 Ethan Lorsung1†
Ethan Lorsung1† Ruifeng Cao
Ruifeng Cao- 1Department of Biomedical Sciences, University of Minnesota Medical School, Duluth, MN, United States
- 2Department of Neuroscience, University of Minnesota Medical School, Minneapolis, MN, United States
Autism spectrum disorders (ASDs) are a spectrum of neurodevelopmental disorders characterized by impaired social interaction and communication, as well as stereotyped and repetitive behaviors. ASDs affect nearly 2% of the United States child population and the worldwide prevalence has dramatically increased in recent years. The etiology is not clear but ASD is thought to be caused by a combination of intrinsic and extrinsic factors. Circadian rhythms are the ∼24 h rhythms driven by the endogenous biological clock, and they are found in a variety of physiological processes. Growing evidence from basic and clinical studies suggest that the dysfunction of the circadian timing system may be associated with ASD and its pathogenesis. Here we review the findings that link circadian dysfunctions to ASD in both experimental and clinical studies. We first introduce the organization of the circadian system and ASD. Next, we review physiological indicators of circadian rhythms that are found disrupted in ASD individuals, including sleep–wake cycles, melatonin, cortisol, and serotonin. Finally, we review evidence in epidemiology, human genetics, and biochemistry that indicates underlying associations between circadian regulation and the pathogenesis of ASD. In conclusion, we propose that understanding the functional importance of the circadian clock in normal and aberrant neurodevelopmental processes may provide a novel perspective to tackle ASD, and clinical treatments for ASD individuals should comprise an integrative approach considering the dynamics of daily rhythms in physical, mental, and social processes.
Introduction
Circadian rhythms are evolved as a result of the axial rotation of the earth and have been observed in almost all living organisms including human beings. The approximately 24 h rhythms are intrinsically driven by circadian clocks but are entrained by environmental cues such as light (Reppert and Weaver, 2002). Many neurophysiological processes exhibit robust daily fluctuations in their functional states. In humans, language, learning, memory, and social behavior adapt to the sleep–wake cycles, and the performance in all these activities exhibits daily fluctuations (Amir and Stewart, 2009). The functional significance of the circadian clock is being increasingly appreciated as circadian dysfunctions have been linked to an increasing number of human diseases including metabolic syndromes, cardiovascular diseases, diabetes, and cancer (Rijo-Ferreira and Takahashi, 2019). Anomalies in timing have been observed in neurological and psychiatric diseases including seasonal affective disorders, bipolar disorder, and schizophrenia, etc. (Wehr et al., 2001; Wulff et al., 2012; Logan and McClung, 2019). In neurodegenerative diseases such as Alzheimer’s disease, the disruption of daily activity rhythms is often associated with or even precedes underlying pathophysiological changes in the brain (Duncan, 2020). In fact, disruption of daily rhythms is the leading cause of institutionalization of individuals with Alzheimer’s disease (Musiek et al., 2015). Thus, a key role for circadian regulation/deregulation in neurological and psychiatric disorders is emerging in recent decades.
Autism spectrum disorders (ASDs) are a compilation of neurodevelopmental disorders defined by behavioral abnormalities (Mughal et al., 2020; American Psychiatric Association DSM-5). Growing evidence indicates dysfunction of the endogenous circadian system is associated with the neural dysfunctions prevalent in the development of ASD. Studies on the circadian clock and sleep in ASD improve our understanding of its pathogenesis and inspire potential chronotherapeutic strategies to treat or prevent the diseases. In this review, we discuss the involvement of the circadian timekeeping system in the development and functionality of the nervous system, and summarize evidence indicating underlying links between the circadian clock and ASD. We first introduce the organization of the circadian system and ASD. Next, we review physiological parameters of endogenous rhythms that are found disrupted in ASD patients, including the sleep/wake cycle, and the daily oscillations of the circadian biomarkers melatonin, cortisol and serotonin. Finally, we review evidence indicating underlying links between circadian dysfunction and ASD pathogenesis, including epidemiology, human genetics, and the mTOR pathway.
The Circadian Timekeeping System
The term “circadian” was originally coined by Franz Halberg from the Latin root circa meaning “around” and diem meaning “day.” Circadian rhythms refer to the approximately 24 h rhythms that are found in a variety of physical, mental, or behavioral processes (Halberg, 1969; Pittendrigh, 1993). The rhythms are endogenously driven by circadian clocks, which are oscillating proteins in cells that are found in nearly all living organisms (Rosbash, 2009). A variety of physiological events are regulated by circadian clocks and exhibit circadian rhythms, including the sleep–wake cycles, core body temperature, blood pressure, hormone secretion, and cognition (Patke et al., 2020). Circadian rhythms are found at every level of the organization of life: cellular, tissue, organ, and organismal level (Takahashi et al., 2008).
The oscillations of the cellular clock are driven by transcriptional-translational feedback loops (TTFLs) (Rosbash, 2009). In mammals, TTFLs are driven by rhythmic oscillations of about a dozen clock genes and their protein products (Figure 1), including two Period genes (Per1 and Per2), two Cryptochrome genes (Cry1 and Cry2), Clock, Bmal1, Rev-erbα/β, Rorα/β/γ, and CkIε/δ (Takahashi et al., 2008). The CLOCK and BMAL1 proteins are activators and form a heterodimer to bind E-box enhancers in the promoters of Per and Cry genes. PER and CRY proteins are synthesized during the day and form a protein complex which accumulates in the cytoplasm during the afternoon and evening. Upon reaching a certain level, the PER-CRY complexes translocate into the cell nucleus during the nighttime and block the activities of the CLOCK: BMAL1 heterodimer to inhibit their own gene transcription (Shearman et al., 2000; Ramanathan et al., 2006; Yan et al., 2020). In addition, the CLOCK: BMAL1 complex also promotes the transcription of Rev-erbα/β and Rorα/β/γ. REV-ERBα/β in turn inhibits Bmal1 transcription whereas RORα/β/γ promotes Bmal1 transcription (Preitner et al., 2002). In this way, the CLOCK: BMAL1 heterodimer is a self-regulator.
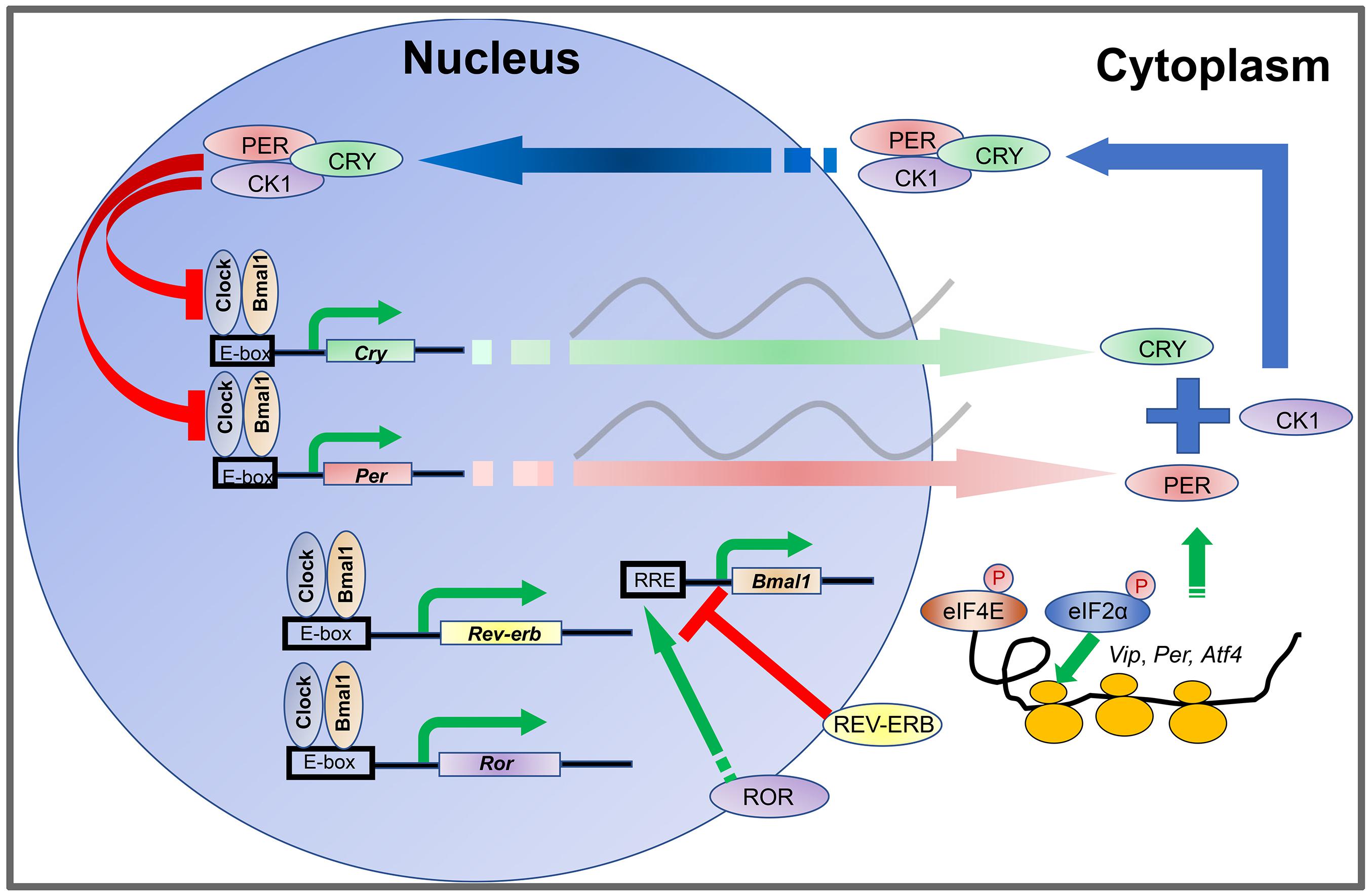
Figure 1. Transcription-translation feedback loops (TTFLs) in the mammalian circadian clock. The CLOCK and BMAL1 proteins are activators and form a heterodimer to bind to E-box enhancers in the promoters of Per and Cry genes. PER and CRY proteins are synthesized during the day and form a protein complex which accumulates in the cytoplasm during the afternoon and evening. Upon reaching certain level, the PER-CRY complexes translocate into the cell nucleus during the nighttime and block the activities of the CLOCK: BMAL1 heterodimer to inhibit their own gene transcription. In addition, the CLOCK: BMAL1 complex also promotes the transcription of Rev-erb and Ror. REV-ERB in turn inhibits Bmal1 transcription whereas ROR promotes Bmal1 transcription. The abundance of PER proteins is controlled at the level of mRNA translation by rhythmic phosphorylation of eIF4E. Phosphorylation of eIF2α promotes translation of Atf4. ATF4 directly activates Per2 transcription. At the posttranslational level, levels of PER and CRY protein are regulated by phosphorylation and ubiquitination-mediated protein degradation CKI phosphorylates PER. Phosphorylation of PER and CRY proteins promotes their degradation and speeds up the clock.
The abundance of PER proteins is also controlled at the level of mRNA translation by an eIF4E-dependent mechanism. Rhythmic phosphorylation of eIF4E by the mitogen-activated protein kinase-interacting kinases (MNKs) promotes mRNA translation of Per1 and Per2 (Cao et al., 2015). At the posttranslational level, levels of PER and CRY proteins are regulated by phosphorylation and ubiquitination-mediated protein degradation (Hirano et al., 2013; Yoo et al., 2013). CKIε and CKIδ phosphorylate PER (Lee et al., 2001; Meng et al., 2008; Etchegaray et al., 2009; Lee et al., 2011), whereas AMPK phosphorylates CRY (Lamia et al., 2009). Phosphorylation of PER and CRY proteins promotes their degradation and speeds up the clock. Although intracellular clock mechanisms are thought to be conserved in different cells, intercellular coupling mechanisms are unique between neurons and glial cells in the suprachiasmatic nucleus (SCN) and confer robustness and precision to the SCN clock (Aton and Herzog, 2005; Hastings et al., 2018). When SCN cells are isolated, the cell autonomous oscillations are poorly organized (Welsh et al., 1995; Herzog et al., 1998; Patton et al., 2016). Numerous body clocks are orchestrated by the SCN pacemaker in the hypothalamus, which is a pair of tear-drop-like structures in the inferior portion of the brain composed of ∼20,000 neurons (Moore et al., 2002; Hastings et al., 2018). The neurons express the neuropeptide vasoactive intestinal polypeptide (VIP) and gastrin-releasing peptide (GRP) in the core (ventral) region of the SCN, and arginine vasopressin (AVP) in the shell (dorsal) region. The astrocytes are regulatory cells to the neurons, and primarily utilize the neuro-excitatory molecule glutamate at night to inhibit SCN neuron activity (Brancaccio et al., 2017).
