 Mei-Gang Ma1,2†
Mei-Gang Ma1,2† Xiao-Rong Liu1†‡
Xiao-Rong Liu1†‡ Yuan Wu2Jie Wang1
Yuan Wu2Jie Wang1 Bing-Mei Li1
Bing-Mei Li1 Yi-Wu Shi1Tao Su1Bin Li1
Yi-Wu Shi1Tao Su1Bin Li1 De-Tian Liu1
De-Tian Liu1 Yong-Hong Yi1*
Yong-Hong Yi1* Wei-Ping Liao1*‡ for the China Epilepsy Gene 1.0 Project
Wei-Ping Liao1*‡ for the China Epilepsy Gene 1.0 Project- 1Institute of Neuroscience and Department of Neurology of the Second Affiliated Hospital of Guangzhou Medical University, Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, Guangzhou, China
- 2Department of Neurology, The First Affiliated Hospital of Guangxi Medical University, Nanning, China
RYR2 encodes ryanodine receptor 2 protein (RYR-2) that is mainly located on endoplasmic reticulum membrane and regulates intracellular calcium concentration. The RYR-2 protein is ubiquitously distributed and highly expressed in the heart and brain. Previous studies have identified the RYR2 mutations in the etiology of arrhythmogenic right ventricular dysplasia 2 and catecholaminergic polymorphic ventricular tachycardia. However, the relationship between RYR2 gene and epilepsy is not determined. In this study, we screened for novel genetic variants in a group of 292 cases (families) with benign epilepsy of childhood with centrotemporal spikes (BECTS) by trio-based whole-exome sequencing. RYR2 mutations were identified in five cases with BECTS, including one heterozygous frameshift mutation (c.14361dup/p.Arg4790Pro fs∗6), two heterozygous missense mutations (c.2353G > A/p.Asp785Asn and c.8574G > A/p.Met2858Ile), and two pairs of compound heterozygous mutations (c.4652A > G/p.Asn1551Ser and c.11693T > C/p.Ile3898Thr, c.7469T > C/p.Val2490Ala and c.12770G > A/p.Arg4257Gln, respectively). Asp785Asn was a de novo missense mutation. All the missense mutations were suggested to be damaging by at least three web-based prediction tools. These mutations do not present or at low minor allele frequency in gnomAD database and present statistically higher frequency in the cohort of BECTS than in the control populations of gnomAD. Asp785Asn, Asn1551Ser, and Ile3898Thr were predicted to affect hydrogen bonds with surrounding amino acids. Three affected individuals had arrhythmia (sinus arrhythmia and occasional atrial premature). The two probands with compound heterozygous missense mutations presented mild cardiac structural abnormalities. Strong evidence from ClinGen Clinical Validity Framework suggested an association between RYR2 variants and epilepsy. This study suggests that RYR2 gene is potentially a candidate pathogenic gene of BECTS. More attention should be paid to epilepsy patients with RYR2 mutations, which were associated with arrhythmia and sudden unexpected death in previous reports.
Introduction
RYR2 gene (OMIM∗180902) resides on chr1q43. It encodes ryanodine receptor 2 protein (RYR-2) that is mainly located on endoplasmic reticulum (ER) membrane. RYR-2, as a subunit of homotetramer, constituted the calcium channel that regulates intracellular calcium concentration. The RYR-2 protein is ubiquitously distributed and highly expressed in the heart and brain, during all stages of human development (Martin et al., 1998). The RYR2 mutations have been established in the etiology of arrhythmogenic right ventricular dysplasia 2 (ARVD2) and catecholaminergic polymorphic ventricular tachycardia (CPVT) (Tiso et al., 2002). A recent study identified an RYR2 mutation in a family with CPVT, in which an affected individual presented epilepsy (Yap and Smyth, 2019). However, the relationship between RYR2 gene and epilepsy is not determined.
Benign epilepsy of childhood with centrotemporal spikes (BECTS), also known as Rolandic epilepsy (OMIM# 245570)1, is a common form of epilepsy in children and estimated to affect approximately 0.2% of the population (Kramer et al., 2002). Although BECTS is generally regarded as autosomal dominant inheritance with age-dependent penetrance, the genetic causes of BECTS are largely undetermined. Recent studies have shown that several genes, including GRIN2A, ELP4, SRPX2, and DEPDC5 (Lemke et al., 2013; Lal et al., 2014; Reinthaler et al., 2014), are associated with BECTS. Approximately 4.9% of the patients with BECTS harbor a variant of the glutamate ionotropic receptor N-methyl-D-aspartate type subunit 2A (GRIN2A) gene (Lemke et al., 2013). In this study, whole-exome sequencing (WES) was performed on a group of patients with BECTS and their parents (trios). RYR2 mutations, including two compound heterozygous missense mutations, were identified in five unrelated BECTS cases with or without arrhythmia. This study suggests that RYR2 gene is potentially a candidate pathogenic gene of BECTS. More attention should be paid to epilepsy patients with RYR2 mutations, which were associated with arrhythmia and sudden unexpected death in previous reports (Bagnall et al., 2016).
Materials and Methods
Patients
A cohort of 292 cases with BECTS was recruited. The patients were from the Epilepsy Center of the Second Affiliated Hospital of Guangzhou Medical University between July 2016 and February 2020.
Clinical characteristics of the affected individuals were collected, including present age, gender, age at seizure onset, seizure course, detailed family history, general and neurological examination results, and effective antiepileptic drugs (AEDs). Magnetic resonance imaging (MRI) scans were performed to detect any brain structure abnormalities. Long-term (24 h) video electroencephalogram (EEG) examination was obtained that included open–close eyes test, intermittent photic stimulation, hyperventilation, and sleeping recording. The results were reviewed by two qualified electroencephalographers. Epileptic seizures and epilepsies were diagnosed according to the criteria of the Commission on Classification and Terminology of the ILAE (1981, 1989, 2001, 2010, and 2017). All of the subjects were followed up for at least 1 year in the epilepsy center.
The studies adhered to the guidelines of the International Committee of Medical Journal Editors with regard to patient consent for research or participation and received approval from the ethics committee of the Second Affiliated Hospital of Guangzhou Medical University. All participants gave written informed consents.
Whole-Exome Sequencing
Blood samples were obtained from the probands, their parents, and available family members to determine the origin of the identified genetic variants. Genomic DNA was extracted from peripheral blood using a QuickGene DNA whole-blood kit (Fujifilm, Tokyo, Japan). Trio-based WES was conducted on the Illumina HiSeq 2500/4000 platform by BGI-Shenzhen (Shenzhen, China) as previously reported (Wang et al., 2018; Shi et al., 2019). To identify candidate causative variants in each trio, we adopted a case-by-case analytical approach. We first prioritized the rare variants with a minor allele frequency (MAF) <0.005 in the 1000 Genomes Projects, Exome Aggregation Consortium, and gnomAD. We retained potentially pathogenic variants containing frameshift, nonsense, canonical splice site, initiation codon, and missense mutations predicted as being damaging by in silico tools2. We then screened the potential disease-causing mutations in each case under five models: (1) epilepsy-associated gene mutations; (2) de novo dominant mutations; (3) autosomal recessive inheritance model, including homozygous and compound heterozygous variants; (4) X-linked model; and (5) cosegregated mutations. After excluding the known epilepsy-associated genes (Wang et al., 2017), the genes with null mutations, de novo mutations, biallelic mutations, hemizygous mutations, and mutations with segregations were considered for further evaluation as possible novel epilepsy genes. Conservation of mutated positions was evaluated using sequence alignment of different species. All RYR2 mutations were annotated based on the transcript NM_001035.2. Variants having potential clinical significance were confirmed by Sanger sequencing.
