 Michelle E. McClements
Michelle E. McClements Federica Staurenghi1
Federica Staurenghi1 Jasmina Cehajic-Kapetanovic
Jasmina Cehajic-Kapetanovic- 1Nuffield Laboratory Ophthalmology, Department of Clinical Neurosciences, NIHR Oxford Biomedical Research Centre, University of Oxford, Oxford, United Kingdom
- 2Oxford Eye Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford, United Kingdom
The degeneration of light-detecting rod and cone photoreceptors in the human retina leads to severe visual impairment and ultimately legal blindness in millions of people worldwide. Multiple therapeutic options at different stages of degeneration are being explored but the majority of ongoing clinical trials involve adeno-associated viral (AAV) vector-based gene supplementation strategies for select forms of inherited retinal disease. Over 300 genes are associated with inherited retinal degenerations and only a small proportion of these will be suitable for gene replacement therapy. However, while the origins of disease may vary, there are considerable similarities in the physiological changes that occur in the retina. When early therapeutic intervention is not possible and patients suffer loss of photoreceptor cells but maintain remaining layers of cells in the neural retina, there is an opportunity for a universal gene therapy approach that can be applied regardless of the genetic origin of disease. Optogenetic therapy offers such a strategy by aiming to restore vision though the provision of light-sensitive molecules to surviving cell types of the retina that enable light perception through the residual neurons. Here we review the recent progress in attempts to restore visual function to the degenerate retina using optogenetic therapy. We focus on multiple pre-clinical models used in optogenetic strategies, discuss their strengths and limitations, and highlight considerations including vector and transgene designs that have advanced the field into two ongoing clinical trials.
Introduction
Optogenetics is a method that allows optical control of neural circuitry by ectopic expression of light-sensitive tools in target cells (Deisseroth et al., 2006). It offers a unique opportunity to treat inherited retinal degenerations of varied genetic origins with a universal therapeutic strategy. In a degenerate retina that has lost the light-sensitive photoreceptor cells, optogenetic therapy is a promising approach that combines neurobiology and genetic engineering techniques to provide light-mediated control over the cell physiology in surviving retinal cells that are normally insensitive to light. In a pioneering study, adeno-associated viral (AAV) vector-based delivery of a transgene encoding light-sensitive protein, channelrhodopsin (ChR2), was shown to be targeted to surviving cells of the retina, whereby its ectopic expression in cellular membranes converted the cells into artificial photoreceptors (Bi et al., 2006; Sahel and Roska, 2013). However, this landmark study highlighted several important challenges that needed to be addressed before optogenetics could be considered for human studies. The light levels needed for activation of the ChR2 sensor were very high and not encountered under normal light conditions. Thus, stimulation via external artificial light source would be necessary for this strategy to work as human therapy, but such high radiation levels are potentially toxic to the retina. In addition, being a microbial opsin, questions regarding safety and immune responses if used in humans needed to be answered. The strategy was also shown to have limited expression of the transgene in in the inner retina of a murine model of retinal degeneration and questions were raised about its translational potential when used in larger animal models and indeed human retina.
In this review we consider the multiple areas of research that have enabled development of optogenetic strategies for the treatment of inherited retinal degeneration, including vector design, transgene and opsin selection across various models of disease. Combined, the work reviewed highlight the great progress achieved in the field to date, which has led to two ongoing clinical trials with further upcoming human trials trials likely in the near future.
Inherited Retinal Disease and the Degenerate Retina
Inherited retinal degeneration can result from different mutations in more than 300 genes (RetNet: https://sph.uth.edu/retnet/) and is the leading cause of blindness in the working age population, affecting approximately 1 in 3,000 people world-wide (Sohocki et al., 2001). Clinical trials have shown encouraging success in recent years and have primarily focused on AAV vector gene supplementation strategies for particular forms of inherited retinal disease (Bainbridge et al., 2015; Russell et al., 2017; Xue et al., 2018; Cehajic-Kapetanovic et al., 2020b). However, such therapies are suitable for only a small subset of people suffering from inherited retinal disease. Many of the identified genetic causes have very low prevalence, which makes it unfeasible to develop gene-specific treatments for all forms and yet, regardless of the differences in genetic origin, the progression of disease occurs similarly in many patients. The process of retinal degeneration in the condition retinitis pigmentosa typically involves loss of rod photoreceptor cells followed by loss of cone photoreceptor cells with subsequent migration of retinal pigment epithelium into the inner retina (Figure 1). A healthy retina is highly structured and enables complex interactions between multiple cell types. When there is significant loss of photoreceptor cells, which form the outer retinal layer, cells of the inner retina continue to survive but over time can undergo remodeling. This refers to a change in the normal structure of the remaining retinal layers and includes synaptic rewiring, retraction of bipolar cell dendrites and changes to protein expression patterns and trafficking (Marc et al., 2003; Puthussery et al., 2009). Despite this, residual cells have been shown to maintain key molecular signatures and morphology, suggesting the surviving retina is a receptive environment for ectopic optogene expression (Jones et al., 2003).
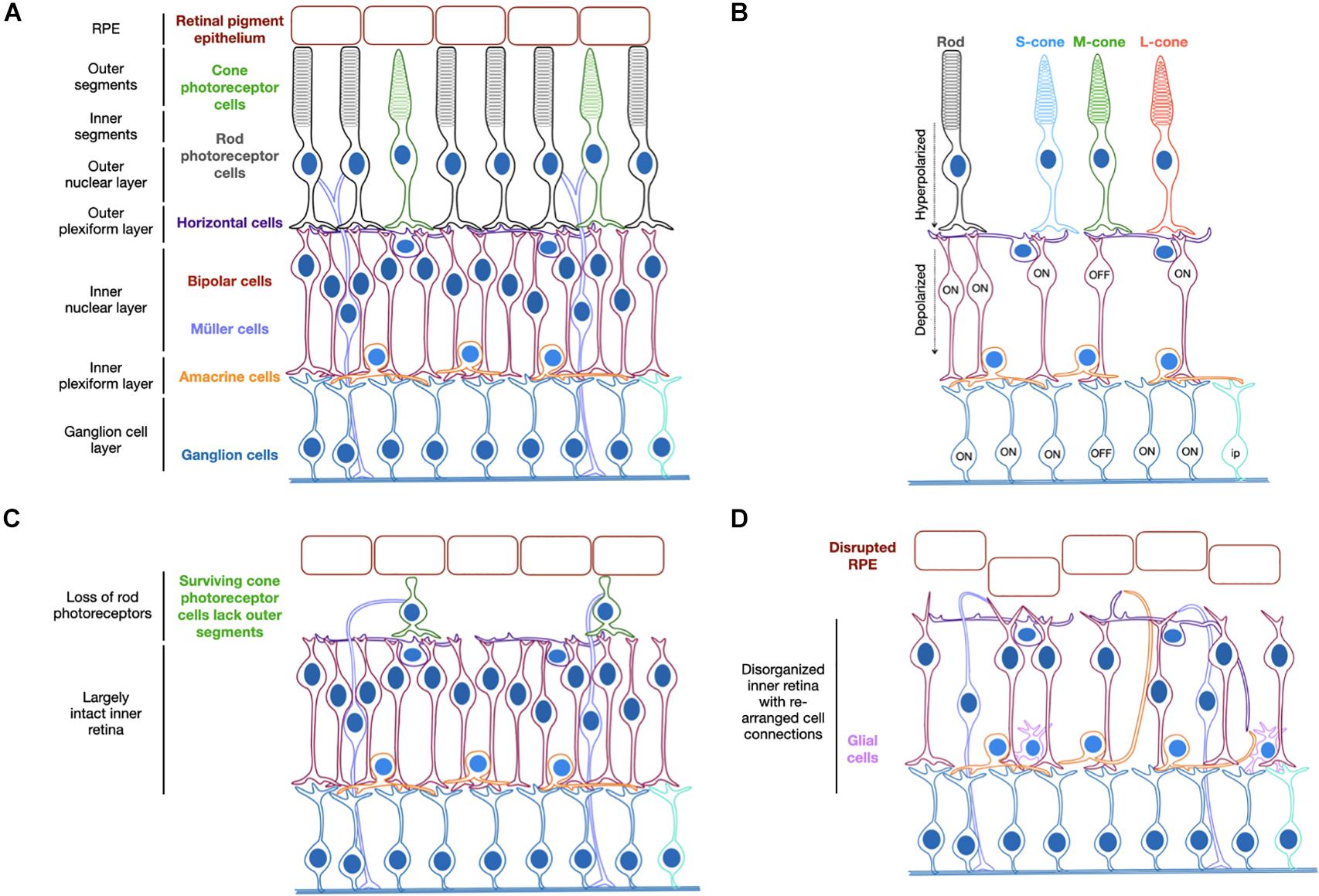
Figure 1. Schematic representation of the organized structure of a healthy retina (A). Light activation of opsins triggers photoreceptor hyperpolarization, which causes depolarization of ON-bipolar cells with cone OFF-bipolar cells in the same receptive field hyperpolarized. The ON-ganglion cells are subsequently activated and the OFF-ganglion cells inhibited (B). Degeneration begins by loss of rod photoreceptor cells and loss of outer segments on residual cone photoreceptor cells (C) followed by further cell loss and structural rearrangements in later stages of disease (D). S/M/L-cone = cone photoreceptor cells containing short, medium or long wavelength-sensitive opsins. Ip = intrinsically photosensitive ganglion cell.
The pattern of retinal degeneration is common across cases of retinitis pigmentosa regardless of the genetic origin and the strategy of optogenetic therapy therefore holds great potential as a universal treatment approach. It aims to target the surviving cell populations of the retina, which remain largely functional despite the structural changes that occur (Jones et al., 2016; Pfeiffer et al., 2020) and convert them to become light-sensitive through the provision of a light-sensitive opsin protein. The disease state of a given patient will determine the cell types that would most benefit from being targeted. For example, loss of rod photoreceptors occurs before cone photoreceptor cells, which can continue to survive for some time despite changes to their structure (Milam et al., 1998; Banin et al., 1999). In this scenario, encouraging light sensitivity in these residual cell types may prove beneficial. However, it may be the case that such cells are too few in number or too diseased to function following gene therapy and therefore the secondary layer of neurons, the bipolar cells, might prove a better target. In a healthy retina, the bipolar cells transfer the light signals received from photoreceptor cells to amacrine and retinal ganglion cells (Figure 1). Targeting these secondary neurons may therefore provide more opportunity for visual processing via the interconnected pathways than targeting the inner most cells of the retinal ganglion layer. Finally, in some instances of severe degeneration and retina remodeling, the retinal ganglion cells may be the desired cell targets. Hence there is justification for targeting different cell types of the degenerate retina for optogenetic therapy, which will be discussed further within this review.
Relevant Models for Optogenetic Therapy
Developing optogenetic therapies requires pre-clinical testing in relevant in vivo and ex vivo models of inherited retinal disease (Table 1). The selection of model when investigating optogenetic therapies is critical to understanding their translational potential and may impact the degree of therapeutic efficacy observed. The efficacy of a particular optogenetic sensor may thus depend on the level of expression achieved in the target cell, which in turn can depend on the study model used. For example, studies with AAV-based reporter vectors indicate that cellular transduction profiles can vary depending on the mouse model used (Charbel Issa et al., 2013; van Wyk et al., 2017). Specifically, there are differences in transduction profiles achieved in healthy, wild-type retinas compared to disease models (Kolstad et al., 2010) or those with compromised barriers (Vacca et al., 2013). In addition, the stages of the disease in humans are mimicked in various animal models, both naturally occurring and transgenic in origin, and whilst preliminary data are often achieved in rd1 mice for testing in eyes that reflect a late-stage degenerate retina, subsequent efficacy and long-term approaches may be better suited in a variety of models with different genetic origins and rates of disease. This would provide an indication on the potential and scope of optogenetic strategies, which are hoped to have universal application across patients with different causative mutations. The progression of retinal disease changes the global structure and function of the retina but at a cellular level, the genetic origin will impact the function of a given cell type. It may be that expanded testing across different mouse models, and indeed larger animal models, will confirm the promise of optogenetic strategies or provide evidence of preferred optogene selection for particular groups of patients. Current studies tend to be limited to testing in a single model when the scope of the field could be expanded by testing across multiple models. Different genes mean different mechanisms for disease and though the principle of optogenetics is to act as a universal approach, it needs to be confirmed that this is feasible.