The circadian clocks are entrained by external signals called zeitgebers to synchronize themselves with the ever-changing environment (Figure 2). The SCN utilizes light as its primary zeitgeber. SCN receives photic information from the intrinsically photosensitive retinal ganglion cells (ipRGCs) in the retina (Berson et al., 2002). The ipRGCs express the photopigment melanopsin and their axons form the retinohypothalamic tract (RHT) that terminates in the SCN. The RHT pathway is separated from the image forming visual pathway (Provencio et al., 2002; Peirson and Foster, 2006). The RHT terminals form direct synaptic connections with the core SCN neurons that express the neuropeptides VIP or GRP. Upon photic stimulation at night, RHT terminals release glutamate and the neuropeptide pituitary adenylate cyclase activating polypeptide (PACAP) that are the neurotransmitters functioning to evoke clock gene expression and reset the SCN clock by regulating intracellular signaling pathways (Obrietan et al., 1998; Butcher et al., 2002; Hannibal, 2002).
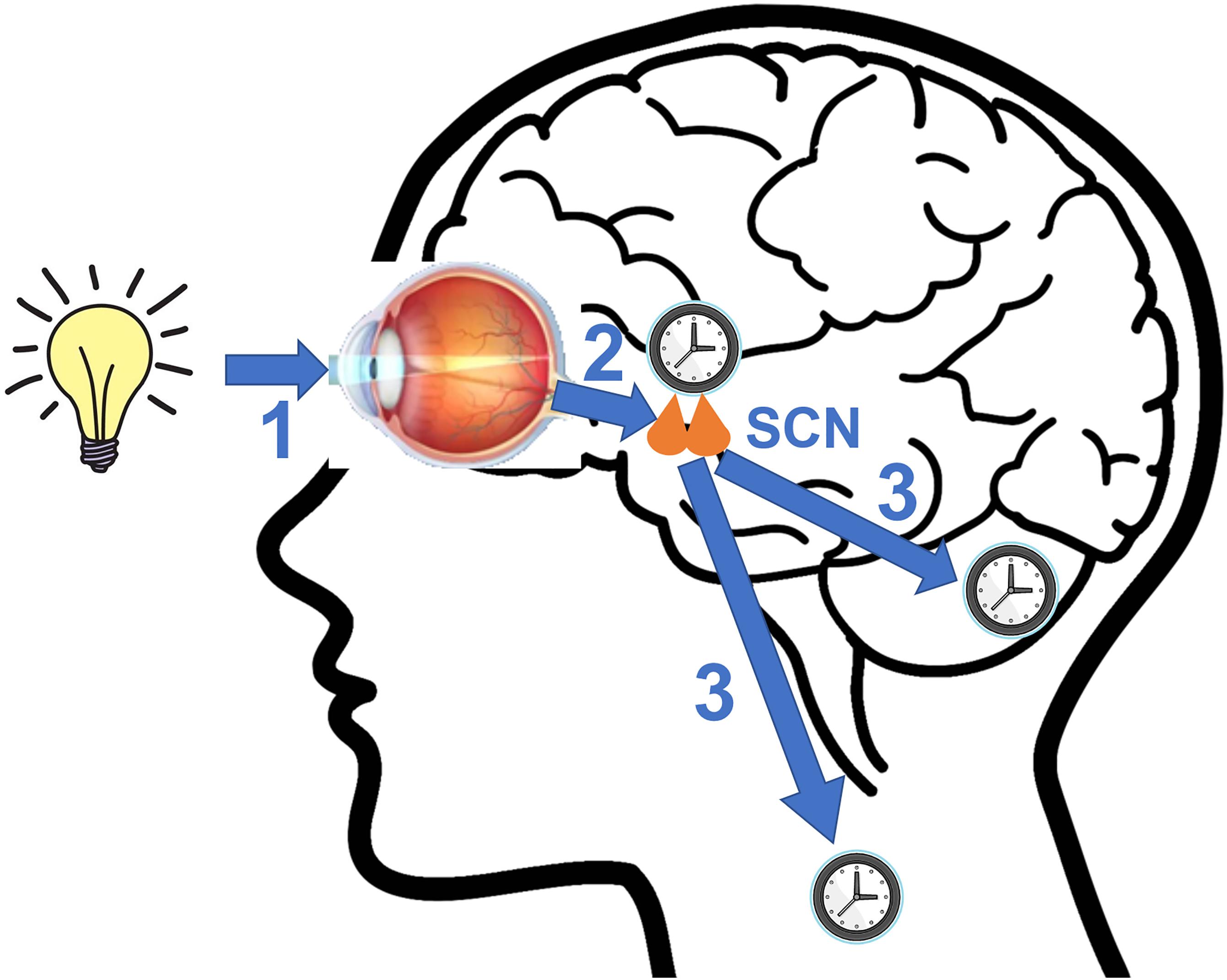
Figure 2. A diagram illustrating key steps involved in photic entrainment of the circadian system. (1) Ambient light stimulates intrinsically photosensitive retinal ganglion cells (ipRGCs) in the retina. (2) The axons of ipRGCs travel via the retinohypothalamic tract (RHT) to form synaptic connections with the core neurons of hypothalamic suprachiasmatic nucleus (SCN). Glutamate and pituitary adenylate cyclase activating polypeptide (PACAP), among other neurotransmitters are released at the synapses of the RHT terminals to the SCN neurons. Synaptic activities induce clock gene expression and reset the SCN clock. (3) SCN sends rhythmic outputs to other brain regions and peripheral oscillators to reset their rhythms.
Besides light, non-photic inputs (electrical stimulation, odor, etc.) can also influence the SCN through two brain regions, the intergeniculate leaflets (IGL) and the dorsal/median raphe nucleus (DRN/MRN) (Rusak et al., 1989; Meyer-Bernstein et al., 1997). The afferent pathway from the IGL is the geniculohypothalamic tract (GHT), and the DRN/MRN communicates with the SCN through serotonergic neurons (Meyer-Bernstein and Morin, 1996; Moga and Moore, 1997). Besides these inputs, other external cues such as social activities, exercise, and temperature have been examined as zeitgebers to the sleep-wake cycle in adult humans, but there are critiques of the role for social zeitgebers beyond their role of light regulation (Korczak et al., 2008). The SCN communicates internally with peripheral tissues through neural and endocrine outputs, i.e., electrical signals, neurotransmitters, and hormones (Kalsbeek et al., 2006). The SCN resets the peripheral clocks via these output signals. The peripheral clocks can also be reset by extrinsic and intrinsic cues that are relevant to their physiological functions. For example, adrenal hormones and feeding schedule are of notable importance to liver clock gene rhythms (Su et al., 2016). The timing of physical activities is a cue for the skeletal muscle clock (Wolff and Esser, 2012). The peripheral clocks regulate local physiology and help to orchestrate the organismal function by synchronizing rhythms in various systems. Thus, by synchronizing with the environmental light-dark cycles, the SCN clock orchestrates rhythms in different systems and coordinates various physiological processes and systemic well-being.
Autism Spectrum Disorders (ASDs)
Autism spectrum disorders are a group of developmental disabilities diagnosed by core behavioral symptoms including trouble with social interaction, abnormal communication skills, and atypically restricted, stereotyped, repetitive behaviors (American Psychiatric Association DSM-5, 2013). Children are now commonly diagnosed by 3 years of age, which is earlier than in the past (Mazurek et al., 2014). Clinical symptoms exhibited by autistic children are not uniform. Children with autism have social developmental problems and exhibit interest toward repetitive behavioral processes (Bodfish et al., 2000). Developmental deficits in ASDs have been confirmed by studies finding abnormalities in both prenatal and postnatal brain development (Carper et al., 2002; Hazlett et al., 2005; Bonnet-Brilhault et al., 2018). Their delay in development of communication and restricted interests is thought to be correlated to the severity of the anomalies in the brain (Anderson et al., 2009). ASD is often accompanied by intellectual disability and hyperactivity. In addition, children with ASD commonly exhibit comorbid medical conditions, including abnormal tactile sensation, food selectivity, and sleep disruption (Baranek et al., 2006; Ben-Sasson et al., 2007; Souders et al., 2009). Currently there is no unified theory to explain all core and comorbid abnormalities in ASD children.
The incidence of ASD has dramatically increased around the globe in the past 50 years (World Health Organization, 2017). For example, according to a British study, autism incidence rate was 4.5 per 10,000 children in the 1960s (Lotter, 1966). In the 1980s, the prevalence of autism was between 5 and 12 in 10,000 persons (Gillberg et al., 1991). In the U.S., the frequency of the autism has risen from 3 per 10,000 individuals in 1991–1992 to 53 per 10,000 children in 2003–2004 (Gurney et al., 2006). According to estimates from CDC’s Autism and Developmental Disabilities Monitoring Network, about 1 in 54 children are diagnosed with ASDs in 20141. Notably, the diagnostic criteria for ASD was changed in the DSM-5 published in 2013, and an expanded group of disorders are now classified as ASDs (American Psychiatric Association DSM-5, 2013). ASDs now include several conditions that used to be diagnosed separately including autistic disorder, pervasive developmental disorder not otherwise specified (PDD-NOS), and Asperger syndrome. The high prevalence of ASDs is accompanied by unprecedented social and economic burdens on the affected families and society (Newschaffer et al., 2007). The yearly total costs for children with ASD were estimated to be between $11.5 and $60.9 billion in the U.S. The clinical expenses of autism children are comparatively higher than normal children and are ten times greater than the costs of normal children’s medical expenditure (Mandell et al., 2006). Children with ASD cost more than those without ASD by $4,110–$6,200 per year. Thus, there is an urgent need to find novel therapeutic strategies to tackle these diseases.
The etiology of ASD remains elusive, but it is thought to be a combination of extrinsic and intrinsic factors (Figure 3). Less than 20% of ASDs have an identifiable genetic origin, whereas over 75% of cases are idiopathic, suggesting a multifactorial etiology (Abrahams and Geschwind, 2008). The discovery of single nucleotide variants (SNVs) and copy number variants (CNVs) in genes associated with ASD supports the claim for a genetic basis of ASD etiology and over 1200 risk genes have been identified (Xiong et al., 20192). In addition, epigenetic and immunological factors are also speculated as possible causes of autism (Lee et al., 2015; Sun et al., 2016). An increased risk of ASD with advanced paternal age coupled to an increased rate of DNA methylation abnormalities in older fathers at multiple imprinted gene loci suggests an epigenetic association (Kong et al., 2012; Smith et al., 2013). Animal models have shown transgenerational aberrant DNA methylation and histone modifications with abnormal neurodevelopment as a result of abnormal nutrition, stress and drugs, as well as transplacental psychiatric medication affecting GABAergic, dopaminergic, serotonergic, and glutamatergic pathways (Franklin et al., 2010; Morgan and Bale, 2011). Besides genetic and epigenetic factors, development of ASD can be influenced by embryonic exposure to detrimental environmental factors including pollution, maternal stressors, etc. (Becerra et al., 2013; Volk et al., 2013; Walder et al., 2014). Perinatal brain injury, especially cerebellar injury, can also contribute to autism development (Singh et al., 2016).
