 Jan O. Aasly1,2*
Jan O. Aasly1,2*- 1Department of Neurology, St. Olavs Hospital, Trondheim, Norway
- 2Department of Neuromedicine and Movement Science (INB), Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
The first families with LRRK2 related Parkinson’s disease (PD) were presented around 15 years ago and numerous papers have described the characteristics of the LRRK2 phenotype. The prevalence of autosomal dominant PD varies around the world mainly depending on local founder effects. The highest prevalence of LRRK2 G2019S PD in Norway is located to the central part of the country and most families could be traced back to common ancestors. The typical Norwegian LRRK2 phenotype is not different from classical PD and similar to that seen in most other LRRK2 families. The discovery of LRRK2 PD has allowed us to follow-up multi-incident families and to study their phenotype longitudinally. In the Norwegian LRRK2 families there has been a significantly higher incidence of inflammatory diseases like multiple sclerosis and rheumatoid arthritis that seen in other PD populations. Recent studies in LRRK2 mechanisms have indicated that this protein may be crucial in initiating disease processes. In this short survey of 100 Norwegian mutation carriers followed through more than 15 years are presented. The prevalence of inflammatory diseases among these cases is highlighted. The role of LRRK2 in the conversion process from carrier status to PD phenotype is still unknown and disease generating mechanisms important for initiating LRRK2 PD are still to be identified.
Introduction
The etiology of Parkinson’s disease, PD, is unknown and for many years it was regarded as a sporadic disease explained by environmental causes. The first PD gene locus was reported in 1996 and 1 year later the gene was located to the SNCA coding for α-synuclein (Polymeropoulos et al., 1997). This protein was later shown to constitute a major part of the Lewy-bodies, the pathoanatomical hallmark of Parkinson’s disease (Spillantini et al., 1997). During the following years three important genes coding for autosomal recessive PD were found, one for parkin, PRKN, the most common gene for young onset parkinsonism, and PINK1 and DJ-1 as autosomal recessive causes of PD (Kitada et al., 1998; Valente et al., 2001; Bonifati et al., 2003). A Japanese group had pointed to an important locus on chromosome 12 and mutations in the LRRK2 gene were finally identified by several other groups in 2004. LRRK2 is probably the most common cause of autosomal dominant PD and the most common monogenic form of PD (Paisan-Ruiz et al., 2004; Zimprich et al., 2004).
The discovery of LRRK2 as a major cause of PD has led to a tremendous race of new biomarkers for PD and new insights on disease pathogenesis (Paisan-Ruiz et al., 2004). The LRRK2 G2019S is the most prevalent risk factor among the LRRK2 mutations. It is most common in the Middle East and there is a very clear south north gradient in distribution. The prevalence of LRRK2 G2019S PD in Scandinavia is low with an exception located to the northwestern coast of Norway. This cohort of PD patients has been followed for many years and were included in many studies for better understanding of clinical and biochemical processes related to PD (Aasly et al., 2005). Although the LRRK2 PD phenotype is rather close to classical PD the long-term follow up of Norwegian LRRK2 cases have shown that inflammatory mechanism may contribute to the disease process.
Neuroinflammation is now considered to play a major role in the pathogenesis of PD. This change in paradigm has come after findings of activated microglia and upregulation of cytokines in PD brains. One of the most important substances seem to be cyclooxygenase-1 and -2, which show increased expression, together with inflammatory cytokines and inflammatory-related substances. The risk for PD is correlated with inflammatory cytokine genes (i.e., tumor necrosis factor-α and interleukin-1β) polymorphisms and with cell-surface human leukocyte antigen2 (Crotty et al., 2020).
The role of the immune system and inflammation and LRRK2 upregulation is also being increasingly explored and coupled to the innate and adaptive immune system. Large multi-center whole exome, WES, -studies have shown that the LRRK2 gene is associated with several chronic inflammatory disorders, including Crohn’s disease and leprosy (Hakimi et al., 2011).
The aims of this survey are to present the long-time follow up of the original Norwegian LRRK2 cohort and to discuss the possible importance of recognizing the high prevalence of inflammatory diseases among these cases.
Clinical Material
The LRRK2 G2019S cohort in central Norway was first presented in Aasly et al. (2005). The families had been followed for several years and more subjects were added to the material within a few years after the discovery of the mutation. The patients were identified and included as part of a screening program of multi-incident PD families living at the coastline of central Norway. When the LRRK2 gene was found and connected to PD, these families were further evaluated and characterized.
In this survey of the first 100 Norwegians known to carry the a heterozygous LRRK2 G2019S mutation, are presented. Twenty-nine patients out of the first 100 cases had developed PD at the time when they were identified through the family screening program. Three more cases converted to PD through the follow-up period. The age of onset of the 32 first PD cases varied substantially, with an average onset of 61.2 years. The majority of cases had converted to PD in their seventh decade. Nine cases, 29%, were in their sixties at disease onset and the mean age at onset and for this small group the mean onset was 64 years. Two patients had disease onset in their late thirties and two cases showed first signs of tremor and bradykinesia up in their eighties (Table 1). The phenotypical features at onset were similar to that commonly found in sporadic PD. All cases had asymmetric signs and symptoms, more than 80% showed rest tremor at onset. All patients diagnosed with PD had well preserved cognition at disease onset. The remaining 68 mutation carriers who had not developed PD and were diagnosed as healthy LRRK2 carriers were all assessed as cognitively well-doing. During the follow-up period three asymptomatic LRRK2 mutation carriers developed dementia of Lewy body type, DLB, without typical PD signs. The mean age of the mutation carriers who did not developed PD was 62 years (range 45–83 years) at the end of the follow-up period. The majority of cases had been evaluated clinically several times and had been to repeated PET- or Datscans.

Table 1. List of 100 LRRK2 G2019S mutation carriers, 15 years-follow-up.
Familial Clustering
The vast majority of Norwegian LRRK2 G2019S mutation carriers known so far, all originate from small settlements along the coastline of Central Norway. The LRRK2 mutation was found in about 15 families or family branches. The first married couple mentioned by name common to all these families, were born around year 1580 (Johansen et al., 2010). Although this is an autosomal dominant disease, multi-incident PD families were only reported in among half of the cases. In some families with clear familiar clustering of cases this knowledge was more or less kept as a secret and was not a topic for open discussions. When unveiling the cause of the disease these families became very motivated for further collaboration in the struggle for finding new therapeutic remedies. All families had been living in the same area since many generations and most families had families that could give precise information on their ancestors through the last century. One fourth of the families had 3 or more affected family members. Our first identified family LRRK2 PD case with tremor and parkinsonism was born at the time when Charcot named the disease. She had developed the disease in her early forties and had to be taken care of by her family for many years. In a family photo from 1911 the very typical parkinsonian features in her face and body are visualized and even the hand tremor is depicted in the grip of one of her daughters (Figure 1).

Figure 1. First identified Norwegian LRRK2 patients, family photo 1911 (with permissions from the family).
Clinical Phenotypes
About a third of the mutation carriers had developed PD at the time when the follow-up period started 15 years ago. Tremor was the initial parkinsonian sign that brought 3 out of four to the neurologists. This percentage of tremor is well in line with reports from that observed in sporadic PD. The last one-fourth of the cases experienced akinesia, gait problems, micrographia or dystonia as initial signs. This does not differ from other PD populations. Many studies have shown that LRRK2 PD usually respond very well to levodopa and to dopamine agonists. The same was seen in Norwegian cases. Four out of the 32 cases have also ended up with severe complications and needed advanced therapy. In all four patients were treated with deep brain stimulation, DBS, and all four had good or excellent effects. Their mean age at PD onset was 46 years (range 39–59). The first one had DBS 19 years ago at age 49 years. She is still in H&Y III, cognitively intact and living in her own home. It is not well understood why LRRK2 patients with mutations in the kinase domain respond very well to DBS (Schupbach et al., 2007; Johansen et al., 2011; Angeli et al., 2013). This impression is based on reports from several centers around the world. It may also be true for those with mutations in the cor domain, while patients with roc domain mutations seem to have a less favorable outcome from DBS (Gomez-Esteban et al., 2008; Hatano et al., 2014). Some reports on LRRK2 families have noticed that atypical phenotypes also occur (Wszolek et al., 2004). There were no multiple system atrophy or other atypical PD phenotypes observed among members of our families who converted to PD. There was one members diagnosed with MSA observed in one of our families but eventually he tested negative for the G2019S mutation.
