 Zhengjie Yang
Zhengjie Yang Xiaona Zhang
Xiaona Zhang Chengqian Li
Chengqian Li Song Chi
Song Chi Anmu Xie
Anmu Xie- Department of Neurology, The Affiliated Hospital of Qingdao University, Qingdao, China
Sleep–wake disruptions are among the most prevalent and burdensome non-motor symptoms of Parkinson’s disease (PD). Clinical studies have demonstrated that these disturbances can precede the onset of typical motor symptoms by years, indicating that they may play a primary function in the pathogenesis of PD. Animal studies suggest that sleep facilitates the removal of metabolic wastes through the glymphatic system via convective flow from the periarterial space to the perivenous space, upregulates antioxidative defenses, and promotes the maintenance of neuronal protein homeostasis. Therefore, disruptions to the sleep–wake cycle have been associated with inefficient metabolic clearance and increased oxidative stress in the central nervous system (CNS). This leads to excessive accumulation of alpha-synuclein and the induction of neuronal loss, both of which have been proposed to be contributing factors to the pathogenesis and progression of PD. Additionally, recent studies have suggested that PD-related pathophysiological alterations during the prodromal phase disrupt sleep and circadian rhythms. Taken together, these findings indicate potential mechanistic interactions between sleep–wake disorders and PD progression as proposed in this review. Further research into the hypothetical mechanisms underlying these interactions would be valuable, as positive findings may provide promising insights into novel therapeutic interventions for PD.
Introduction
Parkinson’s disease (PD) is one of the most prevalent neurodegenerative diseases worldwide and is associated with a significant burden on health care systems. Whereas the defining motor symptoms and pathological features of the disease, such as the loss of dopaminergic neurons and Lewy pathology, are attributed to the accumulation of alpha-synuclein, PD is recognized as a multisystem disorder associated with diverse non-motor symptoms (Munhoz et al., 2015). Disturbances of sleep patterns are among the most burdensome defects and manifest as insomnia, parasomnia, disturbances of wakefulness, or disruption of circadian rhythms, occurring with high prevalence during the course of the disease (Politis et al., 2010; Chahine et al., 2017). Subjective sleep complaints have been found in nearly half of newly diagnosed PD patients and result in prominent adverse effects on their quality of life (Breen et al., 2014). In a multicenter survey assessing the incidence of non-motor complaints in PD patients, sleep problems were present in 64.1% of subjects, making it the second most frequent non-motor symptom (Barone et al., 2009). Other studies have reported the prevalence of sleep disruptions in PD patients to be even higher, up to 73.7% (Maeda et al., 2017). Interestingly, accumulating evidence suggests that the onset of sleep–wake disturbances occurs years before the typical motor manifestations of PD, providing promising insight into the potential disease-modifying effect of sleep disturbances (Hastings and Goedert, 2013; Videnovic et al., 2014; Abbott and Videnovic, 2016). The rapid eye movement (REM) sleep behavior disorder (RBD), characterized by dream enactment behavior and loss of atonia during REM sleep, has been widely investigated over the past decades as a prodromal syndrome for PD which may antecede motor symptoms for years (Reichmann, 2017). Recently, a systematic review of 51 longitudinal studies indicated that an ongoing diagnosis of RBD is associated with a progressively increasing cumulative risk of neurodegenerative disorders (5-year risk: 33.5%; 10.5-year risk: 82.4%; and 14-year risk: 96.6%), with the majority of participants studied developing PD (43.6%) and Lewy body dementia (25.0%) (Galbiati et al., 2019). Recent studies suggest the occurrence of other subtype of sleep disorders may also precedes PD for years. For example, a population-based longitudinal study of 91,273 participants with non-apnea sleep–wake disorders but without PD revealed that sleep disorders might be an independent contributor associated with higher risk of PD, according to analysis by a Cox regression model with multivariate adjustment (Hsiao et al., 2017). Further subtype analysis by the authors revealed that chronic insomnia conferred the highest risk of PD. A population-based study over 64,855 person-years demonstrated 75 incident parkinsonism cases, 62.7% of whom exhibited PD during the 13 years of follow-up (Lysen et al., 2019). Statistical analysis indicated that worse sleep quality and shorter sleep duration were associated with a higher risk of incident parkinsonism within the first 2 years.
Findings from post-mortem and animal experiments corroborate this hypothesis. Elderly individuals without PD suffering from years of sleep fragmentation were found to have a greater risk of PD-related pathology and dopaminergic neuron loss after death (Sohail et al., 2017). Experimentally induced disturbances of sleep patterns have also been shown to impact the efficiency of the metabolic clearance system (Xie et al., 2013), resulting in disruption of protein homeostasis and increased oxidative stress in the central nervous system (CNS) (Naidoo et al., 2008; Singh et al., 2008; Andreazza et al., 2010; Lima et al., 2014; Zhang et al., 2014; Kröller-Schön et al., 2018; Rodrigues et al., 2018); all of these factors have been proposed to contribute to the progression of PD. Furthermore, the disruption of circadian rhythm has also been suggested to relate to neurodegeneration in animal models (Mattis and Sehgal, 2016).
As such, an increasing number of studies suggest that PD-related pathology per se can contribute to the disturbance of sleep and of circadian rhythms. Increased deposition of alpha-synuclein related to PD was found in structures responsible for the regulation of sleep patterns, in line with disruptions to sleep and circadian rhythm (Rothman and Mattson, 2012; Kalaitzakis et al., 2013). Taken together, existing findings broaden our perspective on the potential mechanisms that may underlie reciprocal interactions between PD pathogenesis and sleep–wake disruptions. The purpose of this review, therefore, is to provide a framework and a comprehensive overview of the candidate molecular mechanisms proposed to underlie such a bidirectional relationship (Figure 1). In particular, it focuses on the potential molecular mechanism by which sleep disturbances lead to the pathogenesis and development of PD.
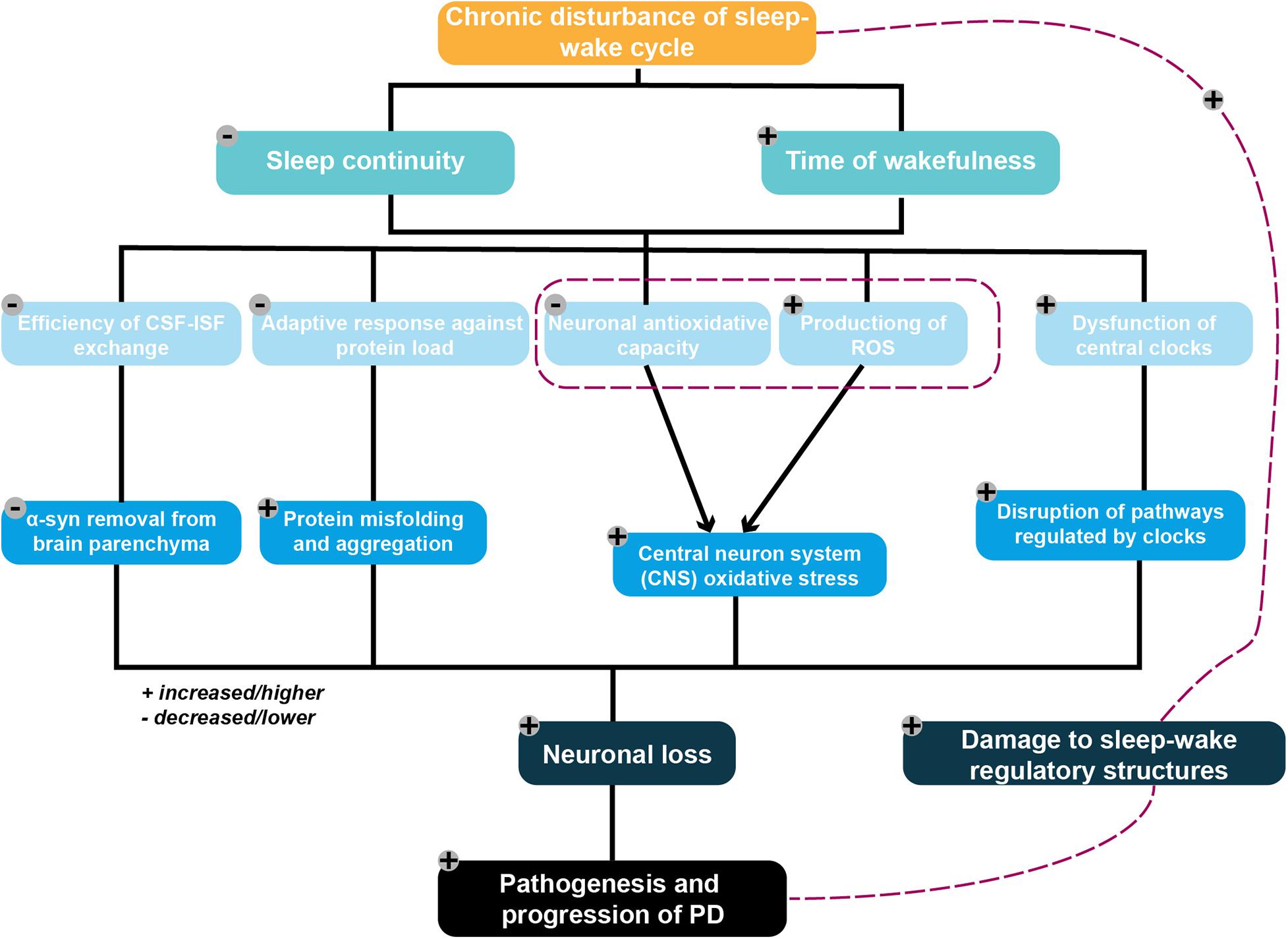
Figure 1. Framework of the potential mechanistic interplay between sleep–wake disruption and Parkinson’s disease (PD) pathogenesis. Chronic disruption of the sleep–wake cycle characterized by an increased waking time and decreased sleep continuity are proposed to serve as contributing factors to neuronal loss. These ultimately result in PD pathogenesis via several potential mechanisms, including the following: (1) diminishing the removal of metabolic wastes from the brain by adversely affecting the efficiency of the glymphatic system, resulting in excessive accumulation of the PD-promoting protein alpha-synuclein; (2) placing an excessive burden on the protein folding and degradation system, which is already overloaded in elderly individuals, resulting in the acceleration of protein aggregation and a positive feedback loop; (3) reducing neuronal antioxidative capacity, with the resulting increased ROS production triggered by excessive neuronal energy consumption subsequently leading to increased oxidative stress in the CNS; and (4) aggravating the disruption of pathways regulating the central circadian clock following dysfunction of these clocks. PD-related pathophysiological processes may also damage sleep–wake regulatory circuits, thereby exacerbating any existing disruptions to the sleep–wake cycle.