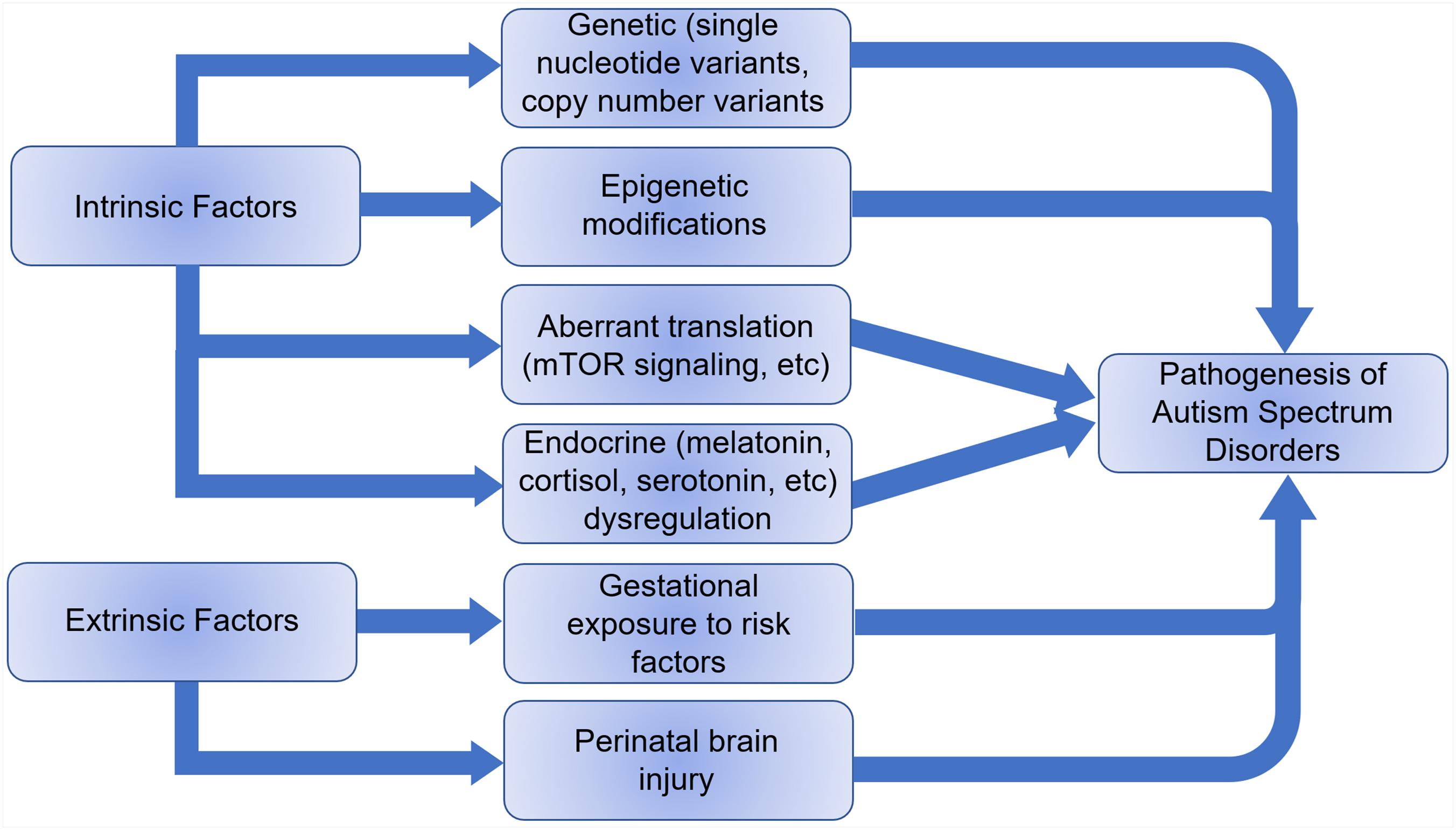
Figure 3. Intrinsic and extrinsic factors that can lead to the pathogenesis of autism spectrum disorders.
Multiple theories have been proposed regarding the neural mechanisms underlying ASD pathogenesis, but no theory has convincingly integrated the diverse behavioral dysfunctions in autism. Aberrant neurotransmission of dopamine, glutamate, serotonin, oxytocin/vasopressin and GABA have all been implicated in the development of ASD (Modahl et al., 1998; Blatt et al., 2001; Chandana et al., 2005). A number of studies suggest that glutamate systems are dysfunctional in ASD (Purcell et al., 2001; Shinohe et al., 2006; Rojas, 2014). The dysfunction of the GABAergic system, synthesized from glutamate, has been suggested to result in impaired cognitive and motor function as well as seizure disorder, a comorbidity of autism (Russo, 2013; Rojas, 2014). Excessive dopamine receptor DRD1a activation has been shown to elicit autistic behaviors in mouse models (Lee et al., 2018). The increased prevalence of ASD in recent decades cannot be simply explained by reclassification and increased diagnosis. Neither can it be completely ascribed to genetic factors that cause the disease. Apparently, a combination of extrinsic and intrinsic factors should be investigated in the development of autism.
Disruption of Sleep and Daily Rhythms in ASD
Sleep Problems in ASD
Sleep is a conserved physiological process in animals that is critical for brain development and maturation. Suboptimal sleep can have adverse effects on children’s cognitive functions including attention, memory, mood regulation, and behavior (Pilcher and Huffcutt, 1996; Belenky et al., 2003). Children with ASD exhibit sleep problems at a higher rate than children with other developmental disorders as well as typically developing children (Owens et al., 2000; Cotton and Richdale, 2006; Johnson and Zarrinnegar, 2021). Repeated sleep disruption adversely affects the process of neural development in ASD children, whereas impaired neurodevelopment further exacerbates the sleep problem in ASD.
It is estimated that 50∼80% ASD children have sleep problems, compared to less than 30% in the general children population (Souders et al., 2017). Prolonged sleep latency, frequent waking at night, alterations in sleep architecture, unusual morning arousal and reduction in total sleep duration are commonly found in children with autism (Richdale, 1999; Polimeni et al., 2005). A reduced percentage of REM sleep and higher percentage of slow-wave sleep has been observed in association with ASD (Buckley et al., 2010). There is also a report of a higher rate of REM sleep behavior disorder in autistic children (Thirumalai et al., 2002). Atypical REM sleep patterns indicate disruption in central nervous system maturation and neuronal network organization, as well as atypical synapse homeostasis involved in sleep–wake function (Buckley et al., 2010). Notably, sleep problems may differ between different types of ASD patients. In a study by Richdale and Prior (1995), it was found that low functioning (IQ < 55) ASD individuals showed increased naps, earlier time going to sleep, increased sleep latency, increased sleeping time at night, and increased total sleeping time over 24 h compared to controls. By contrast, high functioning (IQ > 55) ASD individuals exhibited increased sleep latency, decreased total night sleep, increased length of waking episodes at night, and earlier wake time. A more detailed review on sleep problems in ASD has been published recently (Karthikeyan et al., 2020).
The possible causes for the sleep problems in ASD can be classified into four categories. (1) Synaptic protein abnormalities. Sleep and synaptic functions are tightly interconnected. Sleep relies on normal functionality of complex neural circuitries with synapses as the connectors between neurons (Scammell et al., 2017). Proper sleep is essential for synaptogenesis and synaptic plasticity (Cirelli and Tononi, 2019). The disruption of the Neurexin/Neuroligin/Shank synaptic protein complex has been found to be involved in ASD (Jamain et al., 2003; Durand et al., 2007). Neuroligins and Neurexins are synaptic proteins of excitatory glutamatergic and inhibitory GABAergic synapses. Neuroligin-1/3/4 are confined to glutamatergic synapses whereas neuroligin-2 is specific to GABAergic synapses (Graf et al., 2004; Varoqueaux et al., 2004). Neurexins, encoded by Nrxn 1/2/3, are a family of presynaptic cell adhesion proteins that interact with Neuroligins to connect neurons at the synapse. Mutation of Shank3, a gene encoding a scaffolding protein on the postsynaptic membrane that tethers Neuroligins and regulates dendritic organization, has been shown to be associated with autism (Durand et al., 2007). Furthermore, a de novo deletion on chromosome 2p16 encoding Neurexin-1 was identified in ASD (Szatmari et al., 2007). Interestingly, Neuroligins, Neurexin, and Shank3 have all been shown to regulate the sleep architecture and clock gene expression in mouse models (El Helou et al., 2013; Tong et al., 2016; Seok et al., 2018; Ingiosi et al., 2019). It is possible that dysregulated synaptic proteins link sleep disorders to the development of autism. (2) Sensory dysregulation and increased arousal (Wiggs and Stores, 2004; Souders et al., 2009). This can be explained by two hypotheses, cognitive arousal and physiological arousal. Cognitive arousal, which is caused by increased cognitive activities due to increased anxiety in ASD, can increase the sleep latency. Physiological arousal is caused by increased responses to environmental stimuli due to low sensory thresholds in children with ASD, and can result in difficulty falling or staying asleep (Mazurek and Petroski, 2015). (3) Abnormal sleep-regulating hormones. Abnormal levels of hormones such as melatonin in ASD will be discussed in “Disruption of Circadian Biomarkers in ASD.” (4) Circadian sleep disruptions. Mutations in genes that regulate circadian timing can also cause changes in the timing and duration of sleep in ASD and will be discussed in “Clock Gene Polymorphisms in ASD.” It is important to recognize the possible overlaps in causes, and their additive or amplified effects on the complex sleep problems in ASD.
Disruption of Circadian Biomarkers in ASD
In clinical studies, levels of traditional circadian biomarkers including melatonin and cortisol are measured from biological specimens such as blood, urine, and saliva at different times of day in order to assess the functions of the body clock. Melatonin is a pineal hormone with daily rhythmic synthesis that peaks at night and is suppressed by light during the day (Socaciu et al., 2020). Cortisol is a sterol hormone that peaks in the early morning and falls throughout the day (Russell and Lightman, 2019). Serotonin is a monoamine neurotransmitter and also the intermediate product to synthesize melatonin (Comai et al., 2020). Here we discuss how levels and daily rhythms of these biomarkers are changed in ASD, and how abnormalities in these hormones may in turn contribute to neural dysfunctions in ASD individuals. The abnormalities of these biomarkers have been summarized in Table 1.
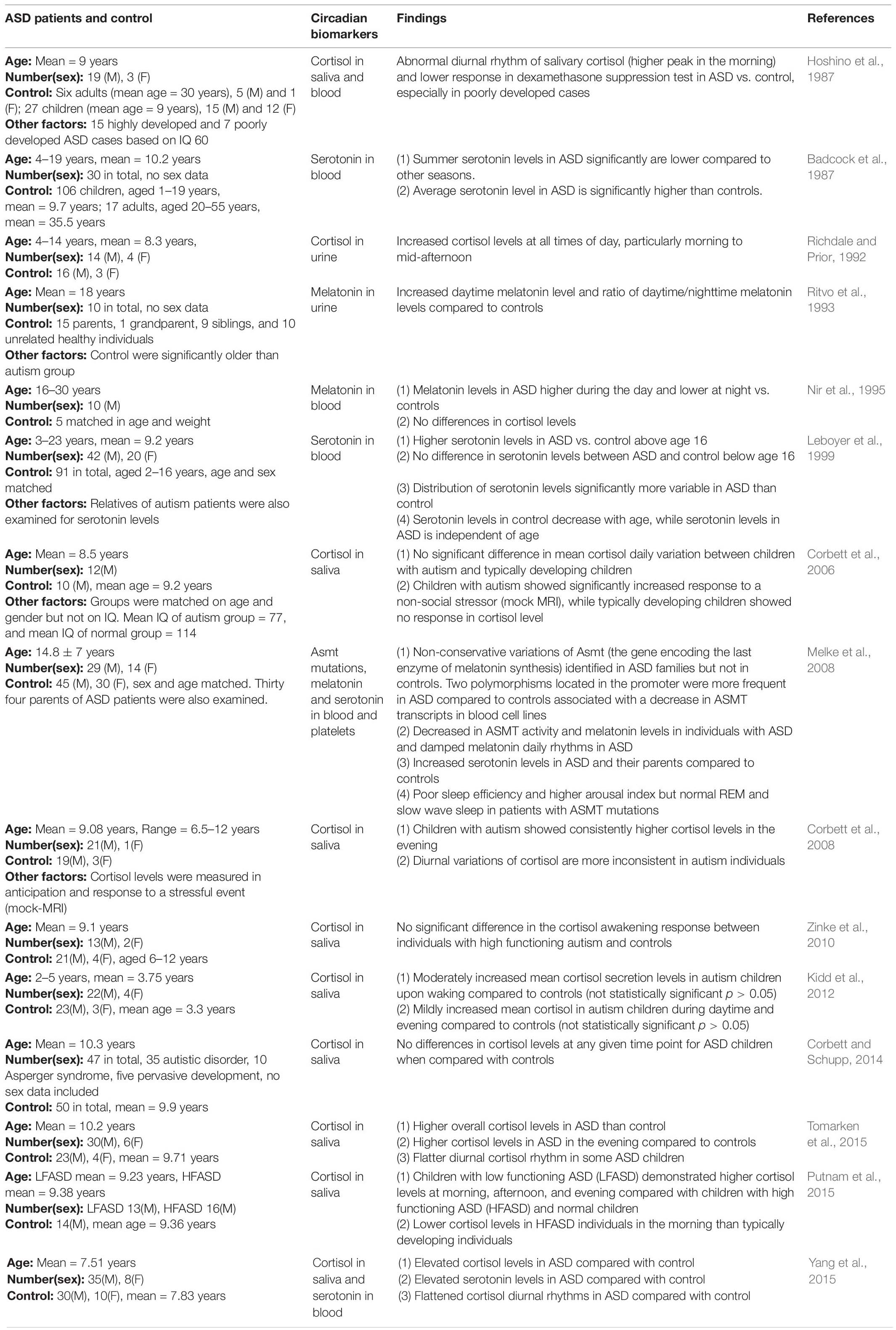
Table 1. Circadian dysfunctions in ASD.
