 Sheng-Yu Zhou
Sheng-Yu Zhou Guo-Zhen Cui
Guo-Zhen Cui Xiu-Li Yan1
Xiu-Li Yan1 Xu Wang
Xu Wang Yang Qu
Yang Qu Hang Jin
Hang Jin- 1Department of Neurology, Stroke Center, The First Hospital of Jilin University, Changchun, China
- 2Department of Hepatology, Cancer Center, The First Hospital of Jilin University, Changchun, China
- 3Clinical Trial and Research Center for Stroke, Department of Neurology, The First Hospital of Jilin University, Changchun, China
Intracerebral hemorrhage (ICH) is a fatal cerebrovascular disease with high morbidity and mortality, for which no effective therapies are currently available. Brain tissue damage caused by ICH is mediated by a newly identified form of non-apoptotic programmed cell death, called ferroptosis. Ferroptosis is characterized by the iron-induced accumulation of lipid reactive oxygen species (ROS), leading to intracellular oxidative stress. Lipid ROS cause damage to nucleic acids, proteins, and cell membranes, eventually resulting in ferroptosis. Numerous biological processes are involved in ferroptosis, including iron metabolism, lipid peroxidation, and glutathione biosynthesis; therefore, iron chelators, lipophilic antioxidants, and other specific inhibitors can suppress ferroptosis, suggesting that these modulators are beneficial for treating brain injury due to ICH. Accumulating evidence indicates that ferroptosis differs from other types of programmed cell death, such as necroptosis, apoptosis, oxytosis, and pyroptosis, in terms of ultrastructural characteristics, signaling pathways, and outcomes. Although several studies have emphasized the importance of ferroptosis due to ICH, the detailed mechanism underlying ferroptosis remains unclear. This review summarizes the available evidence on the mechanism underlying ferroptosis and its relationship with other types of cell death, with the aim to identify therapeutic targets and potential interventions for ICH.
Introduction
Intracerebral hemorrhage (ICH) accounts for 10–15% of all stroke types, which is a catastrophic event associated with high mortality and morbidity rates (Van Asch et al., 2010). Edema in a bleeding brain after ICH contributes to secondary brain injury (SBI). Current ICH treatments include dehydration treatment, antihypertensive therapy, platelet transfusion, and surgery. However, these therapies have some limitations; for example, there is not sufficient evidence to support the safety of platelet transfusion, and dehydration treatment causes a rapid decrease in blood pressure in a specific area (Morotti and Goldstein, 2016). Moreover, the mechanism underlying SBI after ICH remains unclear. However, it is known that hemoglobin in a hematoma after ICH can cause lipid peroxidation and form hydroxyl radicals, which are highly neurotoxic, leading to damage to the membranes, DNA, and proteins (Xi et al., 2006; Cao and Dixon, 2016). As the key element of hemoglobin, iron also plays an important role in SBI. Iron toxicity is mainly based on the Fenton reaction. Through the Fenton reaction, iron forms a large number of hydroxyl radicals, which trigger and aggravate the oxidation of tissues, causing neuronal cell damage (Zhao et al., 2011b; Auriat et al., 2012; He et al., 2020). Some preclinical and clinical studies about ferroptosis in neurodegeneration and stroke indicate that iron released from the hematoma can lead to a newly identified form of programmed cell death, ferroptosis, which may greatly contribute to SBI after ICH (Zhang Z. et al., 2018; Djulbegovic and Uversky, 2019; Zhang et al., 2020). Ferroptosis was first defined in 2012 (Dixon et al., 2012); it refers to an oxidative and iron-dependent form of programmed cell death that differs from other known types of programmed cell death in terms of its morphological and biochemical features, including the ultrastructural characteristics, signaling pathways, and outcomes (Zille et al., 2017; Bobinger et al., 2018; Hirschhorn and Stockwell, 2019). Ferroptosis is associated with iron and amino acid metabolism together with lipid peroxidation, with the iron-dependent accumulation of lipid peroxidation being the key trigger (Dixon et al., 2014; Conrad et al., 2018; Fujii et al., 2019; Hirschhorn and Stockwell, 2019). In this process, inhibition of glutathione peroxidase 4 (GPX4) prevents the appropriate reduction of lipid peroxides, resulting in increased generation of reactive oxygen species (ROS), which in turn leads to oxidative lipid damage, protein aggregation, and ultimately, neuronal cell death (Gao et al., 2015; Bai et al., 2019).
Although the mechanism of ferroptosis is not entirely understood, the use of iron chelators, ferroxidase, and other mediators of ferroptosis may be effective as a potential therapeutic approach for ICH. Strategies to inhibit or delay the rate of iron accumulation, glutathione (GSH) depletion, and lipid peroxidation are currently being tested in trials (Gao et al., 2015; Wang et al., 2016; Conrad et al., 2018). Therefore, in this review, we present the available evidence on the mechanisms underlying ferroptosis to highlight candidate therapeutic targets and potential interventions for ICH. We further discuss the relationships between ferroptosis and other types of cell death. This overview will not only provide a better understanding of the clinical significance of ferroptosis but also promote the progress of research regarding novel aspects, such as the development of potential anti-ferroptotic strategies to prevent SBI after ICH.
Mechanisms Underlying Ferroptosis Following ICH
Iron Metabolism
Iron is essential for excessive lipid peroxidation, which leads to ferroptosis, particularly in ICH (Dixon et al., 2012). Therefore, iron uptake, utilization, export, and storage can affect nearly all ferroptosis processes. ICH occurs upon vessel rupture; thereafter, blood enters the surrounding parts of the brain, causing an intracerebral hematoma, which releases hemoglobin, heme, and iron into the surrounding tissue (Li et al., 2019). Hemoglobin, the most abundant protein in the blood, can act as a neurotoxin released from lysed red blood cells after hematoma formation (Xi et al., 2006). Two forms of iron are involved in this process: ferric (Fe2+) and ferrous (Fe3+) iron. Fe2+ can transfer electrons and has high solubility; thus, Fe2+-containing proteins serve as cofactors and catalysts in several types of oxidation–reduction reactions. In contrast, Fe3+ stores and transports iron and is, therefore, much more stable than the active Fe2+ (Dixon et al., 2012). Under physiological conditions, transferrin (Tf) recognizes Fe3+ and binds two Fe3+ molecules to form diferric Tf, which subsequently binds to the Tf receptor 1 (TfR1) with high-affinity to form the Tf-Fe3+-TfR1 complex on the surface of neurons. The Tf-Fe3+-TfR1 complex is then transported into cells via endocytosis and then into endosomes, where iron is released from the complex because of the acidic environment. Free Fe2+ is then reduced to Fe3+ and stored in the labile iron pool (LIP; Hentze et al., 2010). The Fe2+ in the LIP reacts with hydrogen peroxide, thereby forming hydroxyl radicals via the Fenton reaction (Gao et al., 2015; Feng et al., 2020; He et al., 2020). These highly toxic hydroxyl radicals attack the lipid membrane, DNA, and proteins to cause lipid ROS production, disrupting cellular function and leading to ferroptosis. Moreover, iron can be exported from the cells by ferroportin, which contributes to the reduction of the intracellular Fe2+ concentration. Therefore, ferroportin may serve as a potential therapeutic target for ICH. However, Li et al. (2019) showed that the hemoglobin content and hematoma volume gradually decrease after ICH, and they illustrated that the endogenous clearance system can be activated and eliminates the hematoma after ICH. Several studies have indicated that the decrease in hematoma volume might be related with erythrophagocytosis (Cao et al., 2016; Ni et al., 2016; Chang et al., 2020). Since intraparenchymal hematomas and red blood cells are the major source of free iron in the ICH brain, hematoma resolution and the clearance and phagocytosis of red blood cells might reduce iron-induced ferroptosis. Further studies are warranted to investigate these approaches to tackle iron-induced ferroptosis.
Depletion of GSH
Glutathione biosynthesis is intricately linked to the regulation of ferroptosis. As a physiological defense under normal conditions, ROS can be maintained at a stable level via mitochondrial oxidative phosphorylation and antioxidant mechanisms. However, upon ICH, substantial amounts of ROS are produced, which cause cell damage, ultimately leading to ferroptosis. This event activates GSH in the brain as an antioxidant enzyme. Thus, the inhibition of GSH synthesis is predicted to induce ferroptosis. Intracellular glutamate is exchanged for cysteine, and system Xc- transports cysteine into neurons at a 1:1 ratio. Cysteine, glutamate, and glycine are used in GSH synthesis (Dixon et al., 2014; Gao et al., 2015), whereas erastin can inhibit the expression of system Xc-, causing GSH depletion and ultimately leading to ROS accumulation (Dixon et al., 2012). Therefore, the concentration and balance of these amino acids can impact ferroptosis. For example, high extracellular concentrations of glutamate and low intracellular concentrations of cysteine inhibit the function of system Xc- and induce ferroptosis. This may explain the toxic effects of glutamate following its accumulation at high levels in the nervous system.
