 Sabine M. Ulamec
Sabine M. Ulamec David J. Brockwell
David J. Brockwell Sheena E. Radford
Sheena E. Radford- Astbury Centre for Structural Molecular Biology, School of Molecular and Cellular Biology, Faculty of Biological Sciences, University of Leeds, Leeds, United Kingdom
Amyloid proteins are involved in many neurodegenerative disorders such as Alzheimer’s disease [Tau, Amyloid β (Aβ)], Parkinson’s disease [alpha-synuclein (αSyn)], and amyotrophic lateral sclerosis (TDP-43). Driven by the early observation of the presence of ordered structure within amyloid fibrils and the potential to develop inhibitors of their formation, a major goal of the amyloid field has been to elucidate the structure of the amyloid fold at atomic resolution. This has now been achieved for a wide variety of sequences using solid-state NMR, microcrystallography, X-ray fiber diffraction and cryo-electron microscopy. These studies, together with in silico methods able to predict aggregation-prone regions (APRs) in protein sequences, have provided a wealth of information about the ordered fibril cores that comprise the amyloid fold. Structural and kinetic analyses have also shown that amyloidogenic proteins often contain less well-ordered sequences outside of the amyloid core (termed here as flanking regions) that modulate function, toxicity and/or aggregation rates. These flanking regions, which often form a dynamically disordered “fuzzy coat” around the fibril core, have been shown to play key parts in the physiological roles of functional amyloids, including the binding of RNA and in phase separation. They are also the mediators of chaperone binding and membrane binding/disruption in toxic amyloid assemblies. Here, we review the role of flanking regions in different proteins spanning both functional amyloid and amyloid in disease, in the context of their role in aggregation, toxicity and cellular (dys)function. Understanding the properties of these regions could provide new opportunities to target disease-related aggregation without disturbing critical biological functions.
Introduction
In the 60 years since the first atomic structure of the protein myoglobin was solved using X-ray diffraction of protein crystals (Kendrew et al., 1960), the field of structural biology has been dominated by the study of globular proteins with a well-defined tertiary structure, with more than 160,000 unique structures solved to date using crystallography, NMR or cryoEM (Geraets et al., 2020). Despite this feat, more than 50% of the proteins in eukaryotes are now known to have at least one long (>30 residues) sequence that is intrinsically disordered [intrinsically disordered regions (IDRs)] and also 12% of eukaryotic proteins are completely intrinsically disordered [intrinsically disordered proteins (IDPs)] (Dunker et al., 2000; Ward et al., 2004; Tompa, 2009). IDRs and IDPs are enriched in/for proteins with important regulatory or signaling functions (Ward et al., 2004; Tompa et al., 2006; Coletta et al., 2010) demonstrating the crucial role of non-globular protein structures for biological processes.
Amyloid proteins, some of which are involved in neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington disease (Iadanza et al., 2018a; Benson et al., in press) are commonly IDPs or are proteins that contain IDRs. These diseases share a common fundamental etiology: aberrant self-assembly of their amyloid precursor proteins to form toxic oligomers and highly ordered fibrils with a cross β-sheet structure (Gallardo et al., 2020b; Ulamec and Radford, 2020). A combination of biochemical and biophysical approaches, including limited proteolysis, hydrogen exchange (HX), solution NMR, solid state NMR (ssNMR), cryoEM and EPR have shown that the structured cross-β amyloid core commonly involves only a portion of the amyloid precursor sequence, whilst regions flanking the fibril core (commonly the N- and/or C-terminal regions of the sequence), are flexible and thus are either “invisible” in the structures determined or give rise to only low resolution density in EM images (Gallardo et al., 2020b). Consequently, the high resolution cryoEM and ssNMR structures of fibril architectures determined over the last ∼5 years, have necessarily focused on the conformations and interactions within fibril cores (Gallardo et al., 2020b), whilst the more dynamic flanking regions remain elusive. The structured cores of amyloid fibrils usually contain short peptide sequences, some with high aggregation propensity, shown to be necessary and sufficient for fibril formation (Giasson et al., 2001; Thompson et al., 2006) and reviewed in Eisenberg and Sawaya (2017). Despite the importance of these sequences for the formation and stability of the amyloid fold, several studies have shown that modifying or changing (e.g., by deletion, mutation, or post-translational modification) the regions that flank the amyloid core can affect the fibril growth kinetics (Johnson et al., 2009; Benilova et al., 2014; Doherty et al., 2020), fibril morphology (Guerrero-Ferreira et al., 2020; Scheres et al., 2020) and the formation of crucial contacts with interaction partners (Gao et al., 2015; Burmann et al., 2020; Figure 1). Additionally, the presence of so called “gatekeeper” residues, which surround aggregation-prone regions (APRs) to mitigate their aggregation propensity (Reumers et al., 2009) further underlines the important role of residues not directly involved in forming the amyloid core in the process of aggregation and in the functional consequences of the fibril structure formed. The aim of this review is to highlight the importance of the dynamically disordered flanking regions in amyloid sequences, focusing on their roles in fibril formation, cytotoxicity, and other physiological functions. While more than 50 proteins are known to form amyloid associated with disease (Iadanza et al., 2018a; Benson et al., in press), here we focus on αSyn, TDP-43, Aβ, Tau, β2m, Orb2, and PrP as exemplars of IDPs, natively folded proteins, prions, and functional and disease related proteins.
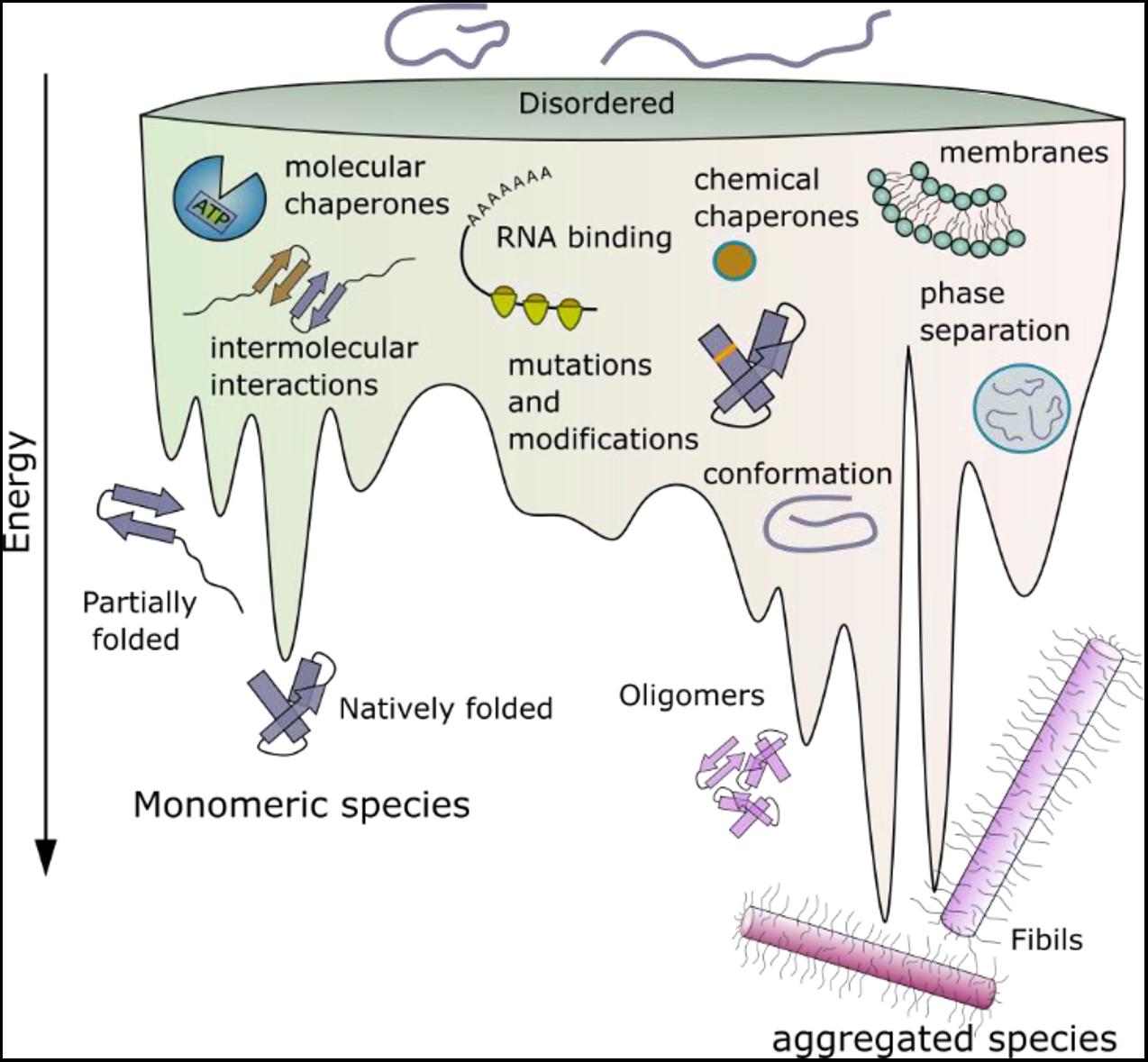
Figure 1. Schematic energy landscape of folding and aggregation of proteins focusing on factors affecting the kinetics of folding and amyloid assembly. The surface shows the conformations that proteins can adopt (monomeric and aggregated species) during folding and aggregation into amyloid. The lowest energy state is the amyloid fibril (Baldwin et al., 2011). Adopting a folded native structure or stabilization of this structure by ligand binding, disfavors amyloid formation, whilst mutations or post-translational modifications that favor inter-molecular interactions and phase separation can destabilize the monomer and tip the balance toward fibril formation. Binding of a protein to a membrane surface can also favor aggregation, by changing the structure of the amyloid precursor and driving inter-molecular interactions. Note that the relative position of each folding- and aggregation-factor within the energy landscape is arbitrary, as these factors can influence folding and aggregation at multiple different points. Their relative position, therefore, does not imply that they act at only one point on the landscape, or interact with a specific folding or aggregation intermediate.
Flanking Regions – What Are They and Why Are They Important?
Decades of studies of protein folding have led to insights into the roles of individual amino acids in a protein sequence in the search for the native fold (they are kinetically important) and in stabilizing the final folded state (they are thermodynamically important) (Rumbley et al., 2001; Baldwin et al., 2011). In addition, some residues may be conserved because they are important in chaperone binding or in the destabilization of incorrect folds (so-called negative design) (Richardson and Richardson, 2002; Berezovsky et al., 2007). By contrast with our the wealth of knowledge about protein folding, predicting which residues in an amyloid precursor sequence could be kinetically important in driving or controlling the rate or mechanism of amyloid fibril formation; which form the stable fibril core; and which may be innocuous passengers during self-assembly, but may play a role in the fibrillar state, is immensely complex. This is a fundamentally important question, since it is now widely appreciated that, by contrast with protein folding wherein the same globular structure is (usually) adopted by a protein sequence independent of mutation or changes in the solution conditions, amyloid fibril formation is under kinetic control, with the structure of the fibril product being determined by the assembly pathway taken (Figure 1). The result is a rugged energy landscape, which can result in potentially many different fibril structures for the same (or very similar) protein sequences (known as fibril polymorphism) (Fitzpatrick et al., 2017; Close et al., 2018; Kollmer et al., 2019; Guerrero-Ferreira et al., 2020). Recent advances in computational methods have provided a suite of algorithms able to define the most aggregation prone region of a protein sequence (Fernandez-Escamilla et al., 2004; Sormanni et al., 2015; Ebo et al., 2020; Figure 2A). These regions are commonly found in the amyloid fibril core, stabilizing the final fibril structure, without necessarily playing a role in the kinetics of amyloid formation. Other residues in amyloidogenic peptides and proteins which flank the APRs, can play kinetic and/or thermodynamic roles in amyloid assembly. Changes in these sequences (mutation or post-translational modification) can dramatically alter the rates of fibril formation and the structures of the fibrils formed, without the sequence necessarily forming part of the stable fibril core. As more fibril structures are solved of full-length protein sequences (rather than short peptide fragments), it is becoming clear that the amyloid core can involve only a minor part of the protein sequence, with substantial regions of the polypeptide chain remaining dynamically disordered in the fibril structure (Figure 2A). Such regions which flank the amyloid core can play functional roles (binding ligands and receptors for functional amyloid), or perturbing proteostasis (for amyloid fibrils associated with disease) (Figures 2B–D).
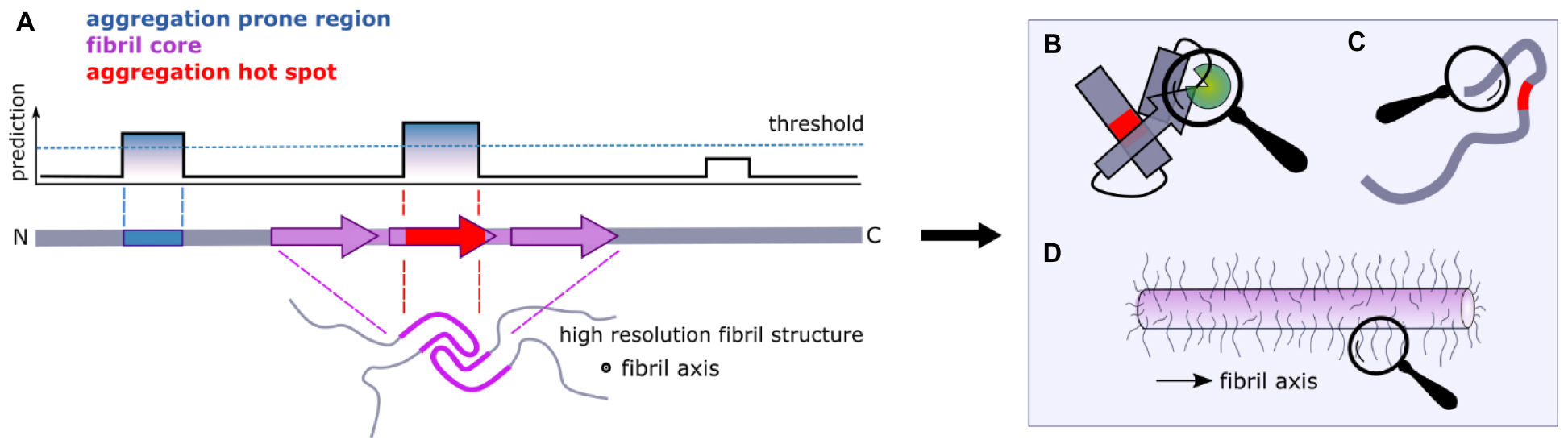
Figure 2. Flanking regions and amyloid formation. (A) Computational approaches can be used to identify aggregation-prone regions (APRs) from sequence data alone (blue). Fibril cores are defined by cryoEM and ssNMR structures of amyloid fibrils and include residues that adopt a stable cross-β structure (purple). When the APRs and the fibril core overlap, we term the region as an “aggregation hotspot” (red). Large segments of a protein sequence can remain dynamically disordered in amyloid structures (lower image). (B) Interaction partners and/or (C) the formation of transient non-local intra-molecular interactions that involve regions that flank the APRs can have significant effects on the aggregation kinetics of amyloidogenic proteins, as well as on their function. (D) Dynamically disordered regions flanking the amyloid core in the fibrillar state form an amyloid “fuzzy coat” that can impart biological functions to the amyloid fold.
In this review we focus on the roles of regions that flank the APRs and/or fibril cores in amyloid formation. We differentiate between (i) sequences that flank the APRs determined by in silico techniques, (ii) sequences that flank the structured cross-β fibril core based on recent cryoEM and NMR structures of fibrils, and (iii) sequences that flank coincident APRs and fibril cores (producing an “aggregation hotspot”) (Figure 2). In 2005 used a similar approach where they differentiated between regions that are predicted to be most important for promoting amyloid growth and experimentally determined sensitive regions Dobson and coworkers (Pawar et al., 2005). It is important to note that different flanking regions can be identified in the same protein using these definitions. For example, the use of different algorithms [e.g., CamSol or TANGO (Figure 3)] as well as posttranslational modifications, might result in different flanking regions for the same protein primary sequence. Similarly, on a structural level, as polymorphs for the same protein have different residues involved in their cores, each will have a different flanking region (Figure 3).