Melatonin
Melatonin is a neurohormone synthesized in the pineal gland. Melatonin levels are normally higher at night than during the day in both nocturnal and diurnal animals. Melatonin induces sleep and resets the SCN circadian clock (McArthur et al., 1991; Zhao et al., 2019). Sleep phase and duration is determined by the phase of the melatonin cycle suggesting a key role for melatonin in regulating the sleep–wake cycle (Lockley et al., 1997). Melatonin has direct effects on the SCN circadian clock through its two G protein coupled receptors MT1 and MT2. MT1 is the high affinity receptor responsible for acute suppression of neuronal firing and MT2 is the low affinity receptor required for efficient phase-shifts (Liu et al., 1997; Jin et al., 2003). Binding of melatonin to MT1 and MT2 indirectly regulates clock gene expression by inhibition of adenylate cyclase, and inhibition of PKA due to reduction of cAMP. The G protein coupled receptors also directly inhibit phosphorylation of cAMP response element-binding protein (CREB) (Ross et al., 1996; von Gall et al., 2002). CREB inhibition causes decreased expression of clock proteins PER1 and PER2 and attenuates photic entrainment of the circadian clock (Lee et al., 2010). Melatonin signaling disruption has been linked to sleep disorders such as insomnia, and has been reported in neurological and psychiatric conditions such as Parkinson’s disease and depression (Claustrat et al., 1984; Adi et al., 2010). There is also evidence that melatonin is involved in neural differentiation, and its dysfunction in ASD individuals could contribute to their non-typical development (Shu et al., 2016).
Melatonin is the most well documented circadian biomarker associated with ASD. Lower levels of melatonin and its major metabolite, urinary 6-sulfatoxymelatonin, have been found in the urine, serum, and plasma of ASD individuals (Nir et al., 1995; Tordjman et al., 2005; Melke et al., 2008). Low melatonin amplitude and a delayed melatonin rhythm have been associated with increased sleep problems in ASD children (Kulman et al., 2000; Melke et al., 2008). Excretory levels of the metabolite 6-sulphatoxymelatonin were decreased in a group of 50 autistic children and these decreased concentrations were associated with their verbal and play abilities (Tordjman et al., 2005). Seizure comorbidities and electroencephalogram (EEG) discrepancies in autism individuals have been associated with the aberrant phase cycles of melatonin (Nir et al., 1995). Increased levels of melatonin have been found during the daytime in small samples of autistic children, whereas no significant difference was reported in nighttime concentration (Ritvo et al., 1993; Kulman et al., 2000). Low overall levels of melatonin and a sharp increase in concentration during the daytime was reported in a study of 14 autistic children (Kulman et al., 2000). Interestingly, 10 of the 14 autistic individuals in the study exhibited no observable daytime rhythmic changes in their blood melatonin levels (Kulman et al., 2000). Unusual patterns of melatonin in both amplitude and phase suggests fundamental impairments of the body circadian clock. As the level of melatonin is in general decreased in ASD individuals, melatonin supplements at night before bedtime may help with the sleep problems in ASD. In one study, melatonin administered 30 min before bedtime improved sleep latency in ASD children (Malow et al., 2012).
The mechanisms underlying melatonin abnormalities in ASD remain elusive and is a topic still under investigation. It is unclear whether the total amount of melatonin is reduced in a circadian period or if the phase has been altered by the abnormal circadian clock (Tordjman et al., 2005). There may also be abnormalities in melatonin synthesis, regulation, or receptor binding and efficacy in ASD. There are three main G-protein coupled receptors (GPCR) receptors involved in melatonin signaling: MNTR1A (MT1), MNTR1B (MT2), and the orphan receptor GPR50, which has no affinity for melatonin, but inhibits melatonin signaling when bound to MT1 (Chaste et al., 2010). When individuals with ASD were screened for mutations in the genes encoding melatonin receptors, no significant difference was found compared to controls, indicating that abnormal melatonin production rather than abnormal receptor function may be involved in ASD. Thus, rectifying melatonin levels using exogenous melatonin is a plausible therapeutic strategy. Indeed, clinical evidence exists demonstrating high efficacy of melatonin treatment for ASD individuals with sleep disruption (Chaste et al., 2010; Malow et al., 2012).
It is likely that melatonin disruption in a significant number of ASD individuals is due to dysfunction of melatonin synthesis. There are two enzymatic steps in the conversion of serotonin into melatonin: the conversion of serotonin to N-acetylserotonin by the enzyme Serotonin N-acetyl transferase (SNAT), and the conversion of N-acetylserotonin into melatonin by the enzyme Acetylserotonin O-methyltransferase (ASMT) (Figure 4). 14-3-3 is a family of conserved regulatory proteins that bind to a variety of signaling proteins. It has been proposed interaction of the protein 14-3-3 with SNAT, and more importantly 14-3-3 with ASMT, is necessary for melatonin synthesis (Obsil et al., 2001; Maronde et al., 2011). The protein miR-451, a known suppressor of 14-3-3, was elevated in ASD individuals (Pagan et al., 2017). An increasing body of evidence supports a disruption of the 14-3-3/ASMT/SNAT ‘melatoninosome’ in ASD individuals (Obsil et al., 2001; Maronde et al., 2011). Slow metabolization of melatonin may lead to accumulation of melatonin in the body and cause sleep problems. The liver cytochrome P450 enzyme, CYP1A2, has been demonstrated to be the primary metabolizing enzyme of melatonin in the liver (Facciolá et al., 2001). In the small number of ASD individuals where exogenous treatment with melatonin loses effectiveness, high level of melatonin was found around noon. It has been hypothesized this is due to a single nucleotide polymorphism (SNP) in CYP1A2 (Braam et al., 2013). While melatonin treatment has been shown to be effective in treating sleep–wake difficulties in ASD individuals, more research is necessary to elucidate the precise role of melatonin and its dysregulation in ASD.
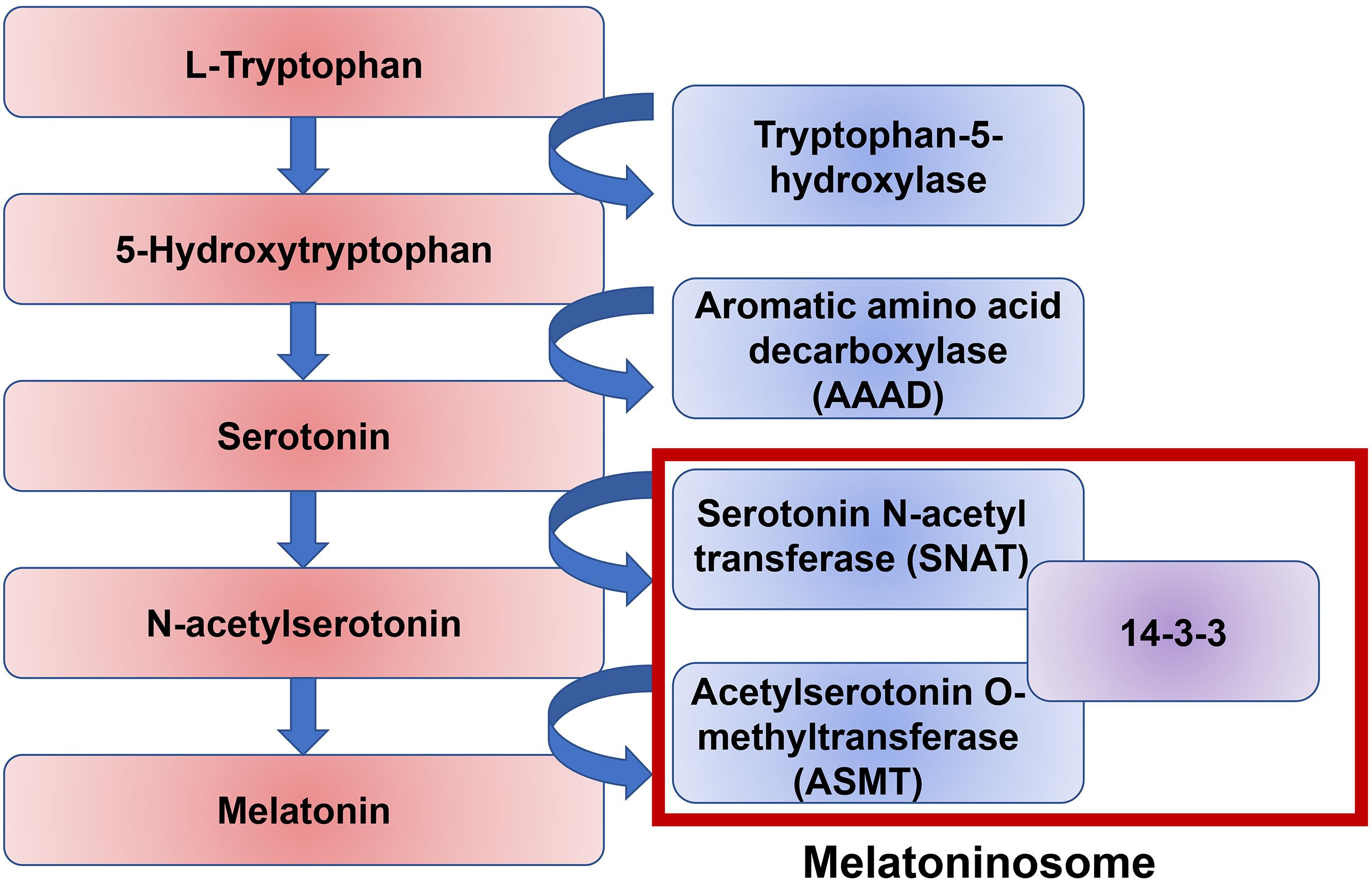
Figure 4. A pathway of melatonin biosynthesis. Melatonin synthesis is the result of the amino acid tryptophan through the intermediate neurotransmitter serotonin. The conversion of serotonin to melatonin is mediated by two enzymes, serotonin N-acetyl transferase (SNAT) and acetylserotonin O-methyltransferase (ASMT). The protein 14-3-3 mediates both of these stepwise interactions.
Cortisol
Cortisol production is a result of a cascade response along the hypothalamic-pituitary-adrenocortical (HPA) axis in three major steps with negative feedback at each step: (1) Corticotropin releasing hormone (CRH) is released by the paraventricular nucleus (PVN) of the hypothalamus; (2) Adrenocorticotropic hormone (ACTH) is released from the anterior pituitary; and (3) Cortisol is released from the adrenal cortex (Figure 5). Each hormone is released in response to the action of the preceding hormones action on its respective target tissue. The HPA axis regulates hormonal stress response. Daily cortisol levels exhibit robust oscillations as PVN is innervated by the SCN. Here we discuss circadian rhythm abnormalities of cortisol in ASD individuals and whether these abnormalities are associated with ASD as a causative factor that may exacerbate traits of the disorder, or simply a result of ASD symptoms.
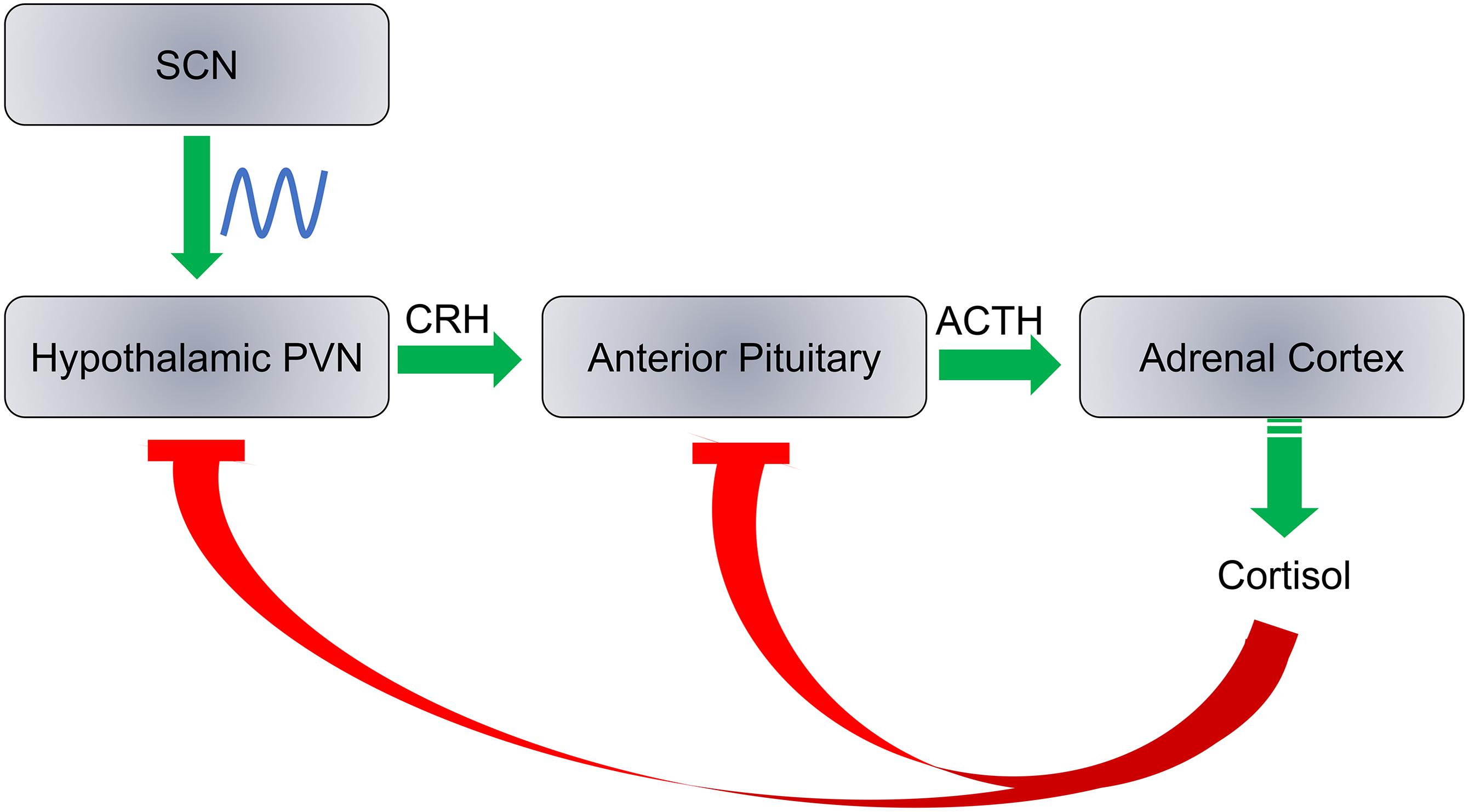
Figure 5. SCN regulates the hypothalamic-pituitary-adrenocortical axis. Cortisol production is regulated by three steps: (1) Release of corticotropin releasing hormone (CRH) by the hypothalamic paraventricular nucleus (PVN); (2) the adrenocorticotropic hormone (ACTH) is released from the anterior pituitary; and (3) Cortisol is synthesized and released from the adrenal cortex. Cortisol inhibits CRH and ACTH release at a feedback mechanism. The SCN has direct neural inputs into PVN and controls the daily rhythms of cortisol levels.