Lipid Peroxidation
Lipid peroxidation is the key process that directly activates ferroptosis. It involves three pathways: (i) the non-enzymatic pathway generates lipid ROS through the Fenton reaction using iron (Bai et al., 2019; He et al., 2020); (ii) the oxygenation and esterification of polyunsaturated fatty acids generate lipid peroxides (Yang et al., 2016; Doll and Conrad, 2017; Stockwell, 2018; Das, 2019); and (iii) iron catalyzes lipid auto-oxidation. Some studies have reported that exogenous arachidonic acid (AA) and AA-OOH-phosphatidylethanolamine (PE) enhance ferroptosis. The conversion of AA-OOH-PE to AA in cells requires lipoxygenase enzymes. AA-OOH-PE is then reduced to AA-OH-PE by GPX4. Ferroptosis occurs when the AA-OH-PE levels are sufficiently high to surpass the cellular threshold (Kagan et al., 2017). Under physiological conditions, toxic lipid peroxides are reduced to non-toxic lipid alcohols by GPX4 to protect cells against oxidative stress (Kagan et al., 2017; Zhang Z. et al., 2018; Vuckovic et al., 2020). However, substantial accumulation of lipid peroxides cannot be effectively eliminated by GPX4, and the excess lipid peroxide damages membrane integrity, leading to cell rupture and ferroptosis, which has been proven in proteomics studies (Forcina and Dixon, 2019; Fujii et al., 2019). Therefore, the processes that inhibit the reduction in lipid peroxide levels and promote their formation result in the accumulation of lipid peroxides and, ultimately, ferroptosis (Stoyanovsky et al., 2019). However, the lipid autoxidation in ICH is not completely clear and should be further examined (Qiu et al., 2020).
Relationships Between Ferroptosis and Other Cell Death Pathways
Table 1 shows an overview and comparison of different neuronal cell death types: necroptosis, apoptosis, oxytosis, and pyroptosis. Each type, along with its characteristics and mechanisms, and their potential roles in brain damage after ICH, are discussed below, and are compared with the corresponding features of ferroptosis.

Table 1. Comparison of different neuronal cell death types.
Ferroptosis and Necroptosis
Numerous reports have suggested that ferroptosis is always accompanied by necroptosis. Necroptosis is activated during inflammation (Oberst, 2016). The activation of necroptosis relies on receptor-interacting kinase 1 (RIPK1), receptor-interacting protein kinase 3 (RIPK3), and mixed lineage kinase domain-like (MLKL; Dondelinger et al., 2014; Conrad et al., 2016). RIPK1 activates RIPK3 and thereby recruits MLKL at the cell membrane, which causes membrane rupture and eventually triggers necroptosis (Cook et al., 2014; Hildebrand et al., 2014). Some studies have demonstrated that the ultrastructure of neurons after ICH manifests as a morphotype, involving ferroptosis and necroptosis. Ferroptosis is characterized by the loss of plasma membrane integrity, disruption and swelling of organelles, and shrunken mitochondria, whereas necroptosis does not involve mitochondrial shrinkage, suggesting that shrunken mitochondria are a characteristic change in ferroptosis after ICH (Li et al., 2018). The major ultrastructural characteristics of hemin-induced neuron death are related to ferroptosis and not necroptosis. In contrast, molecular marker levels of both ferroptosis (Fe3+ iron, GSH, and GPX4) and necroptosis (MLKL and RIPK3) may increase after ICH; however, such studies regarding ICH are lacking (Roderick et al., 2014; Lin et al., 2016; Newton et al., 2016; Minagawa et al., 2020). NADPH might be a link between ferroptosis and necroptosis (Hou et al., 2019). When cells undergo necroptosis or ferroptosis, adjacent cells are more prone to another form of cell death. Moreover, necrostatin-1 was shown to exert its neuroprotective effects by inhibiting the RIPK1 and RIPK3 signal pathways of necroptosis and suppressing apoptosis and autophagy after ICH (Chang et al., 2014; Cook et al., 2014; Su et al., 2015), highlighting its relevance to necroptosis, apoptosis, and autophagy; however, this does not clarify the specific link between ferroptosis and necroptosis, which require further studies.
Ferroptosis and Apoptosis
Apoptosis is one of the most well-studied forms of programmed cell death. It includes two major pathways: extrinsic and intrinsic pathways. The extrinsic pathway is triggered by cell surface receptors, such as tumor necrosis factor (TNF) receptors (Hasegawa et al., 2011; Fricker et al., 2018; Zhao et al., 2018). The TNF receptors can be activated by caspase-1, which is stimulated by the P2X7 receptor (Lee et al., 2016). Upon binding to the TNF receptor, the Fas-associated death domain protein will be recruited and will bind pro-caspase-8 molecules, activating caspase-8 and allowing apoptosis to occur (Micheau and Tschopp, 2003). The intrinsic pathway is activated by the mitochondrial outer membrane permeabilization (MOMP) and the B-cell lymphoma 2 (Bcl-2) family proteins. As a member of Bcl-2 family, Bax can promote apoptosis by its expression and activation (Haase et al., 2008). Furthermore, Zhang et al. showed that the increased Bax/Bcl-2 ratio can promote apoptosis (Zhang W. et al., 2018). Therefore, Bcl-2 can be considered as an inhibitor of apoptosis. The ultrastructural features of an apoptotic neural cell include chromatin condensation, nuclear shrinkage, and DNA fragmentation (Castagna et al., 2016; Li et al., 2018). Compared with other types of cell death, DNA fragmentation can be considered as the distinguishing characteristic of apoptosis. Therefore, the presence of DNA fragments indicates apoptosis, which can occur at a later ICH phase (Kanter et al., 2016). Ferroptosis is an iron-dependent form of programmed cell death, but it is non-apoptotic, non-necroptotic, and non-autophagic. Ferroptosis can also be inhibited by iron chelators and several novel small molecules, such as ferrostatin-1 and liproxstatin-1, but cannot be inhibited by specific inhibitors of apoptosis (e.g., zVAD; Thermozier et al., 2020) or necroptosis (e.g., necrostatin-1; Chang et al., 2014; Chen X. et al., 2019).
Ferroptosis and Oxytosis
Oxytosis is induced by the glutamate-mediated inhibition of system Xc-, which leads to the depletion of GSH; it represents a distinct type of oxidative neuronal cell death after ICH (Landshamer et al., 2008; Grohm et al., 2010). Depletion of GSH causes excessive ROS production, which cannot be eliminated, leading to ROS accumulation in neurons and, ultimately, oxytosis (Tobaben et al., 2010; Reuther et al., 2014; Jelinek et al., 2016). Oxytosis is highly similar to ferroptosis; it has even been regarded as a component of ferroptosis (Lewerenz et al., 2018; Gupta et al., 2020). A previous study suggested that BID protein inhibitors can prevent erastin-induced ferroptosis (Neitemeier et al., 2017), and that inhibitors of ferroptosis, such as ferrostatin-1 and liproxstatin-1, can block glutamate-induced oxytosis. The study further showed that erastin-induced ferroptosis in neuronal cells is accompanied by mitochondrial transactivation of BID, loss of mitochondrial membrane potential, enhanced mitochondrial fragmentation, and reduced ATP levels. These signs of mitochondrial death are distinctive features of oxytosis (Lewerenz et al., 2018; Takashima et al., 2019; Nagase et al., 2020). However, because ferroptosis cannot be readily distinguished from oxytosis, the relationship between ferroptosis and oxytosis remains unclear and controversial. Therefore, further research is needed to clarify the differences between these two types of cell death.
Ferroptosis and Pyroptosis
Pyroptosis is another type of neuronal cell death and is activated by gasdermin D (GSDMD) and caspase-1 or caspase-11 in mice (corresponding to caspase-1, caspase-4, and caspase-5, respectively, in humans), consequently upregulating cytokines [interleukin (IL)-1β, IL-18] and immune system activators (Ruehl and Broz, 2015; Chen K.W. et al., 2019). Upon activation, caspase-1, or caspase-11 (in mice) acts on GSDMD and generates the N-terminal domain and C-terminal domain, and the lipid-selective N-terminal domain acts on the cell membrane by combining with phosphatidylinositol of the lipid plasma membrane, which leads to cell lysis. Cytokines such as IL-1β and IL-18 are secreted through the damaged cell membrane, recruiting immune cells, and activating the immune system, and ultimately resulting in pyroptosis (Shi et al., 2015; Ding et al., 2016; Feng et al., 2018). Pyroptosis is characterized by nuclear condensation coupled with cell swelling and lipid membrane vacuole formation at the plasma membrane, which eventually ruptures without exhibiting DNA fragmentation (Li et al., 2018), making the process distinct from ferroptosis. The activation of caspase-1 relies on the accumulation of a type of protein complex known as the inflammasome. Nucleotide-binding oligomerization domain-like receptor 3 (NLRP3) protein binds to apoptosis-related speck-like protein through its pyridine domain and recruits pro-caspase-1 via CARD–CARD interaction, forming NLRP3 inflammasomes (Li et al., 2020). NLRP3 can also be activated by K+ efflux, which is indirectly induced by caspase-11. In addition to recruiting and activating caspase-1 via apoptosis-related speck-like protein, inflammasomes can also bind with caspase-8 to induce cell death. Some studies also found that other members of the gasdermin family (e.g., GSDMA, GSDMB, and GSDMC) are recruited to the plasma membrane to trigger membrane repair upon gasdermin D activation and act on membrane-disrupting cytotoxicity (Denes et al., 2015; Man and Kanneganti, 2015; Shi et al., 2015; Chen et al., 2016). Therefore, pyroptosis has attracted substantial attention as an ICH therapeutic target, and inhibitors of NLRP3 were shown to have therapeutic efficiency. The biochemical processes of ferroptosis are much simpler than those of pyroptosis.
Potential Interventions Targeting Ferroptosis After ICH
The most promising anti-ICH drugs targeting ferroptosis are summarized in Table 2. Ferroptosis is mainly activated by Fe2+ and ROS; thus, iron chelators and inhibitors of lipid ROS generation can be regarded as candidate therapeutic targets for ICH. Here, we focus on the potential targets, including iron metabolism, GSH biosynthesis, and lipid ROS.
