 Federica Di Cintio1,2
Federica Di Cintio1,2 Michele Dal Bo1Lorena Baboci1
Michele Dal Bo1Lorena Baboci1 Elena De Mattia1
Elena De Mattia1 Maurizio Polano1
Maurizio Polano1 Giuseppe Toffoli1*
Giuseppe Toffoli1*- 1Experimental and Clinical Pharmacology Unit, Centro di Riferimento Oncologico di Aviano, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Aviano, Italy
- 2Department of Life Sciences, University of Trieste, Trieste, Italy
Glioblastoma (GBM) is the most frequent and aggressive primary central nervous system tumor. Surgery followed by radiotherapy and chemotherapy with alkylating agents constitutes standard first-line treatment of GBM. Complete resection of the GBM tumors is generally not possible given its high invasive features. Although this combination therapy can prolong survival, the prognosis is still poor due to several factors including chemoresistance. In recent years, a comprehensive characterization of the GBM-associated molecular signature has been performed. This has allowed the possibility to introduce a more personalized therapeutic approach for GBM, in which novel targeted therapies, including those employing tyrosine kinase inhibitors (TKIs), could be employed. The GBM tumor microenvironment (TME) exerts a key role in GBM tumor progression, in particular by providing an immunosuppressive state with low numbers of tumor-infiltrating lymphocytes (TILs) and other immune effector cell types that contributes to tumor proliferation and growth. The use of immune checkpoint inhibitors (ICIs) has been successfully introduced in numerous advanced cancers as well as promising results have been shown for the use of these antibodies in untreated brain metastases from melanoma and from non-small cell lung carcinoma (NSCLC). Consequently, the use of PD-1/PD-L1 inhibitors has also been proposed in several clinical trials for the treatment of GBM. In the present review, we will outline the main GBM molecular and TME aspects providing also the grounds for novel targeted therapies and immunotherapies using ICIs for GBM.
Introduction
Glioblastoma (GBM) is the most common and aggressive primary CNS tumor (Stupp et al., 2009; Louis et al., 2016; Mendes et al., 2018; Altmann et al., 2019; Ostrom et al., 2019) and it has been included in the group of diffuse astrocytic and oligodendroglial tumors by the 2016 CNS WHO (Louis et al., 2016). It is thought that genetic alterations affecting neuroglial stem or progenitors cells give origin to GBM. The incidence of this tumor seems to increase with age; in fact, 62 years is the median age at diagnosis. Males are affected by GBM tumors 1.7 fold more often than females. According to the presence of mutations in the isocitrate dehydrogenase (IDH) 1 and IDH2 genes GBM is subdivided by the WHO into two major types. More than 90% of GBM cases is constituted by GBM with wild type IDH (Louis et al., 2016). Clinically, grade IV lesions (namely primary GBM) are presented de novo by the majority of patients, while progression from a less aggressive form of WHO grade II diffuse astrocytomas and WHO grade III anaplastic astrocytomas (i.e., secondary GBM) is shown by a small fraction of patients (5–10%; Ohgaki and Kleihues, 2013; Louis et al., 2016). Primary GBM and secondary GBM differ in prognosis and age of onset. As for overall survival (OS) (Doetsch et al., 1999; Louis et al., 2016), primary GBM is typically diagnosed at older age and has a worse prognosis while secondary GBM are less common and affect people under the age of 45; also they develop into low-grade astrocytoma and are associated with better prognosis (Doetsch et al., 1999; Brennan et al., 2013; Ohgaki and Kleihues, 2013; Louis et al., 2016).
Standard of care first-line treatment is constituted by maximal surgical resection (complete resection is performed quite rarely because of the presence of diffuse infiltrations), followed by radiotherapy with concomitant and adjuvant chemotherapy such as the oral alkylating agent, temozolomide (TMZ). Upon this treatment combination GBM show a median OS of about 15 months (Canoll and Goldman, 2008; Stupp et al., 2009; Ohgaki and Kleihues, 2013; Levine et al., 2015).
The increase of patient survival is small and tumors invariably recur after TMZ (Canoll and Goldman, 2008; Stupp et al., 2009; Ohgaki and Kleihues, 2013; Levine et al., 2015). Following the first recurrence, treatment choices can be represented by further surgical resection when possible, or conventional chemotherapy, e.g., TMZ (with different dosing schedules) or nitrosoureas, or treatment with the anti-vascular endothelial growth factor (VEGF) agent, bevacizumab, or the use of the low-intensity alternating electric fields (TTFields). However, these treatments have not achieved significant improvements in survival (Canoll and Goldman, 2008; Stupp et al., 2009; Chamberlain and Johnston, 2010; Stupp et al., 2012; Ohgaki and Kleihues, 2013; Stupp and Hegi, 2013; Chamberlain, 2015). Moreover, the tyrosine kinase inhibitor (TKI) regorafenib has been introduced in the treatment of recurrent GBM (Lombardi et al., 2019).
A detailed characterization of the GBM-associated molecular signatures has made possible the development of novel therapies, including the use of TKIs (Friedman et al., 2009; Quant et al., 2009; Brennan et al., 2013; Wang et al., 2016; Lombardi et al., 2019). On the other hand, based on the results obtained in the context of other tumors (Brahmer et al., 2010; Eder and Kalman, 2014; Larkin et al., 2015; Weber et al., 2015; Kessler et al., 2018; Stathias et al., 2018), the use of programmed cell death protein (PD-1) receptor/programmed death ligand 1 (PD-L1) inhibitors has been suggested for gliomas, including GBM (Motzer et al., 2015; Goldberg et al., 2016; Reck et al., 2016; Schwartz et al., 2016; Reiss et al., 2017; Reardon et al., 2018; Cloughesy et al., 2019; Schalper et al., 2019).
In the present review, we will outline the principal GBM molecular and tumor microenvironment (TME) aspects providing also the grounds for novel targeted therapies and immunotherapy approaches using ICIs for the treatment of GBM affected patients.
Genomic Landscape of GBM
Specific molecular signatures of GBM have been identified through the introduction of next generation sequencing methods, in particular in untreated GBM tumors. It has been found mutations of several genes in GBM including phosphatase and tensin homolog (PTEN), tumor suppressor P53 (TP53), epidermal growth factor receptor (EGFR), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), phosphatidylinositol 3-kinase regulatory subunit alpha (PIK3R1), platelet derived growth factor receptor alpha (PDGFRA), retinoblastoma 1 (RB1), neurofibromin 1 (NF1), gamma-aminobutyric acid receptor subunit alpha-6 (GABRA6), IDH1, mutS homolog 2 (MSH2), mutS homolog 6 (MSH6), mutL homolog 1 (MLH1), and PMS1 homolog 2 (PMS2). Furthermore, several hotspot mutations have been found, like the IDH1 R132H mutation, the B-Raf proto-oncogene (BRAF) V600E mutation (Zhao et al., 2009; Kloosterhof et al., 2011; Kannan et al., 2012; Schwartzentruber et al., 2012; Brennan et al., 2013; Eder and Kalman, 2014; Ceccarelli et al., 2016; Wang et al., 2016; Kessler et al., 2018; Stathias et al., 2018; D’Angelo et al., 2019).
Glioblastoma cases characterized by the presence of mutations in DNA mismatch repair (MMR) genes, e.g., MSH2, MSH6, MLH1, and PMS2 have been suggested to be defined as having a hypermutated profile (Hunter et al., 2006; Cahill et al., 2007; Greenman et al., 2007; Tcga, 2008; Yip et al., 2009; Brennan et al., 2013; Daniel et al., 2019).
Frequent amplification events found in GBM concern chromosome 7 [EGFR/ MET proto-oncogene (MET)/ cyclin dependent kinase 6 (CDK6)], chromosome 12 [cyclin dependent kinase 4 (CDK4)/, mouse double minute 2 homolog (MDM2)], and chromosome 4 (PDGFRA). Gains of the genes SRY-box transcription factor 2 (SOX2), MYCN proto-oncogene (MYCN), cyclin D1 (CCND1), and cyclin E2 (CCNE2) have also been found (Hunter et al., 2006; Cahill et al., 2007; Kuttler and Mai, 2007; Parsons et al., 2008; Tcga, 2008; Yip et al., 2009; Brennan et al., 2013; Sanborn et al., 2013; Zheng et al., 2013; Furgason et al., 2015). Frequent deletions in GBM include deletions in cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B), deletions of 6q26 in which the minimal deleted region seems to include the QKI, KH domain containing RNA binding (QKI) gene, and single gene deletions of low-density lipoprotein receptor-related protein 1B (LRP1B), neuronal PAS domain protein 3 (NPAS3), limbic system associated membrane protein (LSAMP), SET and MYND domain-containing protein 3 (SMYD3) genes (Kamiryo et al., 2002; Hunter et al., 2006; Cahill et al., 2007; Tcga, 2008; Yip et al., 2009; Moreira et al., 2011; Chen et al., 2012; Mizoguchi et al., 2012; Brennan et al., 2013; Nobusawa et al., 2014; Tabouret et al., 2015; Yang et al., 2016).
Repeatedly, EGFR mutations have been found associated with regional gene amplification (Ekstrand et al., 1991; Jaros et al., 1992; Schlegel et al., 1994; Dunn et al., 2012; Brennan et al., 2013; Cominelli et al., 2015). Remarkably, the aberrant exon 1–8 junction of epidermal growth factor receptor variant III (EGFRvIII) was found expressed in a relevant proportion of cases. Additional recurrent non-canonical EGFR transcript forms were also observed (Ekstrand et al., 1991; Jaros et al., 1992; Nishikawa et al., 1994; Tcga, 2008; Brennan et al., 2013; Cominelli et al., 2015). The O-6-methylguanine-DNA methyltransferase (MGMT) locus has been found methylated in about 50% of GBM cases (Esteller et al., 2000; Paz et al., 2004; Hegi et al., 2005; Tcga, 2008; Zawlik et al., 2009; Malmstrom et al., 2012; Reifenberger et al., 2012; Armstrong et al., 2013; Brennan et al., 2013; Wiestler et al., 2013; Wick et al., 2014, 2018; Cominelli et al., 2015).
Recent studies have defined the evolution of tumor cells in GBM cases undergoing therapy as a process of clonal replacement where a fraction of tumor cells is eliminated by the treatment while clones of resistant cells are positively selected (Tcga, 2008; Brennan et al., 2013; Wang et al., 2016). Specifically, intratumoral heterogeneity, with the presence of resistant subclones, both in low grade and high grade glioma is frequently associated with treatment failure. Although a clearly defined pattern of tumor evolution has not yet been described in GBM, TP53 gene mutations have been recently proposed as a marker of subclonal heterogeneity (Tcga, 2008; Brennan et al., 2013; Wang et al., 2016).
Glioblastoma evolution is highly branched, specific alterations and evolutionary patterns frequently occurring depending on the treatment. There is no linear link between the dominant clone at diagnosis and the dominant clone at relapse (Tcga, 2008; Brennan et al., 2013; Wang et al., 2016). Remarkably, genes such as TP53, EGFR, PDGFRA are frequently subjected to a process of mutational switching where a mutated version of a gene, found at diagnosis, is replaced by another mutated version of the same gene at relapse (Tcga, 2008; Brennan et al., 2013; Wang et al., 2016). Hypermutated tumors, which are highly enriched for mutations at CpG dinucleotides, generally harbor mutations in MMR pathway genes, most commonly in MSH6. These MMR alterations have been thought to be associated with putative mutagenic mechanisms of TMZ treatment (Hunter et al., 2006; Cahill et al., 2007; Tcga, 2008; Yip et al., 2009; Brennan et al., 2013; Wang et al., 2016).
A gene expression-based molecular classification has been proposed to integrate somatic mutation and DNA copy number data (Verhaak et al., 2010; Behnan et al., 2019). According to this classification, GBM cases were subdivided in proneural, neural, classical and mesenchymal subtypes. These different subtypes have been associated with gene signatures of normal brain cell types of different neural lineages. Moreover, GBM cases included in the different subtypes have also been associated with a different pathogenesis with GBM clones developing as the result of different causes and/or from different cell type of origin. However, further studies, also investigating glioma stem cells, have been able to identify three subtypes: proneural, mesenchymal and classical subtypes (Verhaak et al., 2010; Behnan et al., 2019).
According to the first proposed classification, GBM cases belonging to the classical subtype show in about the 100% of cases the chromosome 7 amplification paired with chromosome 10 loss. This event is also very frequent in the totality of GBM cases. High-level of EGFR amplification has been observed in 97% of cases belonging to the classical subtype, whereas this alteration has been infrequently found in the other GBM subtypes. Moreover, in association with frequent EGFR alteration, a lack of TP53 mutations has been found in a subset of the classical subtype (Tcga, 2008; Verhaak et al., 2010; Brennan et al., 2013; Wang et al., 2016). The focal 9p21.3 homozygous deletion, targeting the CDKN2A gene, has been also frequently found in the classical subtype, in the 94% of cases belonging to this subtype found associated with EGFR amplification. The homozygous 9p21.3 deletion has been also found mutually exclusive with aberrations in genes belonging to the RB1 pathway such as RB1, CDK4 and cyclin-D2 (CCDN2), thus suggesting that in the cases with focal EGFR amplification the CDKN2A deletion is the sole alteration affecting the RB1 pathway. GBM cases belonging to the classical subtype are also characterized by the high expression of genes belonging to the notch homolog 1, translocation-associated (NOTCH) pathway such as neurogenic locus notch homolog-3 (NOTCH3), jagged-1 (JAG1) and lunatic fringe (LFNG), sonic hedgehog pathway such as smoothened (SMO), growth arrest-specific protein 1 (GAS1) and zinc finger protein GLI1 (GLI1) and the neural precursor and stem cell marker nestin (NES) pathway (Tcga, 2008; Verhaak et al., 2010; Brennan et al., 2013; Wang et al., 2016).