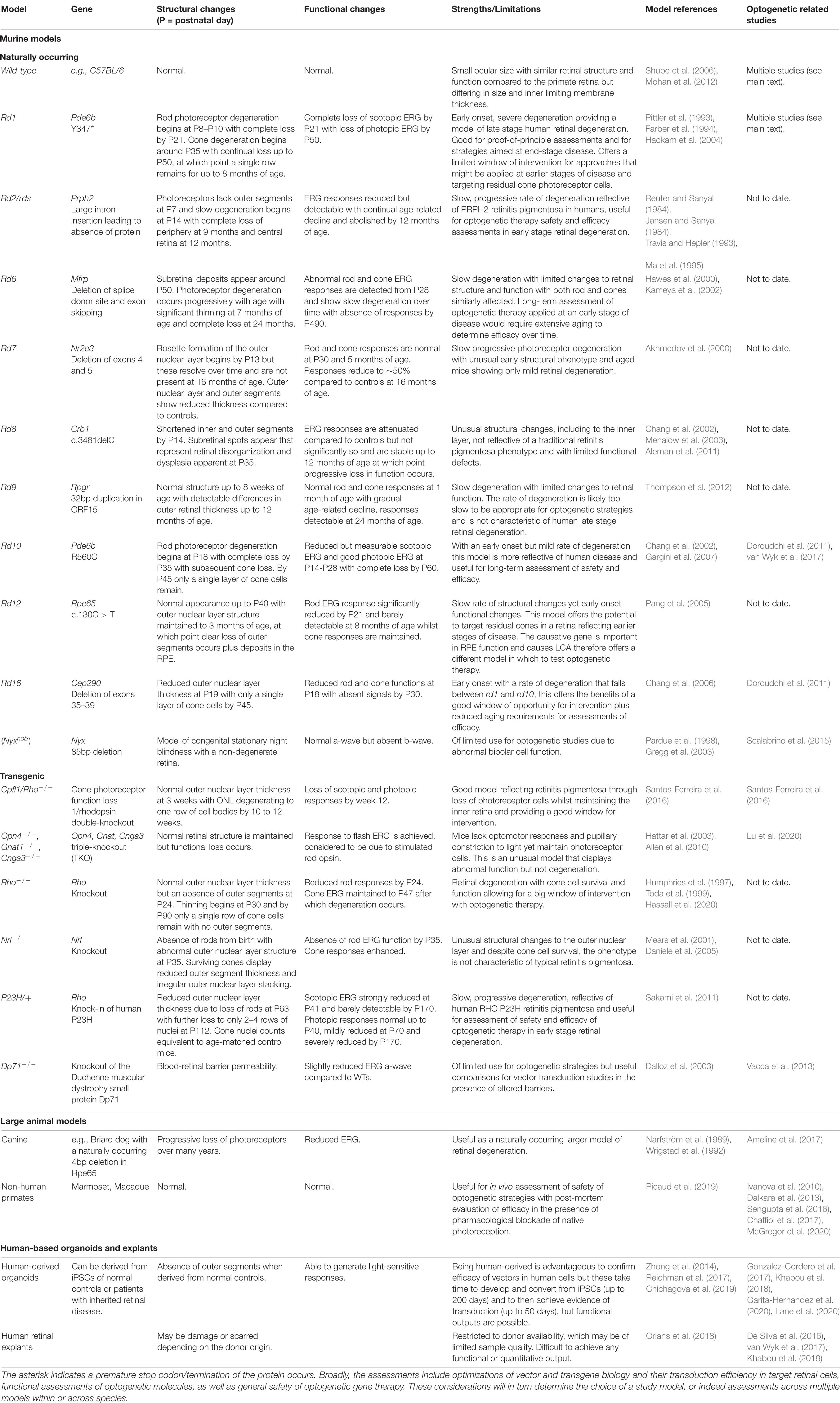
Table 1. Summary of models that can be used in the pre-clinical assessment of optogenetic gene therapies.
Commonly Used Murine Models of Inherited Retinal Degeneration
The most historic model of inherited retinal disease is the naturally occurring rd1 (Farber et al., 1994). Resulting from a mutation in the photoreceptor-specific gene Pde6b, this model is well characterized and displays a fast rate of degeneration in which most rod photoreceptor cells are lost by post-natal day 18 while the inner neural retinal structure is maintained with subsequent functional and morphological changes occurring over time (Strettoi et al., 2002). This rate of change is not equivalent to human disease, yet it is the most commonly used model for pre-clinical testing of optogenetic therapies. This is largely due to the fact that in a short time frame the mouse displays a retina lacking photoreceptor cells and that structurally reflects the later stages of human disease. The drawback of this rapid loss of photoreceptor cells is that it makes the rd1 model less appropriate for testing optogenetic strategies aiming to rescue function from residual cone photoreceptor cells. Whereas cone photoreceptor cells can survive for many years following the loss of rod photoreceptor cells in human presentations of inherited retinal disease (Milam et al., 1998; Banin et al., 1999) and are therefore a potential target of optogenetic therapy, testing such an approach in rd1 mice may be of limited use as they maintain only a single row of cone cells at 3 weeks of age. Although despite this, AAV targeting of residual cone photoreceptors with cone-specific promoters has been achieved with reporter EYFP expression observed in rd1 mice up to 8 months of age (Busskamp et al., 2010). While residual cone photoreceptors were observed to survive in the rd1 model, the opportunity for effective optogenetic intervention of cone photoreceptors may be greater in a slower model of degeneration. The health and function of cones in rd1 mice may be compromised due to the early absence of rods, slower models of retinal degeneration should suffer less from these issues and therefore better reflect the degenerative status of the neural retina in human patients.
The rd10 mouse offers an alternative slower model of retinal degeneration but also results from a mutation in Pde6b, although in this case it is a missense mutation that enables partial activity of Pde6b (Gargini et al., 2007). Though less commonly used than the rd1 model, optogenetic strategies have been tested in rd10 mice (Doroudchi et al., 2011; van Wyk et al., 2017). Whilst this model presents an early onset of retinal changes, the severity of disease is reduced compared to rd1 and the rate of progression slower, providing greater opportunity for optogenetic intervention and long-term assessment. Other models of retinal degeneration offer similar opportunities, such as the naturally occurring rd12, rd16 and the transgenic models Rho–/–, RhoP23H/+, and Cpfl1/Rho–/–. Consideration of the structural and functional features of retinal degeneration and the equivalent stage of human disease are important when assessing optogenetic therapy success. Another factor for consideration in pre-clinical testing in models of retinal degeneration is that one argument for use of optogenetic therapy is that the same vector may be applied to patients with various genetic origins of disease. To this end, initial testing may be best achieved in the rd1 model with subsequent assessments performed in slower models of disease. Presentation of efficacy across different models of retinal disease would provide the strongest evidence of optogenetic success. Table 1 includes various models used in optogenetic studies to date and other potential models that could be explored in future studies.
Large Animal Models
While murine models are commonly used in pre-clinical testing, large animal models such as pigs, dogs and non-human primates, have become increasingly necessary to investigate the safety and efficacy of potential optogenetic therapies prior to use in human trials. There are several important differences in ocular anatomy and physiology that make larger animal models more advantageous (Figure 2). First, the ocular size in a larger model enables investigations of intraocular vector delivery and development of surgical delivery techniques that would not be possible in the much smaller murine eye. In addition, the dimensions of the canine and non-human primate eyes enable intravitreal vector delivery that would have a comparable dilution effect to the pediatric human eye. Importantly, larger animals and specifically the primate retina has a thicker inner limiting membrane, a physical barrier separating the vitreous from the neural retina. In addition, canine and primate immune responses are likely to differ from murine models. These critical inter-species differences need to be taken into consideration when developing optogenetic vectors with ability to penetrate retinal cells from the vitreous.
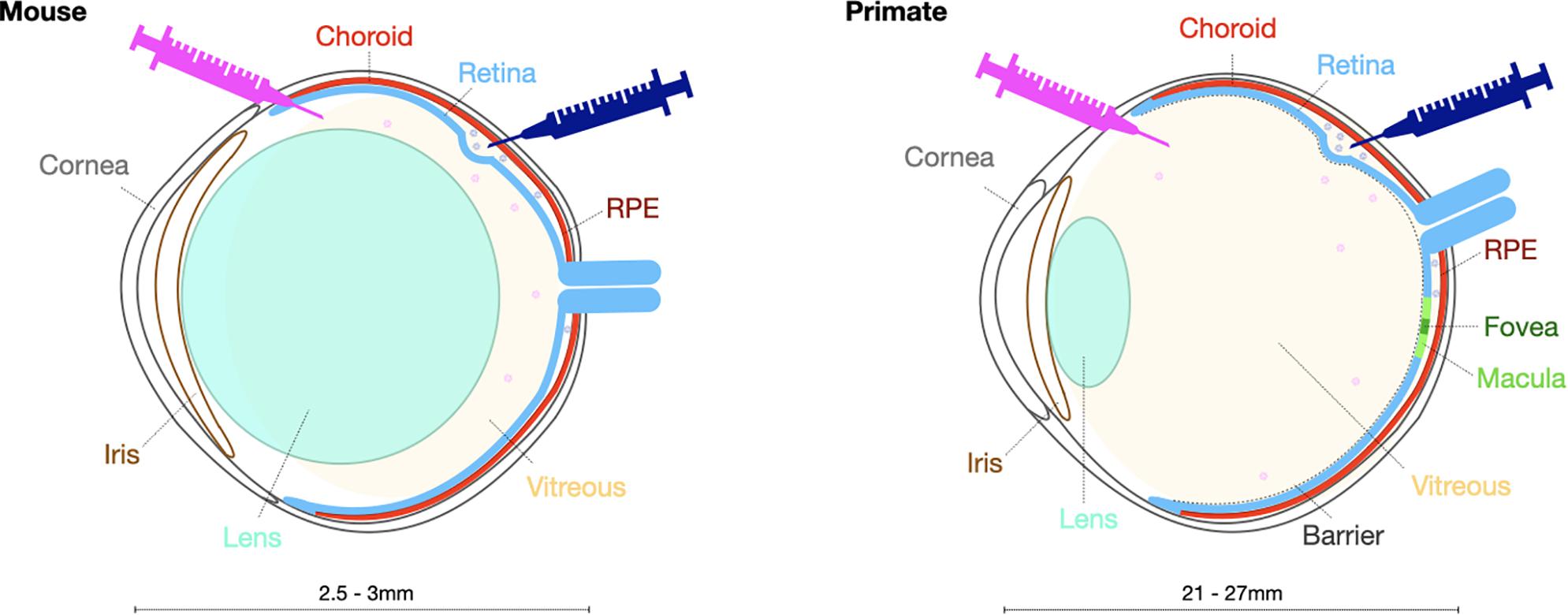
Figure 2. Schematic presentation highlighting key structural differences in the mouse and primate eye to be taken into account when selecting the surgical delivery method.
In contrast to non-human primates, there are naturally occurring canine models of retinal degeneration, where progressive retinal atrophy secondary to mutations in, for example, RPE65, XLPRA1 or PDE6β genes, displays great phenotypic similarities with equivalent retinal degeneration in humans (Beltran, 2009). On the other hand, the macula is only present in primates and has different cellular composition and anatomical arrangement, especially at the cone rich fovea where retinal ganglion cells and bipolar cells are displaced laterally. Canines nevertheless possess “area centralis” a central region of the retina with an increased density of cone photoreceptors. These important differences need to be considered when developing treatments for retinal degenerations that involve the macula, both in terms of cellular transduction profiles and functional assessments of ectopically expressed optogenetic molecules.
Several pre-clinical optogenetic studies have used canine and non-human primate models to assess the safety and expression profile of optogenetic vectors (Ivanova et al., 2010; Sengupta et al., 2016; Ameline et al., 2017) as well as the function and characteristics of optogenetic tools in terms of their ability to restore vision (Chaffiol et al., 2017; McGregor et al., 2020). Compared to murine models, limited in vivo (Ivanova et al., 2010; Chaffiol et al., 2017; McGregor et al., 2020) and ex-vivo (Sengupta et al., 2016) transduction of retinal ganglion cells was demonstrated in marmoset and macaque retinae, respectively, following delivery of AAV encoding microbial opsin-based sensors, with intra-ocular inflammatory responses observed in treated eyes (Chaffiol et al., 2017). In addition, despite long-term expression, similar findings of limited expression were observed in a canine model following treatment with a microbial and a human opsin (Ameline et al., 2017). Even the “state-of-the art” AAV vectors evolved for improved transduction in murine models (as will be discussed later in the review), demonstrate expression that is limited to a central para-foveal ring of retinal ganglion cells in a macaque retina following intraocular application (Dalkara et al., 2013). These results thus highlight the importance of testing optogenetic systems in primate models with outcomes most likely to mimic those of human subjects. However, these primate retinae are wild-type healthy retinae and not affected by retinal degeneration and remodeling that are likely to influence type of cell transduced and level of transgene expression within the cell. Additionally, healthy non-human primate retinae with a full set of photoreceptors make any functional electrophysiological or behavioral assessments of optogenetic therapies very challenging, although in vitro electrophysiology evaluation is achievable following pharmacological blockade of native photoreception (Chaffiol et al., 2017).