Inflammatory Diseases in LRRK2 G2019S Mutation Carriers Before and After Converting to PD
Multiple Sclerosis, MS
Three out of 100 LRRK2 G2019S mutation carriers were diagnosed with MS. Two cases had no parkinsonian signs or symptoms. The third had mild rigidity and bradykinesia.
Case 1: A 69 years old man, his father had PD. He worked as a carpenter and was a moderate smoker. At the age of 45 year he was diagnosed with retrobulbar neuritis. Two years later he suffered a mild central paresis of the right lower limb with increased tendon reflexes and his right sided plantar reflex was clearly abnormal. Lumbar puncture showed increased cerebrospinal fluid, CSF, immunoglobulins with 5–6 bands not found in his serum. Repeated cerebral MRI scans showed typical MS lesions. His disease was quite benign with little progression. He was not on any medication. At age 69, 24 years after first attack, he was still ambulating and had only minor autonomic dysfunctions; his EDSS grade was 2. He had been to regular follow-up as an asymptomatic LRRK2 carrier and he has developed mild rigidity and bradykinesia but no tremor. A Datscan showed mild reduction of uptake in the putamen bilaterally but not in the caudate nuclei. No anti-parkinsonian therapy was needed.
Case 2: A 57 years old woman was diagnosed with MS at the age of 35 years. CSF bands indicated intrathecal synthesis of immunoglobulins and brain MRI demonstrated periventricular high-signal intensity lesions with typical distribution for multiple sclerosis. Initially she suffered 5–6 attacks and later her disease turned to a more chronic progressive pattern. She was on interferon-beta treatment over a period of 5 years. She had moderate autonomic dysfunctions. Her dominating MS-pattern through all these years has been an extreme feeling of fatigue. Twenty-two years after onset she was still ambulating, living in her own apartment and her EDSS grade was 4. She had moderate spasticity and no rigidity. There was no bradykinesia unrelated to MS. A Datscan was not done. In contrast to the male MS patients she had always been a non-smoker.
Case 3: A 59 years old man, his mother developed PD around the age of 60 years and he is the brother of case 2. There was no additional family history of MS. From age 32 years he suffered multiple attacks of optic neuritis and central nervous manifestations. CSF and MRI examinations showed typical MS pathology. He had a rapid disease progression and reached EDSS grade 4 already after 4 years of disease duration. He stopped smoking around age 50, mainly because of his physical condition. At the age of 59 he was non-ambulating, almost quadriplegic with a tiny rest function of motility in one hand. He had no rigidity and no tremor. His speech was unremarkable but talking could trigger attacks of severe trigeminal neuralgia. His cognitive functions were considered as good. His autonomic dysfunctions were affected and regarded as part of his long-lasting MS. At last examination, age 59 years, his EDSS was between 8.0 and 9. He died from an acute abdominal disease 6 months later. At autopsy his brain weight was 1,290 g with focal demyelinated plaques and variable degrees of inflammation, gliosis, and neurodegeneration. There was no neuron loss in the substantia nigra and multiple stains for tau, amyloid and alpha-synuclein pathology were negative.
Achalasia
Case 4: A 52 years old previously healthy teacher presented with swallowing problems. His mother had PD and was genotyped with the LRRK2 G2019S mutation. He was on anti-hypertensives and he was a heavy smoker. His dysphagia progressed over the period of some months and he underwent a barium esophagogram which showed a narrowed part of the lower sphincter of the esophagus (Figure 2). It was supplied with high resolution manometry pressure topography, confirming the diagnosis of achalasia. A pneumatic balloon dilatation was successfully performed. Followed-up visits through 18 years have been unremarkable and he had no swallowing problems. No tests for viral agents or immunological causes were performed. At age 54 he tested positive for the G2019S mutation and was included in the long-term follow-up study. During the last 10 years he has developed mild bradykinesia and rigidity but no tremor. A Datscan showed mainly left-sided abnormalities (Figure 3). He does not need anti-parkinsonian therapy. At age 65 he had an acute episode with a ruptured colon diverticulitis. A biopsy from the lower colon did not show any Lewy bodies. His esophagus problems have not recurred and a control esophagogram was unremarkable.
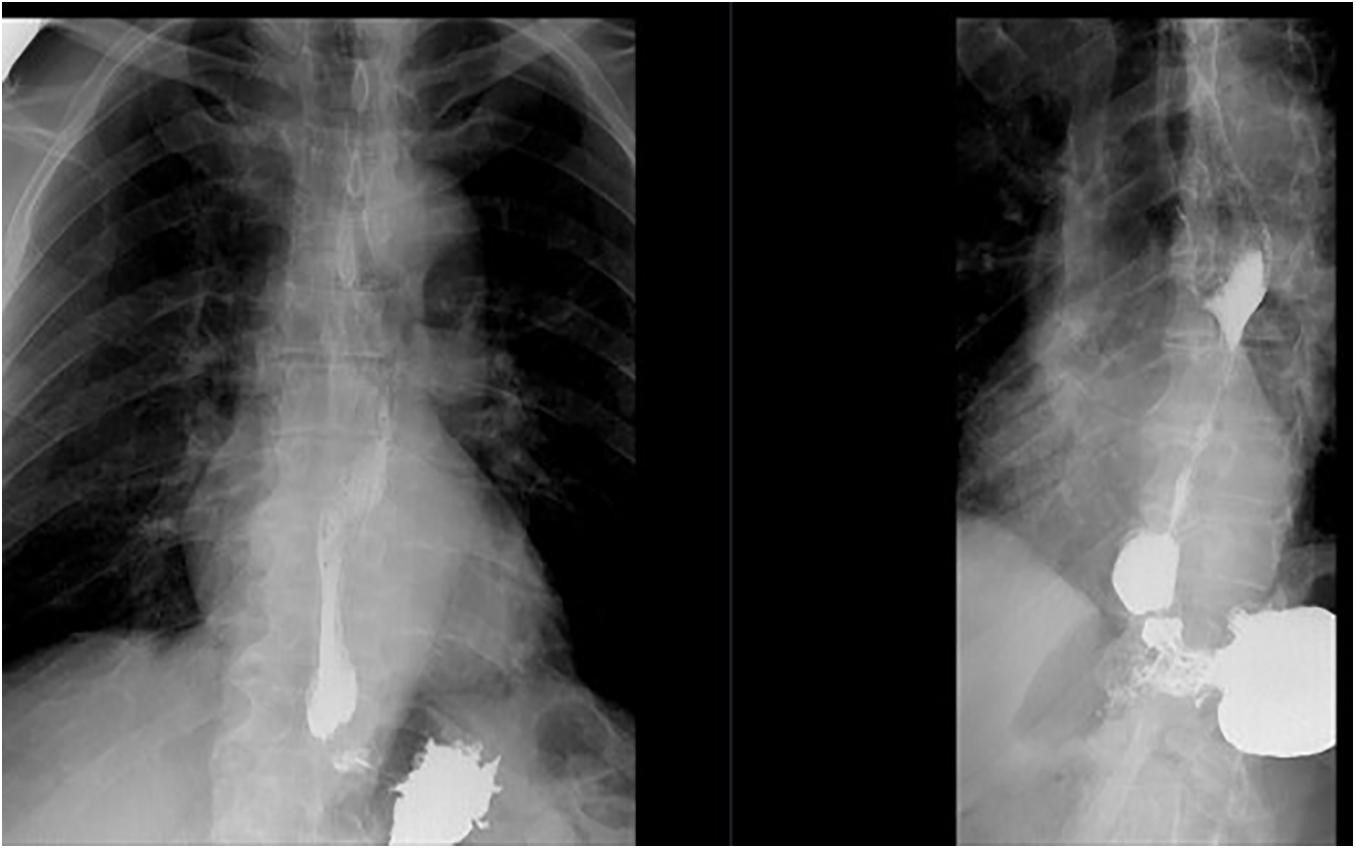
Figure 2. Case 4, achalasia, a barium esophagogram showing a narrowed part of the lower part of the esophagus.