Glioblastoma cases belonging to the proneural subtype were found to be mainly characterized by alterations of PDGFRA and point mutations of IDH1. The focal amplification at the locus 4q12 harboring PDGFRA was associated with high levels of PDGFRA gene expression and the frequent presence of mutations in the PDGFRA gene. The great majority of IDH1 mutations has been found in GBM cases belonging to the proneural subtype. Of note, they have been found to be generally mutually exclusive to PDGFRA alterations. Loss of heterozygosity and mutations of the TP53 gene have been found to be frequent events in the proneural subtype. PIK3CA/PIK3R1 mutations have also been found in the proneural subtypes in cases without PDGFRA abnormalities. The proneural group has been found to be characterized also by the high expression of genes other than PDGFRA that characterize the oligodendrocytic development such as oligodendrocyte transcription factor (OLIG2) and homeobox protein nkx-2.2 (NKX2-2). This group has been found also characterized by the expression of proneural development genes such as SOX family genes and achaete-scute homolog 1 (ASCL1), doublecortin (DCX), delta-like 3 (DLL3), transcription factor 4 (TCF4; Tcga, 2008; Verhaak et al., 2010; Brennan et al., 2013; Wang et al., 2016).
Glioblastoma cases belonging to the neural subtype were characterized by the expression of genes well-known as neuron markers such as GABRA1, neurofilament light chain (NEFL), synaptotagmin-1 (SYT1) and solute carrier family 12 member 5 (SLC12A5). GBM cases belonging to the neural subtype show an enrichment in genes involved in neuron protection and in axon and synaptic transmission (Tcga, 2008; Verhaak et al., 2010; Brennan et al., 2013; Wang et al., 2016).
Glioblastoma cases belonging to the mesenchymal subtype are frequently characterized by the presence of focal hemizygous deletions at 17q11.2 region encompassing the NF1 gene. This has been frequently associated with low NF1 expression levels. Moreover, mutations at the NF1 gene have been found in GBM cases belonging to the mesenchymal subgroup. Concomitant PTEN mutations have also been found in mesenchymal subgroup cases carrying NF1 mutations (Tcga, 2008; Verhaak et al., 2010; Brennan et al., 2013; Wang et al., 2016). GBM cases belonging to the mesenchymal subtype are also characterized by the expression of mesenchymal markers such as chitinase-3-like protein 1 (CHI3L1) and MET. It has been thought that the higher activity of mesenchymal and astrocytic markers such as CD44 and MERKT is linked to an epithelial-to-mesenchymal transition proper of dedifferentiated and transdifferentiated tumors. Finally, GBM cases belonging to the mesenchymal subtype are also characterized by the high expression of genes belonging to the TNF superfamily pathway and NF-kB pathway such as tumor necrosis factor receptor type 1-associated death domain (TRADD), RELB and TNF receptor superfamily member 1A (TNFRSF1A) (Tcga, 2008; Verhaak et al., 2010; Brennan et al., 2013; Wang et al., 2016).
Several clinical features have been associated with the four subtypes. In particular, an association between proneural subtype and age as well as between this subtype and a trend for a longer survival. However, GBM belonging to the proneural subtype have not shown a survival advantage from aggressive treatment protocols. On the other hand, a clear treatment effect has been observed among GBM cases belonging to the classical and mesenchymal subtypes (Tcga, 2008; Verhaak et al., 2010; Brennan et al., 2013; Wang et al., 2016).
The proneural-to-mesenchymal transition upon tumor recurrence has been proposed as a mechanism of treatment resistance for GBM to radiotherapy and/or chemotherapy. GBM patients belonging to the mesenchymal subtype have been associated with survival shorter than the other subtypes, particularly when cases with low transcriptional heterogeneity are considered. Although in the context of poor prognosis patients, GBM cases belonging to the mesenchymal subtype have been found to show favorable response to immunotherapy and intensive radiotherapy and chemotherapy (Verhaak et al., 2010; Behnan et al., 2019).
Long non-coding RNAs (LncRNAs) are RNA transcripts longer than 200 base pairs which do not code for proteins. Although the human genome harbors more than 50,000 LncRNA genes, they are still poor characterized. However, LncRNAs have been found to play key roles in various cell activities related to regulation of gene expression, protein synthesis, stemness, immunity (Schlackow et al., 2017). Moreover, LncRNAs have been found to exert relevant roles in pathogenesis and progression of various cancers including GBM. In particular, a large number of LncRNAs has been found associated with deregulated gene expression and imbalanced biological processes in GBM (Zeng et al., 2018). In this context, the expression of the LncRNA P73 antisense RNA 1T (TP73-AS1) has been associated with poor outcome in GBM patients. GBM patients belonging to less aggressive subgroups have been found to be characterized by hypermethylation and low expression of TP73-AS1. Moreover, it has been found that TP73-AS1 downregulation is associated with the loss of aldehyde dehydrogenase 1 family member A1 (ALDH1A1) expression and the re-sensitivity of the GBM stem cell (GSC) population to TMZ treatment (Mazor et al., 2019). Expression of the LncRNA HOX transcript antisense intergenic RNA (HOTAIR) in GBM has been demonstrated to be significantly higher than in normal tissues and low grade gliomas. Moreover, HOTAIR has been demonstrated to be an independent prognostic factor in GBM associated with proliferation and tumorigenic potential of GBM cells (Zhang et al., 2015; Zhang et al., 2018). LncRNA colorectal neoplasia differentially expressed (CRNDE) has been found highly expressed in GBM and other brain cancers such as astrocytomas. It has also been explained that its overexpression is associated with promotion of tumor cell growth and migration (Ellis et al., 2014; Wang et al., 2016). LncRNA nuclear enriched abundant transcript 1 (NEAT1) has been shown to be a key regulator of nuclear domains implicated in mRNA nuclear retention and splicing. NEAT1 has been found upregulated in human GBM tissues and GBM cell line models and a high NEAT1 expression has been associated with larger tumor size, higher WHO grade, higher recurrence rate and unfavorable overall survival (He et al., 2016). The LncRNA X-inactive specific transcript (XIST) has been found highly expressed in glioma tissues and GSCc. The knockdown of XIST has been shown to suppress proliferation, migration, invasion and tumorigenic potential of GSCs by upregulating miR152 (Yao et al., 2015). The LncRNA SOX2 overlapping transcript (SOX2OT) is characterized by the fact that its transcription genomic region includes the SOX2 gene; a SOX2OT involvement in the transcriptional regulation of SOX2 has also been observed. SOX2OT has been shown to be involved in the proliferation, migration, invasion of GSCs (Su et al., 2017). The LncRNA H19 has been shown to be upregulated in glioma tissues and associated with poor outcome. Moreover, invasion, angiogenesis, stemness and tumorigenicity of GBM cells have been found enhanced when H19 is overexpressed (Jiang et al., 2016). The LncRNA LOC441204 has been found highly expressed in glioma tumor specimens and cell lines. Tumor cell proliferation has been found suppressed by knockdown of LOC441204 in glioma. On the other hand, LOC441204-induced tumor cell growth has been shown to be modulated by the stabilization of the β-catenin pathway (Lin et al., 2017). Regarding the role of other LncRNAs, evidence has also been reported about the fact that the high expression of other LncRNAs such as maternally expressed gene 3 (MEG3), metastasis associated lung adenocarcinoma (MALAT1), cancer susceptibility candidate 2 (CASC2), taurine-upregulated gene 1 (TUG1), DBH antisense RNA 1 (DBH-AS1), AC005035.1, AC010336.2, AC108134.2, AC116351.2, Clorf132, C10orf91, LINC00475, MIR210HG could be associated with poor outcome in GBM cases (Zeng et al., 2018).
Role of the GBM Tumor Microenvironment
The brain is distinguished from the other organs by the presence of the blood-brain-barrier (BBB). The BBB provides a selective barrier between the systemic circulation and the brain, thus representing a limit for the delivery of many therapeutic agents (Chen et al., 2012; Miura et al., 2013). However, a loss of BBB integrity could be displayed in the presence of cancer, in particular during the cancer progression. This seems to be the reason why several agents, including ICIs, that are known to be not capable of penetrating the BBB, have however shown in some extent a clinical efficacy (de Vries et al., 2006; van Tellingen et al., 2015). Specialized endothelial cells, pericytes, and astrocytic foot processes, dictating junctional integrity, are the elements that constitute the BBB. Moreover, BBB integrity can be also regulated by microglia, being these cells capable of repairing the BBB in a purinergic receptor P2RY12-dependent manner in case of injury (de Vries et al., 2006; van Tellingen et al., 2015).
The complex crosstalk of TME components is involved in the regulation of tumor progression (Quail and Joyce, 2013, 2017; Gritsenko et al., 2012; Nakasone et al., 2012; Quail and Joyce, 2013, 2017). The composition of ECM of normal brain is distinctive, with specific tissue-resident cell types such as neurons, astrocytes and microglia. Moreover, the BBB physically protects the ECM from inflammation (Novak and Kaye, 2000; Mahesparan et al., 2003). The most common component of the brain ECM is hyaluronic acid which is localized in the intraparenchymal region (Kim et al., 2018). The haptotactic cues from the vascular basement membrane, the enrichment of vascular derived chemoctatic cues, as well as interconnected axon tracts can determine the therapeutic resistance of GBM cells in the perivascular space further providing haptotactic cues for cellular invasion (Giese and Westphal, 1996; Nimsky et al., 2005; Gritsenko et al., 2012).
A diffuse invasion pattern characterizes GBM (Young et al., 2015). Healthy tissue beyond the tumor margin is infiltrated by the tumor cells, generally enriched in the GSC stem cell fraction, that either migrate individually or collectively practically impeding complete surgical resection (Eyler and Rich, 2008; Sherriff et al., 2013). On the other hand, GBM tumors rarely intravasate and metastasize from the brain to distant organs (Quail and Joyce, 2013, 2017).
Glioblastoma frequently develop in a hypoxic microenvironment which can modify the metabolic pathways of GBM cells. The brain has a high metabolism level in which the glucose is the major energy substrate and lactate, ketone bodies, fatty acids and aminoacids can also be employed. The metabolic homeostasis of the brain is maintained by the interaction among its various constituent cells such as astrocytes, neurons and microglia (Gritsenko et al., 2012; Nakasone et al., 2012; Quail and Joyce, 2013, 2017). This equilibrium can be altered by genomic aberrations and biochemical variations in GBM cells that often metabolize glucose into lactate even when oxygen is present in a process called Warburg effect. GBM cells can also increase intracellular lipid, aminoacid and nucleotide levels. These metabolic adaptations can favor GBM tumor growth (Gritsenko et al., 2012; Nakasone et al., 2012; Quail and Joyce, 2013, 2017).
Hypervascularity is a characteristic of GBM tumors with an increment in angiogenesis compared to healthy brain tissue. This tumor-associated vasculature is not completely formed, with leaky vessels, and associated with an increase in interstitial fluid pressure. A necrotic core softer than the surrounding tissue characterizes the TME of GBM (Brat and Van Meir, 2004; Brat et al., 2004; Persano et al., 2011; Hambardzumyan and Bergers, 2015; Chen and Hambardzumyan, 2018). High density regions called pseudopalisades are formed when cells migrate away from the hypoxic regions. Increased matrix production with respect to both necrotic regions and healthy tissues characterizes these regions (Brat and Van Meir, 2004; Brat et al., 2004; Persano et al., 2011; Hambardzumyan and Bergers, 2015; Chen and Hambardzumyan, 2018). GBM cells are capable of rapidly invading vasculature (Akiyama et al., 2001; Ponta et al., 2003; Zimmermann and Dours-Zimmermann, 2008; Dicker et al., 2014; Schiffer et al., 2018).
Circulatory and immune systems are connected by the lymphatic system that is involved, together with blood vessels, in the exchange of various elements including fluid, waste, debris as well as immune cells (Engelhardt et al., 2017). Together with the absence of a classic lymphatic drainage system, the CNS exhibits several other peculiar features, such as the presence of tight junctions in the BBB, as well as the limited rejection of foreign tissues within the CNS (Louveau et al., 2015; Schiffer et al., 2018).
There are functional lymphatic vessels in the CNS with the presence of different types of antigen-presenting cells (APCs), including microglia, macrophages, astrocytes and canonical APC such as dendritic cells (DCs; Figure 1; Louveau et al., 2015; Schiffer et al., 2018). In the brain, microglia are the predominant APCs whereas DCs carry out a less relevant role (Lowe et al., 1989; Ulvestad et al., 1994; Weiss et al., 2009; Goldmann et al., 2016).