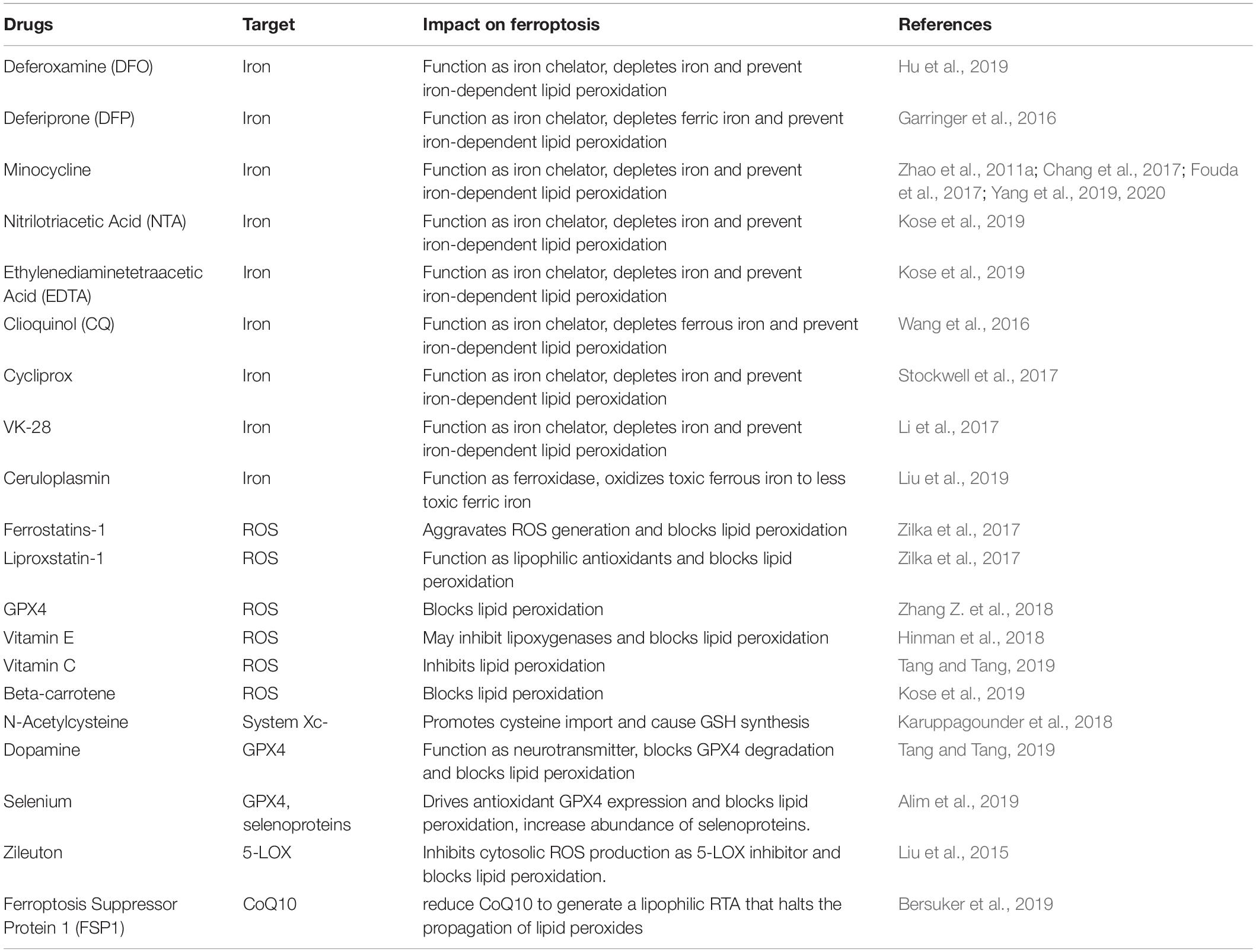
Table 2. Promising anti-ICH drugs that target ferroptosis.
Inhibitors of Iron Metabolism
Iron Chelators
Because ferroptosis is characterized by the iron-induced accumulation of lipid ROS, iron is essential for the accumulation of lipid peroxides and the consequent execution of ferroptosis. As inhibitors of iron metabolism, iron chelators deplete iron by reducing the Fe2+ in LIP to prevent iron-dependent lipid peroxidation (Doll and Conrad, 2017), reduce the lipid ROS, and eventually inhibit ferroptosis. As a result, the potential therapeutic use of iron chelators has attracted substantial attention in recent years (De Domenico et al., 2009). Hu et al. showed that deferoxamine (DFO) treatment decreased hemin release from a hematoma, and demonstrated the potential role of DFO in reducing iron deposition and brain injury in a piglet model (Hu et al., 2019). However, a phase I clinical trial found that DFO led to hypotension, pancytopenia, retinal toxicity, and neurotoxicity (Selim et al., 2011). Li et al. (2017) further identified VK-28 as an iron chelator with similar effects to those of DFO in a rat model. However, compared with DFO, VK-28 improves neurobehavioral performance by polarizing the microglia to an M2-like phenotype, which reduces brain water content, decreases white matter injury, and eventually reduces the overall mortality due to ICH. Selim et al. (2019) showed that DFO was ineffective in improving the clinical outcome on day 90 in ICH patients of the phase II clinical trials. In this context, VK-28 appears to be more effective and safer than DFO. Another iron chelator, deferiprone (DFP), showed efficacy to some extent in humans and experimental neurodegenerative and neurodevelopmental conditions (Garringer et al., 2016; Carboni et al., 2017; Tian et al., 2017; Shao et al., 2020), but a phase II clinical study shows that it caused injury to the surrounding tissue (Klopstock et al., 2019). Therefore, the clinical application of DFO is limited. Wang et al. (2016) found that in rat models, DFP could reduce iron contents after ICH, but it was found to be ineffective against brain edema and lipid ROS and failed to improve the outcome. Furthermore, some studies found that minocycline functions as an iron chelator and can reduce iron-induced neuronal cell death (Zhao et al., 2011a; Chang et al., 2017; Fouda et al., 2017; Yang et al., 2019, 2020). Minocycline is being investigated in the phase II clinical trials of Zhao et al. (2011a), which shows that minocycline inhibited iron overload, reduced iron neurotoxicity and iron-induced brain injury after ICH, which can be regarded as a new treatment option of ICH. Yang et al. (2020) shows that minocycline reduced the brain edema and hematoma volume, prevented iron accumulation, and protected brain from iron-induced injury in ICH minipig models. Several other iron chelators, including nitrilotriacetic acid, ethylenediaminetetraacetic acid, and clioquinol, are effective for treating ICH, as demonstrated in animal models (Wang et al., 2016; Kose et al., 2019). Since iron chelators are mainstream drugs of ferroptosis after ICH, the outcomes of using iron chelators in clinical and preclinical studies are summarized in Table 3.
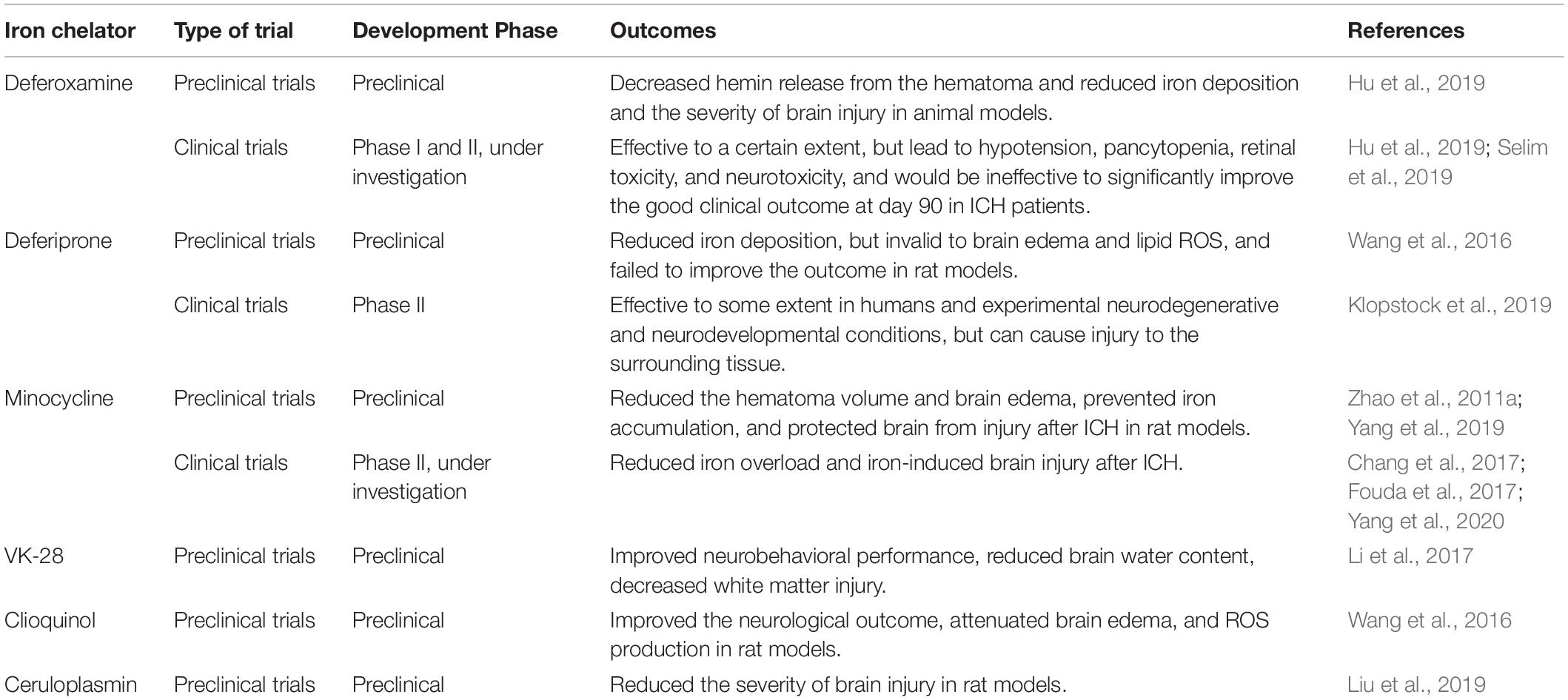
Table 3. Outcomes of some major iron chelators used in ICH in the clinical and preclinical studies.