Sleep–Wake Disruption Is a Contributing Factor to the Pathogenesis and Progression of Parkinson’s Disease
Accumulation of Abnormal Proteins: The Endoplasmic Reticulum Stress Response and the Glymphatic System
Er Stress Response
Protein misfolding, accumulation, and aggregation play a fundamental role in the pathogenesis of neurodegenerative diseases, including Alzheimer’s disease (AD) and PD. Intracellular protein homeostasis, or proteostasis, is maintained by an integrated cellular quality control system based on the proper functioning of the endoplasmic reticulum (ER), which protects cells against stress induced by protein aggregation (Kaushik and Cuervo, 2015). ER stress is triggered by the aggregation of misfolded proteins and leads to activation of the unfolded protein response (UPR; also called the ER stress response) (Schröder and Kaufman, 2005a, b), an adaptive, coordinated response initiated by three transmembrane proteins. The UPR attenuates protein load via several different pathways, including diminishing protein translation via phosphorylation of eukaryotic initiation factor 2α (eIF2α) and enhancing protein modification and maturation via upregulation of the expression of specific genes, in coordination with elevated expression of ER chaperones (Szegezdi et al., 2006).
Given that one important protective function of the UPR is to maintain ER protein hemostasis by attenuating protein load, comprising a homeostatic response to counteract ER stress, it is unsurprising to note that dysfunction of the UPR can therefore give rise to disease (Scheper and Hoozemans, 2015). In neurodegenerative disease, disturbed activation of the UPR is correlated with ER dysfunction, which leads to synaptic dysfunction and neuronal malfunction. In vitro models have provided the majority of the evidence implicating UPR mediators in PD. Analysis of cortical neurons derived from induced pluripotent stem (iPS) cells from PD patients haboring alpha-synuclein (α-syn) mutations were found to show an accumulation of ER-associated degradation (ERAD) substrates, indicating impairment of the UPR and induction of ER stress (Chung et al., 2013). Consistent with these findings, a post-mortem study demonstrated the presence of UPR mediators in substantia nigra obtained from PD patients (Hoozemans et al., 2007). Furthermore, an immunohistochemical study found increased phosphorylation of pancreatic ER kinase (PKR)-like ER kinase (PERK) and eIF2α in dopaminergic neurons in the substantia nigra (SN) of PD patients compared with age-matched controls, and notably, increased α-syn immunoreactivity was shown to be colocalized with these UPR mediators. Taken together, these studies indicate that chronic ER stress caused by the accumulation of misfolded proteins and by disturbance of the UPR may comprise part of the underlying mechanism of PD, as well as contributing to abnormal aggregation of the key PD-related protein α-syn.
Acute sleep deprivation has been shown to cause activation of the ER stress response in the cerebral cortex of young mice (Naidoo et al., 2005). In contrast, aged mice were found to demonstrate significantly suppressed UPR activation in response to sleep disturbance (Naidoo et al., 2008). Whereas sleep deprivation did not increase expression of the major ER chaperone Ig binding protein (BiP) in aged mice, levels of BiP in young mice increased after 6 h of acute sleep deprivation. Additionally, the authors of the study found that levels of pro-apoptotic proteins increased in the cerebral cortex of aged mice, suggesting that the ER stress response is attenuated by aging (Naidoo et al., 2008). Similar to aged mice, patients with PD consistently show well-documented evidence of fragmented sleep (Chahine et al., 2017; Pillai and Leverenz, 2017). Therefore, it is reasonable to hypothesize that sleep disturbances characterized by sleep loss and fragmentation place an excessive burden on the proteostasis system, which is already overloaded in elderly patients. This results in the acceleration of protein aggregation, thereby forming a positive feedback loop. Interestingly, a recent study of BiP heterozygous mice reported a deterioration of the age-related sleep–wake phenotype, which was especially characterized by a fragmented sleep–wake cycle associated with decreased expression of BiP (Naidoo et al., 2018). These results indicate that impairment of the UPR may in turn contribute to damage to sleep architectures. Taken together, these findings suggest that the interaction between sleep disruption and maladaptive proteostatic regulation contributes to the development and progression of PD, but further efforts are needed to clarify how these three aspects interrelate.
Furthermore, increasing evidence has demonstrated that exposure to intermittent cyclical hypoxia/reoxygenation, which models sleep apnea-induced ER stress in specific brainstem motor neurons in adult mice as well as hippocampus and prefrontal cortex in young mice (Zhu et al., 2008; Cai et al., 2014), is ultimately implicated in impaired motor and cognitive function (Cai et al., 2013). This suggests that sleep-related breathing disorders, which are another sleep-disorder phenotype alongside sleep loss and fragmentation, are an aging-independent mechanism for inducing ER stress-mediated neuronal impairment. However, further studies are needed to determine whether the impairment observed in the mammalian models is also evident in patients with sleep apnea.
Evidence from human studies lending further support to the these hypotheses has started to emerge. Recently, a study conducted on 246 elderly patients without PD revealed that greater sleep fragmentation was associated with a higher risk of Lewy pathology and dopaminergic neuron loss (Sohail et al., 2017). Analyses with logistic regression models suggested that each 1 standard deviation greater sleep fragmentation led to a >40% higher likelihood of PD-related pathologic alterations. However, longitudinal studies that use non-invasive imaging techniques to evaluate correlations between sleep disorders and PD pathology still need to determine the directionality of the causal relationship between these two conditions. Additionally, to corroborate the hypotheses presented, assessments of cerebrospinal fluid (CSF) α-syn level in sleep disorder patients without PD are required to quantify α-syn deposition.
Glymphatic System
The glymphatic system is a newly described brain-wide network of perivascular spaces linked by astrocytic aquaporin-4 (AQP-4) water channels that facilitates the clearance of interstitial soluble protein and metabolic wastes from the brain. Cerebrospinal fluid enters the parenchyma via perivascular spaces that surround the penetrating arteries, while brain interstitial fluid exits through perivenous spaces (Iliff et al., 2012). Evidence suggests that impairment of this system is associated with the formation of neurotoxic protein aggregates that are implicated in the progression of neurodegenerative disease (Kress et al., 2014; Verheggen et al., 2018).
Sleeping mice were reported in one study to demonstrate an increased glymphatic clearance efficiency compared with freely behaving awake mice, with enhanced removal of the potentially neurotoxic metabolic products that typically accumulate during wakefulness (Xie et al., 2013). Based on real-time assessment of tetramethylammonium diffusion and two-photon imaging in live mice, the authors demonstrated that the brains of sleeping mice showed a 60% increase in the volume of the interstitial space compared with that of awake mice, resulting in increased convective flow between the CSF and interstitial fluid (ISF). The removal of potentially neurotoxic waste products increased sharply during sleep, as evidenced by a twofold faster removal of Aβ compared with awake mice, consistent with an increased efficiency of CSF–ISF exchange. Interestingly, other sleep-like states have also been found to exhibit an increased clearance efficiency. For example, studies have confirmed that glymphatic transport is enhanced during anesthesia, in line with results from animal studies (Benveniste et al., 2017; Ratner et al., 2017). Given these observations, sleep-like brain activity rather than sleep itself is assumed to determine the volume of the interstitial space, further impacting the efficiency of glymphatic solute clearance (Sundaram et al., 2019). This conclusion implies that factors correlated with the alteration of sleep architecture, including RBD and circadian rhythm, would be expected to subsequently induce or diminish the glymphatic disturbance.