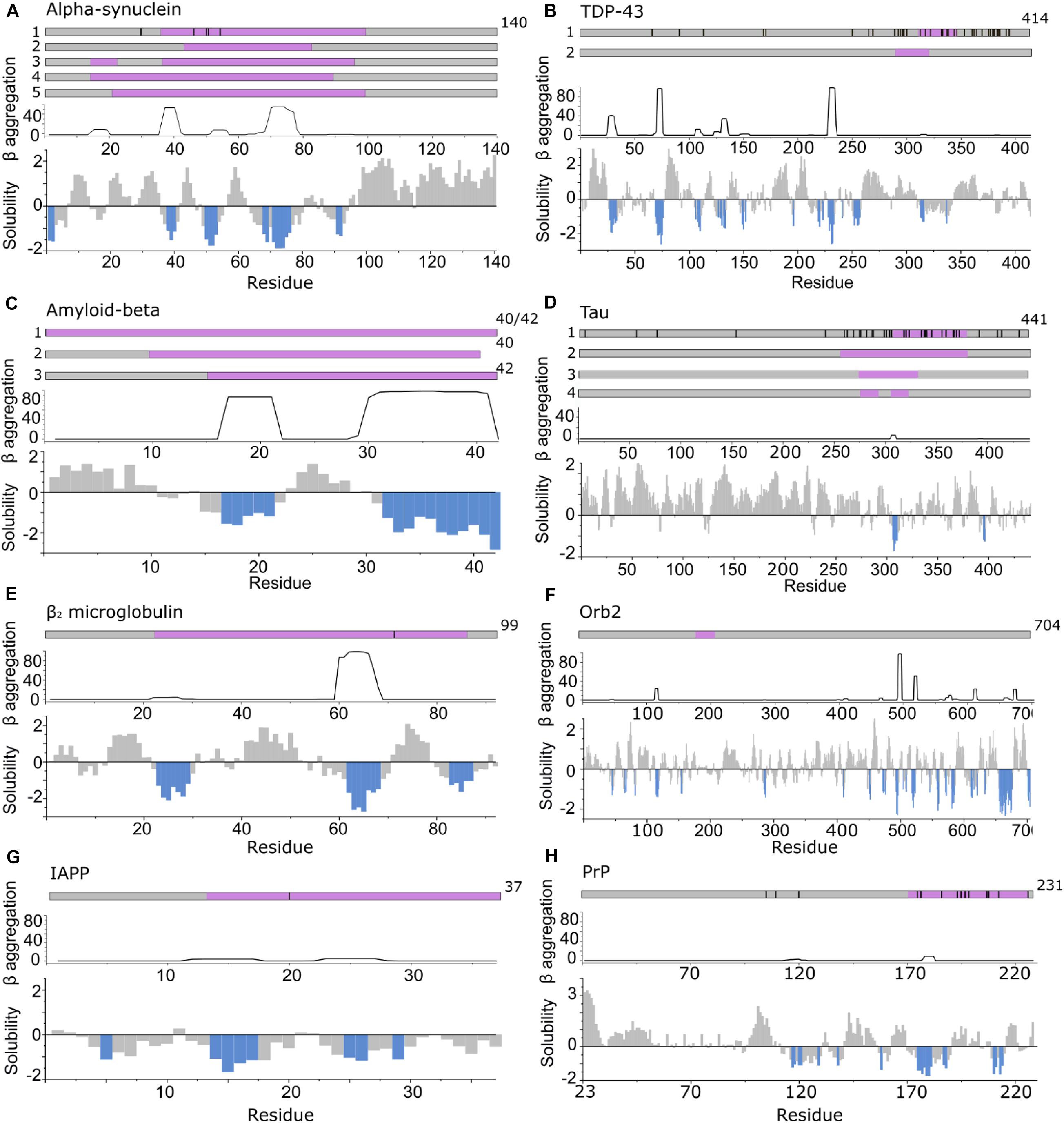
Figure 3. APRs comprise only a small part of the amyloid core. Top in A–H: location of fibril cores of αSyn, TDP-43, Aβ, Tau, β2m, Orb2B, IAPP, and PrP defined by recent cryoEM or ssNMR fibril structures (purple). The positions of familial disease mutations are highlighted where appropriate as black lines. Bottom in A–H: regions with low solubility predicted by CamSol (below –1 is aggregation promoting highlighted in blue) (Sormanni et al., 2015) and the β-aggregation potential of each sequence predicted using TANGO (Fernandez-Escamilla et al., 2004). (A) αSyn including polymorph 1a (1) (Guerrero-Ferreira et al., 2018; Li B. et al., 2018; Li Y. et al., 2018) (core residues 37–99), 1b (2) (Li B. et al., 2018) (core residues 43–83), 2a and b (3) (Guerrero-Ferreira et al., 2019) (core residues 14–24, 36–96) and the MSA ex vivo structures including residues 14–94 (for PF-IA and PF-IIA) (4) or residues 21–99 (for PF-IB and PF-IIB) (5) in the fibril core (Schweighauser et al., 2020). (B) TDP-43 cryoEM structure solved from C-terminal segments forming a dagger shaped core (1) (residues 312–346) or R-shaped core (2) (residues 288–319) (Cao et al., 2019). (C) Aβ structures solved (1) for Aβ42 (Gremer et al., 2017) and Aβ40 in which all residues comprise the core (Lu et al., 2013; Kollmer et al., 2019) (2) fibrils in which the core is formed by residues 10–40 for Aβ40 (including polymorphs 2A and 3Q) (Petkova et al., 2002; Paravastu et al., 2008) and (3) for Aβ42 (core formed by residues 15–42) (Colvin et al., 2016; Wälti et al., 2016). (D) Tau fibril structures PHF and SF from Alzheimer disease patients (1) (Fitzpatrick et al., 2017) (core residues 306–378), NPF and WPF from Pick’s Disease (2) (Falcon et al., 2018) (core residues 254–378) and heparin induced structures 4R-s and 3R formed in vitro (3) (core residues 272–330) and 4R-t and 4R-j (4) (core residues 274–292, 304–321, respectively) (Zhang et al., 2019). (E) The β2m fibril core involves residues 22–85 (Iadanza et al., 2018b; Gallardo et al., 2020a). (F) The Orb2B fibril core consists of residues 176–206 (Hervas et al., 2020). (G) Human IAPP forms fibrils with residues 13–37 (Röder et al., 2020), 14–37 (Cao et al., 2020), or 13–37 (Gallardo et al., 2020a), with its early onset S20G variant adopting fibrils with two- and three filaments involving residues 15–37 in the core (Gallardo et al., 2020a). (H) PrP fibrils form fibril core with residues 170–229 revealed using cryoEM (Wang et al., 2020).
Identification of Sequences Involved in Forming Amyloid
Prior to the development of high-resolution structural methods capable of solving amyloid structures in atomic detail, lower resolution techniques were employed to provide information about the sequences that drive aggregation. These include analysis of the ability of arrays of peptide fragments from different amyloid precursor sequences to form cross-β amyloid-like structures in isolation (Tenidis et al., 2000; von Bergen et al., 2000; Jones et al., 2003a; Ivanova et al., 2004; Nelson et al., 2005), scanning mutagenesis of a sequence followed by analysis of fibril formation using aggregation assays (e.g., Thioflavin T fluorescence) (Williams et al., 2006; Platt et al., 2008), and determination of the sequences that form the stable amyloid core, e.g., using protease digestion followed by mass spectrometry (Miake et al., 2002; Myers et al., 2006; Kushnirov et al., 2020) or HX monitored by 1H-NMR (Hoshino et al., 2002; Cho et al., 2011; Strohäker et al., 2019). These experimental approaches have been complemented by the development of in silico tools (Ebo et al., 2020; Santos et al., 2020), able to identify APRs by calculation of β-sheet propensity (using TANGO) (Fernandez-Escamilla et al., 2004) or solubility (CamSol) (Sormanni et al., 2015) (see Santos et al., 2020 for a recent review of these and other approaches). Most, but not all APRs are found in the fibril core. Residues that are found in fibril cores determined by cryoEM or ssNMR and in the APRs identified using computational methods described above are here defined as “aggregation hotspots” (Figure 3). They often contain motifs crucial for fibril formation [e.g., the Non-Amyloid β-Component (NAC) region of αSyn, the 22NFGAIL27 sequence from human islet amyloid polypeptide (IAPP), 15KLVFF20 for Aβ and 306VQIVYK311 from Tau].
Huge strides have been made in amyloid fibril structure elucidation in the last decade using X-ray diffraction of microcrystals (usually of short, 6–15 residue peptides) (Rodriguez et al., 2015; Guenther et al., 2018), and, more recently, using ssNMR and cryoEM of amyloid fibrils formed in vitro and ex vivo from full-length proteins. These studies have shown that the same sequence can produce fibrils with remarkably different quaternary, tertiary and even secondary structural elements (Gallardo et al., 2020b). For example, more than six different amyloid fibril structures of the 140 residue protein, α-synuclein (αSyn) have been solved to date using cryoEM or ssNMR (Table 1; Guerrero-Ferreira et al., 2020; Schweighauser et al., 2020). Notably, while the fibril cores in all of these structures contain the NAC region known to be necessary and sufficient for amyloid formation (Giasson et al., 2001), the length and location of the sequence involved in the remaining portions of structured amyloid core (or conversely the residues involved in unresolved, dynamically disordered regions) varies depending on the morphology of the amyloid fibril formed (Figure 3; Guerrero-Ferreira et al., 2019, 2020; Schweighauser et al., 2020). Thus, between 50% and 70% of the 140 residues of this protein are not involved in the cross-β amyloid core. A second striking example is the two fibril structures formed from antibody light chains (LCs) that were extracted from two patients with systemic LC amyloidosis (involving different LC sequences). These studies revealed that these proteins with the same initial immunoglobulin (Ig)-containing native structure (Huang et al., 1996; Swuec et al., 2019), form completely different amyloid fibril architectures, with residues 16–23 and 86–93 (Radamaker et al., 2019), or residues 1–37 and 66–105 forming the core (Swuec et al., 2019).
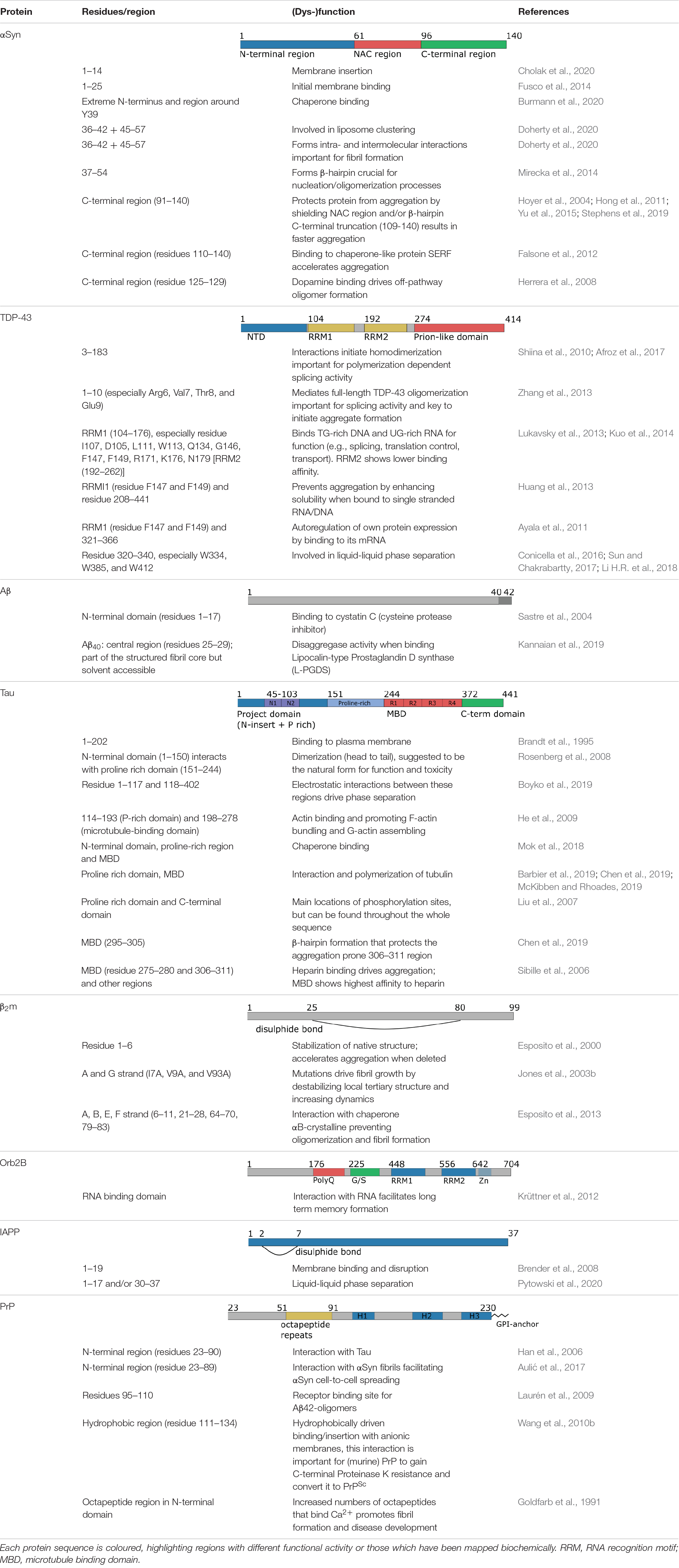
Table 1. Summary of the functional roles of different regions of the amyloid proteins shown in Figure 3.
In general, in silico-identified APRs (which may differ slightly when using different algorithms) are shorter in length compared with the experimentally determined fibril core using cryoEM or ssNMR (Figure 3). It should be noted that in silico methods also identify APRs that can reside outside the structured amyloid core, and so form the flanking region of aggregation hotspots (Figure 2A). A particularly striking example of this is seen in the functional amyloid Orb2, associated with long term memory formation and storage (Keleman et al., 2007; Majumdar et al., 2012; Khan et al., 2015; Hervas et al., 2020), where there is no overlap of sequence between residues in the experimentally derived fibril core and those in APRs identified using in silico methods (Figure 3F). This clearly demonstrates the division of sequence motifs into those which may be kinetically important, those which are thermodynamically important, and some which play neither or both roles.
Similar to the diversity of amyloid flanking regions, it can also be difficult to parse structured and unstructured regions. For the functional amyloid protein Sup35, for example, which is involved in controlling translation in yeast (Lyke et al., 2019), site-directed mutagenesis and fluorescence labeling suggested that the amyloid core comprises amino acids 21–121, flanked by a structurally heterogeneous “transition zone” (residues 1–20 and 122–158), and a very flexible C-terminal region formed by residues 159–250 (Krishnan and Lindquist, 2005). Another example of different residual mobility of flanking regions was seen by performing immunogold labeling and transmission EM, force-volume measurements using atomic force microscopy (AFM) and solution NMR experiments on Tau filaments (Sillen et al., 2005; Wegmann et al., 2013). These experiments suggested that the flanking regions of Tau form a “two layered polyelectrolyte brush” surrounding the fibril core: a dense and mechanically more rigid layer (residues ∼173–243) and an N-terminal, less dense and more dynamic layer (residues ∼1–172). The increased exposure to solvent renders longer unstructured regions, such as those in Tau and αSyn sensitive to environmental conditions: Tau amyloid fibrils exhibit a 50% reduction in the rigidity of flanking regions upon increasing KCl concentration from 50 mM to 200 mM and show increased affinity (70% higher adhesion) to negatively charged membranes when lowering the pH from 7.4 to 4.5 (Wegmann et al., 2013). These changes may be relevant during lysosomal degradation (Wang et al., 2009). Finally, while not part of the structured β-sheet rich fibril core, some sequences in the flanking regions can nonetheless form secondary structure as shown using ssNMR for the 17 amino acid long N-terminal region (residues 4–11) of exon 1 of huntingtin. In this case, the region flanking the aggregation-prone expanded polyQ region adopts a solvent exposed and dynamic α-helical structure (Sivanandam et al., 2011).
Role of Flanking Regions and Aggregation Hotspots in Fibril Formation
Flanking regions have been shown to play roles in modulating the rates and mechanisms of aggregation (Benilova et al., 2014; Doherty et al., 2020). For example, point mutations linked with early onset familial Parkinson’s disease (Mehra et al., 2019) and ALS (Prasad et al., 2019) or post-translational modifications (including phosphorylation, acetylation, sumoylation, methylation, ubiquitination, glycosylation, and truncations that alter aggregation kinetics) are often remote from the APRs within these protein sequences (Figures 3A,B). These regions also bind to other molecules such as chaperones, nucleic acids, and membranes, and hence, can play functionally important roles (Table 1). Consequently, these regions re-sculpt the aggregation energy landscape (Figure 1) enhancing [or in some cases suppressing (Jonsson et al., 2012)] aggregation and its associated cytotoxicity, and can alter the function of the native amyloid precursor. It is thus crucial to analyze these regions when assessing amyloid formation mechanisms, rather than focusing solely on short peptide sequences that constitute the APRs. The latter approach, however, can be incredibly fruitful, leading to novel anti-microbial agents (Khodaparast et al., 2018) and potential cancer treatments (Gallardo et al., 2016). The latter study showed that a de novo designed peptide, vascin, based on an amyloidogenic fragment of vascular endothelial growth factor receptor 2 (VEGFR2) knocked down VEGFR2 as a consequence of VEGFR2-dependent fibril growth (Gallardo et al., 2016). Importantly, aggregation kinetics can be either slowed or accelerated depending on the protein and/or the precise modification of the flanking sequences. This is particularly apposite for IDPs, as the shallow, but rough, energy landscapes of these proteins renders their conformational ensemble sensitive to changes in their sequence and environment. This can result in APRs or binding motifs being exposed or sequestered, which in turn can accelerate, slow or prevent aggregation for the same protein sequence relative to a reference condition. Changes in pH, ionic strength and even being in a different cellular environment (e.g., oligodendrocytes vs neurons) can thus have a significant effect on fibril formation as shown for αSyn (Peng et al., 2018; Stephens et al., 2019). Indeed, far from being passive bystanders, flanking regions may be as important in defining the physiological role and amyloid disease etiology as the canonical APRs themselves (Figure 4). Here we highlight examples where flanking regions are involved in promoting or disfavoring aggregation to draw an overview of the significance of sequence and interaction partners of amyloidogenic proteins and peptides for fibril formation.
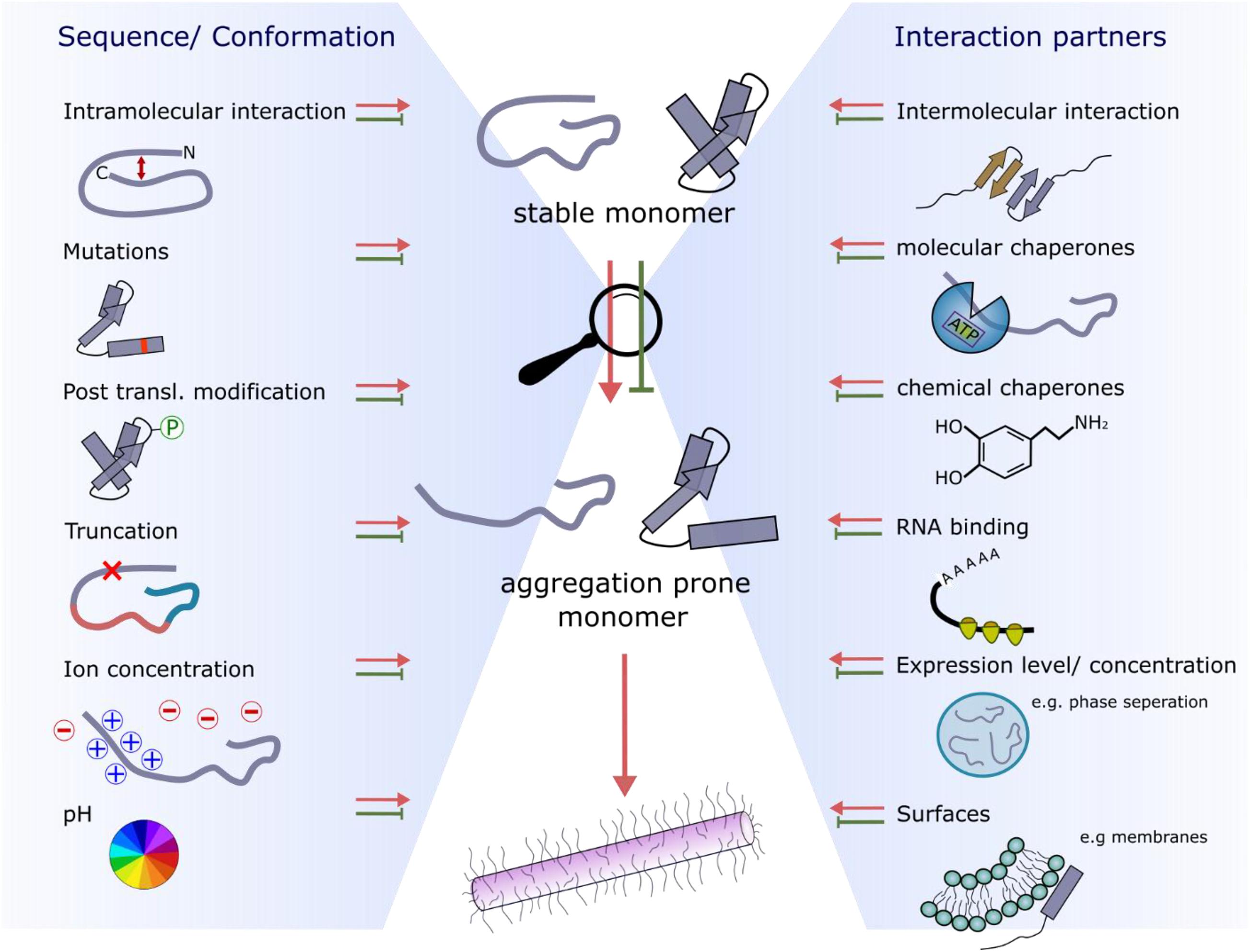
Figure 4. Factors affecting the conformational properties and interactions with amyloid precursors that retard or accelerate fibril growth. The two main influences are sequence/conformation-dependent (left-hand side) or occur as a result of binding to interaction partners (right-hand side).