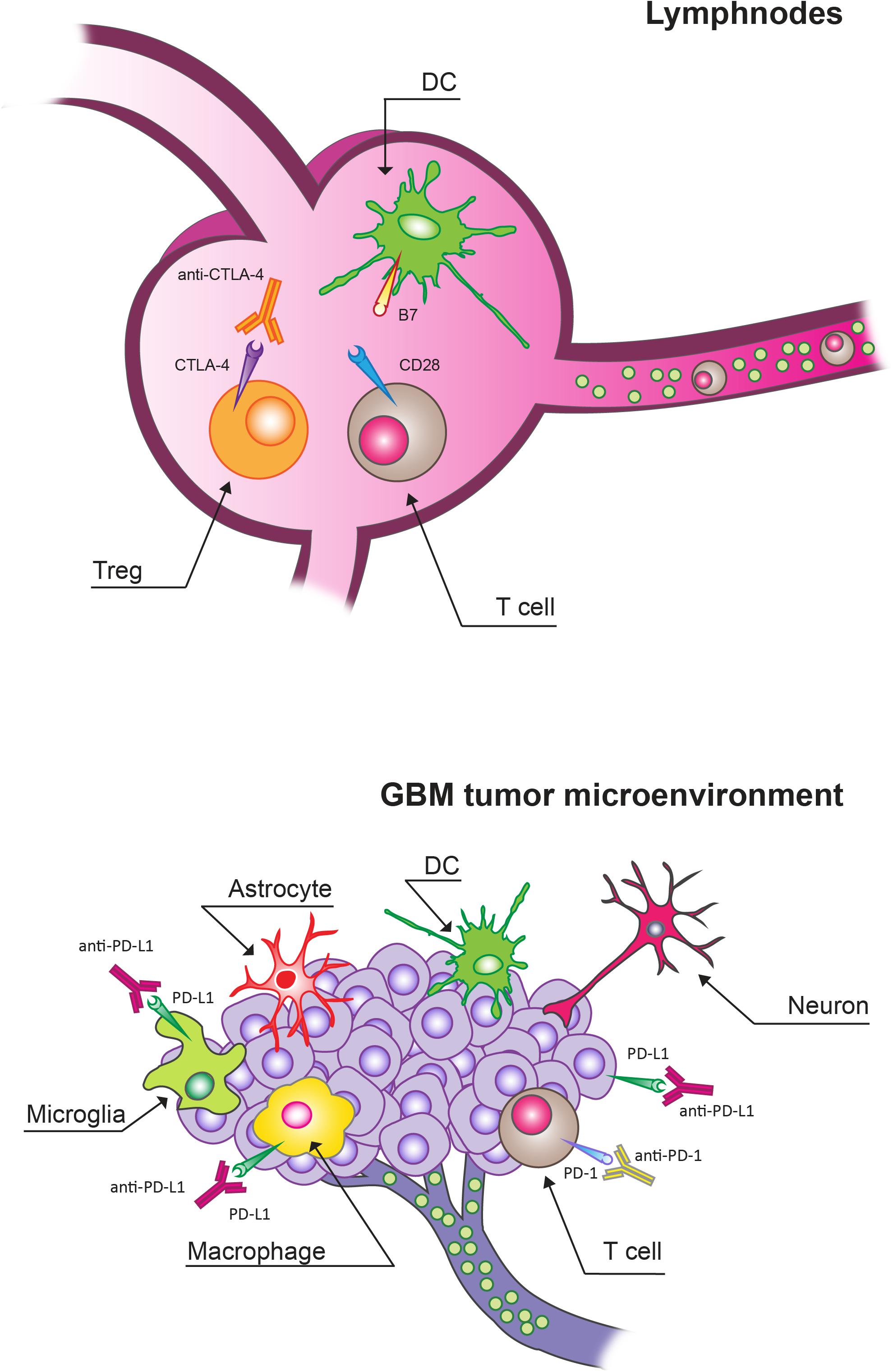
Figure 1. Immune checkpoint inhibitors (ICIs) targets in lymphnodes and in glioblastoma (GBM) tumor microenvironment (TME). Cytotoxic T lymphocyte protein 4 (CTLA-4) blockade mainly acts by targeting Tregs expressing CTLA-4 in lymphnodes. In the context of the GBM TME, programmed cell death protein receptor 1 (PD-1)/programmed death ligand (PD-L1) blockade can overcome the T cell exhaustion and reverse immunosuppression.
Activated T cells can invade the CNS. These activated T cells can cover these compartments in an unrestricted manner. On the other hand, antigens can be presented locally or in the draining cervical lymph nodes. Moreover, the BBB can be compromised, thus allowing a relevant infiltration of multiple immune cell types from the peripheral circulation (Weiss et al., 2009). However, GBM tumors present low numbers of tumor-infiltrating lymphocytes (TILs) and other immune effector cell types compared to other tumor types (Schiffer et al., 2018). The interaction of TILs with the TME can cause their re-education. In particular, the local TME can alter T cell effector function in the process related to anti-tumor immunity even in the CNS, where T cell-mediated inflammatory responses are considered poor under normal physiological contexts. The number of antigen-specific TILs can remain relatively low besides frequently displaying an exhausted phenotype. The peculiar immune environment of the brain can be responsible for this reduced quantity and limited activity of T cells in GBM. In particular, there is a specific need of avoiding unrestrained inflammation in the brain given its solid enclosure and the potential for damage from increased intracranial pressure (Quail and Joyce, 2013, 2017; Gajewski et al., 2017; Keskin et al., 2019). This need is not present with the same extent in peripheral organs. In fact, this environment in which both inflammatory and adaptive immune responses are tightly regulated is specific of the brain; besides there is a variety of immunosuppressive mechanisms at both the molecular and cellular levels (Perng and Lim, 2015). In particular, stromal cells of the brain produce high levels of the classic immunosuppressive cytokines transforming growth factor β (TGFβ), interleukin-10 (IL-10) in response to inflammatory stimuli, including those derived from GBM tumors, in order to maintain homeostasis (Vitkovic et al., 2001; Gong et al., 2012). Furthermore, the accumulation of regulatory T cells (Tregs) is stimulated by IDO which can suppress T cell activity by depleting tryptophan from the microenvironment. Microglia and tumor- infiltrating myeloid cells can also inhibit T cell proliferation and function through the production of high levels of arginase that causes the depletion of tissue arginine levels (Uyttenhove et al., 2003; Fecci et al., 2006a, b; Wainwright et al., 2012).
Immune checkpoints exert a key role in central and peripheral tolerance by counteracting activating signaling (Xu et al., 2018). Under physiological conditions, immune checkpoint molecules represent a negative feedback to regulate inflammatory responses following T cell activation (Krummel and Allison, 1996; Chambers et al., 2001; Collins et al., 2002; Stone et al., 2009; Inarrairaegui et al., 2018). A mechanism used by tumors, including GBM, to inhibit and escape the anti-tumor immune response is represented by the expression of checkpoint molecules, such as cytotoxic T-lymphocyte antigen 4 (CTLA-4) and PD1 (Stone et al., 2009; Francisco et al., 2010; Cheng et al., 2013; Bhandaru and Rotte, 2017; Hui et al., 2017; Wei et al., 2017, 2018, 2019a,b; Rotte et al., 2018; Kalbasi and Ribas, 2020; Sharma and Allison, 2020).
Small Molecules for Targeted Therapies in GBM
The progresses in the molecular classification of GBM have allowed the identification of dysregulated pathways that could represent potential targets for new treatment strategies (Figure 2).
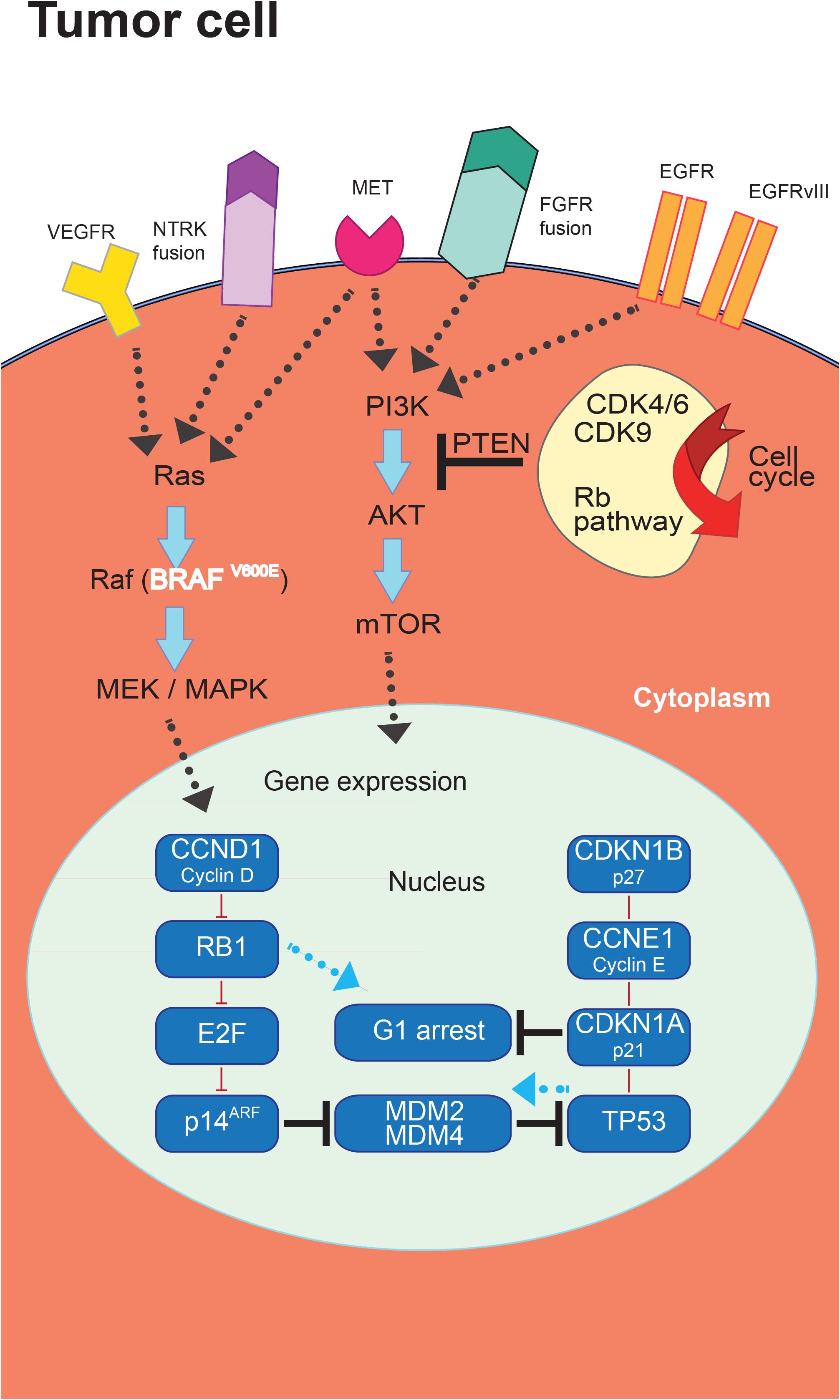
Figure 2. Targeted therapies in glioblastoma (GBM). The introduction of novel targeted therapies has been allowed by the comprehensive characterization of the molecular landscape of somatic genomic alterations identifying a series of mutated genes and abnormal rearrangements potentially utilizable as therapeutic targets.
Glioblastoma is a vascularized tumor which is histologically characterized by the expression of VEGF and other proangiogenic cytokines involved in the stimulation of endothelial cell proliferation, migration and survival (Schiffer et al., 2018). In patients with a relapsed GBM, the TKI regorafenib has received approval in the GBM treatment (Lombardi et al., 2019).
Other TKIs targeting VEGF family components have been proposed for the treatment of GBM besides regorafenib. Of note, vascular normalization has been proposed as an alternative strategy for the employment of antiangiogenic therapies in which the objective is to modulate the tumor vasculature in order to reduce hypoxia, and to support physiological angiogenesis. This process could ultimately improve perfusion and drug delivery. In this context, promising results in reducing angiogenesis and normalizing vascularization have been shown by cediranib and sutinib (Batchelor et al., 2013; Grisanti et al., 2019).
The PI3K/mammalian target of rapamycin (mTOR) pathway is a targetable pathway in GBM. In this context, the mTOR inhibitor temsirolimus did not show a treatment efficacy as single agent in recurrent GBM (Chang et al., 2005). Similarly, the pan-PI3K inhibitor buparlisib did not demonstrate a treatment efficacy (Wen et al., 2019). Also treatment combinations of mTOR pathway inhibitors with radiotherapy and TMZ or in combination with radiotherapy only did not show efficacy (Ma et al., 2015; Wick et al., 2016).
Targeting MDM2 and mouse double minute 4 homolog (MDM4) activity has been suggested for GBM cases carrying MDM2 or MDM4 gene amplification (Wick et al., 2019).
Moreover, the CDK4/6 inhibitor palbociclib failed to demonstrate the efficacy of this treatment in GBM (Taylor et al., 2018). CDK9 is an alternative targetable CDK (Taylor et al., 2018).
The use of TKIs targeting EGFR as single agents did not demonstrate significant activity for GBM treatment (Lassman et al., 2005; Hegi et al., 2011). It has not yet been agreed on the potential use of MET as target for GBM treatment. The use of the TKIs crizotinib and cabozantinib in recurrent GBM has achieved modest efficacy after several attempts (International Cancer Genome Consortium PedBrain Tumor Project, 2016; Wen et al., 2018). Tests have been carried out for larotrectinib and entrectinib in neutrophic tyrosine receptor kinase (NTRK) fusion-positive GBM without any confirmation on their efficacy (Ferguson et al., 2018). Notwithstanding the frequent expression in GBM of fibroblast growth factor receptors (FGFRs), a relevance as potential therapy target seems to be restricted to GBM exhibiting FGFR-transforming acidic coiled-coil containing protein TACC fusions (Singh et al., 2012), as shown by using the pan-FGFR kinase inhibitor erdafitinib (Di Stefano et al., 2015). A modest treatment efficacy has been obtained for the possible targeting of BRAFV600E mutation in GBM (Kaley et al., 2018). Finally, eribulin has been proposed to inhibit TERT activity in GBM (Takahashi et al., 2019). A list of the current clinical trials employing TKIs for GBM treatment is reported in Table 1.