While the clearance of α-syn via the glymphatic system has not yet been investigated, Hoshi and colleagues showed a negative correlation between AQP-4 expression and α-syn deposition, suggesting that a defined population of AQP-4 proteins serves as mediators via which the glymphatic system modifies α-syn deposition in the neocortex of patients with PD (Hoshi et al., 2017). A recent study indicated that glymphatic system dysfunction may be implicated in accelerating brain α-syn aggregation and the progression of α-syn-related pathology (Zou et al., 2019). Ligation of the deep cervical lymph nodes (LDclns) blocks meningeal lymphatic drainage in both wild-type and A53T-ligated mice and is associated with almost complete blockage of the glymphatic influx of a CSF tracer (Zou et al., 2019). The intensities of both α-syn-positive intercellular inclusions in neurons and extracellular α-syn aggregations in the perivascular space were significantly increased in LDclns mice compared with controls (Zou et al., 2019). These results indicate that an inefficiency of glymphatic solute clearance is implicated in the excessive accumulation of α-syn and that such clearance deteriorates after ligation of the lymph nodes. As a cautionary note, the LDclns mice in this study showed an increased number and distribution of gliocytes, which were associated with a fiercer neuroinflammatory response. This adds further support to the existing theory that excessive α-syn aggregation induces reactive gliosis, triggering the production of inflammatory cytokines, resulting in aggravated neuronal degeneration and apoptosis (Kempuraj et al., 2017; Wong and Krainc, 2017). Previous studies have confirmed that reactive astrogliosis leads to an impairment of AQP-4 polarization, subsequently causing glymphatic clearance inefficiency (Iliff and Nedergaard, 2013; Verkhratsky et al., 2015). Based on these findings, we hypothesize a vicious spiral in which dysfunction of glymphatic clearance is a crucial factor in the exacerbation of α-syn accumulation in the brain parenchyma. This accumulation leads to increased levels of reactive astrogliosis and AQP-4 depolarization, aggravating glymphatic system deficits and promoting PD-related pathology as a result.
Dopamine (DA) and noradrenaline receptors appear to drive the brain state-dependent alterations of the interstitial space volume, ultimately modulating CSF–ISF exchange (Benveniste et al., 2017). During waking states, levels of DA and adrenaline increase (Murillo-Rodríguez et al., 2009; España et al., 2016), amplifying both DA and catecholamine volume transmission signaling (Fuxe et al., 2015). DA volume transmission has been shown to be strongly involved in neuron–glia interactions (Fuxe et al., 2015), while suppression of adrenergic signaling in the brain during the waking state results in augmented glymphatic tracer influx (Xie et al., 2013). Therefore, it is reasonable to hypothesize that sleep disorders characterized by an extended waking duration and increased sleep fragmentation may show reduced α-syn removal via the glymphatic clearance system due to enhanced DA and catecholamine volume transmission during waking states.
Promotion of Oxidative Stress
The accumulation of reactive oxygen species (ROS) following cellular redox imbalance leads to mitochondrial dysfunction and oxidative stress in the brain. These processes are also thought to play important roles in the pathogenesis of PD, independent of the effects of aggregation and deposition of misfolded proteins in the CNS (Phillipson, 2017). Data collected from preclinical PD patients indicates that they exhibit an elevated level of cellular oxidative stress, which occurs prior to significant neuronal loss (Trist et al., 2019). Mounting evidence from both in vitro and in vivo studies indicates that redox imbalance is a contributor to the cascade underlying dopaminergic neurodegeneration; moreover, strategies to restore redox homeostasis reverse this neurodegenerative process (Devos et al., 2014; Filograna et al., 2016; Biosa et al., 2018). Excessive accumulation of ROS is thought to adversely affect the chemical modification and oxidation of DNA, lipids, and proteins at the molecular level and contribute to apoptosis, cytoplasmic cell death, and autophagic cell death at the cellular level (Umeno et al., 2017; Morris et al., 2018). Damage to mitochondria, especially inhibition of the electron transport chain leading to chronic production of ROS, has also been observed in the SN of PD patients (Schapira et al., 1990). In addition, electron transport chain inefficiency caused by chronic administration of the complex I inhibitor rotenone was shown to be an important contributing factor to the aggregation of proteinaceous inclusions resembling Lewy bodies, as well as to degeneration of the SN in animal models (Cannon et al., 2009).
Reactive oxygen species and reactive nitrogen species (RNS) are unavoidable byproducts of a variety of essential physiological reactions in all aerobic organisms. An antioxidant system composed of antioxidants and protective enzymes is responsible for providing the organism with an antioxidant defense, which involves scavenging free radicals and transforming them into more stable chemical forms. Once the excessive release of reactive species surpasses the protective capacity of the antioxidant system, oxidative stress occurs. This potentially leads to adverse effects on pivotal cellular constituents, including lipids, proteins, and nucleic acids, affecting both the structure and function of neurons, and finally initiating cell death and hence causing neurodegeneration (Sultana and Butterfield, 2010; Jiang et al., 2016; Trist et al., 2019).
Sleep is thought to promote the removal of excessive reactive species accumulated during the waking state by decreasing oxidant production and upregulating the efficiency of antioxidant mechanisms (Reimund, 1994). Sleep deprivation has been implicated in increased cerebral oxidative stress in a multitude of animal studies (Singh et al., 2008; Andreazza et al., 2010; Lima et al., 2014; Zhang et al., 2014; Kröller-Schön et al., 2018; Rodrigues et al., 2018), indicating a potential mechanistic correlation between sleep deprivation and the pathogenesis of PD. Sleep deprivation for 8 h daily over 3 days in mice was found to increase the extent of oxidative stress and the acetylation of mitochondrial proteins in locus coeruleus neurons, in line with an elevated concentration of superoxide (Zhang et al., 2014). Another study found that mice experiencing either 48 or 72 h paradoxical sleep deprivation (PSD) demonstrated increased lipid peroxidation in striatum neurons, indicating an elevated level of oxidative stress in this brain region. It should be noted that nitrite level was selectively attenuated in the striatum in the 72-h PSD group, while it was reduced in hippocampus and prefrontal cortex in both the 72- and 48-h PSD groups (Lima et al., 2014). The unaltered level of nitrite in the striatum in the 48-h group indicates that this structure is less susceptible to oxidative stress and is only affected by prolonged sleep deprivation. A meta-analysis of data extracted from 44 studies using either total sleep deprivation (TSD) or PSD protocols suggested that the brain regions with elevated rates of reactive species production during periods of extended wakefulness are those which primarily show signs of oxidative stress, namely the hypothalamus and hippocampus (Villafuerte et al., 2015). The results of this study further suggest that REM sleep plays a significant role in the antioxidant function of sleep, while that of slow wave sleep is less important given the discrepant results obtained from the TSD experiments.
Several hypotheses have emerged to explain the potential mechanisms underlying sleep loss and oxidative stress in the CNS. One such hypothesis focuses on the reduced expression of genes that occurs during adaptive response to stress. In line with this theory, 7 days of sleep deprivation was found to attenuate the expression of transcriptor Nrf-2 (Xue et al., 2019), a transcription factor modulating basal and inducible expression of several antioxidant and detoxification enzymes, such as superoxide dismutase and catalase (Rodrigues et al., 2018). Sleep deprivation has also been reported to result in an inefficient response of cellular scavenger mechanisms to reactive species, accompanied by a reduction in the activity of mitochondrial membrane binding protein sirtuin type 3 to upregulate antioxidant defenses (Zhang et al., 2014). Further studies are warranted to clarify the molecular mechanisms underlying the correlation between sleep deprivation and increased oxidative stress in the brain.
Circadian Rhythm Dysfunction
Studies conducted on night-shift workers and delayed sleep phase disorder patients have found that they exhibit altered melatonin profiles, as well as suppressed nocturnal melatonin secretion (Gibbs et al., 2002; Borugian et al., 2005; Marie Hansen et al., 2006; Micic et al., 2015). This reduction of melatonin production may be attributable to reduced rhythmicity of the central circadian pacemaker in the hypothalamic suprachiasmatic nucleus (SCN), which regulates melatonin secretion driven by the light–dark cycle (Phillipson, 2017). In line with this theory, mice treated with continuous light exposure for 24 weeks demonstrated a significant reduction of the rhythm and amplitude of SCN activity, which rapidly recovered upon return to a standard light–dark cycle (Lucassen et al., 2016). Melatonin has been shown to be multifunctional molecule that is pivotal in modulating the antioxidative system of cells, especially by acting as a powerful free-radical scavenger (Reiter et al., 2010). In vitro studies have also revealed that melatonin inhibiting the assembly of α-syn fibrils and destabilizing already formed fibrils, thereby preventing α-syn-induced cytotoxic events leading to neuronal death (Ono et al., 2012). Independent of its direct inhibitory and destabilizatory effects on fibril formation, studies have demonstrated that melatonin participates in multiple mechanisms implicated in neuroprotective processes, such as in the vitagene system, which includes sirtuins, heat shock proteins and heme oxygenase-1 (Gallardo-Fernandez et al., 2019). The vitagene system has been proposed to exert a pronounced neuroprotective effect given its positive role in attenuating α-syn misfolding and oxidative stress that would otherwise adversely affect neurons (Wang et al., 2007; Ebrahimi-Fakhari et al., 2012). In vitro investigations have demonstrated that melatonin significantly increases expression of sirtuin type 2 and nuclear transcription of Nrf-2, adding further support to the idea that melatonin acts as neuroprotective molecule on multiple levels (Gallardo-Fernandez et al., 2019; Yu et al., 2019). In conclusion, long-term delayed sleep phase or shift work has been shown to suppress melatonin secretion, thereby creating a vulnerability to alpha-synucleinopathy.
