 Andreza Fabro de Bem1
Andreza Fabro de Bem1 Rachel Krolow2
Rachel Krolow2 Hémelin Resende Farias2
Hémelin Resende Farias2 Victória Linden de Rezende2
Victória Linden de Rezende2 Daniel Pens Gelain2
Daniel Pens Gelain2 José Cláudio Fonseca Moreira2
José Cláudio Fonseca Moreira2 João Miguel das Neves Duarte3,4*
João Miguel das Neves Duarte3,4* Jade de Oliveira2*
Jade de Oliveira2*- 1Department of Physiological Sciences, Institute of Biology, University of Brasilia, Brazilia, Brazil
- 2Postgraduate Program in Biological Sciences: Biochemistry, Department of Biochemistry, Institute of Basic Health Sciences, Federal University of Rio Grande do Sul, Porto Alegre, Brazil
- 3Department of Experimental Medical Science, Faculty of Medicine, Lund University, Lund, Sweden
- 4Wallenberg Centre for Molecular Medicine, Faculty of Medicine, Lund University, Lund, Sweden
The incidence of metabolic disorders, as well as of neurodegenerative diseases—mainly the sporadic forms of Alzheimer’s and Parkinson’s disease—are increasing worldwide. Notably, obesity, diabetes, and hypercholesterolemia have been indicated as early risk factors for sporadic forms of Alzheimer’s and Parkinson’s disease. These conditions share a range of molecular and cellular features, including protein aggregation, oxidative stress, neuroinflammation, and blood-brain barrier dysfunction, all of which contribute to neuronal death and cognitive impairment. Rodent models of obesity, diabetes, and hypercholesterolemia exhibit all the hallmarks of these degenerative diseases, and represent an interesting approach to the study of the phenotypic features and pathogenic mechanisms of neurodegenerative disorders. We review the main pathological aspects of Alzheimer’s and Parkinson’s disease as summarized in rodent models of obesity, diabetes, and hypercholesterolemia.
Introduction
Alzheimer’s disease (AD) and Parkinson’s disease (PD) are the two most common neurodegenerative diseases. They are chronic and progressive, and are defined by protein abnormalities, neuroinflammation (characterized by glial activation), and neuronal loss (Dugger and Dickson, 2017). Both AD and PD present genetic and sporadic forms, though the majority of cases are of sporadic type (Bekris et al., 2010; Klein and Westenberger, 2012). Vascular risk factors, such as metabolic disorders, have been linked to sporadic forms of AD and PD (Castillo et al., 2019). Several epidemiological studies have demonstrated a connection between hypercholesterolemia, obesity, and neurodegenerative disease development (Kivipelto et al., 2001, 2005; Gustafson et al., 2003; Whitmer et al., 2008; Solomon et al., 2009; Santos et al., 2017). Notably, the metabolic diseases in early life are risk factors for dementia. The epidemiological studies have shown that obesity and hypercholesterolemia in adulthood or in middle age increase the risk of dementia in the future (Kivipelto et al., 2001, 2005; Whitmer et al., 2005; Ariza et al., 2016). In the elderly, obesity and high plasma cholesterol levels are not correlated with a higher occurrence of dementia (Reitz et al., 2008; Anjum et al., 2018).
These findings were confirmed and further explored by experimental studies (Ullrich et al., 2010; de Oliveira et al., 2011; Moreira et al., 2014; Denver et al., 2018). Experimental models of obesity and hypercholesterolemia display similar brain alterations to those present in the brain of patients with neurodegenerative diseases, such as amyloid-β peptide (Aβ) accumulation, as well as abnormal tau protein and α-synuclein (Ullrich et al., 2010; Busquets et al., 2017; Han et al., 2017; Nakandakari et al., 2019). Blood-brain barrier (BBB) disruption and neuroinflammation have also been found in the brain structures of obese and hypercholesterolemic rodents (Ullrich et al., 2010; de Oliveira et al., 2014; Denver et al., 2018). Importantly, behavioral impairments related to neurodegenerative disease, particularly cognitive impairment, are evident in experimental models of obesity and hypercholesterolemia (Ullrich et al., 2010; de Oliveira et al., 2011; Moreira et al., 2014; Denver et al., 2018).
Another critical point is that the molecular mechanisms of AD and PD are still not completely known (Zeng et al., 2018), and, so far, these diseases have no cure. In this regard, experimental studies are needed to understand the structural, functional, and molecular features of these diseases and to propose therapies. The evidence is mounting that the environment has an essential role in the development of neurodegenerative diseases. It is now known that unhealthy lifestyles (including the excessive ingestion of food) combined or otherwise with specific predisposing genes are responsible for most cases of sporadic forms of AD and greatly contribute to sporadic forms of PD (Bhatti et al., 2020; Popa-Wagner et al., 2020). However, the classic models of neurodegenerative diseases, based on genetic alterations, do not encompass all the characteristics of these complex diseases (Ransohoff, 2018).
Given that animal models of metabolic disorders share most of the brain dysfunction associated with neurodegenerative disease, even in early life (Ullrich et al., 2010; Moreira et al., 2014; Paul et al., 2017a; de Oliveira et al., 2020b), they represent an interesting strategy for studying the prior events of neurodegeneration. This review contains the main findings regarding AD and PD in rodent models of metabolic diseases (in particular, obesity and hypercholesterolemia), to highlight their relevance in the study of aspects related to neurodegeneration.
Animal Models of Alzheimer’s Disease
Alzheimer’s disease is clinically characterized by cognitive impairments such as memory deficits (Kelley and Petersen, 2007), as well as the irreversible decline in the number of basal forebrain cholinergic neurons and synaptic loss mainly in the hippocampus and cerebral cortex (Schliebs and Arendt, 2011; Kozlov et al., 2017). Neuropathological components of this disorder include the presence of extracellular Aβ aggregates that precipitate in amyloid plaques, and neurofibrillary tangles mainly formed by hyperphosphorylated tau protein (Hardy and Selkoe, 2002; Selkoe and Hardy, 2016; Figure 1).
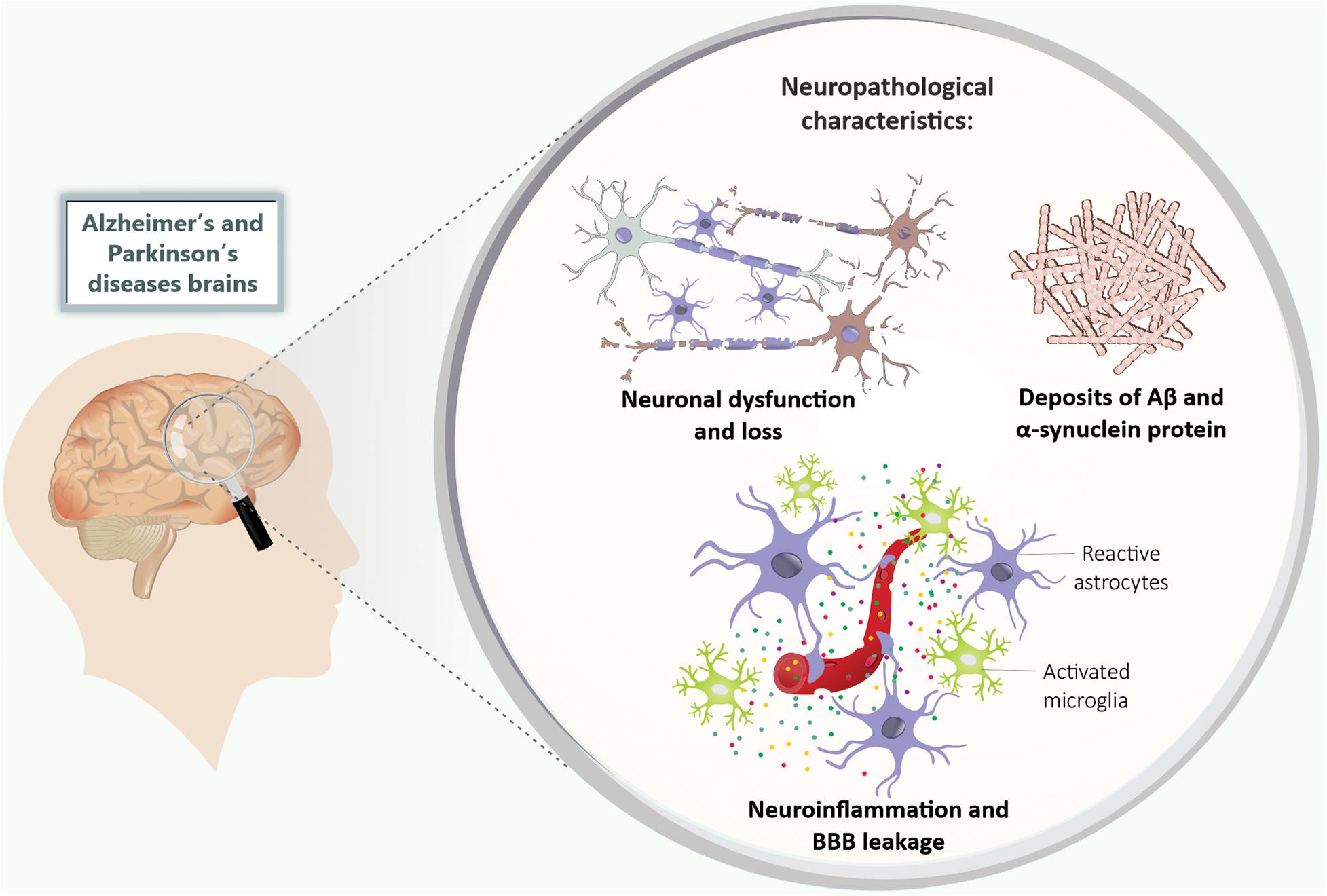
Figure 1. Neuropathological features of Alzheimer’s and Parkinson’s disease. The Alzheimer’s and Parkinson’s disease patients’ brains present neuronal dysfunction and loss, deposits of Aβ and α-synuclein protein, neuroinflammation (mainly characterized by microglia and astrocytes activation), and BBB leakage. Aβ, amyloid-β peptide; BBB, blood-brain barrier.
Alzheimer’s disease research mainly focuses on the hypothesis of the amyloid cascade, which postulates that the characteristic neuronal damage of the disease is partly attributed to changes in Aβ metabolism (Hardy and Selkoe, 2002; Selkoe and Hardy, 2016). Aβ is the product of amyloidogenic metabolism of amyloid precursor protein (APP) by enzymes called secretases (β and γ secretases) (O’Brien and Wong, 2011). Studies indicated that the Aβ becomes toxic by forming oligomers, which ultimately result in amyloid plaques deposition, neurodegeneration, and, consequently, cognitive impairments (Ferreira and Klein, 2011; Hardy and Selkoe, 2002; Selkoe and Hardy, 2016). AD is also characterized by chronic brain inflammation and BBB disruption (Zenaro et al., 2017; Figure 1).
Mutations in genes related to APP processing result in genetic AD (early-onset) (Levy et al., 1990). However, mutations in these genes account for only a small proportion of the disease, with sporadic (late-onset) AD accounting for 99% of the cases. It is also important to highlight that the etiology of Aβ deposits in sporadic AD remains unclear in most cases (Zhang et al., 2018; Tzioras et al., 2019). Sporadic AD is considered a multifactorial and complex neurodegenerative pathology, resulting from the interaction of genetic and environmental risk factors. Several diseases have been considered risk factors for AD, among them are the metabolic diseases that are causative events for cardiovascular diseases (Campos-Peña et al., 2017). Also, the presence of apolipoprotein E (ApoE) ε4 allele (APOE4) is the most important genetic risk factor for sporadic AD (Liu et al., 2013).
Even AD being the most important cause of dementia, until now, no treatment delays the onset or progression of the disease and its pathogenesis is still not elucidated. The availability of experimental models that cover the multifaceted aspects of AD is essential to perform translational studies (Long and Holtzman, 2019; Tiwari et al., 2019). There are several experimental models of AD, including genetic-based models of amyloid pathology (mainly transgenic mice) and those involving rodents exposed to intracerebroventricular (ICV) or intrahippocampal injection of Aβ (Puzzo et al., 2015).
Genetic-Based Mouse Models of Alzheimer’s Disease
Although none of the existing models fully reproduces the complete spectrum of AD, specific critical aspects of AD pathology and disease processes can be experimentally recreated in experimental rodent models (LaFerla and Green, 2012). Several animal models, using mice and rats mainly, have been used to create genetically altered phenocopies of human AD. Transgenic mice overproducing mutant tau and APP proteins (e.g., PDAPP and PS19 mice) and/or some of the enzymes implicated in their metabolic processing have been bred (Games et al., 1995; Hsiao et al., 1996; Oddo et al., 2003a, b; Drummond and Wisniewski, 2017).
For instance, PDAPP mice overexpress different isoforms of APP (695, 751, and 770), presenting Aβ deposits in different brain areas. The brain Aβ deposits start at 6–9 months of age and progress in an age-dependent manner (Games et al., 1995; Johnson-Wood et al., 1997; Chen et al., 2000), while behavioral alterations, such as spatial learning and memory impairments in the radial maze, appear at 3–4 months in PDAPP mice (Dodart et al., 1999; Hartman et al., 2005). This fact is a critical issue in this model, since the memory impairment does not correlate with brain Aβ deposits (Chen et al., 2000; Hartman et al., 2005). Concerning other pathogenic aspects, the PDAPP model displays an increased number of activated microglia and astrogliosis (Games et al., 1995), but the BBB integrity is intact until 16 months of age (Blockx et al., 2016).
Another example is the transgenic mouse Tg2576, which overexpresses a mutant form of APP (695) associated with the Swedish mutation (K670N, M671NL; Hsiao et al., 1996). The Tg2576 mouse better represents the connection between the formation of amyloid plaques and behavioral changes that are characteristic of AD. Around 4–5 months of age, Tg2576 mice present contextual memory deficits and an increase in the fraction of Aβ1–42 relative to Aβ1–40 (Jacobsen et al., 2006). Parenchymal Aβ plaques occur between 11 and 13 months of age (Hsiao et al., 1996). An increase in microglial density was observed in these mice when they turned 10–16-months of age (Frautschy et al., 1998). Before the formation of plaques, at 4 months of age, Tg2576 mice exhibit BBB disruption in some areas of the cerebral cortex (Ujiie et al., 2003) and in others brain areas at 8 months of age, which can be visualized by magnetic resonance imaging (MRI; Elhaik Goldman et al., 2018).
The triple-transgenic mouse model (3xTg) expresses three significant genes associated with familial AD (APPSwe, PSN1M146V, and tauP301L; Oddo et al., 2003a, b, 2005). It was designed to be an animal model for studying plaque and tangle pathology associated with synaptic dysfunction. The intracellular Aβ deposition starts at 3 months of age, while extracellular deposition of Aβ occurs in 6-month-old animals (Oddo et al., 2003b); cognitive impairments start at 4 months old (Billings et al., 2005). Other notable features of AD recreated in the 3xTg mice are neuroinflammation, synaptic dysfunction, and BBB impairment (Parachikova et al., 2010; Do et al., 2014; Belfiore et al., 2019). Although this mouse model is considered the most complete transgenic mouse model of AD available, the widespread presence of plaques and tangles are typically not observed until old age, and are not representative of human AD (Drummond and Wisniewski, 2017). In fact, the limitations to finding the AD molecular and morphology features in this transgenic animal model impair translational comparisons.
The 5xFAD mouse is an experimental model designed to reduce the time before amyloid plaques are formed. This transgenic mouse combines five mutations: Swedish mutation (APP KM670/671NL), London (V717l), Florida (APP I716V), L286V in PSN1, and M146L. The brain Aβ1–42 levels increase as early as 1.5 months of age and an AD-like amyloid pathology occurs at 2 months of age, while other available mouse models display amyloid deposition in the brain parenchyma between 3 and 12 months after birth (Oakley et al., 2006). In addition, these mice present spatial memory and learning deficits at 4–6 months of age (Oakley et al., 2006; Ohno, 2009).
PS19 mice expressing the P301S mutant form of human microtubule-associated protein Tau (MAPT) is also an AD mouse model (Yoshiyama et al., 2007). PS19 mice develop filamentous tau lesions at 6 months of age. Tangle pathology is accompanied by microgliosis and astrocytosis, but not by amyloid plaques (Yoshiyama et al., 2007). Interestingly, hippocampal synaptic dysfunction and loss were detected before fibrillary tau tangles emerged in the brains of these mice (Yoshiyama, 2008).
It is important to mention that metabolic parameters have been investigated in the AD animal models. For example, a glucose homeostasis impairment was demonstrated in the 3xTg-AD mice, which occurred in an age-dependent manner (Vandal et al., 2015).
One critical point in using these transgenic mouse models of AD is that they recapitulate the early-onset (familial) form of AD, which accounts for only 1% of cases. Therefore, these models may still present an incomplete perspective of the pathology (Kitazawa et al., 2012; Zou et al., 2014).
On the other hand, E4FAD and E3FAD mouse models, which are crosses between the 5xFAD mice and mice expressing APOE4 and APOE3 human isoforms, represent an effort to replicate sporadic AD. However, the E4FAD and E3FAD mice display less severe phenotypes compared with the 5xFAD mice. The brain Aβ accumulation appears from 2 to 6 months of age and is more intense in the brains of E4FAD mice (Youmans et al., 2012). Both E4FAD and E3FAD mice showed reactive microglia and dystrophic astrocytes at 6 months of age, but in E4FAD mice, the microglial reactivity was higher than in E3FAD mice (Rodriguez et al., 2014). Moreover, E4FAD mice exhibited more severe age-dependent memory deficits than E3FAD mice (Liu et al., 2015). Table 1 summarizes the main findings regarding AD-like pathology in the main transgenic AD mice models.