Flanking Regions That Protect Against Aggregation
A protein concentration higher than the critical amyloid concentration (Xue et al., 2008), and (sometimes) the presence of surfaces that can act as nucleation sites are required to overcome the energy barrier needed to form amyloid structures (Buell, 2017). Such surfaces include membranes, other proteins, or the air-water interface [so-called heterogeneous primary nucleation (Buell, 2017; Figure 4)] or the amyloid fibril surface itself (secondary nucleation) (Linse, 2017). In the crowded cell, there is a fine-balance of expressing sufficient protein to maintain function, but avoiding high expression levels to disfavor aggregation. This strategy was described as “life on the edge” by Dobson and co-workers (Tartaglia et al., 2007). Interestingly, more recent work has shown that regions surrounding an aggregation hotspot can play a role in maintaining the correct balance between expression level and aggregation potential, such that aggregation is disfavored, whilst the function of the native protein is maintained. A particularly clear example is the TAR DNA-binding protein 43 (TDP-43). The formation of a negative feedback loop involving the RNA-recognition motif 1 (RRM1, especially residues F147 and F149) and the fibril core-forming C-terminal region [residues 321–366 (Figure 3B)] self-regulates protein expression via a mechanism in which TDP-43 binds its own mRNA leading to decreased protein expression (Ayala et al., 2011; Table 1). Single stranded RNA (ssRNA) or DNA (ssDNA) binding to the flanking regions of TDP-43 also functions as a protective mechanism against amyloid formation by increasing the solubility of TDP-43 (Huang et al., 2013). In addition to translational control, the flanking regions of TDP-43 have also been found to affect its aggregation kinetics directly. For example, cells overexpressing Δ1–10 TDP-43 do not form cellular inclusions, by contrast with cells producing the full-length protein (Zhang et al., 2013). Another example where flanking regions protect from aggregation is the homo-tetrameric protein transthyretin (TTR), which is expressed in the liver and cerebrospinal fluid and is associated with amyloidosis. For this protein, the gatekeeper residue K35 [located just at the edge of the fibril core of a recently solved cryoEM structure (Schmidt et al., 2019)] protects the full-length protein from fibril formation (Sant’Anna et al., 2014).
Flanking regions can also limit fibril formation by binding to chaperones, which stabilize and protect the native state of the protein (Wentink et al., 2019; Figure 4). Using in vitro and in cell NMR Burmann et al. (2020) recently showed that αSyn is bound to an array of molecular chaperones. Some of these are physiologically relevant (e.g., hsc70, hsp90) whereas others are not (e.g., the periplasmic bacterial chaperones Skp and SurA). Similarly, the ATP-independent nascent polypeptide associated complex (NAC) has been shown to bind to the flanking regions of αSyn and ataxin 3 and retard their aggregation (Martin et al., 2018; Shen et al., 2019). Despite their diversity, all six chaperones in the study by Burmann et al. (2020) were found to bind the N-terminal 10 residues of αSyn and a segment around residue Y39 (Table 1). Inhibition of chaperones Hsc70 and Hsp90 in mammalian cells resulted in the re-localization of αSyn from the cytosol to the mitochondria, amplified fibril growth and increased membrane binding, which has been shown to be the toxic form of αSyn in yeast (Newberry et al., 2020). Interestingly, phosphorylation of Y39, which is associated with αSyn-dependent neurodegeneration (Brahmachari et al., 2016), decreased the interaction with chaperones. Enhancing the concentration of various heat shock proteins thus disfavors αSyn aggregation, potentially providing a therapeutic approach to treat αSyn-aggregation and its associated cytotoxicity (Jones et al., 2014). Similar aggregation-suppressive effects have been reported for other chaperone:protein pairs: β2-microglobulin (β2m) interacts with αB-crystallin mainly via its flanking regions [residues 6–11, 21–28, 9–83 (Figure 3E and Table 1)] and some residues within the APR (residues 64–70) (Esposito et al., 2013); the N-terminal domain of Tau (residues ∼1–40) (as well as the proline-rich region and repetition motifs [residues ∼160–370 (Figure 3D and Table 1)] binds to the chaperone DnaJA2 (Mok et al., 2018) and the N-terminal 17 residue flanking region of the huntingtin protein forms a complex with the molecular chaperones Hsc70 (Monsellier et al., 2015) or TriC (Tam et al., 2009) reducing its aggregation. This might be explained by the fact that chaperones bind to flanking regions preventing the formation of crucial conformations/interactions required for amyloid formation. For instance, the exon 1 domain of huntingtin has been shown to initiate aggregation by dimerization of its N-terminal 17 residues (Kelley et al., 2009) which is inhibited by chaperone binding to this sequence (Tam et al., 2009; Monsellier et al., 2015).
By contrast with the chaperones discussed above, other chaperone-like proteins have been shown to accelerate aggregation. For example, the binding of the human protein SERF (or MOAG-4, the SERF homolog in Caenorhabditis elegans) has been shown to accelerate aggregation of polyglutamine peptides, huntingtin, Aβ and αSyn in vitro and in vivo (van Ham et al., 2010; Falsone et al., 2012; Meinen et al., 2019). Although the binding site on SERF for all of these amyloidogenic proteins has yet to be identified, αSyn was shown to bind via its C-terminal region (residues 110–140) (Falsone et al., 2012). In other cases, binding of small molecules [chemical chaperones (Figure 4)] has been shown to accelerate or to retard the rate of aggregation and/or the morphology of the fibrils formed. For example, the glycosaminoglycan heparin, has been shown to induce the aggregation of Tau, resulting in fibrils with a different structure from Tau fibrils extracted from the brains of patients with Alzheimer’s, Pick’s or other Tau-associated neurodegenerative disorders (Zhang et al., 2019; Scheres et al., 2020; Figure 3D). NMR studies revealed multiple binding sites on Tau for heparin, with binding affinities ranging from 10 μM to the mM range. The tightest binding is observed within the microtubule binding domain (MBD) (residues 275–280 or 306–311), again highlighting that binding outside the APR modulates aggregation (Sibille et al., 2006). As a final example, the small molecule dopamine binds the C-terminal region of αSyn (residues 125–129), driving off-pathway oligomer formation that does not result in fibril growth (Herrera et al., 2008).
Flanking Regions That Accelerate Aggregation
As several amyloidogenic proteins are IDPs, transient intra- or inter-molecular interactions mediated by flanking regions can play an important role in defining the overall aggregation propensity of a protein sequence by altering the solvent accessibility of key APRs (Figure 4). For example, paramagnetic relaxation enhancement (PRE) NMR and computational modeling experiments revealed that the flexible negatively charged C-terminal region of αSyn (residues 96–140) forms intra- and inter-molecular interactions with the positively charged N-terminal region of the protein, which protect the aggregation-prone NAC region (Hong et al., 2011; Janowska et al., 2015; Yu et al., 2015; Stephens et al., 2019; Table 1). Conditions that disfavor these transient interactions [e.g., high cation concentration (Nath et al., 2011), low pH (Hoyer et al., 2002) or familial Parkinson’s disease mutations (Ranjan and Kumar, 2017)] result in perturbation of the protective long-range contacts and accelerate aggregation (reviewed by Stephens et al., 2019). Likewise, truncation of the protective C-terminal region of αSyn, as found in Lewy bodies in disease-associated brains (Muntané et al., 2012), causes more rapid fibril formation, potentially rationalizing the role of truncation of these regions in the development of disease (Hoyer et al., 2004). Similarly, for TTR the C and D strands (not part of the aggregation hotspot) are involved in the interchain contacts that lead to aggregation (Kelly and Lansbury, 1994; Palaninathan et al., 2008). Also, many disease associated mutations of TTR (e.g., V30M and L55P) are located within the C and D strand region (Murakami et al., 1992; Lashuel et al., 1999).
More recently, studies analyzing the aggregation kinetics of N-terminal deletion variants of αSyn have shown that specific regions of the protein are also required for the aggregation of full-length αSyn, building on an array of previous data that suggested an importance of the N-terminal region of the protein for its function (membrane binding) (Fusco et al., 2016; Cholak et al., 2020) and its aggregation (Kessler et al., 2003; Terada et al., 2018). Perhaps most remarkably, based on prediction of aggregation-prone and insoluble regions (Figure 3A) discrete sequences were identified that form a range of precise interactions with residues in the NAC and C-terminal regions, protecting the protein from aggregation (Doherty et al., 2020). Deleting residues 38–61 (that encapsulates a region (residues 47–56) shown by Eisenberg and Sawaya (2017), to form fibrils in isolation and named the pre-NAC region (Rodriguez et al., 2015), or deleting/replacing an even shorter peptide [named P1, residues 36–42] or P1P2 (residues 36–57) results in significantly slower fibril growth compared with the wild-type protein both in vitro and in C. elegans models (Doherty et al., 2020). The reduced aggregation rate may be a consequence of preventing the formation of inter-molecular contacts between regions of the IDP that form a transient β-hairpin structure (strand 1: residue 37–43 and strand 2: residue 48–54) previously postulated based on Thioflavin T assays and molecular dynamics simulations to drive aggregation (Mirecka et al., 2014; Yu et al., 2015). In accord with this hypothesis, binding of a nanobody known as a β-wrapin to this motif prevents aggregation in vitro and in Drosophila (Mirecka et al., 2014; Agerschou et al., 2019). Similarly, a specific short aggregation-modulating peptide sequence that lies outside its APRs has been observed for another IDP, Tau, but here the formation of a β-hairpin structure involving residues 295–311 protects the aggregation hotspot [residues 306–311 (Figure 3D)] by using the flanking region 295–300 as a protective shield. Familial point mutations or alternative splicing can result in weakening of this secondary structural element, exposing the APR and promoting aggregation (Chen et al., 2019). Finally, for apolipoprotein apoA-I, involved in systemic amyloidosis, the aggregation hotspot (residues 14–22) is protected by a helix bundle formed at the N-terminus of the protein. Familial amyloidosis associated with disease-promoting mutations (such as G26R, W50R, F71Y, or L170P), all of which are located in regions flanking the amyloid hotspot, induce conformational changes that result in exposure of residues 14–22 and lead to amyloid formation (Adachi et al., 2014; Das et al., 2016).
The aggregation of globular proteins can also be affected by their flanking regions. One well-understood example is β2-microglobulin (β2m, a 99-residue protein with an Ig fold when natively folded) which is associated with the disease dialysis-related amyloidosis (DRA) (Gejyo et al., 1986). Approximately 30% of the molecules in fibrils of DRA patients is comprised of an N-terminally truncated variant missing the N-terminal six residues (Esposito et al., 2000). This deletion variant which is significantly more aggregation-prone than the full-length protein, is destabilized in its native state, and exhibits increased dynamics that facilitates amyloid formation (Chiti et al., 2001; Jahn et al., 2006; Eichner et al., 2011; Karamanos et al., 2019). An in vitro study that introduced point mutations into different β-strands of β2m further showed the importance of A- (residues 6–11) and G-strands (residues 91–94), both distant to the aggregation hotspot (Figure 3E). Amino acid substitutions in these strands (I7A, V9A, or V93A) induced aggregation by destabilizing the monomeric structure specifically in these regions, while similar mutations elsewhere in the sequence caused similar loss of thermodynamic stability, yet did not drive aggregation (Jones et al., 2003b). Hence local, rather than global, stability, is important in tailoring the aggregation of natively folded proteins, potentially because of the specific effects this has on fulfilling the aggregation-potential of a sequence’s APRs (Langenberg et al., 2020). In a similar vein, a rare mutation in β2m has recently been discovered in a French family that results in a different amyloid disease in which the variant protein (D76N) forms fibrils that deposit in the viscera without loss of renal function (Valleix et al., 2012). Importantly, the substituted amino acid (D76N) lies in a solvent exposed loop distant to the single APR in the protein (Figure 3E).
Other globular proteins have also been found to have flanking regions that are critically important for aggregation, including polyglutamine expansion (polyQ) proteins such as ataxin-3 (Saunders and Bottomley, 2009). This ∼40 kDa protein has a structured N-terminal protease domain (the Josephin domain) followed by an IDR which contains two (or sometimes three) ubiquitin interacting motifs (UIMs) and an expanded polyQ tract (Paulson, 2012). The aggregation mechanism of ataxin 3 in vitro has been shown to involve two kinetically resolved stages. In the first phase, the aggregation-prone Josephin domain self-associates into worm-like fibrils (that lack the cross-β structure of amyloid), with the slow formation of amyloid involving the polyQ tract occurring in a second phase (Ellisdon et al., 2006). Like all polyQ proteins, aggregation of ataxin 3 is critically dependent on the length of the polyQ tract. Importantly, the presence of a long (disease-causing) poly-glutamine tract changes the conformational dynamics of the Josephin domain, exposing the aggregation-prone N-terminal region and allowing self-association that results in aggregation (Gales et al., 2005; Scarff et al., 2015). Akin to the results for the mutation causing β2m aggregation discussed above, the polyQ tract is believed to diminish the stability of neighboring domains in ataxin 3, creating a local denaturing environment (Ignatova and Gierasch, 2006). The longer the polyQ sequence, the greater its effect in accelerating aggregation (Scarff et al., 2015).
In addition to intra- and inter-molecular homotypic association, interactions of flanking regions with other molecules/surfaces (heterotypic interactions) can also affect the kinetics of fibril formation (Sarell et al., 2013; Figure 4). For several amyloidogenic proteins, including IAPP, Aβ40/42 and αSyn, aggregation is accelerated in the presence of membranes (Terakawa et al., 2018). For example, the N-terminal region of αSyn binds to lipid bilayers (forming an α-helical structure) that leads to extensive surface-induced fibril growth (Fusco et al., 2014; Doherty et al., 2020; Table 1). For IAPP, the N-terminal 19 residues (which flank the core region of amyloid involving residues ∼15–37) bind to membranes and also become helical (Brender et al., 2008). Studies on an N-terminal fragment of IAPP involving residues 1–19 showed that membrane binding and disruption of the bilayer occur independently of fibril formation (Brender et al., 2008). Tau has also been shown to bind membranes, having functional as well as pathogenic effects where the lipid bilayer facilitates protein-protein interactions driving aggregation (Elbaum-Garfinkle et al., 2010). Another example where binding to membranes initiates a critical process can be found for PrP, a GPI anchor protein, as binding and insertion into lipid membranes (in the presence of RNA) initiates the conformational transition to a highly aggregation-prone form of PrP (Wang et al., 2010a). In addition, binding of PrP to anionic phospholipids is mediated by a flanking region outside of the fibril core and comprises the contiguous positively charged (residues 100–110) and “hydrophobic domain” regions (residues 111–134) (Wang et al., 2010b). Surface-induced aggregation is also important in the growing field of nanotechnology where nanoparticles coated with sugars, lipids or proteins, are widely used in drug delivery or diagnostics. These surfaces can also enhance protein aggregation. For example, β2m has been shown to aggregate more rapidly in the presence of copolymer particles, cerium oxide particles, quantum dots, and carbon nanotubes in a manner that is dependent on the surface area and surface modification (Linse et al., 2007). Fibril formation of IAPP is also enhanced in the presence of chiral silica nanoribbons (Faridi et al., 2018).
Fibril formation of one protein can also be affected by interactions with other amyloidogenic proteins. For example, αSyn aggregation is enhanced in the presence of Tau, which is biologically relevant since these two proteins are observed to co-aggregate in inclusions in brains from patients with Dementia with Lewy Bodies (DLB) (Colom-Cadena et al., 2013). Similarly, CsgA, a bacterial functional amyloid, also accelerates αSyn aggregation, possibly explaining clinical and epidemiological data that show an accumulation of aggregated αSyn first being found in olfactory epithelium or gastrointestinal tract, before spreading to the brain (Sampson et al., 2020). Aggregation assays with a C-terminally truncated variant of αSyn using PRE NMR experiments revealed that the C-terminal region of αSyn interacts with Tau (Dasari et al., 2019; Lu et al., 2020; Table 1). This region is also involved in ion binding, most importantly Ca2+, which drives fibril formation probably by changing the conformational dynamics of αSyn to a more extended form (Han et al., 2018). In vivo assays have shown that the native prion protein (PrPC) or protease-resistant isoform (PrPSc) bind Tau with their N-terminal disordered segment (residue 23–90) (Han et al., 2006), and also bind to Aβ oligomers and αSyn fibrils, facilitating cell surface binding and cell-to-cell spreading (Laurén et al., 2009; Aulić et al., 2017). Additionally, single molecule Förster resonance energy transfer experiments (FRET) measuring the conformational ensemble of Tau have shown that heparin induces conformational changes that could be important in promoting amyloid formation (Elbaum-Garfinkle and Rhoades, 2012). These involve a loss of long-range contacts of the N- and C-terminal regions and a compaction of the aggregation-prone MBD (Elbaum-Garfinkle and Rhoades, 2012).