The PVN receives direct neural input from the hippocampus, amygdala, prefrontal cortex, and SCN (Gray et al., 1989; Boudaba et al., 1996; Li and Kirouac, 2012). Cortisol has ubiquitous physiological effects throughout the body, and has been proposed to play a key role in daily cognitive and behavioral functions. Disruption of its rhythms have been implicated in the etiology of a variety of physical and mental health disorders (Adam et al., 2017). The significance of adrenal glucocorticoids to peripheral circadian rhythms have been demonstrated, as elimination of the adrenal glands in rats caused disruption of clock gene expression in the kidneys and corneas (Pezük et al., 2012). Interestingly, the adrenalectomy did not have a significant impact on the SCN, pituitary gland, or lungs of the rats, but introduction of hydrocortisone following adrenalectomy did have a significant impact on all circadian gene expression in each of these tissues (Pezük et al., 2012).
The blood concentration of cortisol, the human glucocorticoid stress hormone, varies across the 24 h day and possesses significant diurnal rhythms. It reaches its peak during the early morning, decreases during the day, and starts to rise at late night (Hoshino et al., 1987; Van Cauter et al., 1996). The phase and levels of cortisol can be used as reliable indicators for the endogenous circadian clock (Désir et al., 1980; James et al., 2007). The circadian clock could also be responsible for regulating the cortisol awakening response, an increase of cortisol within the first hour after awakening that is separate from the cortisol increase during the second half of the night (Vargas and Lopez-Duran, 2020). Circadian misalignment can cause deregulated cortisol production in neurotypical individuals. Even one night of sleep loss can elevate cortisol concentration, notably during the early morning and evening hours (Wright et al., 2015). Interestingly, there is some evidence for the absence of feedback on the SCN from the HPA axis (Oster et al., 2006). An abnormal central pacemaker stimulating the HPA axis at abnormal intervals with no feedback may cause abnormal cortisol profiles.
A number of studies suggest abnormalities in circadian rhythms of cortisol in ASD individuals, but the severity and type vary greatly (Yamazaki et al., 1975; Hill et al., 1977; Corbett et al., 2006). Aberrant rhythmic patterns of cortisol were found to be associated with lower functioning autistic children (Jensen et al., 1985; Hoshino et al., 1987). Additionally, one study found significantly decreased cortisol in ASD children, but elevated ACTH levels compared to typically developing subjects (Curin et al., 2003). Subsequently, the same researchers found a delayed cortisol response to artificial ACTH stimulation in ASD children compared to controls (Marinović-Curin et al., 2008). Another investigation found elevated ACTH and β-Endorphin (a hormone released concurrently with ACTH) in ASD individuals, but no difference in cortisol levels (Tordjman et al., 1997). On the contrary, one study measuring total daily urinary cortisol secretion found no abnormalities between typically developing individuals and ASD individuals (Marinović-Curin et al., 2008). This suggests that while measured cortisol levels and rhythms may be abnormal in ASD individuals, total cortisol output may be similar to typically developing individuals. The variable results across studies could be caused by the differences in investigation methods, size of the sample groups, and variation in determined cognitive function (low functioning vs. high functioning). A comprehensive future examination of the cortisol circadian rhythm in ASD individuals with larger sample sizes, standardized measurement methods, controlled environments, and age/gender diversification is warranted.
There is also evidence of abnormal sensitivity to stress in ASD individuals related to increased variability of cortisol rhythms (Corbett et al., 2009). One study examined cortisol response to amicable social interaction with a confederate among younger and older ASD children compared to neurotypical children. The investigators found a significant increase in cortisol levels for older ASD children compared to younger ASD children; this significant difference was not present in neurotypical individuals (Corbett et al., 2010). This difference could be explained by an awareness of social limitations among older ASD individuals, or a learned negative threat response to what would commonly be perceived as a neutral or positive social stimulus, leading to an increased cortisol response. Regardless of the etiology for the heightened response, the findings suggest increased susceptibility of the cortisol rhythm to external social zeitgebers in ASD children. Given the evidence for ASD symptoms impacting the cortisol circadian rhythm, the question remains if this relationship is unilateral, or can an aberrant circadian system in ASD individuals exert an atypical influence on cortisol levels and exacerbate negative symptoms. There is some intriguing evidence that abnormalities of cortisol circadian rhythms may be a function of ASD symptoms. A study in 2006 investigated salivary cortisol response to a disturbing non-social stimuli (mock MRI) in ASD individuals (IQ mean = 77) and neurotypical individuals, and found a strikingly significant cortisol increase in relation to the controls that exhibited a mean decrease in cortisol levels (Corbett et al., 2006). The finding of increased HPA response to a non-social stressor (blood draw) in ASD individuals compared to matched controls was replicated across variable measurements of cortisol including salivary, urinary, and serum (Spratt et al., 2012). A follow up to Corbett et al. (2006) examined cortisol response to a stressor as well as a subsequent exposure to the same stressor, and found increased circadian variability of cortisol in ASD individuals as well as increased cortisol levels in the evening following stressor exposure. This suggests the aberrant cortisol rhythm of ASD individuals may be more susceptible to entrainment by external non-social zeitgebers (Corbett et al., 2009). While there is evidence of abnormal circadian cortisol profiles in ASD individuals, the extent of the relationship and underlying mechanisms remains unclear and warrants further study.
Serotonin
Serotonin is produced in the central nervous system and duodenum. As serotonin cannot cross the blood-brain barrier, central and peripheral serotonergic systems are thought to be anatomically ad functionally separated (Hery et al., 1977; Ebert-Zavos et al., 2013). Serum serotonin levels exhibit diurnal variations, with a peak early in the morning and a trough in the midafternoon and during sleep (Wirz-Justice et al., 1977; Kwon et al., 2018). The diurnal oscillations of serotonin are affected by meal intake or fasting and are blunted in obese individuals (Ebert-Zavos et al., 2013; Kwon et al., 2018). An earlier study detected another peak of serotonin in the early evening (Sarai and Kayano, 1968).
In the brain, serotonin (5-hydroxytryptamine/5-HT) is synthesized in a stepwise manner from the amino acid tryptophan with two enzymes, tryptophan hydroxylase and aromatic amino acid decarboxylase (AAAD) respectively, through the intermediate, 5-hydroxytryptophan. Following release into the synaptic cleft, serotonin is retaken back into the neuron by its transporter, 5-hydroxytryptamine transporter (5HTT/SERT), or signals via one of 15 known receptors. Serotonin can also signal through monoaminylation that has been described in the monoamines 5-HT, histamine, dopamine, and norepinephrine (Walther et al., 2003; Farrelly et al., 2019). 5-HT neurons innervate into and have regulatory control on both the SCN and the intergeniculate leaflets (IGL) (Meyer-Bernstein and Morin, 1996; Glass et al., 2000, 2003). Serotonin is integral to the regulation and development of neural systems of the brain; including neural cell growth, differentiation and development of synaptic processes (D’Amato et al., 1987; Cases et al., 1995). An imbalance of serotonin negatively affects the neocortical excitation/inhibition balance, sensory stimulus perception and social communication. Abnormal serotonin levels seem to affect the synaptic processes in the sensory cortices during the developmental period (Bennett-Clarke et al., 1994; Cases et al., 1996). Abnormalities in the 5-HT system have also been associated with disruption of the mammalian circadian system and sleep–wake cycles during development (Paulus and Mintz, 2012).
The primary resource of serotonin required for development of the forebrain in the fetus is the tryptophan concentration in the placenta of the mother (Bonnin et al., 2011). Sufficient concentrations of serotonin during the prenatal and perinatal period is a determining factor for the normal regulation of the neural system, and abnormal serotonin concentration might be integral to development of ASD. Differential levels of serotonin synthesis during the stages of development underscores the importance of serotonin in the structural development of the brain in ASD individuals (Chugani et al., 1999). Human and animal studies have found that disruption of the 5-HT system during development is particularly catastrophic to phenotypic behavioral function (Sundström et al., 1993; Anderson et al., 2002; Chen et al., 2015). Proponents of the 5-HT/brainstem theory in ASD pathogenesis have proposed manifestation of ASD as a cascade of events with multiple entry points, rather than a singular devastating event. A stepwise mechanism of this cataclysmic cascade has been proposed, with particular emphasis on a mutual disturbance of the 5-HT system and the mammalian circadian system causing downstream ASD behavioral manifestation and comorbid impairments (Takumi et al., 2020).
Studies using various methodologies from biochemical analysis, genetics, neuroimaging and pharmacology have established the abnormalities of the serotonin system in ASD (Muller et al., 2016). It is well established that serotonin levels are elevated in ASD individuals (Hoshino et al., 1984; Cook et al., 1990; Gabriele et al., 2014). One of the first markers observed in autistic children was excessive levels of serotonin present in the blood plasma and more than 25% of autistic children exhibit this aberrant level of serotonin (Gabriele et al., 2014). In children diagnosed with autism, serotonin secretion is unusual, its synthesis was significantly elevated throughout late development, and the levels of serotonin correlated with the severity of autistic symptoms (Chugani et al., 1999; Abdulamir et al., 2018). However, in another study, seven ASD boys exhibited reduction in their serotonin production in left frontal cortex and thalamus, whereas one girl was unaffected. High levels of serotonin were found in the contralateral dentate nucleus of all the autistic boys (Chugani et al., 1997). High blood serotonin was found in an analysis of studies comparing ASD children to typically developing children (Gabriele et al., 2014). This further reinstates the notion of excessive serotonin in ASD children. There is some evidence of serotonin treatment returning behavior and brain function to a more typical state in an ASD mouse model (Nakai et al., 2017). Maintaining the right concentration of serotonin rescued normal conditions from autistic symptoms in mice (Nakai et al., 2017). Furthermore, a polymorphic variant identified at the site of serotonin transporter gene could result in aberrant serotonin concentration in thalamo-cortical projections (Tordjman et al., 2001).
The serotonin synthesis pathway, its molecular interactions, and the genes responsible for these interactions have been investigated in relation to the development of ASD. There is some evidence of serotonin synthesis capacity abnormalities in developing ASD individuals (Chugani et al., 1997, 1999). Decreased transporter binding of serotonin has also been demonstrated in both ASD children and adults (Makkonen et al., 2008; Nakamura et al., 2010), but there is also contradictory evidence of no significant reduction in individuals with Asperger’s Disorder (Girgis et al., 2011). Receptor binding has also been found to be reduced in ASD individuals (Murphy et al., 2006; Beversdorf et al., 2012), again with contradictory evidence among Asperger’s individuals (Girgis et al., 2011). The genes for the serotonin pathway enzymes (tryptophan hydroxylase and AAAD), its transporter (5HTT/SERT), and its receptors have all been studied as candidates for ASD pathogenesis (Makkonen et al., 2008; Nakamura et al., 2010). The gene SLC6A4 encodes the serotonin transporter (SERT), and significant variation in allele transmission to progeny of the locus HTTLPR within SLC6A4 has been examined in ASD; particularly an increased rate of the short allele relay compared to its long form (Devlin et al., 2005). An interesting amino acid substitution in SLC6A4, Gly56Ala, has been associated with certain behaviors in ASD, including compulsiveness and increased sensory aversion (Sutcliffe et al., 2005). In mice, the Gly56Ala mutation caused inhibition of social behaviors and impaired multisensory processing (Veenstra-VanderWeele et al., 2012; Siemann et al., 2017). Furthermore, there is evidence of an association between high-expressing SERT genotypes and tactile hypersensitivity in ASD individuals (Schauder et al., 2015). Regarding the enzymes responsible for 5-HT synthesis, the brain specific gene for tryptophan hydroxylase, TPH2, has been manipulated in mouse models extensively. When knocked out, TPH2 null mice showed decreased vocalizations and interactions with social odors, deficits in social memory, impaired motor control, and cognitive inflexibility (Alenina et al., 2009; Del’Guidice et al., 2014; Mosienko et al., 2015). Concerning receptors, there is little evidence of malfunction in ASD. However, a few mouse models have found significant social deficits with manipulation of 5-HT1a, 5-HT1b, and 5-HT3a (Saudou et al., 1994; Smit-Rigter et al., 2010). Beyond the enzymes of the 5-HT pathway, its transporter, and its receptors, there is some evidence of malfunction in regulatory molecules and their interaction with serotonin; particularly monoamine oxidase A, the protein responsible for metabolizing 5-HT, and integrin B3 (Carneiro et al., 2008; Bortolato et al., 2013; Whyte et al., 2014). Despite intensive investigation, the mechanism of serotonin pathology in ASD remains unclear and warrants further investigations.