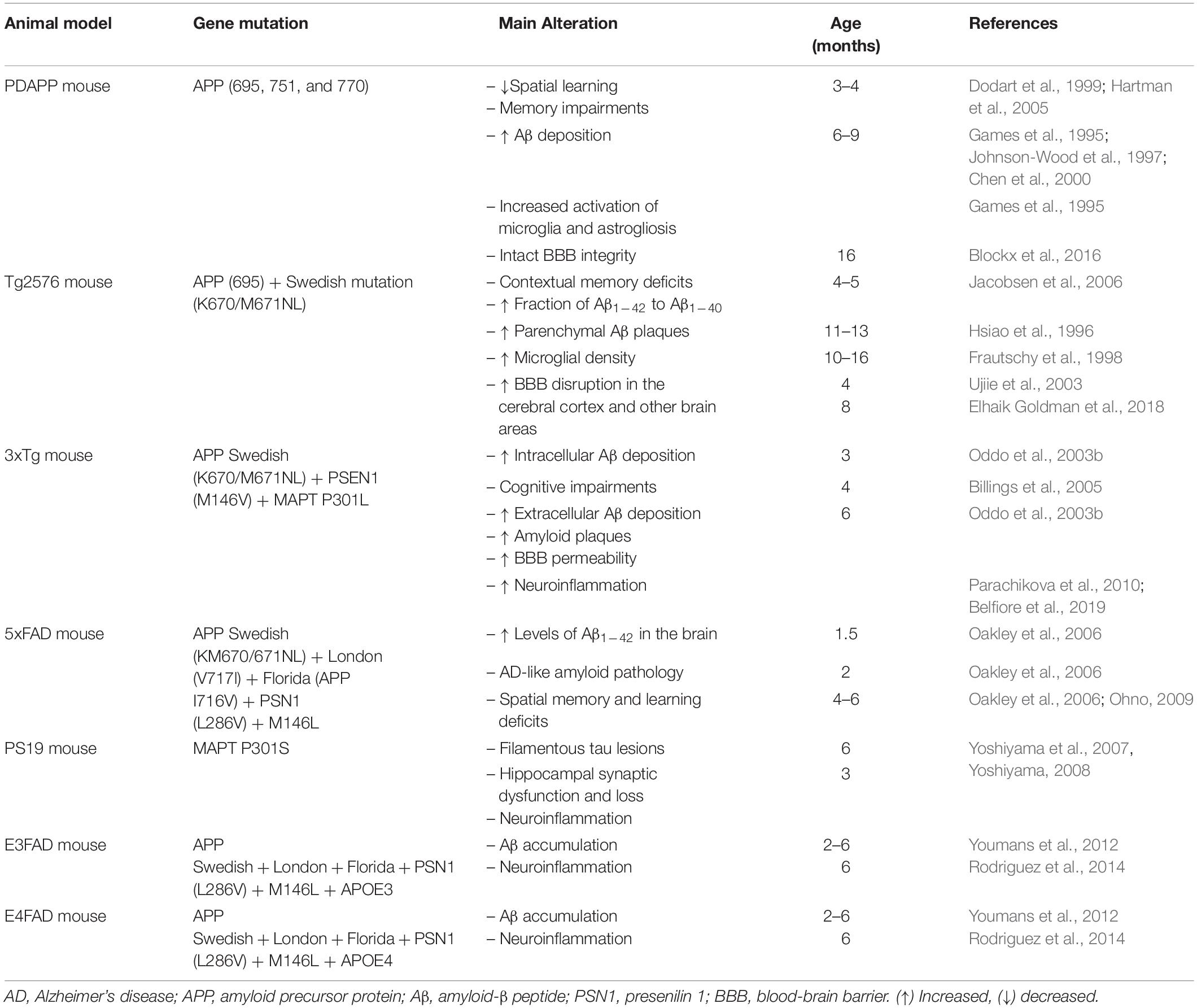
Table 1. Summary of the main alterations observed in the transgenic mouse models of Alzheimer’s disease.
Intracerebroventricular Injection of Aβ in Rodents
It takes some time to recreate the features of AD using transgenic mice models, because the increase in Aβ levels, amyloid plaque formation, and behavioral impairments appear typically from 6 months of age (Games et al., 1995; Hsiao et al., 1996; Sturchler-Pierrat et al., 1997; Dewachter et al., 2000). Therefore, another interesting experimental tool with which to study Aβ toxicity is ICV and intrahippocampal injections of Aβ peptides (Flood et al., 1991; Piermartiri et al., 2010; Ferreira and Klein, 2011). Here, studies have demonstrated spatial learning and memory deficits induced by ICV administration of aggregated Aβ1–40 or Aβ1–42 (Yamada et al., 1999; Yan et al., 2001; Jhoo et al., 2004; Yamaguchi et al., 2006; Prediger et al., 2007) in rodents after only a few days/weeks of administration. Cognitive impairments in rodents exposed to aggregated Aβ1–40 were associated with synaptic loss and cell death in the hippocampus and prefrontal cortex (Prediger et al., 2007; Piermartiri et al., 2010; Figueiredo et al., 2011; Santos et al., 2012). Furthermore, ICV injection of aggregated Aβ1–40 or Aβ1–42 led to an increase in the hippocampal concentration of the proinflammatory cytokine, interleukin (IL)-1β (Yan et al., 2001; Minogue et al., 2003), as well as microglial activation (Clarke et al., 2007; Medeiros et al., 2007; Figueiredo et al., 2011).
Another approach is the ICV administration of soluble Aβ oligomers (AβOs). These are potent neurotoxins derived from Aβ1–42, which can be found in AD brains (Lambert et al., 1998; Ferreira and Klein, 2011). The ICV infusion of AβOs has been shown to cause synaptic loss in the hippocampus and memory impairment related to AD in mice (Ledo et al., 2013; Figueiredo et al., 2013). Moreover, mice injected by the ICV route with AβOs presented hippocampal activation of microglia and astrocytes, as well as brain increased tumor necrosis factor α (TNF-α), IL1β, and IL-6 levels (Ledo et al., 2013; Brkic et al., 2015).
In both models, that is, mice ICV injected with aggregated Aβ or AβOs, BBB integrity was not entirely explored. With regard to the ICV injection of AβOs, a loss of blood – CSF barrier integrity in the choroid plexus was observed in mice (Brkic et al., 2015). On the other hand, C57BL/6 mice injected with an aggregated form of Aβ1–40 did not present changes in the hippocampal immunoreactivity of aquaporin-4 (AQP-4), a putative marker of edema and BBB leakage (de Oliveira et al., 2014). More studies are needed to better describe this aspect in these AD experimental models. Another critical point is that it is difficult to observe amyloid plaques in the brain in this particular model (Kim et al., 2016). The heterogeneity of the peptide samples is also a problem with the application of Aβ in rodent models (Kasza et al., 2017).
Animal Models of Parkinson’s Disease
Parkinson’s disease is the second most prevalent neurodegenerative disease after AD, and the most common movement disorder (Obeso et al., 2017). The main features of PD are dopaminergic neuronal loss in the substantia nigra pars compacta (SNpc) and dopamine depletion in the striatum (Poewe et al., 2017), with the presence of intracytoplasmic inclusions called Lewy bodies, which are composed mainly of misfolded α-synuclein (Spillantini et al., 1998). Neuroinflammation and BBB disruption have also been considered to be pathogenic features of PD (Collins et al., 2012; Wang et al., 2015; Guzman-Martinez et al., 2019; Figure 1).
Clinically, PD is characterized by motor symptoms such as resting tremor, bradykinesia, rigidity, and loss of postural reflex. These result from dopaminergic degeneration of the nigrostriatal pathway (Magrinelli et al., 2016). This neuropathology is also associated with non-motor symptoms, for example anxiety, depression, and cognitive impairments (dementia; Franke and Storch, 2017).
Parkinson’s disease can be genetic or sporadic (Singleton et al., 2013). It is known that the development of the disease occurs due to genetic susceptibility associated with environmental risk factors (Chen and Ritz, 2018). Exposure to heavy metals, fungicides, and pesticides (e.g., rotenone and paraquat) have been associated with the development of the disease (Ball et al., 2019). In addition, in the past few years, it has been suggested that metabolic disorders may be causative events of PD (Limphaibool et al., 2018; Nam et al., 2018; Alecu and Bennett, 2019).
Because PD occurs mainly as a sporadic form, experimental models based on compounds’ neurotoxicity are useful for the study of this neuropathology. For instance, the 6-hydroxydopamine (6-OHDA) animal model of PD has been used in the PD research field since 1968 (Ungerstedt, 1968). The 6-OHDA is an analog of dopamine and norepinephrine and is endogenously produced through the hydroxylation of dopamine (Tieu, 2011). This neurotoxin is unable to cross the BBB; therefore, the only way to expose the brain to neurotoxic actions of this substance is through stereotaxic surgery (Duty and Jenner, 2011). Bilateral injection of 6-OHDA into the substantia nigra (SN) of rats has caused anterograde degeneration of the nigrostriatal dopaminergic system, leading to akinesia and high mortality. The first experimental model of PD was generated thus (Ungerstedt, 1968). Specifically, after stereotaxic injection, 6-OHDA is removed from the extracellular space by dopamine or noradrenaline membrane transporters and stored in catecholaminergic neurons. Inside these neurons, 6-OHDA undergoes both enzymatic degradation by monoamine oxidase A (MAO-A) and auto-oxidation, generating several cytotoxic species that lead to neuronal damage (Soto-Otero et al., 2000). Many studies have demonstrated that brain 6-OHDA injections are associated with decreased locomotor activity, reduced tyrosine-hydroxylase (TH)-positive neurons, and brain oxidative stress in mice (Ungerstedt, 1968; Simola et al., 2007; Ozsoy et al., 2015). The 6-OHDA experimental models of PD are also associated with neuroinflammation, that is, microgliosis and astrogliosis (Walsh et al., 2011; Gasparotto et al., 2017), as well as BBB disruption (Carvey et al., 2005). Unilateral injections of 6-OHDA into the striatum or the medial forebrain bundle induced an increased BBB permeability to FITC-labeled albumin in the SN and striatum (Carvey et al., 2005). One limitation of 6-OHDA-based models is that they do not cause changes in α-synuclein expression or deposition, and for this reason they are more correctly referred to as models of “Parkinson-like” dopaminergic denervation or, simply, dopaminergic denervation.
Other rodent models of PD have been developed through exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). It was demonstrated that MPTP itself is not toxic; however, as a lipophilic compound, it passes through the BBB. Once in the brain, the molecule is rapidly converted into its toxic metabolite, 1-methyl-4-phenylpyridinium or MPP+, by monoamine oxidase B (MAO-B; Langston, 2017). Studies performed with rodents showed a reduction of locomotion and rearing in the open field task for several weeks after the subcutaneous and intraperitoneal administration of MPTP (Fredriksson et al., 1999; Fornai et al., 2005). In contrast, other studies reported no change in locomotion and even hyperactivity in mice after peripheral exposure to MPTP (Tomac et al., 1995; Chia et al., 1996; Luchtman et al., 2009). In a chronic model of intraperitoneal MPTP exposure, mouse dopaminergic neurons presented α-synuclein-positive inclusions and secondary lysosomes filled with proteinaceous debris and lipid droplets, which resemble deposits in the brains of PD patients (Wu et al., 2002). Yazdani et al. (2006) showed that the infusion of MPP+ into the left cerebral ventricle of rats destroyed dopaminergic neurons in the nigrostriatal pathway. Another study indicated that the presence of MPP+ is related to inflammatory reaction along with the infiltration of T-cells into the SN and striatum and activation of the microglia and increased gene expression of proinflammatory cytokines such as IL-1β, interferon γ (INFγ), and TNFα in those brain regions (Kurkowska-Jastrzebska et al., 2009). The inflammation induced by MPTP treatment seems to cause BBB failure. Mice exposed to MPTP intraperitoneal injection presented less TH-positive dopaminergic neurons, which was related to an increase in leakage of Evan’s blue dye and FITC-albumin into the striatum. The striatum BBB disruption in the MPTP mouse model was also characterized by a reduction in the tight junctions’ proteins content (Chen et al., 2008).
Rotenone (pesticide), paraquat (herbicide), and maneb (fungicide) exposure have been considered a possible environmental cause of PD (Hatcher et al., 2008). Epidemiological studies have shown that exposure to agrochemicals increases the risk of PD (Narayan et al., 2017; Pouchieu et al., 2018). The administration of agrochemicals in rodents has been used to study the mechanisms underlying PD pathogenesis. These compounds can cross the BBB and affect the dopaminergic system (Bastías-Candia et al., 2019). For example, rotenone and paraquat are known to cause dopaminergic degeneration in mice (McCormack et al., 2002; Drechsel and Patel, 2008). One advantage of rotenone administration as a PD model is that the agrochemical mimics the chronic progression of PD patients, while other metabolite exposure results only in acute damage of dopaminergic neurons (von Wrangel et al., 2015). Cannon et al. (2009) demonstrated that rotenone-treated animals presented bradykinesia, postural instability, and/or rigidity. The authors also observed the presence of α-synuclein positive aggregates in the dopamine neurons of SN. Rotenone exposure is also associated with neuroinflammation. Martinez et al. (2017) reported that the chronic administration of intragastric rotenone in mice caused progressive nigral degeneration and neuroinflammation.
Another tool used to study PD is the intracerebral administration of α-synuclein pre-formed fibrils (PFFs) in mice. Usually, the administration of PFFs is performed with unilateral and intrastriatal injection. Specifically, this model has been used to study the mechanisms by which α-synuclein aggregates spread throughout the brain. In addition to the broad spread of pathological α-synuclein deposition, neuronal loss, neuroinflammation, and some behavioral deficits were also observed in the mice injected with PFFs (Luk et al., 2012; Gordon et al., 2018; Chung et al., 2019; Earls et al., 2019).
It is also important to mention that genetic mouse models are used to study PD; however, they are heterogeneous, and no perfect model exists (Fleming et al., 2005; Bogaerts et al., 2008). One example is A53T mice that overexpress human α-synuclein with a PD-associated mutation (A53T; Giasson et al., 2002). In an A53T mouse, the human-specific soluble α-synuclein expression increases in the brain between 2 and 6 months of age and remains constant after 12 months, resulting in the dispersal of α-synuclein aggregates throughout the cortex, hippocampus, brain stem, and cerebellum. The onset of motor symptoms is variable and generally appears at 9–10 months of age (Lee et al., 2002; Paumier et al., 2013).
Although there are many animal models of PD, none of them accurately represent all the characteristic events of the pathogenesis of this disease (Dawson et al., 2010). Table 2 summarizes the characteristics of the main PD rodent models.
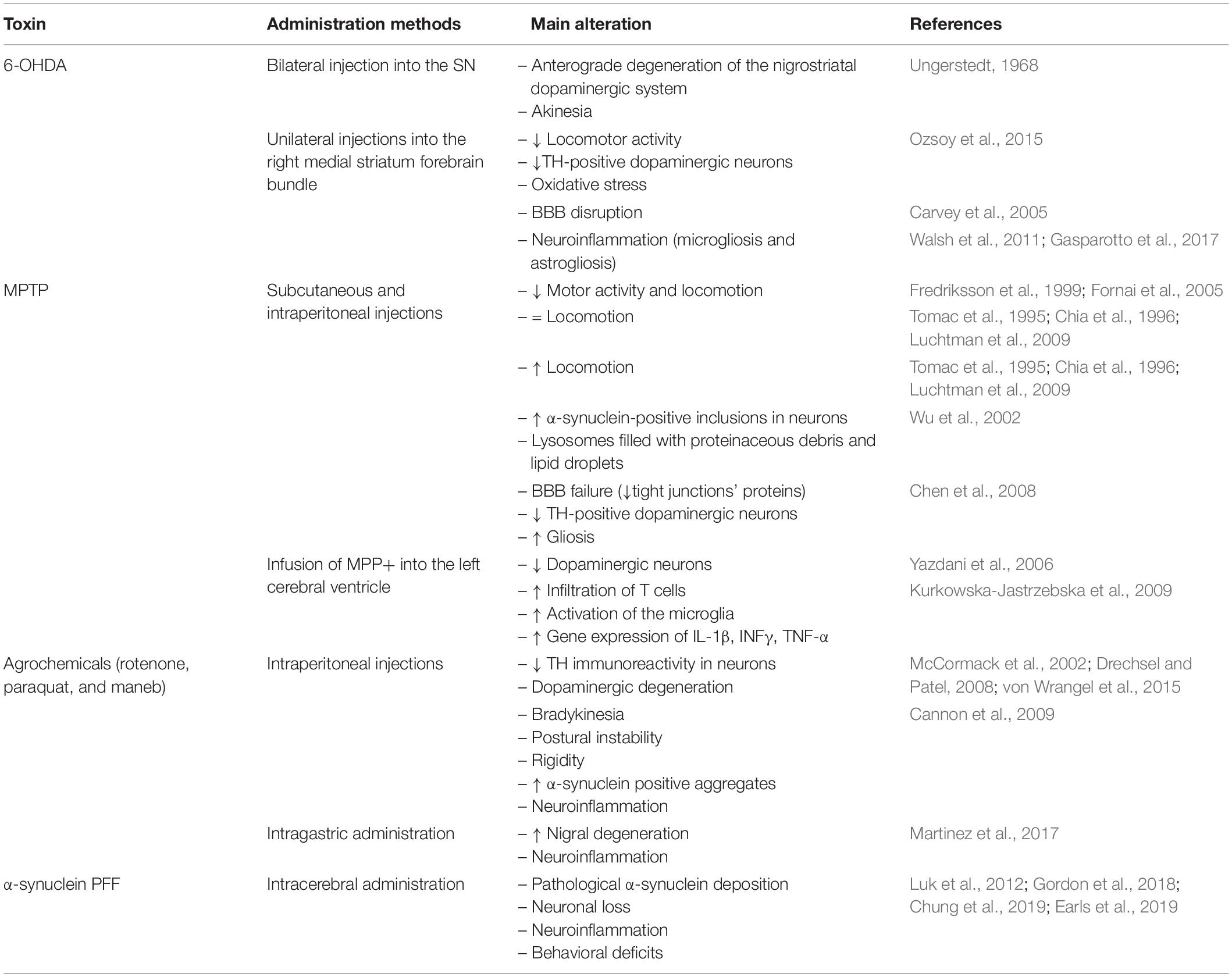
Table 2. A summary of the main findings in toxin-induced rodent models of Parkinson’s disease.
Metabolic Diseases as a Risk Factor for Neurodegenerative Diseases
Metabolic disorders, e.g., obesity, diabetes, hypercholesterolemia, and hypertension, are the main risk factors for cardiovascular disease. The link between metabolic disorders and the future risk of dementia has been reported in several studies (Beydoun et al., 2008; Fitzpatrick et al., 2009; Hassing et al., 2009). In the past few decades, hypercholesterolemia, that is, high levels of blood cholesterol and obesity have been connected to neurodegenerative diseases development (Kivipelto et al., 2001, 2005; Gustafson et al., 2003; Whitmer et al., 2008; Solomon et al., 2009; Santos et al., 2017). It is postulated that one of the mechanisms behind this connection is high levels of blood cholesterol or free fatty acids (FFA) inducing deregulation of lipid metabolism and altering the permeability of the BBB, leading to neuroinflammation and cognitive decline (Takechi et al., 2013; Paul and Borah, 2017; Paul et al., 2017b).