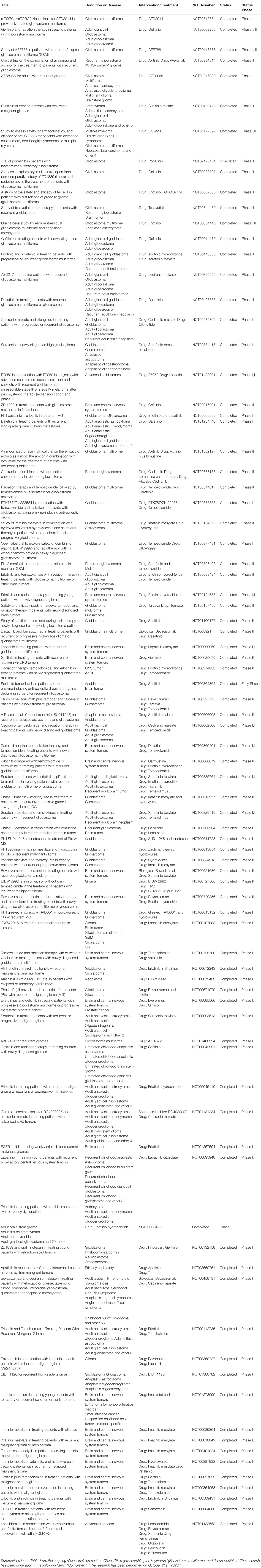
Table 1. Clinical trials in glioblastoma (GBM) using tyrosine kinase inhibitors (TKIs).
Use of Icis for GBM Treatment
Following the results of ICIs use in other cancers, the use of PD-1/PD-L1 inhibitors has been proposed for GBM (Table 2). Clinical trial results have shown that GBM patients with unresectable tumors do not benefit from monotherapy with nivolumab in terms of survival improvement when compared to bevacizumab (Reiss et al., 2017). Moreover, pembrolizumab showed limited activity for GBM (Reardon et al., 2014, 2016; Schwartz et al., 2016; Reardon et al., 2017; Reiss et al., 2017; Wen et al., 2018).
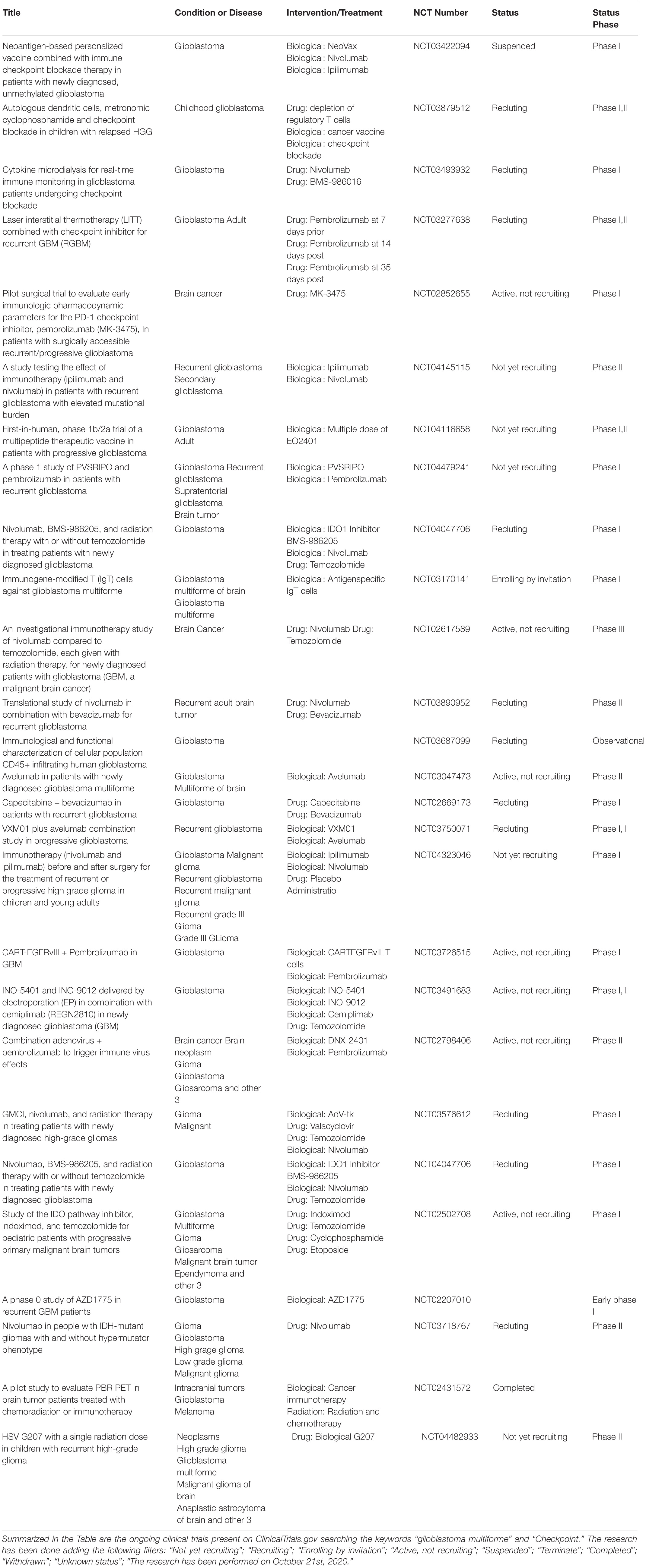
Table 2. Clinical trials in glioblastoma (GBM) using immune checkpoint inhibitors (ICIs).
Recent tests have been carried out involving patients with newly diagnosed or relapsed GBMs for the use of ICIs (e.g., nivolumab or pembrolizumab) in neoadjuvant and/or adjuvant administration, although no straightforward results have been obtained (Cloughesy et al., 2019; Schalper et al., 2019).
Glioblastoma tumors of cases non-responsive to ICIs have shown an enrichment in mutations of the PTEN gene (Zhao et al., 2019) that has been associated with an immunosuppressive TME characterized by the presence of GBM cells expressing CD44. PTEN mutant tumors were characterized by highly clustering tumor cells with a lack of T cell infiltration (Peng et al., 2016; George et al., 2017). Furthermore, the poor responsiveness to ICIs of GBM cases carrying PTEN mutations has been related to a low PD-L1 expression for the involvement of the PI3K-mTOR pathway that is downstream to PTEN (Lastwika et al., 2016).
Responsiveness to ICI was associated with the presence of mutations of BRAF/protein tyrosine phosphatase non-receptor type 11 (PTPN11). In this subset of BRAF/PTPN11 GBM patients, treatment combinations of ICIs and MAPK inhibitors could be introduced (Toso et al., 2014; Toso et al., 2014; Ebert et al., 2016; Wang et al., 2016).
The heterogeneous response rate to ICIs highlights the need of identifying the subgroups of patients who could benefit the most from the use of this immunotherapy treatment. PD-L1 expression was the first marker evaluated as predictor of a clinical response to ICIs (Ansell et al., 2015). PD-L1 expression in gliomas was associated with IDH status (Berghoff and Preusser, 2016; Garber et al., 2016; Berghoff et al., 2017). Importantly, mesenchymal GBM has been found having high levels of PD-L1 expression that may suggest that the expression of immune checkpoint proteins and aggressiveness of GBM tumors may be correlated (Garber et al., 2016). More recently, the tumor mutational burden has been proposed as a predictive marker of responsiveness to ICIs. However, it has not generally been demonstrated that the tumor mutational burden is capable of sufficiently predicting long term clinical benefits (Champiat et al., 2014; Rizvi et al., 2015; Schumacher et al., 2015; Le et al., 2017). Moreover, recent studies have shown that higher somatic mutation and neoepitope loads have not been found in GBM cases responsive to ICIs (Zhao et al., 2019). The infiltration of mutation-reactive class I and class II T cells into the tumor seems not to be precluded by a low mutational load in GBM (Cloughesy et al., 2019; Schalper et al., 2019; Zhao et al., 2019). The presence of alterations in the MMR genes is another proposed biomarker (Cloughesy et al., 2019; Schalper et al., 2019). The expression of MHC class I molecules has been associated to responsiveness to ICIs since it is involved in the presentation of antigens and characterized by highly heterogeneous expression levels in GBM (Indraccolo et al., 2019).
Discussion
Surgery followed by radiotherapy and chemotherapy with alkylating agents constitutes the standard first-line treatment of GBM (Stupp et al., 2005; Canoll and Goldman, 2008; Levine et al., 2015). Complete resection of the GBM tumors is generally not possible given its high invasive features. Although this combination therapy can prolong survival, the prognosis is still poor due to several factors including chemoresistance. Multiple mechanisms appear to be involved in the development of drug resistance in GBM including overexpression of drug efflux transporter pumps such as p-glycoprotein, the presence of a GSC population, a relevant activity of DNA repair mechanisms and dysregulated apotosis processes such as MGMT, the MMR pathway, the base excision repair (BER) pathway and the TP53 pathway (Walker et al., 1992; Bobola et al., 1996; Qian and Brent, 1997; Jaeckle et al., 1998; Chen et al., 1999; Esteller et al., 2000; Middlemas et al., 2000; Paz et al., 2004; Hegi et al., 2005; Helleday et al., 2005; Bryant and Helleday, 2006; Zawlik et al., 2009; van Nifterik et al., 2010; Malmstrom et al., 2012; Reifenberger et al., 2012; Armstrong et al., 2013; Brennan et al., 2013; Wiestler et al., 2013; Wick et al., 2014, 2018; Erasimus et al., 2016; Peng et al., 2016; Sun et al., 2018; Gupta et al., 2018; Zhang et al., 2018; Christmann and Kaina, 2019; Hafner et al., 2019; Mantovani et al., 2019). Tumor/TME interactions also contribute to the development of drug resistance in GBM tumor cells (Hanahan and Weinberg, 2011; Ab and Jn, 2012; Rodriguez-Hernandez et al., 2014; Munoz et al., 2015).
Systemic delivery uses existing vessels to deliver anti-tumor drugs to the tumor. To overcome the impediment of the BBB several strategies have been proposed including chemical modification of the drugs, high dose chemotherapy capable of inducing a transient BBB disruption, nanoparticle-based drug delivery and peptide-based drug delivery. Nevertheless, no straightforward results have still been reached (Siegal, 2013).
Glioblastoma stem cell cell population has been shown to induce a certain degree of radio- chemoresistance given their high expression of anti-apoptotic proteins, ATP-binding cassette pumps, their increased capability of DNA damage repair, as well as their high capacity of migration and invasion (Bao et al., 2006; Calabrese et al., 2007; Eyler and Rich, 2008; Diehn et al., 2009; Pietras et al., 2014). GSCs have been found capable of secreting angiogenic factors which in turn are responsible for an enhancement in the formation of tumor blood vessels, this has been frequently associated with high tumor aggressiveness. Moreover, the TME cell components can promote GSC survival by VEGF secretion (Wada et al., 2006). The interaction of TME with GSCs can facilitate tumor progression and consequently therapeutic resistance (Chen and Liu, 2012; Miura et al., 2013).
Over the past 10 years, the knowledge regarding genomic features of GBM has been greatly increased by comprehensive multiplatform genome-wide analyses. As a result of these analyses, it has emerged that GBM comprises a group of highly heterogeneous tumor types, each with peculiar molecular/genetic features (Tcga, 2008; Brennan et al., 2013; Wang et al., 2016).
In GBM the phase I/II and III trials investigating the use of therapies molecularly targeting oncogenic alterations did not generally show straightforward results and, consequently, their clinical utilization is still limited. However, although limited activity or no therapeutic efficacy has so far been produced by the use of TKIs, improvement in understanding the mechanisms of action of these compounds could help to determine how to better incorporate their use in the existing treatment modalities. Redundancies are frequently present in the molecular pathways that can be targeted which makes the inhibition of any pathway largely ineffective (Tcga, 2008; Brennan et al., 2013; Wang et al., 2016). The failure of targeted therapies can also be ascribed to another possible reason such as the fact that several genomic alterations are important only for the initial stages of tumor progression whereas other molecular mechanisms outweigh their role in the later stages. On the other hand, several genomic alterations in GBM can interfere with GBM cell metabolism. In particular, alterations in the growth factor signaling pathways that can control metabolic flux have been found in high frequency as well as recurrent mutations in IDH1 and IDH2 genes, whose encoded proteins are part of the tricarboxylic acid (TCA) cycle. Alterations of the cellular metabolism, which is controlled also by the biochemical microenvironment, could contribute to the failure of the proposed targeted therapies (Tcga, 2008; Brennan et al., 2013; Wang et al., 2016). A better understanding of the interactions constituting this interplay between altered genome and biochemical microenvironment could contribute to finding more effective treatment strategies in the reverting of altered cellular metabolism of GBM cells.
The TME of GBM is largely immunosuppressive, therefore efficiency of ICI treatments can be strongly affected by this condition (Akiyama et al., 2001; Brat and Van Meir, 2004; Brat et al., 2004; Nimsky et al., 2005; Zimmermann and Dours-Zimmermann, 2008; Persano et al., 2011; Sherriff et al., 2013; Dicker et al., 2014; Hambardzumyan and Bergers, 2015; Young et al., 2015; Chen and Hambardzumyan, 2018). GBM patients frequently present reduced levels of circulating CD4+ and CD8+ lymphocytes as a consequence of chemotherapy treatments (Gustafson et al., 2010; Mirzaei et al., 2017). A clear molecular/immunological signature that can be predictive of response to ICI treatments has not yet been identified (Motzer et al., 2015; Goldberg et al., 2016; Reck et al., 2016; Schwartz et al., 2016; Reiss et al., 2017; Reardon et al., 2018; Cloughesy et al., 2019; Schalper et al., 2019).
The treatment of different cancers has markedly been revolutionized by immunotherapy. Nevertheless, the data obtained so far concerning the use of ICIs for the treatment of GBM patients seem to be not sufficient to propose this type of immunotherapy as a standard treatment for GBM (Reardon et al., 2014, 2016; Motzer et al., 2015; Goldberg et al., 2016; Reck et al., 2016; Schwartz et al., 2016; Reardon et al., 2017; Reiss et al., 2017; Wen et al., 2018; Cloughesy et al., 2019; Schalper et al., 2019).