Human-Derived Retinal Organoids
Use of model organisms serve several purposes and the benefits of in vivo testing are many, particularly in the field of optogenetics where assessment of visual function and processing can be achieved. However, it is important to consider the benefits of other models in pre-clinical testing. In recent years, protocols for generating retinal organoids from human iPSCs have been developed, which enable formation of cultured structures derived from human cells that reflect the cell types and organization of a neural retina (Zhong et al., 2014; Reichman et al., 2017; Quinn et al., 2018). These retinal organoids provide an incredibly useful tool not only for optogenetic therapy pre-clinical testing but also in general for the field of retinal gene therapy. As described above, not all animal models replicate effectively human disease states therefore testing of gene therapy vectors can be challenging. For example, the Rp2–/– mouse model poorly replicates human retinal disease caused by absence of RP2 function. Generation of retinal organoids from patient-derived samples was recently described and these proved a useful in vitro model for assessing RP2 gene therapy rescue (Lane et al., 2020). For optogenetic strategies, retinal organoids also provide an interesting model for pre-clinical testing. Whilst they do not perfectly replicate the neural retina, distinctive layers of different cell types have been shown across different research groups using similar protocols (Zhong et al., 2014; Reichman et al., 2017; Quinn et al., 2018; Chichagova et al., 2019). A consistent feature across all protocols appears to be a lack of photoreceptor outer segment structures, but this does not pose a problem for testing of optogenetic vectors as indeed it mimics the absence of such structures in retinitis pigmentosa.
The initial challenge of testing any gene therapy strategy is to achieve transduction of the desired cell types. Vector tropisms and photoreceptor-specific promoters have been tested in retinal organoids (Gonzalez-Cordero et al., 2017; Garita-Hernandez et al., 2020). Current data suggest a large number of AAV capsid serotypes offer relatively poor transduction efficiencies (2–20%), including AAV2, ShH10, AAV5, AAV8, AAV8 Y733F, and AAV9 with the most encouraging variant appearing to be AAV2(7m8), achieving up to 60% transduction efficiency (Gonzalez-Cordero et al., 2017; Khabou et al., 2018; Garita-Hernandez et al., 2020). Differentiation of retinal organoids is a long process and can take up to 140 days with vector application possible during early stages of the differentiation process (Garita-Hernandez et al., 2020) or at the later stages of differentiation (Lane et al., 2020). The extensive protocols and long experimental time frames required for retinal organoid differentiation and the subsequent transduction reflect the inhibitory aspects of using these for pre-clinical testing, but the benefits of testing in human-derived samples may outweigh such issues.
A successful optogenetic strategy will depend on achieving opsin localization in the membrane of the targeted cell type whilst avoiding build-up inside the cell. It has been indicated that while delivery and expression of a variety of different microbial opsins can be achieved in retinal organoids, not all localized well to the membrane with endoplasmic reticulum accumulation and unfolded protein responses mechanisms observed (Garita-Hernandez et al., 2020). For the opsin variants that trafficked well to the membrane of targeted photoreceptor cells, functional activation was achieved. In addition, any mutagenic changes within protein structures aimed to improve functional properties of optogenetic molecules (e.g., making them more light sensitive) may have deleterious effect on protein trafficking and expression within cell membrane. In general, human cone opsin mutations can lead to accumulation within the cells and an absence of functional response (McClements et al., 2013) and rhodopsin mutations may impair folding and trafficking and lead to cell death (Athanasiou et al., 2017). While pre-clinical testing of optogenetic strategies in retinal organoids is still a relatively new technique, the data achieved so far indicate it offers an exciting addition to the development and assessment of such therapies.
Human Retinal Explants
A final model for consideration when conducting pre-clinical testing involves the use of human retinal explants. These can be obtained and cultured post-mortem (van Wyk et al., 2017; Khabou et al., 2018) or following an emergency retinectomy (De Silva et al., 2016) and may be derived from human or non-human primates (Hickey et al., 2017). Whilst useful as a screening tool to observe successful transduction of human cells, the health of such explants can be problematic and maintaining survival in culture long enough to observe successful transduction can be an issue. Additionally, the cell types that can be transduced in human retinal explants appear to be limited with no detectable expression observed in ON-bipolar cells, most likely due to downregulation of bipolar cell specific gene expression in the explant system (van Wyk et al., 2017). With human retinal explants unlikely to provide any quantitative or functional outputs following gene therapy treatment, retinal organoids may provide a more appropriate model for in vitro testing of optogenetic vectors.
Cell-Specific Targeting in the Degenerate Retina
Vector Considerations
While optogenetic strategies are not intended to be restricted by the original genetic cause of degeneration, the stage of disease will influence the type of suitable vector and hence key features of vector design. To be successful, optogenetic therapy requires expression of a light-sensitive opsin molecule in the membranes of the surviving cells of the retina and achieving this relies on efficient delivery and expression of an optogenetic transgene to target cells. AAV vectors have been the primary vector of choice over the past decade and several AAV vectors have now been used in numerous clinical trials for a wide variety of retinal diseases (Bainbridge et al., 2015; Russell et al., 2017; Xue et al., 2018; Cehajic-Kapetanovic et al., 2020b), the majority being single gene diseases. Indeed, the first approved human AAV gene therapy (Luxturna) is for the treatment of an inherited retinal disease, Leber congenital amaurosis. While systemic delivery of AAV vectors has proved more challenging and recently resulted in the tragic deaths of trial participants (Wilson and Flotte, 2020), their use in the eye has been shown to cause minimal adverse events in clinical trials (Nuzbrokh et al., 2020). While lentiviral vectors can struggle to achieve good transduction efficiency and with transgene expression typically only observed around the site of injection (Balaggan and Ali, 2012; Puppo et al., 2014), AAV delivery can achieve transduction patterns exceeding the area of the bleb (Boye et al., 2016; Yiu et al., 2020). AAV vectors also offer flexibility on the delivery route used, namely intravitreal or subretinal (Figures 2, 3).
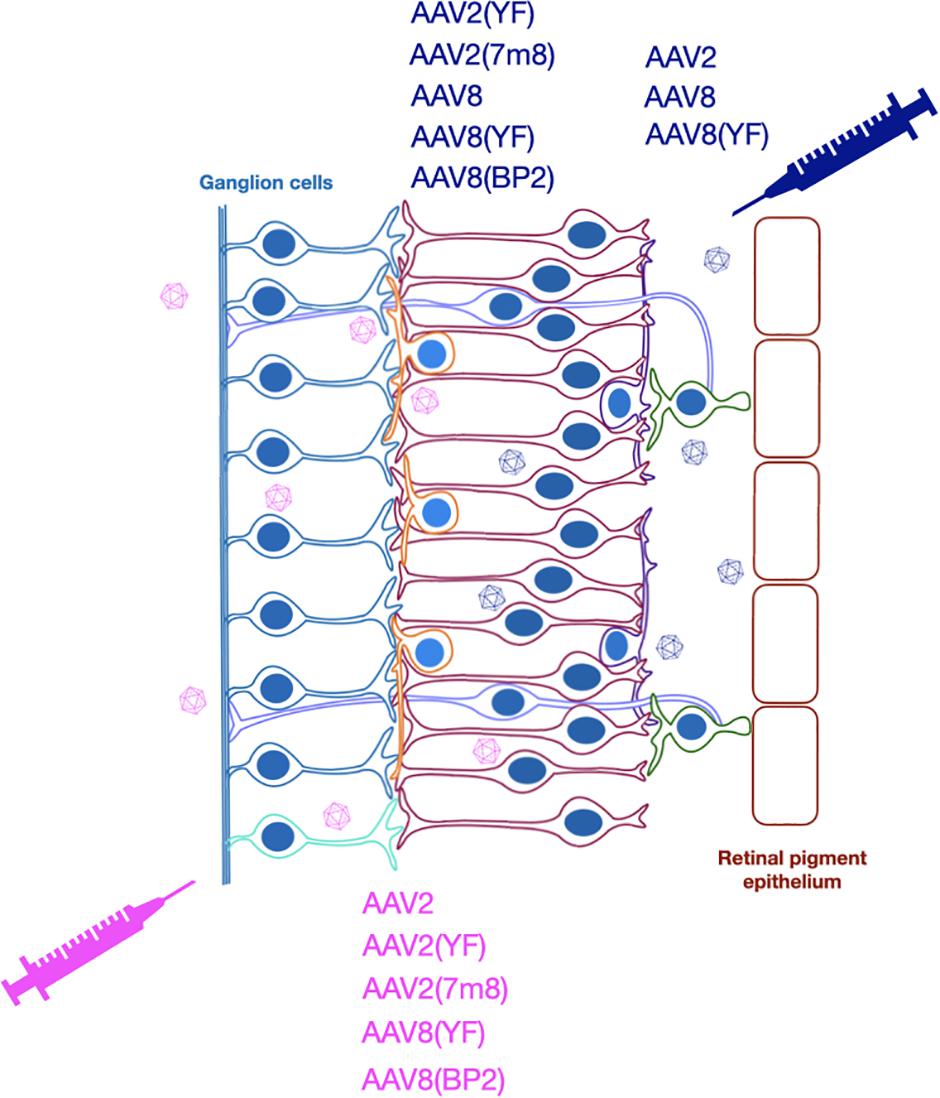
Figure 3. Optogenetic strategies have used subretinal or intravitreal injection to deliver various AAV vectors for expression of different opsins.
Currently, the main limitations of AAV vector use for treating inherited retinal disease appear to be their packaging capacity and ability to favor transduction of particular cell types. For optogenetic transgenes, the packaging capacity is not likely to be an issue as opsin coding sequences are relatively small compared to genes used in gene supplementation strategies, such as RPGR (Fischer et al., 2017). However, AAV vectors for optogenetic strategies have benefited in recent years from development of engineered capsid variants. While unnecessary for current clinical trials targeting photoreceptors or cells of the retinal pigment epithelium, for which AAV8 and AAV2 variants have proven effective (Xue et al., 2018; Cehajic-Kapetanovic et al., 2020b), the requirement of optogenetic therapies to target residual cone photoreceptors, inner retinal layers and the outer ganglion cell layer has required investigation of new capsid variants. As previously discussed, cone photoreceptor cells can survive for many years after the loss of the rod photoreceptor cells and despite the loss of outer segments, and therefore light sensitivity (Milam et al., 1998; Banin et al., 1999), they are nevertheless considered an optogenetic target. Historically, efficient transduction in cones has proven difficult relative to rod photoreceptors (Mussolino et al., 2011; Boye et al., 2012) but recent studies with new capsid variants have increased potency (Khabou et al., 2018). Successful reporter transgene expression was shown in the cone photoreceptors of rd10 mice as well as retinal organoids, human post-mortem explants and in non-human primates (Khabou et al., 2018). However, despite the improvements in cone photoreceptor targeting abilities for pre-clinical testing, these cell types will not be appropriate targets for all cases of late stage retinitis pigmentosa. Although the cones may survive, targeting a cell of compromised health and structure may not provide the best long-term option and indeed in some patients, they may not maintain enough surviving cones for optogenetic therapy of these cells to be worthwhile. Delivery of optogenetic transgenes to residual cone photoreceptors in rd1 mice enabled restoration of visual function (Busskamp et al., 2010) and while this is encouraging, it should be considered that the pattern of cone density in mice is different to humans. In humans, cone photoreceptors are enriched in the macula region and reduce in density outwardly from this point, therefore targeting such cells will make use only of a small central region of the retina.
Optogenetic strategies that target the inner layer of the retina or ganglion cell layer have potential to enable a broader area of induced photosensitivity. Achieving efficient transduction of the inner retinal layers has largely relied on engineered and novel AAV capsid serotypes as opposed to wild type forms that have struggled to transduce the inner retina (Charbel Issa et al., 2013). Increased tropism and transduction success of the retinal layers has been achieved by mutation of multiple surface tyrosine (Y) residues to phenylalanine (F). While single residue Y to F changes do not effectively enable inner retinal targeting (De Silva et al., 2016), multiple residue mutations in AAV2(2–4YF) have shown more success in both in wild type mice (Petrs-Silva et al., 2011) and mice with retinal degeneration (Gaub et al., 2015). Furthermore, a directed evolution approach has created the AAV2(7m8) variant featuring a seven amino acid insertion in capsid protein VP1 (Dalkara et al., 2013). The AAV2(4YF) and AAV2(7m8) variants provided similar transduction profiles when directly compared following subretinal and intravitreal injection in the rd1 mouse and included transduction of the inner retina (Hickey et al., 2017). However, expression in bipolar cells was not robust, indicating that while successful, further improvements in transduction may be required.