Protein modeling was performed to predict the effects of missense variants on molecular structure by using SWISS-MODEL (Shen et al., 2018)3, based on the updated template of 6jiy.1.A.pdb4. PyMOL 1.7 was used for three-dimensional protein structure visualization and analysis.
Statistical Analysis
SPSS Statistics 26.0 was used for statistical analysis. The frequencies of the RYR2 variants in the cohort of BECTS and in the control populations were compared by Fisher exact test (CONVERGE consortium, 2015). A p-value of < 0.05 was considered to be statistically significant.
Evaluating RYR2 Gene as a Novel Candidate Epilepsy Gene
To explore the genotype–phenotype association, we used the Clinical Validity Framework that was developed by Clinical Genome Resource (ClinGen) to evaluate RYR2 as a novel candidate epilepsy gene (Strande et al., 2017).
Results
Identification of RYR2 Mutations
Among the 292 patients with BECTS, RYR2 mutations were identified in five unrelated cases. The mutations included one heterozygous frameshift mutation (c.14361dup/p.Arg4790Pro fs∗6), two heterozygous missense mutations (c.2353G > A/p.Asp785Asn and c.8574G > A/p.Met2858Ile), and two pairs of compound heterozygous mutations (c.4652A > G/p.Asn1551Ser and c.11693T > C/p.Ile3898Thr, c.7469T > C/p.Val2490Ala and c.12770G > A/p.Arg4257Gln, respectively; Figures 1A,B). The heterozygous frameshift mutation Arg4790Pro fs∗6 was inherited from his asymptomatic father. The heterozygous Asp785Asn was a de novo mutation. Segregation of the identified mutations is shown in Figure 1A.
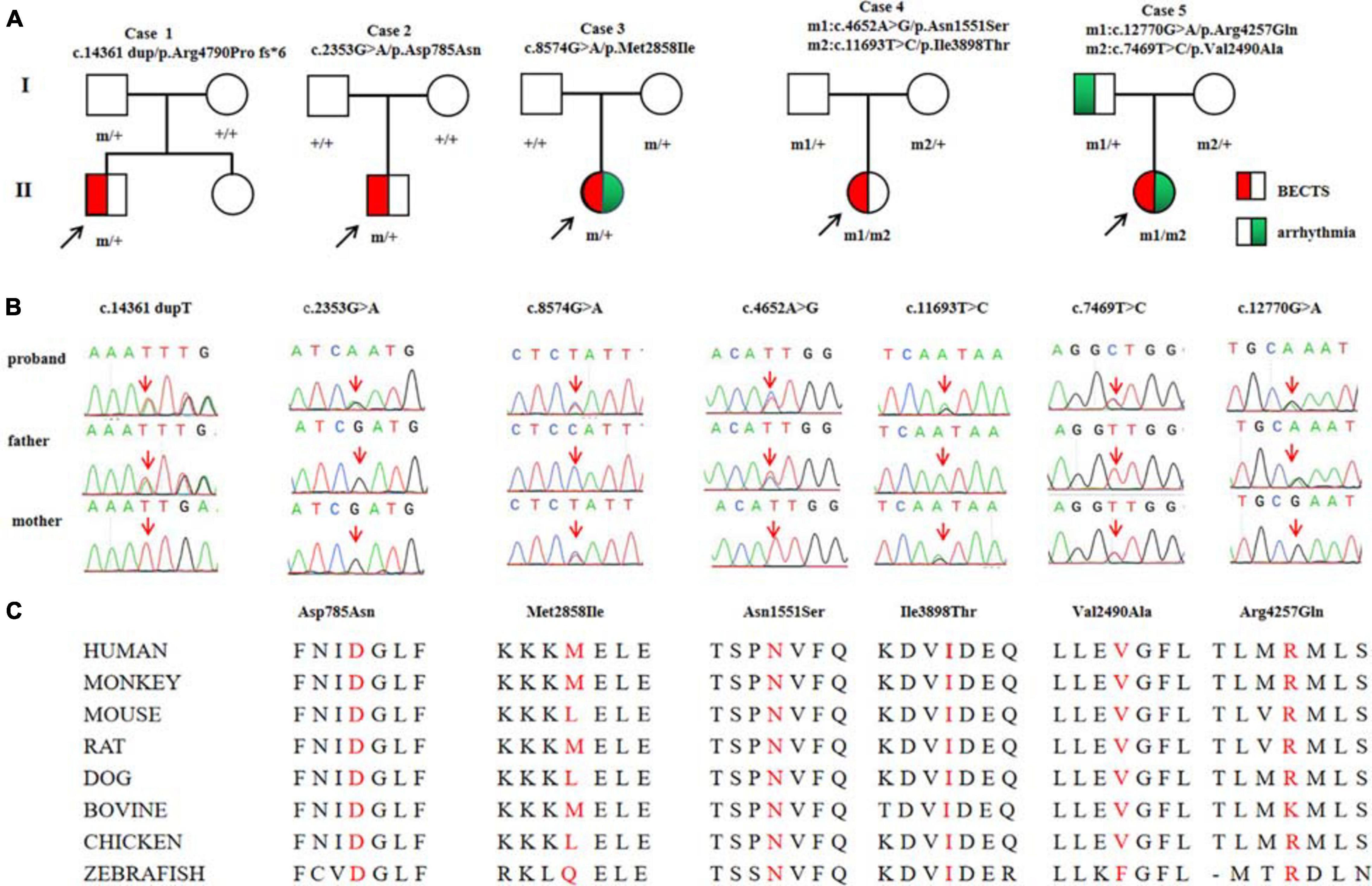
Figure 1. Genetic data on the patients with RYR2 mutations. (A) Pedigrees of the five cases with RYR2 mutations and their corresponding phenotypes. (B) DNA sequence chromatogram of the RYR2 mutations. Arrows indicate the positions of the mutations. (C) The amino acid sequence alignment of the residues of missense mutations shows Asp785, Asn1551, and Ile3898 are highly conserved in various species, whereas Val2490, Met2858, and Arg4257 are less conserved.
Mutation Arg4790Pro fs∗6 was assumed to be deleterious by causing premature termination of transcript and leading to complete loss of RYR-2 function or haploinsufficiency. All the missense mutations were suggested to be damaging by at least three web-based prediction tools (Table 1). These mutations are not present or at low MAF in gnomAD database (the MAF of Asp785Asn and Asn1551Ser were 4.0e-6 and 3.0e-4, respectively). The missense mutations (Asp785Asn, Asn1551Ser, and Ile3898Thr) affected amino acid residues that are highly conserved in various species, whereas the missense mutations (Val2490Ala, Met2858Ile, and Arg4257Gln) were less conserved by sequence alignment (Figure 1C), but were predicted to be conserved by in silico tools (Table 1). All the five cases had no other pathogenic or likely pathogenic mutations in the 977 genes known to be associated with seizure disorders (Wang et al., 2017). A statistical analysis on the frequency of the RYR2 variants showed significant difference between the present cohort and the control populations in gnomAD (7/584 vs. 40/108530 in controls of all populations, p = 6.34e-9; vs. 35/8638 in controls of East-Asian population, p = 0.015) (Table 2).