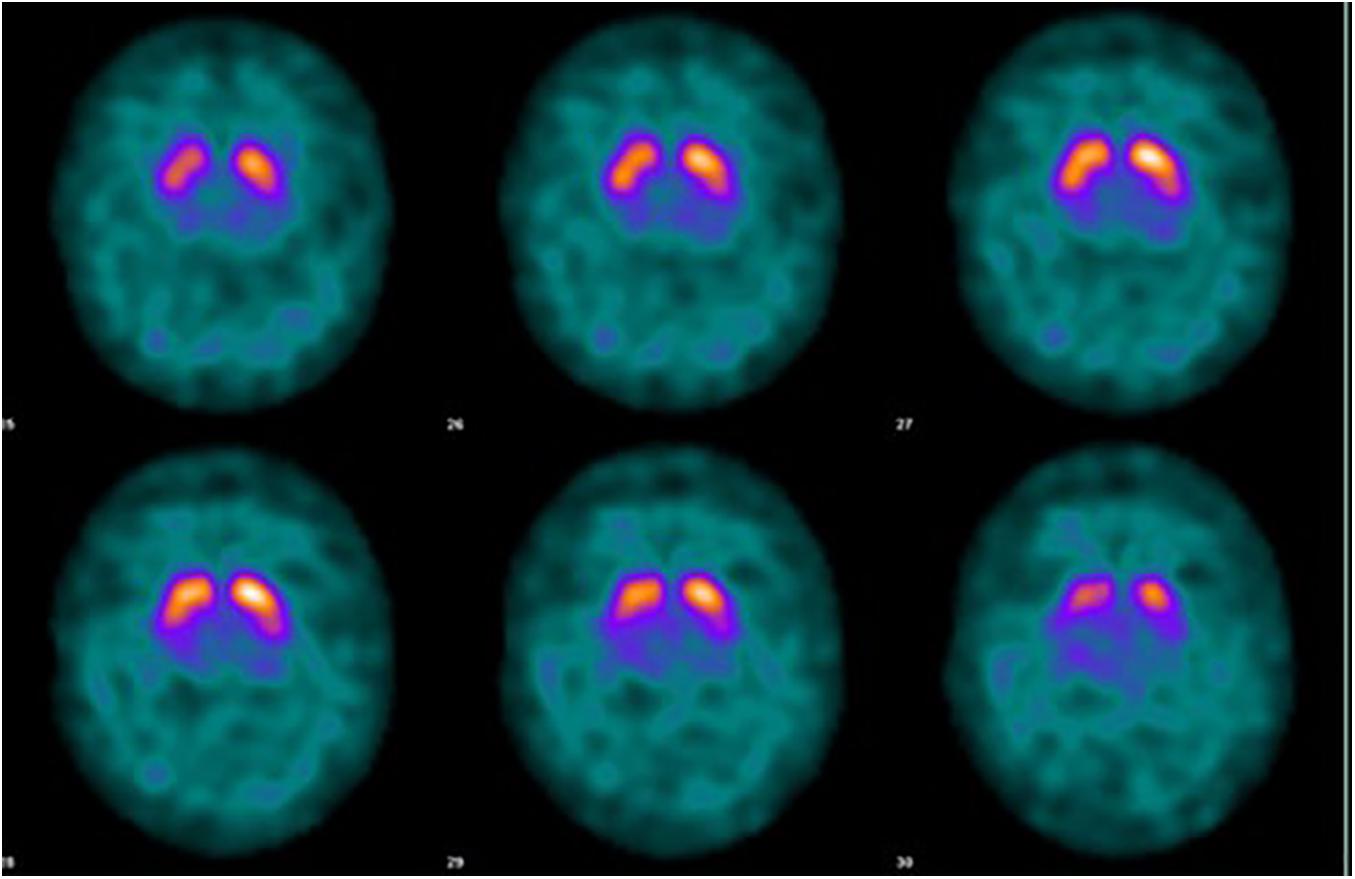
Figure 3. Case 4, Datscan at age 67 years. Bradykinesia and rigidity but no tremor.
Rheumatoid Arthritis
Case 5: A 44 years old nursing home assistant was member of a multi-incident PD family and had been followed as a healthy mutation carrier of a heterozygous LRRK2 G2019S mutation. She was a moderate smoker. Her mother had PD and case 5 was positive for the same mutation. Five years after inclusion she converted to PD. Her most prominent signs were asymmetric rest tremor, bradykinesia and rigidity. She responded well to levodopa and dopamine agonists. In the years before converting to PD she complained of joint stiffness and pain. A diagnostic procedure was performed at the department of rheumatology and their conclusion was sero-positive rheumatoid arthritis. She was treated with local injections and systemic therapy according to current guidelines. Her rheumatic disease progressed and was the main reason for her retirement a few years later. Around the age of 60 years her PD is fluctuating and is in an advanced stage and she has been under consideration for DBS therapy or apomorphine infusions.
Case 6: A 50 years old woman, manager of a small trading company, came to evaluation together with her mother, who, like the grandmother, had been diagnosed with PD. A genetic test showed that they both were carriers of the LRRK2 G2019S mutation. A PET-scan performed the same year revealed marked basal ganglia pathology (Figure 4). She has now been followed annually for 15 years. She gradually developed moderate bradykinesia and rigidity without any other parkinsonian symptoms and she has not converted to PD. The same year as she had her first PET-scan she started to complain of finger stiffness and swollen, painful joints. She was positive for multiple inflammatory markers and were diagnosed with sero-positive rheumatoid arthritis. She has been on methotrexate therapy for more than 10 years, in combination with other anti-inflammatory drugs. She had to stop working at the age of 65, mainly due to her rheumatism and because of the general COVID-19 situation.
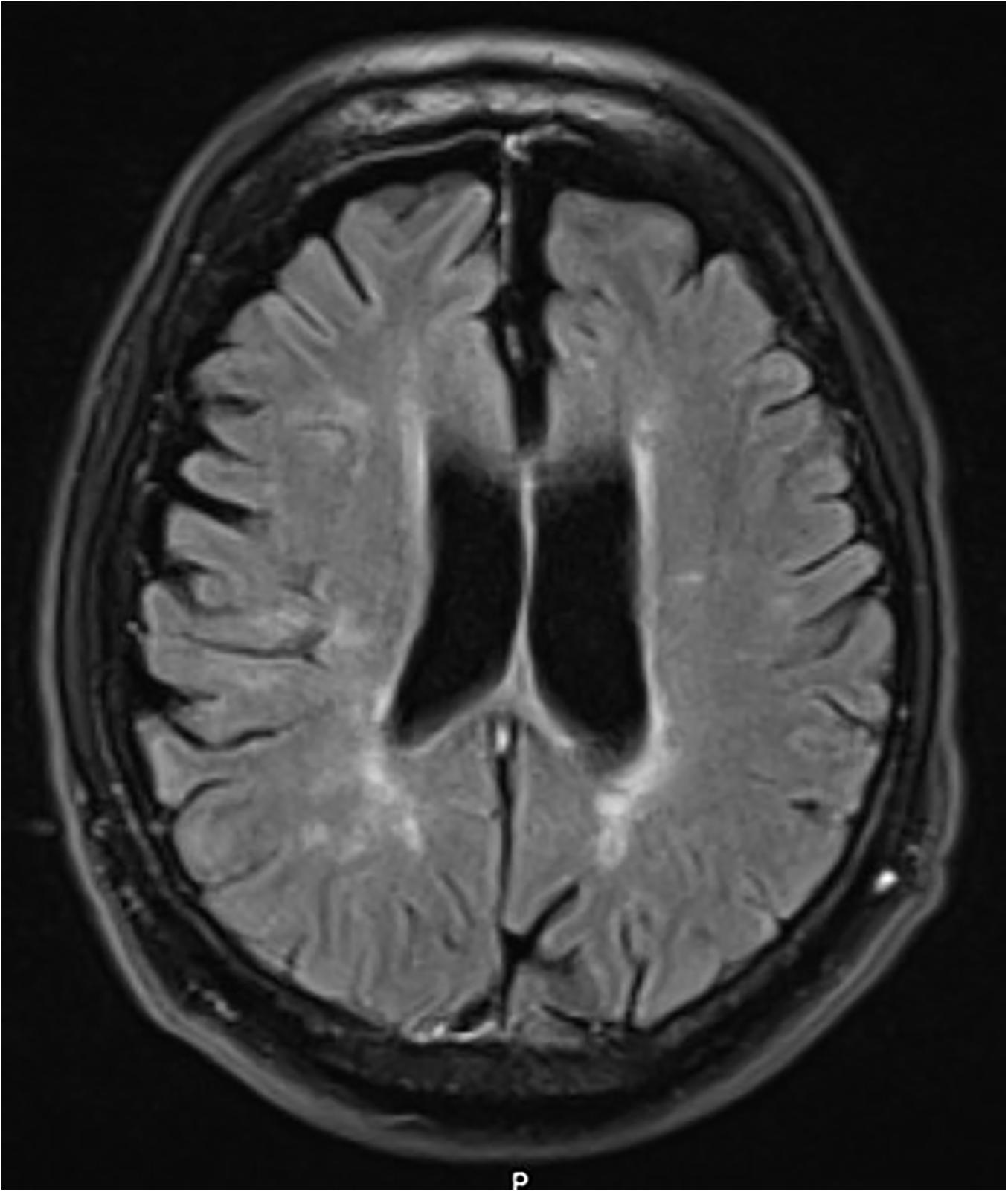
Figure 4. Case 3, LRRK2 mutation carrier, severe MS. MRI at age 50 with white matter lesions and some central and cortical brain atrophy.