A particularly promising AAV capsid option for optogenetic approaches targeting the inner retina is the AAV8(BP2) variant. This was created by directed selection of a library of AAV8 capsid mutants focused on a variable region of nine amino acids, considered important for receptor attachment and transduction (Cronin et al., 2014). This AAV8(BP2) capsid provided good transduction of ON-bipolar cells, including limited expression in non-diseased human retinal explants with a strong pan-neuronal CMV promoter. This was the first demonstration of robust bipolar cell transduction from an AAV vector but transduction in a wild type mouse does not necessarily translate to degenerate retina or indeed non-human primate and human retina. Further confirmation of human bipolar cell transduction was provided in a later study using human retinal explants that also showed targeting of cone ON-bipolar cells by a reporter AAV8(BP2) vector (van Wyk et al., 2017). This study showed comparable human bipolar cell expression patterns were achieved from both AAV8(BP2) and AAV2(7m8) vectors and also performed similarly in the degenerate 11-week-old rd10 mice. Such developments in capsids have advanced the potential of optogenetic strategies by enabling transduction in bipolar cells.
Transduction of the innermost layer of the retina, the retinal ganglion cells has been readily achieved in mouse models following intravitreal injection (Yin et al., 2011; Smith and Chauhan, 2018). Wild type AAV capsid transduction appears limited to the retinal ganglion cells unless applying an additional adjunctive (Cehajic-Kapetanovic et al., 2011, 2018) whereas modified AAV capsids penetrate further following intravitreal injection into the inner retina (Kay et al., 2013; Hickey et al., 2017; van Wyk et al., 2017).
Transgene Considerations
While the first hurdle in any gene therapy strategy is getting the AAV to enter the desired cell type, transgene design can influence the success of subsequent expression. In addition to being difficult to target with AAV, cone photoreceptor cells have also proved difficult in the development of efficient cell-specific promoters (Li et al., 2008) but the development of a 1.7kb version of the human red/green cone opsin promoter, known as PR1.7, was shown to achieve robust and selective expression in cone photoreceptor cells (Ye et al., 2016; Khabou et al., 2018). Utilizing the AAV2(7m8) capsid and an intravitreal injection route, the latter research group found the PR1.7 promoter to be more specific than the mouse cone arrestin (mCAR) promoter, which resulted in expression in both rod and cone photoreceptor cells. Cell-specific promoters are often considered to pose a risk in gene therapy strategies in case of changes in gene expression profiles in degenerating cells. This may be particularly concerning for optogenetic strategies targeting residual cone photoreceptor cells, which have a changing profile of expression, function and morphology over time (Hassall et al., 2020). Despite these concerns, successful expression from the PR1.7 promoter was observed 2 months post-injection in rd10 mice though how this might translate to humans should be approached with caution.
Following loss of photoreceptors in the degenerating retina, the secondary neurons of the retina, the horizontal and the bipolar cells are considered as the next best option for optogenetic approaches. Transducing the distal retinal circuitry has greater chance of preserving upstream retinal processing, and the ON-bipolar cell-specific expression is thought to be particularly important in achieving this. In a normal retina, the bipolar cells transfer the light signals received from photoreceptor cells to amacrine and retinal ganglion cells (Figure 1). Morphological comparisons have determined there to be just one rod-bipolar cell type but multiple cone-bipolar cell types (Wässle et al., 2009) and these fall into two broad functional categories of ON (rod and cone) and OFF (cone only) (Nelson and Connaughton, 2007). The ON-bipolar cells are depolarized by light-stimulated photoreceptor cells whilst OFF-bipolar cells are hyperpolarized in the same receptive field. Selective expression of opsins in ON-bipolar cells is considered important to avoid interfering with the complex interconnected signaling pathways of the remaining neural retina. Making both ON- and OFF-bipolar cells respond to light in the same way could result in ambiguous signals, although cortical plasticity could play a role in filtering out the noise, as seen with subretinal implants (Cehajic-Kapetanovic et al., 2020a). A combination of effective AAV transduction and promoter for selective expression of opsins in ON-bipolar cells are therefore highly desirable features of optogenetic therapies.
To date, 4xGrm6-SV40 is the most studied promoter for optogenetic strategies aiming to achieve opsin expression in ON-bipolar cells. The metabotropic glutamate receptor type 6 (mGluR6) receptor was found to be associated with ON-bipolar cell activity (Nakajima et al., 1993; Nomura et al., 1994). A critical 200bp enhancer region in the mouse Grm6 gene that encodes mGluR6 was subsequently paired with the SV40 promoter to achieve ON-bipolar cell specific expression of a reporter gene (Kim et al., 2008). This has since been used with success by other research groups in their optogenetic approaches (Lagali et al., 2008; Doroudchi et al., 2011; Cehajic-Kapetanovic et al., 2015; Macé et al., 2015). A comparison of multiple copies of the Grm6 enhancer identified that including four copies prior to the SV40 promoter improved reporter gene expression (Cronin et al., 2014). Whilst this promoter has been shown to primarily enable transgene expression in ON-bipolar cells, expression has been identified in other cell types (van Wyk et al., 2015, 2017). The expression profile of an optogenetic transgene with fluorescent reporter revealed predominant expression in amacrine cells and a lack of ON-bipolar cell targeting in rd1 mice. In wild type mice, amacrine cells were also the most targeted cell type when using the AAV2(7m8) yet when the AAV8(BP2) capsid was used, multiple cell types revealed reporter transgene expression, including ON-bipolar cells. Interestingly, a similar expression profile was then observed in rd10 mice yet not in rd1 mice, even when injected at earlier age. This highlights the consideration raised earlier regarding the need to use multiple models of retinal degeneration when assessing a given optogenetic therapy. Furthermore, the difference in expression profile from a given promoter varies between mouse models and is to an extent dependent on other vector selections, such as the capsid.
Use of an extended mGluR6 promoter containing additional intronic sequence enabled enhanced reporter gene expression in ON-bipolar cells in mice and non-human primates (Lu et al., 2016). However, this promoter variant is considerably longer than the commonly used variant and therefore potentially less desirable as this can be limiting to transgene design. Transgenes for AAV vectors cannot exceed the packaging capacity of AAV, which is typically ∼4.8 kb. The majority of opsin coding sequences are relatively small (∼2 kb) enabling room for a larger promoter. However, other transgene elements may be desirable to enhance expression, such as inclusion of a WPRE (Patrício et al., 2017) or fluorescent marker to create a fusion protein. Additional membrane trafficking signals may also be required (discussed later within opsin considerations). Therefore, the larger mGluR6 promoter may not prove to be a limiting factor in its future use in optogenetic transgenes but it will be dependent on other factors of transgene design that may prove critical for a given optogenetic strategy.
A mini-promoter named Ple155 (derived from the PCP2 gene) has also been shown to provide ON-bipolar cell expression in wild-type mice (De Leeuw et al., 2014) and was used to restore vision in a mouse model (Nyx–/–) of congenital stationary night blindness and non-degenerated retina (Scalabrino et al., 2015). Whilst the Nyx–/– mouse model is not of relevance to optogenetic strategies, NYX is a bipolar cell-specific gene and is therefore of interest in its use as an alternative ON-bipolar specific promoter. Interestingly, ON-bipolar cell specific expression was achieved only when using the Ple155 promoter in combination with an AAV2 (and not AAV8) vector and only in very young mice (at P2 and not at P30) with undifferentiated bipolar cells. It therefore remains to be seen whether the Ple155 promoter offers any advantage in ON-bipolar cell targeting over the 4xGrm6-SV40 in degenerate retina and whether ON-bipolar cell transduction can be achieved with either promoter in non-human primate or indeed in human retina.
Targeting of bipolar cells may be preferable in the earlier stages of retinal degeneration but in later stages of disease, these cells can become disordered and also degenerate (Pfeiffer et al., 2020). These changes include alterations in the neural connections made between cell types of the neural retina structure but also their morphology and general structural layout, Figure 1. Not all patients will present in clinics at the earlier stages of disease, therefore some optogenetic approaches aim to target the retinal ganglion cells, which remain most resistant to degeneration retaining their physiological properties and laminar location in the retina. In mouse models, this is achieved relatively easily with multiple AAV serotypes by intravitreal injection and gene expression can be restricted using the synapsin-1 promoter (Syn1) (Sengupta et al., 2016) or the neurofilament heavy polypeptide promoter (Nefh) (Hanlon et al., 2017). Given the preference for transduction of ganglion cells when using the intravitreal injection route, ubiquitous promoters have also been used with success (Bi et al., 2006; Bin Lin et al., 2008; Zhang et al., 2009; Liu et al., 2016). However, the risks of introducing light-sensitive responses to more than one cell type are currently unknown and may interfere with the interconnectivity of the complex neural circuits of the retina.
Opsins for Optogenetic Therapy
The opsins used for optogenetic approaches include both microbial and vertebrate varieties (Figure 4). These have different characteristics and vary in many ways including their sensitivity to light, recovery rates and in the type of response they elicit (Simon et al., 2020). Opsins are transmembrane proteins that absorb light and in the case of vertebrate opsins, they are G-protein coupled receptors whose activation leads to cGMP-gated cation channels closing and a subsequent hyperpolarization of the photoreceptor cell (Figure 4). In contrast, microbial opsins are light-activated ion channels rather than G-protein coupled receptors that can cause depolarization or hyperpolarization depending on the nature of the ion channel. It is for this reason that the microbial opsins have proven popular for optogenetic strategies as they can directly influence the cell polarization in response to light without the need for other G-protein cascade elements. This may prove important in successfully converting cells that were not intended to be light sensitive.
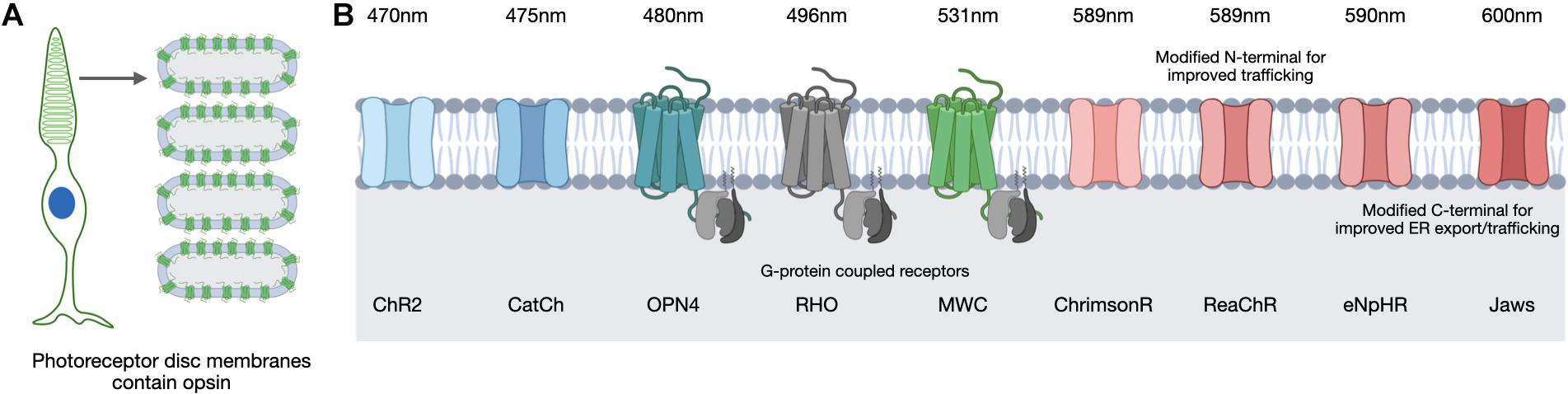
Figure 4. In the human retina, light-sensitive opsins for vision are located in the photoreceptor cells on the membranes of specialized discs (A). Optogenetic strategies use multiple opsin variants of microbial and vertebrate origin that are sensitive to different peak wavelengths (B). Images created using BioRender.com.
Opsin selection requires consideration of various factors related to opsin function, including the conductance rate and ion selectivity. For example, microbial opsins exhibit differences in their conductance of cations or anions and in the rate of ion influx achieved upon light activation. Considerations of opsin kinetics are also important as the timing of a channel opening and closing to enable ion influx will influence the extent of a response. Opsins differ in the intensity of the light required to induce a response and the peak wavelength of light they respond to (Figure 4). The intensity of light necessary to induce stimulation is of particular safety importance as the retina can be damaged by certain light intensities and wavelengths (Youssef et al., 2011). Natural light provides a general exposure range of 10–4 to 105 lux and safety thresholds for humans have been defined (The European Parliament and the Council of the European Union, 2006; ICNIRP, 2013; van Norren and Vos, 2015) therefore an optogenetic therapy would need to be successful within these limits. The position of the opsin is also critical in order to be optimally exposed to the light source and in a great enough proportion to achieve an appropriate response without inducing membrane instability or overexpression. Finally, opsins can suffer desensitization, in which they become less responsive to light after repeated stimulation and therefore require recovery time prior to their next light-activation. This is also referred to as bleaching and opsins vary in the extent to which they suffer and recover from this process, for example, rhodopsin of the rod photoreceptors is particularly susceptible and suffers reduced phototransduction capacity following exposure to bright light (Pepperberg, 2003). These features are all important considerations and will be referred to throughout the following discussion of opsin candidates for optogenetic therapeutic approaches.