Mounting evidence suggests that malfunctioning endogenous clocks are a direct contributing factor to the pathogenesis and progression of neurodegenerative disease through their adverse effects on gene transcription and antioxidative processes. Myriad animal and human studies have reported that mistimed sleep leads to significantly reduced expression of circadian transcripts in the brain (Archer and Oster, 2015). The altered expression of clock genes that are fundamental to maintaining circadian rhythm has been recognized as playing a causal role in the induction of neurodegeneration (Hood and Amir, 2017). For example, one studying using genetic analysis identified the single-nucleotide polymorphisms in the ARNTL and PER1 genes as potential contributors to an increased risk of PD (Gu et al., 2015). Endogenous clock dysfunction in mutant drosophila has been correlated with the loss of dopaminergic neurons and results in early development of age-related locomotor deficits via the activation of proapoptosis pathways and increased oxidative stress (Vaccaro et al., 2017). A multitude of findings add further support to the idea that the suppressed expression of clock genes contributes to accelerated neuronal degeneration, driven by weakened antioxidant defenses and the resulting increased oxidative stress in neurons. Mice with a brain and muscle Arnt-like protein 1 (BMAL1) deletion demonstrated significantly elevated rates of cellular oxidative damage, consistent with reduced expression of Nrf-2, the major antioxidative regulatory factor (Lee et al., 2013). Further chromatin immunoprecipitation analysis in this study revealed that BMAL1 directly regulates the expression of Nrf-2 via a transcriptional mechanism. Supplementation of BMAL1-deficient mice with the antioxidant N-acetyl-L-cysteine significantly reversed their decreased longevity and age-related pathologies, supporting a casual link between clock genes and an increased oxidative stress response, which eventually leads to aging and age-related diseases (Kondratov et al., 2009). A recent study linked suppressed BMAL1 function to a reduced level of silent information regulator, an aldehyde dehydrogenase-dependent deacetylase that deacetylates BMAL1 and PER2 proteins, thereby modulating the expression of clock genes and affecting circadian rhythms (Wang et al., 2018). Rats treated with 6-hydroxydopamine (6-OHDA) demonstrated significant circadian misalignment, augmented ROS accumulation, and attenuated expression of clock-controlled antioxidant genes. An in vitro study of 6-OHDA-treated SH-SY5Y cells reported an elevated level of acetylated BMAL1 and a decreased level of SIRT1 protein, suggesting that the molecular mechanism underlying the circadian dysfunction-induced impairment of antioxidative defenses in brain neurons is closely associated with the SIRT1–BMAL1 pathway (Wang et al., 2018). Studies on transgenic mice also indicate that the circadian disruption contributes to PD pathogenesis via exaggerating 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridinen (MPTP)-induced neurotoxicity. Mice that underwent circadian disruption for 60 days before receiving MPTP treatment exhibited a worsening of their locomotor and cognitive deficits compared with those kept on a regular sleep-wake cycle, secondary to a significant reduced number of tyrosine hydroxylase (TH)-positive dopaminergic neurons and intense glial response (Lauretti et al., 2017). Another study demonstrated that inactivation of BMAL1 in the MPTP-treated mice resulted in significant motor deficit, further in vivo and in vitro study showed a significant reduction of dopaminergic neurons in the substantia nigra pars compacta, decreased level of the TH protein and the dopamine content in the striatum, and increased activation of glial-mediated neuroinflammation (Liu et al., 2020). Taken together, these findings suggest that dysfunction of the molecular clock attenuates the expression of redox defense genes, weakening the antioxidative response and thereby increasing the accumulation of reactive species. Meanwhile, the activation of gial cells and expression level of inflammatory factors will increase responsing to the circadian disruption. These in turn create neurotoxic conditions that may result in mitochondrial damage and microglial overactivation, leading to the loss of dopaminergic neurons via activation of proapoptotic or pro-inflammatory pathways, and ultimately accelerating the pathogenesis and progression of neurodegenerative disease.
Although these studies have provided novel insights into the mechanisms underlying the correlation between circadian disturbance and neurodegenerative disease, there are still several gaps remaining in our knowledge that warrant further investigation. Given that the glymphatic system promotes the effective clearance of accumulated neurotoxic metabolic products from the brain and generally operates during sleep, it is of major importance to clarify whether clock gene deletion impairs the efficiency of such glymphatic clearance. Furthermore, it is important to ascertain to what extent glymphatic clearance efficiency is impacted by disturbances to the circadian system. One proposal is that disturbances to the 24-h circadian cycle involving prolonged nighttime waking triggered by decreased clock gene expression could give rise to malfunction of the glymphatic system, thereby promoting the accumulation of PD-promoting metabolites in the brain parenchyma and speeding disease progression (Sundaram et al., 2019). Additionally, investigations of circadian α-syn dynamics in the CSF and ISF of the human brain are hampered by the lack of accurate measurements of glymphatic operation. Although a multitude of animal studies have shed light on the role of sleep as a regulator of metabolic waste clearance from the brain, further studies should transfer these investigations to human models to explore more reliable and valid methods of measuring glymphatic efficiency (Sundaram et al., 2019).
Sleep and Circadian Rhythm Disturbance Resulting From PD-Related Processes
Mounting lines of evidence have revealed a potential causal link between PD-related processes and circadian disturbance in PD patients. Epidemiological studies conducted on early, untreated PD patients and age-matched controls have demonstrated a tendency for increased sleep latency, decreased sleep efficiency, and reduced REM duration in PD patients (Bušková et al., 2011; Yong et al., 2011; Breen et al., 2014). These results are in line with those of transgenic rodent models overexpressing α-synuclein, which exhibit increased non-REM sleep, decreased REM sleep, and disturbed oscillatory electroencephalographic activity reminiscent of PD (McDowell et al., 2014). Additionally, changes to sleep architecture have been shown to worsen as PD progresses (Diederich et al., 2005). The accumulation of α-syn in brain regions involved in regulating sleep physiology is considered to be highly linked to disrupted sleep and circadian rhythm in PD patients (Rothman and Mattson, 2012; Kalaitzakis et al., 2013). A retrospective clinical pathological study revealed higher levels of α-syn burden in specific sleep-related brain regions in PD patients who also suffered from disrupted sleep compared with those without sleep disturbances (Kalaitzakis et al., 2013). The affected regions included: the locus coeruleus and the raphe nuclei, which are responsible for regulating REM, non-REM sleep, and the transition between them; the posterior hypothalamus, which is associated with somnolence; and the thalamus, which has been shown to be pivotal in organizing the sleep–wake rhythm (Reeves et al., 2006; Boeve et al., 2007). Other brainstem regions involved in regulating the sleep–wake cycle and arousal also showed lesions preceding the onset of motor symptoms in PD patients (Grinberg et al., 2010). Consistent with these clinical findings, accumulating evidence from animal studies lends further support to the notion that PD-related features are associated with changes to sleep parameters. For instance, one study found that mice overexpressing α-synuclein show increased α-syn burden and decreased firing rate in SCN neurons, which attenuates the ability of these neurons to transmit neural and hormonal signals from the SCN pacemaker (Kudo et al., 2011). Additionally, there were no defects in circadian parameters in transgenic MitoPark mice with slow and progressive degeneration of midbrain dopaminergic neurons prior to 19 weeks of age (Fifel and Cooper, 2014). However, after this age, DA levels significantly decreased in the midbrain, eventually declining to nearly 3% of their initial level, and all circadian parameters were markedly deteriorated. One explanation for these results given the known circadian rhythm of dopaminergic activity is that the dopaminergic signaling system transmits the circadian signal originating in the SCN to downstream target neuronal networks (Fifel and Cooper, 2014). Together, α-syn deposition and dopaminergic neuron loss are pivotal in PD-related pathological alteration and have been shown to be contributing factors to the disruption of sleep patterns and disturbances in circadian rhythm.
Melanopsin-containing retinal ganglion cells (mRGCs) are responsible for the non-image vision forming pathways projecting to different CNS regions and mediate several physiological responses, including the entrainment of circadian rhythms to the light–dark cycle, acute control of locomotor activity, sleep regulation, and the pupillary light reflex (Ksendzovsky et al., 2017). It is well documented that mRGCs innervate the SCN, which synchronizes circadian rhythms, as well as the lateral hypothalamus and the ventrolateral preoptic nucleus, which are jointly reponsible for regulating sleep behaviors (Münch and Kawasaki, 2013; La Morgia et al., 2017). Recently, a study analyzing the retinas of 24 donors demonstrated an age-associated reduction in the number of mRGCs and their extent of dendritic degeneration (Esquiva et al., 2017). Given that elderly individuals frequently suffer from sleep–wake disorders, it is reasonable to hypothesize that these changes to mRGC number and morphology may account for the incidence of circadian rhythm disorders in the aged (Esquiva et al., 2017). Post-mortem studies indicate that the retinal melanopsin-positive system degenerates in PD, as revealed by the significantly decreased density and morphological alterations of mRGCs in PD patients compared with controls (Ortuño-Lizarán et al., 2018). The reduction in the number of dopaminergic amacrine cells and the loss of synaptic connections with melanopsin cells in PD pathology may be responsible for the degeneration of mRGCs (Ortuño-Lizarán et al., 2020), given the bidirectional correlation between dopaminergic system and mRGCs. Previous studies have shown that mRGCs are modulated by DA released from dopaminergic cell innervations (Van Hook et al., 2012; Liao et al., 2016), while mRGCs are responsible for the continuous dopaminergic response to light (Prigge et al., 2016).