Non-inflammatory Diseases
Cancer
It has been claimed that LRRK2 PD cases have higher prevalence of cancer compared to sporadic PD (Agalliu et al., 2015). This has also been studied in our local Norwegian PD population. We obtained our data from the national Cancer Registry of Norway and we calculated data and cancer outcomes from 857 sporadic PD patients and 76 LRRK2 mutation carriers. The PD population also included 27 LRRK2 PD cases. These were compared data obtained from the national Cancer Registry of Norway and included cancer type and age at cancer onset. All participants were ethnic Norwegians. The LRRK2 mutation carriers had increased risk of non-skin cancer compared with sporadic PD subjects (OR 2.09; 95% CI 1.16–3.77; p = 0.015). A significant association was found between the mutation and breast cancer in women (OR 4.58; 95% CI 1.45–14.51; p = 0.010). There were no other associations between specific cancer types and the LRRK2 mutation. There was one otherwise healthy LRRK2 mutation carrier who had been threated since age 50 years for hairy cell leukemia. He was still without signs of PD at the end of the 15 years follow-up period. It was concluded that being a LRRK2 mutation carriers included an increased risk of non-skin cancer compared with sporadic PD subjects. The increased risk for cancer among LRRK2 carriers was mainly driven by the association between harboring the mutation and breast cancer, observed in women (Waro and Aasly, 2018).
Longitudinal Clinical Evaluations
Olfaction
Olfaction was tested in PD patients and in healthy LRRK2 mutation carriers using the UPSIT and B-sit tests. The cohort of LRRK2 carriers and PD patients in central Norway showed the same level of impaired olfactory identification as reported from other centers (Marras et al., 2011; Saunders-Pullman et al., 2011; Sierra et al., 2013; Gaig et al., 2014). The impairment seen in our LRRK2 group was significant although less than in subjects with idiopathic Parkinson disease (Johansen et al., 2014). Others have shown that olfactory dysfunction in LRRK2 patients is positively correlated with reduced uptake of (123)I-meta-iodobenzylguanidine (MIBG) on cardiac scintigraphy, a measure of postganglionic sympathetic cardiac innervation (Valldeoriola et al., 2011).
Cognition
Prevalence of Dementia With Lewy Bodies, DLB
All LRRK2 mutation carriers below the age of 60 years had normal cognitive functions, PD patients included. About half of the LRRK2 PD patients developed cognitive decline as the disease progressed. These cases have been reported in previous publications and at autopsy there was a significant association between cognitive impairment/dementia and the presence of Lewy bodies after adjustment for the degree of Alzheimer disease–related pathology (Kalia et al., 2015).
Previous studies have aimed to determine the risk for conversion to PD in LRRK2 mutation carriers. Norwegian LRRK2 mutation carriers seem to have a significant higher age at conversion compared to individuals carrying the same mutation in Tunisia and Israel (Hentati et al., 2014; Trinh et al., 2014). There have been no studies on the prevalence of DLB in asymptomatic LRRK2 mutation carriers. In our rural districts with scattered population, patients with gradual cognitive decline without the combination of obvious movement disorder signs, often ends up in nursing homes and do not undergo further specific diagnostic procedures. There has been less focus on cognitive decline in LRRK2 mutation carriers without motor signs and who do not convert to PD. Patients who gradually develop cognitive decline often desist from long-time follow-up programs and must be retrieved by active calls from the hospitals. Three of our mutation carriers were located to nursing homes and all had an unspecified diagnose of dementia. None had tremor and a neurological examination showed that they all were rigid and bradykinetic thus fulfilling the criteria for DLB (McKeith et al., 2020). These three cases illustrate the problem of ignoring DLB cases as part of the phenoconversion to PD or to DLB. The three mutation carriers in our cohort had all been diagnosed with unspecified dementia by their local physicians. Whether a LRRK2 mutation carrier converts to PD or DLB is equal from a medical point of view although the histopathological distribution of Lewy bodies in the brain may have slightly different patterns.
LRRK2 Mutations Combined With GBA-Mutations
There are 6–7 LRRK2 mutations which are strongly correlated with PD. Other strong PD risk factors are mutations in the gene for Gaucher disease, GBA-mutations. Recently 10 of our patients also had their GBA genes fully sequenced and four cases were shown to carry GBA mutations in addition to the G2019S mutation. One 58 years old asymptomatic woman had two GBA mutations. Only one of our cases had converted to PD, at the age of 47 years. The combination LRRK2 and GBA mutations does not seem to have an additive effect to the phenotype. Some have postulated a possible modifying effect of the G2019S mutation on GBA PD (Omer et al., 2020).
Biomarkers
There have been a large number of studies aiming to find robust biomarkers for PD progression by using LRRK2 pre-clinical cases and compare these to LRRK2 PD, sporadic PD and normal controls. The Norwegian cohort has been part of many of these studies (Shi et al., 2011; Aasly et al., 2012, 2014; Loeffler et al., 2016; Ichinose et al., 2017, 2018). Most studies have been performed in cerebrospinal fluid, CSF, some in blood and others in urine and in saliva (Stewart et al., 2015; Wang et al., 2017).
Most CSF studies have included known metabolites involved in neurodegeneration, like Aβ1-42, tau, α-synuclein, oxidative stress markers, autophagy-related proteins, pteridines, neurotransmitter metabolites, exosomal LRRK2 protein, RNA species, inflammatory cytokines, mitochondrial DNA (mtDNA), and intermediary metabolites. Better technique and smarter machines later added the possibility of studying pteridines, α-synuclein, mtDNA, 5-hydroxyindolacetic acid, β-D-glucose, lamp2, interleukin-8, and vascular endothelial growth factors. Many of the studies suggested to differentiate LRRK2 PD from sporadic PD patients. It was claimed that 8-hydroxy-2′-deoxyguanosine (8-OHdG), 8-isoprostane (8-ISO), 2-hydroxybutyrate, mtDNA, lamp2, and neopterin may differentiate between healthy LRRK2 carriers and LRRK2 PD subjects; and soluble oligomeric α-synuclein, 8-OHdG, and 8-ISO might differentiate healthy LRRK2 carriers from control subjects (Aasly et al., 2012, 2014; Shi et al., 2012; Podlesniy et al., 2016; Vilas et al., 2016; Loeffler et al., 2017; Wang et al., 2017; Ichinose et al., 2018; Klaver et al., 2018). The high number of analytes in combination with the low numbers of investigations of each analyte, and the small sample sizes, together with methodological differences, has limited the conclusions that can be drawn from these studies. There is so far no useful biomarker that can predict PD phenoconversion; not in sporadic PD and not in monogenic PD types. The validity of the analytes identified in these studies needs to be confirmed in larger studies (Aasly, 2020). So far, no robust biomarker for useful PD-specific progression has been found (Loeffler et al., 2019). Neurofilament light chain (NfL), a neuronal cytoplasmic protein highly expressed in large myelinated axons is a well-known marker in a variety of neurological disorders, including inflammatory, neurodegenerative, traumatic and cerebrovascular diseases but it is rather unspecific (Gaetani et al., 2019).