Another immunotherapy approach that can be used also in combinations with ICIs is the chimeric antigen receptor-T (CAR-T) cell therapy targeting specific tumor associated antigens. The introduction of CAR-T cell therapy approaches also in solid tumors including GBM has been favored by the success of this therapy in hematological malignancies (Neelapu et al., 2017; Maude et al., 2018). Concerning GBM treatment, several clinical trials have been proposed showing that there are still substantial obstacles including TME immune suppression (Table 3; Morgan et al., 2010; Brown et al., 2015, 2016; Zah et al., 2016; Ahmed et al., 2017; Walseng et al., 2017; Richman et al., 2018). To increase CAR-T treatment efficacy several CAR-T modifications have been proposed such as the knocking out of genes encoding T cell inhibitory receptors or signaling molecules (e.g., PD-1 or CTLA-4) or the co-expression of activating chimeric switch receptor (CSR; Figure 3; Prosser et al., 2012; Shin et al., 2012; Ankri et al., 2013; Kobold et al., 2015; Liu et al., 2016).
Table 3. Clinical trials in glioblastoma (GBM) using chimeric antigen receptor-T (CAR-T).
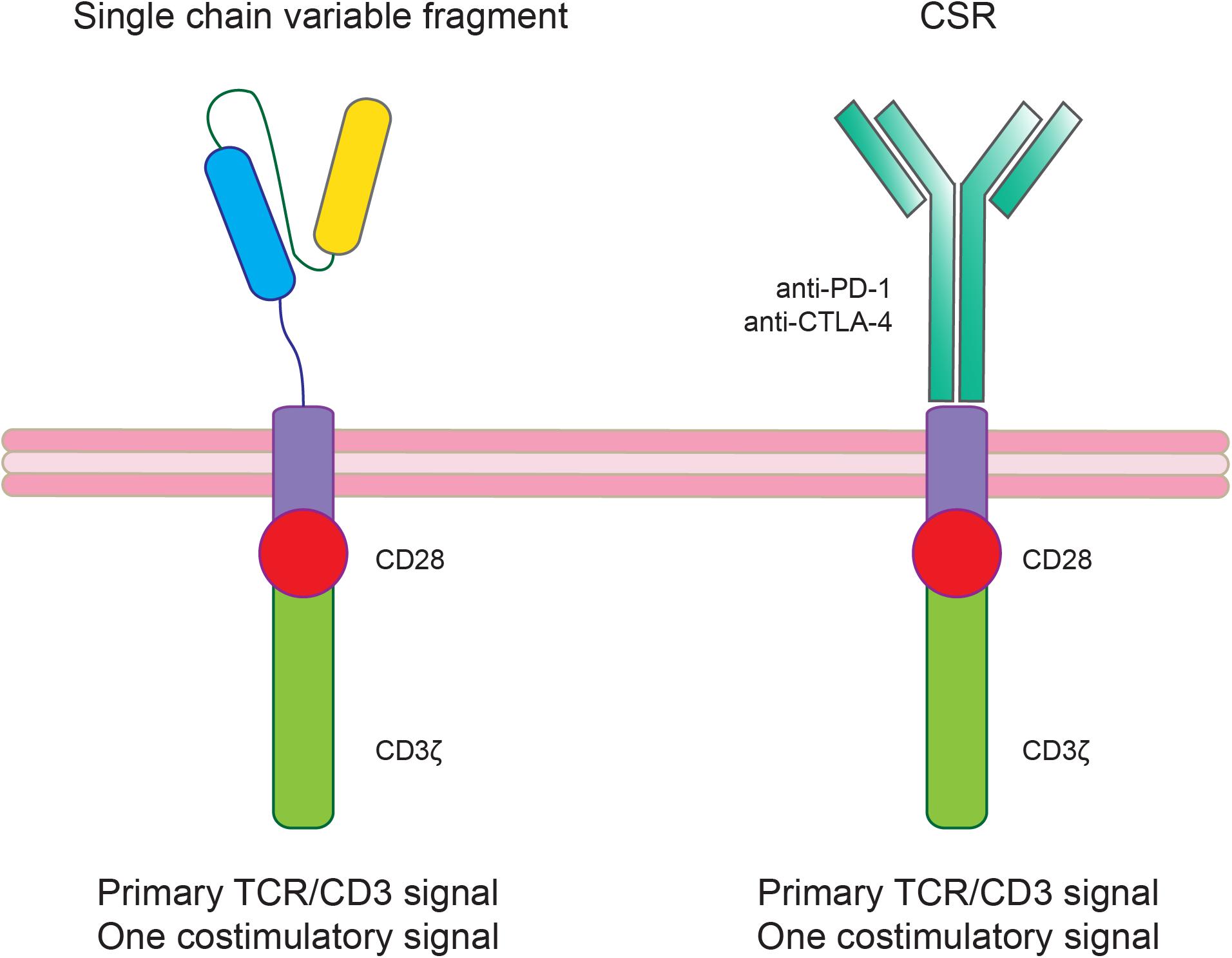
Figure 3. Modified chimeric antigen receptor-T (CAR-T) cells to ameliorate treatment efficacy by counteracting the immunosuppressive glioblastoma (GBM) tumor microenvironment (TME). The co-expression of an activating chimeric switch receptor (CSR), that combines the extracellular ligand-binding domain of an inhibitory receptor (PD-1 or CTLA-4) fused through a transmembrane domain with the cytoplasmic co-stimulatory signaling domain of CD28, could improve CAR-T cell efficacy in GBM.
Understanding the molecular and immunological complexity of GBM more and more could provide the grounds for the introduction of other immunotherapeutic approaches such as the use of CAR-T cell therapy, in combination with ICIs or TKIs, in the treatment paradigm of GBM.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Supported by Progetto Giovani Ricercatori n. GR-2016-02364678, Ministero della Salute, Rome, Italy.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ab, H., and Jn, R. (2012). The quest for self-identity: not all cancer stem cells are the same. Clin. Cancer Res. 18, 3495–3498. doi: 10.1158/1078-0432.ccr-12-1456
Ahmed, N., Brawley, V., Hegde, M., Bielamowicz, K., Kalra, M., Landi, D., et al. (2017). HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 3, 1094–1101.
Akiyama, Y., Jung, S., Salhia, B., Lee, S., Hubbard, S., Taylor, M., et al. (2001). Hyaluronate receptors mediating glioma cell migration and proliferation. J. Neurooncol. 53, 115–127. doi: 10.1023/a:1012297132047
Altmann, C., Keller, S., and Schmidt, M. H. H. (2019). The role of svz stem cells in glioblastoma. Cancers 11:448. doi: 10.3390/cancers11040448
Ankri, C., Shamalov, K., Horovitz-Fried, M., Mauer, S., and Cohen, C. J. (2013). Human T cells engineered to express a programmed death 1/28 costimulatory retargeting molecule display enhanced antitumor activity. J. Immunol. 191, 4121–4129. doi: 10.4049/jimmunol.1203085
Ansell, S. M., Lesokhin, A. M., Borrello, I., Halwani, A., Scott, E. C., Gutierrez, M., et al. (2015). PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 372, 311–319. doi: 10.1056/NEJMoa1411087
Armstrong, T. S., Wefel, J. S., Wang, M., Gilbert, M. R., Won, M., Bottomley, A., et al. (2013). Net clinical benefit analysis of radiation therapy oncology group 0525: a phase III trial comparing conventional adjuvant temozolomide with dose-intensive temozolomide in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 31, 4076–4084.
Bao, S., Wu, Q., McLendon, R. E., Hao, Y., Shi, Q., Hjelmeland, A. B., et al. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760. doi: 10.1038/nature05236
Batchelor, T. T., Mulholland, P., Neyns, B., Nabors, L. B., Campone, M., Wick, A., et al. (2013). Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol. 31, 3212–3218.
Behnan, J., Finocchiaro, G., and Hanna, G. (2019). The landscape of the mesenchymal signature in brain tumours. Brain 142, 847–866. doi: 10.1093/brain/awz044
Berghoff, A. S., Kiesel, B., Widhalm, G., Wilhelm, D., Rajky, O., Kurscheid, S., et al. (2017). Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol. 19, 1460–1468.
Berghoff, A. S., and Preusser, M. (2016). In search of a target: PD-1 and PD-L1 profiling across glioma types. Neuro Oncol. 18, 1331–1332.
Bhandaru, M., and Rotte, A. (2017). Blockade of programmed cell death protein-1 pathway for the treatment of melanoma. J. Dermatol. Res. Ther. 1, 1–11.
Bobola, M. S., Tseng, S. H., Blank, A., Berger, M. S., and Silber, J. R. (1996). Role of O6-methylguanine-DNA methyltransferase in resistance of human brain tumor cell lines to the clinically relevant methylating agents temozolomide and streptozotocin. Clin. Cancer Res. 2, 735–741.
Brahmer, J. R., Drake, C. G., Wollner, I., Powderly, J. D., Picus, J., Sharfman, W. H., et al. (2010). Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 28, 3167–3175.
Brat, D. J., Castellano-Sanchez, A. A., Hunter, S. B., Pecot, M., Cohen, C., Hammond, E. H., et al. (2004). Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 64, 920–927. doi: 10.1158/0008-5472.can-03-2073
Brat, D. J., and Van Meir, E. G. (2004). Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Invest. 84, 397–405.
Brennan, C. W., Verhaak, R. G., McKenna, A., Campos, B., Noushmehr, H., Salama, S. R., et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477.
Brown, C. E., Alizadeh, D., Starr, R., Weng, L., Wagner, J. R., Naranjo, A., et al. (2016). Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 375, 2561–2569. doi: 10.1056/NEJMoa1610497
Brown, C. E., Badie, B., Barish, M. E., Weng, L., Ostberg, J. R., Chang, W. C., et al. (2015). Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 21, 4062–4072.
Bryant, H. E., and Helleday, T. (2006). Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 34, 1685–1691. doi: 10.1093/nar/gkl108
Cahill, D. P., Levine, K. K., Betensky, R. A., Codd, P. J., Romany, C. A., Reavie, L. B., et al. (2007). Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 13, 2038–2045.
Calabrese, C., Poppleton, H., Kocak, M., Hogg, T. L., Fuller, C., Hamner, B., et al. (2007). A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82. doi: 10.1016/j.ccr.2006.11.020
Canoll, P., and Goldman, J. E. (2008). The interface between glial progenitors and gliomas. Acta Neuropathol. 116, 465–477. doi: 10.1007/s00401-008-0432-9
Ceccarelli, M., Barthel, F. P., Malta, T. M., Sabedot, T. S., Salama, S. R., Murray, B. A., et al. (2016). Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse Glioma. Cell 164, 550–563.
Chamberlain, M. C. (2015). Salvage therapy with lomustine for temozolomide refractory recurrent anaplastic astrocytoma: a retrospective study. J. Neurooncol. 122, 329–338. doi: 10.1007/s11060-014-1714-9
Chamberlain, M. C., and Johnston, S. K. (2010). Salvage therapy with single agent bevacizumab for recurrent glioblastoma. J. Neurooncol. 96, 259–269. doi: 10.1007/s11060-009-9957-6
Chambers, C. A., Kuhns, M. S., Egen, J. G., and Allison, J. P. (2001). CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 19, 565–594. doi: 10.1146/annurev.immunol.19.1.565
Champiat, S., Ferte, C., Lebel-Binay, S., Eggermont, A., and Soria, J. C. (2014). Exomics and immunogenics: bridging mutational load and immune checkpoints efficacy. Oncoimmunology 3:e27817.
Chang, S. M., Wen, P., Cloughesy, T., Greenberg, H., Schiff, D., Conrad, C., et al. (2005). Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest. New Drugs 23, 357–361. doi: 10.1007/s10637-005-1444-0
Chen, A.-J., Paik, J.-H., Zhang, H., Shukla, S. A., Mortensen, R., Hu, J., et al. (2012). STAR RNA-binding protein Quaking suppresses cancer via stabilization of specific miRNA. Genes Dev. 26, 1459–1472.
Chen, Y., and Liu, L. (2012). Modern methods for delivery of drugs across the blood-brain barrier. Adv. Drug Deliv. Rev. 64, 640–665.
Chen, Z., and Hambardzumyan, D. (2018). Immune microenvironment in glioblastoma subtypes. Front. Immunol. 9:1004. doi: 10.3389/fimmu.2018.01004
Chen, Z. P., Yarosh, D., Garcia, Y., Tampieri, D., Mohr, G., Malapetsa, A., et al. (1999). Relationship between O6-methylguanine-DNA methyltransferase levels and clinical response induced by chloroethylnitrosourea therapy in glioma patients. Can. J. Neurol. Sci. 26, 104–109.
Cheng, X., Veverka, V., Radhakrishnan, A., Waters, L. C., Muskett, F. W., Morgan, S. H., et al. (2013). Structure and interactions of the human programmed cell death 1 receptor. J. Biol. Chem. 288, 11771–11785.
Christmann, M., and Kaina, B. (2019). Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat. Res. 780, 15–28. doi: 10.1016/j.mrrev.2017.10.001
Cloughesy, T. F., Mochizuki, A. Y., Orpilla, J. R., Hugo, W., Lee, A. H., Davidson, T. B., et al. (2019). Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 25, 477–486. doi: 10.1038/s41591-018-0337-7
Collins, A. V., Brodie, D. W., Gilbert, R. J., Iaboni, A., Manso-Sancho, R., Walse, B., et al. (2002). The interaction properties of costimulatory molecules revisited. Immunity 17, 201–210.