Finally, post-translational modifications including phosphorylation, ubiquitinoylation or acetylation are important regulatory modifications of amyloidogenic proteins that influence cellular mechanisms, such as protein degradation, signaling or protein-protein interactions, and also lead to misfolding and aggregation (Figure 4). Examples include the 441-residue protein Tau, in which 85 residues have been identified as phosphorylation sites mainly located in the aggregation hotspot that flanks the proline rich and C-terminal domains. Hyperphosphorylation of Tau is believed to trigger its dissociation from microtubules and to drive amyloid formation (Liu et al., 2007). However, recent studies on the four repeat region (K18) of Tau suggest an inhibitory effect of phosphorylation on fibril formation (Haj-Yahya et al., 2020). Finally, the protein huntingtin contains three lysines (K6, K9, K15) in its N-terminal 17-residue region which are often ubiquitinylated or SUMOylated, reducing toxicity by protease degradation or monomer stabilization, respectively (Ehrnhoefer et al., 2011).
Roles of Flanking Regions in Protein Function
As proteins associated with amyloidosis have functional roles in their soluble states, aggregation can lead to a loss of function, as well as a gain of toxic function. For example, Tau (Table 1) is known to be present in six different isoforms in the central nervous system formed by alternative splicing processes. This results in deletions in the N-terminal region (45–103) or one of the four repeat regions, R2 (residues 275–305) (Himmler et al., 1989). The expression and translation levels of these isoforms are correlated with different developmental stages (Kosik et al., 1989), pointing to discrete functional roles of each. Indeed, Tau’s main function is to bind microtubules with its aggregation-prone MBD (residues 244–371) which induces assembly of microtubules. Although the MBD is primarily responsible for microtubule binding, its N-terminal and C-terminal flanking regions have been shown to modulate the conformation and accessibility of the MBD as part of its functional activity (Goode et al., 2000; Barbier et al., 2019). Further, the proline rich region (residues 151–243) (Table 1) also contributes to tubulin binding and its polymerization into microtubules (McKibben and Rhoades, 2019). Other amyloidogenic proteins such as αSyn are also considered to be microtubule-associated proteins (MAP). In this case much less is known, with current research suggesting a microtubule-polymerizing activity when αSyn binds tubulin via its C-terminal region (Alim et al., 2004), or supporting microtubule association and dynamics by binding probably via its N-terminal- and NAC-regions adopting a helical conformation (Cartelli et al., 2016). Oligomeric αSyn on the other hand has been shown to inhibit tubulin polymerization, resulting in cell death (Chen et al., 2007).
αSyn not only interacts with tubulin, but it also binds to many different proteins and molecules that are important for function (e.g., interaction with receptors to increase the neuronal levels of dopamine or inhibiting SNARE complex formation) and toxicity (e.g., the binding of αSyn to Parkin contributes to the pathophysiology of Parkinson’s disease) (Emamzadeh, 2016). The aggregation-prone NAC region of αSyn itself has been implicated in its function [e.g., binding to the dopamine receptor in neurons, regulating dopamine concentration (Lee et al., 2001)]. The ability of αSyn to bind to membranes is key to its physiological function of remodeling membrane vesicles within the presynaptic termini (Diao et al., 2013). In vitro NMR studies of αSyn:liposome interactions revealed that the N-terminal 25 residues trigger the interaction with the membrane by acting as an anchor motif, initializing binding of the whole N-terminal and NAC region (membrane sensor region, residues 26–98) with the formation of α-helical structure throughout this region (Fusco et al., 2014). Residues 26–98 are believed to modulate the affinity of αSyn for membranes and are crucial for its function in clustering synaptic vesicles (Fusco et al., 2016). Deleting or replacing residues 36–57 (the P1 and P2 regions discussed above), which flank the aggregation hotspot, showed an inhibition of the membrane remodeling activity, supporting a role of these flanking regions in liposome fusion (Doherty et al., 2020). A study from Cholak et al. (2020) suggests that the very N-terminal region (residues 1–14) of αSyn inserts into membranes to initiate membrane binding, with N-terminal acetylation of αSyn enhancing the lifetime of the membrane-bound state. In accord with the functional importance of these regions, αSyn shows higher sequence similarity to its two known homologs (βSyn and γSyn) in the N-terminal region compared with the C-terminal region (90% and 77% sequence identity in the N-terminal region and 36% and 1% sequence identity in the C-terminal region between αSyn and βSyn and αSyn and γSyn, respectively) (George, 2001). Membrane binding is not only involved in αSyn function, but it also represents a risk factor for αSyn aggregation and cytotoxicity, as association with membranes accelerates αSyn fibril formation (Galvagnion et al., 2016; Fusco et al., 2017). Binding to lipid bilayers via its N-terminal helical region has been suggested to be the pathological conformation of αSyn in yeast, causing slower cell growth and cell death (Newberry et al., 2020). Similarly, binding to mitochondrial membranes resulted in cytotoxicity due to enhanced formation of mitochondrial reactive oxygen species (ROS) and reduced ATP levels (Vicario et al., 2018; Ganjam et al., 2019).
The functional amyloid Orb2B is involved in forming long term memories (Keleman et al., 2007; Majumdar et al., 2012; Khan et al., 2015) by affecting translation within neurons (Krüttner et al., 2012) via its RNA binding domain (RBD). The RBD flanks a glutamine-rich region that drives the formation of fibrils whose formation activates translation (Hervas et al., 2020). TDP-43 is involved in maintaining mRNA stability, maturation and transport via specific RNA-recognition motifs (residues 104–262) (Prasad et al., 2019). For splicing, TDP-43 functions as a dimer stabilized by inter-molecular interactions in the N-terminal region, especially the N-terminal 10 residues (Shiina et al., 2010; Zhang et al., 2013; Afroz et al., 2017). These intermolecular interactions are also involved in initiating aggregation (Zhang et al., 2013), providing a further example of the tug-of-war between sequences involved in function that also enhance aggregation. TDP-43 also exhibits reversible liquid-liquid phase separation, a reversible process of de-mixing fluids into two distinct liquid-phases [reviewed by Alberti et al. (2019) and de Oliveira et al. (2019)] important for the formation of stress granules that store mRNA:protein complexes under cellular stress (e.g., oxidative or thermal stress) (McDonald et al., 2011; Sun and Chakrabartty, 2017). This function is mediated by the C-terminal prion-like domain of TDP-43 (Table 1). Substitution and deletion variants identified Trp334, Trp385, and Trp412 as important drivers of phase separation (Li H.R. et al., 2018). These residues flank the aggregation hotspot of TDP-43 [residues 288–346 (Figure 3B)] and only Trp334 is part of the fibril core in the dagger shaped polymorph (Cao et al., 2019). TDP-43 liquid droplets remain stable for only a short period of time (timescale of hours) before transforming into irreversible aggregates (Conicella et al., 2016). Using PRE NMR experiments, Conicella et al. (2016) identified residues 321–340 as those responsible for the crucial interactions for phase separation, by forming inter-molecular helix-helix self-assemblies that are disrupted by the ALS-associated mutations (A321G, Q331K, and M337V). Tau also undergoes liquid-liquid phase separation prior to the formation of gel-like and then amyloid-like aggregates (Wegmann et al., 2018). A detailed study of deletion variants revealed that electrostatic interactions between the N-terminal region (residues 1–117) and parts of the C-terminal domain (residues 118–402) drive phase separation [whereas residues in the microtubule binding region are thought to be important for amyloid formation (Elbaum-Garfinkle and Rhoades, 2012)]. Deleting either one of the former regions ablated droplet formation (Boyko et al., 2019). Other amyloid-associated proteins also undergo phase separation, at least in vitro (Elbaum-Garfinkle, 2019). For example, liquid-liquid phase separation of IAPP is catalyzed by the air-water-interface (Pytowski et al., 2020). Interestingly, Comparison with the non-fibrillogenic rat IAPP revealed that phase separation does not require the presence of the highly amyloidogenic region (residue 20–29), but hydrogelation and aggregation do (Pytowski et al., 2020). The prion-like functional amyloid Sup35 also undergoes phase separation, generating protein-specific environmental responses. In this protein, the N-terminal prion domain, as well as the flanking M-domain, have a benign role: promoting reversible phase separation and gelation in a pH dependent manner (Franzmann et al., 2018). Finally, αSyn has also been shown to form phase separated droplets which precede aggregation into amyloid in vitro and in cells (Ray et al., 2020) The cellular droplets later transform into perinuclear aggresomes, with the familial mutations and phosphorylation of Y39 as discussed above promoting liquid-liquid phase separation, as well as aggregation into amyloid.
Regions Flanking the Structured Core of Amyloid Fibrils: The Importance of the Invisible Flanking Regions
The biophysical and biochemical properties of each amyloid fibril polymorph and their effect on cells can differ dramatically. As amyloid formation is under kinetic control, changes in the protein sequence and/or the assembly conditions (in vitro or in cells) can affect the structure of the fibrils formed. Accordingly, different fibril structures for Tau isotypes have been observed to form in different amyloid diseases (Scheres et al., 2020). Interestingly, for αSyn amyloid, heterogeneity is also observed for fibrils from patients with Parkinson’s disease or Multiple system atrophy [detected using ssNMR and differences in the fluorescence emission spectra of extrinsic fluorophores (Strohäker et al., 2019)]. The ability to discriminate polymorphs by comparison of fluorescence emission spectra of some extrinsic fluorophores (e.g., Thioflavin T or Congo Red) when intercalated into cross-β fibril structures has been noted previously (reviewed by Bhattacharya and Mukhopadhyay, 2016). The differential binding affinity for some antibodies to oligomers and mature fibrils also highlights the structural conversion that has to occur for amyloid fibrils to form. For example, the Aβ42 antibody anti-Trx(Aβ15)4 recognizes a structural epitope of oligomers and fibrils but not of the monomer, with the same selectivity for other amyloidogenic proteins (Moretto et al., 2007). Consequently, it is perhaps not surprising that fibrils with different structures may induce different effects in vivo. Ferrari et al. (2020), for example, used a pull-down approach with FLAG-tagged Tau to show that different aggregation stages (monomer, oligomer, fibril) have altered reactivity with its cellular environment due to large conformational changes that occur when this natively unfolded protein self-assembles into a β-sheet rich fibrillary protein state. Given that the “fuzzy coat” of disordered peptide regions that flank the cores of amyloid fibrils can vary between fibril polymorphs (Gallardo et al., 2020b), the effect of these dynamic regions on the function, toxicity and further amplification of the fibrils themselves may also vary, perhaps rationalizing the epigenetic difference in disease development in individuals expressing the same aggregation-prone proteins. Below we discuss the importance of these structurally “invisible” flanking regions in amyloid formation and the cellular consequences of these regions in amyloid deposition (Figure 5).
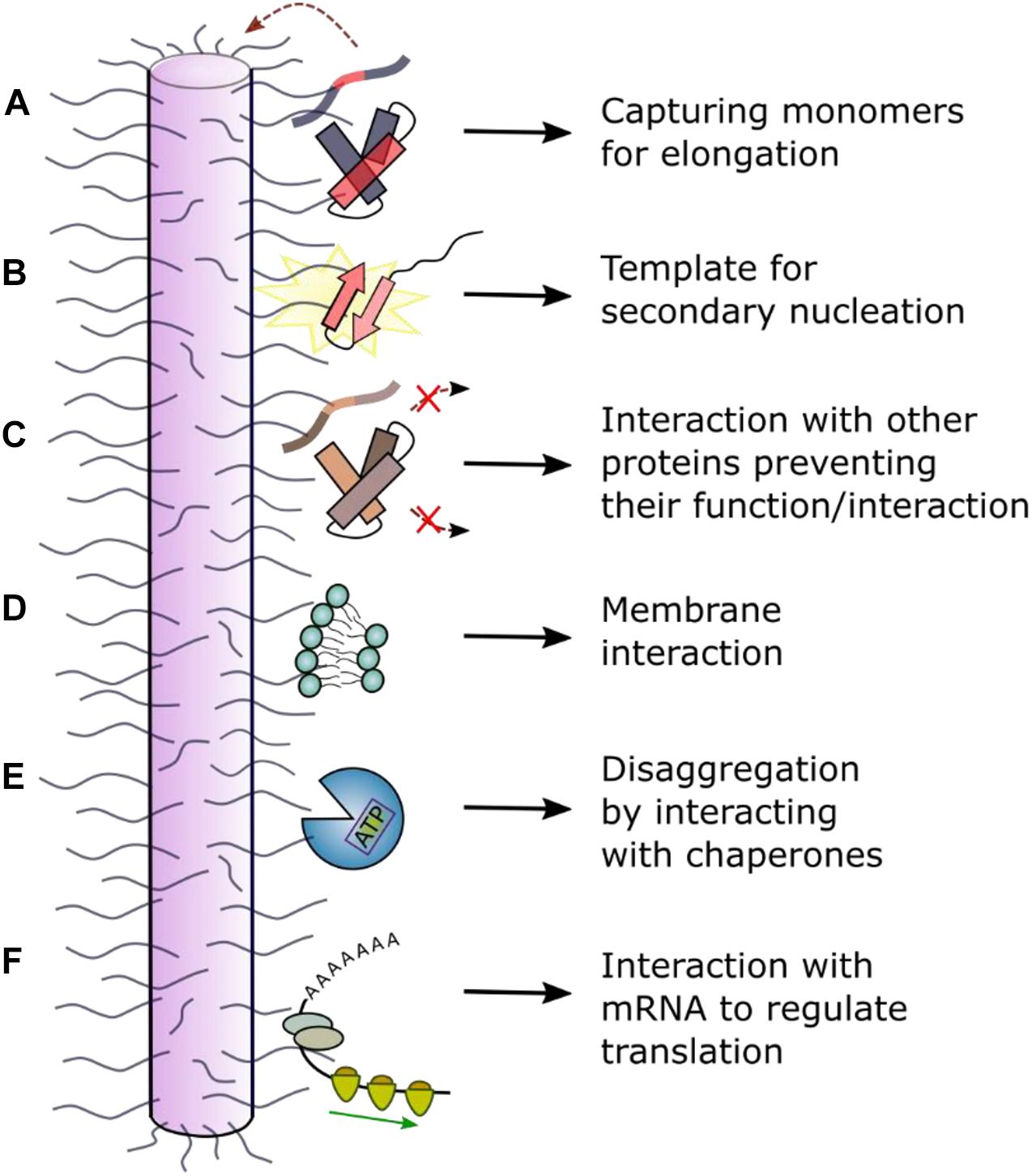
Figure 5. Schematic of interactions between the dynamically disordered regions of fibrils and other molecules. Dynamically disordered regions displayed on the surface of ordered amyloid fibrils can play roles in their function and cellular dysfunction. (A) The fibril “fuzzy coat” could capture amyloid precursors and facilitate their self-assembly into amyloid by (B) secondary nucleation or other molecular events. (C) Interaction with other (non-)amyloidogenic proteins could inhibit or alter their function. (D) Interaction with membranes can result in a toxic mechanism involving membrane disruption. (E) Interaction with chaperones can result in fibril depolymerization. (F) Interaction with RNA has been observed in a functional context for the protein CPEB (cytoplasmic polyadenylation element binding) which regulates long-term memory (Khan et al., 2015; Hervas et al., 2020). In other cases, disruption of cellular RNA could enhance phase separation and lead to cellular toxicity and/or dysfunction.
Fibril Elongation, Secondary Nucleation and Seeding
The extent of protein incorporation into insoluble fibrils is controlled by processes such as seeding (fragments broken off from fibrils, creating new fibril ends), elongation (the addition of monomers or oligomers onto fibril seeds) and secondary nucleation (creation of new nucleation sites on pre-existing fibril surfaces) (Michaels et al., 2018; Shvadchak et al., 2018; Scheidt et al., 2019). These kinetic events that drive amyloid formation could depend on the monomeric amyloid precursor, the structured amyloid core and the nature of the fibril “fuzzy coat.” For example, the flanking regions may be involved in the prion-like ability of αSyn to spread and seed further fibril growth (summarized by Longhena et al., 2017) as secretion of αSyn into the extracellular space occurs in association with membrane vesicles for which binding of the N-terminal region of αSyn is crucial (Emmanouilidou et al., 2010). The elongation rate may be accelerated as the large volume “fuzzy coat” may form transient interactions, “capturing” incoming monomeric protein molecules (Tompa, 2009; Figure 5A). This model is supported by the example of the human Prion Protein (PrP), in which increased numbers of an octapeptide Cu2+-binding motif in the N-terminal IDR (which flanks the folded C-terminal prion domain) promotes fibril formation and disease development (Goldfarb et al., 1991). Additionally, the fibril surface might have a key role in surface-induced secondary nucleation (Figure 5B). This is because the surfaces of all but one (Gremer et al., 2017) amyloid fibril structures solved to date are decorated by potentially large region of dynamically disordered protein flanking regions. Secondary nucleation for αSyn and other amyloid proteins, including Aβ and IAPP, is strongly dependent on pH (Gaspar et al., 2017). This could be explained by fact that the pH affects the dynamics of the solvent exposed flanking regions, as well as the charge along the ordered fibril core (Wegmann et al., 2013). The importance of flanking regions for these processes is illustrated by co-incubation of αSyn and βSyn. NMR analysis of the fibrils formed show increased dynamics in the N-terminal region compared with pure αSyn fibrils, whilst the core structure is unchanged. These co-incubated fibrils exhibited a reduced seeding capacity (Yang et al., 2019). However, the precise molecular mechanism(s) of the elongation and secondary nucleation processes, and how they alter with changes in sequence and fibril structure is not currently understood in detail.
Interaction With Other Amyloidogenic Proteins
Amyloid fibrils can interact with other amyloidogenic precursors and alter their amyloid potential (Figure 5C). αSyn fibrils, for instance, have been shown to bind Tau monomers via the αSyn acidic C-terminal region which is dynamically disordered in all fibril structures determined to date (Guerrero-Ferreira et al., 2020; Schweighauser et al., 2020). Since binding of Tau stabilizes microtubules, αSyn fibrils indirectly affect microtubule stability by removing Tau from microtubule surfaces resulting in neuronal dysfunction (Oikawa et al., 2016). Interaction of αSyn fibrils with Tau monomers further induces a conformational change in Tau, promoting its subsequent assembly into Tau amyloid structures (Oikawa et al., 2016). As reviewed by Luo et al. (2016), Aβ interacts with at least 10 other disease-related amyloidogenic proteins (e.g., IAPP, Tau, αSyn). For example, aggregation of Aβ40/42 is inhibited by cystatin C (Sastre et al., 2004), a protein which also colocalizes with Aβ40/42 in brain amyloid deposits. ELISA assays with an antibody targeting the N-terminal end of Aβ (residues 1–17) defined the binding site to be the first 17 residues, since binding was abolished in the presence of the antibody (Sastre et al., 2004). Aβ40/42 aggregation is also inhibited in the presence of TTR tetramers. NMR experiments revealed the interaction site to be between the thyroxine binding pocket of the TTR tetramer and Aβ residues 18–21 (Li et al., 2013). Finally, amyloid proteins have been observed to interact with other proteins in the context of cross-seeding. For example, pre-formed fibrils of IAPP have been shown to cross-seed Aβ40 monomers, accelerating fibril growth (Moreno-Gonzalez et al., 2017). The exact binding site is not known, but the two proteins have been shown to co-localize in disease (Moreno-Gonzalez et al., 2017).