Autopsies
Five LRRK2 mutation carriers came to autopsy during the 15-year follow-up period, the results have been presented elsewhere (Kalia et al., 2015; Aasly, 2020). Four had developed PD and one was the mutation carrier with MS, without signs of the PD. The presence of LBs were closely correlated to their cognitive functions. An 85-years-old woman died after 25 years of PD. She had no cognitive defects and no LBs. An 80-years-old man died 20 years after disease onset and in a state of very severe dementia. The autopsy showed diffuse LB disease. A 79-years-old male died 20 years after disease onset. His cognitive function was slightly impaired and the autopsy showed only a few LBs. We concluded that there was a clear correlation with the presence of Lewy bodies in the brain and the intellectual performance (Kalia et al., 2015).
Imaging Markers
LRRK2 mutation carriers in the Norwegian LRRK2 cohort have taken part in a number of imaging studies. It was soon shown that asymptomatic mutation carriers may have quite extensive basal ganglia dopaminergic defects with very low UPDRS scores. We did a retrospective evaluation of a cohort of 39 participants who underwent Datscan as part of their follow-up. Our goal was to assess whether a combination of systematic clinical testing and different imaging techniques in familial PD cases could detect subclinical signs in the preclinical and prodromal stages of PD. Our cohort of 39 participants were studied with visual analysis of Datscan imaging to assess patterns of dopaminergic degeneration. They were grouped according to diagnostic criteria suggested by the Movement Disorders Society (MDS) Research Criteria for Prodromal PD (Aasly et al., 2005; Gao et al., 2018).
The imaging studies showed that LRRK2 mutation carriers above the age of 60 all had some kind of Datscan or PET abnormalities, always reflecting subclinical rigidity and bradykinesia (Ichinose et al., 2018). Corresponding defects has been shown for other neurotransmitters. The Norwegian LRRK2 cohort has taken part in three PET-studies aiming at central dopaminergic, serotoninergic and cholinergic activities. These studies all showed a change in transmitter activity years before conversion to PD (Figure 5). This may be interpreted as an early upregulation for LRRK2-dysfunction or a change of transmitter content in non-neuronal cells (Sossi et al., 2010; Wile et al., 2017; Liu et al., 2018).
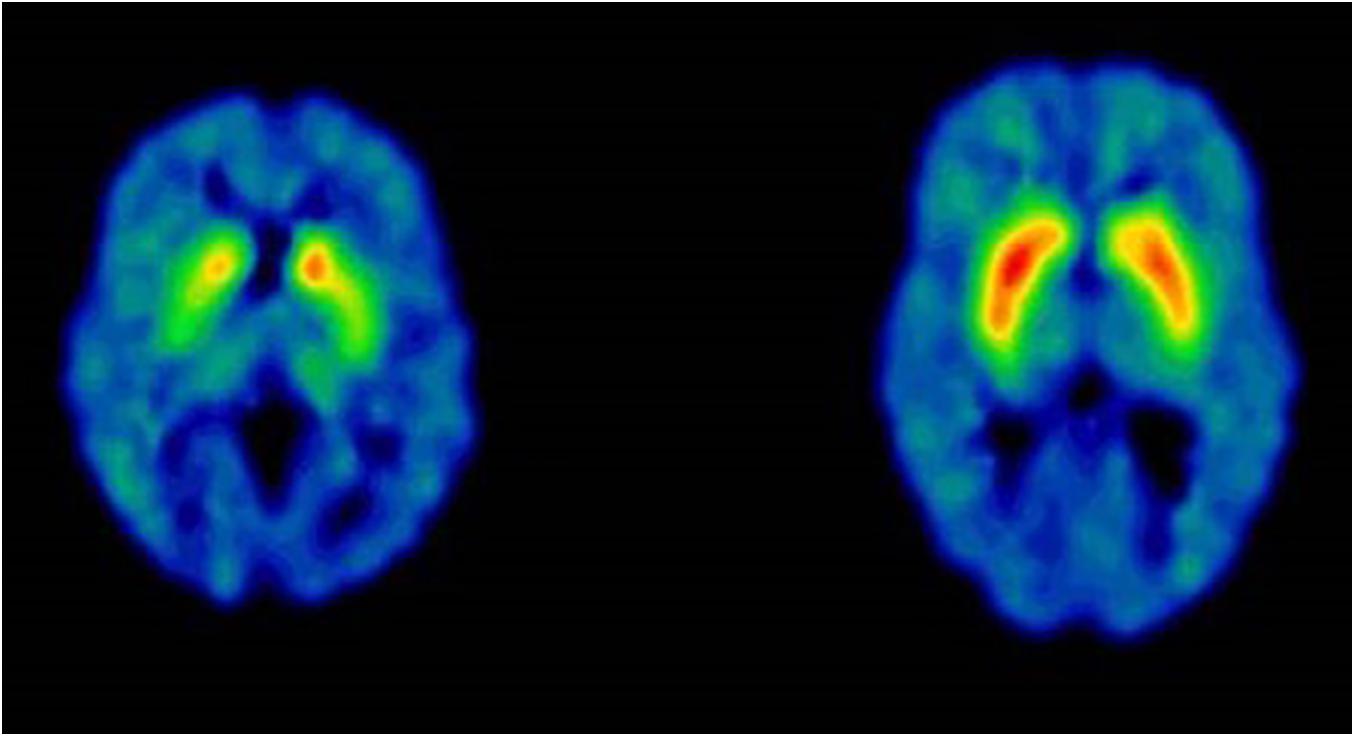
Figure 5. Case 6, 65 years-old healthy LRRK2 mutation carrier with sero-positive rheumatoid arthritis. PET-scan at age 50 DAT 11C-MP, methylphenidate. Left scan: Case 6, at age 50, right scan: 54 years-old normal control (with permission from J Stoessl and V Sossi, Pacific Parkinson’s Research Centre, Vancouver, British Columbia, Canada).
Discussion
This is the first report of the 15 years follow-up of a Norwegian cohort of LRRK2 G2019S mutation carriers. About one third of the original Norwegian LRRK2 cohort had developed PD after 15 years follow-up. In those who converted to PD the phenotype was close to that seen in sporadic PD and did not differ from patients seen in other LRRK2 G2019S cohorts. The motor signs were levodopa responsive, they had better olfaction functions than sporadic PD, had less autonomic deficits and they responded very well to DBS. There was an increased prevalence of inflammatory diseases among members of this material. This has not been observed in previous reports from LRRK2 cohorts. Three mutation carriers without PD developed multiple sclerosis, two had sero-positive rheumatoid arthritis and one needed treatment for severe achalasia. This is a relatively high percentage of inflammatory diseases, not commonly seen in combinations with PD.
Multiple sclerosis is rarely seen in PD patients and vice versa. The three MS cases in this report represent per se different types of the wide specter of MS. They all fulfilled the criteria for MS and they all had in common that they are LRRK2 G2019S mutation carriers. The local prevalence of MS among the population in Central Norway is 160 out of 100,000 (Dahl et al., 2004), or 0.16%. The 3% prevalence in a small cohort of LRRK2 carriers could be a coincidence. So far there has been drawn very few connections between PD and MS in genetic studies. In a large Danish register study there was no increase in incidence of PD among MS patients (Nielsen et al., 2014) and reports on both MS and PD are mostly anecdotal case reports (Valkovic et al., 2007). But the association between the immune system and PD needs to be kept in mind. A recent genome-wide association study systemically investigated pleiotropy between PD and autoimmune diseases. There was an overlap between PD and inflammatory diseases, including rheumatoid arthritis and multiple sclerosis, in 17 novel loci, including LRRK2 (Witoelar et al., 2017). The neuroinflammation in PD may be initiated by activated microglia, upregulated cyclooxygenase-1 and -2-expression, increased inflammatory cytokines and related molecules. In addition, polymorphisms in inflammatory cytokine genes (i.e., tumor necrosis factor-α and interleukin-1β) and cell-surface human leukocyte antigen have been associated with an increased PD risk (Crotty et al., 2020). Recently it was shown that PD patients share a LRRK2 risk variant, N2081D, and a protecting variant, N551K, with Crohn’s disease, CD, patients. This pleiotropic effect of LRRK2 functional variants affect the risk for PD and CD independent of ethnicity (Hui et al., 2018). It is further of interest that CD and MS share common principles for modern treatment. Natalizumab is the most potent drug for MS, mainly by blocking the T lymphocyte intrusion in the central nervous system through the blood-brain barrier, and is effective for CD by blocking cell trafficking into the gut (Pagnini et al., 2017). In both diseases the main effects are achieved through several vascular cell adhesion molecules (Zundler et al., 2019). It is also noted that another very potent drug used for MS, cladribine, is a drug of choice for hairy cell leukemia (Paillassa et al., 2020). Maybe similar treatments should be explored in future LRRK2 PD studies.