Cominelli, M., Grisanti, S., Mazzoleni, S., Branca, C., Buttolo, L., Furlan, D., et al. (2015). EGFR amplified and overexpressing glioblastomas and association with better response to adjuvant metronomic temozolomide. J. Natl. Cancer Inst. 107:djv041. doi: 10.1093/jnci/djv041
D’Angelo, F., Ceccarelli, M., Tala, Garofano, L., Zhang, J., Frattini, V., et al. (2019). The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat. Med. 25, 176–187. doi: 10.1038/s41591-018-0263-8
Daniel, P., Sabri, S., Chaddad, A., Meehan, B., Jean-Claude, B., Rak, J., et al. (2019). Temozolomide induced hypermutation in glioma: evolutionary mechanisms and therapeutic opportunities. Front. Oncol. 9:41. doi: 10.3389/fonc.2019.00041
de Vries, N. A., Beijnen, J. H., Boogerd, W., and van Tellingen, O. (2006). Blood-brain barrier and chemotherapeutic treatment of brain tumors. Expert Rev. Neurother. 6, 1199–1209. doi: 10.1586/14737175.6.8.1199
Di Stefano, A. L., Fucci, A., Frattini, V., Labussiere, M., Mokhtari, K., Zoppoli, P., et al. (2015). Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-type Glioma. Clin. Cancer Res. 21, 3307–3317. doi: 10.1158/1078-0432.CCR-14-2199
Dicker, K. T., Gurski, L. A., Pradhan-Bhatt, S., Witt, R. L., Farach-Carson, M. C., and Jia, X. (2014). Hyaluronan: a simple polysaccharide with diverse biological functions. Acta Biomater. 10, 1558–1570.
Diehn, M., Cho, R. W., Lobo, N. A., Kalisky, T., Dorie, M. J., Kulp, A. N., et al. (2009). Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780–783. doi: 10.1038/nature07733
Doetsch, F., Caille, I., Lim, D. A., Garcia-Verdugo, J. M., and Alvarez-Buylla, A. (1999). Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97, 703–716.
Dunn, G. P., Rinne, M. L., Wykosky, J., Genovese, G., Quayle, S. N., Dunn, I. F., et al. (2012). Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 26, 756–784.
Ebert, P. J. R., Cheung, J., Yang, Y., McNamara, E., Hong, R., Moskalenko, M., et al. (2016). MAP Kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 44, 609–621. doi: 10.1016/j.immuni.2016.01.024
Eder, K., and Kalman, B. (2014). Molecular heterogeneity of glioblastoma and its clinical relevance. Pathol. Oncol. Res. 20, 777–787. doi: 10.1007/s12253-014-9833-3
Ekstrand, A. J., James, C. D., Cavenee, W. K., Seliger, B., Pettersson, R. F., and Collins, V. P. (1991). Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 51, 2164–2172.
Ellis, B. C., Graham, L. D., and Molloy, P. L. (2014). CRNDE, a long non-coding RNA responsive to insulin/IGF signaling, regulates genes involved in central metabolism. Biochim. Biophys. Acta 1843, 372–386. doi: 10.1016/j.bbamcr.2013.10.016
Engelhardt, B., Vajkoczy, P., and Weller, R. O. (2017). The movers and shapers in immune privilege of the CNS. Nat. Immunol. 18, 123–131. doi: 10.1038/ni.3666
Erasimus, H., Gobin, M., Niclou, S., and Van Dyck, E. (2016). DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res. 769, 19–35.
Esteller, M., Garcia-Foncillas, J., Andion, E., Goodman, S. N., Hidalgo, O. F., Vanaclocha, V., et al. (2000). Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 343, 1350–1354. doi: 10.1056/NEJM200011093431901
Eyler, C. E., and Rich, J. N. (2008). Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 26, 2839–2845.
Fecci, P. E., Mitchell, D. A., Whitesides, J. F., Xie, W., Friedman, A. H., Archer, G. E., et al. (2006a). Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 66, 3294–3302. doi: 10.1158/0008-5472.can-05-3773
Fecci, P. E., Sweeney, A. E., Grossi, P. M., Nair, S. K., Learn, C. A., Mitchell, D. A., et al. (2006b). Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin. Cancer Res. 12, 4294–4305. doi: 10.1158/1078-0432.CCR-06-0053
Ferguson, S. D., Zhou, S., Huse, J. T., de Groot, J. F., Xiu, J., Subramaniam, D. S., et al. (2018). Targetable gene fusions associate with the IDH wild-type astrocytic lineage in adult gliomas. J. Neuropathol. Exp. Neurol. 77, 437–442.
Francisco, L. M., Sage, P. T., and Sharpe, A. H. (2010). The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242.
Friedman, H. S., Prados, M. D., Wen, P. Y., Mikkelsen, T., Schiff, D., Abrey, L. E., et al. (2009). Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 27, 4733–4740.
Furgason, J. M., Koncar, R. F., Michelhaugh, S. K., Sarkar, F. H., Mittal, S., Sloan, A. E., et al. (2015). Whole genome sequence analysis links chromothripsis to EGFR, MDM2, MDM4, and CDK4 amplification in glioblastoma. Oncoscience 2, 618–628.
Gajewski, T. F., Corrales, L., Williams, J., Horton, B., Sivan, A., and Spranger, S. (2017). Cancer immunotherapy targets based on understanding the T cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv. Exp. Med. Biol. 1036, 19–31. doi: 10.1007/978-3-319-67577-0_2
Garber, S. T., Hashimoto, Y., Weathers, S. P., Xiu, J., Gatalica, Z., Verhaak, R. G., et al. (2016). Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro Oncol. 18, 1357–1366. doi: 10.1093/neuonc/now132
George, S., Miao, D., Demetri, G. D., Adeegbe, D., Rodig, S. J., Shukla, S., et al. (2017). Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 46, 197–204. doi: 10.1016/j.immuni.2017.02.001
Giese, A., and Westphal, M. (1996). Glioma invasion in the central nervous system. Neurosurgery 39, 235–250. doi: 10.1097/00006123-199608000-00001
Goldberg, S. B., Gettinger, S. N., Mahajan, A., Chiang, A. C., Herbst, R. S., Sznol, M., et al. (2016). Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 17, 976–983. doi: 10.1016/S1470-2045(16)30053-5
Goldmann, T., Wieghofer, P., Jordão, M. J. C., Prutek, F., Hagemeyer, N., Frenzel, K., et al. (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805. doi: 10.1038/ni.3423
Gong, D., Shi, W., Yi, S. J., Chen, H., Groffen, J., and Heisterkamp, N. (2012). TGFbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 13:31. doi: 10.1186/1471-2172-13-31
Greenman, C., Stephens, P., Smith, R., Dalgliesh, G. L., Hunter, C., Bignell, G., et al. (2007). Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158.
Grisanti, S., Ferrari, V. D., Buglione, M., Agazzi, G. M., Liserre, R., Poliani, L., et al. (2019). Second line treatment of recurrent glioblastoma with sunitinib: results of a phase II study and systematic review of literature. J. Neurosurg. Sci. 63, 458–467. doi: 10.23736/S0390-5616.16.03874-1
Gritsenko, P. G., Ilina, O., and Friedl, P. (2012). Interstitial guidance of cancer invasion. J. Pathol. 226, 185–199. doi: 10.1002/path.3031
Gupta, S. K., Smith, E. J., Mladek, A. C., Tian, S., Decker, P. A., Kizilbash, S. H., et al. (2018). PARP inhibitors for sensitization of alkylation chemotherapy in glioblastoma: impact of blood-brain barrier and molecular heterogeneity. Front. Oncol. 8:670. doi: 10.3389/fonc.2018.00670
Gustafson, M. P., Lin, Y., New, K. C., Bulur, P. A., O’Neill, B. P., Gastineau, D. A., et al. (2010). Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 12, 631–644.
Hafner, A., Bulyk, M. L., Jambhekar, A., and Lahav, G. (2019). The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 20, 199–210.
Hambardzumyan, D., and Bergers, G. (2015). Glioblastoma: defining tumor niches. Trends Cancer 1, 252–265. doi: 10.1016/j.trecan.2015.10.009
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
He, C., Jiang, B., Ma, J., and Li, Q. (2016). Aberrant NEAT1 expression is associated with clinical outcome in high grade glioma patients. APMIS 124, 169–174. doi: 10.1111/apm.12480
Hegi, M. E., Diserens, A. C., Bady, P., Kamoshima, Y., Kouwenhoven, M. C., Delorenzi, M., et al. (2011). Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib–a phase II trial. Mol. Cancer Ther. 10, 1102–1112. doi: 10.1158/1535-7163.MCT-11-0048
Hegi, M. E., Diserens, A.-C., Gorlia, T., Hamou, M.-F., Tribolet, N. D., Weller, M., et al. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. Engl. J. Med. 352, 997–1003.
Helleday, T., Bryant, H. E., and Schultz, N. (2005). Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle 4, 1176–1178.
Hui, E., Cheung, J., Zhu, J., Su, X., Taylor, M. J., Wallweber, H. A., et al. (2017). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433. doi: 10.1126/science.aaf1292
Hunter, C., Smith, R., Cahill, D. P., Stephens, P., Stevens, C., Teague, J., et al. (2006). A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 66, 3987–3991.
Inarrairaegui, M., Melero, I., and Sangro, B. (2018). Immunotherapy of hepatocellular carcinoma: facts and hopes. Clin. Cancer Res. 24, 1518–1524. doi: 10.1158/1078-0432.CCR-17-0289
Indraccolo, S., Lombardi, G., Fassan, M., Pasqualini, L., Giunco, S., Marcato, R., et al. (2019). Genetic, epigenetic, and immunologic profiling of MMR-deficient relapsed glioblastoma. Clin. Cancer Res. 25, 1828–1837. doi: 10.1158/1078-0432.CCR-18-1892
International Cancer Genome Consortium PedBrain Tumor Project. (2016). Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 22, 1314–1320.
Jaeckle, K. A., Eyre, H. J., Townsend, J. J., Schulman, S., Knudson, H. M., Belanich, M., et al. (1998). Correlation of tumor O6 methylguanine-DNA methyltransferase levels with survival of malignant astrocytoma patients treated with bis-chloroethylnitrosourea: a Southwest Oncology Group study. J. Clin. Oncol. 16, 3310–3315.
Jaros, E., Perry, R. H., Adam, L., Kelly, P. J., Crawford, P. J., Kalbag, R. M., et al. (1992). Prognostic implications of p53 protein, epidermal growth factor receptor, and Ki-67 labelling in brain tumours. Br. J. Cancer 66, 373–385. doi: 10.1038/bjc.1992.273
Jiang, X., Yan, Y., Hu, M., Chen, X., Wang, Y., Dai, Y., et al. (2016). Increased level of H19 long noncoding RNA promotes invasion, angiogenesis, and stemness of glioblastoma cells. J. Neurosurg. 2016, 129–136. doi: 10.3171/2014.12.JNS1426.test
Kalbasi, A., and Ribas, A. (2020). Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 20, 25–39. doi: 10.1038/s41577-019-0218-4
Kaley, T., Touat, M., Subbiah, V., Hollebecque, A., Rodon, J., Lockhart, A. C., et al. (2018). BRAF Inhibition in BRAF(V600)-Mutant Gliomas: results From the VE-BASKET Study. J. Clin. Oncol. 36, 3477–3484. doi: 10.1200/JCO.2018.78.9990
Kamiryo, T., Tada, K., Shiraishi, S., Shinojima, N., Nakamura, H., Kochi, M., et al. (2002). Analysis of homozygous deletion of the p16 gene and correlation with survival in patients with glioblastoma multiforme. J. Neurosurg. 96, 815–822.
Kannan, K., Inagaki, A., Silber, J., Gorovets, D., Zhang, J., Kastenhuber, E. R., et al. (2012). Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 3, 1194–1203. doi: 10.18632/oncotarget.689
Keskin, D. B., Anandappa, A. J., Sun, J., Tirosh, I., Mathewson, N. D., Li, S., et al. (2019). Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239. doi: 10.1038/s41586-018-0792-9
Kessler, T., Sahm, F., Sadik, A., Stichel, D., Hertenstein, A., Reifenberger, G., et al. (2018). Molecular differences in IDH wildtype glioblastoma according to MGMT promoter methylation. Neuro Oncol. 20, 367–379.
Kim, Y., Kang, H., Powathil, G., Kim, H., Trucu, D., Lee, W., et al. (2018). Role of extracellular matrix and microenvironment in regulation of tumor growth and LAR-mediated invasion in glioblastoma. PLoS One 4:e0117296. doi: 10.1371/journal.pone.0204865
Kloosterhof, N. K., Bralten, L. B., Dubbink, H. J., French, P. J., and van den Bent, M. J. (2011). Isocitrate dehydrogenase-1 mutations: a fundamentally new understanding of diffuse glioma? Lancet Oncol. 12, 83–91. doi: 10.1016/S1470-2045(10)70053-X
Kobold, S., Grassmann, S., Chaloupka, M., Lampert, C., Wenk, S., Kraus, F., et al. (2015). Impact of a new fusion receptor on PD-1-mediated immunosuppression in adoptive T cell therapy. J. Natl. Cancer Inst. 107:djv146. doi: 10.1093/jnci/djv146
Krummel, M. F., and Allison, J. P. (1996). CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 183, 2533–2540. doi: 10.1084/jem.183.6.2533
Kuttler, F., and Mai, S. (2007). Formation of non-random extrachromosomal elements during development, differentiation and oncogenesis. Semin. Cancer Biol. 17, 56–64. doi: 10.1016/j.semcancer.2006.10.007
Larkin, J., Chiarion-Sileni, V., Gonzalez, R., Grob, J. J., Cowey, C. L., Lao, C. D., et al. (2015). Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34. doi: 10.1056/NEJMoa1504030
Lassman, A. B., Rossi, M. R., Raizer, J. J., Abrey, L. E., Lieberman, F. S., Grefe, C. N., et al. (2005). Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin. Cancer Res. 11, 7841–7850.