In their pioneering evidence, Sparks et al. (1990) pointed out that the brains of non-demented individuals with coronary artery disease, a condition strictly related to hypercholesterolemia, presented amyloid plaques. The same research group observed that hypercholesterolemia induced increased intracellular Aβ deposition in the hippocampus and cerebral cortex of rabbits fed a high cholesterol diet for 4, 6, and 8 weeks (Sparks et al., 1994). Furthermore, Refolo et al. (2000) showed a positive correlation between the plasma cholesterol levels and the content of the Aβ brain when treating a transgenic mouse model of AD with a hypercholesterolemic diet. Hypercholesterolemic rabbits and individuals with cardiovascular diseases also displayed neuroinflammation, and BBB increased permeability (Streit and Sparks, 1997; Sparks et al., 2000).
It has been demonstrated that obesity contributes to impaired cognitive performance and dementia (Beydoun et al., 2008; Nguyen et al., 2014; Pegueroles et al., 2018; Hou et al., 2019). An earlier 18-year follow-up conducted by Gustafson et al. (2003) suggested that overweight in old age is a risk factor for dementia, particularly AD. Moreover, Whitmer et al. (2008) reported that central obesity in midlife increases the risk of dementia regardless of diabetes and cardiovascular comorbidities. It is worth noting that the vascular and inflammatory effects of obesity may play a role in the development of neurodegenerative diseases such as AD (Naderali et al., 2009; Businaro et al., 2012; Walker and Harrison, 2015). Relatedly, obese women presented a decrease in BBB function (Gustafson et al., 2007) while bariatric surgery reversed obesity and reduced hypothalamic gliosis in women (van de Sande-Lee et al., 2020).
Insulin is mostly known for its role in clearing glucose from the circulation. The absence of insulin production and release in type 1 diabetes (T1D) and the poor action of insulin on target cells (insulin resistance) in type 2 diabetes (T2D) result in hyperglycemia. Poorly controlled glycemia in diabetes impacts the cerebrovascular system and BBB integrity and is a risk factor for AD (Vagelatos and Eslick, 2013). However, the brains of AD patients showed downregulated insulin receptors, pointing toward a role of neuronal insulin resistance in AD etiology (Steculorum et al., 2014; de la Monte, 2017), and so several clinical trials have focused on the administration of insulin to treat or prevent dementia (Lee et al., 2018).
Metabolic disorders are also related to PD. Some findings have suggested a prospective association between plasma cholesterol or a history of hypercholesterolemia and PD risk (de Lau et al., 2006; Simon et al., 2007; Hu et al., 2008; Hu, 2010). Some evidence has also pointed to obesity, diabetes, and cerebrovascular risk factors as contributors to PD development (Chen et al., 2014; De Pablo-Fernandez et al., 2018; Kummer et al., 2019), though the correlation is not yet well established; epidemiological and clinical studies present controversial data. For instance, one study found an inverse association between plasma cholesterol levels and PD clinical progression (Huang et al., 2011).
Taking into account the fact that (i) metabolic conditions such as obesity, diabetes, and hypercholesterolemia are factors that increase the individual risk of developing AD and PD; and (ii) rodent models of obesity, diabetes, and hypercholesterolemia present all the main hallmarks of AD and PD, we propose these experimental animal models as strategic tools to study neurodegeneration. The overlap of pathological features between metabolic and neurodegenerative disorders supports a mechanistic connection among these conditions that needs to be better understood.
Animal Models of Hypercholesterolemia and Brain Dysfunction
Diet-Induced Hypercholesterolemia in Rodents and Brain Effects
Hypercholesterolemia can occur by genetic origin or due to a high intake of cholesterol (Cha and Park, 2019). There are many different types of experimental models of diet-induced hypercholesterolemia. Studies have used different diets and periods of exposure (Ullrich et al., 2010; Moreira et al., 2014; Paul et al., 2017a).
Diet-induced hypercholesterolemia appears to be associated with memory damage. Swiss mice fed a high cholesterol diet (1.25% cholesterol and 20% fat) for 2 months displayed short-term memory impairment (Moreira et al., 2014). Also, exposure to a high cholesterol diet has led to severe spatial learning and long-term memory deficits in rats (Ullrich et al., 2010).
The cholinergic system is critically important for memory, learning, attention, and other higher brain functions. Evidence has indicated that hypercholesterolemia has an impact on cholinergic functions in the CNS. Ullrich et al. (2010) showed that memory decline was associated with loss of choline acetyltransferase (ChAT)-positive neurons (i.e., cholinergic neurons) in the basal nucleus of Meynert, reduction of acetylcholine levels in the cerebral cortex, and an increase in cortical Aβ1–42 levels in hypercholesterolemic rats. In line with this, we previous reported increased acetylcholinesterase (AChE) activity in the hippocampus and prefrontal cortex of mice fed a high cholesterol diet (Moreira et al., 2014).
Mice exposed to a high cholesterol diet also presented motor alterations characteristic of PD. High levels of plasma cholesterol in mice caused akinesia, catalepsy, and reduced swimming performance, and were associated with a decrease in TH-positive neurons and dopamine levels in the striatum (Paul et al., 2017a). Interestingly, hypercholesterolemia increased the neurotoxicity induced by MPTP in mice. Hypercholesterolemic mice treated with MPTP exhibited a more severe loss of dopaminergic neurons in the SN and reduced striatal levels of dopamine than those treated with MPTP only (Paul et al., 2017b).
Experimental evidence supports the notion that BBB disruption and further neuroinflammation underlying hypercholesterolemia trigger brain dysfunction (Ullrich et al., 2010; de Oliveira et al., 2014; Paul and Borah, 2017; Chen et al., 2018). Ullrich et al. (2010) observed BBB disturbance in the hypercholesterolemic rats’ cortex that was visualized by increased leakage of IgG and microgliosis. Mice fed a high cholesterol diet also displayed BBB disruption in brain regions (e.g., the hippocampus, cerebral cortex, SN, and striatum) associated with neuroinflammation (astrogliosis and microgliosis; Paul and Borah, 2017; Paul et al., 2017b). The brain inflammation in rodents fed a high cholesterol diet was also related to increased levels of cytokines such as TNF-α, IL1-α, IL-1β, and IL-6 (Ullrich et al., 2010; Chen et al., 2018).
Additionally, a previous experimental study indicated that exposure to a high cholesterol diet worsened the outcomes and accelerated the disease course in an animal model of AD. Specifically, Refolo et al. (2000) observed that exposure to a high cholesterol diet increased Aβ accumulation and accelerated the AD-related pathology in a double-mutant PSAPP. The authors demonstrated a positive correlation between the levels of plasma cholesterol and cerebral Aβ content.
Notably, rodents exposed to a high cholesterol diet present innumerable pathogenic characteristics of neurodegenerative diseases. Notably, the brain dysfunction in these rodent models of hypercholesterolemia occurred early in life (Table 3). Therefore, we propose that rodents fed a high cholesterol diet are a useful model with which study the mechanisms that lead to neurodegeneration.
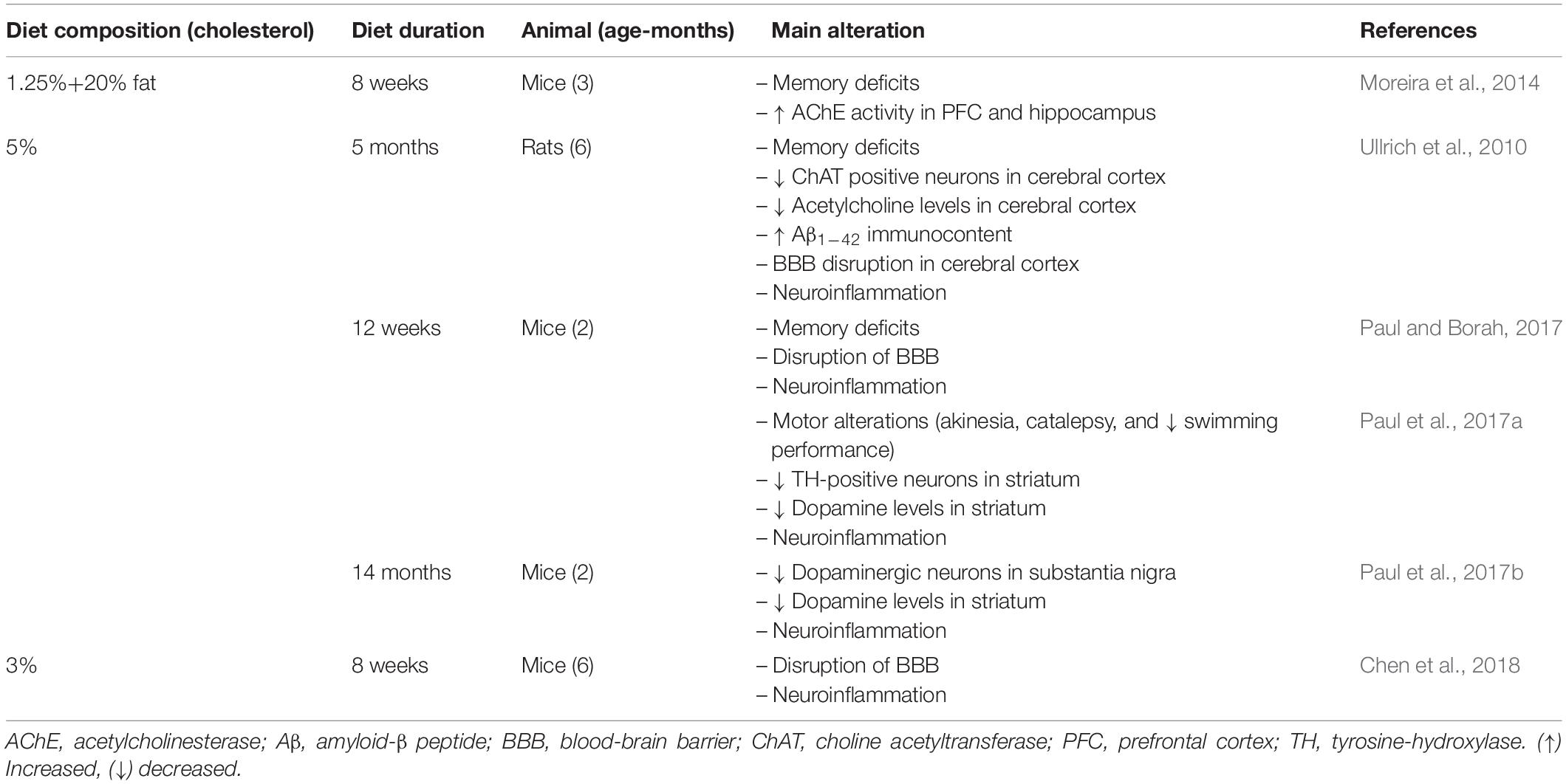
Table 3. A summary of evidence linking high cholesterol diets exposure in rodents to cerebral alterations associated with neurodegenerative diseases.
LDLr–/– Mice as a Model for the Study of Neurodegeneration and Memory Impairments
The main form of genetic hypercholesterolemia is familial hypercholesterolemia, which is very common in the general population (Santos et al., 2016; Fairoozy et al., 2017). This metabolic disorder is caused by mutations in the low-density lipoprotein receptor (LDLr) gene, an important molecule in cholesterol metabolism (Brown and Goldstein, 1986, 2006; Soutar and Naoumova, 2007). Familial hypercholesterolemia is closely related to the development of atherosclerotic cardiovascular disease (Austin et al., 2004). Individuals suffering from familial hypercholesterolemia also display a high incidence of cognitive impairments (Zambon et al., 2010; Ariza et al., 2016).
Similarly, LDLr knockout (LDLr–/–) mice, a mouse model of human familial hypercholesterolemia, display learning and memory deficits (Mulder et al., 2004; de Oliveira et al., 2011). Mulder et al. (2004) reported that 11-month-old LDLr–/– display spatial memory impairment in the Morris water maze task and working memory damage in T-maze spontaneous alternation analysis. We have observed that LDLr–/– present spatial and working memory decline as early as 3 months of age (de Oliveira et al., 2011, 2020a).
Memory deficits in LDLr–/– mice have been linked to synaptic and neuronal dysfunction. Synaptic density reduction in the hippocampus of 11-month-old LDLr–/– mice has been demonstrated (Mulder et al., 2007). We recently showed a high immunoreactivity of caspase-3 protein in the hippocampal and prefrontal cortex neurons of 3-month-old LDLr–/– mice (de Oliveira et al., 2020a). Neuronal damage and synaptic dysfunction have also been associated with impaired hippocampal neurogenesis in LDLr–/– mice (Mulder et al., 2007; Engel et al., 2019). However, these neuronal changes were not linked with the overproduction of Aβ, since LDLr–/– mice did not exhibit alterations in Aβ1–42 content in the hippocampus and prefrontal cortex (de Oliveira et al., 2020a). On the other hand, LDLr–/– mice were more susceptible to Aβ ICV neurotoxicity (de Oliveira et al., 2014).
We have revealed increased activity of AChE and antioxidant disturbance in the brain areas of 3-month-old LDLr–/– mice (Moreira et al., 2012; de Oliveira et al., 2014), while treatment with donepezil, an anticholinergic drug, reversed the memory decline in these hypercholesterolemic mice (Lopes et al., 2015). Importantly, neuroinflammation characterized by astrogliosis was visualized in the hippocampus of 3-month-old LDLr–/– mice. The increased number of astrocytes in the hippocampus of LDLr–/– mice was associated with the increased immunoreactivity of AQP-4, which indicates BBB dysfunction (de Oliveira et al., 2014). Specifically, the increased expression and content of AQP-4, a bidirectional water channel found in astroglial foot processes, and endothelial cells can indicate edema and neurotoxicity (Thomas-Camardiel et al., 2005).
We also submitted 3-month-old wild-type and LDLr–/– mice to a high cholesterol diet for 30 days, and we observed that the BBB leakage was even more intense. The hippocampus and prefrontal cortex BBB dysfunction in LDLr–/– mice was associated with cognitive decline, while C57BL/6 wild-types fed a high cholesterol diet exhibited impairments in the BBB but not in cognition. In addition, LDLr–/– mice displayed intense astrogliosis, increased microvessel content, and decreased levels of IL-6 in the hippocampus (de Oliveira et al., 2020b). Figure 2 presents the main brain alterations found in the LDLr–/– mice.
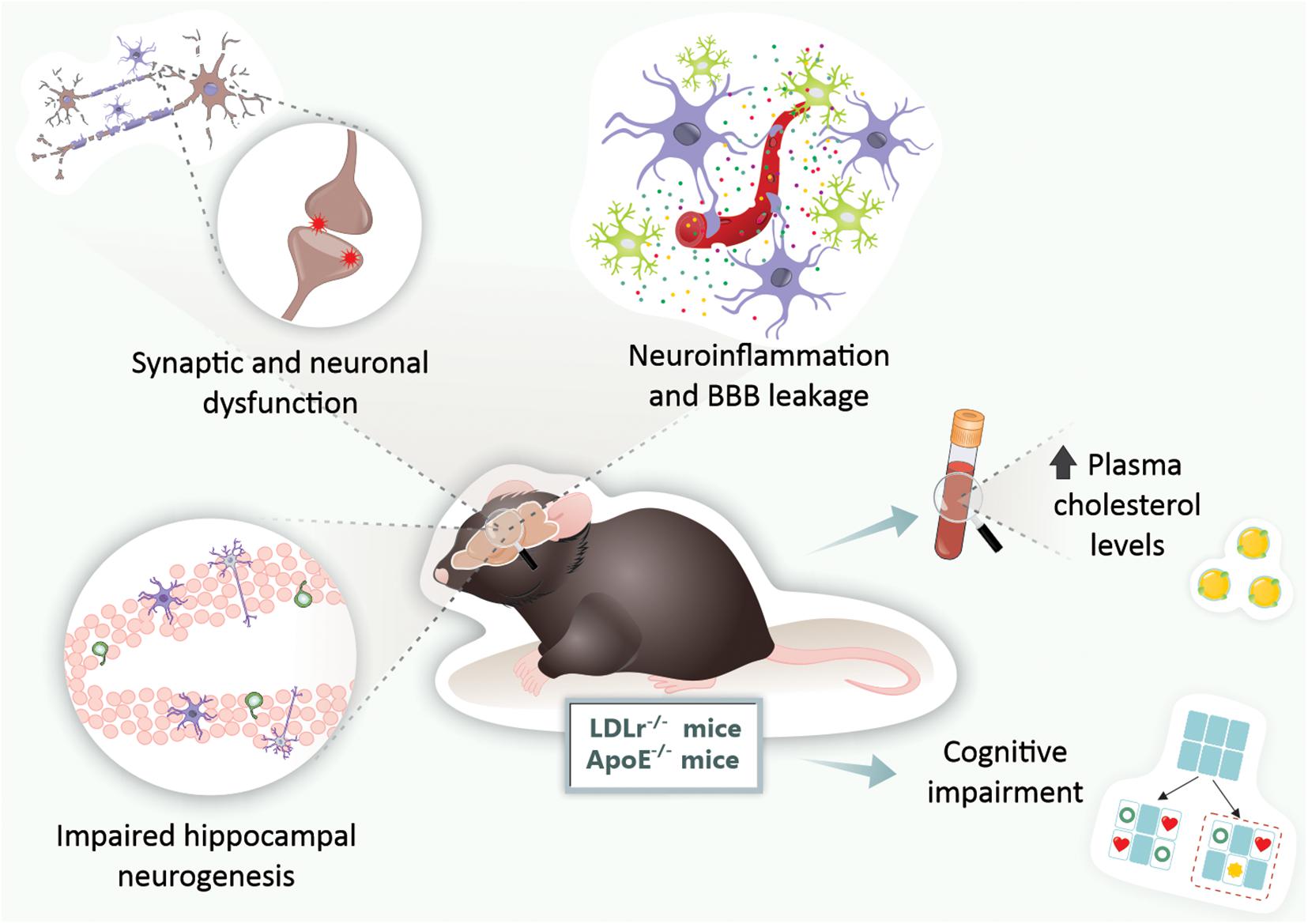
Figure 2. Brain alterations found in genetic mouse models of hypercholesterolemia. In LDLr–/– and ApoE–/– mice, both genetic mouse models of hypercholesterolemia, were observed increased levels of plasma cholesterol, BBB disruption, neuroinflammation, synaptic and neuronal dysfunction, impaired neurogenesis, and ultimately, cognitive impairments. ApoE–/–, apolipoprotein E knockout mice; BBB, blood-brain barrier; LDLr–/–, low-density lipoprotein receptor knockout mice.
ApoE–/– Mice as a Model for the Study of Neurodegenerative Diseases
Apolipoprotein E was first discovered by Shore and Shore (1974) in very-low-density lipoprotein (VLDL). ApoE in the periphery is principally produced by the hepatocytes but is also expressed by other cells (Kurano et al., 2011). In the brain, ApoE is synthesized mainly by astrocytes, and it plays a vital role in neuronal repair and maintenance (Huang et al., 2004). In the CNS, ApoE serves as the primary carrier protein of lipids, redistributing and mobilizing cholesterol between cells. These ApoE functions in cholesterol transport are essential for maintaining myelin and neuronal membranes (Leduc et al., 2010).