The prevalence of rheumatoid arthritis varies between countries and it is highest in high-income western countries. The local prevalence of sero-positive rheumatoid arthritis is 0.35% compared to 2% in this small cohort (Videm et al., 2017). In a survey from Taiwan the cumulative incidence of PD was 2.42% lower in a large RA cohort than in the non-RA cohort (Sung et al., 2016). The lower risk for developing PD in patients affected with RA was not correlated to treatment or use of anti-rheumatic drugs. Other studies have shown that ibuprofen, a non-steroidal anti-inflammatory drug, NSAID, lower the risk for PD while other NSAIDs may not have the same effect (Gao et al., 2011). However, the non-association between treatment and outcome may be differ within PD subgroups. It has been shown that regular NSAID use may be associated with reduced penetrance in LRRK2-associated PD (San Luciano et al., 2020), and that the LRRK2 protein is involved in inflammatory pathways and appears to be modulated by regular anti-inflammatory use. The authors postulate that if LRRK2 set the fire, can non-steroidal anti-inflammatory drug wet the flames? (Crotty et al., 2020).
Achalasia has been connected to PD mainly through case reports. In our LRRK2 case there was also a parallel between start of esophageal symptoms and the clinical manifestation of subclinical parkinsonism. Given its relatively common prevalence (10.82/100,000) achalasia seen in a patient could be a coincidental finding (Sadowski et al., 2010). The etiology of achalasia is unknown but genetic or immune factors may be involved. A number of genes have been shown to increase the risk for achalasia. Polymorphisms of genes for enzymes catalyzing the production of nitric oxide, NO, from L-arginine have been associated with a higher risk for achalasia (Gao et al., 2018). Large genome wide association studies are underway, which may shed further light into genetic predisposition of the disease. Secondly, ample evidence suggests that achalasia is an autoimmune disorder, where an antibody response to a common antigen, perhaps a virus, selectively knocks out esophageal autonomic control mechanisms at the myenteric plexus ganglia and the neuronal level (Villanacci et al., 2010). This theory has been further supported by antibodies that target enteric ganglia which have been identified in the sera of achalasia patients (Moses et al., 2003). Achalasia may be cause by a virus and both Herpes simplex virus type I, measles and human papilloma virus has been suggested as infectious triggers. It has been shown that the nerve plexi/ganglia involved in the motor responses in the distal esophagus show an antigen–antibody response to these agents corresponding to degree of damage contributing greatly to the clinical and smooth muscle findings. This include both the lower esophageal sphincter and esophageal body. Inflammatory responses are mainly seen in type 1 and type 2 with ganglion cells with cell death (Ghoshal et al., 2012).
Biomarker for LRRK2 PD
Traditional CSF biomarkers in PD patients have not shown any significant change in protein fractions related to neuroimmunological disease mechanisms. It could be more relevant to study small (40–100 nm) extracellular membranous vesicles, exosomes, because they may be carriers of more relevant disease markers which also may include the immune system. Exosomes may been isolated from several body substances like urine, CSF or plasma.
It has been shown that the protein pS1292-LRRK2 protein is robustly expressed in CSF exosomes. In a cohort of Norwegian subjects with and without the G2019S-LRRK2 mutations, with and without PD, we quantified levels of pS1292-LRRK2, total LRRK2, and other exosome proteins in urine from 132 subjects and in CSF from 82 subjects. These results provided insights into the effects of LRRK2 mutations in both the periphery and brain in a well-characterized clinical population and showed that LRRK2 protein in brain exosomes may be much more active than in the periphery in most subjects (Wang et al., 2017). In a similar study plasma-derived extracellular vesicles or exosomes, were isolated from PD, matched healthy controls, and atypical parkinsonism with tauopathies.
Specific groups of markers related to inflammatory and immune cells are located to the surface of exosomes. These markers have been analyzed and correlated to movement disorder patients according to the clinical diagnosis. PD and MSA patients had considerably larger parts of immune markers, indicating that neuroimmune regulation in PD and MSA is different from that observed in atypical parkinsonism. The true positive rate for compound exosome markers showed optimal diagnostic performance for PD. The exome marker curves for PD and MSA were rather congruent and different from those of corticobasal degeneration and progressive supranuclear palsy. In the panels shared by PD and MSA was a transcription factor playing, SP1, which is an important regulator of neuroinflammation in multiple sclerosis (Vacchi et al., 2020).
Conclusion
The aim of this survey of 15 years follow-up of the original Norwegian LRRK2 cohort was to emphasize the presence of neuroinflammation in a group of LRRK2 mutation carriers. The role of LRRK2 in inflammation and immune system regulation is being increasingly explored. It is expressed in the cells of the innate and adaptive immune system. The LRRK2 gene is associated with several chronic inflammatory disorders, including Crohn’s disease and leprosy but these results have originated from vast genetic studies like GWAS in heterogenous PD populations. LRRK2 may play a crucial part in the complex interactions of neuroinflammation. The combination of PD and inflammatory diseases is rare. Multiple sclerosis and rheumatoid arthritis are rarely seen with PD. Achalasia may have been reported in early PD but its significance has been debated. The high percentage of inflammatory cases among LRRK2 carriers could indicate that anti-inflammatory drugs may be recommended in risk populations to reduce inflammation and subsequent neurodegeneration.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work has been supported by the Michael J. Fox Foundation, NY, New York, United States, the Norwegian Parkinson’s Disease Association and by Reberg’s Legacy, Oslo, Norway.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aasly, J. O. (2020). Long-term outcomes of genetic Parkinson’s disease. J. Mov. Disord. 13, 81–96. doi: 10.14802/jmd.19080
Aasly, J. O., Johansen, K. K., Bronstad, G., Waro, B. J., Majbour, N. K., Varghese, S., et al. (2014). Elevated levels of cerebrospinal fluid alpha-synuclein oligomers in healthy asymptomatic LRRK2 mutation carriers. Front. Aging Neurosci. 6:248. doi: 10.3389/fnagi.2014.00248
Aasly, J. O., Shi, M., Sossi, V., Stewart, T., Johansen, K. K., Wszolek, Z. K., et al. (2012). Cerebrospinal fluid amyloid beta and tau in LRRK2 mutation carriers. Neurology 78, 55–61. doi: 10.1212/wnl.0b013e31823ed101
Aasly, J. O., Toft, M., Fernandez-Mata, I., Kachergus, J., Hulihan, M., White, L. R., et al. (2005). Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann. Neurol. 57, 762–765.