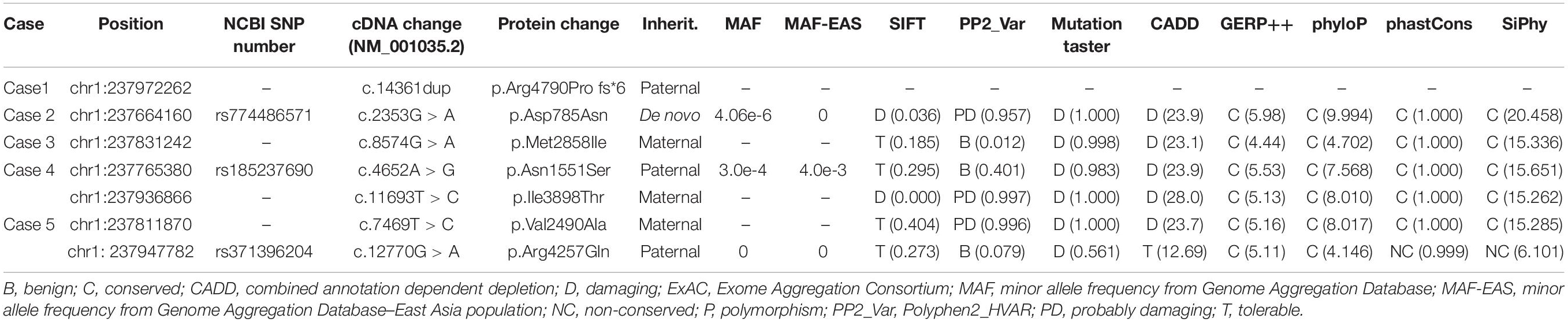
Table 1. Genetic features of the individuals with RYR2 mutations.
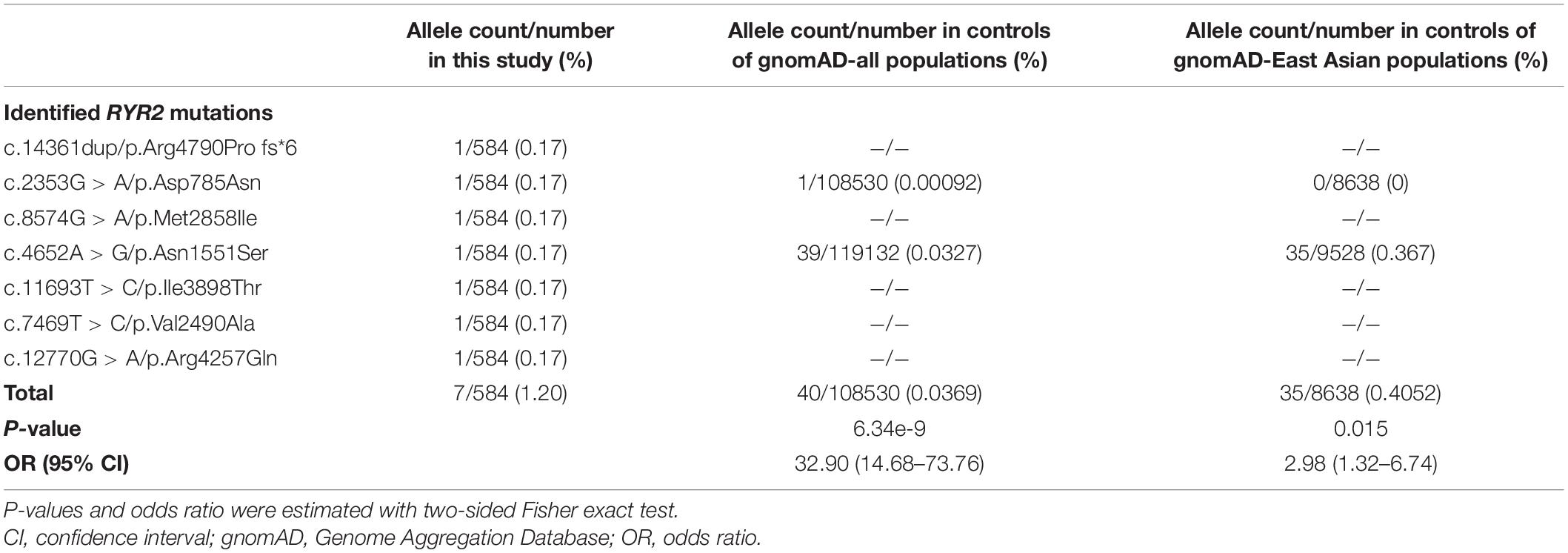
Table 2. Gene-based burden analysis for RYR2 mutations identified in this study.
Molecular Effect of RYR2 Mutations
As shown schematically in Figure 2, the molecular length of RYR-2 protein includes 4,967 amino acids5. Topological domains (1–4,281) and (4,871–4,967) are located within the cytoplasm. Six helical domains (4,282–4,302, 4,504–4,524, 4,580–4,600, 4,730–4,750, 4,769–4,789, and 4,850–4,870) and one pore-forming domain (4,820–4,829) are transmembrane. The cytoplasmic parts include MIR1-5 domains (110–408), SPRY1-3 domains (599–809, 1,025–1,222, and 1,337–1,562), and a domain of interaction with CALM (3,581–3,610). All the missense mutations were located in the cytoplasmic part of RYR-2 protein. Asp785Asn was located in SPRY1 domain, and Asn1551Ser was located in SPRY3 domain (Figure 2). The molecular effect of the missense mutations was further analyzed by protein modeling using SWISS-MODEL. Three missense mutations changed the hydrogen bands. Residue Asp785 formed hydrogen bonds with Gly786 and Ser699 originally. When aspartic acid was replaced by asparagine at residue Asp785, the hydrogen bond with Gly786 was destroyed. Residue Asn1551 originally formed two hydrogen bonds with Phe1290 and one hydrogen bond with Phe1443. When asparagine was replaced by serine at residue Asn1551, one of the hydrogen bonds with Phe1290 disappeared. Residue Ile3898 had no hydrogen bond with surrounding residues. When isoleucine was replaced by threonine at residue Ile 3898, two new hydrogen bonds with Tyr3892 and Gln3964 were formed. The mutations Met2858Ile, Val2490Ala, and Arg4257Gln did not change the hydrogen bonds, of which Val2490Ala and Arg4257Gln were a pair of compound heterozygous mutation (Figure 3).