Recent studies using transgenic animals have offered promising insights into how the hypothetical factors contributing to PD-related pathophysiological processes drive circadian misalignment. The decreased expression levels of Bmal1 and Bmal2 have been reported in PD patients, indicating a potential correlation underlying PD-related processes and desynchronization of the circadian system (Cai et al., 2010; Ding et al., 2011). In support of this hypothesis, results from animal experiments also show that the PD-related genetic phenotype could impact sleep parameters. Mutations in mul1 and parkin in Drosophila are sufficient to induce mitochondrial impairment and mitophagy, which are molecular symptoms observed in PD models, as well as being typical motor symptoms of PD (Kitada et al., 1998; Yun et al., 2014). Altered circadian rhythm and disrupted expression of clock genes were observed in a recent study conducted on mutants carrying mutations of both mul1 and parkin, indicating a bidirectional correlation between PD-related pathology and circadian dysfunction (Ortuño-Lizarán et al., 2018). The level of ROS in the mutants was found to be higher than that in controls, consistent with decreased expression levels of the clock genes clock and tim. Notably, no significant difference was found in the expression rhythm of the per gene between mutants and controls, whereas the rhythm of the PER protein was completely disturbed (Doktór et al., 2019). Potential mechanisms underlying the difference between per transcription and PER translation could be ROS-induced alterations to the expression profiles of translational factors such as eIF2α, which enhance the expression of the PER protein (Lee et al., 2018). It is reasonable to propose that mutations in mul1 and PARKIN which have a causal link with mitochondrial dysfunction and which have been shown to be involved in the development of PD impact circadian rhythms and the clock via multiple molecular mechanisms at both the transcriptional and translational level.
A study conducted on familial PD models indicated that dysfunction of the ER could also be a contributing factor to defects in sleep patterns and circadian rhythm (Lee et al., 2018). Further research has demonstrated that impaired neuropeptide distribution in specific neurons is responsible for disrupted circadian rhythm and sleep patterns in PD mutant Drosophila and has mapped these defects to specific neuronal clusters, including ventral lateral neurons and insulin-producing cells (Valadas et al., 2018). The LNvs are analogous to neurons in the human hypothalamus (Kunst et al., 2015), and secrete the vasoactive intestinal peptide (VIP)-like neuropeptide pigment dispersing factor (PDF), which is involved in regulating the circadian cycle (Muraro et al., 2013; Jones et al., 2018; Hamnett et al., 2019). Further investigations have revealed that the excessive ER–mitochondrial contact involved in phosphatidylserine transport interfere with the production of PDF-loaded dense core vesicles via effects on lipid membrane composition, resulting in a defect in the release of PDF and subsequent sleep pattern defects in flies (Lee et al., 2018). Notably, supplementation with PtdSer was shown to rescue sleep defects in PD models, indicating that the disturbed sleep and circadian rhythmicity can be attributed to neuronal dysfunction rather than neurodegeneration, and may therefore be reversible.
In conclusion, findings from both clinical and in vivo testing have lent strong support to the notion that PD-related processes are causally linked to poor sleep and disrupted circadian rhythms. In turn, disturbed sleep and circadian rhythms might act as possible early markers of neuropathology during the prodromal period of PD. However, further clinical studies investigating sleep parameters should be conducted on normal elderly individuals. These would help to ascertain whether PD-related pathological alterations, such as increased α-syn burden, are pivotal in the induction of sleep disturbance independent of the adverse effects of typical PD motor symptoms and of dopamine supplements taken by PD patients.
Discussion
Clinical trials have demonstrated that patients with disturbed sleep patterns or circadian misalignment in old age more likely to exhibit pathological changes related to PD, along with an increased risk of PD. Evidence from further experimental studies has supported these findings with reports of adverse effects induced by sleep disruption, including increased levels of biomarkers related to PD, increased CNS oxidative stress, and inefficient removal of the PD-promoting metabolite α-syn. Additionally, patients with increased levels of α-syn burden in specific brain regions show disrupted sleep. These findings are in line with those from animal models overexpressing α-syn or demonstrating a progressive degeneration of DA neurons, which show gradual deterioration in sleep and circadian rhythm, indicating a direct causal link between PD-related processes and sleep disturbances. Taken together, these results suggest a bidirectional relationship between PD and sleep disruption, and as such, sleep has been widely considered to serve a disease-modifying role in the management of PD.
Further efforts should be made to disentangle the contribution of sleep disturbance and PD-related pathological alterations to their bidirectional correlation. For instance, longitudinal studies conducted on the general populationare warranted. These would preferably examine groups of patients with sleep complaints initiated preceding the clinical onset of PD across multiple clinical centers. Developments in non-invasive brain imaging methods and the successful identification of biomarkers may bring great benefits to advancing the assessment of sleep, circadian parameters, and the pathological progression of PD in human studies. Additionally, animal models incorporating broadly affected regions in the CNS are needed, given that current animal models selectively targeting the dopaminergic system fail to demonstrate the holistic sleep and circadian defects typical of the disease (Fifel et al., 2016).
Therapeutic managements targeting circadian rhythm have been widely proposed as being conducive to alleviating symptoms and disease burden in patients with PD. The utilization of bright light therapy has been shown to offer short-lived benefits with regard to consolidating activity rhythms or sleep patterns in patients with AD and PD (McCurry et al., 2011; Videnovic et al., 2017). The potential therapeutic effect of timed melatonin supplements has also been studied in PD patients, based on its antioxidant and apoptotic properties in in vitro and animal models. However, at present, no firm conclusion has been reached in terms of the therapeutic effects of light exposure or melatonin administration on motor symptoms, which can likely be attributed to methodological inconsistencies across trials. Therefore, it is important to carefully consider the methodological details of such studies and future longitudinal investigations evaluating the long-term therapeutic effects of circadian-oriented therapy on PD progression are warranted.
Author Contributions
SC and AX planned the study. ZY, XZ, and CL analyzed the data and edited the manuscript. ZY wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Natural Science Foundation of China (No. 81971192). We would like to acknowledge the investigators for their helpful comments regarding this manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbott, S. M., and Videnovic, A. (2016). Chronic sleep disturbance and neural injury: links to neurodegenerative disease. Nat. Sci. Sleep 8, 55–61. doi: 10.2147/NSS.S78947
Andreazza, A. C., Andersen, M. L., Alvarenga, T. A., de-Oliveira, M. R., Armani, F., Ruiz, F. S., et al. (2010). Impairment of the mitochondrial electron transport chain due to sleep deprivation in mice J. Psychiatr. Res. 44, 775–780. doi: 10.1016/j.jpsychires.2010.01.015
Archer, S. N., and Oster, H. (2015). How sleep and wakefulness influence circadian rhythmicity: effects of insufficient and mistimed sleep on the animal and human transcriptome. J. Sleep Res. 24, 476–493. doi: 10.1111/jsr.12307
Barone, P., Antonini, A., Colosimo, C., Marconi, R., Morgante, L., Avarello, T. P., et al. (2009). The PRIAMO study: a multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov. Disord. 24, 1641–1649. doi: 10.1002/mds.22643