Apolipoprotein E knockout (ApoE–/–) mice were first designed for atherosclerosis pathogenesis studies, because they exhibit a 5- to 10-fold increase in plasma cholesterol levels (Zhang et al., 1992). Currently, ApoE–/– mice have also been used for studying the pathophysiology of neurological diseases, including neurodegenerative ones (Yin and Wang, 2018).
Previous studies have demonstrated that ApoE–/– mice display a disruption in spatial learning as early as 3 months and working memory impairment at 6–8 months. Both were Morris water maze protocols (Gordon et al., 1995; Masliah et al., 1997; Champagne et al., 2002). Spatial learning and working memory, tested in the octagonal-arm radial maze, was impaired in ApoE–/– mice at the age of 9–10 months (Evola et al., 2010). It is worth mentioning that ApoE–/– and control mice were submitted to a rotarod test at 5–6 and 12–14 months of age, and both strains presented decreased average latency in the time they remained in the apparatus (because of aging), which means that ApoE deficiency was not associated with motor alterations (Fuentes et al., 2018).
The cholinergic system also seems to be affected in ApoE–/– mice. The ChAT activity was reduced in the hippocampus and frontal cortex of 6-month-old ApoE–/– mice (Gordon et al., 1995) and a significant decrease in AChE activity in the cortex, hippocampus, and septum in 14-week-old ApoE–/– mice (Fisher et al., 1998).
Neuronal cell death markers (caspase-1 positive cells) increase in the hippocampus of ApoE–/– mice when they are fed a high cholesterol diet (Rahman et al., 2005). The amyloid cascade is also disturbed in the brains of ApoE–/– mice. The clearance of synthetic Aβ, which was injected directly into the brain parenchyma of ApoE–/– mice, was impaired (Ji et al., 2001). On the other hand, the elderly ApoE–/– mice did not have their brain Aβ deposition measured by Congo red staining, unlike in a traditional AD mouse model (Shnerb Ganor et al., 2018). The association between Aβ levels and ApoE deficiency needs to be studied further.
Another hallmark of neuroinflammation found in ApoE–/– mice was an increase in GFAP in the hippocampus and corpus callosum (Crisby et al., 2004). Moreover, the disruption of BBB integrity was showed in very young ApoE–/– mice (8 weeks of age) by the BBB extravasation of Evans blue dye (Methia et al., 2001). Hafezi-Moghadam et al. (2007) suggested that ApoE deficient mice have a progressive, age-dependent BBB leakage in the cortex and cerebellum.
Therefore, both genetic mouse models of hypercholesterolemia (LDLr–/– mice and ApoE–/– mice) present cognitive impairments early in life. These are associated with neuroinflammation, BBB disruption, and neurodegeneration, but not with increased brain Aβ deposition (Figure 2). As the models induced by hypercholesterolemic diet consumption, these genetic models present cerebral dysfunction as early as 3 months. Given that genetic forms of hypercholesterolemia (e.g., familial hypercholesterolemia) are highly prevalent in the general population, these animals are suitable for the study of neurodegenerative diseases, especially AD (Figure 2).
Animal Models of Obesity and Its Comorbidities and Features of Neurodegenerative Diseases
Obesity is associated with chronic low-grade systemic inflammation (Gregor and Hotamisligil, 2011; Lumeng and Saltiel, 2011). The pathophysiological effects of obesity are observed not only in adipose tissue but also in other organs, including the brain (Alford et al., 2018). Evidence has suggested that obesity in midlife is a risk factor for AD in later life (Kivipelto et al., 2005; Beydoun et al., 2008; Whitmer et al., 2008; Chuang et al., 2016; Gottesman et al., 2017).
Neurodegenerative Diseases and Rodents Fed a High-Fat Diet
Increased consumption of high energy/high-fat food (i.e., over-nutrition), is considered a critical environmental causative factor of obesity (van Baak, 2013). Taking this into account, the most common experimental models to study the consequences of obesity are rodents fed high-fat diets (HFDs; Buettner et al., 2006, 2007). Nowadays, rodents exposed to HFDs are also widely used to evaluate the impact of obesity on the brain (Arnold et al., 2014; Underwood and Thompson, 2016; Nakandakari et al., 2019; Garcia-Serrano and Duarte, 2020).
Several studies have shown that the consumption of a HFD impairs critical brain areas that are involved in cognition, which are affected in AD (Arcego et al., 2016; Lizarbe et al., 2018; Nakandakari et al., 2019). There are several possible reasons why a HFD may lead to memory impairment. However, neuroinflammation seems to play a central role, because it is present in brain tissues involved with memory (Johnston et al., 2011; Verri et al., 2012; Kao et al., 2019). These changes in inflammatory markers are directly involved in the pathogenesis of AD. Thus, the study of rodents fed a HFD becomes an interesting approach for investigating many aspects of neurodegenerative diseases.
Experimental studies demonstrated impaired working memory, that is, decreased spontaneous alternation in the T-maze in 2-month-old mice fed an extremely HFD (60% fat for 17 days), as well as moderate HFD (45% fat for 8 weeks; Arnold et al., 2014). Young adult rats exposed to a HFD (58% fat) for 12–15 weeks presented spatial memory deficits in the spatial object recognition test (Underwood and Thompson, 2016). More recently, Denver et al. (2018) published a study where 7–14 weeks-old mice fed a HFD (45% fat) for different periods (from 18 days to 21 weeks) presented a sustained recognition memory impairment, evaluated at the novel object recognition task. Also, 24-month-old F344xBN F1 rats who were fed a HFD (60.3% fat) for just 4 days showed impaired long-term memory and partially impaired spatial memory (Spencer et al., 2017). McLean et al. (2018) pointed out that HFD caused a rapid decline in mice’s episodic memory. Specifically, memory deficits appeared to occur after 1 day’s exposure to a HFD (60% fat).
Memory impairments in rodents fed a HFD are associated with synaptic dysfunction and neuronal death. For instance, 3-month-old mice fed diets containing 45 or 60% fat for 6 months present synaptic degeneration, characterized by a reduction in the content of proteins located in synapses in the hippocampus and cortex (Lizarbe et al., 2018). Moreover, the exposure of young mice to a HFD (around 30% fat) for only 3 days caused an increase in hippocampal apoptotic molecular signals (i.e., decreased expression of Bcl-2 and increased expression of Bax; Nakandakari et al., 2019). AChE activity was reduced in brain areas such as the prefrontal cortex of rats fed a HFD (Morganstern et al., 2012).
Notably, Nakandakari et al. (2019) pointed out that a few days’ exposure to a HFD induced alterations in AD markers in the mice’s hippocampus. Mice fed a HFD for 3 days exhibited an increase in Aβ levels and an elevation of tau phosphorylation and total tau content in the hippocampus. Six-week-old C57BL/6NHsd mice fed a HFD supplemented with sugar in drinking water (42g/L, 12 weeks), presented increased expressions of both 4G8 and 6E10, indicating Aβ deposition, as well as an increase in the insulin-degrading enzyme (IDE), an endopeptidase responsible for Aβ degradation, resulting in decreased Aβ clearance. Furthermore, HFD supplemented with sugar in drinking water augmented phosphorylation of tau compared with the control diet (Kothari et al., 2017). Busquets et al. (2017) indicated that chronic exposure (weaning until 16 months of age) to a HFD led to the appearance of amyloid depositions in the brain of C57BL/6J mice, which therefore pointed to a potential model of sporadic AD.
High-fat diet consumption is also associated with motor abnormalities in rodents. After being given a HFD for 20 weeks, mice displayed reduced locomotion in the open field test and increased missteps in a vertical grid test. These changes were associated with TH depletion in the SN and striatum (Jang et al., 2017). Kao et al. (2019) recently demonstrated that mice fed a HFD (60% fat) for 20 weeks presented decreased locomotor function, loss of dopaminergic neurons in the SN, and dendritic spine density reduction. We observed that rats fed a HFD for 25 weeks presented a reduction in ventral tegmental area (VTA) TH levels and non-motor features such as depressive-like behavior (Bittencourt et al., 2020). Moreover, α- synuclein mRNA expression was significantly increased in C57BL/6J mice fed a HFD compared with the control group (Han et al., 2017).
Another critical point is that behavioral alterations in mice and rats fed a HFD were accompanied by neuroinflammation and BBB disruption. Levels of NF-κB, a major transcription factor that regulates inflammatory genes, were more expressed in the brains of mice fed a HFD (45% fat for 34 days) at 7–14 weeks of age (Denver et al., 2018). Additionally, IL-1β protein levels increased in the hippocampus of 4-week-old mice after being fed a HFD for 3 days (Nakandakari et al., 2019). Furthermore, the expression of TLR4 was elevated in the brains of HFD compared with the control group (Denver et al., 2018). The density of astrocytes and microglia was increased in the dentate gyrus and cortex of HFD-fed mice (45% fat for 18 days) (Denver et al., 2018). HFD exposure for 25 weeks in rats induced increased astrocytes and microglia density in the SN and VTA (Bittencourt et al., 2020).
The HFD also caused neuroinflammation in the nigrostriatal pathway, that is, astrogliosis and microgliosis in the SN and striatum (Kao et al., 2019). Finally, the increased neuroinflammatory process in HFD animals is related to enhanced permeability of BBB. An increased extravasation of Evans blue dye through the BBB and a disturbance of brain tight junction proteins were observed in mice fed a HFD for 8 weeks (Zhan et al., 2018). It was recently demonstrated that HFD feeding induced the higher entry of 14C-sucrose and 99mTc-albumin into the brains of mice, which indicates BBB disruption (Salameh et al., 2019). It is also important to note that a HFD also caused an increased neuroinflammatory response, increased brain concentration of Aβ species, and exacerbated behavioral deficits in APP/PS1 mice (Bracko et al., 2020).
Taking all these findings into consideration, it can be verified that rodents exposed to a HFD exhibit the main hallmarks of both AD and PD. Indeed, just a short period of exposure to a HFD leads to brain alterations related to neurodegenerative diseases (Figure 3).
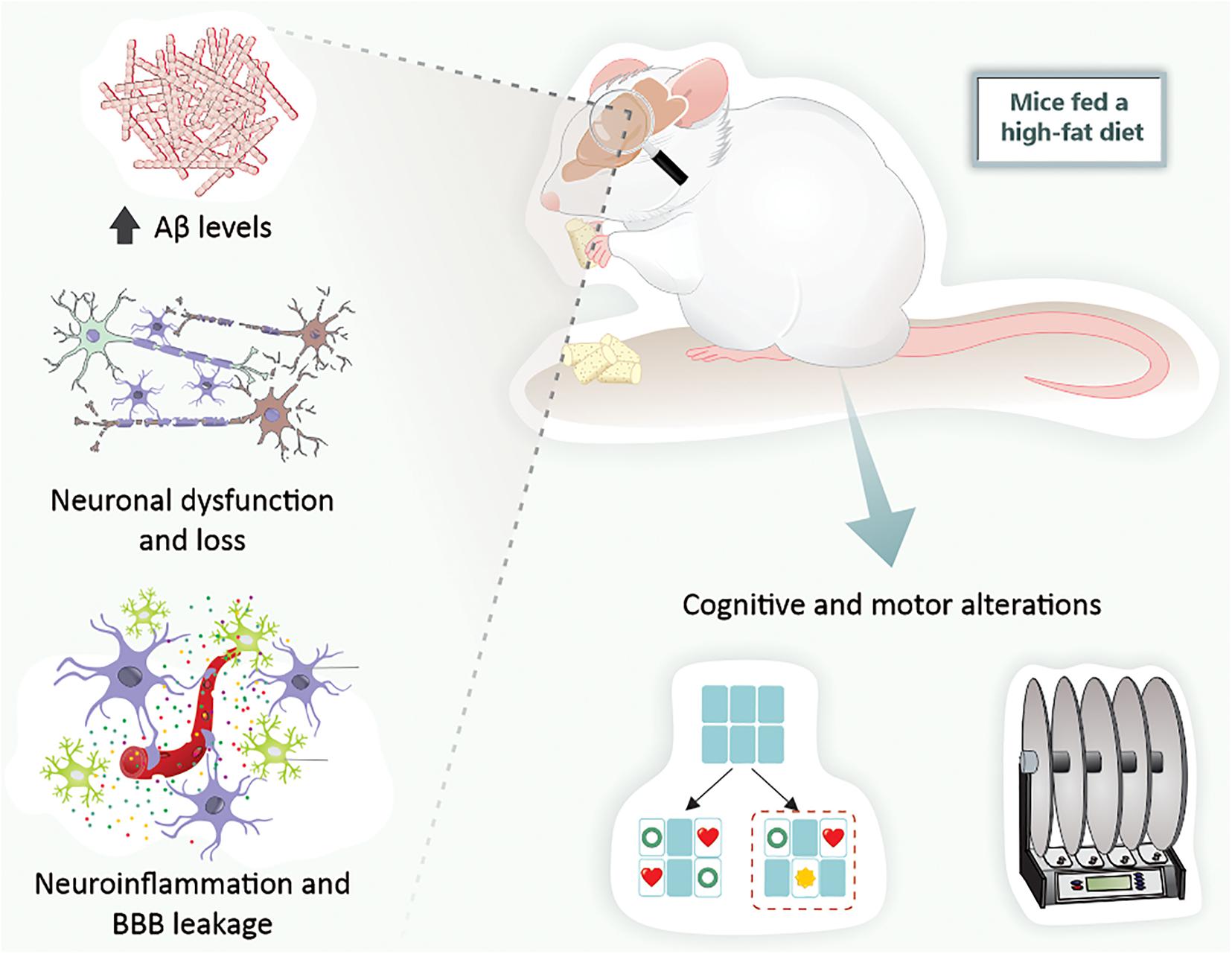
Figure 3. High-fat diet exposure causes cerebral alterations associated with neurodegenerative diseases in mice. Mice fed a high-fat diet present increased Aβ levels, neuronal dysfunction and loss, BBB leakage, neuroinflammation, and, ultimately, cognitive and motor alterations. Aβ, amyloid-β peptide; BBB, blood-brain barrier.
Animal Models of Insulin Resistance and Type 2 Diabetes
Alzheimer’s disease is thought to involve insulin resistance and glucose hypo-metabolism (de la Monte, 2017; Mullins et al., 2017), but it is debatable whether these factors are triggers for neurodegeneration (Stanley et al., 2016). Human imaging studies demonstrate that glucose utilization by the brain declines with age and is notably impaired in subjects with early AD, which may be related to insulin action in key memory and cognition areas in the brain (Lee et al., 2018). Work on animal models of AD and T2D did show an association between dysfunctional insulin signaling in brain cells and AD-like pathology (Duarte, 2015; Lee et al., 2018). However, since glucose transport across the BBB into the brain parenchyma is not dependent on insulin (Duarte and Gruetter, 2012), it seems unlikely that typical glucose hypo-metabolism in AD is directly related to poor insulin sensitivity. On the other hand, insulin receptor activation stimulates signaling cascades for brain function regulation (Mullins et al., 2017). Insulin regulates the expression of genes necessary for memory consolidation (the MAPK/ERK pathway; Kelly et al., 2003; Dou et al., 2005), and also contributes to the control of the cellular metabolic sensor AMPK (Hardie, 2004; Marinangeli et al., 2016).
Diabetes mellitus takes two main forms: T1D results from inadequate insulin secretion, and T2D results from poor insulin action on target cells, that is, insulin resistance. Since T2D is highly heterogeneous, a refined classification of diabetes into five groups with different characteristics and risks of complications has been proposed (Ahlqvist et al., 2018). In any diabetic condition, insulin signaling perturbation impacts the brain, since insulin is involved in key processes such as metabolic regulation and synaptic plasticity (as reviewed in Duarte, 2015). Therefore, in addition to other metabolic syndrome factors such as hyperglycemia, dyslipidemia, hypertension, and vascular complications, impaired insulin signaling in diabetes likely contributes to the development of neurodegenerative disorders.
Several studies aiming to understand how diabetes impacts the brain have employed T1D models characterized by impaired insulin secretion and chronic hyperglycemia. The most extensively used T1D model is based on the administration of streptozotocin, which results in the destruction of β-cells and the halting of insulin production (Rees and Alcolado, 2005). Such treatment to either rats or mice results in a model with chronic severe hyperglycemia, causing neurotoxicity triggering memory deficits and impaired synaptic plasticity (Biessels et al., 1996), synaptic degeneration (Duarte et al., 2006, 2009), increased astrocyte reactivity and proliferation (Duarte et al., 2009), oxidative stress (Silva-Rodrigues et al., 2020), and altered brain metabolism (Duarte et al., 2009; Wang et al., 2012; Ruegsegger et al., 2019). Some of these findings have been reproduced in other T1D models that spontaneously develop diabetes due to the auto-immune destruction of β-cells, namely the non-obese diabetic (NOD) mouse (Saravia et al., 2002) and the BioBreeding/Worcester (BB/Wor) rat (Sima and Li, 2005).
Most available T2D models are associated with obesity (King, 2012). As is the case in diet-induced obesity models, spontaneous T2D rodent models with obesity display overt memory impairment and synaptic dysfunction as a result of a neurodegenerative process. This is so in polygenic strains such as the NONcNZO10/LTJ mouse (Duarte et al., 2012), the Otsuka Long-Evans Tokushima Fatty (OLETF) rat (Cho et al., 2020), and the Kuo Kondo mouse with agouti yellow (Ay) mutation (KK-Ay; Yin et al., 2017), as well as monogenic strains, namely the obese Zucker rat (fa/fa) that carries an autosomal recessive mutation of the fa-gene that encodes for the leptin receptor (Kamal et al., 2013), the leptin-deficient ob/ob mouse (Lepob/ob; Jeon et al., 2016), and the widely used db/db mouse that carries a spontaneous mutation in the leptin receptor gene (Leprdb/db; Chen et al., 2016; Zheng et al., 2016). Most relevant for understanding neurodegenerative pathologies is insulin resistance. When studying insulin resistance one can employ transgenic mice bearing gene deletions or mutations in genes required for insulin action and/or insulin secretion (Nandi et al., 2004). Such models, however, do not recreate a complete T2D phenotype.
There are very few non-obese T2D models available for research (King, 2012), and only the insulin-resistant Goto-Kakizaki (GK) rat has been probed for brain function (Duarte, 2015).