Ferroxidase
Ferroxidase is another type of iron metabolism inhibitor. Ferroxidase can oxidize toxic Fe2+ iron to the less toxic Fe3+ iron and reduce the Fe2+ in LIP, reduce lipid ROS, and inhibit ferroptosis. In a series of studies, Liu et al. (2019) showed that ceruloplasmin is an essential ferroxidase, with the potential to reduce the severity of brain injury due to ferroptosis after ICH in a rat model. Although numerous previous studies showed that ceruloplasmin deficiency is associated with Alzheimer’s disease, Parkinson’s disease, and other neurodegenerative diseases in patients and rat models (Jiang et al., 2015; Stelten et al., 2019; Diouf et al., 2020; Liu et al., 2020; Shang et al., 2020), little is known about the therapeutic function of ceruloplasmin in ferroptosis after ICH. Moreover, clinical studies on ferroxidase in ICH patients are limited. Thus, additional studies are required to identify the specific types and roles of ferroxidase.
Promoters of GSH Biosynthesis
Glutamate and cysteine are important regulators of ferroptosis. As intracellular glutamate is pumped out of the cell and exchanged for extracellular cysteine by system Xc- to synthesize GSH and inhibit ferroptosis (Dixon et al., 2012, 2014); increasing cysteine concentration is a potential therapeutic strategy for ICH. N-acetylcysteine (NAC) is a cysteine prodrug that increases the accumulation of cysteine in neurons. Karuppagounder et al. (2018) showed that NAC has therapeutic effects as a precursor for GSH, which acts on system Xc- to promote cysteine transfer into neuronal cells and targets lipid-derived reactive electrophilic species resulting from increased arachidonate 5-lipoxygenase, along with GSH-dependent enzymes (e.g., GSH S-transferases). Simultaneously, exogenous infusion of a clinically approved protective lipid species, prostaglandin E2, reduces the NAC concentration required to stimulate protection and functional recovery in vitro and in vivo in mice (Zhao et al., 2015; Karuppagounder et al., 2018). Although nearly all of these studies used safe doses of NAC, the dose required to induce a therapeutic effect is unclear. Thus, further studies are required to determine the precise dose of NAC required for the efficient control of brain injury after ICH.
Inhibitors of Lipid ROS Generation
Reactive oxygen species production is the immediate cause of ferroptosis after ICH; thus, inhibiting ROS production is important to prevent ferroptosis in neurons after ICH (Ursini and Maiorino, 2020). During lipid oxidation, GPX4 is the primary enzyme that prevents ferroptosis (Stockwell et al., 2017; Seibt et al., 2019). Many studies have shown that ROS inhibitors (e.g., ferrostatins-1, liproxstatin-1, vitamin E, vitamin C, and beta-carotene), along with GPX4 and its promoters (e.g., dopamine and selenium), are effective in preventing ferroptosis in animal models (Imai et al., 2017; Stockwell, 2018; Bai et al., 2019; Chen X. et al., 2019; Tang and Tang, 2019; Stockwell and Jiang, 2020).
For example, the ROS inhibitors ferrostatin-1 and vitamin C aggravate ROS generation and block lipid peroxidation, and vitamin E may inhibit lipoxygenases (Zilka et al., 2017; Hinman et al., 2018; Chen B. et al., 2019). Dopamine and selenium are GPX4 inducers. Dopamine functions as a neurotransmitter to block GPX4 degradation, whereas selenium drives the expression of GPX4 to increase the abundance of selenoproteins (Alim et al., 2019; Tang and Tang, 2019), and both GPX4 inducers eventually block lipid peroxidation. Moreover, inhibitors targeting 5-lipoxygenase (e.g., zileuton) inhibit AA from oxidizing to AA-OOH-PE, suppress cytosolic ROS production, and block lipid peroxidation (Liu et al., 2015). Several recent reports have also indicated that ferroptosis suppressor protein 1, a GSH-independent ferroptosis suppressor, is recruited to the plasma membrane, where it reduces coenzyme Q10 to produce lipophilic free radicals and capture antioxidants via oxidoreductase, thereby preventing the production of lipid peroxides (Bersuker et al., 2019; Doll et al., 2019; Chen and Xie, 2020; Hadian, 2020). Another regulator of potential therapeutic significance is nuclear factor erythroid 2-related factor 2 (Dodson et al., 2019; Anandhan et al., 2020; Song and Long, 2020), which regulates hundreds of genes, including several genes directly or indirectly involved in modulating ferroptosis. Therefore, nuclear factor erythroid 2-related factor 2 regulates not only lipid metabolism, but also GSH and iron metabolism, and continues its mitochondrial function (Abdalkader et al., 2018).
Conclusion
The mechanisms underlying ferroptosis and its potential modulators after ICH are reviewed and are schematically summarized in Table 2 and Figure 1. The mechanisms of other types of programmed cell death are summarized in Figure 2. As a newly recognized form of programmed cell death, ferroptosis has attracted substantial attention for the development of new strategies to treat ICH and prevent SBI. Since 2012, rapid improvements have been made in elucidating the mechanism underlying ferroptosis and its regulation. Ferroptosis involves intracellular iron accumulation, GSH depletion, and lipid peroxidation. Thus, in theory, ferroptosis can be suppressed by iron chelators (e.g., DFO and DPF), lipophilic antioxidants (e.g., ferrostatin, liproxstatin, and vitamin E), and GSH biosynthesis promoters (e.g., NAC). Because iron plays a critical role in activating ferroptosis after ICH, iron chelators are regarded as mainstream drugs. However, several studies have indicated that DFO and DFP failed to improve the outcome of ICH, and the roles of iron and lipid autoxidation in ICH are not completely clear and should be further examined. Moreover, hematoma resolution and the clearance and phagocytosis of red blood cells might also reduce iron-induced ferroptosis, and they might therefore be considered potential therapeutic targets, warranting further clinical studies.
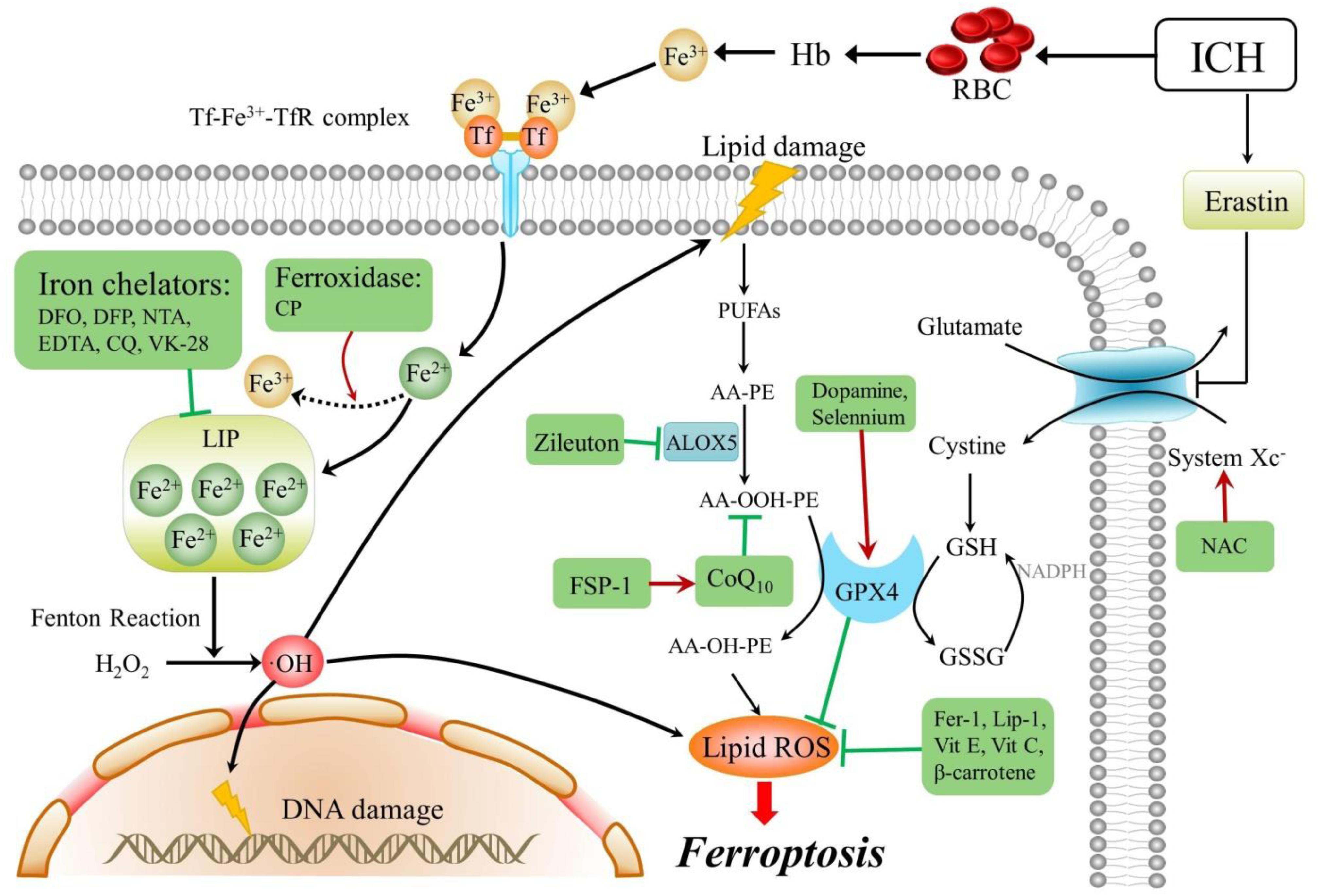
Figure 1. Mechanisms and modulators of ferroptosis after ICH. Arrows indicate promotion, blunt- ended lines indicate inhibition, and the drugs in a green box are ferroptosis inhibitors. ICH, intracerebral hemorrhage; RBC, red blood cell; Tf, transferrin; TfR, transferrin receptor; AA, arachidonic acid; PE, phosphatidylethanolamine; PUFAs, plasma membrane polyunsaturated fatty acids; CoQ 10, coenzyme Q10; DFO, deferoxamine; DFP, deferiprone; NTA, nitrilotriacetic acid; EDTA, ethylenediaminetetraacetic acid; CQ, clioquinol; CP, ceruloplasmin; H2O2, hydrogen peroxide; OH, hydroxyl radical; Fer-1, ferrostatin-1; GPX4, glutathione peroxidase 4; Vit E, vitamin E; Vit C, vitamin C; FSP-1, ferroptosis suppressor protein 1; GSH, glutathione; GSSG, oxidized glutathione; Hb, hemoglobin; Lip-1, liproxstatin-1; NAC, N-acetylcysteine; LOX, lipoxygenase; and ROS, reactive oxygen species.