Circadian Dysfunction and ASD Pathogenesis
It has been long recognized that deficits in temporal processing are fundamental in autism (Hermelin, 1972; Ornitz et al., 1972). Individuals with autism have trouble perceiving the passage of time (Martin et al., 2010; Brenner et al., 2015). Even high-functioning autism patients have a poor intuitive sense of time, and temporal information processing is disrupted (Boucher, 2001; Doenyas et al., 2019). The “weak coherence” hypothesis proposes there is a deficit or alternate pathway for neural information processing in ASD children. In typical brain development, coherence is present in the timing system, whereas in autism, coherence is out of phase and possibly responsible for social behavior deficits (Happé and Frith, 2006). The “temporal binding deficit” hypothesis proposes abnormal visual processing in ASD is due to the aberrant pattern of gamma waves which could partially explain abnormal neurobehavioral function (Brock et al., 2002). The “social timing hypothesis” proposes that biological oscillators are essential for neural information processing, and impairments in any of these oscillators would have physiological and psychological consequences. The timing deficits in ASD could be derived from pathological variations in the structure and function of clock-related genes (Wimpory et al., 2002). In support of this hypothesis, several lines of evidence indicate the dysfunction of circadian timing is associated with ASD. As aforementioned, abnormal diurnal profiles of cortisol, melatonin and abnormal sleep–wake cycles indicate underlying impairments of the circadian system in the ASD patients (Geoffray et al., 2016). In addition, here we discuss epidemiological studies linking the incidence of autism to birth seasons, clock gene polymorphisms in ASD and the role of the mTOR pathway as a common regulator of circadian rhythms and ASD pathogenesis.
Epidemiological Evidence Indicating the Involvement of Birth Timing Factors in ASD
Epidemiological studies have linked the incidence of autism to birth seasons. In Canada, children born in spring and summer are more susceptible to development of autism than children born in winter (Konstantareas et al., 1986). In Israel, higher frequencies of ASD are found in babies born in March (18%) and August (20.2%) than babies born in February (7.6%) (Barak et al., 1995). In a more recent study in Israel, the highest incidence of ASD was found in children born during the month of May (10.3%) (Shalev et al., 2017). Another study in Italy found that some ASD children exhibited a higher degree of sleep problems when the season changed from winter to spring (Giannotti et al., 2006). The correlation between birth months and the development of autism may indicate a role for photoperiod in determining the ontogeny of the individual (Castrogiovanni et al., 1998). Many factors change with seasons. For example, the changing weather (temperature) in different seasons may be associated with different incidence of viral infection during pregnancy.
Among the many factors changing with seasons, a major factor is photoperiod (day length), which has a significant impact on the circadian clocks (Porcu et al., 2018). The durations of light and darkness in a 24-h cycle significantly influence the dynamics of circadian gene expression in different systems (Sumová et al., 2002). Rhythmic gene expression in the SCN are sculpted by the length of photoperiods. Significant differences in synchronization of clock cells and patterns of spatial clock gene expression are found between longer and shorter photoperiods (Sumová et al., 1995; Evans et al., 2013). The secretion of neuroendocrine hormones according to the biological day and night is aligned by the circadian oscillator to the changes in the environmental photoperiod (Wehr, 1998). In addition to fine-tuned circadian output by the SCN, the photoperiod also regulates the phase and levels of cortisol, melatonin and prolactin. Prolonged duration of nighttime is characterized by increased synthesis of cortisol, melatonin and prolactin and shorter nighttime periods are characterized by decreased synthesis (Wehr, 1998). Duration of the photoperiod also affects neural development and functions of offspring. Variation in the photoperiod moderates the function and electrical properties of the serotonin neurons present in the dorsal raphe nuclei of the mouse brain (Green et al., 2015). Functional properties of serotonin neurons are regulated by melatonin signaling. Firing rate and levels of the neurotransmitters serotonin and norepinephrine are altered by the duration of the light/dark cycle (Green et al., 2015). Environmental signaling of light dictates the synthesis of glucocorticoids, and timing of light exposure influences functioning of the HPA axis as well as subsequent levels of stress (Dijk et al., 2012).
Availability of nutrition, inadequate vitamin supply and infection rates vary between seasons, and could also be partially responsible for the seasonal discrepancies in ASD birth rates (Gillberg, 1990). Low birth weight, a risk factor of ASD, has been associated with season of birth (Losh et al., 2012). The birth weight of infants varies according to the season (Doblhammer and Vaupel, 2001; Day et al., 2015). Individuals born in summer had higher mean birth weight, later pubertal development and taller adult height compared to those born in all other seasons. Concordantly, those born in winter showed directionally opposite differences in these outcomes. One interesting hypothesis proposed to explain the variation in ASD rates is the availability of vitamin D to the mother (Grant and Soles, 2009). The photoperiod during the post-natal period mediates the metabolic profile and increases body weight of adults in rat models (Uchiwa et al., 2016). Regarding light-exposure, the percentage of prevalence of autism is higher in congenitally blind children (more than 30%) (Jure et al., 2016) than children with auditory impairments (1 in 59) (Szymanski et al., 2012). These findings demonstrate the association between light and its timing with the development of the neural communication system.
Clock Gene Polymorphisms in ASD
As aforementioned, the molecular circadian clock is driven by TTFLs consisting of about a dozen clock genes in mammals (Figure 1). These clock genes are increasingly found to play fundamental roles in different physiological systems beyond their timing functions. In a mouse model, Npas2 (–/–) caused impairments in complex emotional memory, but not non-emotional memory (Garcia et al., 2000). Another Npas2 (–/–) mouse model showed NPAS2 is critical for non-REM sleep homeostasis and caused a reduction in total sleep time in male mice, an interesting comorbidity noted in ASD investigations (Franken et al., 2006). In humans, the protein variant NPAS2 471 Leu/Ser has been implicated in seasonal affective disorder (SAD) and diurnal preference (Johansson et al., 2003). In the repressing limb of the TTFL, PER, CRY, and CK1e form a complex in the cytoplasm, translocate across the nucleus to inhibit binding of CLOCK:BMAL1 or NPAS2:BMAL1, and downregulate transcription of both the period and cryptochrome genes (Ye et al., 2014). PER1 has been shown to have an instrumental role in cell growth and DNA damage control in human cancer cells (Gery et al., 2006). The PER1 protein also interacts with the checkpoint proteins, ATM and CHK2, regulating DNA repair and cellular apoptosis (Gery et al., 2006). Two rare variants in PER3 in humans with familial advanced sleep phase are associated with seasonal depressive traits (Zhang et al., 2016).
Increasing evidence supports the association between clock gene variants and ASD. Evidence for a genetic basis of timing in communication was originally provided by Drosophila studies. The Drosophila Per gene was the first identified clock gene and Per mutations disrupt the fly’s circadian rhythms (Konopka and Benzer, 1971). In addition to circadian disruption, Per mutations also affect the rate of sound production of the male fly’s courtship song, a primary way of communication that leads to mating (Konopka et al., 1996). In recent decades, human genetic studies of autism have identified single-nucleotide polymorphisms and de novo loss-of-function variants of multiple clock genes, indicating functional abnormality of these genes (Table 2). There is evidence that genes with direct influence on the mammalian circadian rhythm are highly variable in ASD individuals (Yang et al., 2016). A number of polymorphisms located within Npas2 were identified in ASD individuals; however, only a cytosine/thymine SNP in intron 3 (NPAS2_X3_C_T) remained significant following statistical analysis (Nicholas et al., 2007). A few mutations in Per1 were identified in ASD individuals; however, only a cytosine → guanine SNP (Per1_rs885747), and a cytosine/adenine SNP (Per1_rs6416892), remained significant following statistical analysis (Nicholas et al., 2007). A proline/alanine substitution at amino acid 1228 in PER2 and an arginine/glutamine substitution at amino acid 366 in PER3 were shown to negatively affect gene function; implicating PER2 and PER3 in the pathogenesis of ASD via gene expression control through the E-box (Yang et al., 2016). Also, in the repressing limb of the TTFL, REV-ERBα/β (NR1D1/2), encoded from Nr1d1/2, respectively, inhibits transcription of the activating genes Bmal1 and Nfil3. Nfil3 encodes NFIL3, a protein that upregulates production of RORα/β, and in turn RORα/β activates transcription of Bmal1 and Nfil3 (Preitner et al., 2002; Ueda et al., 2005). Aberrant function of RORα, possibly as a result of mutations in Nr1d1, has been implicated in abnormal ASD brain development (Goto et al., 2017). While the above findings are interesting and warrant investigation, polymorphisms in clock genes can only explain a small number of the abnormalities found concurrently between dysfunctional circadian related proteins and ASD phenotypes.
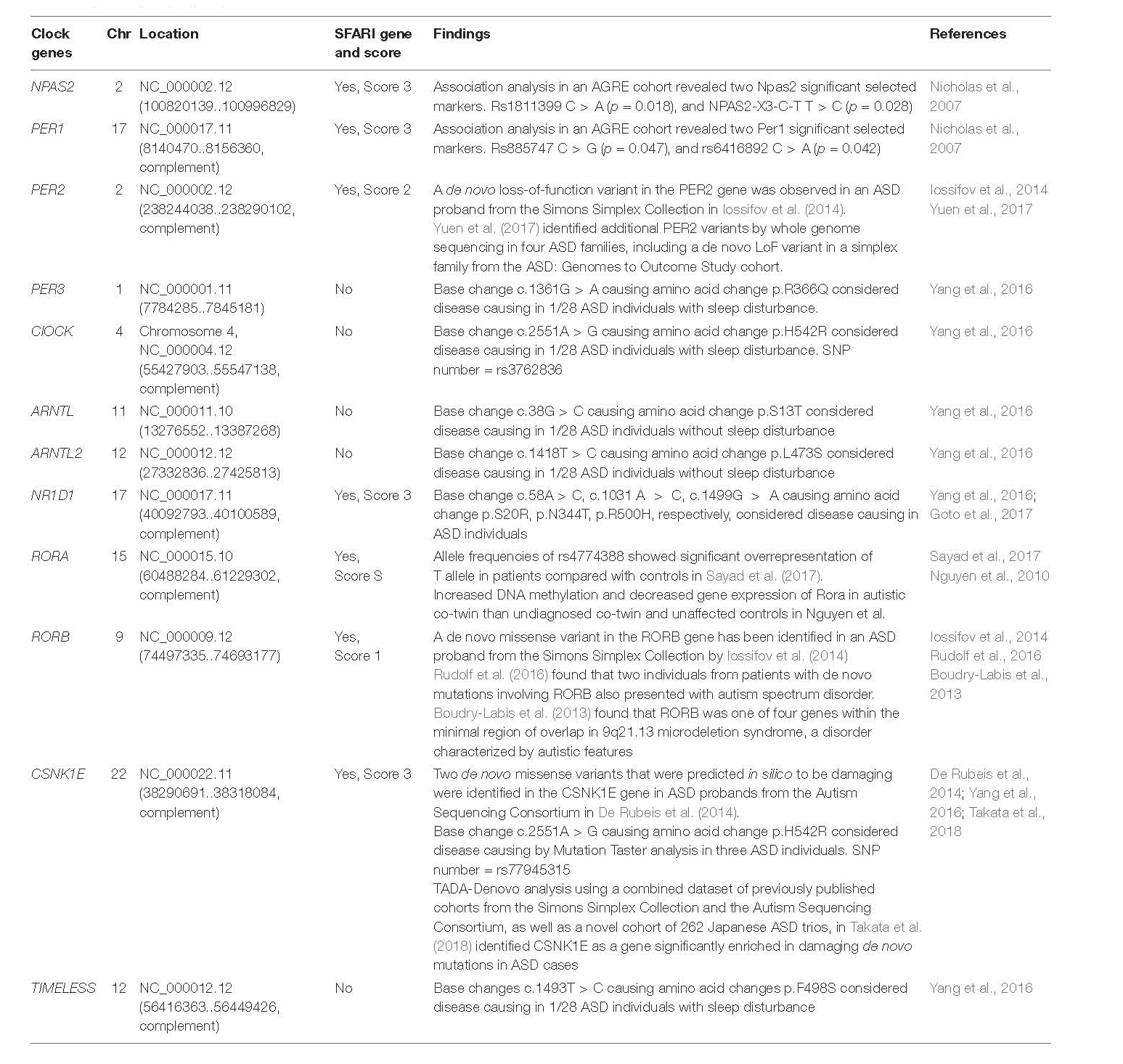
Table 2. Clock gene polymorphisms in ASD.