Goto-Kakizaki rats do not seem to show the overt deposition of Aβ in plaques or tau pathology that are typical of AD (Pereira et al., 2000; Candeias et al., 2017). However, their brains have an increased susceptibility to damage by stressors such as oxidative stress or aging (Duarte, 2015). Moreover, they display reduced neuronal glucose utilization and impaired glutamatergic neurotransmission, together with exacerbated mitochondrial astrocyte metabolism (Girault et al., 2019). In addition, glycogen metabolism in astrocytes, which is crucial for fueling glutamatergic neurotransmission and memory, was found to be impaired in insulin-resistant GK rats (Soares et al., 2019). Accordingly, it has been proposed that glycogen in cultured astrocytes is under insulin and IGF-1 regulation (Muhič et al., 2015).
These metabolic alterations in GK rats are accompanied by the development of synaptic dysfunction and increased astrocyte reactivity in the hippocampus, as well as spatial memory impairment (Duarte et al., 2019). In keeping with synaptic dysfunction in non-obese insulin resistance, activity between synapses was shown to trigger the mobilization of GLUT4 (the insulin-sensitive glucose carrier) from intracellular sources into axonal plasma membranes, a process that is mediated by the metabolic sensor AMPK. This is necessary to support the energy demands of active synapses (Ashrafi et al., 2017). Interestingly, it has been shown that toxic Aβ oligomers impair insulin signaling and decrease plasma membrane translocation of the insulin-sensitive GLUT4 in the hippocampus (Pearson-Leary and McNay, 2012). This might result in poor energy supply to neurons.
Despite astrogliosis (Duarte et al., 2019), neuroinflammatory microglia have not been yet reported in non-obese T2D models. Also, BBB leakage has not been confirmed in insulin resistance models, even though there have been reports of endothelial dysfunction (which impacts cerebral perfusion; see the discussion in Garcia-Serrano and Duarte, 2020).
Conclusion
We can conclude that rodent models of obesity, diabetes, and hypercholesterolemia are useful tools for studying neurodegenerative disease development and characteristics. The main features of AD and PD, which include behavioral alterations such as memory and motor impairments, have been observed in hypercholesterolemic and obese rodents. The behavioral alterations in obese and diet-induced hypercholesterolemic mice are associated with Aβ content and α-synuclein changes, neuroinflammation, BBB dysfunction, and ultimately neuronal death in the brain regions affected by AD and PD (Figures 4A,B). In addition, genetic models of hypercholesterolemia have presented behavioral alterations (mainly those associated with AD) related to neurodegeneration, brain inflammation, and BBB disruption, but not modification in Aβ deposits (Figure 4A). Finally, brain alterations in mice and rats submitted to metabolic disorders occur earlier than in the classic rodent models of neurodegenerative diseases. The rodent models of metabolic disorders represent primarily the sporadic forms of neurodegenerative diseases.

Figure 4. Comparison between animal models of metabolic disorders, Alzheimer’s disease mouse model and Parkinson’s disease mouse model. The features of Alzheimer’s disease (A), including memory alterations, are also observed in hypercholesterolemic and obese rodents. The cognitive decline in obese and diet-induced hypercholesterolemic mice is associated with changes in Aβ levels, neuroinflammation, BBB dysfunction, and neuronal dysfunction, in the brain regions affected in Alzheimer’s disease. On the other hand, the genetic models of hypercholesterolemia also presented memory alterations, which are related to neurodegeneration, brain inflammation, BBB disruption, and neuronal dysfunction, but not modification in the Aβ deposits. Parkinson’s disease (B) features, including motor alterations, are also observed in hypercholesterolemic and obese rodents. Moreover, the hypercholesterolemic and 6-OHDA mouse model did not present an increase in α-synuclein mRNA expression while mice fed a high-fat diet presented. The motor alterations in obese, diet-induced hypercholesterolemic and 6-OHDA mice are associated with neuroinflammation, BBB dysfunction, and neuronal dysfunction in the brain regions affected by Parkinson’s disease. 6-OHDA, 6-hydroxydopamine; AD, Alzheimer disease; Aβ, amyloid-β peptide; BBB, blood-brain barrier; LDLr–/–, low-density lipoprotein receptor knockout mice. (+) There is alteration, (–) there is no alteration.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This research was supported by the Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF grant 00193-00001324/2019-27), Fundação de Apoio à Pesquisa de Santa Catarina (FAPESC grant 16802017 06/2016), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq grant 424809-2018-4), Brazilian National Institute of Science and Technology on Neuromodulation (485489/2014-1), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). JD received research support from the Knut and Alice Wallenberg Foundation, the Tage Blücher Foundation, Dementiafonden, Swedish Foundation for International Cooperation in Research and Higher Education (BR2019-8508), Swedish Research Council (2019-01130), Swedish Diabetes Foundation (Dia2019-440), Crafoord Foundation (20200564), Direktör Albert Påhlssons Foundation, and the Lund University Diabetes Centre, which was funded by the Swedish Research Council (Strategic Research Area EXODIAB, grant 2009-1039) and the Swedish Foundation for Strategic Research (grant IRC15-0067).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahlqvist, E., Storm, P., Käräjämäki, A., Martinell, M., Dorkhan, M., Carlsson, A., et al. (2018). Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 6, 361–369. doi: 10.1016/S2213-8587(18)30051-2
Alecu, I., and Bennett, S. A. L. (2019). Dysregulated lipid metabolism and its role in α-synucleinopathy in Parkinson’s disease. Front. Neurosci. 13:328. doi: 10.3389/fnins.2019.00328
Alford, S., Patel, D., Perakakis, N., and Mantzoros, C. S. (2018). Obesity as a risk factor for Alzheimer’s disease: weighing the evidence. Obes. Rev. 19, 269–280. doi: 10.1111/obr.12629
Anjum, I., Fayyaz, M., Wajid, A., Sohail, W., and Ali, A. (2018). Does obesity increase the risk of dementia: a literature review. Cureus 10:e2660. doi: 10.7759/cureus.2660
Arcego, D. M., Krolow, R., Lampert, C., Toniazzo, A. P., Berlitz, C., Lazzaretti, C., et al. (2016). Early life adversities or high fat diet intake reduce cognitive function and alter BDNF signaling in adult rats: interplay of these factors changes these effects. Int. J. Dev. Neurosci. 50, 16–25. doi: 10.1016/j.ijdevneu.2016.03.001
Ariza, M., Cuenca, N., Mauri, M., Jurado, M. A., and Garolera, M. (2016). Neuropsychological performance of young familial hypercholesterolemia patients. Eur. J. Intern. Med. 34, e29–e31. doi: 10.1016/j.ejim.2016.05.009
Arnold, S. E., Lucki, I., Brookshire, B. R., Carlson, G. C., Browne, C. A., Kazi, H., et al. (2014). High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis. 67, 79–87. doi: 10.1016/j.nbd.2014.03.011
Ashrafi, G., Wu, Z., Farrell, R. J., and Ryan, T. A. (2017). GLUT4 mobilization supports energetic demands of active synapses. Neuron 93, 606.e3–615.e3. doi: 10.1016/j.neuron.2016.12.020
Austin, M. A., Hutter, C. M., Zimmern, R. L., and Humphries, S. E. (2004). Familial hypercholesterolemia and coronary heart disease: a HuGE association review. Am. J. Epidemiol. 160, 421–429. doi: 10.1093/aje/kwh237
Ball, N., Teo, W. P., Chandra, S., and Chapman, J. (2019). Parkinson’s disease and the environment. Front. Neurol. 10:218. doi: 10.3389/fneur.2019.00218
Bastías-Candia, S., Zolezzi, J. M., and Inestrosa, N. C. (2019). Revisiting the paraquat-induced sporadic parkinson’s disease-like model. Mol. Neurobiol. 56, 1044–1055. doi: 10.1007/s12035-018-1148-z
Bekris, L. M., Yu, C. E., Bird, T. D., and Tsuang, D. W. (2010). Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 23, 213–227. doi: 10.1177/0891988710383571
Belfiore, R., Rodin, A., Ferreira, E., Velazquez, R., Branca, C., Caccamo, A., et al. (2019). Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging Cell 18:e12873. doi: 10.1111/acel.12873
Beydoun, M. A., Beydoun, H. A., and Wang, Y. (2008). Obesity and central obesity as risk factors for incident dementia and its subtypes: a systematic review and meta-analysis. Obes. Rev. 9, 204–218. doi: 10.1111/j.1467-789X.2008.00473.x
Bhatti, G. K., Reddy, A. P., Reddy, P. H., and Bhatti, J. S. (2020). Lifestyle modifications and nutritional interventions in aging-associated cognitive decline and Alzheimer’s disease. Front. Aging Neurosci. 11:369. doi: 10.3389/fnagi.2019.00369
Biessels, G. J., Kamal, A., Ramakers, G. M., Urban, I. J., Spruijt, B. M., Erkelens, D. W., et al. (1996). Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes Metab. Res. Rev. 45, 1259–1266. doi: 10.2337/diab.45.9.1259
Billings, L. M., Oddo, S., Green, K. N., McGaugh, J. L., and LaFerla, F. M. (2005). Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688. doi: 10.1016/j.neuron.2005.01.040
Bittencourt, A., Brum, P. O., Ribeiro, C. T., Gasparotto, J., Bortolin, R. C., de Vargas, A., et al. (2020). High fat diet-induced obesity causes a reduction in brain tyrosine hydroxylase levels and non-motor features in rats through metabolic dysfunction, neuroinflammation and oxidative stress. Nutr. Neurosci. [Epub ahead of print]. doi: 10.1080/1028415X.2020.1831261
Blockx, I., Einstein, S., Guns, P. J., Van Audekerke, J., Guglielmetti, C., Zago, W., et al. (2016). Monitoring blood-brain barrier integrity following amyloid-β immunotherapy using gadolinium-enhanced MRI in a PDAPP mouse model. J. Alzheimers. Dis. 54, 723–735. doi: 10.3233/jad-160023
Bogaerts, V., Theuns, J., and van Broeckhoven, C. (2008). Genetic findings in Parkinson’s disease and translation into treatment: a leading role for mitochondria? Genes Brain Behav. 7, 129–151. doi: 10.1111/j.1601-183X.2007.00342.x
Bracko, O., Vinarcsik, L. K., Cruz Hernández, J. C., Ruiz-Uribe, N. E., Haft-Javaherian, M., Falkenhain, K., et al. (2020). High fat diet worsens Alzheimer’s disease-related behavioral abnormalities and neuropathology in APP/PS1 mice, but not by synergistically decreasing cerebral blood flow. Sci. Rep. 10:9884. doi: 10.1038/s41598-020-65908-y
Brkic, M., Balusu, S., Van Wonterghem, E., Gorle, N., Benilova, I., Kremer, A., et al. (2015). Amyloid beta oligomers disrupt blood-CSF barrier integrity by activating matrix metalloproteinases. J. Neurosci. 35, 12766–12778. doi: 10.1523/JNEUROSCI.0006-15.2015
Brown, M. S., and Goldstein, J. L. (1986). A receptor-mediated pathway for cholesterol homeostasis. Science 232, 34–47. doi: 10.1126/science.3513311
Brown, M. S., and Goldstein, J. L. (2006). Biomedicine. Lowering LDL–not only how low, but how long? Science 311, 1721–1723. doi: 10.1126/science.1125884
Buettner, R., Parhofer, K. G., Woenckhaus, M., Wrede, C. E., Kunz-Schughart, L. A., Scholmerich, J., et al. (2006). Defining high-fat-diet rat models: metabolic and molecular effects of different fat types. J. Mol. Endocrinol. 36, 485–501. doi: 10.1677/jme.1.01909
Buettner, R., Scholmerich, J., and Bollheimer, L. C. (2007). High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity 15, 798–808. doi: 10.1038/oby.2007.608
Businaro, R., Ippoliti, F., Ricci, S., Canitano, N., and Fuso, A. (2012). Alzheimer’s disease promotion by obesity: induced mechanisms-molecular links and perspectives. Curr. Gerontol. Geriatr. Res. 2012:986823. doi: 10.1155/2012/986823
Busquets, O., Ettcheto, M., Pallas, M., Beas-Zarate, C., Verdaguer, E., Auladell, C., et al. (2017). Long-term exposition to a high fat diet favors the appearance of beta-amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer’s disease. Mech. Ageing Dev. 162, 38–45. doi: 10.1016/j.mad.2016.11.002
Campos-Peña, V., Toral-Rios, D., Becerril-Pérez, F., Sánchez-Torres, C., Delgado-Namorado, Y., Torres-Ossorio, E., et al. (2017). Metabolic syndrome as a risk factor for Alzheimer’s disease: is Aβ a crucial factor in both pathologies? Antioxid. Redox Signal. 26, 542–560. doi: 10.1089/ars.2016.6768
Candeias, E., Duarte, A. I., Sebastiao, I., Fernandes, M. A., Placido, A. I., Carvalho, C., et al. (2017). Middle-aged diabetic females and males present distinct susceptibility to Alzheimer disease-like pathology. Mol. Neurobiol. 54, 6471–6489. doi: 10.1007/s12035-016-0155-1
Cannon, J. R., Tapias, V., Na, H. M., Honick, A. S., Drolet, R. E., and Greenamyre, J. T. (2009). A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 34, 279–290. doi: 10.1016/j.nbd.2009.01.016
Carvey, P. M., Zhao, C. H., Hendey, B., Lum, H., Trachtenberg, J., Desai, B. S., et al. (2005). 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur. J. Neurosci. 22, 1158–1168. doi: 10.1111/j.1460-9568.2005.04281.x
Castillo, X., Castro-Obregon, S., Gutierrez-Becker, B., Gutierrez-Ospina, G., Karalis, N., Khalil, A. A., et al. (2019). Re-thinking the etiological framework of neurodegeneration. Front. Neurosci. 13:728. doi: 10.3389/fnins.2019.00728
Cha, D., and Park, Y. (2019). Association between dietary cholesterol and their food sources and risk for hypercholesterolemia: the 2012(-)2016 Korea National Health and Nutrition Examination Survey. Nutrients 11:846. doi: 10.3390/nu11040846
Champagne, D., Dupuy, J. B., Rochford, J., and Poirier, J. (2002). Apolipoprotein E knockout mice display procedural deficits in the Morris water maze: analysis of learning strategies in three versions of the task. Neuroscience 114, 641–654. doi: 10.1016/s0306-4522(02)00313-5
Chen, F., Dong, R. R., Zhong, K. L., Ghosh, A., Tang, S. S., Long, Y., et al. (2016). Antidiabetic drugs restore abnormal transport of amyloid-β across the blood-brain barrier and memory impairment in db/db mice. Neuropharmacology 101, 123–136. doi: 10.1016/j.neuropharm.2015.07.023
Chen, G., Chen, K. S., Knox, J., Inglis, J., Bernard, A., Martin, S. J., et al. (2000). A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer’s disease. Nature 408, 975–979. doi: 10.1038/35050103
Chen, H., Huang, X., Guo, X., and Peddada, S. (2014). Individual and joint prevalence of three nonmotor symptoms of PD in the US general population. Mov. Disord. 29, 1316–1319. doi: 10.1002/mds.25950
Chen, H., and Ritz, B. (2018). The search for environmental causes of parkinson’s disease: moving forward. J. Parkinsons Dis. 8, S9–S17. doi: 10.3233/JPD-181493
Chen, X., Lan, X., Roche, I., Liu, R., and Geiger, J. D. (2008). Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J. Neurochem. 107, 1147–1157. doi: 10.1111/j.1471-4159.2008.05697.x
Chen, Y., Yin, M., Cao, X., Hu, G., and Xiao, M. (2018). Pro- and anti-inflammatory effects of high cholesterol diet on aged brain. Aging Dis. 9, 374–390. doi: 10.14336/AD.2017.0706
Chia, L. G., Ni, D. R., Cheng, L. J., Kuo, J. S., Cheng, F. C., and Dryhurst, G. (1996). Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 5,7-dihydroxytryptamine on the locomotor activity and striatal amines in C57BL/6 mice. Neurosci. Lett. 218, 67–71. doi: 10.1016/0304-3940(96)13091-3
Cho, J. A., Park, S. H., Cho, J., Kim, J. O., Yoon, J. H., and Park, E. (2020). Exercise and curcumin in combination improves cognitive function and attenuates ER stress in diabetic rats. Nutrients 12:1309. doi: 10.3390/nu12051309
Chuang, Y. F., An, Y., Bilgel, M., Wong, D. F., Troncoso, J. C., O’Brien, R. J., et al. (2016). Midlife adiposity predicts earlier onset of Alzheimer’s dementia, neuropathology and presymptomatic cerebral amyloid accumulation. Mol. Psychiatry 21, 910–915. doi: 10.1038/mp.2015.129
Chung, H. K., Ho, H. A., Pérez-Acuña, D., and Lee, S. J. (2019). Modeling α-synuclein propagation with preformed fibril injections. J. Mov. Disord. 12, 139–151. doi: 10.14802/jmd.19046
Clarke, R. M., O’Connell, F., Lyons, A., and Lynch, M. A. (2007). The HMG-CoA reductase inhibitor, atorvastatin, attenuates the effects of acute administration of amyloid-beta1-42 in the rat hippocampus in vivo. Neuropharmacology 52, 136–145. doi: 10.1016/j.neuropharm.2006.07.031
Collins, L. M., Toulouse, A., Connor, T. J., and Nolan, Y. M. (2012). Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology 62, 2154–2168. doi: 10.1016/j.neuropharm.2012.01.028
Crisby, M., Rahman, S. M., Sylven, C., Winblad, B., and Schultzberg, M. (2004). Effects of high cholesterol diet on gliosis in apolipoprotein E knockout mice. Implications for Alzheimer’s disease and stroke. Neurosci. Lett. 369, 87–92. doi: 10.1016/j.neulet.2004.05.057
Dawson, T. M., Ko, H. S., and Dawson, V. L. (2010). Genetic animal models of Parkinson’s disease. Neuron 66, 646–661. doi: 10.1016/j.neuron.2010.04.034
de la Monte, S. M. (2017). Insulin resistance and neurodegeneration: progress towards the development of new therapeutics for Alzheimer’s disease. Drugs 77, 47–65. doi: 10.1007/s40265-016-0674-0
de Lau, L. M., Koudstaal, P. J., Hofman, A., and Breteler, M. M. (2006). Serum cholesterol levels and the risk of Parkinson’s disease. Am. J. Epidemiol. 164, 998–1002. doi: 10.1093/aje/kwj283
de Oliveira, J., Engel, D. F., de Paula, G. C., Melo, H. M., Lopes, S. C., Ribeiro, C. T., et al. (2020a). LDL Receptor Deficiency Does not Alter Brain Amyloid-beta Levels but Causes an Exacerbation of Apoptosis. J. Alzheimers. Dis. 73, 585–596. doi: 10.3233/JAD-190742
de Oliveira, J., Engel, D., Paula, G. C., dos Santos, D. B., Lopes, J. B., Farina, M., et al. (2020b). High cholesterol diet exacerbates blood-brain barrier disruption in LDLr-/- mice: impact on cognitive function. J. Alzheimers Dis. [Epub ahead of print]. doi: 10.3233/JAD-200541
de Oliveira, J., Hort, M. A., Moreira, E. L., Glaser, V., Ribeiro-do-Valle, R. M., Prediger, R. D., et al. (2011). Positive correlation between elevated plasma cholesterol levels and cognitive impairments in LDL receptor knockout mice: relevance of cortico-cerebral mitochondrial dysfunction and oxidative stress. Neuroscience 197, 99–106. doi: 10.1016/j.neuroscience.2011.09.009
de Oliveira, J., Moreira, E. L., dos Santos, D. B., Piermartiri, T. C., Dutra, R. C., Pinton, S., et al. (2014). Increased susceptibility to amyloid-beta-induced neurotoxicity in mice lacking the low-density lipoprotein receptor. J. Alzheimers. Dis. 41, 43–60. doi: 10.3233/JAD-132228
De Pablo-Fernandez, E., Goldacre, R., Pakpoor, J., Noyce, A. J., and Warner, T. T. (2018). Association between diabetes and subsequent Parkinson disease: a record-linkage cohort study. Neurology 91, e139–e142. doi: 10.1212/WNL.0000000000005771
Denver, P., Gault, V. A., and McClean, P. L. (2018). Sustained high-fat diet modulates inflammation, insulin signalling and cognition in mice and a modified xenin peptide ameliorates neuropathology in a chronic high-fat model. Diabetes. Obes. Metab. 20, 1166–1175. doi: 10.1111/dom.13210
Dewachter, I., Van Dorpe, J., Smeijers, L., Gilis, M., Kuiperi, C., Laenen, I., et al. (2000). Aging increased amyloid peptide and caused amyloid plaques in brain of old APP/V717I transgenic mice by a different mechanism than mutant presenilin1. J. Neurosci. 20, 6452–6458.