Membrane Binding
Although many studies have shown that oligomers can be cytotoxic and possibly a major culprit of amyloid diseases (Fändrich, 2012; Verma et al., 2015; Fusco et al., 2017; Karamanos et al., 2019), the surface-induced fibril growth process on membranes (Engel et al., 2008; Qiang et al., 2015) and the interaction of mature fibrils with membranes (Figure 5D) have both been shown to disrupt cellular function and homeostasis (Martins et al., 2008; Xue et al., 2009; Pieri et al., 2012). In vitro experiments on αSyn have shown that its kinetics of aggregation are strongly affected by the presence of liposomes, highlighting the important role of lipid bilayers for fibril formation (Galvagnion et al., 2015). αSyn fibrils (and oligomers) bind negative, but not neutrally charged, liposomes (Grey et al., 2011; Pieri et al., 2012), pointing to an interaction with the N-terminal positively charged flanking region, which is dynamically disordered in the fibrillar state, similar to the interaction that is observed in the monomeric state. Striking work from Bäuerlein et al. (2017) used cryo-electron tomography in situ to show that polyQ fibrils (from huntingtin-exon 1) interact with membranes of the endoplasmic reticulum, changing the dynamics and structural organization of the organelle. These studies also showed that the sides of the fibril, as well as fibril ends, interact with membranes, although which regions of the fibril are involved in the interaction (the core or dynamic sequences) is not currently known. Similar vesicle membrane disruption was observed in vitro for β2m fibrils, with the majority of fibrils binding via their ends (Milanesi et al., 2012). Interestingly, the most severe membrane damage was observed for membranes containing the lipid BMP [bis(monoacylglycero)phosphate] which is enriched in lysosomal membranes, rationalizing the role of this organelle in the etiology of many amyloid diseases (Goodchild et al., 2014).
Interaction With Chaperones
Amyloid fibrils have been shown to interact with chaperones in different contexts (Figure 5E) of which neuroprotection is one of the foremost (Kannaian et al., 2019). Chaperone binding functions as an inhibitor for primary nucleation (when binding monomers), as well as for secondary nucleation and elongation, and some chaperone systems can even induce disaggregation of fibrils in the presence of ATP (Kannaian et al., 2019). Chaperones such as αB-crystallin have been shown to bind along the length of the fibril surface, perturbing secondary nucleation and elongation for both Aβ40/Aβ42 and αSyn (Waudby et al., 2010; Shammas et al., 2011). However, despite the authors revealing fascinating images of the chaperone in situ using immunogold labeling and immunoelectron microscopy, the exact binding site could not be determined due to the low resolution of this technique. In the case of Hsp27, which binds on the surface of αSyn fibrils causing decreased cytotoxicity and inhibited elongation, total internal reflection fluorescence (TIRF)-based imaging suggested that Hsp27 preferentially binds to hydrophobic patches along the fibril surface (Cox et al., 2018). These hydrophobic motifs involved in the interaction could involve the dynamic regions [residues 1–6 or 36–42 (Figure 3A)] as well as the fibril core itself. A higher resolution analysis, e.g., using HX methods, or more directly using ssNMR, is required to identify the exact chaperone binding sites.
Given the high thermodynamic stability of fibrils, it is perhaps a remarkable feat that chaperones can induce their depolymerization. For αSyn fibrils, for example, Bukau and coworkers have shown that the human chaperone Hsc70, specifically in complex with DNAJB1 (Hsp40 family) and Apg2 is a hsp110 family member (but not other co-chaperones from these families), induces ATP-dependent fibril fragmentation and depolymerization (Gao et al., 2015). Experiments using deletion variants allowed the interaction site of the fibrils with the chaperones to be identified, involving residues 1–30 and 111–140: the flanking regions of the αSyn amyloid core. A similar disaggregase activity has been observed for the Lipocalin-type Prostaglandin D synthase (L-PGDS) and Aβ40 fibrils (Kannaian et al., 2019). L-PGDS binds to the central region of the Aβ40 sequence (G25–G29). As this sequence forms a bend connecting two β-strands (Petkova et al., 2002) it is part of the structured fibril core (Figure 3C), yet is solvent accessible. L-PGDS fulfils its chaperone activity without ATP consumption or any co-chaperones. Also, the protease HTRA1S 328A can act as a chaperone and disassemble pre-formed 4R Tau filaments (Poepsel et al., 2015). Such observations may provide exciting new strategies to reduce the fibril load in amyloid diseases involving intracellular amyloid deposition.
Interaction With RNA
Several amyloidogenic proteins and prions have been identified in the context of RNA-modulating functions (Nizhnikov et al., 2016). Usually, as in the case of TDP-43, the native monomeric protein is involved in binding the RNA (see above). However, fibrils can also interact with RNA, as shown for the functional amyloid CPEB, that is involved in long term memory formation (Keleman et al., 2007; Majumdar et al., 2012; Khan et al., 2015). A study on the Drosophila melanogaster CPEB homolog, Orb2B, demonstrated that RNA binding to the monomeric protein represses protein translation of some genes, whilst binding to oligomeric states and fibrils activates protein production by stabilizing and elongating the poly(A) tail of mRNA in neurons in complex with other proteins (e.g., CG4612) (Khan et al., 2015; Hervas et al., 2020; Figure 5F). Translation activation changes the synthesis of specific synaptic proteins involved in memory formation. A high resolution cryoEM fibril structure demonstrated that only a small part of the Orb2 sequence (residues 176–206) forms the fibril core (Figure 3F). The RNA-recognition motif and protein interaction domain are located in the long flanking regions of this 704 residue protein (Hervas et al., 2020).
Discussion
In this review we have discussed the roles of regions that flank the APR sequences in monomeric amyloid precursors, and the role of the dynamically disordered regions that flank the structured core of amyloid fibrils in the interaction with other molecules and how this impacts cellular (dys)function. The importance of APRs is well recognized and their prediction using various computational tools is now straightforward. How the sequences that flank the APRs affect the kinetics and mechanisms of fibril growth, the structures of fibrils that ultimately form, and the extent and chemical identity of the dynamically disordered “fuzzy coat” regions of fibrils is still not well understood. This understanding is important as flanking regions play vital roles in the formation and interaction of amyloid with the cell and hence in disease.
In several native amyloid precursors, whether initially disordered or structured, regions surrounding the APRs or aggregation hotspot have been shown to be crucial for modulating amyloid formation. Intrinsic interactions in IDPs, including the APR flanking regions, can either promote or disfavor aggregation, by altering the conformational landscape of the IDP and “switching” amyloid formation on or off, as clearly shown for αSyn, Aβ, and Tau (Elbaum-Garfinkle and Rhoades, 2012; Stephens et al., 2019; Doherty et al., 2020). Also, for initially natively folded proteins, such as β2m, regions that flank the APRs often stabilize the native conformation. Mutations or truncations in these regions can consequently release the aggregation potential of the APR by locally destabilizing the protein fold leading to fibril formation (Langenberg et al., 2020). Advanced biophysical methods, such as NMR PRE experiments and single molecule FRET analysis can be used to identify transient long-range interactions in IDPs and IDRs in all-residue, if not all-atom, detail. Complemented by molecular dynamics simulations and experiments using deletion or substitution variants, these approaches have helped to identify the role of these “master controller” (Doherty et al., 2020) motifs for amyloid formation and provide targets to develop new strategies to combat amyloid formation and disease (Mirecka et al., 2014; Agerschou et al., 2019; Chen et al., 2019).
In addition to homotypic intra- and inter-molecular events, interactions with other molecules can be vital for amyloid function and pathology. Determining the interaction site of an amyloidogenic protein, or an amyloid fibril, with other molecules can be challenging, given the dynamic nature of the proteins involved. However, NMR experiments using PREs or chemical shifts, or binding assays with deletion variants can enable the identification of the residues that are involved in these binding processes. Such experiments can be used to clarify whether the aggregation hotspot or its flanking regions are required for the interaction, and potentially provide an evolutionary explanation for the development of such high-risk sequences.
Proteins undergo dramatic conformational changes on the pathway from initial precursor to amyloid states. More focus on these structural changes and which parts of the protein sequence drive these transformations might further demonstrate a crucial role of flanking regions in the amyloid cascade. Flanking regions of the fibril core are relatively straightforward to identify (e.g., using HX or protease protection experiments) but are especially hard to analyze structurally since their dynamic properties displayed on a static high molecular weight fibrillar particle provide significant experimental and computational challenges. All high-resolution structural information on amyloid fibrils formed from intact proteins [rather than short peptides and peptide fragments (Guenther et al., 2018; Rodriguez et al., 2015)] has been gained using ssNMR or cryoEM (Fitzpatrick and Saibil, 2019; Gallardo et al., 2020b; Guerrero-Ferreira et al., 2020; Scheres et al., 2020). These techniques, however, cannot provide atomic resolution information about the structure of the flexible regions which can comprise the large majority of the protein sequence in some amyloid states (Figure 3; Gallardo et al., 2020b). Nevertheless, some studies have shown the importance of the amyloid “fuzzy coat” for interactions with other amyloid precursors, cellular membranes, RNA and chaperones (Figures 4, 5). Assays with protease-treated fibrils, where the “fuzzy coat” is shaved off, would allow a better understanding of which part of the fibril is involved in the binding process and how the cellular consequences of fibril formation depend on the dynamically disordered regions displayed on the cross-β amyloid fold.
Fibril polymorphism has been shown to be responsible for the development of different diseases caused by the same protein [e.g., αSyn causing Parkinson’s disease or Multiple System Atrophy or Dementia with Lewy Bodies (Shahnawaz et al., 2020) or Tau causing Alzheimer’s disease, Pick’s disease, chronic traumatic encephalitis, and corticobasal degeneration (Fitzpatrick et al., 2017; Falcon et al., 2018; Scheres et al., 2020)]. Polymorphs, so far, have been defined by a fibril possessing a different core: but the flanking regions to the core could also be different in fibrils with the same (or similar) core structures. This adds a further, currently unexplored, dimension to amyloid polymorphism and its consequences for disease, since the dynamically disordered regions could have different interactions with cellular components (Wegmann et al., 2013). A more detailed analysis of the extent of conformational fluctuation of these amyloid flanking regions, for instance using EPR, cross-linking, or other techniques able to tackle dynamic heterogeneity, in the future might reveal “polymorphism” in the “fuzzy coat” and how this is related to the development of disease.
It should not be forgotten that flanking regions can have very different lengths. In the case of Aβ40/42, (nearly) the whole protein forms the fibril core (Figure 3C) (Lu et al., 2013; Gremer et al., 2017), whilst in other proteins, e.g., Orb2, flanking regions >500 residues in length are observed (Figure 3F; Hervas et al., 2020). Tompa (2009) hypothesized that the longer the dynamically disordered region, the more likely it can interact with other molecules by a “fly fishing mechanism.” This might explain why functional amyloid fibrils such as Orb2 have long flanking regions able to interact with other proteins, RNA or surfaces, whilst pathological amyloid fibrils, the sequences of which have not evolved for functional reasons, may present shorter flanking regions.
Conclusion
In summary, this review highlights the crucial role of regions that flank the APRs in amyloidogenic protein sequences and the dynamic regions that flank the amyloid fibril core for function, fibril formation and cellular dysfunction for a few example proteins. While beautiful fibril structures are now emerging from cryoEM and ssNMR studies, the often poorly visible, but functionally important flanking regions must not be forgotten. Focusing more on these regions with a broad range of biophysical and cellular techniques might help to gain a better understanding of the molecular mechanism of fibril formation and to identify new targets for drug development that do not involve the ordered amyloid core and the aggregation hotspots.
Author Contributions
SMU, DJB, and SER wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
We acknowledge, with thanks, funding from the Wellcome Trust [SU (215062/Z/18/Z) and SR (204963)].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Radford and Brockwell Amyloid group and our collaborators for many helpful and stimulating discussions. We are also grateful to David Klebl for critically reading this review and providing many helpful comments.
References
Adachi, E., Kosaka, A., Tsuji, K., Mizuguchi, C., Kawashima, H., Shigenaga, A., et al. (2014). The extreme N−terminal region of human apolipoprotein A−I has a strong propensity to form amyloid fibrils. FEBS Lett. 588, 389–394. doi: 10.1016/j.febslet.2013.11.031
Afroz, T., Hock, E.-M., Ernst, P., Foglieni, C., Jambeau, M., Gilhespy, L. A., et al. (2017). Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 8, 1–15. doi: 10.1038/s41467-017-00062-0
Agerschou, E. D., Flagmeier, P., Saridaki, T., Galvagnion, C., Komnig, D., Heid, L., et al. (2019). An engineered monomer binding-protein for α-synuclein efficiently inhibits the proliferation of amyloid fibrils. eLife 8:e46112. doi: 10.7554/eLife.46112
Alberti, S., Gladfelter, A., and Mittag, T. (2019). Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176, 419–434. doi: 10.1016/j.cell.2018.12.035
Alim, M. A., Ma, Q.-L., Takeda, K., Aizawa, T., Matsubara, M., Nakamura, M., et al. (2004). Demonstration of a role for α-synuclein as a functional microtubule-associated protein. J. Alzheimer’s Dis. 6, 435–442. doi: 10.3233/JAD-2004-6412
Aulić, S., Masperone, L., Narkiewicz, J., Isopi, E., Bistaffa, E., Ambrosetti, E., et al. (2017). α-Synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci. Rep. 7, 1–12. doi: 10.1038/s41598-017-10236-x
Ayala, Y. M., De Conti, L., Avendaño-Vázquez, S. E., Dhir, A., Romano, M., D’ambrogio, A., et al. (2011). TDP−43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–288. doi: 10.1038/emboj.2010.310
Baldwin, A. J., Knowles, T. P., Tartaglia, G. G., Fitzpatrick, A. W., Devlin, G. L., Shammas, S. L., et al. (2011). Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 133, 14160–14163. doi: 10.1021/ja2017703
Barbier, P., Zejneli, O., Martinho, M., Lasorsa, A., Belle, V., Smet-Nocca, C., et al. (2019). Role of Tau as a microtubule associated protein: structural and functional aspects. Front. Aging Neurosci. 11:204. doi: 10.3389/fnagi.2019.00204
Bäuerlein, F. J., Saha, I., Mishra, A., Kalemanov, M., Martínez-Sánchez, A., Klein, R., et al. (2017). In situ architecture and cellular interactions of PolyQ inclusions. Cell 171, 179–187. doi: 10.1016/j.cell.2017.08.009
Benilova, I., Gallardo, R., Ungureanu, A.-A., Cano, V. C., Snellinx, A., Ramakers, M., et al. (2014). The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J. Biol. Chem. 289, 30977–30989. doi: 10.1074/jbc.M114.599027
Benson, M. D., Buxbaum, J. N., Eisenberg, D. S., Merlini, G., Saraiva, M. J., Sekijima, Y., et al. (in press). Amyloid nomenclature 2020: update and recommendations by the international society of amyloidosis (ISA)nomenclature committee. Amyloid 1–6. doi: 10.1080/13506129.2020.1835263
Berezovsky, I. N., Zeldovich, K. B., and Shakhnovich, E. I. (2007). Positive and negative design in stability and thermal adaptation of natural proteins. PLoS Comput. Biol. 3:e52. doi: 10.1371/journal.pcbi.0030052
Bhattacharya, M., and Mukhopadhyay, S. (2016). “Studying protein misfolding and aggregation by fluorescence spectroscopy,” in Reviews in Fluorescence 2015, ed. C. Geddes (Cham: Springer), 1–27. doi: 10.1007/978-3-319-24609-3_1
Boyko, S., Qi, X., Chen, T.-H., Surewicz, K., and Surewicz, W. K. (2019). Liquid–liquid phase separation of tau protein: the crucial role of electrostatic interactions. J. Biol. Chem. 294, 11054–11059. doi: 10.1074/jbc.AC119.009198
Brahmachari, S., Ge, P., Lee, S. H., Kim, D., Karuppagounder, S. S., Kumar, M., et al. (2016). Activation of tyrosine kinase c-Abl contributes to α-synuclein–induced neurodegeneration. J. Clin. Invest. 126, 2970–2988. doi: 10.1172/JCI85456
Brandt, R., Léger, J., and Lee, G. (1995). Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 131, 1327–1340. doi: 10.1083/jcb.131.5.1327
Brender, J. R., Lee, E. L., Cavitt, M. A., Gafni, A., Steel, D. G., and Ramamoorthy, A. (2008). Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J. Am. Chem. Soc. 130, 6424–6429. doi: 10.1021/ja710484d
Buell, A. K. (2017). The nucleation of protein aggregates-from crystals to amyloid fibrils. Int. Rev. Cell Mol. Biol. 329, 187–226. doi: 10.1016/bs.ircmb.2016.08.014
Burmann, B. M., Gerez, J. A., Matečko-Burmann, I., Campioni, S., Kumari, P., Ghosh, D., et al. (2020). Regulation of α-synuclein by chaperones in mammalian cells. Nature 577, 127–132. doi: 10.1038/s41586-019-1808-9
Cao, Q., Boyer, D. R., Sawaya, M. R., Ge, P., and Eisenberg, D. S. (2019). Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 26, 619–627. doi: 10.1038/s41594-019-0248-4
Cao, Q., Boyer, D. R., Sawaya, M. R., Ge, P., and Eisenberg, D. S. (2020). Cryo-EM structure and inhibitor design of human IAPP (amylin) fibrils. Nat. Struct. Mol. Biol. 27, 653–659. doi: 10.1038/s41594-020-0435-3
Cartelli, D., Aliverti, A., Barbiroli, A., Santambrogio, C., Ragg, E. M., Casagrande, F. V., et al. (2016). α-Synuclein is a novel microtubule dynamase. Sci. Rep. 6, 1–13. doi: 10.1038/srep33289
Chen, D., Drombosky, K. W., Hou, Z., Sari, L., Kashmer, O. M., Ryder, B. D., et al. (2019). Tau local structure shields an amyloid-forming motif and controls aggregation propensity. Nat. Commun. 10, 1–14. doi: 10.1038/s41467-019-10355-1
Chen, L., Jin, J., Davis, J., Zhou, Y., Wang, Y., Liu, J., et al. (2007). Oligomeric α-synuclein inhibits tubulin polymerization. Biochem. Biophys. Res. Commun. 356, 548–553. doi: 10.1016/j.bbrc.2007.02.163
Chiti, F., De Lorenzi, E., Grossi, S., Mangione, P., Giorgetti, S., Caccialanza, G., et al. (2001). A partially structured species of β2-microglobulin is significantly populated under physiological conditions and involved in fibrillogenesis. J. Biol. Chem. 276, 46714–46721. doi: 10.1074/jbc.M107040200