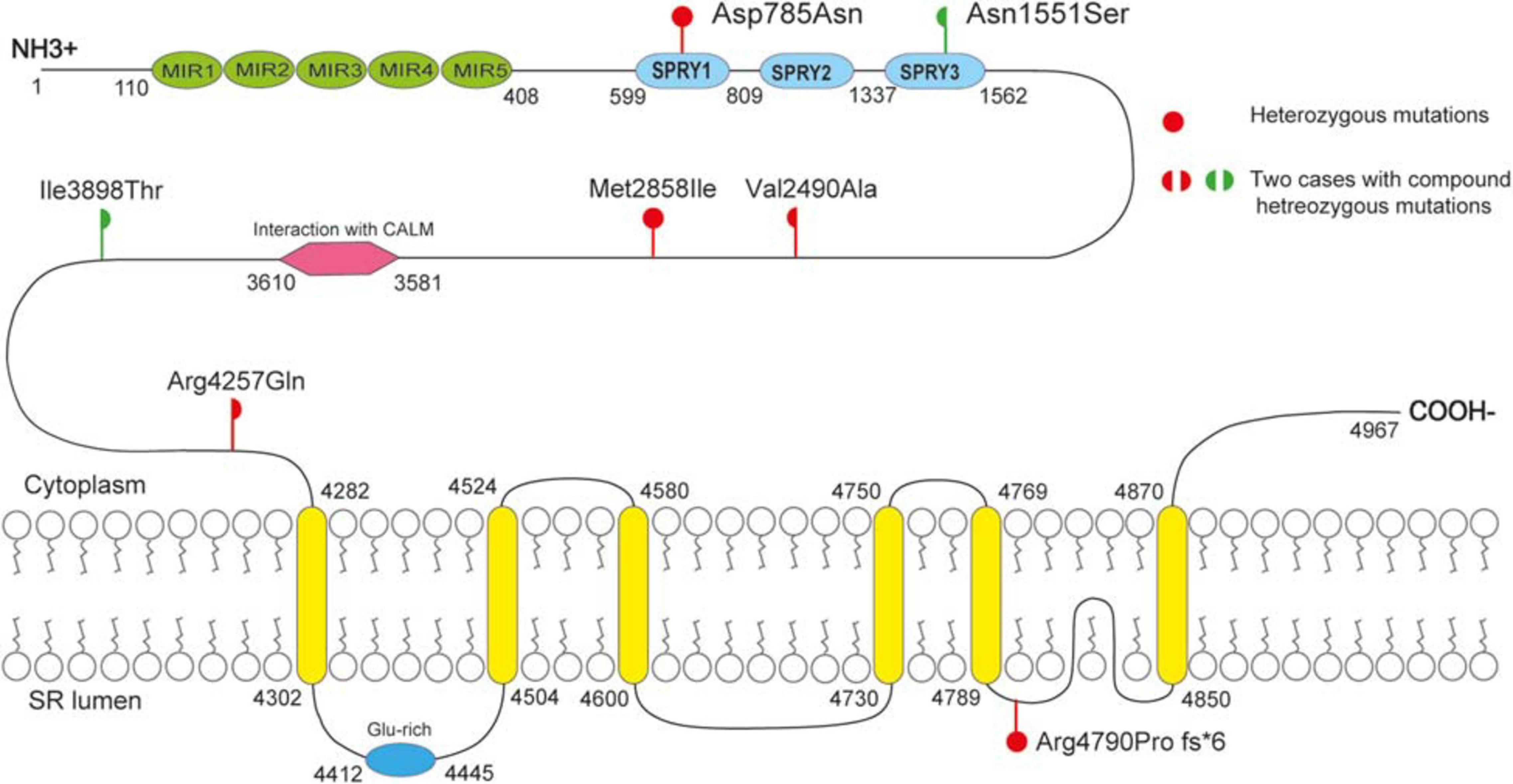
Figure 2. Schematic illustration of mutations. All the missense mutations were located in the cytoplasmic part of RYR-2 protein. The de novo missense mutation Asp785Asn was located in SPRY1 domain; mutation Asn551 was located in SPRY3 domain.
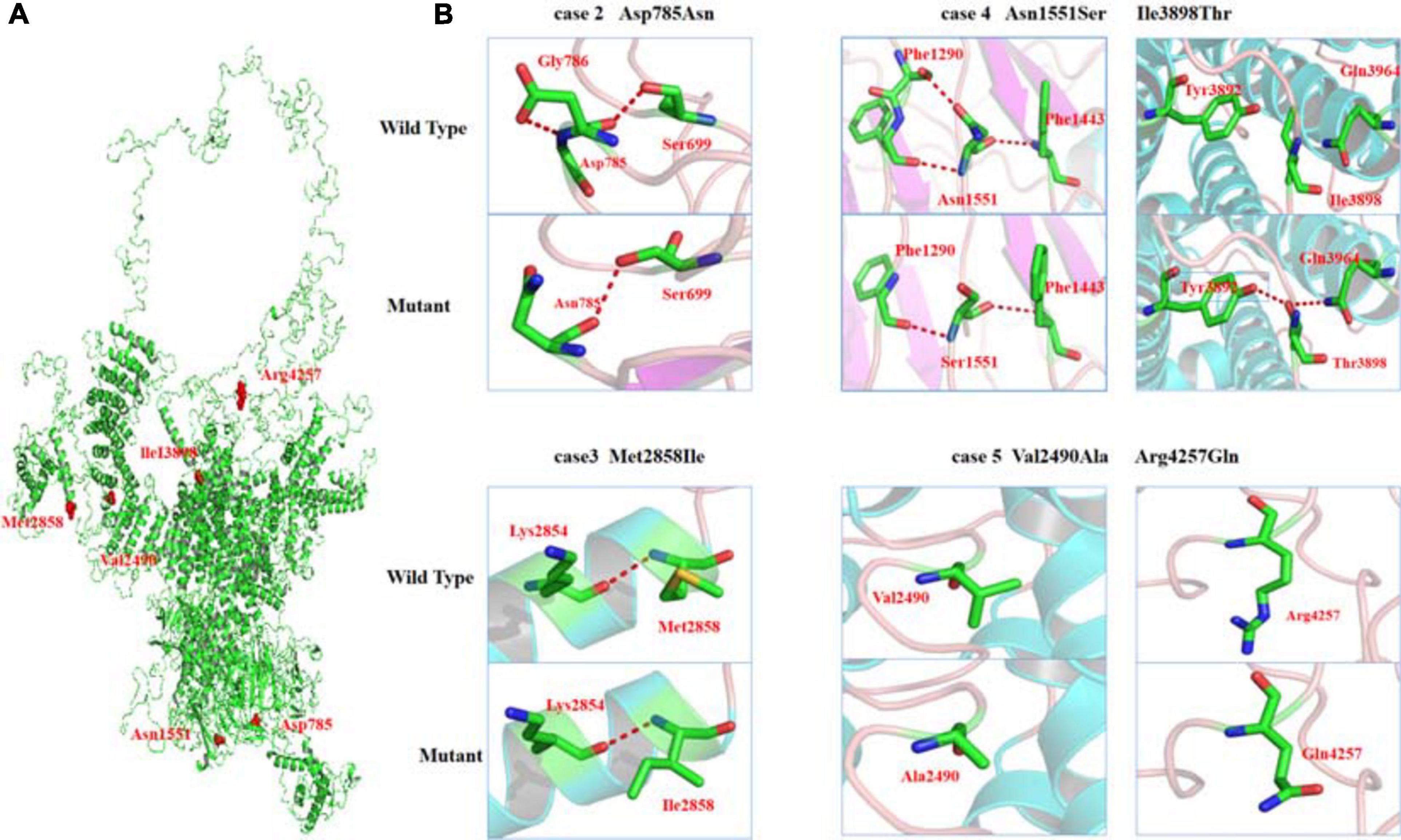
Figure 3. Crystal structure of RYR-2 (PDB: 6jiy) and hydrogen bond changes of the mutants. (A) Overview of RYR2 and positions of the mutations. (B) Asp785Asn, hydrogen bond with Gly786 disappeared; Asn1551Ser, hydrogen bond with Phe1290 disappeared; Ile3898Thr, formed new hydrogen bonds with Tyr3892 and Gln3964.
Clinical Features of the Cases With RYR2 Mutations
All probands, including two boys and three girls, showed childhood-onset focal epilepsy (Figure 1A). Table 3 summarized the main clinical features of the cases with RYR2 mutations. The onset age of seizures ranged from 4 to 12 years. All the cases had infrequent generalized tonic–clonic seizure (GTCS) and/or complex partial seizure. All the cases had normal development, intelligence, and speech ability. They did not have any other neurological symptoms, either. The EEGs showed unilateral or bilateral independent centrotemporal discharges, predominantly during sleep in all probands (representatives in Figures 4A,B). Arrhythmia was found in two families (three individuals in cases 3 and 5). In case 3, arrhythmias in interictal stage were recorded; her ictal EEG showed a seizure originated from the left central area, which was not secondary to cardiac attack (Figure 4C). The probands in case 5 had sinus arrhythmia with occasional atrial premature beats (Figure 4D). Her father presented the symptom of palpitations. His ECG examination also showed sinus arrhythmia. Additionally, light tricuspid regurgitation was detected in case 4, and light tricuspid and pulmonary valve regurgitations were detected in case 5 by cardiac ultrasound. The brain MRI scans in all patients were normal. All the cases got seizure-free with monotherapy of valproate or lamotrigine, or combination of valproate and levetiracetam.