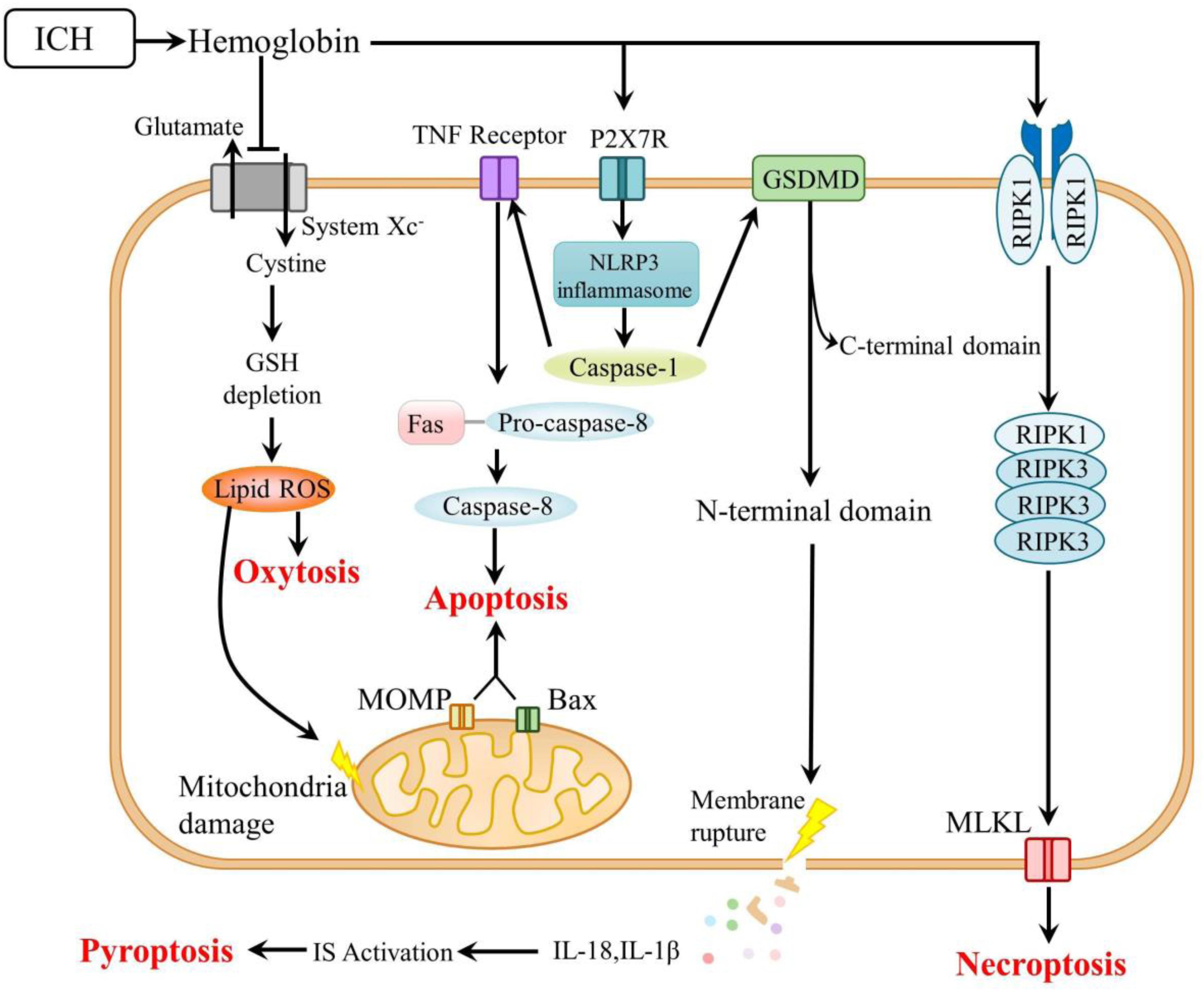
Figure 2. Mechanisms of other types of cell death after ICH. ICH, intracerebral hemorrhage; GSH, glutathione; ROS, reactive oxygen species; TNF, tumor necrosis factor; MOMP, mitochondrial outer membrane permeabilization; P2X7R, P2X7 receptor; NLRP3, nucleotide-binding oligomerization domain-like receptor 3; GSDMD, gasdermin D; RIPK, receptor-interacting protein kinase; MLKL, mixed lineage kinase domain-like; and IS, immune system.
Currently, several molecules are known to regulate ferroptosis by directly or indirectly targeting iron metabolism, GSH biosynthesis, and lipid peroxidation. Some of these regulators (e.g., BID and NADPH) have also been implicated in other types of programmed cell death. For example, BID is involved in ferroptosis and oxytosis, and NADPH links ferroptosis and necroptosis. Moreover, these various forms of cell death are not independent but are rather intricately connected, forming a network to mediate cell damage after ICH. Therefore, ferroptosis is always accompanied by other types of programmed cell death. Accordingly, an important objective for further research on ferroptosis is to identify the signaling pathways, executors of iron-dependent ROS metabolism, and ultrastructural features of each type of programmed cell death. Thus, ferroptosis can be distinguished from the other types of cell death and improved therapeutic effects against each type of cell death can be achieved.
However, several issues remain to be addressed. First, it is essential to determine whether the regulation of ferroptosis is influenced by different types of brain cells, physiological conditions, individual lifestyles, and other factors. Second, more clinical trials should be conducted to test the effects of ferroptosis inhibitors, which represent the most promising strategy to prevent SBI after ICH. Thus, further studies and clinical trials are expected to improve the understanding of ferroptosis and aid the development of new strategies for effective treatment of ICH.