Agalliu, I., San Luciano, M., Mirelman, A., Giladi, N., Waro, B., Aasly, J., et al. (2015). Higher frequency of certain cancers in LRRK2 G2019S mutation carriers with Parkinson disease: a pooled analysis. JAMA Neurol. 72, 58–65. doi: 10.1001/jamaneurol.2014.1973
Angeli, A., Mencacci, N. E., Duran, R., Aviles-Olmos, I., Kefalopoulou, Z., Candelario, J., et al. (2013). Genotype and phenotype in Parkinson’s disease: lessons in heterogeneity from deep brain stimulation. Mov. Disord. 28, 1370–1375. doi: 10.1002/mds.25535
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Crotty, G. F., Lo, R. Y., and Schwarzschild, M. A. (2020). If LRRK2 set the fire, can nonsteroidal anti-inflammatory drugs wet the flames? Mov. Disord. 35, 1727–1730. doi: 10.1002/mds.28240
Dahl, O. P., Aarseth, J. H., Myhr, K. M., Nyland, H., and Midgard, R. (2004). Multiple sclerosis in Nord-Trondelag county, Norway: a prevalence and incidence study. Acta Neurol. Scand. 109, 378–384. doi: 10.1111/j.1600-0404.2004.00244.x
Gaetani, L., Blennow, K., Calabresi, P., Di Filippo, M., Parnetti, L., and Zetterberg, H. (2019). Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 90, 870–881. doi: 10.1136/jnnp-2018-320106
Gaig, C., Vilas, D., Infante, J., Sierra, M., Garcia-Gorostiaga, I., Buongiorno, M., et al. (2014). Nonmotor symptoms in LRRK2 G2019S associated Parkinson’s disease. PLoS One. 9:e108982. doi: 10.1371/journal.pone.0108982
Gao, X., Chen, H., Schwarzschild, M. A., and Ascherio, A. (2011). Use of ibuprofen and risk of Parkinson disease. Neurology 76, 863–869. doi: 10.1212/wnl.0b013e31820f2d79
Gao, Y., Liu, J. F., He, X., Liu, X. B., Zhang, L. L., Zhao, L. M., et al. (2018). Calcium receptor and nitric oxide synthase expression in circular muscle of lower esophagus from patients with Achalasia. Chin. Med. J. (Engl.) 131, 2882–2885.
Ghoshal, U. C., Daschakraborty, S. B., and Singh, R. (2012). Pathogenesis of achalasia cardia. World J. Gastroenterol. 18, 3050–3057. doi: 10.3748/wjg.v18.i24.3050
Gomez-Esteban, J. C., Lezcano, E., Zarranz, J. J., Gonzalez, C., Bilbao, G., Lambarri, I., et al. (2008). Outcome of bilateral deep brain subthalamic stimulation in patients carrying the R1441G mutation in the LRRK2 dardarin gene. Neurosurgery 62, 857–862 discussion 62–3.
Hakimi, M., Selvanantham, T., Swinton, E., Padmore, R. F., Tong, Y., Kabbach, G., et al. (2011). Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J. Neural. Transm. (Vienna) 118, 795–808. doi: 10.1007/s00702-011-0653-2
Hatano, T., Funayama, M., Kubo, S. I., Mata, I. F., Oji, Y., Mori, A., et al. (2014). Identification of a Japanese family with LRRK2 p.R1441G-related Parkinson’s disease. Neurobiol. Aging 35, 2656 e17–e23.
Hentati, F., Trinh, J., Thompson, C., Nosova, E., Farrer, M. J., and Aasly, J. O. (2014). LRRK2 parkinsonism in Tunisia and Norway: a comparative analysis of disease penetrance. Neurology 83, 568–569. doi: 10.1212/wnl.0000000000000675
Hui, K. Y., Fernandez-Hernandez, H., Hu, J., Schaffner, A., Pankratz, N., Hsu, N. Y., et al. (2018). Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med. 10:eaai7795.
Ichinose, H., Inoue, K. I., Arakawa, S., Watanabe, Y., Kurosaki, H., Koshiba, S., et al. (2017). Alterations in the reduced pteridine contents in the cerebrospinal fluids of LRRK2 mutation carriers and patients with Parkinson’s disease. J. Neural. Transm. (Vienna) 125, 45–52. doi: 10.1007/s00702-017-1784-x
Ichinose, H., Inoue, K. I., Arakawa, S., Watanabe, Y., Kurosaki, H., Koshiba, S., et al. (2018). Alterations in the reduced pteridine contents in the cerebrospinal fluids of LRRK2 mutation carriers and patients with Parkinson’s disease. J. Neural. Transm. (Vienna) 125, 45–52. doi: 10.1007/s00702-017-1784-x
Johansen, K. K., Hasselberg, K., White, L. R., Farrer, M. J., and Aasly, J. O. (2010). Genealogical studies in LRRK2-associated Parkinson’s disease in central Norway. Parkinsonism Relat. Disord. 16, 527–530. doi: 10.1016/j.parkreldis.2010.05.005
Johansen, K. K., Jorgensen, J. V., White, L. R., Farrer, M. J., and Aasly, J. O. (2011). Parkinson-related genetics in patients treated with deep brain stimulation. Acta Neurol. Scand. 123, 201–206. doi: 10.1111/j.1600-0404.2010.01387.x
Johansen, K. K., Waro, B. J., and Aasly, J. O. (2014). Olfactory dysfunction in sporadic Parkinson’s disease and LRRK2 carriers. Acta Neurol. Scand. 129, 300–306. doi: 10.1111/ane.12172
Kalia, L. V., Lang, A. E., Hazrati, L. N., Fujioka, S., Wszolek, Z. K., Dickson, D. W., et al. (2015). Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 72, 100–105.
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Klaver, A. C., Coffey, M. P., Aasly, J. O., and Loeffler, D. A. (2018). CSF lamp2 concentrations are decreased in female Parkinson’s disease patients with LRRK2 mutations. Brain Res. 1683, 12–16. doi: 10.1016/j.brainres.2018.01.016
Liu, S. Y., Wile, D. J., Fu, J. F., Valerio, J., Shahinfard, E., McCormick, S., et al. (2018). The effect of LRRK2 mutations on the cholinergic system in manifest and premanifest stages of Parkinson’s disease: a cross-sectional PET study. Lancet Neurol. 17, 309–316. doi: 10.1016/s1474-4422(18)30032-2
Loeffler, D. A., Aasly, J. O., LeWitt, P. A., and Coffey, M. P. (2019). What have we learned from cerebrospinal fluid studies about biomarkers for detecting LRRK2 Parkinson’s disease patients and healthy subjects with Parkinson’s-associated LRRK2 mutations? J. Parkinsons Dis. 9, 467–488. doi: 10.3233/jpd-191630
Loeffler, D. A., Klaver, A. C., Coffey, M. P., Aasly, J. O., and LeWitt, P. A. (2016). Age-related decrease in heat shock 70-kDa protein 8 in cerebrospinal fluid is associated with increased oxidative stress. Front. Aging Neurosci. 8:178. doi: 10.3389/fnagi.2016.00178
Loeffler, D. A., Klaver, A. C., Coffey, M. P., Aasly, J. O., and LeWitt, P. A. (2017). Increased oxidative stress markers in cerebrospinal fluid from healthy subjects with Parkinson’s disease-associated LRRK2 gene mutations. Front. Aging Neurosci. 9:89. doi: 10.3389/fnagi.2017.00089
Marras, C., Schule, B., Munhoz, R. P., Rogaeva, E., Langston, J. W., Kasten, M., et al. (2011). Phenotype in parkinsonian and nonparkinsonian LRRK2 G2019S mutation carriers. Neurology 77, 325–333. doi: 10.1212/wnl.0b013e318227042d
McKeith, I. G., Ferman, T. J., Thomas, A. J., Blanc, F., Boeve, B. F., Fujishiro, H., et al. (2020). Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology 94, 743–755.
Moses, P. L., Ellis, L. M., Anees, M. R., Ho, W., Rothstein, R. I., Meddings, J. B., et al. (2003). Antineuronal antibodies in idiopathic achalasia and gastro-oesophageal reflux disease. Gut 52, 629–636. doi: 10.1136/gut.52.5.629
Nielsen, N. M., Pasternak, B., Stenager, E., Koch-Henriksen, N., and Frisch, M. (2014). Multiple sclerosis and risk of Parkinson’s disease: a Danish nationwide cohort study. Eur. J. Neurol. 21, 107–111. doi: 10.1111/ene.12255
Omer, N., Giladi, N., Gurevich, T., Bar-Shira, A., Gana-Weisz, M., Goldstein, O., et al. (2020). A possible modifying effect of the G2019S mutation in the LRRK2 gene on GBA Parkinson’s disease. Mov. Disord. 35, 1249–1253. doi: 10.1002/mds.28066
Pagnini, C., Arseneau, K. O., and Cominelli, F. (2017). Natalizumab in the treatment of Crohn’s disease patients. Expert Opin. Biol. Ther. 17, 1433–1438.