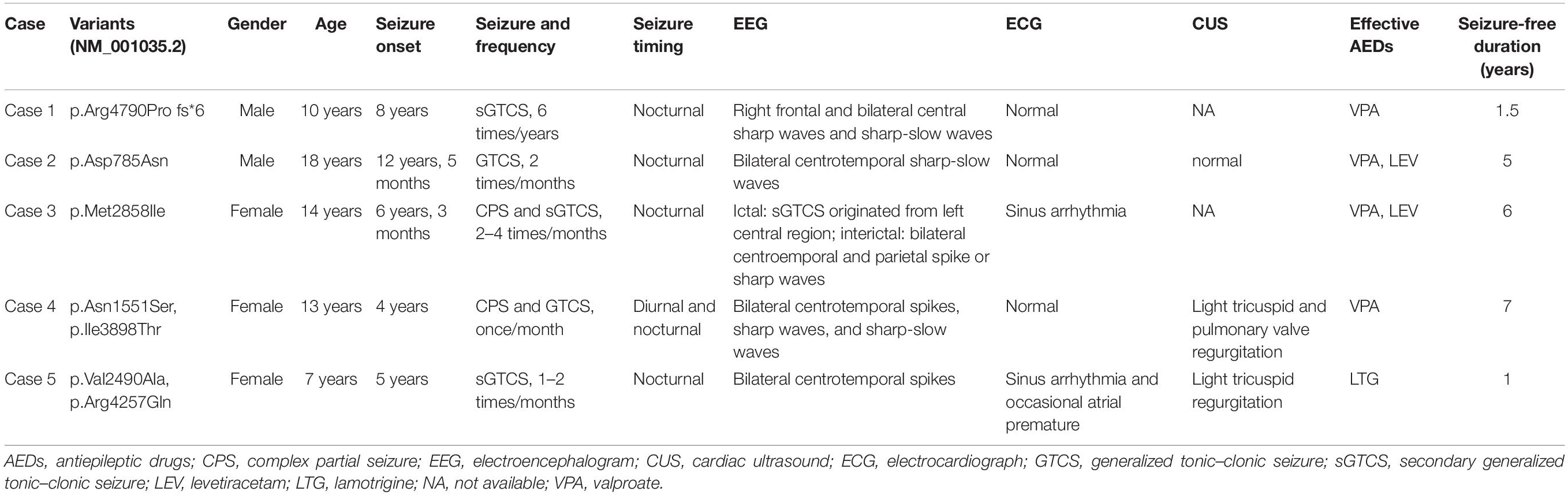
Table 3. Clinical features of the individuals with RYR2 mutations.
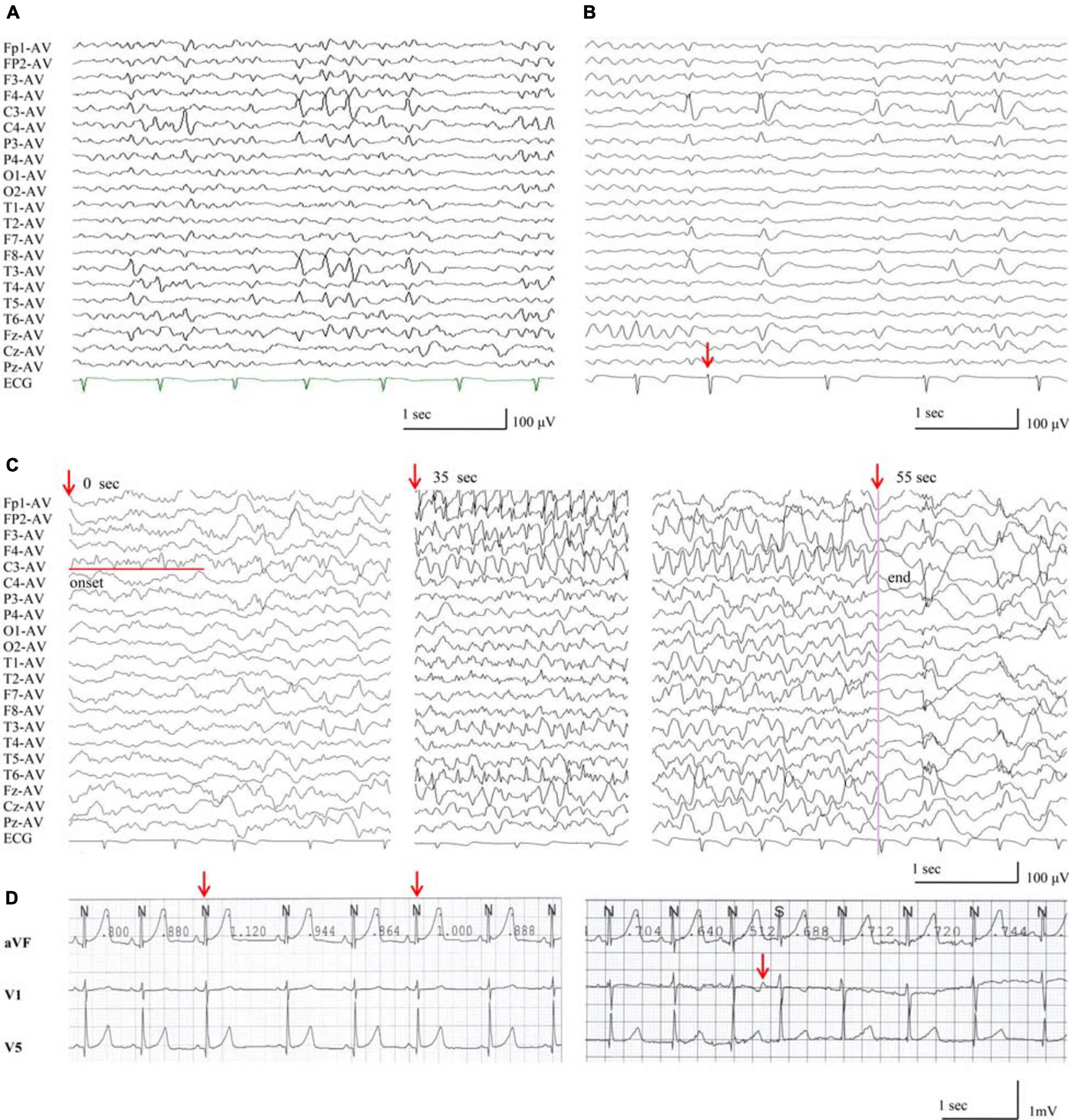
Figure 4. EEG discharges in the patients with RYR2 mutations. (A) Interictal EEG of case 4 showed left or right centrotemporal spike and slow waves (obtained at the age of 11 years). (B) Interictal EEG of case 3, the red arrow indicates electrodes recording ECG of arrhythmia during NREMIII sleep (at the age of 7 years). (C) Ictal EEG of case 3, the red arrows indicate timing of seizure (at the age of 7 years). (D) The ECG of case 5 showed sinus arrhythmia and atrial premature beats.
Evaluation of Epilepsy as a Novel Phenotype of RYR2 Variants
We evaluated the RYR2 variants-epilepsy correlation using ClinGen Clinical-Validity Framework. Both total allowable points for genetic aspect and experimental aspect were 6 points. The results of clinical validity summary matrix were 12 points that was categorized as “strong,” supporting the association between RYR2 variants and epilepsy (Table 4).