A Role for the mTOR Pathway in Circadian Regulation and ASD Pathogenesis
The mTOR (mammalian target of rapamycin) signaling cascade integrates various intracellular signals to regulate cell growth and metabolism (Wullschleger et al., 2006). mTOR is a serine/threonine protein kinase that forms two multiprotein complexes in cells, mTORC1 and mTORC2. mTORC1 is composed of six components, mTOR, PRAS40, DEPTOR, mLST8 (Mammalian lethal with sec13 protein 8), Raptor, and the Tti1/Tel2 complex (Brown et al., 1994). mTORC2 is composed of seven components, four of which are shared with mTORC1: mTOR, DEPTOR, mLST8, and the Tti1/Tel2 complex. The other three, Rictor (rapamycin insensitive companion of mTOR), mSin1 (mammalian stress-activated map kinase-interacting protein 1), and Proctor are unique for mTORC2 (Jacinto et al., 2006; Pearce et al., 2007). The upstream regulators of mTORC1 are diverse, but can be generally grouped into four activators, which are oxygen, growth factors (e.g., insulin), amino acids (e.g., leucine and arginine), and energy (e.g., ATP), and one inhibitor, which is stress (Laplante and Sabatini, 2012). Notably, the GTPase activating protein Tuberous Sclerosis Complex (TSC) is the key negative regulator of mTORC1 by an intermediary effect on the GTPase, Rheb, which directly binds and activates mTORC1. The downstream effects of mTORC1 are also diverse but can be generally grouped into three categories, regulation of protein synthesis, regulation of lipid and nucleotide synthesis, and inhibition of autophagy. The regulatory and signaling pathways of mTORC2 are not as well defined, but generally it is regulated by growth factors (e.g., insulin or insulin-like growth factor-1) and has the downstream effect of cell survival and proliferation (Saxton and Sabatini, 2017). mTOR signaling regulates a variety of fundamental biological processes. During brain development, it regulates cell growth and differentiation, neuronal migration and differentiation, axonogenesis, axonal navigation and regeneration, dendrite growth and spine development, myelination by oligodendrocytes and Schwann cells, and autophagy (Cao et al., 2009). In the mature brain, it regulates synaptic plasticity, learning, memory, and feeding (Lipton and Sahin, 2014). Disruption of mTOR signaling has been implicated in a number of human brain diseases (Costa-Mattioli and Monteggia, 2013).
mTOR is emerging as a conserved circadian regulator (Cao, 2018). The mTORC1/eIF4E (eukaryotic translation initiation factor 4E) pathway regulates fundamental functions of the circadian clock such as entrainment, synchrony, and timing (Cao et al., 2013, 2015; Liu et al., 2018). In mammals, mTOR regulates the SCN circadian clock in three facets. First, mTORC1 signaling is part of the photic entrainment pathway in the SCN. In the SCN, light activates S6K1 by phosphorylating Thr389. S6K1 then phosphorylates ribosomal protein S6, a component of the 40S ribosomal subunit and regulates mRNA translation (Cao et al., 2010). S6K1 also phosphorylates the clock protein BMAL1 and activates translation (Lipton et al., 2015). On another branch, light-induced mTORC1 activation increases phosphorylation of eIF4E-binding proteins (4E-BPs) in the SCN, causing disinhibition of eIF4E-dependent translational initiation (Cao et al., 2008). Phosphorylation of both S6K1 and 4E-BP1 is mTORC1-dependent, because rapamycin eliminates the phosphorylation of both these targets in the SCN and regulates photic entrainment of the clock in animals (Cao and Obrietan, 2010; Cao et al., 2010). Second, mTORC1 regulates network properties of coupled circadian oscillators in the SCN by translational control of Vip (Vasoactive intestinal peptide). By phosphorylating and inhibiting the eIF4E repressor protein 4E-BP1, mTORC1 upregulates mRNA translation of Vip (Cao et al., 2013). VIP is synthesized by core SCN neurons, and following their photic input and entrainment, entrain and reset the shell SCN neurons that typically express arginine vasopressin (AVP). VIP signaling promotes synchrony of SCN cells, and increases the robustness of clock gene oscillations and clock functionality (Harmar et al., 2002; Aton and Herzog, 2005; Maywood et al., 2006). Conditional mTOR deletion in VIP neurons disrupts SCN cell synchrony and impairs circadian rhythms in mice, in a way largely similar to Vip mutation (Liu et al., 2018). Third, mTOR regulates autonomous clock properties in a variety of cellular circadian oscillators. Effects of pharmacological and genetic mTOR manipulation on autonomous circadian clock properties have been examined in various cellular and tissue oscillators including the SCN, fibroblasts, hepatocytes, and adipocytes. mTOR inhibition reduces amplitudes of oscillation and increases circadian period of the clock gene Per2 expression, whereas mTOR activation shortens circadian period and augments amplitudes (Ramanathan et al., 2018), indicating the mTOR pathway regulates both central and peripheral clock properties.
Abnormal mTOR activities have been associated with several genetic forms of ASDs, including Tuberous Sclerosis Complex (TSC), Phosphatase and tensin homolog (PTEN), Hamartoma Tumor syndrome, Fragile X syndrome, RASopathies, Angelman Syndrome, Rett Syndrome, and Phelan-McDermid syndrome (Bhattacharya et al., 2012; Costa-Mattioli and Monteggia, 2013; Jülich and Sahin, 2014; Winden et al., 2018). Mutations in negative regulators of mTORC1, such as TSC1, TSC2, and PTEN are found in monogenic ASD (Buxbaum et al., 2007; O’Roak et al., 2012; Lipton and Sahin, 2014). In laboratory studies, mTOR dysregulation has been found in ASD derived neural progenitor cells (Alsaqati et al., 2020). Deletion of the Tsc1 gene in Purkinje cells leads to mTORC1 hyperactivation and autism-like behaviors in mice (Tsai et al., 2012). Mice lacking the repressor of eIF4E, 4E-BP2, demonstrate increased translation of neuroligins, which are causally linked to ASD (Gkogkas et al., 2013). The increased levels of eIF4E also increase the ratio of excitatory: inhibitory synaptic inputs, social interaction deficits, and repetitive/stereotyped behaviors (Santini et al., 2013). A model has been proposed describing the relationship between synaptic proteins and translational control in ASD. The model includes proteins and protein complexes implicated in circadian control discussed in this paper such as Neurexins, Neuroligins, Shank, mTOR/4E-BP, and eIF4E (Santini and Klann, 2014). Thus, the circadian clock and autism are both regulated by mTOR signaling pathways. Dysregulation of the mTORC1/eIF4E axis disrupts the circadian clock and engenders ASD-like phenotypes in animal models, indicating potential crosstalk between the circadian clock and ASD via the mTORC1/eIF4E axis.
Conclusion
In an era of rapidly increased prevalence of ASD, there is an urgent need to understand the mechanisms underlying ASD pathogenesis and develop new therapeutic strategies. Various physiological parameters such as circadian biomarkers, sleep/wake rhythms, neurotransmitters, language and communication, information processing and brain rhythms are associated with circadian clock function and are altered in ASD patients. Mounting evidence exists demonstrating malfunctions of the endogenous circadian timing system in ASD. Correlations exist between clock gene polymorphisms, seasonal discrepancies, and ASD. Understanding the functional importance of the circadian clock in neurodevelopment and its dysregulation in neurodevelopmental disorders may provide a novel approach to tackle ASD. Clinical treatments for ASD children can comprise an integrated approach considering physical, mental and social strategies based on highly dynamic daily rhythms in neurophysiology and behavior. The associations between circadian dysfunction and ASD can be bidirectional. Circadian clock malfunctions may be one of the many pathophysiological aspects underlying ASD pathogenesis, whereas experimental evidence demonstrating that circadian disruption can lead to neurodevelopmental disorders is still lacking. We propose it is necessary to comprehensively investigate the altered circadian patterns of the sleep/wake cycle, cortisol, melatonin and clock gene polymorphisms in ASD. The findings would not only reveal intrinsic connections between aberrant circadian timing and ASD development, but also be instrumental for applying chronotherapy-based strategies to treat the diseases.
Author Contributions
EL and RK created the figures and tables. EL, RK, and RC wrote the manuscript. All the authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from NIH (NS118026), a Faculty Start-Up Grant from the University of Minnesota Medical School, a grant from the Winston and Maxine Wallin Neuroscience Discovery Fund, a grant from the University of Minnesota Foundation, and a grant from Whiteside Institute for Clinical Research to RC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Abdulamir, H. A., Abdul-Rasheed, O. F., and Abdulghani, E. A. (2018). Serotonin and serotonin transporter levels in autistic children. Saudi Med. J. 39, 487–494. doi: 10.15537/smj.2018.5.21751
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355. doi: 10.1038/nrg2346
Adam, E. K., Quinn, M. E., Tavernier, R., Mcquillan, M. T., Dahlke, K. A., and Gilbert, K. E. (2017). Diurnal cortisol slopes and mental and physical health outcomes: a systematic review and meta-analysis. Psychoneuroendocrinology 83, 25–41. doi: 10.1016/j.psyneuen.2017.05.018
Adi, N., Mash, D. C., Ali, Y., Singer, C., Shehadeh, L., and Papapetropoulos, S. (2010). Melatonin MT1 and MT2 receptor expression in Parkinson’s disease. Med. Sci. Monit. 16, Br61–Br67.
Alenina, N., Kikic, D., Todiras, M., Mosienko, V., Qadri, F., Plehm, R., et al. (2009). Growth retardation and altered autonomic control in mice lacking brain serotonin. Proc. Natl. Acad. Sci. U.S.A. 106, 10332–10337. doi: 10.1073/pnas.0810793106
Alsaqati, M., Heine, V. M., and Harwood, A. J. (2020). Pharmacological intervention to restore connectivity deficits of neuronal networks derived from ASD patient iPSC with a TSC2 mutation. Mol. Autism. 11:80.
American Psychiatric Association DSM-5 (2013). Diagnostic and Statistical Manual of Mental Disorders (DSM-5®). Washington, DC: American Psychiatric Publication.
Amir, S., and Stewart, J. (2009). Motivational modulation of rhythms of the expression of the clock protein PER2 in the limbic forebrain. Biol. Psychiatry 65, 829–834. doi: 10.1016/j.biopsych.2008.12.019
Anderson, D. K., Oti, R. S., Lord, C., and Welch, K. (2009). Patterns of growth in adaptive social abilities among children with autism spectrum disorders. J. Abnorm. Child Psychol. 37, 1019–1034. doi: 10.1007/s10802-009-9326-0
Anderson, G. M., Gutknecht, L., Cohen, D. J., Brailly-Tabard, S., Cohen, J. H., Ferrari, P., et al. (2002). Serotonin transporter promoter variants in autism: functional effects and relationship to platelet hyperserotonemia. Mol. Psychiatry 7, 831–836.
Aton, S. J., and Herzog, E. D. (2005). Come together, right.now: synchronization of rhythms in a mammalian circadian clock. Neuron 48, 531–534.