Do, T. M., Alata, W., Dodacki, A., Traversy, M.-T., Chacun, H., Pradier, L., et al. (2014). Altered cerebral vascular volumes and solute transport at the blood–brain barriers of two transgenic mouse models of Alzheimer’s disease. Neuropharmacology 81, 311–317.
Dodart, J. C., Meziane, H., Mathis, C., Bales, K. R., Paul, S. M., and Ungerer, A. (1999). Behavioral disturbances in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Behav. Neurosci. 113, 982–990. doi: 10.1037//0735-7044.113.5.982
Dou, J. T., Chen, M., Dufour, F., Alkon, D. L., and Zhao, W. Q. (2005). Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn. Mem. 12, 646–655. doi: 10.1101/lm.88005
Drechsel, D. A., and Patel, M. (2008). Role of reactive oxygen species in the neurotoxicity of environmental agents implicated in Parkinson’s disease. Free Radic. Biol. Med. 44, 1873–1886. doi: 10.1016/j.freeradbiomed.2008.02.008
Drummond, E., and Wisniewski, T. (2017). Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 133, 155–175. doi: 10.1007/s00401-016-1662-x
Duarte, J. M. N. (2015). Metabolic alterations associated to brain dysfunction in diabetes. Aging Dis. 6, 304–321. doi: 10.14336/AD.2014.1104
Duarte, J. M. N., and Gruetter, R. (2012). Cerebral Glucose Transport and Homeostasis Neural Metabolism In Vivo. Cham: Springer, 655–673.
Duarte, J. M. N., Agostinho, P. M., Carvalho, R. A., and Cunha, R. A. (2012). Caffeine consumption prevents diabetes-induced memory impairment and synaptotoxicity in the hippocampus of NONcZNO10/LTJ mice. PLoS One 7:e21899. doi: 10.1371/journal.pone.0021899
Duarte, J. M. N., Carvalho, R. A., Cunha, R. A., and Gruetter, R. (2009). Caffeine consumption attenuates neurochemical modifications in the hippocampus of streptozotocin-induced diabetic rats. J. Neurochem. 111, 368–379. doi: 10.1111/j.1471-4159.2009.06349.x
Duarte, J. M. N., Oliveira, C. R., Ambrósio, A. F., and Cunha, R. A. (2006). Modification of adenosine A1 and A2A receptor density in the hippocampus of streptozotocin-induced diabetic rats. Neurochem. Int. 48, 144–150. doi: 10.1016/j.neuint.2005.08.008
Duarte, J. M. N., Skoug, C., Silva, H. B., Carvalho, R. A., Gruetter, R., and Cunha, R. A. (2019). Impact of caffeine consumption on type 2 diabetes-induced spatial memory impairment and neurochemical alterations in the hippocampus. Front. Neurosci. 12:1015. doi: 10.3389/fnins.2018.01015
Dugger, B. N., and Dickson, D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 9:a028035. doi: 10.1101/cshperspect.a028035
Duty, S., and Jenner, P. (2011). Animal models of Parkinson’s disease: a source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 164, 1357–1391. doi: 10.1111/j.1476-5381.2011.01426.x
Earls, R. H., Menees, K. B., Chung, J., Barber, J., Gutekunst, C. A., Hazim, M. G., et al. (2019). Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J. Neuroinflammation 16:250. doi: 10.1186/s12974-019-1636-8
Elhaik Goldman, S., Goez, D., Last, D., Naor, S., Liraz Zaltsman, S., Sharvit-Ginon, I., et al. (2018). High-fat diet protects the blood-brain barrier in an Alzheimer’s disease mouse model. Aging Cell 17:e12818. doi: 10.1111/acel.12818
Engel, D. F., Grzyb, A. N., de Oliveira, J., Pötzsch, A., Walker, T. L., Brocardo, P. S., et al. (2019). Impaired adult hippocampal neurogenesis in a mouse model of familial hypercholesterolemia: a role for the LDL receptor and cholesterol metabolism in adult neural precursor cells. Mol. Metab. 30, 1–15. doi: 10.1016/j.molmet.2019.09.002
Evola, M., Hall, A., Wall, T., Young, A., and Grammas, P. (2010). Oxidative stress impairs learning and memory in apoE knockout mice. Pharmacol. Biochem. Behav. 96, 181–186. doi: 10.1016/j.pbb.2010.05.003
Fairoozy, R. H., Futema, M., Vakili, R., Abbaszadegan, M. R., Hosseini, S., Aminzadeh, M., et al. (2017). The genetic spectrum of familial hypercholesterolemia (FH) in the iranian population. Sci. Rep. 7:17087. doi: 10.1038/s41598-017-17181-9
Ferreira, S. T., and Klein, W. L. (2011). The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 96, 529–543. doi: 10.1016/j.nlm.2011.08.003
Figueiredo, C. P., Bicca, M. A., Latini, A., Prediger, R. D., Medeiros, R., and Calixto, J. B. (2011). Folic acid plus alpha-tocopherol mitigates amyloid-beta-induced neurotoxicity through modulation of mitochondrial complexes activity. J. Alzheimers. Dis. 24, 61–75. doi: 10.3233/JAD-2010-101320
Figueiredo, C. P., Clarke, J. R., Ledo, J. H., Ribeiro, F. C., Costa, C. V., Melo, H. M., et al. (2013). Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 33, 9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013
Fisher, A., Brandeis, R., Chapman, S., Pittel, Z., and Michaelson, D. M. (1998). M1 muscarinic agonist treatment reverses cognitive and cholinergic impairments of apolipoprotein E-deficient mice. J. Neurochem. 70, 1991–1997. doi: 10.1046/j.1471-4159.1998.70051991.x
Fitzpatrick, A. L., Kuller, L. H., Lopez, O. L., Diehr, P., O’Meara, E. S., Longstreth, W. T., et al. (2009). Midlife and late-life obesity and the risk of dementia: cardiovascular health study. Arch. Neurol. 66, 336–342. doi: 10.1001/archneurol.2008.582
Fleming, S. M., Fernagut, P. O., and Chesselet, M. F. (2005). Genetic mouse models of parkinsonism: strengths and limitations. NeuroRx 2, 495–503. doi: 10.1602/neurorx.2.3.495
Flood, J. F., Morley, J. E., and Roberts, E. (1991). Amnestic effects in mice of four synthetic peptides homologous to amyloid beta protein from patients with Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 88, 3363–3366. doi: 10.1073/pnas.88.8.3363
Fornai, F., Schluter, O. M., Lenzi, P., Gesi, M., Ruffoli, R., Ferrucci, M., et al. (2005). Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 102, 3413–3418. doi: 10.1073/pnas.0409713102
Franke, C., and Storch, A. (2017). Nonmotor fluctuations in Parkinson’s disease. Int. Rev. Neurobiol. 134, 947–971. doi: 10.1016/bs.irn.2017.05.021
Frautschy, S. A., Yang, F., Irrizarry, M., Hyman, B., Saido, T. C., Hsiao, K., et al. (1998). Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 152, 307–317.
Fredriksson, A., Palomo, T., and Archer, T. (1999). Effects of co-administration of anticonvulsant and putative anticonvulsive agents and sub/suprathreshold doses of L-dopa upon motor behaviour of MPTP-treated mice. J. Neural Transm. 106, 889–909. doi: 10.1007/s007020050209
Fuentes, D., Fernandez, N., Garcia, Y., Garcia, T., Morales, A. R., and Menendez, R. (2018). Age-related changes in the behavior of apolipoprotein E knockout mice. Behav. Sci. 8:33. doi: 10.3390/bs8030033
Games, D., Adams, D., Alessandrini, R., Barbour, R., Berthelette, P., Blackwell, C., et al. (1995). Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373, 523–527. doi: 10.1038/373523a0
Garcia-Serrano, A. M., and Duarte, J. M. N. (2020). Brain metabolism alterations in type 2 diabetes: what did we learn from diet-induced diabetes models? Front. Neurosci. 14:229. doi: 10.3389/fnins.2020.00229
Gasparotto, J., Ribeiro, C. T., Bortolin, R. C., Somensi, N., Rabelo, T. K., Kunzler, A., et al. (2017). Targeted inhibition of RAGE in substantia nigra of rats blocks 6-OHDA-induced dopaminergic denervation. Sci. Rep. 7:8795. doi: 10.1038/s41598-017-09257-3
Giasson, B. I., Duda, J. E., Quinn, S. M., Zhang, B., Trojanowski, J. Q., and Lee, V. M.-Y. (2002). Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 34, 521–533. doi: 10.1016/s0896-6273(02)00682-7
Girault, F. M., Sonnay, S., Gruetter, R., and Duarte, J. M. N. (2019). Alterations of brain energy metabolism in type 2 diabetic goto-kakizaki rats measured in vivo by (13)C magnetic resonance spectroscopy. Neurotox. Res. 36, 268–278. doi: 10.1007/s12640-017-9821-y
Gordon, I., Grauer, E., Genis, I., Sehayek, E., and Michaelson, D. M. (1995). Memory deficits and cholinergic impairments in apolipoprotein E-deficient mice. Neurosci. Lett. 199, 1–4. doi: 10.1016/0304-3940(95)12006-p
Gordon, R., Albornoz, E. A., Christie, D. C., Langley, M. R., Kumar, V., Mantovani, S., et al. (2018). Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 10:eaah4066. doi: 10.1126/scitranslmed.aah4066
Gottesman, R. F., Schneider, A. L., Zhou, Y., Coresh, J., Green, E., Gupta, N., et al. (2017). Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 317, 1443–1450. doi: 10.1001/jama.2017.3090
Gregor, M. F., and Hotamisligil, G. S. (2011). Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 29, 415–445. doi: 10.1146/annurev-immunol-031210-101322
Gustafson, D., Rothenberg, E., Blennow, K., Steen, B., and Skoog, I. (2003). An 18-year follow-up of overweight and risk of Alzheimer disease. Arch. Intern. Med. 163, 1524–1528. doi: 10.1001/archinte.163.13.1524
Gustafson, D. R., Karlsson, C., Skoog, I., Rosengren, L., Lissner, L., and Blennow, K. (2007). Mid-life adiposity factors relate to blood-brain barrier integrity in late life. J. Intern. Med. 262, 643–650. doi: 10.1111/j.1365-2796.2007.01869.x
Guzman-Martinez, L., Maccioni, R. B., Andrade, V., Navarrete, L. P., Pastor, M. G., and Ramos-Escobar, N. (2019). Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 10:1008. doi: 10.3389/fphar.2019.01008
Hafezi-Moghadam, A., Thomas, K. L., and Wagner, D. D. (2007). ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am. J. Physiol. Cell Physiol. 292, C1256–C1262. doi: 10.1152/ajpcell.00563.2005
Han, J., Plummer, J., Liu, L., Byrd, A., Aschner, M., and Erikson, K. M. (2017). The impact of obesity on brain iron levels and α-synuclein expression is regionally dependent. Nutr. Neurosci. 22, 335–343. doi: 10.1080/1028415x.2017.1387720
Hardie, D. G. (2004). The AMP-activated protein kinase pathway–new players upstream and downstream. J. Cell Sci. 117(Pt 23), 5479–5487. doi: 10.1242/jcs.01540
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Hartman, R. E., Izumi, Y., Bales, K. R., Paul, S. M., Wozniak, D. F., and Holtzman, D. M. (2005). Treatment with an amyloid-beta antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer’s disease. J. Neurosci. 25, 6213–6220. doi: 10.1523/JNEUROSCI.0664-05.2005
Hassing, L. B., Dahl, A. K., Thorvaldsson, V., Berg, S., Gatz, M., Pedersen, N. L., et al. (2009). Overweight in midlife and risk of dementia: a 40-year follow-up study. Int. J. Obes. 33, 893–898. doi: 10.1038/ijo.2009.104
Hatcher, J. M., Pennell, K. D., and Miller, G. W. (2008). Parkinson’s disease and pesticides: a toxicological perspective. Trends Pharmacol. Sci. 29, 322–329. doi: 10.1016/j.tips.2008.03.007
Hou, Q., Guan, Y., Yu, W., Liu, X., Wu, L., Xiao, M., et al. (2019). Associations between obesity and cognitive impairment in the Chinese elderly: an observational study. Clin. Interv. Aging 14, 367–373. doi: 10.2147/CIA.S192050
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., et al. (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. doi: 10.1126/science.274.5284.99
Hu, G. (2010). Total cholesterol and the risk of Parkinson’s disease: a review for some new findings. Parkinsons Dis. 2010:836962. doi: 10.4061/2010/836962
Hu, G., Antikainen, R., Jousilahti, P., Kivipelto, M., and Tuomilehto, J. (2008). Total cholesterol and the risk of Parkinson disease. Neurology 70, 1972–1979. doi: 10.1212/01.wnl.0000312511.62699.a8
Huang, X., Auinger, P., Eberly, S., Oakes, D., Schwarzschild, M., Ascherio, A., et al. (2011). Serum cholesterol and the progression of Parkinson’s disease: results from DATATOP. PLoS One 6:e22854. doi: 10.1371/journal.pone.0022854
Huang, Y., Weisgraber, K. H., Mucke, L., and Mahley, R. W. (2004). Apolipoprotein E: diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer’s disease. J. Mol. Neurosci. 23, 189–204. doi: 10.1385/JMN:23:3:189
Jacobsen, J. S., Wu, C. C., Redwine, J. M., Comery, T. A., Arias, R., Bowlby, M., et al. (2006). Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5161–5166. doi: 10.1073/pnas.0600948103
Jang, Y., Lee, M. J., Han, J., Kim, S. J., Ryu, I., Ju, X., et al. (2017). A high-fat diet induces a loss of midbrain dopaminergic neuronal function that underlies motor abnormalities. Exp. Neurobiol. 26, 104–112. doi: 10.5607/en.2017.26.2.104
Jeon, B. T., Heo, R. W., Jeong, E. A., Yi, C. O., Lee, J. Y., and Kim, K. E. (2016). Effects of caloric restriction on O-GlcNAcylation, Ca(2+) signaling, and learning impairment in the hippocampus of ob/ob mice. Neurobiol. Aging 44, 127–137. doi: 10.1016/j.neurobiolaging.2016.05.002
Jhoo, J. H., Kim, H. C., Nabeshima, T., Yamada, K., Shin, E. J., Jhoo, W. K., et al. (2004). Beta-amyloid (1-42)-induced learning and memory deficits in mice: involvement of oxidative burdens in the hippocampus and cerebral cortex. Behav. Brain Res. 155, 185–196. doi: 10.1016/j.bbr.2004.04.012
Ji, Y., Permanne, B., Sigurdsson, E. M., Holtzman, D. M., and Wisniewski, T. (2001). Amyloid beta40/42 clearance across the blood-brain barrier following intra-ventricular injections in wild-type, apoE knock-out and human apoE3 or E4 expressing transgenic mice. J. Alzheimers. Dis. 3, 23–30. doi: 10.3233/jad-2001-3105
Johnson-Wood, K., Lee, M., Motter, R., Hu, K., Gordon, G., Barbour, R., et al. (1997). Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 94, 1550–1555. doi: 10.1073/pnas.94.4.1550
Johnston, H., Boutin, H., and Allan, S. M. (2011). Assessing the contribution of inflammation in models of Alzheimer’s disease. Biochem. Soc. Trans. 39, 886–890. doi: 10.1042/BST0390886
Kamal, A., Ramakers, G. M., Gispen, W. H., and Biessels, G. J. (2013). Hyperinsulinemia in rats causes impairment of spatial memory and learning with defects in hippocampal synaptic plasticity by involvement of postsynaptic mechanisms. Exp. Brain Res. 226, 45–51. doi: 10.1007/s00221-013-3409-4
Kao, Y. C., Wei, W. Y., Tsai, K. J., and Wang, L. C. (2019). High fat diet suppresses peroxisome proliferator-activated receptors and reduces dopaminergic neurons in the substantia nigra. Int. J. Mol. Sci. 21:207. doi: 10.3390/ijms21010207
Kasza, A., Penke, B., Frank, Z., Bozso, Z., Szegedi, V., Hunya, A., et al. (2017). Studies for improving a rat model of Alzheimer’s disease: icv administration of well-characterized beta-amyloid 1-42 oligomers induce dysfunction in spatial memory. Molecules 22:2007. doi: 10.3390/molecules22112007
Kelley, B. J., and Petersen, R. C. (2007). Alzheimer’s disease and mild cognitive impairment. Neurol. Clin. 25, 577–609. doi: 10.1016/j.ncl.2007.03.008
Kelly, A., Laroche, S., and Davis, S. (2003). Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. J. Neurosci. 23, 5354–5360.