Cho, M. K., Kim, H. Y., Fernandez, C. O., Becker, S., and Zweckstetter, M. (2011). Conserved core of amyloid fibrils of wild type and A30P mutant α−synuclein. Protein Sci. 20, 387–395. doi: 10.1002/pro.570
Cholak, E., Bugge, K., Khondker, A., Gauger, K., Pedraz-Cuesta, E., Pedersen, M. E., et al. (2020). Avidity within the N−terminal anchor drives α−synuclein membrane interaction and insertion. FEBS J. 34, 7462–7482. doi: 10.1096/fj.202000107R
Close, W., Neumann, M., Schmidt, A., Hora, M., Annamalai, K., Schmidt, M., et al. (2018). Physical basis of amyloid fibril polymorphism. Nat. Commun. 9, 1–7. doi: 10.1038/s41467-018-03164-5
Coletta, A., Pinney, J. W., Solís, D. Y. W., Marsh, J., Pettifer, S. R., and Attwood, T. K. (2010). Low-complexity regions within protein sequences have position-dependent roles. BMC Syst. Biol. 4:43. doi: 10.1186/1752-0509-4-43
Colom-Cadena, M., Gelpi, E., Charif, S., Belbin, O., Blesa, R., Martí, M. J., et al. (2013). Confluence of α-synuclein, tau, and β-amyloid pathologies in dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 72, 1203–1212. doi: 10.1097/NEN.0000000000000018
Colvin, M. T., Silvers, R., Ni, Q. Z., Can, T. V., Sergeyev, I., Rosay, M., et al. (2016). Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc. 138, 9663–9674. doi: 10.1021/jacs.6b05129
Conicella, A. E., Zerze, G. H., Mittal, J., and Fawzi, N. L. (2016). ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24, 1537–1549. doi: 10.1016/j.str.2016.07.007
Cox, D., Whiten, D. R., Brown, J. W., Horrocks, M. H., San Gil, R., Dobson, C. M., et al. (2018). The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 293, 4486–4497. doi: 10.1074/jbc.M117.813865
Das, M., Wilson, C. J., Mei, X., Wales, T. E., Engen, J. R., and Gursky, O. (2016). Structural stability and local dynamics in disease-causing mutants of human apolipoprotein AI: what makes the protein amyloidogenic? J. Mol. Biol. 428, 449–462. doi: 10.1016/j.jmb.2015.10.029
Dasari, A. K., Kayed, R., Wi, S., and Lim, K. H. (2019). Tau interacts with the C-terminal region of α-synuclein, promoting formation of toxic aggregates with distinct molecular conformations. Biochemistry 58, 2814–2821. doi: 10.1021/acs.biochem.9b00215
de Oliveira, G. A., Cordeiro, Y., Silva, J. L., and Vieira, T. C. (2019). Liquid-liquid phase transitions and amyloid aggregation in proteins related to cancer and neurodegenerative diseases. Adv. Protein Chem. Struct. Biol. 118, 289–331. doi: 10.1016/bs.apcsb.2019.08.002
Diao, J., Burré, J., Vivona, S., Cipriano, D. J., Sharma, M., Kyoung, M., et al. (2013). Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife 2:e00592. doi: 10.7554/eLife.00592
Doherty, C. P., Ulamec, S. M., Maya-Martinez, R., Good, S. C., Makepeace, J., Khan, G. N., et al. (2020). A short motif in the N-terminal region of α-synuclein is critical for both aggregation and function. Nat. Struct. Mol. Biol. 27, 249–259. doi: 10.1038/s41594-020-0384-x
Dunker, A. K., Romero, P., Obradovic, Z., Garner, E. C., and Brown, C. J. (2000). Intrinsic protein disorder in complete genomes. Genome Inform. 11, 161–171. doi: 10.11234/gi1990.11.161
Ebo, J. S., Guthertz, N., Radford, S. E., and Brockwell, D. J. (2020). Using protein engineering to understand and modulate aggregation. Curr. Opin. Struct. Biol. 60, 157–166. doi: 10.1016/j.sbi.2020.01.005
Ehrnhoefer, D. E., Sutton, L., and Hayden, M. R. (2011). Small changes, big impact: posttranslational modifications and function of huntingtin in Huntington disease. Neuroscientist 17, 475–492. doi: 10.1177/1073858410390378
Eichner, T., Kalverda, A. P., Thompson, G. S., Homans, S. W., and Radford, S. E. (2011). Conformational conversion during amyloid formation at atomic resolution. Mol. Cell 41, 161–172. doi: 10.1016/j.molcel.2010.11.028
Eisenberg, D. S., and Sawaya, M. R. (2017). Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 86, 69–95. doi: 10.1146/annurev-biochem-061516-045104
Elbaum-Garfinkle, S. (2019). Matter over mind: liquid phase separation and neurodegeneration. J. Biol. Chem. 294, 7160–7168. doi: 10.1074/jbc.REV118.001188
Elbaum-Garfinkle, S., Ramlall, T., and Rhoades, E. (2010). The role of the lipid bilayer in tau aggregation. Biophys. J. 98, 2722–2730. doi: 10.1016/j.bpj.2010.03.013
Elbaum-Garfinkle, S., and Rhoades, E. (2012). Identification of an aggregation-prone structure of tau. J. Am. Chem. Soc. 134, 16607–16613. doi: 10.1021/ja305206m
Ellisdon, A. M., Thomas, B., and Bottomley, S. P. (2006). The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J. Biol. Chem. 281, 16888–16896. doi: 10.1074/jbc.M601470200
Emamzadeh, F. N. (2016). Alpha-synuclein structure, functions, and interactions. J. Res. Med. Sci. 21:29. doi: 10.4103/1735-1995.181989
Emmanouilidou, E., Melachroinou, K., Roumeliotis, T., Garbis, S. D., Ntzouni, M., Margaritis, L. H., et al. (2010). Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010
Engel, M. F., Khemtémourian, L., Kleijer, C. C., Meeldijk, H. J., Jacobs, J., Verkleij, A. J., et al. (2008). Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. U.S.A. 105, 6033–6038. doi: 10.1073/pnas.0708354105
Esposito, G., Garvey, M., Alverdi, V., Pettirossi, F., Corazza, A., Fogolari, F., et al. (2013). Monitoring the interaction between β2-Microglobulin and the molecular chaperone αB-crystallin by NMR and mass spectrometry αB-crystallin dissociates β2-microglobulin oligomers. J. Biol. Chem. 288, 17844–17858. doi: 10.1074/jbc.M112.448639
Esposito, G., Michelutti, R., Verdone, G., Viglino, P., Hernandez, H., Robinson, C., et al. (2000). Removal of the N-terminal hexapeptide from human β2-microglobulin facilitates protein aggregation and fibril formation. Protein Sci. 9, 831–845. doi: 10.1110/ps.9.5.831
Falcon, B., Zhang, W., Murzin, A. G., Murshudov, G., Garringer, H. J., Vidal, R., et al. (2018). Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561, 137–140. doi: 10.1038/s41586-018-0454-y
Falsone, S. F., Meyer, N. H., Schrank, E., Leitinger, G., Pham, C. L., Fodero-Tavoletti, M. T., et al. (2012). SERF protein is a direct modifier of amyloid fiber assembly. Cell Reports 2, 358–371. doi: 10.1016/j.celrep.2012.06.012
Fändrich, M. (2012). Oligomeric intermediates in amyloid formation: structure determination and mechanisms of toxicity. J. Mol. Biol. 421, 427–440. doi: 10.1016/j.jmb.2012.01.006
Faridi, A., Sun, Y., Okazaki, Y., Peng, G., Gao, J., Kakinen, A., et al. (2018). Mitigating human IAPP amyloidogenesis in vivo with chiral silica nanoribbons. Small 14:1802825. doi: 10.1002/smll.201802825
Fernandez-Escamilla, A.-M., Rousseau, F., Schymkowitz, J., and Serrano, L. (2004). Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 22, 1302–1306. doi: 10.1038/nbt1012
Ferrari, L., Stucchi, R., Konstantoulea, K., van de Kamp, G., Kos, R., Geerts, W. J., et al. (2020). Arginine π-stacking drives binding to fibrils of the Alzheimer protein Tau. Nat. Commun. 11, 1–13. doi: 10.1038/s41467-019-13745-7
Fitzpatrick, A. W., Falcon, B., He, S., Murzin, A. G., Murshudov, G., Garringer, H. J., et al. (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. doi: 10.1038/nature23002
Fitzpatrick, A. W., and Saibil, H. R. (2019). Cryo-EM of amyloid fibrils and cellular aggregates. Curr. Opin. Struct. Biol. 58, 34–42. doi: 10.1016/j.sbi.2019.05.003
Franzmann, T. M., Jahnel, M., Pozniakovsky, A., Mahamid, J., Holehouse, A. S., Nüske, E., et al. (2018). Phase separation of a yeast prion protein promotes cellular fitness. Science 359:eaao5654. doi: 10.1126/science.aao5654
Fusco, G., Chen, S. W., Williamson, P. T., Cascella, R., Perni, M., Jarvis, J. A., et al. (2017). Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 358, 1440–1443. doi: 10.1126/science.aan6160
Fusco, G., De Simone, A., Gopinath, T., Vostrikov, V., Vendruscolo, M., Dobson, C. M., et al. (2014). Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 5, 1–8. doi: 10.1038/ncomms4827
Fusco, G., Pape, T., Stephens, A. D., Mahou, P., Costa, A. R., Kaminski, C. F., et al. (2016). Structural basis of synaptic vesicle assembly promoted by α-synuclein. Nat. Commun. 7, 1–12. doi: 10.1038/ncomms12563
Gales, L., Cortes, L., Almeida, C., Melo, C. V., do Carmo Costa, M., Maciel, P., et al. (2005). Towards a structural understanding of the fibrillization pathway in Machado-Joseph’s disease: trapping early oligomers of non-expanded ataxin-3. J. Mol. Biol. 353, 642–654. doi: 10.1016/j.jmb.2005.08.061
Gallardo, R., Iadanza, M. G., Xu, Y., Heath, G. R., Foster, R., Radford, S. E., et al. (2020a). Fibril structures of diabetes-related amylin variants reveal a basis for surface-templated assembly. Nat. Struct. Mol. Biol. 27, 1048–1056. doi: 10.1038/s41594-020-0496-3
Gallardo, R., Ramakers, M., De Smet, F., Claes, F., Khodaparast, L., Khodaparast, L., et al. (2016). De novo design of a biologically active amyloid. Science 354:aah4949. doi: 10.1126/science.aah4949
Gallardo, R., Ranson, N. A., and Radford, S. E. (2020b). Amyloid structures: much more than just a cross-β fold. Curr. Opin. Struct. Biol. 60, 7–16. doi: 10.1016/j.sbi.2019.09.001
Galvagnion, C., Brown, J. W., Ouberai, M. M., Flagmeier, P., Vendruscolo, M., Buell, A. K., et al. (2016). Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 113, 7065–7070. doi: 10.1073/pnas.1601899113
Galvagnion, C., Buell, A. K., Meisl, G., Michaels, T. C., Vendruscolo, M., Knowles, T. P., et al. (2015). Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 11, 229–234. doi: 10.1038/nchembio.1750
Ganjam, G. K., Bolte, K., Matschke, L. A., Neitemeier, S., Dolga, A. M., Höllerhage, M., et al. (2019). Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 10, 1–16. doi: 10.1038/s41419-019-2091-2
Gao, X., Carroni, M., Nussbaum-Krammer, C., Mogk, A., Nillegoda, N. B., Szlachcic, A., et al. (2015). Human Hsp70 disaggregase reverses Parkinson’s-linked α-synuclein amyloid fibrils. Mol. Cell 59, 781–793. doi: 10.1016/j.molcel.2015.07.012
Gaspar, R., Meisl, G., Buell, A. K., Young, L., Kaminski, C. F., Knowles, T. P., et al. (2017). Secondary nucleation of monomers on fibril surface dominates α-synuclein aggregation and provides autocatalytic amyloid amplification. Q. Rev. Biophys. 50:e6. doi: 10.1017/S0033583516000172
Gejyo, F., Odani, S., Yamada, T., Honma, N., Saito, H., Suzuki, Y., et al. (1986). β2-microglobulin: a new form of amyloid protein associated with chronic hemodialysis. Kidney Int. 30, 385–390. doi: 10.1038/ki.1986.196
Geraets, J. A., Pothula, K. R., and Schröder, G. F. (2020). Integrating cryo-EM and NMR data. Curr. Opin. Struct. Biol. 61, 173–181. doi: 10.1016/j.sbi.2020.01.008
Giasson, B. I., Murray, I. V., Trojanowski, J. Q., and Lee, V. M.-Y. (2001). A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J. Biol. Chem. 276, 2380–2386. doi: 10.1074/jbc.M008919200
Goldfarb, L. G., Brown, P., McCombie, W. R., Goldgaber, D., Swergold, G. D., Wills, P. R., et al. (1991). Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc. Natl. Acad. Sci. U.S.A. 88, 10926–10930. doi: 10.1073/pnas.88.23.10926
Goodchild, S. C., Sheynis, T., Thompson, R., Tipping, K. W., Xue, W.-F., Ranson, N. A., et al. (2014). β 2-Microglobulin amyloid fibril-induced membrane disruption is enhanced by endosomal lipids and acidic pH. PLoS One 9:e104492. doi: 10.1371/journal.pone.0104492
Goode, B. L., Chau, M., Denis, P. E., and Feinstein, S. C. (2000). Structural and functional differences between 3-repeat and 4-repeat tau isoforms implications for normal tau function and the onset of neurodegenerative disease. J. Biol. Chem. 275, 38182–38189. doi: 10.1074/jbc.M007489200
Gremer, L., Schölzel, D., Schenk, C., Reinartz, E., Labahn, J., Ravelli, R. B., et al. (2017). Fibril structure of amyloid-β (1–42) by cryo–electron microscopy. Science 358, 116–119. doi: 10.1126/science.aao2825
Grey, M., Linse, S., Nilsson, H., Brundin, P., and Sparr, E. (2011). Membrane interaction of α-synuclein in different aggregation states. J. Parkinsons Dis. 1, 359–371. doi: 10.3233/JPD-2011-11067
Guenther, E. L., Cao, Q., Trinh, H., Lu, J., Sawaya, M. R., Cascio, D., et al. (2018). Atomic structures of TDP-43 LCD segments and insights into reversible or pathogenic aggregation. Nat. Struct. Mol. Biol. 25, 463–471. doi: 10.1038/s41594-018-0064-2
Guerrero-Ferreira, R., Kovacik, L., Ni, D., and Stahlberg, H. (2020). New insights on the structure of alpha-synuclein fibrils using cryo-electron microscopy. Curr. Opin. Neurobiol. 61, 89–95. doi: 10.1016/j.conb.2020.01.014
Guerrero-Ferreira, R., Taylor, N. M., Arteni, A.-A., Kumari, P., Mona, D., Ringler, P., et al. (2019). Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. eLife 8:e48907. doi: 10.7554/eLife.48907
Guerrero-Ferreira, R., Taylor, N. M., Mona, D., Ringler, P., Lauer, M. E., Riek, R., et al. (2018). Cryo-EM structure of alpha-synuclein fibrils. eLife 7:e36402. doi: 10.7554/eLife.36402
Haj-Yahya, M., Gopinath, P., Rajasekhar, K., Mirbaha, H., Diamond, M. I., and Lashuel, H. A. (2020). Site−specific hyperphosphorylation inhibits, rather than promotes, Tau fibrillization, seeding capacity, and its microtubule binding. Angew. Chem. 132, 4088–4096. doi: 10.1002/ange.201913001
Han, J., Zhang, J., Yao, H., Wang, X., Li, F., Chen, L., et al. (2006). Study on interaction between microtubule associated protein tau and prion protein. Sci. China Life Sci. 49, 473–479. doi: 10.1007/s11427-006-2019-9
Han, J. Y., Choi, T. S., and Kim, H. I. (2018). Molecular role of Ca 2+ and hard divalent metal cations on accelerated fibrillation and interfibrillar aggregation of α-Synuclein. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-20320-5
He, H. J., Wang, X. S., Pan, R., Wang, D. L., Liu, M. N., and He, R. Q. (2009). The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol. 10:81. doi: 10.1186/1471-2121-10-81
Herrera, F. E., Chesi, A., Paleologou, K. E., Schmid, A., Munoz, A., Vendruscolo, M., et al. (2008). Inhibition of α-synuclein fibrillization by dopamine is mediated by interactions with five C-terminal residues and with E83 in the NAC region. PLoS One 3:e3394. doi: 10.1371/journal.pone.0003394
Hervas, R., Rau, M. J., Park, Y., Zhang, W., Murzin, A. G., Fitzpatrick, J. A., et al. (2020). Cryo-EM structure of a neuronal functional amyloid implicated in memory persistence in Drosophila. Science 367, 1230–1234. doi: 10.1126/science.aba3526
Himmler, A., Drechsel, D., Kirschner, M. W., and Martin, D. (1989). Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell. Biol. 9, 1381–1388. doi: 10.1128/MCB.9.4.1381
Hong, D.-P., Xiong, W., Chang, J.-Y., and Jiang, C. (2011). The role of the C-terminus of human α-synuclein: intra-disulfide bonds between the C-terminus and other regions stabilize non-fibrillar monomeric isomers. FEBS Lett. 585, 561–566. doi: 10.1016/j.febslet.2011.01.009
Hoshino, M., Katou, H., Hagihara, Y., Hasegawa, K., Naiki, H., and Goto, Y. (2002). Mapping the core of the β 2-microglobulin amyloid fibril by H/D exchange. Nat. Struct. Biol. 9, 332–336. doi: 10.1038/nsb792
Hoyer, W., Antony, T., Cherny, D., Heim, G., Jovin, T. M., and Subramaniam, V. (2002). Dependence of α-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 322, 383–393. doi: 10.1016/S0022-2836(02)00775-1
Hoyer, W., Cherny, D., Subramaniam, V., and Jovin, T. M. (2004). Impact of the acidic C-terminal region comprising amino acids 109- 140 on α-synuclein aggregation in vitro. Biochemistry 43, 16233–16242. doi: 10.1021/bi048453u
Huang, D.-B., Ainsworth, C. F., Stevens, F. J., and Schiffer, M. (1996). Three quaternary structures for a single protein. Proc. Natl. Acad. Sci. U.S.A. 93, 7017–7021. doi: 10.1073/pnas.93.14.7017
Huang, Y.-C., Lin, K.-F., He, R.-Y., Tu, P.-H., Koubek, J., Hsu, Y.-C., et al. (2013). Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS One 8:e64002. doi: 10.1371/journal.pone.0064002
Iadanza, M. G., Jackson, M. P., Hewitt, E. W., Ranson, N. A., and Radford, S. E. (2018a). A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 19, 755–773. doi: 10.1038/s41580-018-0060-8
Iadanza, M. G., Silvers, R., Boardman, J., Smith, H. I., Karamanos, T. K., Debelouchina, G. T., et al. (2018b). The structure of a β 2-microglobulin fibril suggests a molecular basis for its amyloid polymorphism. Nat. Commun. 9, 1–10. doi: 10.1038/s41467-018-06761-6
Ignatova, Z., and Gierasch, L. M. (2006). Extended polyglutamine tracts cause aggregation and structural perturbation of an adjacent β barrel protein. J. Biol. Chem. 281, 12959–12967. doi: 10.1074/jbc.M511523200
Ivanova, M. I., Sawaya, M. R., Gingery, M., Attinger, A., and Eisenberg, D. (2004). An amyloid-forming segment of β2-microglobulin suggests a molecular model for the fibril. Proc. Natl. Acad. Sci. U.S.A. 101, 10584–10589. doi: 10.1073/pnas.0403756101
Jahn, T. R., Parker, M. J., Homans, S. W., and Radford, S. E. (2006). Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nat. Struct. Mol. Biol. 13, 195–201. doi: 10.1038/nsmb1058
Janowska, M. K., Wu, K.-P., and Baum, J. (2015). Unveiling transient protein-protein interactions that modulate inhibition of alpha-synuclein aggregation by beta-synuclein, a pre-synaptic protein that co-localizes with alpha-synuclein. Sci. Rep. 5, 15164–15174. doi: 10.1038/srep15164
Johnson, B. S., Snead, D., Lee, J. J., McCaffery, J. M., Shorter, J., and Gitler, A. D. (2009). TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339. doi: 10.1074/jbc.M109.010264
Jones, D. R., Moussaud, S., and McLean, P. (2014). Targeting heat shock proteins to modulate α-synuclein toxicity. Ther. Adv. Neurol. Disord. 7, 33–51. doi: 10.1177/1756285613493469
Jones, S., Manning, J., Kad, N. M., and Radford, S. E. (2003a). Amyloid-forming peptides from β2-microglobulin—insights into the mechanism of fibril formation in vitro. J. Mol. Biol. 325, 249–257. doi: 10.1016/S0022-2836(02)01227-5
Jones, S., Smith, D. P., and Radford, S. E. (2003b). Role of the N and C-terminal strands of beta 2-microglobulin in amyloid formation at neutral pH. J. Mol. Biol. 330, 935–941. doi: 10.1016/S0022-2836(03)00688-0
Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., et al. (2012). A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99. doi: 10.1038/nature11283
Kannaian, B., Sharma, B., Phillips, M., Chowdhury, A., Manimekalai, M. S., Adav, S. S., et al. (2019). Abundant neuroprotective chaperone Lipocalin-type prostaglandin D synthase (L-PGDS) disassembles the Amyloid-β fibrils. Sci. Rep. 9, 1–17. doi: 10.1038/s41598-019-48819-5
Karamanos, T. K., Jackson, M. P., Calabrese, A. N., Goodchild, S. C., Cawood, E. E., Thompson, G. S., et al. (2019). Structural mapping of oligomeric intermediates in an amyloid assembly pathway. eLife 8:e46574. doi: 10.7554/eLife.46574
Keleman, K., Krüttner, S., Alenius, M., and Dickson, B. J. (2007). Function of the Drosophila CPEB protein Orb2 in long-term courtship memory. Nat. Neurosci. 10, 1587–1593. doi: 10.1038/nn1996
Kelley, N. W., Huang, X., Tam, S., Spiess, C., Frydman, J., and Pande, V. S. (2009). The predicted structure of the headpiece of the Huntingtin protein and its implications on Huntingtin aggregation. J. Mol. Biol. 388, 919–927. doi: 10.1016/j.jmb.2009.01.032
Kelly, J. W., and Lansbury, P. T. (1994). A chemical approach to elucidatetin mechanism of transthyretin and β-protein amyloid fibril formation. Amyloid 1, 186–205. doi: 10.3109/13506129409148451
Kendrew, J. C., Dickerson, R. E., Strandberg, B. E., Hart, R. G., Davies, D. R., Phillips, D. C., et al. (1960). Structure of myoglobin: a three-dimensional Fourier synthesis at 2 Å. resolution. Nature 185, 422–427.