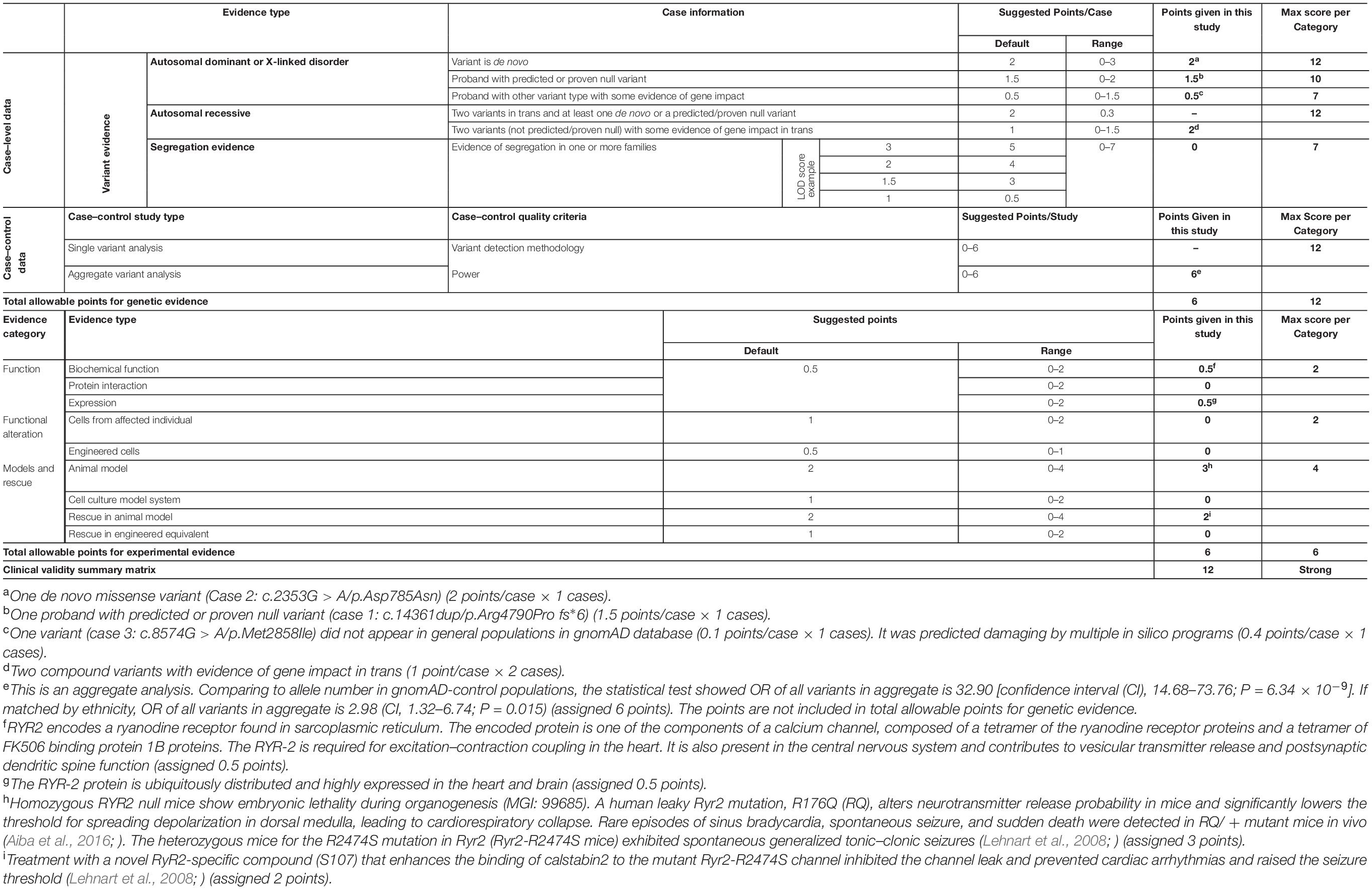
Table 4. Evaluating the clinical validity of RYR2-epilepsy associations based on the framework developed by the clinical genome resource.
Discussion
In the present study, RYR2 mutations were identified in five unrelated cases with BECTS, including one case with heterozygous frameshift mutation, one case with de novo heterozygous missense mutation, and two cases with compound heterozygous mutations. As a subunit of homotetramer, RYR-2 constitutes the calcium channel that regulates intracellular calcium concentration (Marx et al., 2000). The RYR-2 protein is mainly distributed on the ER membrane and is ubiquitously expressed with predominance in the heart and brain during all stages of human development6. In RYR2-deficiency mice, homozygotes showed embryonic lethality during organogenesis and altered cardiomyocyte morphology (Choy et al., 2016), suggesting that RYR2 is vital for fetal development. The heterozygous deficiency mice presented increased startle reflex, as well as cardiovascular abnormities, such as abnormal myocardial fiber physiology, irregular heartbeat, and ventricular premature beat (Loaiza et al., 2013; Liu et al., 2014; Choy et al., 2016; Dickinson et al., 2016). The RYR2 mutations have been established in the etiology of ARVD2 and CPVT (Tiso et al., 2002). RYR-2 mediates intracellular Ca2+ release to trigger cardiomyocyte contraction through L-type voltage gated channels. Abnormal functioning of RYR-2 will result in imbalance of intracellular calcium ion homeostasis, leading to arrhythmia, cardiac hypertrophy, heart failure, and other heart abnormalities (Seidel et al., 2020). Patients with ARVD2 or CPVT occasionally had seizures. However, the relationship between RYR2 and epilepsy is unclear. In the present study, RYR2 mutations were identified in five cases with BECTS. The frequency of the RYR2 variants in the cohort of epilepsy was significantly higher than that in control populations in gnomAD. Taken together the facts that RYR-2 is highly expressed in the brain (Martin et al., 1998) and that RYR2 knockout animals presented increased cellular excitability (Loaiza et al., 2013; Liu et al., 2014; Choy et al., 2016; Dickinson et al., 2016; Seidel et al., 2020), it is considered that RYR2 gene is potentially a candidate pathogenic gene of epilepsy. The evidence from ClinGen Clinical-Validity Framework also supports a strong association between RYR2 mutations and epilepsy.
Generally, calcium homeostasis is critical for neuronal stability, and RYR-2 plays a key role in the regulation of calcium homeostasis (Lin et al., 2018). Intracellular Ca2+ is an important secondary messenger for signal transduction and essential for cellular processes such as excitatory contraction coupling. The major sources of intracellular Ca2+ include the sarcoplasmic reticulum (SR) in striated muscle and the ER in other cell types (Otsu et al., 1990). There are two major Ca2+ release channels localized in the SR/ER, the ryanodine receptors (RyRs) (Lanner et al., 2010), and inositol 1,4,5-triphosphate receptors (IP3Rs) (Nixon et al., 1994). RyRs, encoded by RYR1, RYR2, and RYR3, are widely distributed in the central nervous system of human beings; RYR-2 is the most abundant isoform in the brain, especially in hippocampus (Martin et al., 1998), suggesting a central role in Ca2+ signaling and Ca2+ homeostasis in the human central nervous system. Previous studies in animals have indicated that the RYR2 deficiency is associated with epilepsy or alterations of electrical conduction in nerve cells. GTCSs were observed in knock-in mouse model of heterozygous RYR2 missense mutation Arg2474Ser, which was identified in the patients with CPVT (Lehnart et al., 2008). RYR2 Arg176Gln mutant mice altered neurotransmitter releasing probability and lowered the threshold for spreading depolarization significantly, leading to spreading depolarization across the neocortex and brainstem dorsal medulla autonomic microcircuits (Aiba et al., 2016). Clinically, in a family with RYR2 mutation (c.229G > A/p.Ala77Thr) that caused CPVT, one affected individual presented unprovoked generalized seizures and epileptiform activities (Yap and Smyth, 2019). A compound heterozygous missense mutation (c.3248A > G/p.Glu1083Gly and c. 6779 C > T/p.Arg2260Leu) had been identified in a child with early infantile epileptic encephalopathy (Peng et al., 2018). The ictal EEG with focal originated discharges presented in this study provided direct evidence of epileptic seizure, instead of attacks due to heart abnormalities. These evidences suggest RYR2 mutations were potentially associated with epilepsy. All cases in this study were diagnosed as BECTS, suggesting RYR2 was potentially a causative gene of BECTS.