Microbial Opsins
The first microbial opsin identified for use as an optogenetic tool was channelrhodopsin-2 (ChR2) (Boyden et al., 2005), which causes entry of cations into the cell resulting in depolarization. It has been the most commonly used microbial opsin to date and is induced by blue light (470 nm). It was used in the first pre-clinical optogenetic strategy for rescuing visual function to the rd1 mouse (Bi et al., 2006) and a subsequent humanized version (containing an H143R mutation) fused to GFP has been stably expressed using the Grm6-SV40 promoter in rd1 mice following subretinal injection (Doroudchi et al., 2011) and intravitreal injection (Macé et al., 2015). In the latter approach, both ON and OFF visual responses were detected at 4–10 weeks post-injection, indicating its potential for optogenetic therapy.
An engineered variant of ChR2 carrying an L132C mutation was developed and named CatCh as it is a calcium translocating channelrhodopsin and has nearly six-fold enhanced Ca2+ permeability relative to ChR2, which improved its sensitivity to light and kinetics (Kleinlogel et al., 2011). An optogenetic transgene containing an EGFP fusion of this protein was expressed in ON-bipolar cells in rd1 mice and incurred light-induced ganglion cell responses 4 weeks post-injection (Cronin et al., 2014). A red-light activated depolarizing ChR (ReaChR) was generated with a peak spectral sensitivity of 590nm and this ReaChR included an N-terminal sequence to aid membrane trafficking (Lin et al., 2013). When expressed specifically in retinal ganglion cells using the SYN1 promoter following AAV2 intravitreal delivery in rd1 mice, visual and behavioral responses to light were observed at light intensities within safety thresholds (Sengupta et al., 2016). An algal-derived variant similar to ReaChR is ChrimsonR (Klapoetke et al., 2014). Comparisons of ReaChR and ChrimsonR localization and function in human retinal organoids revealed better trafficking of ReaChR to the cell membrane than ChrimsonR but both provided detectable responses to light stimulation (Garita-Hernandez et al., 2018). Despite the reduced trafficking efficiency, ChrimsonR did not accumulate excessively in the cell organelles or co-localize with markers of endoplasmic reticulum retention or the unfolded protein response.
Other microbial opsins being used for optogenetic strategies are light-activated hyperpolarizing chloride pumps, of which halorhodopsin (NpHR) was the first to be reported and is sensitive to yellow light (Zhang et al., 2007). Hyperpolarizing opsins are of particular interest in optogenetic strategies targeting surviving cone photoreceptor cells, which as described above, can continue to exist in a dormant state in the degenerate retina despite loss of cone-mediated light responses (Lin et al., 2009; Busskamp et al., 2010). NpHR has a peak wavelength sensitivity of 589 nm and light activation leads to hyperpolarization of the cell, mimicking the native response of a photoreceptor cell. However, this protein was found to accumulate and form unwanted aggregates when expressed at high levels (Gradinaru et al., 2007) so an enhanced version, eNpHR, was created (Gradinaru et al., 2008). The addition of an N-terminal signal peptide and a C-terminal endoplasmic reticulum export signal improved the membrane localization of this hyperpolarizing opsin. AAV delivery of eNpHR using photoreceptor-specific promoters achieved electrophysiological responses to light in rd1 mice equivalent to those in wild type mice (Busskamp et al., 2010). Further improvements to eNpHR were made by way of additional trafficking signals to provide variant eNpHR 3.0 (Gradinaru et al., 2010).
Despite the addition of these signals, testing in vitro did not reveal membrane localization, however, testing in human retinal organoids did achieve membrane localization and subsequent light responses (Garita-Hernandez et al., 2018). However, despite the addition of export and localization signals, these comparisons in human retinal organoids indicated the hyperpolarizing opsin Jaws showed better membrane localization relative to eNpHR 3.0. Jaws is an engineered chloride pump cruxhalorhodopsin that is red-shifted by 14 nm relative to the eNpHR variants and therefore has a peak sensitivity of 600 nm (Chuong et al., 2014). AAV delivery in rd1 mice of a Jaws transgene driven by the mCAR promoter achieved expression 4 weeks post-injection and achieved spike responses from isolated ON- and OFF-retinal ganglion cells following light stimulation at 600 nm (Chuong et al., 2014). It was also observed that responses could be achieved from 550 nm (green) and 470 nm (blue) wavelengths and that Jaws provided more retinal ganglion cell spiking than eNpHR over a broader light spectrum, which suggest Jaws may be more suitable for use in human optogenetic therapy.
The eNpHR 2.0 variant has been used in combination with a human rhodopsin promoter to achieve expression in rod photoreceptors of wild type mice, which were then harvested and the eNpHR 2.0-expressing rod photoreceptor cells subsequently isolated and injected into Cpfl1/Rho–/– female mice (Garita-Hernandez et al., 2019). Responses to 580 nm light were achieved as were spike recordings of ON- and OFF-retinal ganglion cells, indicating connections were made between the transplanted cells and the surviving secondary neurons of the host retina. Encouragingly, light intensities within the safe limits defined for the human eye successfully stimulated responses and light avoidance behavior in treated mice was also observed. This same study also induced Jaws expression from the mCAR promoter in human iPSC-derived cone photoreceptor cells, which were then injected into Cpfl1/Rho–/– and rd1 mice. Robust photocurrents were achieved, which peaked when stimulated with a wavelength of 575 nm. As for eNpHR stimulation, responses were achieved from light intensities below safety thresholds and both ON- and OFF-retinal ganglion cell responses were achieved. Jaws has also been shown to have good membrane localization in cone photoreceptor cells of non-human primates following delivery of an AAV9(7m8) vector with the PR1.7 promoter (Khabou et al., 2018). These studies highlight the potential for both eNpHR and, in particular, Jaws to be used in future human clinical trials for optogenetic stimulation of residual cone cells.
Vertebrate Opsins
Despite the encouraging data from studies using microbial opsins use of a native human opsin may be more desirable as it poses less risk of immune reaction. Both human and mouse retinal cells produce five main opsin variants that respond to different wavelengths of light: rhodopsin, expressed by rod photoreceptor cells (RHO); short-wave cone opsin (SWC), medium-wave cone opsin (MWC), and long-wave cone opsin (LWC), expressed by cone photoreceptor cells; and finally, melanopsin (OPN4). This latter opsin is not essential for vision but instead is an important sensor for the circadian clock and is expressed by a sub-population of intrinsically photosensitive retinal ganglion cells (ipRGCs). One of the first optogenetic studies to use a native opsin for pre-clinical testing in rd1 mice boosted the melanopsin expression with rescue of basic visual functions (Bin Lin et al., 2008). Subsequently, human melanopsin expression in cells of the inner retina of rd1 mice has also been achieved (Bin Lin et al., 2008; Liu et al., 2016; De Silva et al., 2017). These studies reported greater light sensitivity compared to the microbial opsins with rescue of visual responses achieved up to 12 months post-injection (De Silva et al., 2017). A further study created a chimera of mouse melanopsin and the mGluR6 receptor, Opto-mGluR6, under the control of the Grm6-SV40 promoter in order to test the melanopsin kinetics in ON-bipolar cells in a transgenic mouse model (van Wyk et al., 2015). Encouragingly, transduction of only a small fraction of ON-bipolar cells (∼12%) using AAV2(4YF) vector delivery, was sufficient to induce light responses in retinal ganglion cells.
Success in restoring responses to light in the degenerate retina with human opsins have also been achieved with ectopic expression of native rod opsin, rhodopsin. Intravitreal delivery of AAV2 achieved human rhodopsin expression in ON-bipolar cells of rd1 mice and improved visual and behavioral responses 8–12 weeks post-injection including resolution of flicker, coarse spatial patterns and elements of natural scene (Cehajic-Kapetanovic et al., 2015; Eleftheriou et al., 2017). Encouraging data were also achieved with the same transgene in rd1 mice but using an AAV2(4YF) vector (Gaub et al., 2015). The responses of rhodopsin-treated mice were compared to those treated with an identical vector delivering humanized ChR2, and rhodopsin was found to offer greater and more sensitive responses than ChR2. The same study group recently expressed vertebrate MWC in the retinal ganglion cells of rd1 mice (Berry et al., 2019) with behavioral tests showing signs of functional rescue. The speed and sensitivity of response to light of MWC was improved compared to rhodopsin function in ganglion cells, but it remains to be seen how the function of cone photopigments compares to rhodopsin kinetics in ON-bipolar cells. Ultimately, human trials are needed to determine how these ectopically expressed human opsins compare to visual function achieved by native rod and cone opsins in photoreceptor cells.
Discussion
The pre-clinical work to date demonstrates that optogenetic strategies are able to restore vision to the degenerate retina using a multitude of transgene and vector combinations. Development of new capsid varieties has greatly aided transduction success of the inner retina and improved cell-specific expression profiles. Progress has been made in understanding visual and behavioral responses following delivery of a wide variety of opsins in various cell types of the surviving retina in animal models. The major barrier to translation of many of these optogenetic therapies has been the lack of evidence of efficient optogene expression in healthy non-human primate, or indeed degenerate human retina, using the available tools.
None-the-less, both ChR2 and ChrimsonR are currently being tested in Phase I/IIa clinical trials. The NCT02556736 trial describes delivering ChR2 primarily to retinal ganglion cells using AAV2 intravitreal injection in patients with advanced retinitis pigmentosa. This trial was initiated by Retrosense Therapeutics (now part of Allergan) and is an open-label, dose escalation study that began recruiting in 2015 but to date, no data have been released. A more recent trial initiated by GenSight Biologics in 2017 (NCT03326336) also involves intravitreal delivery but of an AAV2(7m8) vector aiming to express the ChrimsonR-tdTomato fusion protein. This PIONEER trial by GenSight is not only of interest because it is the first to use the capsid variant AAV2(7m8) for expression of ChrimsonR-tdTomato, but also because it combines the gene therapy with a specialized wearable visual stimulation device. Given the considerations discussed earlier in this review regarding the microbial opsins, the selection of ChrimsonR instead of ChR2 offers a safer and potentially more sensitive opsin than the first trial using ChR2. The inclusion of tdTomato is intriguing as this is a fluorescent marker used to confirm transgene expression and provides no functional benefit. Typically, such markers are used for pre-clinical studies and are then removed prior to use in human clinical trials. In this particular study design, the confirmation of ChrimsonR expression by detection of tdTomato enables researchers to be confident successful transduction has occurred and that the visual stimulation device can subsequently be used. This is an interesting step forward in gene therapy transgene design for human clinical trials and it may be that such implementation of fluorescent reporters may be used more often in the future. However, caution should always be applied in expressing proteins in human cells if they are not necessarily required to achieve a therapeutic outcome. This year, press releases of preliminary clinical trial safety data have also been provided, suggesting good tolerance of the vector so far (gensight-biologics.com). However, it remains to be seen what the long-term effects of expressing an algal-derived protein on the membranes of retinal cells are and whether these may be detected by the immune system and therefore lead to an undesired immune response against the transgene product. It is clear though that the current dose-escalation study is a vital step forward in providing such treatments to individuals with severe retinal degeneration and limited light perception.
Given the limited light sensitivity of current microbial opsins, the need for an additional device and the potential for immune responses triggered by foreign proteins, it could be that future therapies with human opsins provide a better opportunity for success. It is worth noting that Acucela Inc (now Kubota Vision) are currently developing human rhodopsin optogenetic therapy as are Applied Genetic Technologies Corporation (AGTC) in collaboration with Bionic Sight but the details of the latter treatment are currently unavailable.
In summary, studies to date indicate optogenetic therapies show great therapeutic potential for restoring vision in patients with advanced inherited retinal disease and more human trials are necessary as the next major step in advancing the field. It remains to be seen how the human visual system, affected by degeneration and remodeling, responds to optogenetic stimulation at the level of retinal photoreception and by means of cortical plasticity for improved and meaningful visual perception.