Kessler, J. C., Rochet, J.-C., and Lansbury, P. T. (2003). The N-terminal repeat domain of α-synuclein inhibits β-sheet and amyloid fibril formation. Biochemistry 42, 672–678. doi: 10.1021/bi020429y
Khan, M. R., Li, L., Pérez-Sánchez, C., Saraf, A., Florens, L., Slaughter, B. D., et al. (2015). Amyloidogenic oligomerization transforms Drosophila Orb2 from a translation repressor to an activator. Cell 163, 1468–1483. doi: 10.1016/j.cell.2015.11.020
Khodaparast, L., Khodaparast, L., Gallardo, R., Louros, N. N., Michiels, E., Ramakrishnan, R., et al. (2018). Aggregating sequences that occur in many proteins constitute weak spots of bacterial proteostasis. Nat. Commun. 9, 1–15. doi: 10.1038/s41467-018-03131-0
Kollmer, M., Close, W., Funk, L., Rasmussen, J., Bsoul, A., Schierhorn, A., et al. (2019). Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 10, 1–8. doi: 10.1038/s41467-019-12683-8
Kosik, K. S., Orecchio, L. D., Bakalis, S., and Neve, R. L. (1989). Developmentally regulated expression of specific tau sequences. Neuron 2, 1389–1397. doi: 10.1016/0896-6273(89)90077-9
Krishnan, R., and Lindquist, S. L. (2005). Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 435, 765–772. doi: 10.1038/nature03679
Krüttner, S., Stepien, B., Noordermeer, J. N., Mommaas, M. A., Mechtler, K., Dickson, B. J., et al. (2012). Drosophila CPEB Orb2A mediates memory independent of Its RNA-binding domain. Neuron 76, 383–395. doi: 10.1016/j.neuron.2012.08.028
Kuo, P.-H., Chiang, C.-H., Wang, Y.-T., Doudeva, L. G., and Yuan, H. S. (2014). The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG-and TG-rich nucleic acids. Nucleic Acids Res. 42, 4712–4722. doi: 10.1093/nar/gkt1407
Kushnirov, V. V., Dergalev, A. A., and Alexandrov, A. I. (2020). Proteinase K resistant cores of prions and amyloids. Prion 14, 11–19. doi: 10.1080/19336896.2019.1704612
Langenberg, T., Gallardo, R., van der Kant, R., Louros, N., Michiels, E., Duran-Romaña, R., et al. (2020). Thermodynamic and evolutionary coupling between the native and amyloid state of globular proteins. Cell Reports 31, 107512–107534. doi: 10.1016/j.celrep.2020.03.076
Lashuel, H. A., Wurth, C., Woo, L., and Kelly, J. W. (1999). The mostpathogenic transthyretin variant, L55P, forms amyloid fibrils under acidicconditions and protofilaments under physiological conditions. Biochemistry 38, 13560–13573. doi: 10.1021/bi991021c
Laurén, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Lee, F. J., Liu, F., Pristupa, Z. B., and Niznik, H. B. (2001). Direct binding and functional coupling of α-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FEBS J. 15, 916–926. doi: 10.1096/fsb2fj000334com
Li, B., Ge, P., Murray, K. A., Sheth, P., Zhang, M., Nair, G., et al. (2018). Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 9, 1–10. doi: 10.1038/s41467-018-05971-2
Li, H.-R., Chiang, W.-C., Chou, P.-C., Wang, W.-J., and Huang, J.-R. (2018). TAR DNA-binding protein 43 (TDP-43) liquid–liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 293, 6090–6098. doi: 10.1074/jbc.AC117.001037
Li, X., Zhang, X., Ladiwala, A. R. A., Du, D., Yadav, J. K., Tessier, P. M., et al. (2013). Mechanisms of transthyretin inhibition of β-amyloid aggregation in vitro. J. Neurosci. 33, 19423–19433. doi: 10.1523/JNEUROSCI.2561-13.2013
Li, Y., Zhao, C., Luo, F., Liu, Z., Gui, X., Luo, Z., et al. (2018). Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 28, 897–903. doi: 10.1038/s41422-018-0075-x
Linse, S. (2017). Monomer-dependent secondary nucleation in amyloid formation. Biophys. Rev. 9, 329–338. doi: 10.1007/s12551-017-0289-z
Linse, S., Cabaleiro-Lago, C., Xue, W.-F., Lynch, I., Lindman, S., Thulin, E., et al. (2007). Nucleation of protein fibrillation by nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 104, 8691–8696. doi: 10.1073/pnas.0701250104
Liu, F., Li, B., Tung, E. J., Grundke-Iqbal, I., Iqbal, K., and Gong, C. X. (2007). Site−specific effects of tau phosphorylation on its microtubule assembly activity and self−aggregation. Eur. J. Neurosci. 26, 3429–3436. doi: 10.1111/j.1460-9568.2007.05955.x
Longhena, F., Faustini, G., Missale, C., Pizzi, M., Spano, P., and Bellucci, A. (2017). The contribution of α-synuclein spreading to Parkinson’s disease synaptopathy. Neural Plast. 2017, 1–15. doi: 10.1155/2017/5012129
Lu, J., Zhang, S., Ma, X., Jia, C., Liu, Z., Huang, C., et al. (2020). Structural basis of the interplay between α-synuclein and Tau in regulating pathological amyloid aggregation. J. Biol. Chem. 295, 7470–7480. doi: 10.1074/jbc.RA119.012284
Lu, J.-X., Qiang, W., Yau, W.-M., Schwieters, C. D., Meredith, S. C., and Tycko, R. (2013). Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 154, 1257–1268. doi: 10.1016/j.cell.2013.08.035
Lukavsky, P. J., Daujotyte, D., Tollervey, J. R., Ule, J., Stuani, C., Buratti, E., et al. (2013). Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20, 1443–1449. doi: 10.1038/nsmb.2698
Luo, J., Wärmländer, S. K., Gräslund, A., and Abrahams, J. P. (2016). Cross-interactions between the Alzheimer disease amyloid-β peptide and other amyloid proteins: a further aspect of the amyloid cascade hypothesis. J. Biol. Chem. 291, 16485–16493. doi: 10.1074/jbc.R116.714576
Lyke, D. R., Dorweiler, J. E., and Manogaran, A. L. (2019). The three faces of Sup35. Yeast 36, 465–472. doi: 10.1002/yea.3392
Majumdar, A., Cesario, W. C., White-Grindley, E., Jiang, H., Ren, F., Li, L., et al. (2012). Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell 148, 515–529. doi: 10.1016/j.cell.2012.01.004
Martin, E. M., Jackson, M. P., Gamerdinger, M., Gense, K., Karamonos, T. K., Humes, J. R., et al. (2018). Conformational flexibility within the nascent polypeptide–associated complex enables its interactions with structurally diverse client proteins. J. Biol. Chem. 293, 8554–8568. doi: 10.1074/jbc.RA117.001568
Martins, I. C., Kuperstein, I., Wilkinson, H., Maes, E., Vanbrabant, M., Jonckheere, W., et al. (2008). Lipids revert inert Aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 27, 224–233. doi: 10.1038/sj.emboj.7601953
McDonald, K. K., Aulas, A., Destroismaisons, L., Pickles, S., Beleac, E., Camu, W., et al. (2011). TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 20, 1400–1410. doi: 10.1093/hmg/ddr021
McKibben, K. M., and Rhoades, E. (2019). Independent tubulin binding and polymerization by the proline-rich region of Tau is regulated by Tau’s N-terminal domain. J. Biol. Chem. 294, 19381–19394. doi: 10.1074/jbc.RA119.010172
Mehra, S., Sahay, S., and Maji, S. K. (2019). α-Synuclein misfolding and aggregation: implications in Parkinson’s disease pathogenesis. BBA Proteins Proteom. 1867, 890–908. doi: 10.1016/j.bbapap.2019.03.001
Meinen, B. A., Gadkari, V. V., Stull, F., Ruotolo, B. T., and Bardwell, J. C. (2019). SERF engages in a fuzzy complex that accelerates primary nucleation of amyloid proteins. Proc. Natl. Acad. Sci. U.S.A. 116, 23040–23049. doi: 10.1073/pnas.1913316116
Miake, H., Mizusawa, H., Iwatsubo, T., and Hasegawa, M. (2002). Biochemical characterization of the core structure of α-synuclein filaments. J. Biol. Chem. 277, 19213–19219. doi: 10.1074/jbc.M110551200
Michaels, T. C., Šarić, A., Habchi, J., Chia, S., Meisl, G., Vendruscolo, M., et al. (2018). Chemical kinetics for bridging molecular mechanisms and macroscopic measurements of amyloid fibril formation. Annu. Rev. Phys. 69, 273–298. doi: 10.1146/annurev-physchem-050317-021322
Milanesi, L., Sheynis, T., Xue, W.-F., Orlova, E. V., Hellewell, A. L., Jelinek, R., et al. (2012). Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 20455–20460. doi: 10.1073/pnas.1206325109
Mirecka, E. A., Shaykhalishahi, H., Gauhar, A., Akgül, Ş, Lecher, J., Willbold, D., et al. (2014). Sequestration of a β−Hairpin for Control of α−Synuclein Aggregation. Angew. Chem. Int. 53, 4227–4230. doi: 10.1002/anie.201309001
Mok, S.-A., Condello, C., Freilich, R., Gillies, A., Arhar, T., Oroz, J., et al. (2018). Mapping interactions with the chaperone network reveals factors that protect against tau aggregation. Nat. Struct. Mol. Biol. 25, 384–393. doi: 10.1038/s41594-018-0057-1
Monsellier, E., Redeker, V., Ruiz-Arlandis, G., Bousset, L., and Melki, R. (2015). Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J. Biol. Chem. 290, 2560–2576. doi: 10.1074/jbc.M114.603332
Moreno-Gonzalez, I., Edwards, I. I. I. G., Salvadores, N., Shahnawaz, M., Diaz-Espinoza, R., and Soto, C. (2017). Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol. Psychiatry 22, 1327–1334. doi: 10.1038/mp.2016.230
Moretto, N., Bolchi, A., Rivetti, C., Imbimbo, B. P., Villetti, G., Pietrini, V., et al. (2007). Conformation-sensitive antibodies against Alzheimer amyloid-β by immunization with a thioredoxin-constrained B-cell epitope peptide. J. Biol. Chem. 282, 11436–11445. doi: 10.1074/jbc.M609690200
Muntané, G., Ferrer, I., and Martinez-Vicente, M. (2012). α-synuclein phosphorylation and truncation are normal events in the adult human brain. Neuroscience 200, 106–119. doi: 10.1016/j.neuroscience.2011.10.042
Murakami, T., Atsumi, T., Maeda, S., Tanase, S., Ishikawa, K., Mita, S., et al. (1992). A novel transthyretin mu-tation at position 30 (Leu for Val) associated with familial amyloidoticpolyneuropathy. Biochem. Biophys. Res. Commun. 187, 397–403. doi: 10.1016/S0006-291X(05)81506-2
Myers, S. L., Thomson, N. H., Radford, S. E., and Ashcroft, A. E. (2006). Investigating the structural properties of amyloid−like fibrils formed in vitro from β2−microglobulin using limited proteolysis and electrospray ionisation mass spectrometry. Rapid Comm. Mass Spec. 20, 1628–1636. doi: 10.1002/rcm.2482
Nath, S., Goodwin, J., Engelborghs, Y., and Pountney, D. (2011). Raised calcium promotes α-synuclein aggregate formation. Mol. Cell. Neurosci. 46, 516–526. doi: 10.1016/j.mcn.2010.12.004
Nelson, R., Sawaya, M. R., Balbirnie, M., Madsen, A. Ø, Riekel, C., Grothe, R., et al. (2005). Structure of the cross-β spine of amyloid-like fibrils. Nature 435, 773–778. doi: 10.1038/nature03680
Newberry, R. W., Leong, J. T., Chow, E. D., Kampmann, M., and DeGrado, W. F. (2020). Deep mutational scanning reveals the structural basis for α-synuclein activity. Nat. Chem. Biol. 9, 1–7. doi: 10.1038/s41589-020-0480-6
Nizhnikov, A. A., Antonets, K. S., Bondarev, S. A., Inge-Vechtomov, S. G., and Derkatch, I. L. (2016). Prions, amyloids, and RNA: pieces of a puzzle. Prion 10, 182–206. doi: 10.1080/19336896.2016.1181253
Oikawa, T., Nonaka, T., Terada, M., Tamaoka, A., Hisanaga, S.-I., and Hasegawa, M. (2016). α-Synuclein fibrils exhibit gain of toxic function, promoting tau aggregation and inhibiting microtubule assembly. J. Biol. Chem. 291, 15046–15056. doi: 10.1074/jbc.M116.736355
Palaninathan, S. K., Mohamedmohaideen, N. N., Snee, W. C., Kelly, J. W., and Sacchettini, J. C. (2008). Structural insight into pH-induced conforma-tional changes within the native human transthyretin tetramer. J. Mol. Biol. 382, 1157–1167. doi: 10.1016/j.jmb.2008.07.029
Paravastu, A. K., Leapman, R. D., Yau, W.-M., and Tycko, R. (2008). Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354. doi: 10.1073/pnas.0806270105
Paulson, H. (2012). Machado–Joseph disease/spinocerebellar ataxia type 3. Handb. Clin. Neurol. 103, 437–449. doi: 10.1016/B978-0-444-51892-7.00027-9
Pawar, A. P., Dubay, K. F., Zurdo, J., Chiti, F., Vendruscolo, M., and Dobson, C. M. (2005). Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 350, 379–392. doi: 10.1016/j.jmb.2005.04.016
Peng, C., Gathagan, R. J., Covell, D. J., Medellin, C., Stieber, A., Robinson, J. L., et al. (2018). Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 557, 558–563. doi: 10.1038/s41586-018-0104-4
Petkova, A. T., Ishii, Y., Balbach, J. J., Antzutkin, O. N., Leapman, R. D., Delaglio, F., et al. (2002). A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747. doi: 10.1073/pnas.262663499
Pieri, L., Madiona, K., Bousset, L., and Melki, R. (2012). Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 102, 2894–2905. doi: 10.1016/j.bpj.2012.04.050
Platt, G. W., Routledge, K. E., Homans, S. W., and Radford, S. E. (2008). Fibril growth kinetics reveal a region of β2-microglobulin important for nucleation and elongation of aggregation. J. Mol. Biol. 378, 251–263. doi: 10.1016/j.jmb.2008.01.092
Poepsel, S., Sprengel, A., Sacca, B., Kaschani, F., Kaiser, M., Gatsogiannis, C., et al. (2015). Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat. Chem. Biol. 11, 862–869. doi: 10.1038/nchembio.1931
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12:25. doi: 10.3389/fnmol.2019.00025
Pytowski, L., Lee, C. F., Foley, A. C., Vaux, D. J., and Jean, L. (2020). Liquid–liquid phase separation of type II diabetes-associated IAPP initiates hydrogelation and aggregation. Proc. Natl. Acad. Sci. U.S.A. 117, 12050–12061. doi: 10.1073/pnas.1916716117
Qiang, W., Yau, W.-M., and Schulte, J. (2015). Fibrillation of β amyloid peptides in the presence of phospholipid bilayers and the consequent membrane disruption. Biochim. Biophys. Acta Biomembr. 1848, 266–276. doi: 10.1016/j.bbamem.2014.04.011
Radamaker, L., Lin, Y.-H., Annamalai, K., Huhn, S., Hegenbart, U., Schönland, S. O., et al. (2019). Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat. Commun. 10, 1–8. doi: 10.1038/s41467-019-09032-0
Ranjan, P., and Kumar, A. (2017). Perturbation in long-range contacts modulates the kinetics of amyloid formation in α-synuclein familial mutants. ACS Chem. Neurosci. 8, 2235–2246. doi: 10.1021/acschemneuro.7b00149