Lastwika, K. J., Wilson, W., Li, Q. K., Norris, J., Xu, H., Ghazarian, S. R., et al. (2016). Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 76, 227–238. doi: 10.1158/0008-5472.CAN-14-3362
Le, D. T., Durham, J. N., Smith, K. N., Wang, H., Bartlett, B. R., Aulakh, L. K., et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413. doi: 10.1126/science.aan6733
Levine, J. H., Simonds, E. F., Bendall, S. C., Davis, K. L., Amir, el-A. D., Tadmor, M. D., et al. (2015). Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197. doi: 10.1016/j.cell.2015.05.047
Lin, T.-K., Chang, C.-N., Tsai, C.-S., Huang, Y.-C., Lu, Y.-J., Chen, W.-J., et al. (2017). The long non-coding RNA LOC441204 enhances cell growth in human glioma. Sci. Rep. 7:5603. doi: 10.1038/s41598-017-05688-0
Liu, X., Ranganathan, R., Jiang, S., Fang, C., Sun, J., Kim, S., et al. (2016). A Chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 76, 1578–1590.
Lombardi, G., De Salvo, G. L., Brandes, A. A., Eoli, M., Ruda, R., Faedi, M., et al. (2019). Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 20, 110–119.
Louis, D. N., Perry, A., Reifenberger, G., von Deimling, A., Figarella-Branger, D., Cavenee, W. K., et al. (2016). The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820. doi: 10.1007/s00401-016-1545-1
Louveau, A., Smirnov, I., Keyes, T. J., Eccles, J. D., Rouhani, S. J., Peske, J. D., et al. (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341. doi: 10.1038/nature14432
Lowe, J., MacLennan, K. A., Powe, D. G., Pound, J. D., and Palmer, J. B. (1989). Microglial cells in human brain have phenotypic characteristics related to possible function as dendritic antigen presenting cells. J. Pathol. 159, 143–149. doi: 10.1002/path.1711590209
Ma, D. J., Galanis, E., Anderson, S. K., Schiff, D., Kaufmann, T. J., Peller, P. J., et al. (2015). A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro Oncol. 17, 1261–1269.
Mahesparan, R., Read, T. A., Lund-Johansen, M., Skaftnesmo, K. O., Bjerkvig, R., and Engebraaten, O. (2003). Expression of extracellular matrix components in a highly infiltrative in vivo glioma model. Acta Neuropathol. 105, 49–57. doi: 10.1007/s00401-002-0610-0
Malmstrom, A., Gronberg, B. H., Marosi, C., Stupp, R., Frappaz, D., Schultz, H., et al. (2012). Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 13, 916–926. doi: 10.1016/S1470-2045(12)70265-6
Mantovani, F., Collavin, L., and Del Sal, G. (2019). Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 26, 199–212.
Maude, S. L., Laetsch, T. W., Buechner, J., Rives, S., Boyer, M., Bittencourt, H., et al. (2018). Tisagenlecleucel in children and young adults with B-cell lymphoblastic Leukemia. N. Engl. J. Med. 378, 439–448. doi: 10.1056/NEJMoa1709866
Mazor, G., Levin, L., Picard, D., Ahmadov, U., Carén, H., Borkhardt, A., et al. (2019). The lncRNA TP73-AS1 is linked to aggressiveness in glioblastoma and promotes temozolomide resistance in glioblastoma cancer stem cells. Cell Death Dis. 10:246. doi: 10.1038/s41419-019-1477-5
Mendes, M., Sousa, J., Pais, A., and Vitorino, C. (2018). Targeted theranostic nanoparticles for brain tumor treatment. – Abstract – Europe PMC. Pharmaceutics. Available at: https://europepmc.org/article/pmc/pmc6321593 (accessed September 4, 2020).
Middlemas, D. S., Stewart, C. F., Kirstein, M. N., Poquette, C., Friedman, H. S., Houghton, P. J., et al. (2000). Biochemical correlates of temozolomide sensitivity in pediatric solid tumor xenograft models. Clin. Cancer Res. 6, 998–1007.
Mirzaei, R., Sarkar, S., and Yong, V. W. T. (2017). Cell exhaustion in glioblastoma: intricacies of immune checkpoints. Trends Immunol. 38, 104–115.
Miura, Y., Takenaka, T., Toh, K., Wu, S., Nishihara, H., Kano, M. R., et al. (2013). Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS Nano 7, 8583–8592. doi: 10.1021/nn402662d
Mizoguchi, M., Yoshimoto, K., Ma, X., Guan, Y., Hata, N., Amano, T., et al. (2012). Molecular characteristics of glioblastoma with 1p/19q co-deletion. Brain Tumor Pathol. 29, 148–153.
Moreira, F., Kiehl, T.-R., So, K., Ajeawung, N. F., Honculada, C., Gould, P., et al. (2011). NPAS3 demonstrates features of a tumor suppressive role in driving the progression of Astrocytomas. Am. J. Pathol. 179, 462–476.
Morgan, R. A., Yang, J. C., Kitano, M., Dudley, M. E., Laurencot, C. M., and Rosenberg, S. A. (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843–851.
Motzer, R. J., Escudier, B., McDermott, D. F., George, S., Hammers, H. J., Srinivas, S., et al. (2015). Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 373, 1803–1813. doi: 10.1056/NEJMoa1510665
Munoz, J. L., Walker, N. D., Scotto, K. W., and Rameshwar, P. (2015). Temozolomide competes for P-glycoprotein and contributes to chemoresistance in glioblastoma cells. Cancer Lett. 367, 69–75. doi: 10.1016/j.canlet.2015.07.013
Nakasone, E. S., Askautrud, H. A., Kees, T., Park, J. H., Plaks, V., Ewald, A. J., et al. (2012). Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 21, 488–503. doi: 10.1016/j.ccr.2012.02.017
Neelapu, S. S., Locke, F. L., Bartlett, N. L., Lekakis, L. J., Miklos, D. B., Jacobson, C. A., et al. (2017). Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544.
Nimsky, C., Ganslandt, O., Hastreiter, P., Wang, R., Benner, T., Sorensen, A. G., et al. (2005). Preoperative and intraoperative diffusion tensor imaging-based fiber tracking in glioma surgery. Neurosurgery 56, 130–137. doi: 10.1227/01.neu.0000144842.18771.30
Nishikawa, R., Ji, X. D., Harmon, R. C., Lazar, C. S., Gill, G. N., Cavenee, W. K., et al. (1994). A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 91, 7727–7731.
Nobusawa, S., Hirato, J., Kurihara, H., Ogawa, A., Okura, N., Nagaishi, M., et al. (2014). Intratumoral heterogeneity of genomic imbalance in a case of epithelioid glioblastoma with BRAF V600E mutation. Brain Pathol. 24, 239–246. doi: 10.1111/bpa.12114
Novak, U., and Kaye, A. H. (2000). Extracellular matrix and the brain: components and function. J. Clin. Neurosci. 7, 280–290. doi: 10.1054/jocn.1999.0212
Ohgaki, H., and Kleihues, P. (2013). The definition of primary and secondary glioblastoma. Clin. Cancer Res. 19, 764–772. doi: 10.1158/1078-0432.CCR-12-3002
Ostrom, Q. T., Cioffi, G., Gittleman, H., Patil, N., Waite, K., Kruchko, C., et al. (2019). CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012-2016. Neuro Oncol. 21, v1–v100.
Parsons, D. W., Jones, S., Zhang, X., Lin, J. C.-H., Leary, R. J., Angenendt, P., et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812.
Paz, M. F., Yaya-Tur, R., Rojas-Marcos, I., Reynes, G., Pollan, M., Aguirre-Cruz, L., et al. (2004). CpG island hypermethylation of the DNA repair enzyme methyltransferase predicts response to temozolomide in primary gliomas. Clin. Cancer Res. 10, 4933–4938. doi: 10.1158/1078-0432.CCR-04-0392
Peng, W., Chen, J. Q., Liu, C., Malu, S., Creasy, C., Tetzlaff, M. T., et al. (2016). Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 6, 202–216. doi: 10.1158/2159-8290.CD-15-0283
Perng, P., and Lim, M. (2015). Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS Sites. Front. Oncol. 5:153. doi: 10.3389/fonc.2015.00153
Persano, L., Rampazzo, E., Della Puppa, A., Pistollato, F., and Basso, G. (2011). The three-layer concentric model of glioblastoma: cancer stem cells, microenvironmental regulation, and therapeutic implications. ScientificWorldJournal 11, 1829–1841. doi: 10.1100/2011/736480
Pietras, A., Katz, A. M., Ekström, E. J., Wee, B., Halliday, J. J., Pitter, K. L., et al. (2014). Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 14, 357–369. doi: 10.1016/j.stem.2014.01.005
Ponta, H., Sherman, L., and Herrlich, P. A. (2003). CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 4, 33–45.
Prosser, M. E., Brown, C. E., Shami, A. F., Forman, S. J., and Jensen, M. C. (2012). Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol. Immunol. 51, 263–272. doi: 10.1016/j.molimm.2012.03.023
Qian, X. C., and Brent, T. P. (1997). Methylation hot spots in the 5’ flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res. 57, 3672–3677.
Quail, D. F., and Joyce, J. A. (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437.
Quail, D. F., and Joyce, J. A. (2017). The microenvironmental landscape of brain tumors. Cancer Cell 31, 326–341. doi: 10.1016/j.ccell.2017.02.009
Quant, E. C., Norden, A. D., Drappatz, J., Muzikansky, A., Doherty, L., Lafrankie, D., et al. (2009). Role of a second chemotherapy in recurrent malignant glioma patients who progress on bevacizumab. Neuro Oncol. 11, 550–555. doi: 10.1215/15228517-2009-006
Reardon, D. A., Freeman, G., Wu, C., Chiocca, E. A., Wucherpfennig, K. W., Wen, P. Y., et al. (2014). Immunotherapy advances for glioblastoma. Neuro Oncol. 16, 1441–1458.
Reardon, D. A., Gokhale, P. C., Klein, S. R., Ligon, K. L., Rodig, S. J., Ramkissoon, S. H., et al. (2016). Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol. Res. 4, 124–135. doi: 10.1158/2326-6066.CIR-15-0151
Reardon, D. A., Nayak, L., Peters, K. B., Clarke, J. L., Jordan, J. T., De Groot, J. F., et al. (2018). Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. J. Clin. Oncol. 36, 2006–2006. doi: 10.1200/JCO.2018.36.15_suppl.2006
Reardon, D. A., Omuro, A., Brandes, A. A., Rieger, J., Wick, A., Sepúlveda, J. M., et al. (2017). OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: CheckMate 143. Neuro Oncol. 19:iii21. doi: 10.1093/neuonc/nox036.071
Reck, M., Rodriguez-Abreu, D., Robinson, A. G., Hui, R., Csoszi, T., Fulop, A., et al. (2016). Pembrolizumab versus Chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833. doi: 10.1056/NEJMoa1606774
Reifenberger, G., Hentschel, B., Felsberg, J., Schackert, G., Simon, M., Schnell, O., et al. (2012). Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int. J. Cancer 131, 1342–1350. doi: 10.1002/ijc.27385
Reiss, S. N., Yerram, P., Modelevsky, L., and Grommes, C. (2017). Retrospective review of safety and efficacy of programmed cell death-1 inhibitors in refractory high grade gliomas. J. Immunother. Cancer 5:99. doi: 10.1186/s40425-017-0302-x
Richman, S. A., Nunez-Cruz, S., Moghimi, B., Li, L. Z., Gershenson, Z. T., Mourelatos, Z., et al. (2018). High-Affinity GD2-Specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol. Res. 6, 36–46. doi: 10.1158/2326-6066.CIR-17-0211
Rizvi, N. A., Hellmann, M. D., Snyder, A., Kvistborg, P., Makarov, V., Havel, J. J., et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. doi: 10.1126/science.aaa1348
Rodriguez-Hernandez, I., Perdomo, S., Santos-Briz, A., Garcia, J. L., Gomez-Moreta, J. A., Cruz, J. J., et al. (2014). Analysis of DNA repair gene polymorphisms in glioblastoma. Gene 536, 79–83. doi: 10.1016/j.gene.2013.11.077
Rotte, A., Jin, J. Y., and Lemaire, V. (2018). Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol. 29, 71–83.
Sanborn, J. Z., Salama, S. R., Grifford, M., Brennan, C. W., Mikkelsen, T., Jhanwar, S., et al. (2013). Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons. Cancer Res. 73, 6036–6045.
Schalper, K. A., Rodriguez-Ruiz, M. E., Diez-Valle, R., Lopez-Janeiro, A., Porciuncula, A., Idoate, M. A., et al. (2019). Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 25, 470–476. doi: 10.1038/s41591-018-0339-5
Schiffer, D., Annovazzi, L., Casalone, C., Corona, C., and Mellai, M. (2018). Glioblastoma: microenvironment and niche concept. Cancers 11:E5.
Schlackow, M., Nojima, T., Gomes, T., Dhir, A., Carmo-Fonseca, M., and Proudfoot, N. J. (2017). Distinctive patterns of transcription and RNA processing for human lincRNAs. Mol. Cell 65, 25–38. doi: 10.1016/j.molcel.2016.11.029
Schlegel, J., Stumm, G., Brändle, K., Merdes, A., Mechtersheimer, G., Hynes, N. E., et al. (1994). Amplification and differential expression of members of the erbB-gene family in human glioblastoma. J. Neurooncol. 22, 201–207. doi: 10.1007/bf01052920
Schumacher, T. N., Kesmir, C., and van Buuren, M. M. (2015). Biomarkers in cancer immunotherapy. Cancer Cell 27, 12–14. doi: 10.1016/j.ccell.2014.12.004
Schwartz, L. H., Litiere, S., de Vries, E., Ford, R., Gwyther, S., Mandrekar, S., et al. (2016). RECIST 1.1-Update and clarification: from the RECIST committee. Eur. J. Cancer 62, 132–137. doi: 10.1016/j.ejca.2016.03.081
Schwartzentruber, J., Korshunov, A., Liu, X. Y., Jones, D. T., Pfaff, E., Jacob, K., et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231.