Paillassa, J., Cornet, E., Noel, S., Tomowiak, C., Lepretre, S., Vaudaux, S., et al. (2020). Analysis of a cohort of 279 patients with hairy-cell leukemia (HCL): 10 years of follow-up. Blood Cancer J. 10:62.
Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., van der Brug, M., et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. doi: 10.1016/j.neuron.2004.10.023
Podlesniy, P., Vilas, D., Taylor, P., Shaw, L. M., Tolosa, E., and Trullas, R. (2016). Mitochondrial DNA in CSF distinguishes LRRK2 from idiopathic Parkinson’s disease. Neurobiol. Dis. 94, 10–17. doi: 10.1016/j.nbd.2016.05.019
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Sadowski, D. C., Ackah, F., Jiang, B., and Svenson, L. W. (2010). Achalasia: incidence, prevalence and survival. A population-based study. Neurogastroenterol. Motil. 22, e256–e261.
San Luciano, M., Tanner, C. M., Meng, C., Marras, C., Goldman, S. M., Lang, A. E., et al. (2020). Nonsteroidal anti-inflammatory use and LRRK2 Parkinson’s disease penetrance. Mov. Disord. 35, 1755–1764.
Saunders-Pullman, R., Stanley, K., Wang, C., San Luciano, M., Shanker, V., Hunt, A., et al. (2011). Olfactory dysfunction in LRRK2 G2019S mutation carriers. Neurology 77, 319–324. doi: 10.1212/wnl.0b013e318227041c
Schupbach, M., Lohmann, E., Anheim, M., Lesage, S., Czernecki, V., Yaici, S., et al. (2007). Subthalamic nucleus stimulation is efficacious in patients with Parkinsonism and LRRK2 mutations. Mov. Disord. 22, 119–122. doi: 10.1002/mds.21178
Shi, M., Bradner, J., Hancock, A. M., Chung, K. A., Quinn, J. F., Peskind, E. R., et al. (2011). Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann. Neurol. 69, 570–580.
Shi, M., Furay, A. R., Sossi, V., Aasly, J. O., Armaly, J., Wang, Y., et al. (2012). DJ-1 and alphaSYN in LRRK2 CSF do not correlate with striatal dopaminergic function. Neurobiol. Aging 33, 836 e5–7.
Sierra, M., Sanchez-Juan, P., Martinez-Rodriguez, M. I., Gonzalez-Aramburu, I., Garcia-Gorostiaga, I., Quirce, M. R., et al. (2013). Olfaction and imaging biomarkers in premotor LRRK2 G2019S-associated Parkinson disease. Neurology 80, 621–626. doi: 10.1212/wnl.0b013e31828250d6
Sossi, V., de la Fuente-Fernandez, R., Nandhagopal, R., Schulzer, M., McKenzie, J., Ruth, T. J., et al. (2010). Dopamine turnover increases in asymptomatic LRRK2 mutations carriers. Mov. Disord. 25, 2717–2723. doi: 10.1002/mds.23356
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840.
Stewart, T., Sossi, V., Aasly, J. O., Wszolek, Z. K., Uitti, R. J., Hasegawa, K., et al. (2015). Phosphorylated alpha-synuclein in Parkinson’s disease: correlation depends on disease severity. Acta Neuropathol Commun. 3:7. doi: 10.1097/00132985-200401000-00006
Sung, Y. F., Liu, F. C., Lin, C. C., Lee, J. T., Yang, F. C., Chou, Y. C., et al. (2016). Reduced risk of Parkinson disease in patients with rheumatoid arthritis: a nationwide population-based study. Mayo Clin. Proc. 91, 1346–1353.
Trinh, J., Guella, I., and Farrer, M. J. (2014). Disease penetrance of late-onset parkinsonism: a meta-analysis. JAMA Neurol. 71, 1535–1539. doi: 10.1001/jamaneurol.2014.1909
Vacchi, E., Burrello, J., Di Silvestre, D., Burrello, A., Bolis, S., Mauri, P., et al. (2020). Immune profiling of plasma-derived extracellular vesicles identifies Parkinson disease. Neurol. Neuroimmunol. Neuroinflamm. 7:6.
Valente, E. M., Bentivoglio, A. R., Dixon, P. H., Ferraris, A., Ialongo, T., Frontali, M., et al. (2001). Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet. 68, 895–900. doi: 10.1086/319522
Valkovic, P., Krastev, G., Mako, M., Leitner, P., and Gasser, T. (2007). A unique case of coincidence of early onset Parkinson’s disease and multiple sclerosis. Mov. Disord. 22, 2278–2281. doi: 10.1002/mds.21642
Valldeoriola, F., Gaig, C., Muxi, A., Navales, I., Paredes, P., Lomena, F., et al. (2011). 123I-MIBG cardiac uptake and smell identification in parkinsonian patients with LRRK2 mutations. J. Neurol. 258, 1126–1132. doi: 10.1007/s00415-010-5896-6
Videm, V., Thomas, R., Brown, M. A., and Hoff, M. (2017). Self-reported diagnosis of rheumatoid arthritis or ankylosing spondylitis has low accuracy: data from the Nord-Trondelag health study. J. Rheumatol. 44, 1134–1141. doi: 10.3899/jrheum.161396
Vilas, D., Shaw, L. M., Taylor, P., Berg, D., Brockmann, K., Aasly, J., et al. (2016). Cerebrospinal fluid biomarkers and clinical features in leucine-rich repeat kinase 2 (LRRK2) mutation carriers. Mov. Disord. 31, 906–914. doi: 10.1002/mds.26591
Villanacci, V., Annese, V., Cuttitta, A., Fisogni, S., Scaramuzzi, G., De Santo, E., et al. (2010). An immunohistochemical study of the myenteric plexus in idiopathic achalasia. J. Clin. Gastroenterol. 44, 407–410.
Wang, S., Liu, Z., Ye, T., Mabrouk, O. S., Maltbie, T., Aasly, J., et al. (2017). Elevated LRRK2 autophosphorylation in brain-derived and peripheral exosomes in LRRK2 mutation carriers. Acta Neuropathol. Commun. 5:86.
Waro, B. J., and Aasly, J. O. (2018). Exploring cancer in LRRK2 mutation carriers and idiopathic Parkinson’s disease. Brain Behav. 8:e00858. doi: 10.1002/brb3.858
Wile, D. J., Agarwal, P. A., Schulzer, M., Mak, E., Dinelle, K., Shahinfard, E., et al. (2017). Serotonin and dopamine transporter PET changes in the premotor phase of LRRK2 parkinsonism: cross-sectional studies. Lancet Neurol. 16, 351–359. doi: 10.1016/s1474-4422(17)30056-x
Witoelar, A., Jansen, I. E., Wang, Y., Desikan, R. S., Gibbs, J. R., Blauwendraat, C., et al. (2017). Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol. 74, 780–792. doi: 10.1001/jamaneurol.2017.0469
Wszolek, Z. K., Pfeiffer, R. F., Tsuboi, Y., Uitti, R. J., McComb, R. D., Stoessl, A. J., et al. (2004). Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 62, 1619–1622. doi: 10.1212/01.wnl.0000125015.06989.db
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., et al. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. doi: 10.1016/j.neuron.2004.11.005
Keywords: Parkinson’s disease, LRRK2, inflammation, multiple sclerosis, rheumatoid arthritis, achalasia, dementia
Citation: Aasly JO (2021) Inflammatory Diseases Among Norwegian LRRK2 Mutation Carriers. A 15-Years Follow-Up of a Cohort. Front. Neurosci. 15:634666. doi: 10.3389/fnins.2021.634666
Received: 28 November 2020; Accepted: 06 January 2021;
Published: 28 January 2021.
Edited by:
Hardy Rideout, Biomedical Research Foundation of the Academy of Athens (BRFAA), GreeceReviewed by:
Rubén Fernández-Santiago, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), SpainRoy Alcalay, Columbia University, United States
Copyright © 2021 Aasly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan O. Aasly, SmFuLkFhc2x5QG50bnUubm8=