Benveniste, H., Lee, H., Ding, F., Sun, Q., Al-Bizri, E., Makaryus, R., et al. (2017). Anesthesia with dexmedetomidine and low-dose isoflurane increases solute transport via the glymphatic pathway in rat brain when compared with high-dose isoflurane. Anesthesiology 127, 976–988. doi: 10.1097/ALN.0000000000001888
Biosa, A., Sanchez-Martinez, A., Filograna, R., Terriente-Felix, A., Alam, S. M., Beltramini, M., et al. (2018). Superoxide dismutating molecules rescue the toxic effects of PINK1 and parkin loss. Hum. Mol. Genet. 27, 1618–1629. doi: 10.1093/hmg/ddy069
Boeve, B. F., Silber, M. H., Saper, C. B., Ferman, T. J., Dickson, D. W., Parisi, J. E., et al. (2007). Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 130, 2770–2788. doi: 10.1093/brain/awm056
Borugian, M. J., Gallagher, R. P., Friesen, M. C., Switzer, T. F., and Aronson, K. J. (2005). Twenty-four-hour light exposure and melatonin levels among shift workers. J. Occup. Environ. Med. 47, 1268–1275. doi: 10.1097/01.jom.0000184855.87223.77
Breen, D. P., Vuono, R., Nawarathna, U., Fisher, K., Shneerson, J. M., Reddy, A. B., et al. (2014). Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 71, 589–595. doi: 10.1001/jamaneurol.2014.65
Bušková, J., Klempíø, J., Majerová, V., Picmausová, J., Sonka, K., Jech, R., et al. (2011). Sleep disturbances in untreated Parkinson’s disease. J. Neurol. 258, 2254–2259. doi: 10.1007/s00415-011-6109-7
Cai, X.-H., Li, X.-C., Hu, Q.-Q., Yu, C. Y., Zhou, Y. H., Su, M. S., et al. (2013). Multiple system morbidities associated with children with snore symptom. Pediatr. Pulmonol. 48, 381–389. doi: 10.1002/ppul.22653
Cai, X. H., Li, X. C., Jin, S. W., Liang, D. S., Wen, Z. W., Cao, H. C., et al. (2014). Endoplasmic reticulum stress plays critical role in brain damage after chronic intermittent hypoxia in growing rats. Exp. Neurol. 257, 148–156. doi: 10.1016/j.expneurol.2014.04.029
Cai, Y., Liu, S., Sothern, R. B., Xu, S., and Chan, P. (2010). Expression of clock genes Per1 and Bmal1 in total leukocytes in health and Parkinson’s disease. Eur. J. Neurol. 17, 550–554. doi: 10.1111/j.1468-1331.2009.02848.x
Cannon, J. R., Tapias, V., Na, H. M., Honick, A. S., Drolet, R. E., and Greenamyre, J. T. (2009). A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 34, 279–290. doi: 10.1016/j.nbd.2009.01.016
Chahine, L. M., Amara, A. W., and Videnovic, A. (2017). A systematic review of the literature on disorders of sleep and wakefulness in Parkinson’s disease from 2005 to 2015. Sleep Med. Rev. 35, 33–50. doi: 10.1016/j.smrv.2016.08.001
Chung, C. Y., Khurana, V., Auluck, P. K., Tardiff, D. F., Mazzulli, J. R., Soldner, F., et al. (2013). Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 342, 983–987. doi: 10.1126/science.1245296
Devos, D., Moreau, C., Devedjian, J. C., Kluza, J., Petrault, M., Laloux, C., et al. (2014). Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox. Signal. 21, 195–210. doi: 10.1089/ars.2013.5593
Diederich, N. J., Vaillant, M., Mancuso, G., Lyen, P., and Tiete, J. (2005). Progressive sleep ‘destructuring’ in Parkinson’s disease. A polysomnographic study in 46 patients. Sleep Med. 6, 313–318. doi: 10.1016/j.sleep.2005.03.011
Ding, H., Liu, S., Yuan, Y., Lin, Q., Chan, P., and Cai, Y. (2011). Decreased expression of Bmal2 in patients with Parkinson’s disease. Neurosci. Lett. 499, 186–188. doi: 10.1016/j.neulet.2011.05.058
Doktór, B., Damulewicz, M., and Pyza, E. (2019). Effects of MUL1 and PARKIN on the circadian clock, brain and behaviour in Drosophila Parkinson’s disease models. BMC Neurosci. 20:24. doi: 10.1186/s12868-019-0506-8
Ebrahimi-Fakhari, D., Wahlster, L., and McLean, P. J. (2012). Protein degradation pathways in Parkinson’s disease: curse or blessing. Acta Neuropathol. 124, 153–172. doi: 10.1007/s00401-012-1004-6
España, R. A., Schmeichel, B. E., and Berridge, C. W. (2016). Norepinephrine at the nexus of arousal, motivation and relapse. Brain Res. 1641, 207–216. doi: 10.1016/j.brainres.2016.01.002
Esquiva, G., Lax, P., Pérez-Santonja, J. J., García-Fernández, J. M., and Cuenca, N. (2017). Loss of melanopsin-expressing ganglion cell subtypes and dendritic degeneration in the aging human retina. Front. Aging Neurosci. 9:79. doi: 10.3389/fnagi.2017.00079
Fifel, K., and Cooper, H. M. (2014). Loss of dopamine disrupts circadian rhythms in a mouse model of Parkinson’s disease. Neurobiol. Dis. 71, 359–369. doi: 10.1016/j.nbd.2014.08.024
Fifel, K., Piggins, H., and Deboer, T. (2016). Modeling sleep alterations in Parkinson’s disease: how close are we to valid translational animal models? Sleep Med. Rev. 25, 95–111. doi: 10.1016/j.smrv.2015.02.005
Filograna, R., Beltramini, M., Bubacco, L., and Bisaglia, M. (2016). Anti-oxidants in Parkinson’s disease yherapy: a critical point of view. Curr. Neuropharmacol. 14, 260–271. doi: 10.2174/1570159X13666151030102718
Fuxe, K., Agnati, L. F., Marcoli, M., and Borroto-Escuela, D. O. (2015). Volume transmission in central dopamine and noradrenaline neurons and its astroglial targets. Neurochem. Res. 40, 2600–2614. doi: 10.1007/s11064-015-1574-5
Galbiati, A., Verga, L., Giora, E., Zucconi, M., and Ferini-Strambi, L. (2019). The risk of neurodegeneration in REM sleep behavior disorder: a systematic review and meta-analysis of longitudinal studies. Sleep Med. Rev. 43, 37–46. doi: 10.1016/j.smrv.2018.09.008
Gallardo-Fernandez, M., Hornedo-Ortega, R., Cerezo, A. B., Troncoso, A. M., and Garcia-Parrilla, M. C. (2019). Melatonin, protocatechuic acid and hydroxytyrosol effects on vitagenes system against alpha-synuclein toxicity. Food Chem. Toxicol. 134:110817. doi: 10.1016/j.fct.2019.110817
Gibbs, M., Hampton, S., Morgan, L., and Arendt, J. (2002). Adaptation of the circadian rhythm of 6-sulphatoxymelatonin to a shift schedule of seven nights followed by seven days in offshore oil installation workers. Neurosci. Lett. 325, 91–94. doi: 10.1016/S0304-3940(02)00247-1
Grinberg, L. T., Rueb, U., Alho, A. T. D. L., and Heinsen, H. (2010). Brainstem pathology and non-motor symptoms in PD. J. Neurol. Sci. 289, 81–88. doi: 10.1016/j.jns.2009.08.021
Gu, Z., Wang, B., Zhang, Y. B., Ding, H., Zhang, Y., Yu, J., et al. (2015). Association of ARNTL and PER1 genes with Parkinson’s disease: a case-control study of Han Chinese. Sci. Rep. 5:15891. doi: 10.1038/srep15891
Hamnett, R., Crosby, P., Chesham, J. E., and Hastings, M. H. (2019). Vasoactive intestinal peptide controls the suprachiasmatic circadian clock network via ERK1/2 and DUSP4 signalling. Nat. Commun. 10:542. doi: 10.1038/s41467-019-08427-3
Hastings, M. H., and Goedert, M. (2013). Circadian clocks and neurodegenerative diseases: time to aggregate? Curr. Opin. Neurobiol. 23, 880–887. doi: 10.1016/j.conb.2013.05.004
Hood, S., and Amir, S. (2017). Neurodegeneration and the circadian clock. Front. Aging Neurosci. 9:170. doi: 10.3389/fnagi.2017.00170
Hoozemans, J. J. M., van Haastert, E. S., Eikelenboom, P., de Vos, R. A. I., Rozemuller, J. M., and Scheper, W. (2007). Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 354, 707–711. doi: 10.1016/j.bbrc.2007.01.043
Hoshi, A., Tsunoda, A., Tada, M., Nishizawa, M., Ugawa, Y., and Kakita, A. (2017). Expression of aquaporin 1 and aquaporin 4 in the temporal neocortex of patients with Parkinson’s disease. Brain Pathol. 27, 160–168. doi: 10.1111/bpa.12369
Hsiao, Y.-H., Chen, Y.-T., Tseng, C.-M., Wu, L. A., Perng, D. W., Chen, Y. M., et al. (2017). Sleep disorders and an increased risk of Parkinson’s disease in individuals with non-apnea sleep disorders: a population-based cohort study. J. Sleep Res. 26, 623–628. doi: 10.1111/jsr.12545
Iliff, J. J., and Nedergaard, M. (2013). Is there a cerebral lymphatic system? Stroke 44, S93–S95. doi: 10.1161/STROKEAHA.112.678698
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4:147ra111. doi: 10.1126/scitranslmed.3003748
Jiang, T., Sun, Q., and Chen, S. (2016). Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 147, 1–19. doi: 10.1016/j.pneurobio.2016.07.005
Jones, J. R., Simon, T., Lones, L., and Herzog. (2018). SCN vip neurons are essential for normal light-mediated resetting of the circadian system. J. Neurosci. 38, 7986–7995. doi: 10.1523/JNEUROSCI.1322-18.2018