Author Contributions
S-YZ and G-ZC searched the literature and drafted the manuscript. X-LY, XW, YQ, Z-NG, and HJ critically revised the manuscript. All authors have made contributions to the work and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (81971109) to HJ.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdalkader, M., Lampinen, R., Kanninen, K. M., Malm, T. M., Liddell, J. R. (2018). Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 12:466. doi: 10.3389/fnins.2018.00466
Alim, I., Caulfield, J. T., Chen, Y., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 177:1262. doi: 10.1016/j.cell.2019.03.032
Anandhan, A., Dodson, M., Schmidlin, C. J., Liu, P., Zhang, D. D. (2020). Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell Chem. Biol. 27(4):436–447. doi: 10.1016/j.chembiol.2020.03.011
Auriat, A. M., Silasi, G., Wei, Z., Paquette, R., Paterson, P., Nichol, H., et al. (2012). Ferric iron chelation lowers brain iron levels after intracerebral hemorrhage in rats but does not improve outcome. Exp. Neurol. 234, 136–143. doi: 10.1016/j.expneurol.2011.12.030
Bai, Y., Meng, L., Han, L., Jia, Y., Zhao, Y., Gao, H., et al. (2019). Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 508, 997–1003. doi: 10.1016/j.bbrc.2018.12.039
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575(7784):688. doi: 10.1038/s41586-019-1705-2
Bobinger, T., Burkardt, P., Huttner, H. B., and Manaenko, A. (2018). Programmed Cell Death after Intracerebral Hemorrhage. Curr. Neuropharmacol. 16, 1267–1281. doi: 10.2174/1570159x15666170602112851
Cao, J. Y., and Dixon, S. J. (2016). Mechanisms of ferroptosis. Cell. Mole. Life Sci. 73, 2195–2209. doi: 10.1007/s00018-016-2194-1
Cao, S., Zheng, M., Hua, Y., Chen, G., Keep, R. F., and Xi, G. (2016). Hematoma Changes During Clot Resolution After Experimental Intracerebral Hemorrhage. Stroke 47, 1626–1631. doi: 10.1161/strokeaha.116.013146
Carboni, E., Tatenhorst, L., Toenges, L., Barski, E., Dambeck, V., Baehr, M., et al. (2017). Deferiprone Rescues Behavioral Deficits Induced by Mild Iron Exposure in a Mouse Model of Alpha-Synuclein Aggregation. Neuromole. Med. 19(2-3), 309–321. doi: 10.1007/s12017-017-8447-9
Castagna, C., Merighi, A., and Lossi, L. (2016). Cell death and neurodegeneration in the postnatal development of cerebellar vermis in normal and Reeler mice. Anna. Anat. Anatomischer Anzeiger 207, 76–90. doi: 10.1016/j.aanat.2016.01.010
Chang, C. F., Massey, J., Osherov, A., Angenendt da Costa, L. H., and Sansing, L. H. (2020). Bexarotene Enhances Macrophage Erythrophagocytosis and Hematoma Clearance in Experimental Intracerebral Hemorrhage. Stroke 51, 612–618. doi: 10.1161/strokeaha.119.027037
Chang, J. J., Kim-Tenser, M., Emanuel, B. A., Jones, G. M., Chapple, K., Alikhani, A., et al. (2017). Minocycline and matrix metalloproteinase inhibition in acute intracerebral hemorrhage: a pilot study. Eur. J. Neurol. 24(11), 1384-1391. doi: 10.1111/ene.13403
Chang, P., Dong, W., Zhang, M., Wang, Z., Wang, Y., Wang, T., et al. (2014). Anti-Necroptosis Chemical Necrostatin-1 Can Also Suppress Apoptotic and Autophagic Pathway to Exert Neuroprotective Effect in Mice Intracerebral Hemorrhage Model. J. Mole. Neurosci. 52, 242–249. doi: 10.1007/s12031-013-0132-3
Chen, B., Chen, Z., Liu, M., Gao, X., Cheng, Y., Wei, Y., et al. (2019). Inhibition of neuronal ferroptosis in the acute phase of intracerebral hemorrhage shows long-term cerebroprotective effects. Brain Res. Bull. 153, 122–132. doi: 10.1016/j.brainresbull.2019.08.013
Chen, K. W., Demarco, B., Heilig, R., Shkarina, K., Boettcher, A., Farady, C. J., et al. (2019). Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. Embo. J. 38:e101638. doi: 10.15252/embj.2019101638
Chen, L., and Xie, J. (2020). Ferroptosis-Suppressor-Protein 1: A Potential Neuroprotective Target for Combating Ferroptosis. Movement Disor. 35(3):400. doi: 10.1002/mds.27990
Chen, X., He, W. T., Hu, L., Li, J., Fang, Y., Wang, X., et al. (2016). Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 26(9), 1007–1020. doi: 10.1038/cr.2016.100
Chen, X., Zhang, B., Liu, T., Feng, M., Zhang, Y., Zhang, C., et al. (2019). Liproxstatin-1 Attenuates Morphine Tolerance through Inhibiting Spinal Ferroptosis-like Cell Death. Acs Chem. Neurosci. 10, 4824–4833. doi: 10.1021/acschemneuro.9b00539
Conrad, M., Angeli, J. P., Vandenabeele, P., and Stockwell, B. R. (2016). Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 15, 348–366. doi: 10.1038/nrd.2015.6
Conrad, M., Kagan, V. E., Bayir, H., Pagnussat, G. C., Head, B., Traber, M. G., et al. (2018). Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Devel. 32, 602–619. doi: 10.1101/gad.314674.118
Cook, W. D., Moujalled, D. M., Ralph, T. J., Lock, P., Young, S. N., Murphy, J. M., et al. (2014). RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ. 21, 1600–1612. doi: 10.1038/cdd.2014.70
Das, U. N. (2019). Saturated Fatty Acids, MUFAs and PUFAs Regulate Ferroptosis. Cell Chem. Biol. 26, 309–311. doi: 10.1016/j.chembiol.2019.03.001
De Domenico, I., Ward, D. M., and Kaplan, J. (2009). Specific iron chelators determine the route of ferritin degradation. Blood 114(20), 4546–4551. doi: 10.1182/blood-2009-05-224188
Denes, A., Coutts, G., Lenart, N., Cruickshank, S. M., Pelegrin, P., Skinner, J., et al. (2015). AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. U S A 112(13), 4050–4055. doi: 10.1073/pnas.1419090112
Ding, J., Wang, K., Liu, W., She, Y., Sun, Q., Shi, J., et al. (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535(7610), 111–116. doi: 10.1038/nature18590
Diouf, I., Bush, A. I., and Ayton, S. (2020). Cerebrospinal fluid ceruloplasmin levels predict cognitive decline and brain atrophy in people with underlying beta-amyloid pathology. Neurobiol. Dis. 139:104810. doi: 10.1016/j.nbd.2020.104810
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dixon, S. J., Patel, D., Welsch, M., Skouta, R., Lee, E., Hayano, M., et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3:e02523. doi: 10.7554/eLife.02523
Djulbegovic, M. B., and Uversky, V. N. (2019). Ferroptosis - An iron- and disorder-dependent programmed cell death. Int. J. Biol. Macromole. 135, 1052–1069. doi: 10.1016/j.ijbiomac.2019.05.221
Dodson, M., Castro-Portuguez, R., Zhang, D. D. (2019). NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox. Biol. 23:101107. doi: 10.1016/j.redox.2019.101107
Doll, S., and Conrad, M. (2017). Iron and Ferroptosis: A Still Ill-Defined Liaison. Iubmb Life 69, 423–434. doi: 10.1002/iub.1616
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., da Silva, M. C., Ingold, I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575(7784):693. doi: 10.1038/s41586-019-1707-0
Dondelinger, Y., Declercq, W., Montessuit, S., Roelandt, R., Goncalves, A., Bruggeman, I., et al. (2014). MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7, 971–981. doi: 10.1016/j.celrep.2014.04.026
Feng, H., Schorpp, K., Jin, J., Yozwiak, C. E., Hoffstrom, B. G., Decker, A. M., et al. (2020). Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 30:3411. doi: 10.1016/j.celrep.2020.02.049
Feng, S., Fox, D., and Man, S. M. (2018). Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J. Mole. Biol 430(18 Pt B), 3068–80. doi: 10.1016/j.jmb.2018.07.002
Forcina, G. C., and Dixon, S. J. (2019). GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 19:e1800311. doi: 10.1002/pmic.201800311
Fouda, A. Y., Newsome, A. S., Spellicy, S., Waller, J. L., Zhi, W., Hess, D. C., et al. (2017). Minocycline in Acute Cerebral Hemorrhage: An Early Phase Randomized Trial. Stroke 48(10), 2885–2887. doi: 10.1161/strokeaha.117.018658
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal Cell Death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Fujii, J., Homma, T., and Kobayashi, S. (2019). Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Rad. Res. 23, 1–12. doi: 10.1080/10715762.2019.1666983
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and Transferrin Regulate Ferroptosis. Mole. Cell 59, 298–308. doi: 10.1016/j.molcel.2015.06.011
Garringer, H. J., Irimia, J. M., Li, W., Goodwin, C. B., Richine, B., Acton, A., et al. (2016). Effect of Systemic Iron Overload and a Chelation Therapy in a Mouse Model of the Neurodegenerative Disease Hereditary Ferritinopathy. PLoS One 11:e0161341. doi: 10.1371/journal.pone.0161341
Grohm, J., Plesnila, N., and Culmsee, C. (2010). Bid mediates fission, membrane permeabilization and peri-nuclear accumulation of mitochondria as a prerequisite for oxidative neuronal cell death. Brain Behav. Immun. 24, 831–838. doi: 10.1016/j.bbi.2009.11.015
Gupta, G., Gliga, A., Hedberg, J., Serra, A., Greco, D., Odnevall Wallinder, I., et al. (2020). Cobalt nanoparticles trigger ferroptosis-like cell death (oxytosis) in neuronal cells: Potential implications for neurodegenerative disease. Faseb J. 34(4), 5262–5281. doi: 10.1096/fj.201902191RR
Haase, G., Pettmann, B., Raoul, C., and Henderson, C. E. (2008). Signaling by death receptors in the nervous system. Curr. Opin. Neurobiol. 18, 284–291. doi: 10.1016/j.conb.2008.07.013
Hadian, K. (2020). Ferroptosis Suppressor Protein 1 (FSP1) and Coenzyme Q(10) Cooperatively Suppress Ferroptosis. Biochemistry 59(5):637–638. doi: 10.1021/acs.biochem.0c00030
Hasegawa, Y., Suzuki, H., Sozen, T., Altay, O., and Zhang, J. H. (2011). Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neuroch. Suppl. 110(Pt 1), 43–48. doi: 10.1007/978-3-7091-0353-1_8
He, Y.-J., Liu, X.-Y., Xing, L., Wan, X., Chang, X., and Jiang, H.-L. (2020). Fenton reaction-independent ferroptosis therapy via glutathione and iron redox couple sequentially triggered lipid peroxide generator. Biomaterials 241:119911. doi: 10.1016/j.biomaterials.2020.119911
Hentze, M. W., Muckenthaler, M. U., Galy, B., and Camaschella, C. (2010). Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 142, 24–38. doi: 10.1016/j.cell.2010.06.028
Hildebrand, J. M., Tanzer, M. C., Lucet, I. S., Young, S. N., Spall, S. K., Sharma, P., et al. (2014). Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. U S A 111, 15072–15077. doi: 10.1073/pnas.1408987111
Hinman, A., Holst, C. R., Latham, J. C., Bruegger, J. J., Ulas, G., McCusker, K. P., et al. (2018). Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS One 13:e0201369. doi: 10.1371/journal.pone.0201369
Hirschhorn, T., and Stockwell, B. R. (2019). The development of the concept of ferroptosis. Free Rad. Biol. Med. 133, 130–143. doi: 10.1016/j.freeradbiomed.2018.09.043
Hou, L., Huang, R., Sun, F., Zhang, L., and Wang, Q. N. A. D. P. H. (2019). oxidase regulates paraquat and maneb-induced dopaminergic neurodegeneration through ferroptosis. Toxicology 417, 64–73. doi: 10.1016/j.tox.2019.02.011
Hu, S., Hua, Y., Keep, R. F., Feng, H., and Xi, G. (2019). Deferoxamine therapy reduces brain hemin accumulation after intracerebral hemorrhage in piglets. Exp. Neurol. 318, 244–250. doi: 10.1016/j.expneurol.2019.05.003
Imai, H., Matsuoka, M., Kumagai, T., Sakamoto, T., Koumura, T. (2017). Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 403, 143–70.