Recent studies have shown close relationships between epilepsy and heart disorders that were caused by several genetic abnormalities (Parisi et al., 2013; Partemi et al., 2015; Coll et al., 2016, 2017). These abnormalities were also associated with sudden unexpected death in epilepsy (SUDEP), which is the leading cause of epilepsy-related premature mortality. Three individuals with RYR2 mutations in the present study showed arrhythmia. The two probands with compound heterozygous missense mutations presented mild cardiac structural abnormalities. The RYR2 mutations have been established in the etiology of ARVD2 and CPVT (Tiso et al., 2002), in which arrhythmia was common. So far, 547 RYR2 mutations have been identified, mostly in patients with cardiac disorders (HGMD)7. A recent study showed that one RYR2 mutation was associated with SUDEP, and the patient had occasionally nocturnal seizures (Bagnall et al., 2016). Risk of triggering cardiac arrhythmias by AEDs has also been reported (Shmuely et al., 2017; Yap and Smyth, 2019; Banfi et al., 2020). Thus, special attention should be paid to the patients with RYR2 mutations, who may present benign epilepsy such as BECTS but potentially require careful follow-up and specialized health consultation due to the coexistence of epilepsy and heart disease.
This study has several limitations. The direct functional effects of the mutations identified in the present study were not examined. The mutations associated with heart disorders presented functional abnormalities of loss of function or gain of function. Further studies are required to characterize the functional effects of the mutations associated with epilepsy. In the present study, two pairs of compound heterozygous mutations (p.Asn1551Ser and p.Ile3898Thr, and p.Val2490Ala and p.Arg4257Gln, respectively) were identified in the patients with epilepsy. RYR2 is usually regarded as a dominant gene. Biallelic pathogenic variants have been reported in dominant genes, such as GRIN1 gene (Lemke et al., 2016). Generally, gene variants differ in damage effect, which varies from mild to severe, depending on factors such as the molecular subregional effect as shown in our recent study (Tang et al., 2020). It is possible that these RYR2 mutations were of less damage impact. Each of them alone seemed not pathogenic, whereas biallelic variants compounding together would be pathogenic. Two individuals, the father with p.Arg4790Profs∗6 and the mother with p.Met2858Ile, were asymptomatic, suggesting incomplete penetrance. The patients with RYR2 mutations presented BECTS, which was generally a mild form of epilepsy, reflecting relatively weak pathogenicity of the RYR2 mutations and potentially explaining the incomplete penetrance. However, it is unknown whether RYR2 mutations were associated with more severe forms of epilepsy, and the whole spectrum of phenotype of RYR2 mutations warrants further studies. Several patients presented cardiac abnormalities. The occurrence and significance of cardiac phenotype in epilepsy patients with RYR2 mutations and their long-term outcome need further observation and follow-up.
Conclusion
In conclusion, we identified RYR2 mutations in five unrelated cases with BECTS. The genetic and molecular evidence implied that the mutations led to the defect of the RYR2 gene. Taken together the data from gene expression profile and RYR2 deficiency animal model, it is suggested that RYR2 was potentially a causative gene epilepsy. Special attention should be paid to the patients with RYR2 mutations due to the coexistence of epilepsy and heart disease, which potentially were potentially associated with SUDEP.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Second Affiliated Hospital of Guangzhou Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
Y-HY and W-PL designed and conceptualized the study. M-GM, X-RL, JW, B-ML, BL, and D-TL collected and analyzed the data. M-GM and X-RL drafted the manuscript for intellectual content. M-GM, X-RL, YW, Y-WS, TS, Y-HY, and W-PL revised the manuscript for intellectual content. BL and D-TL provided software support for 3D structure model. M-GM and D-TL prepared the figures. All authors have read and approved the final draft of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81870903 and 81871015), the Natural Science Foundation of Guangdong Province (2020A1515010108), the Science and Technology Project of Guangdong Province (2017B090904036 and 2017B030314159), the Science and Technology Project of Guangzhou (201904020028), and the Multi-center Clinical Research Fund Project of the Second Affiliated Hospital of Guangzhou Medical University (Grant Nos. DZX-002 and 2020-LCYJ-DZX-03).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are deeply grateful to the patients and clinicians who participated in this work.
Footnotes
- ^ http://www.omim.org
- ^ http://varcards.biols.ac.cn/
- ^ https://swissmodel.expasy.org/
- ^ http://www.rcsb.org
- ^ https://www.uniprot.org
- ^ http://www.gdap.org.cn/geneinfo.php
- ^ http://www.hgmd.cf.ac.uk/ac/search.php
References
Aiba, I., Wehrens, X. H., and Noebels, J. L. (2016). Leaky RyR2 channels unleash a brainstem spreading depolarization mechanism of sudden cardiac death. Proc. Natl. Acad. Sci. U.S.A. 113, E4895–E4903. doi: 10.1073/pnas.1605216113
Bagnall, R. D., Crompton, D. E., Petrovski, S., Lam, L., Cutmore, C., Garry, S. I., et al. (2016). Exome-based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann. Neurol. 79, 522–534. doi: 10.1002/ana.24596
Banfi, P., Coll, M., Oliva, A., Alcalde, M., Striano, P., Mauri, M., et al. (2020). Lamotrigine induced Brugada-pattern in a patient with genetic epilepsy associated with a novel variant in SCN9A. Gene 754:144847. doi: 10.1016/j.gene.2020.144847
Choy, L., Yeo, J. M., Tse, V., Chan, S. P., and Tse, G. (2016). Cardiac disease and arrhythmogenesis: mechanistic insights from mouse models. Int. J. Cardiol. Heart Vasc. 12, 1–10. doi: 10.1016/j.ijcha.2016.05.005
Coll, M., Allegue, C., Partemi, S., Mates, J., Del Olmo, B., Campuzano, O., et al. (2016). Genetic investigation of sudden unexpected death in epilepsy cohort by panel target resequencing. Int. J. Legal. Med. 130, 331–339. doi: 10.1007/s00414-015-1269-0
Coll, M., Striano, P., Ferrer-Costa, C., Campuzano, O., Mates, J., Del Olmo, B., et al. (2017). Targeted next-generation sequencing provides novel clues for associated epilepsy and cardiac conduction disorder/SUDEP. PLoS One 12:e0189618. doi: 10.1371/journal.pone.0189618
CONVERGE consortium (2015). Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591. doi: 10.1038/nature14659
Dickinson, M. E., Flenniken, A. M., Ji, X., Teboul, L., Wong, M. D., White, J. K., et al. (2016). High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514. doi: 10.1038/nature19356