Kalaitzakis, M. E., Gentleman, S. M., and Pearce, R. K. (2013). Disturbed sleep in Parkinson’s disease: anatomical and pathological correlates. Neuropathol. Appl. Neurobiol. 39, 644–653. doi: 10.1111/nan.12024
Kaushik, S., and Cuervo, A. M. (2015). Proteostasis and aging. Nat. Med. 21, 1406–1415. doi: 10.1038/nm.4001
Kempuraj, D., Thangavel, R., Selvakumar, G. P., Zaheer, S., Ahmed, M. E., Raikwar, S. P., et al. (2017). Brain and peripheral atypical inflammatory mediators potentiate neuroinflammation and neurodegeneration. Front. Cell. Neurosci. 11:216. doi: 10.3389/fncel.2017.00216
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Kondratov, R. V., Vykhovanets, O., Kondratova, A. A., and Antoch, M. P. (2009). Antioxidant N-acetyl-L-cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging 1, 979–987. doi: 10.18632/aging.100113
Kress, B. T., Iliff, J. J., Xia, M., Wang, M., Wei, H. S., Zeppenfeld, D., et al. (2014). Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 76, 845–861. doi: 10.1002/ana.24271
Kröller-Schön, S., Daiber, A., Steven, S., Oelze, M., Frenis, K., Kalinovic, S., et al. (2018). Crucial role for Nox2 and sleep deprivation in aircraft noise-induced vascular and cerebral oxidative stress, inflammation, and gene regulation. Eur. Heart J. 39, 3528–3539. doi: 10.1093/eurheartj/ehy333
Ksendzovsky, A., Pomeraniec, I. J., Zaghloul, K. A., Provencio, J. J., and Provencio, I. (2017). Clinical implications of the melanopsin-based non-image-forming visual system. Neurology 88, 1282–1290. doi: 10.1212/WNL.0000000000003761
Kudo, T., Loh, D. H., Truong, D., Wu, Y., and Colwell, C. S. (2011). Circadian dysfunction in a mouse model of Parkinson’s disease. Exp. Neurol. 232, 66–75. doi: 10.1016/j.expneurol.2011.08.003
Kunst, M., Tso, M. C. F., Ghosh, D. D., Herzog, and Nitabach, M. N. (2015). Rhythmic control of activity and sleep by class B1 GPCRs. Crit. Rev. Biochem. Mol. Biol. 50, 18–30. doi: 10.3109/10409238.2014.985815
La Morgia, C., Ross-Cisneros, F. N., Sadun, A. A., and Carelli, V. (2017). Retinal ganglion cells and circadian rhythms in Alzheimer’s disease, Parkinson’s disease, and beyond. Front. Neurol. 8:162. doi: 10.3389/fneur.2017.00162
Lauretti, E., Di Meco, A., Merali, S., and Praticò, D. (2017). Circadian rhythm dysfunction: a novel environmental risk factor for Parkinson’s disease. Mol. Psychiatry 22, 280–286. doi: 10.1038/mp.2016.47
Lee, I. C., Ho, X. Y., George, S. E., Goh, C. W., Sundaram, J. R., Pang, K. K. L., et al. (2018). Oxidative stress promotes SIRT1 recruitment to the GADD34/PP1α complex to activate its deacetylase function. Cell Death Differ. 25, 255–267. doi: 10.1038/cdd.2017.152
Lee, J., Moulik, M., Fang, Z., Saha, P., Zou, F., Xu, Y., et al. (2013). Bmal1 and β-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced β-cell failure in mice. Mol. Cell Biol. 33, 2327–2338. doi: 10.1128/MCB.01421-12
Liao, H. W., Ren, X., Peterson, B. B., Marshak, D. W., Yau, K. W., Gamlin, P. D., et al. (2016). Melanopsin-expressing ganglion cells on macaque and human retinas form two morphologically distinct populations. J. Comp. Neurol. 524, 2845–2872. doi: 10.1002/cne.23995
Lima, A. M. A., de Bruin, V. M. S., Rios, E. R. V., and de Bruin, P. F. C. (2014). Differential effects of paradoxical sleep deprivation on memory and oxidative stress. Naunyn Schmiedebergs Arch. Pharmacol. 387, 399–406. doi: 10.1007/s00210-013-0955-z
Liu, W. W., Wei, S. Z., Huang, G. D., Liu, L. B., Gu, C., Shen, Y., et al. (2020). BMAL1 regulation of microglia-mediated neuroinflammation in MPTP-induced Parkinson’s disease mouse model. FASEB J. 34, 6570–6581. doi: 10.1096/fj.201901565RR
Lucassen, E. A., Coomans, C. P., van Putten, M., de Kreij, S. R., van Genugten, J. H., Sutorius, R. P., et al. (2016). Environmental 24-hr cycles are essential for health. Curr. Biol. 26, 1843–1853. doi: 10.1016/j.cub.2016.05.038
Lysen, T. S., Darweesh, S. K. L., Ikram, M. K., Luik, A. I., and Ikram, M. A. (2019). Sleep and risk of parkinsonism and Parkinson’s disease: a population-based study. Brain 142, 2013–2022. doi: 10.1093/brain/awz113
Maeda, T., Shimo, Y., Chiu, S. W., Yamaguchi, T., Kashihara, K., Tsuboi, Y., et al. (2017). Clinical manifestations of nonmotor symptoms in 1021 Japanese Parkinson’s disease patients from 35 medical centers. Parkinson. Relat. Disord. 38, 54–60. doi: 10.1016/j.parkreldis.2017.02.024
Marie Hansen, A., Helene Garde, A., and Hansen, J. (2006). Diurnal urinary 6-sulfatoxymelatonin levels among healthy Danish nurses during work and leisure time. Chronobiol. Int. 23, 1203–1215. doi: 10.1080/07420520601100955
Mattis, J., and Sehgal, A. (2016). Circadian rhythms, sleep, and disorders of aging. Trends Endocrinol. Metab. 27, 192–203. doi: 10.1016/j.tem.2016.02.003
McCurry, S. M., Pike, K. C., Vitiello, M. V., Logsdon, R. G., Larson, E. B., and Teri, L. (2011). Increasing walking and bright light exposure to improve sleep in community-dwelling persons with Alzheimer’s disease: results of a randomized, controlled trial. J. Am. Geriatr. Soc. 59, 1393–1402. doi: 10.1111/j.1532-5415.2011.03519.x
McDowell, K. A., Shin, D., Roos, K. P., and Chesselet, M.-F. (2014). Sleep dysfunction and EEG alterations in mice overexpressing alpha-synuclein. J. Parkins. Dis. 4, 531–539. doi: 10.3233/JPD-140374
Micic, G., Lovato, N., Gradisar, M., Burgess, H. J., Ferguson, S. A., Kennaway, D. J., et al. (2015). Nocturnal melatonin profiles in patients with delayed sleep-wake phase disorder and control sleepers. J. Biol. Rhythms 30, 437–448. doi: 10.1177/0748730415591753
Morris, G., Walker, A. J., Berk, M., Maes, M., and Puri, B. K. (2018). Cell death pathways: a novel therapeutic approach for neuroscientists. Mol. Neurobiol. 55, 5767–5786. doi: 10.1007/s12035-017-0793-y
Münch, M., and Kawasaki, A. (2013). Intrinsically photosensitive retinal ganglion cells: classification, function and clinical implications. Curr. Opin. Neurol. 26, 45–51. doi: 10.1097/WCO.0b013e32835c5e78
Munhoz, R. P., Moro, A., Silveira-Moriyama, L., and Teive, H. A. (2015). Non-motor signs in Parkinson’s disease: a review. Arq. Neuropsiquiatr. 73, 454–462. doi: 10.1590/0004-282X20150029
Muraro, N. I., Pírez, N., and Ceriani, M. F. (2013). The circadian system: plasticity at many levels. Neuroscience 247, 280–293. doi: 10.1016/j.neuroscience.2013.05.036
Murillo-Rodríguez, E., Arias-Carrión, O., Sanguino-Rodríguez, K., González-Arias, M., and Haro, R. (2009). Mechanisms of sleep-wake cycle modulation. CNS Neurol. Disord. Drug Targets 8, 245–253. doi: 10.2174/187152709788921654
Naidoo, N., Ferber, M., Master, M., Zhu, Y., and Pack, A. I. (2008). Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 28, 6539–6548. doi: 10.1523/JNEUROSCI.5685-07.2008
Naidoo, N., Giang, W., Galante, R. J., and Pack, A. I. (2005). Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J. Neurochem. 92, 1150–1157. doi: 10.1111/j.1471-4159.2004.02952.x
Naidoo, N., Zhu, J., Galante, R. J., Lian, J., Strus, E., Lee, A., et al. (2018). Reduction of the molecular chaperone binding immunoglobulin protein (BiP) accentuates the effect of aging on sleep-wake behavior. Neurobiol. Aging 69, 10–25. doi: 10.1016/j.neurobiolaging.2018.04.011
Ono, K., Mochizuki, H., Ikeda, T., Nihira, T., Takasaki, J., Teplow, D. B., et al. (2012). Effect of melatonin on α-synuclein self-assembly and cytotoxicity. Neurobiol. Aging 33, 2172–2185. doi: 10.1016/j.neurobiolaging.2011.10.015
Ortuño-Lizarán, I., Esquiva, G., Beach, T. G., Serrano, G. E., Adler, C. H., Lax, P., et al. (2018). Degeneration of human photosensitive retinal ganglion cells may explain sleep and circadian rhythms disorders in Parkinson’s disease. Acta Neuropathol. Commun. 6:90. doi: 10.1186/s40478-018-0596-z
Ortuño-Lizarán, I., Sánchez-Sáez, X., Lax, P., Serrano, G. E., Beach, T. G., Adler, C. H., et al. (2020). Dopaminergic retinal cell loss and visual dysfunction in Parkinson disease. Ann Neurol. 88, 893–906 doi: 10.1002/ana.25897
Phillipson, O. T. (2017). Alpha-synuclein, epigenetics, mitochondria, metabolism, calcium traffic, & circadian dysfunction in Parkinson’s disease. An integrated strategy for management. Ageing Res. Rev. 40, 149–167. doi: 10.1016/j.arr.2017.09.006
Pillai, J. A., and Leverenz, J. B. (2017). Sleep and neurodegeneration: a critical appraisal. Chest 151, 1375–1386. doi: 10.1016/j.chest.2017.01.002
Politis, M., Wu, K., Molloy, S. G., Bain, P., Chaudhuri, K. R., and Piccini, P. (2010). Parkinson’s disease symptoms: the patient’s perspective. Mov. Disord. 25, 1646–1651. doi: 10.1002/mds.23135