Jelinek, A., Neitemeier, S., and Culmsee, C. (2016). A key role for Bid-mediated mitochondrial damage in paradigms of oxytosis and ferroptosis. Naunyn Schmiedebergs Archiv. Pharmacol. 389, S19–S.
Jiang, R., Hua, C., Wan, Y., Jiang, B., Hu, H., Zheng, J., et al. (2015). Hephaestin and Ceruloplasmin Play Distinct but Interrelated Roles in Iron Homeostasis in Mouse Brain. J. Nutr. 145(5), 1003–1009. doi: 10.3945/jn.114.207316
Kagan, V. E., Mao, G. W., Qu, F., Angeli, J. P. F., Doll, S., St Croix, C., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. doi: 10.1038/nchembio.2238
Kanter, M., Unsal, C., Aktas, C., and Erboga, M. (2016). Neuroprotective effect of quercetin against oxidative damage and neuronal apoptosis caused by cadmium in hippocampus. Toxicol. Industr. Health 32, 541–550. doi: 10.1177/0748233713504810
Karuppagounder, S. S., Alin, L., Chen, Y., Brand, D., Bourassa, M. W., Dietrich, K., et al. (2018). N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E-2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann. Neurol. 84(6), 854–872. doi: 10.1002/ana.25356
Klopstock, T., Tricta, F., Neumayr, L., Karin, I., Zorzi, G., Fradette, C., et al. (2019). Safety and efficacy of deferiprone for pantothenate kinase-associated neurodegeneration: a randomised, double-blind, controlled trial and an open-label extension study. Lancet Neurol. 18(7), 631–642. doi: 10.1016/s1474-4422(19)30142-5
Kose, T., Vera-Aviles, M., Sharp, P. A., and Latunde-Dada, G. O. (2019). Curcumin and (-)- Epigallocatechin-3-Gallate Protect Murine MIN6 Pancreatic Beta-Cells against Iron Toxicity and Erastin-Induced Ferroptosis. Pharmaceuticals 12:26. doi: 10.3390/ph12010026
Landshamer, S., Hoehn, M., Barth, N., Duvezin-Caubet, S., Schwake, G., Tobaben, S., et al. (2008). Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell Death Differ. 15, 1553–1563. doi: 10.1038/cdd.2008.78
Lee, M. S., Kwon, H., Lee, E. Y., Kim, D. J., Park, J. H., Tesh, V. L., et al. (2016). Shiga Toxins Activate the NLRP3 Inflammasome Pathway To Promote Both Production of the Proinflammatory Cytokine Interleukin-1β and Apoptotic Cell Death. Infect. Immun. 84, 172–186. doi: 10.1128/iai.01095-15
Lewerenz, J., Ates, G., Methner, A., Conrad, M., Maher, P. (2018). Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System. Front. Neurosci. 12:214. doi: 10.3389/fnins.2018.00214
Li, D. X., Wang, C. N., Wang, Y., Ye, C. L., Jiang, L., Zhu, X. Y, et al. (2020). NLRP3 inflammasome-dependent pyroptosis and apoptosis in hippocampus neurons mediates depressive-like behavior in diabetic mice. Behav. Brain Res. 391:112684. doi: 10.1016/j.bbr.2020.112684
Li, Q., Wan, J., Lan, X., Han, X., Wang, Z., and Wang, J. (2017). Neuroprotection of brain-permeable iron chelator VK-28 against intracerebral hemorrhage in mice. J. Cereb. Blood Flow Metabol. 37(9), 3110–3123. doi: 10.1177/0271678x17709186
Li, Q., Weiland, A., Chen, X., Lan, X., Han, X., Durham, F., et al. (2018). Ultrastructural Characteristics of Neuronal Death and White Matter Injury in Mouse Brain Tissues After Intracerebral Hemorrhage: Coexistence of Ferroptosis, Autophagy, and Necrosis. Front. Neurol. 9:581. doi: 10.3389/fneur.2018.00581
Li, R., Liu, Z., Wu, X., Yu, Z., Zhao, S., and Tang, X. (2019). Lithium chloride promoted hematoma resolution after intracerebral hemorrhage through GSK-3β-mediated pathways-dependent microglia phagocytosis and M2-phenotype differentiation, angiogenesis and neurogenesis in a rat model. Brain Res. Bull. 152, 117–127. doi: 10.1016/j.brainresbull.2019.07.019
Lin, J., Kumari, S., Kim, C., Van, T.-M., Wachsmuth, L., Polykratis, A., et al. (2016). RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540:124. doi: 10.1038/nature20558
Liu, H., Hua, Y., Keep, R. F., and Xi, G. (2019). Brain Ceruloplasmin Expression After Experimental Intracerebral Hemorrhage and Protection Against Iron-Induced Brain Injury. Transl. Stroke Res. 10(1), 112–119. doi: 10.1007/s12975-018-0669-0
Liu, X., Zhong, S., Yan, L., Zhao, H., Wang, Y., Hu, Y., et al. (2020). Correlations among mRNA expression levels of ATP7A, serum ceruloplasmin levels and neuronal metabolism in unmedicated major depressive disorder. Int. J. Neuropsychopharmacol. 19:pyaa038. doi: 10.1093/ijnp/pyaa038
Liu, Y., Wang, W., Li, Y., Xiao, Y., Cheng, J., Jia, J. (2015). The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biol. Pharmac. Bull. 38(8), 1234–1239. doi: 10.1248/bpb.b15-00048
Man, S. M., and Kanneganti, T. D. (2015). Gasdermin D: the long-awaited executioner of pyroptosis. Cell Res. 25(11), 1183–1184. doi: 10.1038/cr.2015.124
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190. doi: 10.1016/s0092-8674(03)00521-x
Minagawa, S., Yoshida, M., Araya, J., Hara, H., Imai, H., and Kuwano, K. (2020). Regulated Necrosis in Pulmonary Disease. A Focus on Necroptosis and Ferroptosis. Am. J. Respirat. Cell Mole. Biol. 62, 554–562. doi: 10.1165/rcmb.2019-0337TR
Morotti, A., and Goldstein, J. N. (2016). Diagnosis and Management of Acute Intracerebral Hemorrhage. Emerg. Med. Clin. North Am. 34:883. doi: 10.1016/j.emc.2016.06.010
Nagase, H., Katagiri, Y., Oh-hashi, K., Geller, H. M., Hirata, Y. (2020). Reduced Sulfation Enhanced Oxytosis and Ferroptosis in Mouse Hippocampal HT22 Cells. Biomolecules 10:92. doi: 10.3390/biom10010092
Neitemeier, S., Jelinek, A., Laino, V., Hoffmann, L., Eisenbach, I., Eying, R., et al. (2017). BID links ferroptosis to mitochondrial cell death pathways. Redox. Biol. 12, 558–570. doi: 10.1016/j.redox.2017.03.007
Newton, K., Dugger, D. L., Maltzman, A., Greve, J. M., Hedehus, M., Martin-McNulty, B., et al. (2016). RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Different. 23, 1565–1576. doi: 10.1038/cdd.2016.46
Ni, W., Mao, S., Xi, G., Keep, R. F., and Hua, Y. (2016). Role of Erythrocyte CD47 in Intracerebral Hematoma Clearance. Stroke 47, 505–511. doi: 10.1161/strokeaha.115.010920
Oberst, A. (2016). Death in the fast lane: what’s next for necroptosis? Febs J. 283, 2616–2625. doi: 10.1111/febs.13520
Qiu, Y., Cao, Y., Cao, W., Jia, Y., and Lu, N. (2020). The Application of Ferroptosis in Diseases. Pharmacolo. Res. 159:104919. doi: 10.1016/j.phrs.2020.104919
Reuther, C., Ganjam, G. K., Dolga, A. M., Culmsee, C. (2014). The serine protease inhibitor TLCK attenuates intrinsic death pathways in neurons upstream of mitochondrial demise. Apoptosis 19(11), 1545–1558. doi: 10.1007/s10495-014-1027-7
Roderick, J. E., Hermance, N., Zelic, M., Simmons, M. J., Polykratis, A., Pasparakis, M., et al. (2014). Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc. Nat. Acad. Sci. U S A 111, 14436–14441. doi: 10.1073/pnas.1409389111
Ruehl, S., and Broz, P. (2015). Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+ efflux. Europ. J. Immunol 45(10), 2927–2936. doi: 10.1002/eji.201545772
Seibt, T. M., Proneth, B., and Conrad, M. (2019). Role of GPX4 in ferroptosis and its pharmacological implication. Free Rad. Biol. Med. 133, 144–152. doi: 10.1016/j.freeradbiomed.2018.09.014
Selim, M., Foster, L. D., Moy, C. S., Xi, G., Hill, M. D., Morgenstern, L. B., et al. (2019). Deferoxamine mesylate in patients with intracerebral haemorrhage (i-DEF): a multicentre, randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 18(5), 428–438. doi: 10.1016/s1474-4422(19)30069-9
Selim, M., Yeatts, S., Goldstein, J. N., Gomes, J., Greenberg, S., Morgenstern, L. B., et al. (2011). Safety and tolerability of deferoxamine mesylate in patients with acute intracerebral hemorrhage. Stroke 42(11), 3067–3074. doi: 10.1161/strokeaha.111.617589