Author Contributions
MM drafted, edited, and created figures for the manuscript. FS reviewed and edited the manuscript. RM provided funding and critical review of the manuscript. JC-K initiated the manuscript and provided critical review. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the NIHR Oxford Biomedical Research Centre.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akhmedov, N. B., Piriev, N. I., Chang, B., Rapoport, A. L., Hawes, N. L., Nishina, P. M., et al. (2000). A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc. Natl. Acad. Sci. U.S.A. 97, 5551–5556. doi: 10.1073/pnas.97.10.5551
Aleman, T. S., Cideciyan, A. V., Aguirre, G. K., Huang, W. C., Mullins, C. L., Roman, A. J., et al. (2011). Human CRB1-associated retinal degeneration: comparison with the rd8 Crb1-mutant mouse model. Invest. Ophthalmol. Vis. Sci. 52, 6898–6910. doi: 10.1167/iovs.11-7701
Allen, A. E., Cameron, M. A., Brown, T. M., Vugler, A. A., and Lucas, R. J. (2010). Visual responses in mice lacking critical components of all known retinal phototransduction cascades. PLoS One 5:e15063. doi: 10.1371/journal.pone.0015063
Ameline, B., Tshilenge, K.-T., Weber, M., Biget, M., Libeau, L., Caplette, R., et al. (2017). Long-term expression of melanopsin and channelrhodopsin causes no gross alterations in the dystrophic dog retina. Gene Ther. 24, 735–741. doi: 10.1038/gt.2017.63
Athanasiou, D., Aguila, M., Bellingham, J., Li, W., McCulley, C., Reeves, P. J., et al. (2017). The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retinal Eye Res. 17, 1–84. doi: 10.1016/j.preteyeres.2017.10.002
Bainbridge, J. W. B., Mehat, M. S., Sundaram, V., Robbie, S. J., Barker, S. E., Ripamonti, C., et al. (2015). Long-term effect of gene therapy on leber’s congenital amaurosis. Engl. J. Med. 372, 1887–1897. doi: 10.1056/NEJMoa1414221
Balaggan, K. S., and Ali, R. R. (2012). Ocular gene delivery using lentiviral vectors. Gene Ther. 19, 145–153. doi: 10.1038/gt.2011.153
Banin, E., Cideciyan, A. V., Aleman, T. S., Petters, R. M., Wong, F., Milam, A. H., et al. (1999). Retinal rod photoreceptor-specific gene mutation perturbs cone pathway development. Neuron 23, 549–557. doi: 10.1016/s0896-6273(00)80807-7
Beltran, W. A. (2009). The use of canine models of inherited retinal degeneration to test novel therapeutic approaches. Vet. Ophthalmol. 12, 192–204. doi: 10.1111/j.1463-5224.2009.00694.x
Berry, M. H., Holt, A., Salari, A., Veit, J., Visel, M., Levitz, J., et al. (2019). Restoration of high-sensitivity and adapting vision with a cone opsin. Nat. Commun. 10, 1–12. doi: 10.1038/s41467-019-09124-x
Bi, A., Cui, J., Ma, Y.-P., Olshevskaya, E., Pu, M., Dizhoor, A. M., et al. (2006). Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron 50, 23–33. doi: 10.1016/j.neuron.2006.02.026
Bin Lin, Koizumi, A., Tanaka, N., Panda, S., and Masland, R. H. (2008). Restoration of visual function in retinal degeneration mice by ectopic expression of melanopsin. Proc. Natl. Acad. Sci. U.S.A. 105, 16009–16014. doi: 10.1073/pnas.0806114105
Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G., and Deisseroth, K. (2005). Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268. doi: 10.1038/nn1525
Boye, S. E., Alexander, J. J., Boye, S. L., Witherspoon, C. D., Sandefer, K., Conlon, T., et al. (2012). The human rhodopsin kinase promoter in an AAV5 vector confers rod and cone specific expression in the primate retina. Hum. Gene Ther. 23, 1101–1115. doi: 10.1089/hum.2012.125
Boye, S. E., Alexander, J. J., Witherspoon, C. D., Boye, S. L., Peterson, J. J., Clark, M. E., et al. (2016). Highly efficient delivery of adeno-associated viral vectors to the primate retina. Hum. Gene Ther. 27, 580–597. doi: 10.1089/hum.2016.085
Busskamp, V., Duebel, J., Balya, D., Fradot, M., Viney, T. J., Siegert, S., et al. (2010). Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science 329, 413–417. doi: 10.1126/science.1190897
Cehajic-Kapetanovic, J., Eleftheriou, C., Allen, A. E., Milosavljevic, N., Pienarr, A., Bedford, R., et al. (2015). Restoration of vision with ectopic expression of human rod opsin. Curr. Biol. 25, 2111–2122. doi: 10.1016/j.cub.2015.07.029
Cehajic-Kapetanovic, J., Le Goff, M. M., Allen, A., Lucas, R. J., and Bishop, P. N. (2011). Glycosidic enzymes enhance retinal transduction following intravitreal delivery of AAV2. Mol. Vis. 17, 1771–1783.
Cehajic-Kapetanovic, J., Milosavljevic, N., Bedford, R. A., Lucas, R. J., and Bishop, P. N. (2018). Efficacy and safety of glycosidic enzymes for improved gene delivery to the retina following intravitreal injection in mice. Mol. Ther. Methods Clin. Dev. 9, 192–202. doi: 10.1016/j.omtm.2017.12.002
Cehajic-Kapetanovic, J., Troelenberg, N., Edwards, T. L., Xue, K., Ramsden, J. D., Stett, A., et al. (2020a). Highest reported visual acuity after electronic retinal implantation. Acta Ophthalmol. 43, 1–5. doi: 10.1111/aos.14443
Cehajic-Kapetanovic, J., Xue, K., La Camara De, C. M.-F., Nanda, A., Davies, A., Wood, L. J., et al. (2020b). Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med. 26, 354–359. doi: 10.1038/s41591-020-0763-1
Chaffiol, A., Caplette, R., Jaillard, C., Brazhnikova, E., Desrosiers, M., Dubus, E., et al. (2017). A new promoter allows optogenetic vision restoration with enhanced sensitivity in macaque retina. Mol. Ther. 25, 2546–2560. doi: 10.1016/j.ymthe.2017.07.011
Chang, B., Hawes, N. L., Hurd, R. E., Davisson, M. T., Nusinowitz, S., and Heckenlively, J. R. (2002). Retinal degeneration mutants in the mouse. Vision Res. 42, 517–525. doi: 10.1016/S0042-6989(01)00146-8
Chang, B., Khanna, H., Hawes, N., Jimeno, D., Shirley, H., Lillo, C., et al. (2006). In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse | Human Molecular Genetics | Oxford Academic. Hum. Mol. Genet. 15:107. doi: 10.1093/hmg/ddl107
Charbel Issa, P., De Silva, S. R., Lipinski, D. M., Singh, M. S., Mouravlev, A., You, Q., et al. (2013). Assessment of tropism and effectiveness of new primate-derived hybrid recombinant AAV serotypes in the mouse and primate retina. PLoS One 8:e60361. doi: 10.1371/journal.pone.0060361
Chichagova, V., Dorgau, B., Felemban, M., Georgiou, M., Armstrong, L., and Lako, M. (2019). Differentiation of retinal organoids from human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 50, 4975–4911. doi: 10.1002/cpsc.95
Chuong, A. S., Miri, M. L., Busskamp, V., Matthews, G. A. C., Acker, L. C., Sørensen, A. T., et al. (2014). Noninvasive optical inhibition with a red-shifted microbial rhodopsin. Nat. Neurosci. 17, 1123–1129. doi: 10.1038/nn.3752
Cronin, T., Vandenberghe, L. H., Hantz, P., Juttner, J., Reimann, A., Kacsó, A.-E., et al. (2014). Efficient transduction and optogenetic stimulation of retinal bipolar cells by a synthetic adeno-associated virus capsid and promoter. EMBO Mol. Med. 6, 1175–1190. doi: 10.15252/emmm.201404077
Dalkara, D., Byrne, L. C., Klimczak, R. R., Visel, M., Yin, L., Merigan, W. H., et al. (2013). In vivo–directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 5:189ra76. doi: 10.1126/scitranslmed.3005708
Dalloz, C., Sarig, R., Fort, P., Yaffe, D., Bordais, A., Pannicke, T., et al. (2003). Targeted inactivation of dystrophin gene product Dp71: phenotypic impact in mouse retina | Human Molecular Genetics | Oxford Academic. Hum. Mol. Genet. 12, 1543–1554. doi: 10.1093/hmg/ddg170
Daniele, L. L., Lillo, C., Lyubarsky, A. L., Nikonov, S. S., Philp, N., Mears, A. J., et al. (2005). Cone-like morphological, molecular, and electrophysiological features of the photoreceptors of the Nrl knockout mouse. Invest Ophthalmol. Vis. Sci. 46, 2156–2167. doi: 10.1167/iovs.04-1427
De Leeuw, C. N., Dyka, F. M., Boye, S. L., Zhou, M., Chou, A. Y., Borretta, L., et al. (2014). Targeted CNS delivery using human MiniPromoters and demonstrated compatibility with adeno-associated viral vectors. Mol. Ther. Methods Clin. Dev. 1, 1–15. doi: 10.1038/mtm.2013.5
De Silva, S. R., Barnard, A. R., Hughes, S., Tam, S. K. E., Martin, C., Singh, M. S., et al. (2017). Long-term restoration of visual function in end-stage retinal degeneration using subretinal human melanopsin gene therapy. Proc. Natl. Acad. Sci. U.S.A. 114, 11211–11216. doi: 10.1073/pnas.1701589114
De Silva, S. R., Issa, P. C., Singh, M. S., Lipinski, D. M., Barnea-Cramer, A. O., Walker, N. J., et al. (2016). Single residue AAV capsid mutation improves transduction of photoreceptors in the Abca4(-/-) mouse and bipolar cells in the rd1 mouse and human retina ex-vivo. Gene Ther. 23, 767–774. doi: 10.1038/gt.2016.54
Deisseroth, K., Feng, G., Majewska, A. K., Miesenböck, G., Ting, A., and Schnitzer, M. J. (2006). Next-generation optical technologies for illuminating genetically targeted brain circuits. J. Neurosci. 26, 10380–10386. doi: 10.1523/JNEUROSCI.3863-06.2006
Doroudchi, M. M., Greenberg, K. P., Liu, J., Silka, K. A., Boyden, E. S., Lockridge, J. A., et al. (2011). Virally delivered channelrhodopsin-2 safely and effectively restores visual function in multiple mouse models of blindness. Mol. Ther. 19, 1220–1229. doi: 10.1038/mt.2011.69
Eleftheriou, C. G., Cehajic-Kapetanovic, J., Martial, F. P., Milosavljevic, N., Bedford, R. A., and Lucas, R. J. (2017). Meclofenamic acid improves the signal to noise ratio for visual responses produced by ectopic expression of human rod opsin. Mol. Vis. 23, 334–345.