Prigge, C. L., Yeh, P. T., Liou, N. F., Lee, C. C., You, S. F., Liu, L. L., et al. (2016). M1 ipRGCs influence visual function through retrograde signaling in the retina. J. Neurosci. 36, 7184–7197. doi: 10.1523/JNEUROSCI.3500-15.2016
Ratner, V., Gao, Y., Lee, H., Elkin, R., Nedergaard, M., Benveniste, H., et al. (2017). Cerebrospinal and interstitial fluid transport via the glymphatic pathway modeled by optimal mass transport. Neuroimage 152, 530–537. doi: 10.1016/j.neuroimage.2017.03.021
Reeves, W. C., Heim, C., Maloney, E. M., Youngblood, L. S., Unger, E. R., Decker, M. J., et al. (2006). Sleep characteristics of persons with chronic fatigue syndrome and non-fatigued controls: results from a population-based study. BMC Neurol. 6:41. doi: 10.1186/1471-2377-6-41
Reichmann, H. (2017). Premotor diagnosis of Parkinson’s disease. Neurosci. Bull. 33, 526–534. doi: 10.1007/s12264-017-0159-5
Reimund, E. (1994). The free radical flux theory of sleep. Med. Hypothes. 43, 231–233. doi: 10.1016/0306-9877(94)90071-X
Reiter, R. J., Tan, D.-X., and Fuentes-Broto, L. (2010). Melatonin: a multitasking molecule. Prog. Brain Res. 181, 127–151. doi: 10.1016/S0079-6123(08)81008-4
Rodrigues, N. R., Macedo, G. E., Martins, I. K., Gomes, K. K., de Carvalho, N. R., Posser, T., et al. (2018). Short-term sleep deprivation with exposure to nocturnal light alters mitochondrial bioenergetics in Drosophila. Free Radic. Biol. Med. 120, 395–406. doi: 10.1016/j.freeradbiomed.2018.04.549
Rothman, S. M., and Mattson, M. P. (2012). Sleep disturbances in Alzheimer’s and Parkinson’s diseases. Neuromol. Med. 14, 194–204. doi: 10.1007/s12017-012-8181-2
Schapira, A. H., Mann, V. M., Cooper, J. M., Dexter, D., Daniel, S. E., Jenner, P., et al. (1990). Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem. 55, 2142–2145. doi: 10.1111/j.1471-4159.1990.tb05809.x
Scheper, W., and Hoozemans, J. J. (2015). The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 130, 315–331. doi: 10.1007/s00401-015-1462-8
Schröder, M., and Kaufman, R. J. (2005a). ER stress and the unfolded protein response. Mutat. Res. 569, 29–63. doi: 10.1016/j.mrfmmm.2004.06.056
Schröder, M., and Kaufman, R. J. (2005b). The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789. doi: 10.1146/annurev.biochem.73.011303.074134
Singh, R., Kiloung, J., Singh, S., and Sharma, D. (2008). Effect of paradoxical sleep deprivation on oxidative stress parameters in brain regions of adult and old rats. Biogerontology 9, 153–162. doi: 10.1007/s10522-008-9124-z
Sohail, S., Yu, L., Schneider, J. A., Bennett, D. A., Buchman, A. S., and Lim, A. S. P. (2017). Sleep fragmentation and Parkinson’s disease pathology in older adults without Parkinson’s disease. Mov. Disord. 32, 1729–1737. doi: 10.1002/mds.27200
Sultana, R., and Butterfield, D. A. (2010). Role of oxidative stress in the progression of Alzheimer’s disease. J Alzheimers Dis. 19, 341–353. doi: 10.3233/JAD-2010-1222
Sundaram, S., Hughes, R. L., Peterson, E., Müller-Oehring, E. M., Brontë-Stewart, H. M., Poston, K. L., et al. (2019). Establishing a framework for neuropathological correlates and glymphatic system functioning in Parkinson’s disease. Neurosci. Biobehav. Rev. 103, 305–315. doi: 10.1016/j.neubiorev.2019.05.016
Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. Embo Rep. 7, 880–885. doi: 10.1038/sj.embor.7400779
Trist, B. G., Hare, D. J., and Double, K. L. (2019). Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 18:e13031. doi: 10.1111/acel.13031
Umeno, A., Biju, V., and Yoshida, Y. (2017). In vivo ROS production and use of oxidative stress-derived biomarkers to detect the onset of diseases such as Alzheimer’s disease, Parkinson’s disease, and diabetes. Free Radic. Res. 51, 413–427. doi: 10.1080/10715762.2017.1315114
Vaccaro, A., Issa, A. R., Seugnet, L., Birman, S., and Klarsfeld, A. (2017). Drosophila clock Is required in brain pacemaker neurons to prevent premature locomotor aging independently of its circadian function. PLoS Genet. 13:e1006507. doi: 10.1371/journal.pgen.1006507
Valadas, J. S., Esposito, G., Vandekerkhove, D., et al. (2018). ER Lipid defects in neuropeptidergic neurons impair sleep patterns in Parkinson’s disease. Neuron 98, 1155.e6–1169.e6. doi: 10.1016/j.neuron.2018.05.022
Van Hook, M. J., Wong, K. Y., and Berson, D. M. (2012). Dopaminergic modulation of ganglion-cell photoreceptors in rat. Eur. J. Neurosci. 35, 507–518. doi: 10.1111/j.1460-9568.2011.07975.x
Verheggen, I. C. M., Van Boxtel, M. P. J., Verhey, F. R. J., Jansen, J. F. A., and Backes, W. H. (2018). Interaction between blood-brain barrier and glymphatic system in solute clearance. Neurosci. Biobehav. Rev. 90, 26–33. doi: 10.1016/j.neubiorev.2018.03.028
Verkhratsky, A., Nedergaard, M., and Hertz, L. (2015). Why are astrocytes important? Neurochem. Res. 40, 389–401. doi: 10.1007/s11064-014-1403-2
Videnovic, A., Klerman, E. B., Wang, W., Marconi, A., Kuhta, T., and Zee, P. C. (2017). Timed light therapy for sleep and daytime sleepiness associated with Parkinson disease: a randomized clinical trial. JAMA Neurol. 74, 411–418. doi: 10.1001/jamaneurol.2016.5192
Videnovic, A., Lazar, A. S., Barker, R. A., and Overeem, S. (2014). ‘The clocks that time us’–circadian rhythms in neurodegenerative disorders. Nat. Rev. Neurol. 10, 683–693. doi: 10.1038/nrneurol.2014.206
Villafuerte, G., Miguel-Puga, A., Rodriguez, E. M., Machado, S., Manjarrez, E., and Arias-Carrion, O. (2015). Sleep deprivation and oxidative stress in animal models: a systematic review. Oxid. Med. Cell. Longev. 2015:234952. doi: 10.1155/2015/234952
Wang, F., Nguyen, M., Qin, F. X.-F., and Tong, Q. (2007). SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6, 505–514. doi: 10.1111/j.1474-9726.2007.00304.x
Wang, Y., Lv, D., Liu, W., Li, S., Chen, J., Shen, Y., et al. (2018). Disruption of the circadian clock alters antioxidative defense via the SIRT1-BMAL1 pathway in 6-OHDA-induced models of Parkinson’s disease. Oxid. Med. Cell. Longev. 2018, 1–11. doi: 10.1155/2018/4854732
Wong, Y. C., and Krainc, D. (2017). α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 23, 1–13. doi: 10.1038/nm.4269
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. doi: 10.1126/science.1241224
Xue, R., Wan, Y., Sun, X., Zhang, X., Gao, W., and Wu, W. (2019). Nicotinic mitigation of neuroinflammation and oxidative stress after chronic sleep deprivation. Front. Immunol. 10:2546. doi: 10.3389/fimmu.2019.02546
Yong, M.-H., Fook-Chong, S., Pavanni, R., Lim, L.-L., and Tan, E.-K. (2011). Case control polysomnographic studies of sleep disorders in Parkinson’s disease. PLoS One 6:e22511. doi: 10.1371/journal.pone.0022511
Yu, H., Zhang, J., Ji, Q., Yu, K., Wang, P., Song, M., et al. (2019). Melatonin alleviates aluminium chloride-induced immunotoxicity by inhibiting oxidative stress and apoptosis associated with the activation of Nrf2 signaling pathway. Ecotoxicol. Environ. Saf. 173, 131–141. doi: 10.1016/j.ecoenv.2019.01.095
Yun, J., Puri, R., Yang, H., Lizzio, M. A., Wu, C., Sheng, Z. H., et al. (2014). MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. eLife 3:e01958. doi: 10.7554/eLife.01958.022
Zhang, J., Zhu, Y., Zhan, G., Fenik, P., Panossian, L., Wang, M. M., et al. (2014). Extended wakefulness: compromised metabolics in and degeneration of locus ceruleus neurons. J. Neurosci. 34, 4418–4431. doi: 10.1523/JNEUROSCI.5025-12.2014
Zhu, Y., Fenik, P., Zhan, G., Sanfillipo-Cohn, B., Naidoo, N., and Veasey, S. C. (2008). Eif-2a protects brainstem motoneurons in a murine model of sleep apnea. J. Neurosci. 28, 2168–2178. doi: 10.1523/JNEUROSCI.5232-07.2008
Keywords: Parkinson’s disease, glymphatic system, unfolded protein response, oxidative stress, sleep disruption
Citation: Yang Z, Zhang X, Li C, Chi S and Xie A (2021) Molecular Mechanisms Underlying Reciprocal Interactions Between Sleep Disorders and Parkinson’s Disease. Front. Neurosci. 14:592989. doi: 10.3389/fnins.2020.592989
Received: 09 August 2020; Accepted: 27 November 2020;
Published: 10 February 2021.
Edited by:
Aideen M. Sullivan, University College Cork, IrelandReviewed by:
Lies De Groef, KU Leuven, BelgiumErik Steven Musiek, Saint Louis University, United States
Copyright © 2021 Yang, Zhang, Li, Chi and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anmu Xie, eGllYW5tdUAxNjMuY29t