Shang, Y., Luo, M., Yao, F., Wang, S., Yuan, Z., and Yang, Y. (2020). Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cellular Sign. 72:109633. doi: 10.1016/j.cellsig.2020.109633
Shao, C., Yuan, J., Liu, Y., Qin, Y., Wang, X., Gu, J., et al. (2020). Epileptic brain fluorescent imaging reveals apigenin can relieve the myeloperoxidase-mediated oxidative stress and inhibit ferroptosis. Proc. Natl. Acad. Sci. U S A 117(19), 10155–10164. doi: 10.1073/pnas.1917946117
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526(7575), 660–665. doi: 10.1038/nature15514
Song, X. and Long D. (2020). Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 14:267. doi: 10.3389/fnins.2020.00267
Stelten, B. M. L., van Ommen, W., and Keizer, K. (2019). Neurodegeneration With Brain Iron Accumulation A Novel Mutation in the Ceruloplasmin Gene. JAMA Neurol. 76(2), 229–230. doi: 10.1001/jamaneurol.2018.3230
Stockwell, B. R. (2018). Ferroptosis: Death by lipid peroxidation. Free Rad. Biol. Med. 120, S7–S. doi: 10.1016/j.freeradbiomed.2018.04.034
Stockwell, B. R., Angeli, J. P. F., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171(2), 273–285. doi: 10.1016/j.cell.2017.09.021
Stockwell, B. R., and Jiang, X. (2020). The Chemistry and Biology of Ferroptosis. Cell Chem. Biol. 27(4), 365–375. doi: 10.1016/j.chembiol.2020.03.013
Stoyanovsky, D. A., Tyurina, Y. Y., Shrivastava, I., Bahar, I., Tyurin, V. A., Protchenko, O., et al. (2019). Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Rad. Biol. Med. 133, 153–161. doi: 10.1016/j.freeradbiomed.2018.09.008
Su, X., Wang, H., Kang, D., Zhu, J., Sun, Q., Li, T., et al. (2015). Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting RIP1/RIP3 pathway. Neurochem. Res. 40, 643–650. doi: 10.1007/s11064-014-1510-0
Takashima, M., Ichihara, K., Hirata, Y. (2019). Neuroprotective effects of Brazilian green propolis on oxytosis/ferroptosis in mouse hippocampal HT22 cells. Food Chem. Toxicol. 132:110669. doi: 10.1016/j.fct.2019.110669
Tang, H. M., and Tang, H. L. (2019). Cell recovery by reversal of ferroptosis. Biol. Open 8:bio043182. doi: 10.1242/bio.043182
Thermozier, S., Hou, W., Zhang, X., Shields, D., Fisher, R., Bayir, H., et al. (2020). Anti-Ferroptosis Drug Enhances Total-Body Irradiation Mitigation by Drugs that Block Apoptosis and Necroptosis. Radiat. Res. 193, 435–450. doi: 10.1667/rr15486.1
Tian, Y., He, Y., Song, W., Zhang, E., Xia, X. (2017). Neuroprotective effect of deferoxamine on N-methyl-D-aspartate-induced excitotoxicity in RGC-5 cells. Acta Biochim. Et Biophys. Sin. 49(9), 827–834. doi: 10.1093/abbs/gmx082
Tobaben, S., Grohm, J., Seiler, A., Conrad, M., Plesnila, N., and Culmsee, C. (2010). Bid-induced damage of mitochondrial integrity plays a key role in neuronal oxytosis. Naunyn Schmiedebergs Archiv. Pharmacol. 381:22.
Ursini, F., and Maiorino, M. (2020). Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Rad. Biol. Med. 152, 175–185. doi: 10.1016/j.freeradbiomed.2020.02.027
Van Asch, C. J. J., Luitse, M. J. A., Rinkel, G. E., van der Tweel, I., Algra, A., and Klijn, C. J. M. (2010). Incidence, case fatality, and functional outcome of intracerebral haemorrhage overtime, according to age, sex, and ethnic origin: a systematic review and meta-analysis. Lancet Neurol. 9, 167–176. doi: 10.1016/s1474-4422(09)70340-0
Vuckovic, A.-M., Travain, V. B., Bordin, L., Cozza, G., Miotto, G., Rossetto, M., et al. (2020). Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3 epsilon. Febs Lett. 594, 611–624. doi: 10.1002/1873-3468.13631
Wang, G., Hu, W., Tang, Q., Wang, L., Sun, X.-G., Chen, Y., et al. (2016). Effect Comparison of Both Iron Chelators on Outcomes, Iron Deposit, and Iron Transporters After Intracerebral Hemorrhage in Rats. Mole. Neurobiol. 53, 3576–3585. doi: 10.1007/s12035-015-9302-3
Xi, G. H., Keep, R. F., and Hoff, J. T. (2006). Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 5, 53–63. doi: 10.1016/s1474-4422(05)70283-0
Yang, H., Gao, X. J., Li, Y. J., Su, J. B., Zhang, X., et al. (2019). Minocycline reduces intracerebral hemorrhage-induced white matter injury in piglets. CNS Neurosci. Ther. 25(10), 1195–1206. doi: 10.1111/cns.13220
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., and Stockwell, B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Nat. Acad. Sci. U S A. 113, E4966–E4975. doi: 10.1073/pnas.1603244113
Yang, Y., Zhang, K., Yin, X., Lei, X., Chen, X., Wang, J., et al. (2020). Quantitative Iron Neuroimaging Can Be Used to Assess the Effects of Minocycline in an Intracerebral Hemorrhage Minipig Model. Transl. Stroke Res. 11(3), 503–516. doi: 10.1007/s12975-019-00739-2
Zhang, P., Chen, L., Zhao, Q., Du, X., Bi, M., Li, Y., et al. (2020). Ferroptosis was more initial in cell death caused by iron overload and its underlying mechanism in Parkinson’s disease. Free Rad. Biol. Med. 152, 227–234. doi: 10.1016/j.freeradbiomed.2020.03.015
Zhang, W., Cui, Y., Gao, J., Li, R., Jiang, X., Tian, Y., et al. (2018). Recombinant Osteopontin Improves Neurological Functional Recovery and Protects Against Apoptosis via PI3K/Akt/GSK-3β Pathway Following Intracerebral Hemorrhage. Med. Sci. Mon. 24, 1588–1596. doi: 10.12659/msm.905700
Zhang, Z., Wu, Y., Yuan, S., Zhang, P., Zhang, J., Li, H., et al. (2018). Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 1701, 112–125. doi: 10.1016/j.brainres.2018.09.012
Zhao, F., Hua, Y., He, Y., Keep, R. F., and Xi, G. (2011a). Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke 42(12), 3587–3593. doi: 10.1161/strokeaha.111.623926
Zhao, F., Song, S., Liu, W., Keep, R. F., Xi, G., and Hua, Y. (2011b). Red blood cell lysis and brain tissue-type transglutaminase upregulation in a hippocampal model of intracerebral hemorrhage. Acta Neurochirurgica Suppl. 111, 101–105. doi: 10.1007/978-3-7091-0693-8_16
Zhao, H., Chen, Y., and Feng, H. (2018). P2X7 Receptor-Associated Programmed Cell Death in the Pathophysiology of Hemorrhagic Stroke. Curr. Neuropharmacol. 16, 1282–1295. doi: 10.2174/1570159x16666180516094500
Zhao, X., Wu, T., Chang, C. F., Wu, H., Han, X., Li, Q., et al. (2015). Toxic role of prostaglandin E-2 receptor EP1 after intracerebral hemorrhage in mice. Brain Behav. Immun. 46, 293–310. doi: 10.1016/j.bbi.2015.02.011
Zilka, O., Shah, R., Li, B., Angeli, J. PF., Griesser, M., Conrad, M., et al. (2017). On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Centr. Sci. 3(3), 232–243. doi: 10.1021/acscentsci.7b00028
Keywords: ferroptosis, intracerebral hemorrhage, iron, lipid peroxides, glutathione, programmed cell death
Citation: Zhou S-Y, Cui G-Z, Yan X-L, Wang X, Qu Y, Guo Z-N and Jin H (2020) Mechanism of Ferroptosis and Its Relationships With Other Types of Programmed Cell Death: Insights for Potential Interventions After Intracerebral Hemorrhage. Front. Neurosci. 14:589042. doi: 10.3389/fnins.2020.589042
Received: 30 July 2020; Accepted: 05 October 2020;
Published: 13 November 2020.
Edited by:
Gang Chen, First Affiliated Hospital of Soochow University, ChinaReviewed by:
Shilei Hao, Chongqing University, ChinaChe-Feng Chang, National Taiwan University, Taiwan
Copyright © 2020 Zhou, Cui, Yan, Wang, Qu, Guo and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hang Jin, ZG9jdG9yamluaGFuZ0Bob3RtYWlsLmNvbQ==; Zhen-Ni Guo, emhlbjFuaTJAMTYzLmNvbQ==
†These authors have contributed equally to this work