Sharma, P., and Allison, J. P. (2020). Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 20, 75–76. doi: 10.1038/s41577-020-0275-8
Sherriff, J., Tamangani, J., Senthil, L., Cruickshank, G., Spooner, D., Jones, B., et al. (2013). Patterns of relapse in glioblastoma multiforme following concomitant chemoradiotherapy with temozolomide. Br. J. Radiol. 86:20120414. doi: 10.1259/bjr.20120414
Shin, J. H., Park, H. B., Oh, Y. M., Lim, D. P., Lee, J. E., Seo, H. H., et al. (2012). Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood 119, 5678–5687. doi: 10.1182/blood-2011-09-380519
Siegal, T. (2013). Which drug or drug delivery system can change clinical practice for brain tumor therapy? Neuro Oncol. 15, 656–669. doi: 10.1093/neuonc/not016
Singh, D., Chan, J. M., Zoppoli, P., Niola, F., Sullivan, R., Castano, A., et al. (2012). Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337, 1231–1235.
Stathias, V., Jermakowicz, A. M., Maloof, M. E., Forlin, M., Walters, W., Suter, R. K., et al. (2018). Drug and disease signature integration identifies synergistic combinations in glioblastoma. Nat. Commun. 9:5315. doi: 10.1038/s41467-018-07659-z
Stone, J. D., Chervin, A. S., and Kranz, D. M. (2009). T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology 126, 165–176.
Stupp, R., and Hegi, M. E. (2013). Brain cancer in 2012: molecular characterization leads the way. Nat. Rev. Clin. Oncol. 10, 69–70.
Stupp, R., Hegi, M. E., Mason, W. P., van den Bent, M. J., Taphoorn, M. J., Janzer, R. C., et al. (2009). Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466. doi: 10.1016/S1470-2045(09)70025-7
Stupp, R., Mason, W. P., van den Bent, M. J., Weller, M., Fisher, B., Taphoorn, M. J., et al. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996. doi: 10.1056/NEJMoa043330
Stupp, R., Wong, E. T., Kanner, A. A., Steinberg, D., Engelhard, H., Heidecke, V., et al. (2012). NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur. J. Cancer 48, 2192–2202. doi: 10.1016/j.ejca.2012.04.011
Su, R., Cao, S., Ma, J., Liu, Y., Liu, X., Zheng, J., et al. (2017). Knockdown of SOX2OT inhibits the malignant biological behaviors of glioblastoma stem cells via up-regulating the expression of miR-194-5p and miR-122. Mol. Cancer 16:171. doi: 10.1186/s12943-017-0737-1
Sun, Q., Pei, C., Li, Q., Dong, T., Dong, Y., Xing, W., et al. (2018). Up-regulation of MSH6 is associated with temozolomide resistance in human glioblastoma. Biochem. Biophys. Res. Commun. 496, 1040–1046.
Tabouret, E., Labussière, M., Alentorn, A., Schmitt, Y., Marie, Y., and Sanson, M. (2015). LRP1B deletion is associated with poor outcome for glioblastoma patients. J. Neurol. Sci. 358, 440–443.
Takahashi, M., Miki, S., Fujimoto, K., Fukuoka, K., Matsushita, Y., Maida, Y., et al. (2019). Eribulin penetrates brain tumor tissue and prolongs survival of mice harboring intracerebral glioblastoma xenografts. Cancer Sci. 110, 2247–2257. doi: 10.1111/cas.14067
Taylor, J. W., Parikh, M., Phillips, J. J., James, C. D., Molinaro, A. M., Butowski, N. A., et al. (2018). Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J. Neurooncol. 140, 477–483.
Tcga. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. doi: 10.1038/nature07385
Toso, A., Revandkar, A., Di Mitri, D., Guccini, I., Proietti, M., Sarti, M., et al. (2014). Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 9, 75–89. doi: 10.1016/j.celrep.2014.08.044
Ulvestad, E., Williams, K., Bjerkvig, R., Tiekotter, K., Antel, J., and Matre, R. (1994). Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J. Leukoc. Biol. 56, 732–740. doi: 10.1002/jlb.56.6.732
Uyttenhove, C., Pilotte, L., Theate, I., Stroobant, V., Colau, D., Parmentier, N., et al. (2003). Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 9, 1269–1274. doi: 10.1038/nm934
van Nifterik, K. A., van den Berg, J., van der Meide, W. F., Ameziane, N., Wedekind, L. E., Steenbergen, R. D. M., et al. (2010). Absence of the MGMT protein as well as methylation of the MGMT promoter predict the sensitivity for temozolomide. Br. J. Cancer 103, 29–35.
van Tellingen, O., Yetkin-Arik, B., de Gooijer, M. C., Wesseling, P., Wurdinger, T., and de Vries, H. E. (2015). Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 19, 1–12. doi: 10.1016/j.drup.2015.02.002
Verhaak, R. G. W., Hoadley, K. A., Purdom, E., Wang, V., Qi, Y., Wilkerson, M. D., et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. doi: 10.1016/j.ccr.2009.12.020
Vitkovic, L., Maeda, S., and Sternberg, E. (2001). Anti-inflammatory cytokines: expression and action in the brain. Neuroimmunomodulation 9, 295–312. doi: 10.1159/000059387
Wada, T., Haigh, J. J., Ema, M., Hitoshi, S., Chaddah, R., Rossant, J., et al. (2006). Vascular endothelial growth factor directly inhibits primitive neural stem cell survival but promotes definitive neural stem cell survival. J. Neurosci. 26, 6803–6812. doi: 10.1523/JNEUROSCI.0526-06.2006
Wainwright, D. A., Balyasnikova, I. V., Chang, A. L., Ahmed, A. U., Moon, K. S., Auffinger, B., et al. (2012). IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 18, 6110–6121.
Walker, M. C., Masters, J. R., and Margison, G. P. (1992). O6-alkylguanine-DNA-alkyltransferase activity and nitrosourea sensitivity in human cancer cell lines. Br. J. Cancer 66, 840–843.
Walseng, E., Koksal, H., Sektioglu, I. M., Fane, A., Skorstad, G., Kvalheim, G., et al. (2017). A TCR-based Chimeric Antigen Receptor. Sci. Rep. 7:10713. doi: 10.1038/s41598-017-11126-y
Wang, J., Cazzato, E., Ladewig, E., Frattini, V., Rosenbloom, D. I., Zairis, S., et al. (2016). Clonal evolution of glioblastoma under therapy. Nat. Genet. 48, 768–776. doi: 10.1038/ng.3590
Weber, J. S., D’Angelo, S. P., Minor, D., Hodi, F. S., Gutzmer, R., Neyns, B., et al. (2015). Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 16, 375–384. doi: 10.1016/S1470-2045(15)70076-8
Wei, S. C., Anang, N. A. S., Sharma, R., Andrews, M. C., Reuben, A., Levine, J. H., et al. (2019a). Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. U.S.A. 116, 22699–22709. doi: 10.1073/pnas.1821218116
Wei, S. C., Sharma, R., Anang, N. A. S., Levine, J. H., Zhao, Y., Mancuso, J. J., et al. (2019b). Negative co-stimulation constrains T Cell differentiation by imposing boundaries on possible cell states. Immunity 50, 1084. doi: 10.1016/j.immuni.2019.03.004
Wei, S. C., Duffy, C. R., and Allison, J. P. (2018). Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8, 1069–1086. doi: 10.1158/2159-8290.CD-18-0367
Wei, S. C., Levine, J. H., Cogdill, A. P., Zhao, Y., Anang, N. A. S., Andrews, M. C., et al. (2017). Distinct cellular mechanisms underlie anti-CTLA-4 and Anti-PD-1 checkpoint blockade. Cell 170, 1120–1133.e17. doi: 10.1016/j.cell.2017.07.024
Weiss, N., Miller, F., Cazaubon, S., and Couraud, P.-O. (2009). The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta 1788, 842–857. doi: 10.1016/j.bbamem.2008.10.022
Wen, P. Y., Drappatz, J., de Groot, J., Prados, M. D., Reardon, D. A., Schiff, D., et al. (2018). Phase II study of cabozantinib in patients with progressive glioblastoma: subset analysis of patients naive to antiangiogenic therapy. Neuro Oncol. 20, 249–258. doi: 10.1093/neuonc/nox154
Wen, P. Y., Touat, M., Alexander, B. M., Mellinghoff, I. K., Ramkissoon, S., McCluskey, C. S., et al. (2019). Buparlisib in patients with recurrent glioblastoma harboring phosphatidylinositol 3-kinase pathway activation: an open-label, multicenter, multi-arm, phase II trial. J. Clin. Oncol. 37, 741–750. doi: 10.1200/JCO.18.01207
Wick, W., Dettmer, S., Berberich, A., Kessler, T., Karapanagiotou-Schenkel, I., Wick, A., et al. (2019). N2M2 (NOA-20) phase I/II trial of molecularly matched targeted therapies plus radiotherapy in patients with newly diagnosed non-MGMT hypermethylated glioblastoma. Neuro Oncol. 21, 95–105. doi: 10.1093/neuonc/noy161
Wick, W., Gorlia, T., Bady, P., Platten, M., van den Bent, M. J., Taphoorn, M. J., et al. (2016). Phase II Study of Radiotherapy and Temsirolimus versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation (EORTC 26082). Clin. Cancer Res. 22, 4797–4806.
Wick, W., Osswald, M., Wick, A., and Winkler, F. (2018). Treatment of glioblastoma in adults. Ther. Adv. Neurol. Disord. 11, 1756286418790452. doi: 10.1177/1756286418790452
Wick, W., Weller, M., van den Bent, M., Sanson, M., Weiler, M., von Deimling, A., et al. (2014). MGMT testing–the challenges for biomarker-based glioma treatment. Nat. Rev. Neurol. 10, 372–385. doi: 10.1038/nrneurol.2014.100
Wiestler, B., Claus, R., Hartlieb, S. A., Schliesser, M. G., Weiss, E. K., Hielscher, T., et al. (2013). Malignant astrocytomas of elderly patients lack favorable molecular markers: an analysis of the NOA-08 study collective. Neuro Oncol. 15, 1017–1026.
Xu, F., Jin, T., Zhu, Y., and Dai, C. (2018). Immune checkpoint therapy in liver cancer. J. Exp. Clin. Cancer Res. 37:110. doi: 10.1186/s13046-018-0777-4
Yang, D., Zhang, W., Padhiar, A., Yue, Y., Shi, Y., Zheng, T., et al. (2016). NPAS3 regulates transcription and expression of VGF: implications for neurogenesis and psychiatric disorders. Front. Mol. Neurosci. 9:109. doi: 10.3389/fnmol.2016.00109
Yao, Y., Ma, J., Xue, Y., Wang, P., Li, Z., Liu, J., et al. (2015). Knockdown of long non-coding RNA XIST exerts tumor-suppressive functions in human glioblastoma stem cells by up-regulating miR-152. Cancer Lett. 359, 75–86. doi: 10.1016/j.canlet.2014.12.051
Yip, S., Miao, J., Cahill, D. P., Iafrate, A. J., Aldape, K., Nutt, C. L., et al. (2009). MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 15, 4622–4629.
Young, R. M., Jamshidi, A., Davis, G., and Sherman, J. H. (2015). Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 3:121.
Zah, E., Lin, M. Y., Silva-Benedict, A., Jensen, M. C., and Chen, Y. Y. (2016). T Cells Expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res. 4, 498–508.
Zawlik, I., Vaccarella, S., Kita, D., Mittelbronn, M., Franceschi, S., and Ohgaki, H. (2009). Promoter methylation and polymorphisms of the MGMT gene in glioblastomas: a population-based study. Neuroepidemiology 32, 21–29.
Zeng, T., Li, L., Zhou, Y., and Gao, L. (2018). Exploring long noncoding RNAs in glioblastoma: regulatory mechanisms and clinical potentials. Int. J. Genomics 2018:2895958. doi: 10.1155/2018/2895958
Zhang, K., Sun, X., Zhou, X., Han, L., Chen, L., Shi, Z., et al. (2015). Long non-coding RNA HOTAIR promotes glioblastoma cell cycle progression in an EZH2 dependent manner. Oncotarget 6, 537–546. doi: 10.18632/oncotarget.2681
Zhang, Y., Dube, C., Gibert, M. Jr., Cruickshanks, N., Wang, B., Coughlan, M., et al. (2018). The p53 Pathway in Glioblastoma. Cancers 10:297.
Zhao, J., Chen, A. X., Gartrell, R. D., Silverman, A. M., Aparicio, L., Chu, T., et al. (2019). Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 25, 462–469. doi: 10.1038/s41591-019-0349-y
Zhao, S., Lin, Y., Xu, W., Jiang, W., Zha, Z., Wang, P., et al. (2009). Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324, 261–265.
Zheng, S., Fu, J., Vegesna, R., Mao, Y., Heathcock, L. E., Torres-Garcia, W., et al. (2013). A survey of intragenic breakpoints in glioblastoma identifies a distinct subset associated with poor survival. Genes Dev. 27, 1462–1472.
Keywords: GBM, tumor microenvironment, immune checkpoint inhibitors, tyrosine kinase inhibitors, CAR-T, treatment resistance
Citation: Di Cintio F, Dal Bo M, Baboci L, De Mattia E, Polano M and Toffoli G (2020) The Molecular and Microenvironmental Landscape of Glioblastomas: Implications for the Novel Treatment Choices. Front. Neurosci. 14:603647. doi: 10.3389/fnins.2020.603647
Received: 07 September 2020; Accepted: 03 November 2020;
Published: 25 November 2020.
Edited by:
Elisa Roda, Clinical Scientific Institutes Maugeri (ICS Maugeri), ItalyReviewed by:
Floriana Volpicelli, University of Naples Federico II, ItalyDipongkor Saha, Texas Tech University Health Sciences Center, Abilene, United States
Copyright © 2020 Di Cintio, Dal Bo, Baboci, De Mattia, Polano and Toffoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Toffoli, Z3RvZmZvbGlAY3JvLml0