. J. Neurol. Sci. 86, 215–230. doi: 10.1016/0022-510X(88)90100-1
Williams, K. L., Topp, S., Yang, S., Smith, B., Fifita, J. A., Warraich, S. T., et al. (2016). CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 7:11253. doi: 10.1038/ncomms11253
Wong, P. C., Pardo, C. A., Borchelt, D. R., Lee, M. K., Copeland, N. G., Jenkins, N. A., et al. (1995). An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105–1116. doi: 10.1016/0896-6273(95)90259-7
Yang, C., Wang, H., Qiao, T., Yang, B., Aliaga, L., Qiu, L., et al. (2014). Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 111, E1121–E1129. doi: 10.1073/pnas.1322641111
Yedavalli, V. S., Patil, A., and Shah, P. (2018). Amyotrophic lateral sclerosis and its mimics/variants: a comprehensive review. J. Clin. Imaging Sci. 8:53. doi: 10.4103/jcis.JCIS_40_18
Ying, H., Shen, X., Park, B., and Yue, B. Y. J. T. (2010). Posttranslational modifications, localization, and protein interactions of optineurin, the product of a glaucoma gene. PLoS One 5:e9168. doi: 10.1371/journal.pone.0009168
Yu, Y., Hayashi, S., Cai, X., Fang, C., Shi, W., Tsutsui, H., et al. (2014). Pu-erh tea extract induces the degradation of FET family proteins involved in the pathogenesis of amyotrophic lateral sclerosis. Biomed Res. Int. 2014:254680. doi: 10.1155/2014/254680
Zarei, S., Carr, K., Reiley, L., Diaz, K., Guerra, O., Altamirano, P. F., et al. (2015). A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 6:171. doi: 10.4103/2152-7806.169561
Zhang, Q., Mao, C., Jin, J., Niu, C., Bai, L., Dang, J., et al. (2014). Side of limb-onset predicts laterality of gray matter loss in amyotrophic lateral sclerosis. Biomed Res. Int. 2014:473250. doi: 10.1155/2014/473250
Zhao, W., Beers, D. R., Bell, S., Wang, J., Wen, S., Baloh, R. H., et al. (2015). TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp. Neurol. 273, 24–35. doi: 10.1016/j.expneurol.2015.07.019
Zhao, W., Beers, D. R., Henkel, J. S., Zhang, W., Urushitani, M., Julien, J. P., et al. (2010). Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 58, 231–243. doi: 10.1002/glia.20919
Zhong, Z., Deane, R., Ali, Z., Parisi, M., Shapovalov, Y., O’Banion, M. K., et al. (2008). ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 11, 420–422. doi: 10.1038/nn2073
Zhou, H., Chen, G., Chen, C., Yu, Y., and Xu, Z. (2012). Association between extremely low-frequency electromagnetic fields occupations and amyotrophic lateral sclerosis: a meta-analysis. PLoS One 7:e48354. doi: 10.1371/journal.pone.0048354
Zhu, G., Wu, C. J., Zhao, Y., and Ashwell, J. D. (2007). Optineurin Negatively Regulates TNFα- Induced NF-κB Activation by Competing with NEMO for Ubiquitinated RIP. Curr. Biol. 17, 1438–1443. doi: 10.1016/j.cub.2007.07.041
Keywords: ALS, motor neuron, neurodegenerative diseases, retina, optic nerve, eye, neuroinflammation, biomarker
Citation: Rojas P, Ramírez AI, Fernández-Albarral JA, López-Cuenca I, Salobrar-García E, Cadena M, Elvira-Hurtado L, Salazar JJ, de Hoz R and Ramírez JM (2020) Amyotrophic Lateral Sclerosis: A Neurodegenerative Motor Neuron Disease With Ocular Involvement. Front. Neurosci. 14:566858. doi: 10.3389/fnins.2020.566858
Received: 28 May 2020; Accepted: 24 August 2020;
Published: 25 September 2020.
Edited by:
Nicolás Cuenca, University of Alicante, SpainReviewed by:
Jose-Alberto Palma, New York University, United StatesInigo Gabilondo, Biocruces Bizkaia Health Research Institute, Spain
Copyright © 2020 Rojas, Ramírez, Fernández-Albarral, López-Cuenca, Salobrar-García, Cadena, Elvira-Hurtado, Salazar, de Hoz and Ramírez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa de Hoz, cmRlaG96QG1lZC51Y20uZXM=; José M. Ramírez, cmFtaXJlenNAbWVkLnVjbS5lcw==
†These authors have contributed equally to this work