Kramer, U., Zelnik, N., Lerman-Sagie, T., and Shahar, E. (2002). Benign childhood epilepsy with centrotemporal spikes: clinical characteristics and identification of patients at risk for multiple seizures. J. Child. Neurol. 17, 17–19. doi: 10.1177/088307380201700104
Lal, D., Reinthaler, E. M., Schubert, J., Muhle, H., Riesch, E., Kluger, G., et al. (2014). DEPDC5 mutations in genetic focal epilepsies of childhood. Ann. Neurol. 75, 788–792. doi: 10.1002/ana.24127
Lanner, J. T., Georgiou, D. K., Joshi, A. D., and Hamilton, S. L. (2010). Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2:a003996. doi: 10.1101/cshperspect.a003996
Lehnart, S. E., Mongillo, M., Bellinger, A., Lindegger, N., Chen, B. X., Hsueh, W., et al. (2008). Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Invest. 118, 2230–2245. doi: 10.1172/JCI35346
Lemke, J. R., Geider, K., Helbig, K. L., Heyne, H. O., Schutz, H., Hentschel, J., et al. (2016). Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology 86, 2171–2178. doi: 10.1212/WNL.0000000000002740
Lemke, J. R., Lal, D., Reinthaler, E. M., Steiner, I., Nothnagel, M., Alber, M., et al. (2013). Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 45, 1067–1072. doi: 10.1038/ng.2728
Lin, Y., He, S., Liao, Z., Feng, R., Liu, R., Peng, Y., et al. (2018). Whole exome sequencing identified a pathogenic mutation in RYR2 in a Chinese family with unexplained sudden death. J. Electrocardiol. 51, 309–315. doi: 10.1016/j.jelectrocard.2017.10.002
Liu, Y., Wang, R., Sun, B., Mi, T., Zhang, J., Mu, Y., et al. (2014). Generation and characterization of a mouse model harboring the exon-3 deletion in the cardiac ryanodine receptor. PLoS One 9:e95615. doi: 10.1371/journal.pone.0095615
Loaiza, R., Benkusky, N. A., Powers, P. P., Hacker, T., Noujaim, S., Ackerman, M. J., et al. (2013). Heterogeneity of ryanodine receptor dysfunction in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 112, 298–308. doi: 10.1161/CIRCRESAHA.112.274803
Martin, C., Chapman, K. E., Seckl, J. R., and Ashley, R. H. (1998). Partial cloning and differential expression of ryanodine receptor/calcium-release channel genes in human tissues including the hippocampus and cerebellum. Neuroscience 85, 205–216. doi: 10.1016/s0306-4522(97)00612-x
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/s0092-8674(00)80847-8
Nixon, G. F., Mignery, G. A., and Somlyo, A. V. (1994). Immunogold localization of inositol 1,4,5-trisphosphate receptors and characterization of ultrastructural features of the sarcoplasmic reticulum in phasic and tonic smooth muscle. J. Muscle Res. Cell Motil. 15, 682–700. doi: 10.1007/BF00121075
Otsu, K., Willard, H. F., Khanna, V. K., Zorzato, F., Green, N. M., and MacLennan, D. H. (1990). Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J. Biol. Chem. 265, 13472–13483. doi: 10.1016/s0021-9258(18)77371-7
Parisi, P., Oliva, A., Coll Vidal, M., Partemi, S., Campuzano, O., Iglesias, A., et al. (2013). Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. 105, 415–418. doi: 10.1016/j.eplepsyres.2013.02.024
Partemi, S., Vidal, M. C., Striano, P., Campuzano, O., Allegue, C., Pezzella, M., et al. (2015). Genetic and forensic implications in epilepsy and cardiac arrhythmias: a case series. Int. J. Legal. Med. 129, 495–504. doi: 10.1007/s00414-014-1063-4
Peng, J., Wang, Y., He, F., Chen, C., Wu, L. W., Yang, L. F., et al. (2018). Novel West syndrome candidate genes in a Chinese cohort. CNS Neurosci Ther. 24, 1196–1206. doi: 10.1111/cns.12860
Reinthaler, E. M., Lal, D., Jurkowski, W., Feucht, M., Steinbock, H., Gruber-Sedlmayr, U., et al. (2014). Analysis of ELP4, SRPX2, and interacting genes in typical and atypical rolandic epilepsy. Epilepsia 55, e89–e93. doi: 10.1111/epi.12712
Seidel, M., de Meritens, C. R., Johnson, L., Parthimos, D., Bannister, M., Thomas, N. L., et al. (2020). Identification of an amino-terminus determinant critical for ryanodine receptor/Ca2+ release channel function. Cardiovasc. Res. 117, 780–791. doi: 10.1093/cvr/cvaa043
Shen, K., Huang, R. K., Brignole, E. J., Condon, K. J., Valenstein, M. L., Chantranupong, L., et al. (2018). Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature 556, 64–69. doi: 10.1038/nature26158
Shi, Y. W., Zhang, Q., Cai, K., Poliquin, S., Shen, W., Winters, N., et al. (2019). Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes. Brain 142, 3028–3044. doi: 10.1093/brain/awz250
Shmuely, S., van der Lende, M., Lamberts, R. J., Sander, J. W., and Thijs, R. D. (2017). The heart of epilepsy: current views and future concepts. Seizure 44, 176–183. doi: 10.1016/j.seizure.2016.10.001
Strande, N. T., Riggs, E. R., Buchanan, A. H., Ceyhan-Birsoy, O., DiStefano, M., Dwight, S. S., et al. (2017). Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am. J. Hum. Genet. 100, 895–906. doi: 10.1016/j.ajhg.2017.04.015
Tang, B., Li, B., Gao, L. D., He, N., Liu, X. R., Long, Y. S., et al. (2020). Optimization of in silico tools for predicting genetic variants: individualizing for genes with molecular sub-regional stratification. Brief. Bioinform. 21, 1776–1786. doi: 10.1093/bib/bbz115
Tiso, N., Salamon, M., Bagattin, A., Danieli, G. A., Argenton, F., and Bortolussi, M. (2002). The binding of the RyR2 calcium channel to its gating protein FKBP12.6 is oppositely affected by ARVD2 and VTSIP mutations. Biochem. Biophys. Res. Commun. 299, 594–598. doi: 10.1016/s0006-291x(02)02689-x
Wang, J., Lin, Z. J., Liu, L., Xu, H. Q., Shi, Y. W., Yi, Y. H., et al. (2017). Epilepsy-associated genes. Seizure 44, 11–20. doi: 10.1016/j.seizure.2016.11.030
Wang, J. Y., Zhou, P., Wang, J., Tang, B., Su, T., Liu, X. R., et al. (2018). ARHGEF9 mutations in epileptic encephalopathy/intellectual disability: toward understanding the mechanism underlying phenotypic variation. Neurogenetics 19, 9–16. doi: 10.1007/s10048-017-0528-2
Keywords: RYR2, mutations, epilepsy, BECTS, arrhythmia
Citation: Ma M-G, Liu X-R, Wu Y, Wang J, Li B-M, Shi Y-W, Su T, Li B, Liu D-T, Yi Y-H and Liao W-P (2021) RYR2 Mutations Are Associated With Benign Epilepsy of Childhood With Centrotemporal Spikes With or Without Arrhythmia. Front. Neurosci. 15:629610. doi: 10.3389/fnins.2021.629610
Received: 15 November 2020; Accepted: 19 February 2021;
Published: 07 April 2021.
Edited by:
Oscar Campuzano, University of Girona, SpainReviewed by:
Monica Coll, Institute of Biomedical Research of Girona, SpainPasquale Striano, University of Genoa, Italy
Copyright © 2021 Ma, Liu, Wu, Wang, Li, Shi, Su, Li, Liu, Yi and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Hong Yi, eXloMTY4QHNpbmEuY29t; Wei-Ping Liao, d3BsaWFvQDE2My5uZXQ=
†These authors have contributed equally to this work
‡ORCID: Xiao-Rong Liu, orcid.org/0000-0002-7769-9564; Wei-Ping Liao, orcid.org/0000-0001-9929-9185