Kim, H. Y., Lee, D. K., Chung, B. R., Kim, H. V., and Kim, Y. (2016). Intracerebroventricular injection of amyloid-beta peptides in normal mice to acutely induce alzheimer-like cognitive deficits. J. Vis. Exp. 109:53308. doi: 10.3791/53308
King, A. J. (2012). The use of animal models in diabetes research. Br. J. Pharmacol. 166, 877–894. doi: 10.1111/j.1476-5381.2012.01911.x
Kitazawa, M., Medeiros, R., and Laferla, F. M. (2012). Transgenic mouse models of Alzheimer disease: developing a better model as a tool for therapeutic interventions. Curr. Pharm. Des. 18, 1131–1147. doi: 10.2174/138161212799315786
Kivipelto, M., Helkala, E. L., Hanninen, T., Laakso, M. P., Hallikainen, M., Alhainen, K., et al. (2001). Midlife vascular risk factors and late-life mild cognitive impairment: a population-based study. Neurology 56, 1683–1689. doi: 10.1212/wnl.56.12.1683
Kivipelto, M., Ngandu, T., Fratiglioni, L., Viitanen, M., Kareholt, I., Winblad, B., et al. (2005). Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 62, 1556–1560. doi: 10.1001/archneur.62.10.1556
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a008888. doi: 10.1101/cshperspect.a008888
Kothari, V., Luo, Y., Tornabene, T., O’Neill, A. M., Greene, M. W., Geetha, T., et al. (2017). High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 499–508. doi: 10.1016/j.bbadis.2016.10.006
Kozlov, S., Afonin, A., Evsyukov, I., and Bondarenko, A. (2017). Alzheimer’s disease: as it was in the beginning. Rev. Neurosci. 28, 825–843. doi: 10.1515/revneuro-2017-0006
Kummer, B. R., Diaz, I., Wu, X., Aaroe, A. E., Chen, M. L., Iadecola, C., et al. (2019). Associations between cerebrovascular risk factors and parkinson disease. Ann. Neurol. 86, 572–581. doi: 10.1002/ana.25564
Kurano, M., Iso, O. N., Hara, M., Ishizaka, N., Moriya, K., Koike, K., et al. (2011). LXR agonist increases apoE secretion from HepG2 spheroid, together with an increased production of VLDL and apoE-rich large HDL. Lipids Health Dis. 10:134. doi: 10.1186/1476-511X-10-134
Kurkowska-Jastrzebska, I., Balkowiec-Iskra, E., Ciesielska, A., Joniec, I., Cudna, A., Zaremba, M. M., et al. (2009). Decreased inflammation and augmented expression of trophic factors correlate with MOG-induced neuroprotection of the injured nigrostriatal system in the murine MPTP model of Parkinson’s disease. Int. Immunopharmacol. 9, 781–791. doi: 10.1016/j.intimp.2009.03.003
LaFerla, F. M., and Green, K. N. (2012). Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006320. doi: 10.1101/cshperspect.a006320
Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Ledo, J. H., Azevedo, E. P., Clarke, J. R., Ribeiro, F. C., Figueiredo, C. P., Foguel, D., et al. (2013). Amyloid-beta oligomers link depressive-like behavior and cognitive deficits in mice. Mol. Psychiatry 18, 1053–1054. doi: 10.1038/mp.2012.168
Leduc, V., Jasmin-Belanger, S., and Poirier, J. (2010). APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol. Med. 16, 469–477. doi: 10.1016/j.molmed.2010.07.008
Lee, J. H., Jahrling, J. B., Denner, L., and Dineley, K. T. (2018). Targeting insulin for Alzheimer’s disease: mechanisms, status and potential directions. J. Alzheimers. Dis. 64, S427–S453. doi: 10.3233/JAD-179923
Lee, M. K., Stirling, W., Xu, Y., Xu, X., Qui, D., Mandir, A. S., et al. (2002). Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 –> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 8968–8973. doi: 10.1073/pnas.132197599
Levy, E., Carman, M. D., Fernandez-Madrid, I. J., Power, M. D., Lieberburg, I., van Duinen, S. G., et al. (1990). Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248, 1124–1126. doi: 10.1126/science.2111584
Limphaibool, N., Iwanowski, P., Holstad, M. J. V., and Perkowska, K. (2018). Parkinsonism in inherited metabolic disorders: key considerations and major features. Front. Neurol. 9:857. doi: 10.3389/fneur.2018.00857
Liu, C. C., Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, D. S., Pan, X. D., Zhang, J., Shen, H., Collins, N. C., Cole, A. M., et al. (2015). APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol. Neurodegener. 10:7. doi: 10.1186/s13024-015-0002-2
Lizarbe, B., Soares, A. F., Larsson, S., and Duarte, J. M. N. (2018). Neurochemical modifications in the hippocampus, cortex and hypothalamus of mice exposed to long-term high-fat diet. Front. Neurosci. 12:985. doi: 10.3389/fnins.2018.00985
Long, J. M., and Holtzman, D. M. (2019). Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339. doi: 10.1016/j.cell.2019.09.001
Lopes, J. B., de Oliveira, J., Engel, D. F., de Paula, G. C., Moreira, E. L., and de Bem, A. F. (2015). Efficacy of donepezil for cognitive impairments in familial hypercholesterolemia: preclinical proof of concept. CNS Neurosci. Ther. 21, 964–966. doi: 10.1111/cns.12479
Luchtman, D. W., Shao, D., and Song, C. (2009). Behavior, neurotransmitters and inflammation in three regimens of the MPTP mouse model of Parkinson’s disease. Physiol. Behav. 98, 130–138. doi: 10.1016/j.physbeh.2009.04.021
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J. Q., et al. (2012). Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Lumeng, C. N., and Saltiel, A. R. (2011). Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 121, 2111–2117. doi: 10.1172/JCI57132
Magrinelli, F., Picelli, A., Tocco, P., Federico, A., Roncari, L., Smania, N., et al. (2016). Pathophysiology of motor dysfunction in Parkinson’s disease as the rationale for drug treatment and rehabilitation. Parkinsons Dis. 2016:9832839. doi: 10.1155/2016/9832839
Marinangeli, C., Didier, S., and Vingtdeux, V. (2016). AMPK in neurodegenerative diseases: implications and therapeutic perspectives. Curr. Drug Targets 17, 890–907. doi: 10.2174/1389450117666160201105645
Martinez, E. M., Young, A. L., Patankar, Y. R., Berwin, B. L., Wang, L., von Herrmann, K. M., et al. (2017). Editor’s highlight: Nlrp3 is required for inflammatory changes and nigral cell loss resulting from chronic intragastric rotenone exposure in mice. Toxicol. Sci. 159, 64–75. doi: 10.1093/toxsci/kfx117
Masliah, E., Samuel, W., Veinbergs, I., Mallory, M., Mante, M., and Saitoh, T. (1997). Neurodegeneration and cognitive impairment in apoE-deficient mice is ameliorated by infusion of recombinant apoE. Brain Res. 751, 307–314. doi: 10.1016/s0006-8993(96)01420-5
McCormack, A. L., Thiruchelvam, M., Manning-Bog, A. B., Thiffault, C., Langston, J. W., Cory-Slechta, D. A., et al. (2002). Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 10, 119–127. doi: 10.1006/nbdi.2002.0507
McLean, F. H., Grant, C., Morris, A. C., Horgan, G. W., Polanski, A. J., Allan, K., et al. (2018). Rapid and reversible impairment of episodic memory by a high-fat diet in mice. Sci. Rep. 8:11976. doi: 10.1038/s41598-018-30265-4
Medeiros, R., Prediger, R. D., Passos, G. F., Pandolfo, P., Duarte, F. S., Franco, J. L., et al. (2007). Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of Alzheimer’s disease: relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J. Neurosci. 27, 5394–5404. doi: 10.1523/JNEUROSCI.5047-06.2007
Methia, N., Andre, P., Hafezi-Moghadam, A., Economopoulos, M., Thomas, K. L., and Wagner, D. D. (2001). ApoE deficiency compromises the blood brain barrier especially after injury. Mol. Med. 7, 810–815.
Minogue, A. M., Schmid, A. W., Fogarty, M. P., Moore, A. C., Campbell, V. A., Herron, C. E., et al. (2003). Activation of the c-Jun N-terminal kinase signaling cascade mediates the effect of amyloid-beta on long term potentiation and cell death in hippocampus: a role for interleukin-1beta? J. Biol. Chem. 278, 27971–27980. doi: 10.1074/jbc.M302530200
Moreira, E. L., de Oliveira, J., Engel, D. F., Walz, R., de Bem, A. F., Farina, M., et al. (2014). Hypercholesterolemia induces short-term spatial memory impairments in mice: up-regulation of acetylcholinesterase activity as an early and causal event? J. Neural Transm. 121, 415–426. doi: 10.1007/s00702-013-1107-9
Moreira, E. L., de Oliveira, J., Nunes, J. C., Santos, D. B., Nunes, F. C., Vieira, D. S., et al. (2012). Age-related cognitive decline in hypercholesterolemic LDL receptor knockout mice (LDLr-/-): evidence of antioxidant imbalance and increased acetylcholinesterase activity in the prefrontal cortex. J. Alzheimers. Dis. 32, 495–511. doi: 10.3233/JAD-2012-120541
Morganstern, I., Ye, Z., Liang, S., Fagan, S., and Leibowitz, S. F. (2012). Involvement of cholinergic mechanisms in the behavioral effects of dietary fat consumption. Brain Res. 1470, 24–34. doi: 10.1016/j.brainres.2012.06.004
Muhič, M., Vardjan, N., Chowdhury, H. H., Zorec, R., and Kreft, M. (2015). Insulin and insulin-like growth factor 1 (IGF-1) modulate cytoplasmic glucose and glycogen levels but not glucose transport across the membrane in astrocytes. J. Biol. Chem. 290, 11167–11176. doi: 10.1074/jbc.M114.629063
Mulder, M., Jansen, P. J., Janssen, B. J., van de Berg, W. D., van der Boom, H., Havekes, L. M., et al. (2004). Low-density lipoprotein receptor-knockout mice display impaired spatial memory associated with a decreased synaptic density in the hippocampus. Neurobiol. Dis. 16, 212–219. doi: 10.1016/j.nbd.2004.01.015
Mulder, M., Koopmans, G., Wassink, G., Al Mansouri, G., Simard, M. L., Havekes, L. M., et al. (2007). LDL receptor deficiency results in decreased cell proliferation and presynaptic bouton density in the murine hippocampus. Neurosci. Res. 59, 251–256. doi: 10.1016/j.neures.2007.07.004
Mullins, R. J., Diehl, T. C., Chia, C. W., and Kapogiannis, D. (2017). Insulin resistance as a link between amyloid-beta and tau pathologies in Alzheimer’s disease. Front. Aging Neurosci. 9:118. doi: 10.3389/fnagi.2017.00118
Naderali, E. K., Ratcliffe, S. H., and Dale, M. C. (2009). Obesity and Alzheimer’s disease: a link between body weight and cognitive function in old age. Am. J. Alzheimers Dis. Other Demen. 24, 445–449. doi: 10.1177/1533317509348208
Nakandakari, S. C. B. R., Munoz, V. R., Kuga, G. K., Gaspar, R. C., Sant’Ana, M. R., Pavan, I. C. B., et al. (2019). Short-term high-fat diet modulates several inflammatory, ER stress, and apoptosis markers in the hippocampus of young mice. Brain Behav. Immun. 79, 284–293.
Nam, G. E., Kim, S. M., Han, K., Kim, N. H., Chung, H. S., Kim, J. W., et al. (2018). Metabolic syndrome and risk of Parkinson disease: a nationwide cohort study. PLoS Med. 15:e1002640. doi: 10.1371/journal.pmed.1002640
Nandi, A., Kitamura, Y., Kahn, C. R., and Accili, D. (2004). Mouse models of insulin resistance. Physiol. Ver. 84, 623–647. doi: 10.1152/physrev.00032.2003
Narayan, S., Liew, Z., Bronstein, J. M., and Ritz, B. (2017). Occupational pesticide use and Parkinson’s disease in the Parkinson Environment Gene (PEG) study. Environ. Int. 107, 266–273. doi: 10.1016/j.envint.2017.04.010
Nguyen, J. C., Killcross, A. S., and Jenkins, T. A. (2014). Obesity and cognitive decline: role of inflammation and vascular changes. Front. Neurosci. 8:375. doi: 10.3389/fnins.2014.00375
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
Obeso, J. A., Stamelou, M., Goetz, C. G., Poewe, W., Lang, A. E., Weintraub, D., et al. (2017). Past, present, and future of Parkinson’s disease: a special essay on the 200th anniversary of the shaking palsy. Mov. Disord. 32, 1264–1310. doi: 10.1002/mds.27115
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Oddo, S., Caccamo, A., Green, K. N., Liang, K., Tran, L., Chen, Y., et al. (2005). Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 102, 3046–3051. doi: 10.1073/pnas.0408500102
Oddo, S., Caccamo, A., Kitazawa, M., Tseng, B. P., and LaFerla, F. M. (2003a). Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 24, 1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003b). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/s0896-6273(03)00434-3
Ohno, M. (2009). Failures to reconsolidate memory in a mouse model of Alzheimer’s disease. Neurobiol. Learn. Mem. 92, 455–459. doi: 10.1016/j.nlm.2009.05.001
Ozsoy, O., Yildirim, F. B., Ogut, E., Kaya, Y., Tanriover, G., Parlak, H., et al. (2015). Melatonin is protective against 6-hydroxydopamine-induced oxidative stress in a hemiparkinsonian rat model. Free Radic. Res. 49, 1004–1014. doi: 10.3109/10715762.2015.1027198
Parachikova, A., Vasilevko, V., Cribbs, D. H., LaFerla, F. M., and Green, K. N. (2010). Reductions in amyloid-beta-derived neuroinflammation, with minocycline, restore cognition but do not significantly affect tau hyperphosphorylation. J. Alzheimers. Dis. 21, 527–542. doi: 10.3233/JAD-2010-100204
Paul, R., and Borah, A. (2017). Global loss of acetylcholinesterase activity with mitochondrial complexes inhibition and inflammation in brain of hypercholesterolemic mice. Sci. Rep. 7:17922. doi: 10.1038/s41598-017-17911-z
Paul, R., Choudhury, A., Chandra Boruah, D., Devi, R., Bhattacharya, P., Choudhury, M. D., et al. (2017a). Hypercholesterolemia causes psychomotor abnormalities in mice and alterations in cortico-striatal biogenic amine neurotransmitters: relevance to Parkinson’s disease. Neurochem. Int. 108, 15–26. doi: 10.1016/j.neuint.2017.01.021
Paul, R., Choudhury, A., Kumar, S., Giri, A., Sandhir, R., and Borah, A. (2017b). Cholesterol contributes to dopamine-neuronal loss in MPTP mouse model of Parkinson’s disease: involvement of mitochondrial dysfunctions and oxidative stress. PLoS One 12:e0171285. doi: 10.1371/journal.pone.0171285
Paumier, K. L., Sukoff Rizzo, S. J., Berger, Z., Chen, Y., Gonzales, C., Kaftan, E., et al. (2013). Behavioral characterization of A53T mice reveals early and late stage deficits related to Parkinson’s disease. PLoS One 8:e70274. doi: 10.1371/journal.pone.0070274
Pearson-Leary, J., and McNay, E. C. (2012). Intrahippocampal administration of amyloid-beta(1-42) oligomers acutely impairs spatial working memory, insulin signaling, and hippocampal metabolism. J. Alzheimers. Dis. 30, 413–422. doi: 10.3233/JAD-2012-112192
Pegueroles, J., Jimenez, A., Vilaplana, E., Montal, V., Carmona-Iragui, M., Pane, A., et al. (2018). Obesity and Alzheimer’s disease, does the obesity paradox really exist? A magnetic resonance imaging study. Oncotarget 9, 34691–34698. doi: 10.18632/oncotarget.26162
Pereira, C., Moreira, P., Seica, R., Santos, M. S., and Oliveira, C. R. (2000). Susceptibility to beta-amyloid-induced toxicity is decreased in goto-kakizaki diabetic rats: involvement of oxidative stress. Exp. Neurol. 161, 383–391. doi: 10.1006/exnr.1999.7270
Piermartiri, T. C., Figueiredo, C. P., Rial, D., Duarte, F. S., Bezerra, S. C., Mancini, G., et al. (2010). Atorvastatin prevents hippocampal cell death, neuroinflammation and oxidative stress following amyloid-beta(1-40) administration in mice: evidence for dissociation between cognitive deficits and neuronal damage. Exp. Neurol. 226, 274–284. doi: 10.1016/j.expneurol.2010.08.030
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:17013. doi: 10.1038/nrdp.2017.13
Popa-Wagner, A., Dumitrascu, D. I., Capitanescu, B., Petcu, E. B., Surugiu, R., Fang, W. H., et al. (2020). Dietary habits, lifestyle factors and neurodegenerative diseases. Neural Regen. Res. 15, 394–400. doi: 10.4103/1673-5374.266045
Pouchieu, C., Piel, C., Carles, C., Gruber, A., Helmer, C., Tual, S., et al. (2018). Pesticide use in agriculture and Parkinson’s disease in the AGRICAN cohort study. Int. J. Epidemiol. 47, 299–310. doi: 10.1093/ije/dyx225
Prediger, R. D., Franco, J. L., Pandolfo, P., Medeiros, R., Duarte, F. S., Di Giunta, G., et al. (2007). Differential susceptibility following beta-amyloid peptide-(1-40) administration in C57BL/6 and Swiss albino mice: evidence for a dissociation between cognitive deficits and the glutathione system response. Behav. Brain Res. 177, 205–213. doi: 10.1016/j.bbr.2006.11.032
Puzzo, D., Gulisano, W., Palmeri, A., and Arancio, O. (2015). Rodent models for Alzheimer’s disease drug discovery. Expert Opin. Drug Discov. 10, 703–711. doi: 10.1517/17460441.2015.1041913
Rahman, S. M., Van Dam, A. M., Schultzberg, M., and Crisby, M. (2005). High cholesterol diet results in increased expression of interleukin-6 and caspase-1 in the brain of apolipoprotein E knockout and wild type mice. J. Neuroimmunol. 169, 59–67. doi: 10.1016/j.jneuroim.2005.07.018
Ransohoff, R. M. (2018). All (animal) models (of neurodegeneration) are wrong. Are they also useful? J. Exp. Med. 215, 2955–2958. doi: 10.1084/jem.20182042
Rees, D. A., and Alcolado, J. C. (2005). Animal models of diabetes mellitus. Diabet Med. 22, 359–370. doi: 10.1111/j.1464-5491.2005.01499.x
Refolo, L. M., Malester, B., LaFrancois, J., Bryant-Thomas, T., Wang, R., Tint, G. S., et al. (2000). Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 7, 321–331. doi: 10.1006/nbdi.2000.0304
Reitz, C., Tang, M. X., Manly, J., Schupf, N., Mayeux, R., and Luchsinger, J. A. (2008). Plasma lipid levels in the elderly are not associated with the risk of mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 25, 232–237. doi: 10.1159/000115847
Rodriguez, G. A., Tai, L. M., LaDu, M. J., and Rebeck, G. W. (2014). Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflammation 11:111. doi: 10.1186/1742-2094-11-111
Ruegsegger, G. N., Manjunatha, S., Summer, P., Gopala, S., Zabeilski, P., Dasari, S., et al. (2019). Insulin deficiency and intranasal insulin alter brain mitochondrial function: a potential factor for dementia in diabetes. FASEB J. 33, 4458–4472. doi: 10.1096/fj.201802043R
Salameh, T. S., Mortell, W. G., Logsdon, A. F., Butterfield, D. A., and Banks, W. A. (2019). Disruption of the hippocampal and hypothalamic blood-brain barrier in a diet-induced obese model of type II diabetes: prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate. Fluids Barriers CNS 16:1. doi: 10.1186/s12987-018-0121-6
Santos, C. Y., Snyder, P. J., Wu, W. C., Zhang, M., Echeverria, A., and Alber, J. (2017). Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: a review and synthesis. Alzheimers Dement. 7, 69–87. doi: 10.1016/j.dadm.2017.01.005
Santos, D. B., Peres, K. C., Ribeiro, R. P., Colle, D., dos Santos, A. A., Moreira, E. L., et al. (2012). Probucol, a lipid-lowering drug, prevents cognitive and hippocampal synaptic impairments induced by amyloid beta peptide in mice. Exp. Neurol. 233, 767–775. doi: 10.1016/j.expneurol.2011.11.036
Santos, R. D., Gidding, S. S., Hegele, R. A., Cuchel, M. A., Barter, P. J., Watts, G. F., et al. (2016). Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe familial hypercholesterolemia panel. Lancet Diabetes Endocrinol. 4, 850–861. doi: 10.1016/S2213-8587(16)30041-9
Saravia, F. E., Revsin, Y., Gonzalez Deniselle, M. C., Gonzalez, S. L., Roig, P., Lima, A., et al. (2002). Increased astrocyte reactivity in the hippocampus of murine models of type 1 diabetes: the nonobese diabetic (n.d.) and streptozotocin-treated mice. Brain Res. 957, 345–353. doi: 10.1016/s0006-8993(02)03675-2
Schliebs, R., and Arendt, T. (2011). The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 221, 555–563. doi: 10.1016/j.bbr.2010.11.058
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Shnerb Ganor, R., Harats, D., Schiby, G., Rosenblatt, K., Lubitz, I., Shaish, A., et al. (2018). Elderly apolipoprotein E/mice with advanced atherosclerotic lesions in the aorta do not develop Alzheimer’s disease-like pathologies. Mol. Med. Rep. 17, 2488–2492. doi: 10.3892/mmr.2017.8127
Shore, B., and Shore, V. (1974). An apolipoprotein preferentially enriched in cholesteryl ester-rich very low density lipoproteins. Biochem. Biophys. Res. Commun. 58, 1–7. doi: 10.1016/0006-291x(74)90882-1
Silva-Rodrigues, T., de-Souza-Ferreira, E., Machado, C. M., Cabral-Braga, B., Rodrigues-Ferreira, C., and Galina, A. (2020). Hyperglycemia in a type 1 Diabetes Mellitus model causes a shift in mitochondria coupled-glucose phosphorylation and redox metabolism in rat brain. Free Radic. Biol. Med. 160, 796–806. doi: 10.1016/j.freeradbiomed.2020.09.017
Sima, A. A., and Li, Z. G. (2005). The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetic rats. Diabetes Metab. Res. Rev. 54, 1497–1505. doi: 10.2337/diabetes.54.5.1497
Simola, N., Morelli, M., and Carta, A. R. (2007). The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 11, 151–167.