Farber, D. B., Flannery, J. G., and Rickman, C. B. (1994). The rd mouse story: Seventy years of research on an animal model of inherited retinal degeneration. Prog. Retinal Res. 13, 31–64. doi: 10.1016/1350-9462(94)90004-3
Fischer, M. D., McClements, M. E., Martinez-Fernandez de la Camara, C., Bellingrath, J.-S., Dauletbekov, D., Ramsden, S. C., et al. (2017). Codon-optimized RPGR improves stability and efficacy of AAV8 Gene therapy in two mouse models of X-linked retinitis pigmentosa. Mol. Ther. 25, 1854–1865. doi: 10.1016/j.ymthe.2017.05.005
Gargini, C., Terzibasi, E., Mazzoni, F., and Strettoi, E. (2007). Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: a morphological and ERG study. J. Comp. Neurol. 500, 222–238. doi: 10.1002/cne.21144
Garita-Hernandez, M., Guibbal, L., Toualbi, L., Routet, F., Chaffiol, A., Winckler, C., et al. (2018). Optogenetic Light sensors in human retinal organoids. Front. Neurosci. 12:232. doi: 10.3389/fnins.2018.00789
Garita-Hernandez, M., Lampič, M., Chaffiol, A., Guibbal, L., Routet, F., Santos-Ferreira, T., et al. (2019). Restoration of visual function by transplantation of optogenetically engineered photoreceptors. Nat. Commun. 10, 1–13. doi: 10.1038/s41467-019-12330-2
Garita-Hernandez, M., Routet, F., Guibbal, L., Khabou, H., Toualbi, L., Riancho, L., et al. (2020). AAV-mediated gene delivery to 3D retinal organoids derived from human induced pluripotent stem cells. Int. J. Mol. Sci. 20:994. doi: 10.3390/ijms21030994
Gaub, B. M., Berry, M. H., Holt, A. E., Isacoff, E. Y., and Flannery, J. G. (2015). Optogenetic vision restoration using rhodopsin for enhanced sensitivity. Mol. Ther. 23, 1562–1571. doi: 10.1038/mt.2015.121
Gonzalez-Cordero, A., Goh, D., Kruczek, K., Naeem, A., Fernando, M., Kleine Holthaus, S.-M., et al. (2017). Assessment of AAV Vector tropisms for mouse and human pluripotent stem cell–derived RPE and photoreceptor cells. Hum. Gene Ther. 2018:27. doi: 10.1089/hum.2018.027
Gradinaru, V., Thompson, K. R., and Deisseroth, K. (2008). eNpHR: a Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 36, 129–139. doi: 10.1007/s11068-008-9027-6.pdf
Gradinaru, V., Thompson, K. R., Zhang, F., Mogri, M., Kay, K., Schneider, M. B., et al. (2007). Targeting and readout strategies for fast optical neural control in vitro and In Vivo. J. Neurosci. 27, 14231–14238. doi: 10.1523/JNEUROSCI.3578-07.2007
Gradinaru, V., Zhang, F., Ramakrishnan, C., Mattis, J., Prakash, R., Diester, I., et al. (2010). Molecular and cellular approaches for diversifying and extending optogenetics. Cell 141, 154–165. doi: 10.1016/j.cell.2010.02.037
Gregg, R. G., Mukhopadhyay, S., Candille, S. I., Ball, S. L., Pardue, M. T., McCall, M. A., et al. (2003). Identification of the gene and the mutation responsible for the mouse nob phenotype. Invest Ophthalmol. Vis. Sci. 44, 378–384. doi: 10.1167/iovs.02-0501
Hackam, A. S., Strom, R., Liu, D., Qian, J., Wang, C., Otteson, D., et al. (2004). Identification of gene expression changes associated with the progression of retinal degeneration in the rd1 mouse. Invest. Ophthalmol. Vis. Sci. 45, 2929–2942. doi: 10.1167/iovs.03-1184
Hanlon, K. S., Chadderton, N., Palfi, A., Blanco-Fernandez, A., Humphries, P., Kenna, P. F., et al. (2017). A Novel retinal ganglion cell promoter for utility in AAV vectors. Front. Neurosci. 11:521. doi: 10.3389/fnins.2017.00521
Hassall, M. M., McClements, M. E., Barnard, A. R., Patrício, M. I., Aslam, S. A., and MacLaren, R. E. (2020). Analysis of early cone dysfunction in an in vivo model of rod-cone dystrophy. Int. J. Mol. Sci. 20, 6055–6017. doi: 10.3390/ijms21176055
Hattar, S., Lucas, R. J., Mrosovsky, N., Thompson, S., Douglas, R. H., Hankins, M. W., et al. (2003). Melanopsin and rod–cone photoreceptive systems account for all major accessory visual functions in mice. Nature 424, 75–81. doi: 10.1038/nature01761
Hawes, N. L., Chang, B., Hageman, G. S., Nusinowitz, S., Nishina, P. M., Schneider, B. S., et al. (2000). Retinal degeneration 6 (rd6): a new mouse model for human retinitis punctata albescens. Invest. Ophthalmol. Vis. Sci. 41, 3149–3157.
Hickey, D. G., Edwards, T. L., Barnard, A. R., Singh, M. S., De Silva, S. R., McClements, M. E., et al. (2017). Tropism of engineered and evolved recombinant AAV serotypes in the rd1 mouse and ex vivo primate retina. Gene Ther. 24, 787–800. doi: 10.1038/gt.2017.85
Humphries, M. M., Rancourt, D., Farrar, G. J., Kenna, P., Hazel, M., Bush, R. A., et al. (1997). Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat. Genet. 15, 216–219. doi: 10.1038/ng0297-216
ICNIRP (2013). ICNIRP guidelines on limits of exposure to incoherent visible and infrared radiation. Health Phys. 13, 74–96.
Ivanova, E., Hwang, G.-S., Pan, Z.-H., and Troilo, D. (2010). Evaluation of AAV-mediated expression of Chop2-GFP in the marmoset retina. Invest Ophthalmol. Vis. Sci. 51, 5288–5296. doi: 10.1167/iovs.10-5389
Jansen, H. G., and Sanyal, S. (1984). Development and degeneration of retina inrds mutant mice: electron microscopy. J. Comp. Neurol. 224, 71–84. doi: 10.1002/cne.902240107
Jones, B. W., Pfeiffer, R. L., Ferrell, W. D., Watt, C. B., Marmor, M., and Marc, R. E. (2016). Retinal remodeling in human retinitis pigmentosa. Exp. Eye Res. 150, 149–165. doi: 10.1016/j.exer.2016.03.018
Jones, B. W., Watt, C. B., Frederick, J. M., Baehr, W., Chen, C.-K., Levine, E. M., et al. (2003). Retinal remodeling triggered by photoreceptor degenerations. J. Comp. Neurol. 464, 1–16. doi: 10.1002/cne.10703
Kameya, S., Hawes, N. L., Chang, B., Heckenlively, J. R., Naggert, J. K., and Nishina, P. M. (2002). Mfrp, a gene encoding a frizzled related protein, is mutated in the mouse retinal degeneration 6. Hum. Mol. Genet. 11, 1879–1886. doi: 10.1093/hmg/11.16.1879
Kay, C. N., Ryals, R. C., Aslanidi, G. V., Min, S.-H., Ruan, Q., Sun, J., et al. (2013). Targeting photoreceptors via intravitreal delivery using novel, capsid-mutated AAV vectors. PLoS One 8:e62097. doi: 10.1371/journal.pone.0062097
Khabou, H., Garita-Hernandez, M., Chaffiol, A., Reichman, S., Jaillard, C., Brazhnikova, E., et al. (2018). Noninvasive gene delivery to foveal cones for vision restoration. JCI Insight 3:D358. doi: 10.1172/jci.insight.96029
Kim, D. S., Matsuda, T., and Cepko, C. L. (2008). A core paired-type and POU homeodomain-containing transcription factor program drives retinal bipolar cell gene expression. J. Neurosci. 28, 7748–7764. doi: 10.1523/JNEUROSCI.0397-08.2008
Klapoetke, N. C., Murata, Y., Kim, S. S., Pulver, S. R., Birdsey-Benson, A., Cho, Y. K., et al. (2014). Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346. doi: 10.1038/nmeth.2836
Kleinlogel, S., Feldbauer, K., Dempski, R. E., Fotis, H., Wood, P. G., Bamann, C., et al. (2011). Ultra light-sensitive and fast neuronal activation with the Ca 2+ -permeable channelrhodopsin CatCh. Nat. Neurosci. 14, 513–518. doi: 10.1038/nn.2776
Kolstad, K. D., Dalkara, D., Guerin, K., Visel, M., Hoffmann, N., Schaffer, D. V., et al. (2010). Changes in adeno-associated virus-mediated gene delivery in retinal degeneration. Hum. Gene Ther. 21, 571–578. doi: 10.1089/hum.2009.194
Lagali, P. S., Balya, D., Awatramani, G. B., Münch, T. A., Kim, D. S., Busskamp, V., et al. (2008). Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat. Neurosci. 11, 667–675. doi: 10.1038/nn.2117
Lane, A., Jovanovic, K., Shortall, C., Ottaviani, D., Brugulat Panes, A., Schwarz, N., et al. (2020). Modeling and Rescue of RP2 retinitis pigmentosa using iPSC-derived retinal organoids. Stem. Cell Rep. 15, 67–79. doi: 10.1016/j.stemcr.2020.05.007
Li, Q., Timmers, A. M., Guy, J., Pang, J., and Hauswirth, W. W. (2008). Cone-specific expression using a human red opsin promoter in recombinant AAV. Vision Res. 48, 332–338. doi: 10.1016/j.visres.2007.07.026
Lin, B., Masland, R. H., and Strettoi, E. (2009). Remodeling of cone photoreceptor cells after rod degeneration in rd mice. Exp. Eye Res. 88, 589–599. doi: 10.1016/j.exer.2008.11.022
Lin, J. Y., Knutsen, P. M., Muller, A., Kleinfeld, D., and Tsien, R. Y. (2013). ReaChR: a red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat. Neurosci. 16, 1499–1508. doi: 10.1038/nn.3502
Liu, M.-M., Dai, J.-M., Liu, W.-Y., Zhao, C.-J., Lin, B., and Yin, Z.-Q. (2016). Human melanopsin-AAV2/8 transfection to retina transiently restores visual function in rd1 mice. Int. J. Ophthalmol. 9, 655–661. doi: 10.18240/ijo.2016.05.03
Lu, Q., Ganjawala, T. H., Ivanova, E., Cheng, J. G., Troilo, D., and Pan, Z.-H. (2016). AAV-mediated transduction and targeting of retinal bipolar cells with improved mGluR6 promoters in rodents and primates. Gene Ther. 23, 680–689. doi: 10.1038/gt.2016.42
Lu, Q., Ganjawala, T. H., Krstevski, A., Abrams, G. W., and Pan, Z.-H. (2020). Comparison of AAV-Mediated optogenetic vision restoration between retinal ganglion cell expression and ON bipolar cell targeting. Mol. Ther. Methods Clin. Dev. 18, 15–23. doi: 10.1016/j.omtm.2020.05.009
Ma, J., Norton, J. C., Allen, A. C., Burns, J. B., Hasel, K. W., Burns, J. L., et al. (1995). Retinal degeneration slow (rds) in mouse results from simple insertion of a t haplotype-specific element into protein-coding exon II. Genomics 28, 212–219. doi: 10.1006/geno.1995.1133
Macé, E., Caplette, R., Marre, O., Sengupta, A., Chaffiol, A., Barbe, P., et al. (2015). Targeting channelrhodopsin-2 to ON-bipolar cells with vitreally administered AAV restores ON and OFF visual responses in blind mice. Mol. Ther. 23, 7–16. doi: 10.1038/mt.2014.154
Marc, R. E., Jones, B. W., Watt, C. B., and Strettoi, E. (2003). Neural remodeling in retinal degeneration. Prog. Retinal Eye Res. 22, 607–655. doi: 10.1016/S1350-9462(03)00039-9
McClements, M., Davies, W. I. L., Michaelides, M., Young, T., Neitz, M., MacLaren, R. E., et al. (2013). Variations in opsin coding sequences cause x-linked cone dysfunction syndrome with myopia and dichromacy. Invest Ophthalmol. Vis. Sci. 54, 1361–1369. doi: 10.1167/iovs.12-11156
McGregor, J. E., Godat, T., Dhakal, K. R., Parkins, K., Strazzeri, J. M., Bateman, B. A., et al. (2020). Optogenetic restoration of retinal ganglion cell activity in the living primate. Nat. Commun. 11, 1–9. doi: 10.1038/s41467-020-15317-6
Mears, A. J., Kondo, M., Swain, P. K., Takada, Y., Bush, R. A., Saunders, T. L., et al. (2001). Nrl is required for rod photoreceptor development. Nat. Genet. 29, 447–452. doi: 10.1038/ng774
Mehalow, A. K., Kameya, S., Smith, R. S., Hawes, N. L., Denegre, J. M., Young, J. A., et al. (2003). CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum. Mol. Genet. 12, 2179–2189. doi: 10.1093/hmg/ddg232
Milam, A. H., Li, Z. Y., and Fariss, R. N. (1998). Histopathology of the human retina in retinitis pigmentosa. Prog. Retinal Eye Res. 17, 175–205.
Mohan, K., Harper, M. M., Kecova, H., Ye, E. A., Lazic, T., Sakaguchi, D. S., et al. (2012). Characterization of structure and function of the mouse retina using pattern electroretinography, pupil light reflex, and optical coherence tomography. Vet. Ophthalmol. 15, 94–104. doi: 10.1111/j.1463-5224.2012.01034.x
Mussolino, C., Corte, Della, M., Rossi, S., Viola, F., Di Vicino, U., et al. (2011). AAV-mediated photoreceptor transduction of the pig cone-enriched retina. Gene Ther. 18, 637–645. doi: 10.1038/gt.2011.3
Nakajima, Y., Iwakabe, H., Akazawa, C., Nawa, H., Shigemoto, R., Mizuno, N., et al. (1993). Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for L-2-amino-4-phosphonobutyrate. J. Biol. Chem. 268, 11868–11873.
Narfström, K., Wrigstad, A., and Nilsson, S. E. (1989). The Briard dog: a new animal model of congenital stationary night blindness. Br. J. Ophthalmol. 73, 750–756. doi: 10.1136/bjo.73.9.750
Nelson, R., and Connaughton, V. (2007). “Bipolar Cell pathways in the vertebrate retina,” in The Organization of the Retina and Visual System Webvision: The Organization of the Retina and Visual System, eds E. Fernandez and R. Nelson (Salt Lake City, UT: University of Utah Health Sciences Center), 1–48.