Ray, S., Singh, N., Kumar, R., Patel, K., Pandey, S., Datta, D., et al. (2020). α-Synuclein aggregation nucleates through liquid–liquid phase separation. Nat. Chem. 12, 705–716. doi: 10.1038/s41557-020-0465-9
Reumers, J., MaurerStroh, S., Schymkowitz, J., and Rousseau, F. (2009). Protein sequences encode safeguards against aggregation. Hum. Mutat. 30, 431–437. doi: 10.1002/humu.20905
Richardson, J. S., and Richardson, D. C. (2002). Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. U.S.A. 99, 2754–2759. doi: 10.1073/pnas.052706099
Röder, C., Kupreichyk, T., Gremer, L., Schäfer, L. U., Pothula, K. R., Ravelli, R. B., et al. (2020). Cryo-EM structure of islet amyloid polypeptide fibrils reveals similarities with amyloid-β fibrils. Nat. Struct. Mol. Biol. 27, 660–667. doi: 10.1038/s41594-020-0442-4
Rodriguez, J. A., Ivanova, M. I., Sawaya, M. R., Cascio, D., Reyes, F. E., Shi, D., et al. (2015). Structure of the toxic core of α-synuclein from invisible crystals. Nature 525, 486–490. doi: 10.1038/nature15368
Rosenberg, K. J., Ross, J. L., Feinstein, H. E., Feinstein, S. C., and Israelachvili, J. (2008). Complementary dimerization of microtubule-associated tau protein: implications for microtubule bundling and tau-mediated pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 7445–7450. doi: 10.1073/pnas.0802036105
Rumbley, J., Hoang, L., Mayne, L., and Englander, S. W. (2001). An amino acid code for protein folding. Proc. Natl. Acad. Sci. U.S.A. 98, 105–112. doi: 10.1073/pnas.98.1.105
Sampson, T. R., Challis, C., Jain, N., Moiseyenko, A., Ladinsky, M. S., Shastri, G. G., et al. (2020). A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. eLife 9:e53111. doi: 10.7554/eLife.53111
Sant’Anna, R., Braga, C., Varejão, N., Pimenta, K. M., Graña-Montes, R., Alves, A., et al. (2014). The importance of a gatekeeper residue on the aggregation of transthyretin implications for transthyretin-related amyloidoses. J. Biol. Chem. 289, 28324–28337. doi: 10.1074/jbc.M114.563981
Santos, J., Pujols, J., Pallarès, I., Iglesias, V., and Ventura, S. (2020). Computational prediction of protein aggregation: advances in proteomics, conformation-specific algorithms and biotechnological applications. Comput. Struct. Biotechnol. 18, 1403–1413. doi: 10.1016/j.csbj.2020.05.026
Sarell, C. J., Stockley, P. G., and Radford, S. E. (2013). Assessing the causes and consequences of co-polymerization in amyloid formation. Prion 7, 359–368. doi: 10.4161/pri.26415
Sastre, M., Calero, M., Pawlik, M., Mathews, P. M., Kumar, A., Danilov, V., et al. (2004). Binding of cystatin C to Alzheimer’s amyloid β inhibits in vitro amyloid fibril formation. Neurobiol. Aging 25, 1033–1043. doi: 10.1016/j.neurobiolaging.2003.11.006
Saunders, H. M., and Bottomley, S. P. (2009). Multi-domain misfolding: understanding the aggregation pathway of polyglutamine proteins. Protein Eng. Des. Sel. 22, 447–451. doi: 10.1093/protein/gzp033
Scarff, C. A., Almeida, B., Fraga, J., Macedo-Ribeiro, S., Radford, S. E., and Ashcroft, A. E. (2015). Examination of ataxin-3 (atx-3) aggregation by structural mass spectrometry techniques: a rationale for expedited aggregation upon polyglutamine (polyQ) expansion. Mol. Cell. Proteomics 14, 1241–1253. doi: 10.1074/mcp.M114.044610
Scheidt, T., Łapińska, U., Kumita, J. R., Whiten, D. R., Klenerman, D., Wilson, M. R., et al. (2019). Secondary nucleation and elongation occur at different sites on Alzheimer’s amyloid-β aggregates. Sci. Adv. 5:eaau3112. doi: 10.1126/sciadv.aau3112
Scheres, S. H., Zhang, W., Falcon, B., and Goedert, M. (2020). Cryo-EM structures of tau filaments. Curr. Opin. Struct. Biol. 64, 17–25. doi: 10.1016/j.sbi.2020.05.011
Schmidt, M., Wiese, S., Adak, V., Engler, J., Agarwal, S., Fritz, G., et al. (2019). Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 10, 1–9. doi: 10.1038/s41467-019-13038-z
Schweighauser, M., Shi, Y., Tarutani, A., Kametani, F., Murzin, A. G., Ghetti, B., et al. (2020). Structures of α-synuclein filaments from multiple system atrophy. Nature 585, 464–469. doi: 10.1038/s41586-020-2317-6
Shahnawaz, M., Mukherjee, A., Pritzkow, S., Mendez, N., Rabadia, P., Liu, X., et al. (2020). Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 578, 273–277. doi: 10.1038/s41586-020-1984-7
Shammas, S. L., Waudby, C. A., Wang, S., Buell, A. K., Knowles, T. P., Ecroyd, H., et al. (2011). Binding of the molecular chaperone αB-crystallin to Aβ amyloid fibrils inhibits fibril elongation. Biophys. J. 101, 1681–1689. doi: 10.1016/j.bpj.2011.07.056
Shen, K., Gamerdinger, M., Chan, R., Gense, K., Martin, E. M., Sachs, N., et al. (2019). Dual role of ribosome-binding domain of NAC as a potent suppressor of protein aggregation and aging-related proteinopathies. Mol. Cell 74, 729–741. doi: 10.1016/j.molcel.2019.03.012
Shiina, Y., Arima, K., Tabunoki, H., and Satoh, J.-I. (2010). TDP-43 dimerizes in human cells in culture. Cell. Mol. Neurobiol. 30, 641–652. doi: 10.1007/s10571-009-9489-9
Shvadchak, V. V., Afitska, K., and Yushchenko, D. A. (2018). Inhibition of α−Synuclein amyloid fibril elongation by blocking fibril ends. Angew. Chem. Int. 57, 5690–5694. doi: 10.1002/anie.201801071
Sibille, N., Sillen, A., Leroy, A., Wieruszeski, J.-M., Mulloy, B., Landrieu, I., et al. (2006). Structural impact of heparin binding to full-length Tau as studied by NMR spectroscopy. Biochemistry 45, 12560–12572. doi: 10.1021/bi060964o
Sillen, A., Leroy, A., Wieruszeski, J. M., Loyens, A., Beauvillain, J. C., Buée, L., et al. (2005). Regions of tau implicated in the paired helical fragment core as defined by NMR. Chembiochem 6, 1849–1856. doi: 10.1002/cbic.200400452
Sivanandam, V., Jayaraman, M., Hoop, C. L., Kodali, R., Wetzel, R., and van der Wel, P. C. (2011). The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 133, 4558–4566. doi: 10.1021/ja110715f
Sormanni, P., Aprile, F. A., and Vendruscolo, M. (2015). The CamSol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 427, 478–490. doi: 10.1016/j.jmb.2014.09.026
Stephens, A. D., Zacharopoulou, M., and Schierle, G. S. K. (2019). The cellular environment affects monomeric α-synuclein structure. Trends Biochem. Sci. 44, 453–466. doi: 10.1016/j.tibs.2018.11.005
Strohäker, T., Jung, B. C., Liou, S.-H., Fernandez, C. O., Riedel, D., Becker, S., et al. (2019). Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts. Nat. Commun. 10, 1–12. doi: 10.1038/s41467-019-13564-w
Sun, Y., and Chakrabartty, A. (2017). Phase to phase with TDP-43. Biochemistry 56, 809–823. doi: 10.1021/acs.biochem.6b01088
Swuec, P., Lavatelli, F., Tasaki, M., Paissoni, C., Rognoni, P., Maritan, M., et al. (2019). Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 10, 1–9. doi: 10.1038/s41467-019-09133-w
Tam, S., Spiess, C., Auyeung, W., Joachimiak, L., Chen, B., Poirier, M. A., et al. (2009). The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1287. doi: 10.1038/nsmb.1700
Tartaglia, G. G., Pechmann, S., Dobson, C. M., and Vendruscolo, M. (2007). Life on the edge: a link between gene expression levels and aggregation rates of human proteins. Trends Biochem. Sci. 32, 204–206. doi: 10.1016/j.tibs.2007.03.005
Tenidis, K., Waldner, M., Bernhagen, J., Fischle, W., Bergmann, M., Weber, M., et al. (2000). Identification of a penta-and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J. Mol. Biol. 295, 1055–1071. doi: 10.1006/jmbi.1999.3422
Terada, M., Suzuki, G., Nonaka, T., Kametani, F., Tamaoka, A., and Hasegawa, M. (2018). The effect of truncation on prion-like properties of α-synuclein. J. Biol. Chem. 293, 13910–13920. doi: 10.1074/jbc.RA118.001862
Terakawa, M. S., Lin, Y., Kinoshita, M., Kanemura, S., Itoh, D., Sugiki, T., et al. (2018). Impact of membrane curvature on amyloid aggregation. Biochem. Biophys. Acta. Biomembr. 1860, 1741–1764. doi: 10.1016/j.bbamem.2018.04.012
Thompson, M. J., Sievers, S. A., Karanicolas, J., Ivanova, M. I., Baker, D., and Eisenberg, D. (2006). The 3D profile method for identifying fibril-forming segments of proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 4074–4078. doi: 10.1073/pnas.0511295103
Tompa, P. (2009). Structural disorder in amyloid fibrils: its implication in dynamic interactions of proteins. FEBS J. 276, 5406–5415. doi: 10.1111/j.1742-4658.2009.07250.x
Tompa, P., Dosztányi, Z., and Simon, I. (2006). Prevalent structural disorder in E. coli and S. c erevisiae proteomes. J. Proteome Res. 5, 1996–2000. doi: 10.1021/pr0600881
Ulamec, S. M., and Radford, S. E. (2020). Spot the difference: function versus toxicity in amyloid fibrils. Trends Biochem. Sci. 45, 635–636. doi: 10.1016/j.tibs.2020.04.007
Valleix, S., Gillmore, J. D., Bridoux, F., Mangione, P. P., Dogan, A., Nedelec, B., et al. (2012). Hereditary systemic amyloidosis due to Asp76Asn variant β2-microglobulin. N. Engl. J. Med. 366, 2276–2283. doi: 10.1056/NEJMoa1201356
van Ham, T. J., Holmberg, M. A., van der Goot, A. T., Teuling, E., Garcia-Arencibia, M., Kim, H.-E., et al. (2010). Identification of MOAG-4/SERF as a regulator of age-related proteotoxicity. Cell 142, 601–612. doi: 10.1016/j.cell.2010.07.020
Verma, M., Vats, A., and Taneja, V. (2015). Toxic species in amyloid disorders: oligomers or mature fibrils. Ann. Indian Acad. Neurol. 18, 138–145. doi: 10.4103/0972-2327.144284
Vicario, M., Cieri, D., Brini, M., and Calì, T. (2018). The close encounter between alpha-synuclein and mitochondria. Front. Neurosci. 12:388. doi: 10.3389/fnins.2018.00388
von Bergen, M., Friedhoff, P., Biernat, J., Heberle, J., Mandelkow, E.-M., and Mandelkow, E. (2000). Assembly of τ protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134. doi: 10.1073/pnas.97.10.5129
Wälti, M. A., Ravotti, F., Arai, H., Glabe, C. G., Wall, J. S., Böckmann, A., et al. (2016). Atomic-resolution structure of a disease-relevant Aβ (1–42) amyloid fibril. Proc. Natl. Acad. Sci. U.S.A. 113, E4976–E4984. doi: 10.1073/pnas.1600749113
Wang, F., Wang, X., Yuan, C.-G., and Ma, J. (2010a). Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135. doi: 10.1126/science.1183748
Wang, F., Yin, S., Wang, X., Zha, L., Sy, M.-S., and Ma, J. (2010b). Role of the highly conserved middle region of prion protein (PrP) in PrP- lipid interaction. Biochemistry 49, 8169–8176. doi: 10.1021/bi101146v
Wang, L.-Q., Zhao, K., Yuan, H.-Y., Wang, Q., Guan, Z., Tao, J., et al. (2020). Cryo-EM structure of an amyloid fibril formed by full-length human prion protein. Nat. Struct. Mol. Biol. 27, 1–5. doi: 10.1038/s41594-020-0441-5
Wang, Y., Martinez-Vicente, M., Krüger, U., Kaushik, S., Wong, E., Mandelkow, E.-M., et al. (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170. doi: 10.1093/hmg/ddp367
Ward, J. J., Sodhi, J. S., McGuffin, L. J., Buxton, B. F., and Jones, D. T. (2004). Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337, 635–645. doi: 10.1016/j.jmb.2004.02.002
Waudby, C. A., Knowles, T. P., Devlin, G. L., Skepper, J. N., Ecroyd, H., Carver, J. A., et al. (2010). The interaction of αB-crystallin with mature α-synuclein amyloid fibrils inhibits their elongation. Biophys. J. 98, 843–851. doi: 10.1016/j.bpj.2009.10.056
Wegmann, S., Eftekharzadeh, B., Tepper, K., Zoltowska, K. M., Bennett, R. E., Dujardin, S., et al. (2018). Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 37:e98049. doi: 10.15252/embj.201798049
Wegmann, S., Medalsy, I. D., Mandelkow, E., and Müller, D. J. (2013). The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectrolyte brush. Proc. Natl. Acad. Sci. U.S.A. 110, E313–E321. doi: 10.1073/pnas.1212100110
Wentink, A., Nussbaum-Krammer, C., and Bukau, B. (2019). Modulation of amyloid states by molecular chaperones. Cold Spring Harb. Perspect. Biol. 11:a033969. doi: 10.1101/cshperspect.a033969
Williams, A. D., Shivaprasad, S., and Wetzel, R. (2006). Alanine scanning mutagenesis of Aβ (1-40) amyloid fibril stability. J. Mol. Biol. 357, 1283–1294. doi: 10.1016/j.jmb.2006.01.041
Xue, W.-F., Hellewell, A. L., Gosal, W. S., Homans, S. W., Hewitt, E. W., and Radford, S. E. (2009). Fibril fragmentation enhances amyloid cytotoxicity. J. Biol. Chem. 284, 34272–34282. doi: 10.1074/jbc.M109.049809
Xue, W.-F., Homans, S. W., and Radford, S. E. (2008). Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc. Natl. Acad. Sci. U.S.A. 105, 8926–8931. doi: 10.1073/pnas.0711664105
Yang, X., Williams, J. K., Yan, R., Mouradian, M. M., and Baum, J. (2019). Increased dynamics of α-synuclein fibrils by β-synuclein leads to reduced seeding and cytotoxicity. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-019-54063-8
Yu, H., Han, W., Ma, W., and Schulten, K. (2015). Transient β-hairpin formation in α-synuclein monomer revealed by coarse-grained molecular dynamics simulation. J. Chem. Phys. 143:243142. doi: 10.1063/1.4936910
Zhang, W., Falcon, B., Murzin, A. G., Fan, J., Crowther, R. A., Goedert, M., et al. (2019). Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife 8:e43584. doi: 10.7554/eLife.43584
Keywords: flanking region, amyloid, synuclein, fuzzy coat, aggregation, Tau, TDP-43, Orb2
Citation: Ulamec SM, Brockwell DJ and Radford SE (2020) Looking Beyond the Core: The Role of Flanking Regions in the Aggregation of Amyloidogenic Peptides and Proteins. Front. Neurosci. 14:611285. doi: 10.3389/fnins.2020.611285
Received: 28 September 2020; Accepted: 02 November 2020;
Published: 01 December 2020.
Edited by:
Wolfgang Hoyer, Heinrich Heine University Düsseldorf, GermanyReviewed by:
Anita L. Manogaran, Marquette University, United StatesFuyuki Kametani, Tokyo Metropolitan Institute of Medical Science, Japan
Copyright © 2020 Ulamec, Brockwell and Radford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheena E. Radford, Uy5FLlJhZGZvcmRAbGVlZHMuYWMudWs=