Simon, K. C., Chen, H., Schwarzschild, M., and Ascherio, A. (2007). Hypertension, hypercholesterolemia, diabetes, and risk of Parkinson disease. Neurology 69, 1688–1695. doi: 10.1212/01.wnl.0000271883.45010.8a
Singleton, A. B., Farrer, M. J., and Bonifati, V. (2013). The genetics of Parkinson’s disease: progress and therapeutic implications. Mov. Disord. 28, 14–23. doi: 10.1002/mds.25249
Soares, A. F., Nissen, J. D., Garcia-Serrano, A. M., Nussbaum, S. S., Waagepetersen, H. S., and Duarte, J. M. N. (2019). Glycogen metabolism is impaired in the brain of male type 2 diabetic Goto-Kakizaki rats. J. Neurosci. Res. 97, 1004–1017. doi: 10.1002/jnr.24437
Solomon, A., Kivipelto, M., Wolozin, B., Zhou, J., and Whitmer, R. A. (2009). Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement. Geriatr. Cogn. Disord. 28, 75–80. doi: 10.1159/000231980
Soto-Otero, R., Mendez-Alvarez, E., Hermida-Ameijeiras, A., Munoz-Patino, A. M., and Labandeira-Garcia, J. L. (2000). Autoxidation and neurotoxicity of 6-hydroxydopamine in the presence of some antioxidants: potential implication in relation to the pathogenesis of Parkinson’s disease. J. Neurochem. 74, 1605–1612. doi: 10.1046/j.1471-4159.2000.0741605.x
Soutar, A. K., and Naoumova, R. P. (2007). Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 4, 214–225. doi: 10.1038/ncpcardio0836
Sparks, D. L., Hunsaker, J. C. III, Scheff, S. W., Kryscio, R. J., Henson, J. L., and Markesbery, W. R. (1990). Cortical senile plaques in coronary artery disease, aging and Alzheimer’s disease. Neurobiol. Aging 11, 601–607. doi: 10.1016/0197-4580(90)90024-t
Sparks, D. L., Kuo, Y. M., Roher, A., Martin, T., and Lukas, R. J. (2000). Alterations of Alzheimer’s disease in the cholesterol-fed rabbit, including vascular inflammation. Preliminary observations. Ann. N. Y. Acad. Sci. 903, 335–344. doi: 10.1111/j.1749-6632.2000.tb06384.x
Sparks, D. L., Scheff, S. W., Hunsaker, J. C. III, Liu, H., Landers, T., and Gross, D. R. (1994). Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 126, 88–94. doi: 10.1006/exnr.1994.1044
Spencer, S. J., D’Angelo, H., Soch, A., Watkins, L. R., Maier, S. F., and Barrientos, R. M. (2017). High-fat diet and aging interact to produce neuroinflammation and impair hippocampal- and amygdalar-dependent memory. Neurobiol. Aging 58, 88–101. doi: 10.1016/j.neurobiolaging.2017.06.014
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Stanley, M., Macauley, S. L., and Holtzman, D. M. (2016). Changes in insulin and insulin signaling in Alzheimer’s disease: cause or consequence? J. Exp. Med. 213, 1375–1385. doi: 10.1084/jem.20160493
Steculorum, S. M., Solas, M., and Bruning, J. C. (2014). The paradox of neuronal insulin action and resistance in the development of aging-associated diseases. Alzheimers Dement. 10(1 Suppl.), S3–S11. doi: 10.1016/j.jalz.2013.12.008
Streit, W. J., and Sparks, D. L. (1997). Activation of microglia in the brains of humans with heart disease and hypercholesterolemic rabbits. J. Mol. Med. 75, 130–138. doi: 10.1007/s001090050097
Sturchler-Pierrat, C., Abramowski, D., Duke, M., Wiederhold, K. H., Mistl, C., Rothacher, S., et al. (1997). Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U.S.A. 94, 13287–13292. doi: 10.1073/pnas.94.24.13287
Takechi, R., Pallebage-Gamarallage, M. M., Lam, V., Giles, C., and Mamo, J. C. (2013). Aging-related changes in blood-brain barrier integrity and the effect of dietary fat. Neurodegener. Dis. 12, 125–135. doi: 10.1159/000343211
Thomas-Camardiel, M., Venero, J. L., Herrera, A. J., De Pablos, R. M., Pintor-Toro, J. A., Machado, A., et al. (2005). Blood-brain barrier disruption highly induces aquaporin-4 mRNA and protein in perivascular and parenchymal astrocytes: protective effect by estradiol treatment in ovariectomized animals. J. Neurosci. Res. 80, 235–246. doi: 10.1002/jnr.20443
Tieu, K. (2011). A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 1:a009316. doi: 10.1101/cshperspect.a009316
Tiwari, S., Atluri, V., Kaushik, A., Yndart, A., and Nair, M. (2019). Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int. J. Nanomedicine 14, 5541–5554. doi: 10.2147/IJN.S200490
Tomac, A., Lindqvist, E., Lin, L. F., Ogren, S. O., Young, D., Hoffer, B. J., et al. (1995). Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature 373, 335–339. doi: 10.1038/373335a0
Tzioras, M., Davies, C., Newman, A., Jackson, R., and Spires-Jones, T. (2019). Invited Review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 45, 327–346. doi: 10.1111/nan.12529
Ujiie, M., Dickstein, D. L., Carlow, D. A., and Jefferies, W. A. (2003). Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 10, 463–470. doi: 10.1038/sj.mn.7800212
Ullrich, C., Pirchl, M., and Humpel, C. (2010). Hypercholesterolemia in rats impairs the cholinergic system and leads to memory deficits. Mol. Cell. Neurosci. 45, 408–417. doi: 10.1016/j.mcn.2010.08.001
Underwood, E. L., and Thompson, L. T. (2016). A high-fat diet causes impairment in hippocampal memory and sex-dependent alterations in peripheral metabolism. Neural Plast. 2016:7385314. doi: 10.1155/2016/7385314
Ungerstedt, U. (1968). 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur. J. Pharmacol. 5, 107–110. doi: 10.1016/0014-2999(68)90164-7
Vagelatos, N. T., and Eslick, G. D. (2013). Type 2 diabetes as a risk factor for Alzheimer’s disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol. Rev. 35, 152–160. doi: 10.1093/epirev/mxs012
van Baak, M. A. (2013). Nutrition as a link between obesity and cardiovascular disease: how can we stop the obesity epidemic? Thromb. Haemost 110, 689–696. doi: 10.1160/TH13-01-0045
van de Sande-Lee, S., Melhorn, S. J., Rachid, B., Rodovalho, S., De-Lima-Junior, J. C., Campos, B. M., et al. (2020). Radiologic evidence that hypothalamic gliosis is improved after bariatric surgery in obese women with type 2 diabetes. Int. J. Obes. 44, 178–185. doi: 10.1038/s41366-019-0399-8
Vandal, M., White, P. J., Chevrier, G., Tremblay, C., St-Amour, I., Planel, E., et al. (2015). Age-dependent impairment of glucose tolerance in the 3xTg-AD mouse model of Alzheimer’s disease. FASEB J. 29, 4273–4284. doi: 10.1096/fj.14-268482
Verri, M., Pastoris, O., Dossena, M., Aquilani, R., Guerriero, F., Cuzzoni, G., et al. (2012). Mitochondrial alterations, oxidative stress and neuroinflammation in Alzheimer's disease. Int. J. Immunopathol. Pharmacol. 25, 345–353. doi: 10.1177/039463201202500204
von Wrangel, C., Schwabe, K., John, N., Krauss, J. K., and Alam, M. (2015). The rotenone-induced rat model of Parkinson’s disease: behavioral and electrophysiological findings. Behav. Brain Res. 279, 52–61. doi: 10.1016/j.bbr.2014.11.002
Walker, J. M., and Harrison, F. E. (2015). Shared neuropathological characteristics of obesity, type 2 diabetes and alzheimer’s disease: impacts on cognitive decline. Nutrients 7, 7332–7357. doi: 10.3390/nu7095341
Walsh, S., Finn, D. P., and Dowd, E. (2011). Time-course of nigrostriatal neurodegeneration and neuroinflammation in the 6-hydroxydopamine-induced axonal and terminal lesion models of Parkinson’s disease in the rat. Neuroscience 175, 251–261. doi: 10.1016/j.neuroscience.2010.12.005
Wang, Q., Liu, Y., and Zhou, J. (2015). Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 4:19. doi: 10.1186/s40035-015-0042-0
Wang, W. T., Lee, P., Yeh, H. W., Smirnova, I. V., and Choi, I. Y. (2012). Effects of acute and chronic hyperglycemia on the neurochemical profiles in the rat brain with streptozotocin-induced diabetes detected using in vivo 1H MR spectroscopy at 9.4 T. J. Neurochem. 121, 407–417. doi: 10.1111/j.1471-4159.2012.07698.x
Whitmer, R. A., Gunderson, E. P., Barrett-Connor, E., Quesenberry, C. P. Jr., and Yaffe, K. (2005). Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ 330:1360. doi: 10.1136/bmj.38446.466238.E0
Whitmer, R. A., Gustafson, D. R., Barrett-Connor, E., Haan, M. N., Gunderson, E. P., and Yaffe, K. (2008). Central obesity and increased risk of dementia more than three decades later. Neurology 71, 1057–1064. doi: 10.1212/01.wnl.0000306313.89165.ef
Wu, D. C., Jackson-Lewis, V., Vila, M., Tieu, K., Teismann, P., Vadseth, C., et al. (2002). Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 22, 1763–1771.
Yamada, K., Tanaka, T., Mamiya, T., Shiotani, T., Kameyama, T., and Nabeshima, T. (1999). Improvement by nefiracetam of beta-amyloid-(1-42)-induced learning and memory impairments in rats. Br. J. Pharmacol. 126, 235–244. doi: 10.1038/sj.bjp.0702309
Yamaguchi, Y., Miyashita, H., Tsunekawa, H., Mouri, A., Kim, H. C., Saito, K., et al. (2006). Effects of a novel cognitive enhancer, spiro[imidazo-[1,2-a]pyridine-3,2-indan]-2(3H)-one (ZSET1446), on learning impairments induced by amyloid-beta1-40 in the rat. J. Pharmacol. Exp. Ther. 317, 1079–1087. doi: 10.1124/jpet.105.098640
Yan, J. J., Cho, J. Y., Kim, H. S., Kim, K. L., Jung, J. S., Huh, S. O., et al. (2001). Protection against beta-amyloid peptide toxicity in vivo with long-term administration of ferulic acid. Br. J. Pharmacol. 133, 89–96. doi: 10.1038/sj.bjp.0704047
Yazdani, U., German, D. C., Liang, C. L., Manzino, L., Sonsalla, P. K., and Zeevalk, G. D. (2006). Rat model of Parkinson’s disease: chronic central delivery of 1-methyl-4-phenylpyridinium (MPP+). Exp. Neurol. 200, 172–183. doi: 10.1016/j.expneurol.2006.02.002
Yin, H., Wang, W., Yu, W., Li, J., Feng, N., and Wang, L. (2017). Changes in synaptic plasticity and glutamate receptors in type 2 diabetic KK-Ay mice. J. Alzheimers Dis. 57, 1207–1220. doi: 10.3233/JAD-160858
Yin, Y., and Wang, Z. (2018). ApoE and neurodegenerative diseases in aging. Adv. Exp. Med. Biol. 1086, 77–92. doi: 10.1007/978-981-13-1117-8_5
Yoshiyama, Y. (2008). Neurodegeneration and inflammation: analysis of a FTDP-17 model mouse. Rinsho Shinkeigaku 48, 910–902. doi: 10.5692/clinicalneurol.48.910
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S.-M., Iwata, N., Saido, T. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. doi: 10.1016/j.neuron.2007.01.010x
Youmans, K. L., Tai, L. M., Nwabuisi-Heath, E., Jungbauer, L., Kanekiyo, T., Gan, M., et al. (2012). APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem. 287, 41774–41786. doi: 10.1074/jbc.M112.407957
Zambon, D., Quintana, M., Mata, P., Alonso, R., Benavent, J., Cruz-Sanchez, F., et al. (2010). Higher incidence of mild cognitive impairment in familial hypercholesterolemia. Am. J. Med. 123, 267–274. doi: 10.1016/j.amjmed.2009.08.015
Zenaro, E., Piacentino, G., and Constantin, G. (2017). The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 107, 41–56. doi: 10.1016/j.nbd.2016.07.007
Zeng, X. S., Geng, W. S., Jia, J. J., Chen, L., and Zhang, P. P. (2018). Cellular and molecular basis of neurodegeneration in Parkinson disease. Front. Aging Neurosci. 10:109. doi: 10.3389/fnagi.2018.00109
Zhan, R., Zhao, M., Zhou, T., Chen, Y., Yu, W., Zhao, L., et al. (2018). Dapsone protects brain microvascular integrity from high-fat diet induced LDL oxidation. Cell Death Dis. 9, 1–15.
Zhang, S. H., Reddick, R. L., Piedrahita, J. A., and Maeda, N. (1992). Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 258, 468–471. doi: 10.1126/science.1411543
Zhang, X., Fu, Z., Meng, L., He, M., and Zhang, Z. (2018). The early events that initiate beta-amyloid aggregation in Alzheimer’s disease. Front. Aging Neurosci. 10:359. doi: 10.3389/fnagi.2018.00359
Zheng, Y., Yang, Y., Dong, B., Zheng, H., Lin, X., and Du, Y. (2016). Metabonomic profiles delineate potential role of glutamate-glutamine cycle in db/db mice with diabetes-associated cognitive decline. Mol. Brain 18, 9–40. doi: 10.1186/s13041-016-0223-5
Keywords: Alzheimer’s disease, Parkinson’s disease, hypercholesterolemia, obesity, diabetes, rodent models, neurodegeneration
Citation: de Bem AF, Krolow R, Farias HR, de Rezende VL, Gelain DP, Moreira JCF, Duarte JMN and de Oliveira J (2021) Animal Models of Metabolic Disorders in the Study of Neurodegenerative Diseases: An Overview. Front. Neurosci. 14:604150. doi: 10.3389/fnins.2020.604150
Received: 08 September 2020; Accepted: 24 December 2020;
Published: 18 January 2021.
Edited by:
Nicola B. Mercuri, University of Rome Tor Vergata, ItalyReviewed by:
António Francisco Ambrósio, University of Coimbra, PortugalMonica Renee Langley, Mayo Clinic, United States
Copyright © 2021 de Bem, Krolow, Farias, de Rezende, Gelain, Moreira, Duarte and de Oliveira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jade de Oliveira, ZGVvbGl2ZWlyYWphZGUxMEBnbWFpbC5jb20=; João Miguel das Neves Duarte, am9hby5kdWFydGVAbWVkLmx1LnNl