Nomura, A., Shigemoto, R., Nakamura, Y., Okamoto, N., Mizuno, N., and Nakanishi, S. (1994). Developmentally regulated postsynaptic localization of a metabotropic glutamate receptor in rat rod bipolar cells. Cell 77, 361–369. doi: 10.1016/0092-8674(94)90151-1
Nuzbrokh, Y., Kassotis, A. S., Ragi, S. D., Jauregui, R., and Tsang, S. H. (2020). Treatment-emergent adverse events in gene therapy trials for inherited retinal diseases: a narrative review. Ophthalmol. Ther. 20, 1–16. doi: 10.1007/s40123-020-00287-1
Orlans, H. O., Edwards, T. L., De Silva, S. R., Patrício, M. I., and MacLaren, R. E. (2018). Human Retinal Explant Culture for Ex Vivo Validation of AAV Gene Therapy: Retinal Gene Therapy. New York, NY: Humana Press, 289–303. doi: 10.1007/978-1-4939-7522-8_21
Pang, J., Chang, B., Hawes, N. L., Hurd, R. E., Davisson, M. T., Li, J., et al. (2005). Retinal degeneration 12 (rd12): a new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA). Mol. Vis. 11, 152–162.
Pardue, M. T., McCall, M. A., LaVail, M. M., Gregg, R. G., and Peachey, N. S. (1998). A naturally occurring mouse model of X-linked congenital stationary night blindness. Invest. Ophthalmol. Vis. Sci. 39, 2443–2449.
Patrício, M. I., Barnard, A. R., Orlans, H. O., McClements, M. E., and MacLaren, R. E. (2017). Inclusion of the woodchuck hepatitis virus posttranscriptional regulatory element enhances AAV2-driven transduction of mouse and human retina. Mol. Ther. Nucleic Acids 6, 198–208. doi: 10.1016/j.omtn.2016.12.006
Pepperberg, D. R. (2003). Bleaching desensitization: background and current challenges. Vision Res. 43, 3011–3019. doi: 10.1016/S0042-6989(03)00484-X
Petrs-Silva, H., Dinculescu, A., Li, Q., Deng, W.-T., Pang, J.-J., Min, S.-H., et al. (2011). Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol. Ther. 19, 293–301. doi: 10.1038/mt.2010.234
Pfeiffer, R. L., Marc, R. E., and Jones, B. W. (2020). Persistent remodeling and neurodegeneration in late-stage retinal degeneration. Prog. Retinal Eye Res. 74:100771. doi: 10.1016/j.preteyeres.2019.07.004
Picaud, S., Dalkara, D., Marazova, K., Goureau, O., Roska, B., and Sahel, J.-A. (2019). The primate model for understanding and restoring vision. Proc. Natl. Acad. Sci. U.S.A. 116, 26280–26287. doi: 10.1073/pnas.1902292116
Pittler, S. J., Keeler, C. E., Sidman, R. L., and Baehr, W. (1993). PCR analysis of DNA from 70-year-old sections of rodless retina demonstrates identity with the mouse rd defect. Proc. Natl. Acad. Sci. U.S.A. 90, 9616–9619. doi: 10.1073/pnas.90.20.9616
Puppo, A., Cesi, G., Marrocco, E., Piccolo, P., Jacca, S., Shayakhmetov, D. M., et al. (2014). Retinal transduction profiles by high-capacity viral vectors. Gene Ther. 21, 855–865. doi: 10.1038/gt.2014.57
Puthussery, T., Primo, J. G., Pandey, S., Duvoisin, R. M., and Taylor, W. R. (2009). Differential loss and preservation of glutamate receptor function in bipolar cells in the rd10 mouse model of retinitis pigmentosa. Eur. J. Neurosci. 29, 1533–1542. doi: 10.1111/j.1460-9568.2009.06728.x
Quinn, P. M., Buck, T. M., Ohonin, C., Mikkers, H. M. M., and Wijnholds, J. (2018). Production of iPS-Derived Human Retinal Organoids for Use in Transgene Expression Assays: Retinal Gene Therapy Methods and Protocols. New York, NY: Humana Press, 261–273. doi: 10.1007/978-1-4939-7522-8_19
Reichman, S., Slembrouck, A., Gagliardi, G., Chaffiol, A., Terray, A., Nanteau, C., et al. (2017). Generation of storable retinal organoids and retinal pigmented epithelium from adherent human iPS cells in xeno-free and feeder-free conditions. Stem Cells 35, 1176–1188. doi: 10.1002/stem.2586
Reuter, J. H., and Sanyal, S. (1984). Development and degeneration of retina in rds mutant mice: The electroretinogram. Neurosci. Lett. 48, 231–237. doi: 10.1016/0304-3940(84)90024-7
Russell, S., Bennett, J., Wellman, J. A., Chung, D. C., Yu, Z.-F., Tillman, A., et al. (2017). Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860. doi: 10.1016/S0140-6736(17)31868-8
Sahel, J.-A., and Roska, B. (2013). Gene therapy for blindness. Annu. Rev. Neurosci. 36, 467–488. doi: 10.1146/annurev-neuro-062012-170304
Sakami, S., Maeda, T., Bereta, G., Okano, K., Golczak, M., Sumaroka, A., et al. (2011). Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J. Biol. Chem. 286, 10551–10567. doi: 10.1074/jbc.M110.209759
Santos-Ferreira, T., Voelkner, M., Borsch, O., Haas, J., Cimalla, P., Vasudevan, P., et al. (2016). Stem cell-derived photoreceptor transplants differentially integrate into mouse models of cone-rod dystrophy. Invest. Ophthalmol. Vis. Sci. 57, 3509–3520. doi: 10.1167/iovs.16-19087
Scalabrino, M. L., Boye, S. L., Fransen, K. M. H., Noel, J. M., Dyka, F. M., Min, S.-H., et al. (2015). Intravitreal delivery of a novel AAV vector targets ON bipolar cells and restores visual function in a mouse model of complete congenital stationary night blindness. Hum. Mol. Genet. 24, 6229–6239. doi: 10.1093/hmg/ddv341
Sengupta, A., Chaffiol, A., Macé, E., Caplette, R., Desrosiers, M., Lampič, M., et al. (2016). Red-shifted channelrhodopsin stimulation restores light responses in blind mice, macaque retina, and human retina. EMBO Mol. Med. 8, 1248–1264. doi: 10.15252/emmm.201505699
Shupe, J. M., Kristan, D. M., Austad, S. N., and Stenkamp, D. L. (2006). The Eye of the laboratory mouse remains anatomically adapted for natural conditions. BBE 67, 39–52. doi: 10.1159/000088857
Simon, C.-J., Sahel, J.-A., Duebel, J., and Herlitze, S. (2020). Opsins for vision restoration. Biochem. Biophys. Res. Commun. 20, 1–6. doi: 10.1016/j.bbrc.2019.12.117
Smith, C. A., and Chauhan, B. C. (2018). In vivo imaging of adeno-associated viral vector labelled retinal ganglion cells. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-19969-9
Sohocki, M. M., Daiger, S. P., Bowne, S. J., Rodriquez, J. A., Northrup, H., Heckenlively, J. R., et al. (2001). Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 17, 42–51. doi: 10.1002/1098-1004200117:1<42::AID-HUMU5<3.0.CO;2-K
Strettoi, E., Porciatti, V., Falsini, B., Pignatelli, V., and Rossi, C. (2002). Morphological and functional abnormalities in the inner retina of the rd/rd mouse. J. Neurosci. 22, 5492–5504. doi: 10.1523/jneurosci.22-13-05492.2002
The European Parliament and the Council of the European Union (2006). European parliament and council of the european union. directive 2006/25/EC on artificial optical radiation. Off. J. Eur.Union 6, 38–59.
Thompson, D. A., Khan, N. W., Othman, M. I., Chang, B., Jia, L., Grahek, G., et al. (2012). Rd9 is a naturally occurring mouse model of a common form of retinitis pigmentosa caused by mutations in RPGR-ORF15. PLoS One 7:e35865. doi: 10.1371/journal.pone.0035865
Toda, K., Bush, R. A., Humphries, P., and Sieving, P. A. (1999). The electroretinogram of the rhodopsin knockout mouse. Vis. Neurosci. 16, 391–398. doi: 10.1017/S0952523899162187
Travis, G. H., and Hepler, J. E. (1993). A medley of retinal dystrophies. Nat. Genet. 3, 191–192. doi: 10.1038/ng0393-191
Vacca, O., Darche, M., Schaffer, D. V., Flannery, J. G., Sahel, J.-A., Rendon, A., et al. (2013). AAV-mediated gene delivery in Dp71-null mouse model with compromised barriers. Glia 62, 468–476. doi: 10.1002/glia.22617
van Norren, D., and Vos, J. J. (2015). Light damage to the retina: an historical approach. Eye (Lond.) 30, 169–172. doi: 10.1038/eye.2015.218
van Wyk, M., Hulliger, E. C., Girod, L., Ebneter, A., and Kleinlogel, S. (2017). Present molecular limitations of ON-bipolar cell targeted gene therapy. Front. Neurosci. 11:11372. doi: 10.3389/fnins.2017.00161
van Wyk, M., Pielecka-Fortuna, J., Löwel, S., and Kleinlogel, S. (2015). Restoring the ON switch in blind retinas: opto-mGluR6, a next-generation, cell-tailored optogenetic tool. PLoS Biol. 13:e1002143. doi: 10.1371/journal.pbio.1002143
Wässle, H., Puller, C., Müller, F., and Haverkamp, S. (2009). Cone contacts, mosaics, and territories of bipolar cells in the mouse retina. J. Neurosci. 29, 106–117. doi: 10.1523/JNEUROSCI.4442-08.2009
Wilson, J. M., and Flotte, T. R. (2020). Moving forward after two deaths in a gene therapy trial of myotubular myopathy. Hum. Gene Ther. 2020:182. doi: 10.1089/hum.2020.182
Wrigstad, A., Nilsson, S. E. G., and Narfström, K. (1992). Ultrastructural changes of the retina and the retinal pigment epithelium in briard dogs with hereditary congenital night blindness and partial day blindness. Exp. Eye Res. 55, 805–818. doi: 10.1016/0014-4835(92)90007-F
Xue, K., Jolly, J. K., Barnard, A. R., Rudenko, A., Salvetti, A. P., Patrício, M. I., et al. (2018). Beneficial effects on vision in patients undergoing retinal gene therapy for choroideremia. Nat. Med. 24, 1507–1512. doi: 10.1038/s41591-018-0185-5
Ye, G.-J., Budzynski, E., Sonnentag, P., Nork, T. M., Sheibani, N., Gurel, Z., et al. (2016). Cone-specific promoters for gene therapy of achromatopsia and other retinal diseases. Hum. Gene Ther. 27, 72–82. doi: 10.1089/hum.2015.130
Yin, L., Greenberg, K., Hunter, J. J., Dalkara, D., Kolstad, K. D., Masella, B. D., et al. (2011). Intravitreal injection of AAV2 transduces macaque inner retina. Invest. Ophthalmol. Vis. Sci. 52, 2775–2783. doi: 10.1167/iovs.10-6250
Yiu, G., Chung, S. H., Mollhoff, I. N., Nguyen, U. T., Thomasy, S. M., Yoo, J., et al. (2020). Suprachoroidal and Subretinal Injections of AAV Using transscleral microneedles for retinal gene delivery in nonhuman primates. Mol. Ther. Methods Clin. Dev. 16, 179–191. doi: 10.1016/j.omtm.2020.01.002
Youssef, P. N., Sheibani, N., and Albert, D. M. (2011). Retinal light toxicity. Eye (Lond.) 25, 1–14. doi: 10.1038/eye.2010.149
Zhang, F., Wang, L.-P., Brauner, M., Liewald, J. F., Kay, K., Watzke, N., et al. (2007). Multimodal fast optical interrogation of neural circuitry. Nature 446, 633–639. doi: 10.1038/nature05744
Zhang, Y., Ivanova, E., Bi, A., and Pan, Z.-H. (2009). Ectopic expression of multiple microbial rhodopsins restores ON and OFF light responses in retinas with photoreceptor degeneration. J. Neurosci. 29, 9186–9196. doi: 10.1523/JNEUROSCI.0184-09.2009
Keywords: optogenetics, gene therapy, AAV, retinal degeneration, opsins
Citation: McClements ME, Staurenghi F, MacLaren RE and Cehajic-Kapetanovic J (2020) Optogenetic Gene Therapy for the Degenerate Retina: Recent Advances. Front. Neurosci. 14:570909. doi: 10.3389/fnins.2020.570909
Received: 09 June 2020; Accepted: 23 October 2020;
Published: 11 November 2020.
Edited by:
Jacqueline Reinhard, Ruhr-University, GermanyReviewed by:
Jose R. Hombrebueno, University of Birmingham, United KingdomIlaria Piano, University of Pisa, Italy
Copyright © 2020 McClements, Staurenghi, MacLaren and Cehajic-Kapetanovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michelle E. McClements, ZW5xdWlyaWVzQGV5ZS5veC5hYy51aw==