Badcock, N. R., Spence, J. G., and Stern, L. M. (1987). Blood serotonin levels in adults, autistic and non-autistic children—with a comparison of different methodologies. Anna. Clin. Biochem. 24, 625–634. doi: 10.1177/000456328702400613
Barak, Y., Ring, A., Sulkes, J., Gabbay, U., and Elizur, A. (1995). Season of birth and autistic disorder in Israel. Am. J. Psychiatry 152, 798–800. doi: 10.1176/ajp.152.5.798
Baranek, G. T., David, F. J., Poe, M. D., Stone, W. L., and Watson, L. R. (2006). Sensory experiences questionnaire: discriminating sensory features in young children with autism, developmental delays, and typical development. J. Child Psychol. Psychiatry 47, 591–601. doi: 10.1111/j.1469-7610.2005.01546.x
Becerra, T. A., Wilhelm, M., Olsen, J., Cockburn, M., and Ritz, B. (2013). Ambient air pollution and autism in Los Angeles county, California. Environ. Health Perspect. 121, 380–386. doi: 10.1289/ehp.1205827
Belenky, G., Wesensten, N. J., Thorne, D. R., Thomas, M. L., Sing, H. C., Redmond, D. P., et al. (2003). Patterns of performance degradation and restoration during sleep restriction and subsequent recovery: a sleep dose-response study. J. Sleep Res. 12, 1–12. doi: 10.1046/j.1365-2869.2003.00337.x
Bennett-Clarke, C. A., Leslie, M. J., Lane, R. D., and Rhoades, R. W. (1994). Effect of serotonin depletion on vibrissa-related patterns of thalamic afferents in the rat’s somatosensory cortex. J. Neurosci. 14, 7594–7607. doi: 10.1523/jneurosci.14-12-07594.1994
Ben-Sasson, A., Cermak, S. A., Orsmond, G. I., Tager-Flusberg, H., Carter, A. S., Kadlec, M. B., et al. (2007). Extreme sensory modulation behaviors in toddlers with autism spectrum disorders. Am. J. Occup. Ther. 61, 584–592. doi: 10.5014/ajot.61.5.584
Berson, D. M., Dunn, F. A., and Takao, M. (2002). Phototransduction by retinal ganglion cells that set the circadian clock. Science 295, 1070–1073. doi: 10.1126/science.1067262
Beversdorf, D. Q., Nordgren, R. E., Bonab, A. A., Fischman, A. J., Weise, S. B., Dougherty, D. D., et al. (2012). 5-HT2 receptor distribution shown by [18F] setoperone PET in high-functioning autistic adults. J. Neuropsychiatry Clin. Neurosci. 24, 191–197. doi: 10.1176/appi.neuropsych.11080202
Bhattacharya, A., Kaphzan, H., Alvarez-Dieppa, A. C., Murphy, J. P., Pierre, P., and Klann, E. (2012). Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 76, 325–337. doi: 10.1016/j.neuron.2012.07.022
Blatt, G. J., Fitzgerald, C. M., Guptill, J. T., Booker, A. B., Kemper, T. L., and Bauman, M. L. (2001). Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J. Autism Dev. Disord. 31, 537–543.
Bodfish, J. W., Symons, F. J., Parker, D. E., and Lewis, M. H. (2000). Varieties of repetitive behavior in autism: comparisons to mental retardation. J. Autism Dev. Disord. 30, 237–243.
Bonnet-Brilhault, F., Rajerison, T. A., Paillet, C., Guimard-Brunault, M., Saby, A., Ponson, L., et al. (2018). Autism is a prenatal disorder: evidence from late gestation brain overgrowth. Autism Res. 11, 1635–1642. doi: 10.1002/aur.2036
Bonnin, A., Goeden, N., Chen, K., Wilson, M. L., King, J., Shih, J. C., et al. (2011). A transient placental source of serotonin for the fetal forebrain. Nature 472, 347–350. doi: 10.1038/nature09972
Bortolato, M., Godar, S. C., Alzghoul, L., Zhang, J., Darling, R. D., Simpson, K. L., et al. (2013). Monoamine oxidase A and A/B knockout mice display autistic-like features. Int. J. Neuropsychopharmacol. 16, 869–888. doi: 10.1017/s1461145712000715
Boucher, J. (2001). Time-Parsing and Autism. Time and Memory: Issues in Philosophy and Psychology. Oxford: Clarendon Press, 111–135.
Boudaba, C., Szabó, K., and Tasker, J. G. (1996). Physiological mapping of local inhibitory inputs to the hypothalamic paraventricular nucleus. J. Neurosci. 16, 7151–7160. doi: 10.1523/jneurosci.16-22-07151.1996
Boudry-Labis, E., Demeer, B., Le Caignec, C., Isidor, B., Mathieu-Dramard, M., Plessis, G., et al. (2013). A novel microdeletion syndrome at 9q21.13 characterised by mental retardation, speech delay, epilepsy and characteristic facial features. Eur. J. Med. Genet. 56, 163–170. doi: 10.1016/j.ejmg.2012.12.006
Braam, W., Keijzer, H., Struijker Boudier, H., Didden, R., Smits, M., and Curfs, L. (2013). CYP1A2 polymorphisms in slow melatonin metabolisers: a possible relationship with autism spectrum disorder? J. Intellect. Disabil. Res. 57, 993–1000.
Brancaccio, M., Patton, A. P., Chesham, J. E., Maywood, E. S., and Hastings, M. H. (2017). Astrocytes control circadian timekeeping in the suprachiasmatic nucleus via glutamatergic signaling. Neuron 93, 1420.e5–1435.e5.
Brenner, L. A., Shih, V. H., Colich, N. L., Sugar, C. A., Bearden, C. E., and Dapretto, M. (2015). Time reproduction performance is associated with age and working memory in high-functioning youth with autism spectrum disorder. Autism Res. 8, 29–37. doi: 10.1002/aur.1401
Brock, J., Brown, C. C., Boucher, J., and Rippon, G. (2002). The temporal binding deficit hypothesis of autism. Dev. Psychopathol, 14, 209–224. doi: 10.1017/s0954579402002018
Brown, E. J., Albers, M. W., Shin, T. B., Ichikawa, K., Keith, C. T., Lane, W. S., et al. (1994). A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369, 756–758. doi: 10.1038/369756a0
Buckley, A. W., Rodriguez, A. J., Jennison, K., Buckley, J., Thurm, A., Sato, S., et al. (2010). Rapid eye movement sleep percentage in children with autism compared with children with developmental delay and typical development. Arch. Pediatr. Adolesc. Med. 164, 1032–1037.
Butcher, G. Q., Dziema, H., Collamore, M., Burgoon, P. W., and Obrietan, K. (2002). The p42/44 mitogen-activated protein kinase pathway couples photic input to circadian clock entrainment. J. Biol. Chem. 277, 29519–29525. doi: 10.1074/jbc.m203301200
Buxbaum, J. D., Cai, G., Chaste, P., Nygren, G., Goldsmith, J., Reichert, J., et al. (2007). Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144b, 484–491.
Cao, R. (2018). mTOR signaling, translational control, and the circadian clock. Front. Genet. 9:367. doi: 10.3389/fgene.2018.00367
Cao, R., Gkogkas, C. G., De Zavalia, N., Blum, I. D., Yanagiya, A., Tsukumo, Y., et al. (2015). Light-regulated translational control of circadian behavior by eIF4E phosphorylation. Nat. Neurosci. 18, 855–862. doi: 10.1038/nn.4010
Cao, R., Lee, B., Cho, H. Y., Saklayen, S., and Obrietan, K. (2008). Photic regulation of the mTOR signaling pathway in the suprachiasmatic circadian clock. Mol. Cell Neurosci. 38, 312–324. doi: 10.1016/j.mcn.2008.03.005
Cao, R., Li, A., and Cho, H. Y. (2009). mTOR signaling in epileptogenesis: too much of a good thing? J. Neurosci. 29, 12372–12373. doi: 10.1523/jneurosci.3486-09.2009
Cao, R., Li, A., Cho, H. Y., Lee, B., and Obrietan, K. (2010). Mammalian target of rapamycin signaling modulates photic entrainment of the suprachiasmatic circadian clock. J. Neurosci. 30, 6302–6314.
Cao, R., and Obrietan, K. (2010). mTOR signaling and entrainment of the mammalian circadian clock. Mol. Cell Pharmacol. 2, 125–130.
Cao, R., Robinson, B., Xu, H., Gkogkas, C., Khoutorsky, A., Alain, T., et al. (2013). Translational control of entrainment and synchrony of the suprachiasmatic circadian clock by mTOR/4E-BP1 signaling. Neuron 79, 712–724.
Carneiro, A. M., Cook, E. H., Murphy, D. L., and Blakely, R. D. (2008). Interactions between integrin alphaIIbbeta3 and the serotonin transporter regulate serotonin transport and platelet aggregation in mice and humans. J. Clin. Invest. 118, 1544–1552.
Carper, R. A., Moses, P., Tigue, Z. D., and Courchesne, E. (2002). Cerebral lobes in autism: early hyperplasia and abnormal age effects. Neuroimage 16, 1038–1051.
Cases, O., Seif, I., Grimsby, J., Gaspar, P., Chen, K., Pournin, S., et al. (1995). Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science 268, 1763–1766. doi: 10.1126/science.7792602
Cases, O., Vitalis, T., Seif, I., De Maeyer, E., Sotelo, C., and Gaspar, P. (1996). Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron 16, 297–307. doi: 10.1016/s0896-6273(00)80048-3
Castrogiovanni, P., Iapichino, S., Pacchierotti, C., and Pieraccini, F. (1998). Season of birth in psychiatry. A review. Neuropsychobiology 37, 175–181.
Chandana, S. R., Behen, M. E., Juhász, C., Muzik, O., Rothermel, R. D., Mangner, T. J., et al. (2005). Significance of abnormalities in developmental trajectory and asymmetry of cortical serotonin synthesis in autism. Int. J. Dev. Neurosci. 23, 171–182. doi: 10.1016/j.ijdevneu.2004.08.002
Chaste, P., Clement, N., Mercati, O., Guillaume, J. L., Delorme, R., Botros, H. G., et al. (2010). Identification of pathway-biased and deleterious melatonin receptor mutants in autism spectrum disorders and in the general population. PLoS One 5:e11495. doi: 10.1371/journal.pone.0011495
Chen, X., Ye, R., Gargus, J. J., Blakely, R. D., Dobrenis, K., and Sze, J. Y. (2015). Disruption of transient serotonin accumulation by non-serotonin-producing neurons impairs cortical map development. Cell Rep. 10, 346–358. doi: 10.1016/j.celrep.2014.12.033
Chugani, D. C., Muzik, O., Behen, M., Rothermel, R., Janisse, J. J., Lee, J., et al. (1999). Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann. Neurol. 45, 287–295. doi: 10.1002/1531-8249(199903)45:3<287::aid-ana3>3.0.co;2-9
Chugani, D. C., Muzik, O., Rothermel, R., Behen, M., Chakraborty, P., Mangner, T., et al. (1997). Altered serotonin synthesis in the dentatothalamocortical pathway in autistic boys. Ann. Neurol. 42, 666–669. doi: 10.1002/ana.410420420
Cirelli, C., and Tononi, G. (2019). Linking the need to sleep with synaptic function. Science 366, 189–190. doi: 10.1126/science.aay5304
Claustrat, B., Chazot, G., Brun, J., Jordan, D., and Sassolas, G. (1984). A chronobiological study of melatonin and cortisol secretion in depressed subjects: plasma melatonin, a biochemical marker in major depression. Biol. Psychiatry 19, 1215–1228.
Comai, S., Bertazzo, A., Brughera, M., and Crotti, S. (2020). Tryptophan in health and disease. Adv. Clin. Chem. 95, 165–218.
Cook, E. H. Jr., Leventhal, B. L., Heller, W., Metz, J., Wainwright, M., and Freedman, D. X. (1990). Autistic children and their first-degree relatives: relationships between serotonin and norepinephrine levels and intelligence. J. Neuropsychiatry Clin. Neurosci. 2, 268–274. doi: 10.1176/jnp.2.3.268
Corbett, B. A., Mendoza, S., Abdullah, M., Wegelin, J. A., and Levine, S. (2006). Cortisol circadian rhythms and response to stress in children with autism. Psychoneuroendocrinology 31, 59–68. doi: 10.1016/j.psyneuen.2005.05.011
Corbett, B. A., Mendoza, S., Wegelin, J. A., Carmean, V., and Levine, S. (2008). Variable cortisol circadian rhythms in children with autism and anticipatory stress. J. Psychiatry Neurosci. 33, 227–234.
Corbett, B. A., and Schupp, C. W. (2014). The cortisol awakening response (CAR) in male children with autism spectrum disorder. Horm. Behav. 65, 345–350. doi: 10.1016/j.yhbeh.2014.01.012
Corbett, B. A., Schupp, C. W., Levine, S., and Mendoza, S. (2009). Comparing cortisol, stress, and sensory sensitivity in children with autism. Autism Res. 2, 39–49. doi: 10.1002/aur.64
Corbett, B. A., Schupp, C. W., Simon, D., Ryan, N., and Mendoza, S. (2010). Elevated cortisol during play is associated with age and social engagement in children with autism. Mol. Autism 1:13. doi: 10.1186/2040-2392-1-13
Costa-Mattioli, M., and Monteggia, L. M. (2013). mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat. Neurosci. 16, 1537–1543. doi: 10.1038/nn.3546
Cotton, S., and Richdale, A. (2006). Brief report: parental descriptions of sleep problems in children with autism, Down syndrome, and Prader–Willi syndrome. Res. Dev. Disabil. 27, 151–161. doi: 10.1016/j.ridd.2004.12.003
Curin, J. M., Terzić, J., Petković, Z. B., Zekan, L., Terzić, I. M., and Susnjara, I. M. (2003). Lower cortisol and higher ACTH levels in individuals with autism. J. Autism Dev. Disord. 33, 443–448.
D’Amato, R. J., Blue, M. E., Largent, B. L., Lynch, D. R., Ledbetter, D. J., Molliver, M. E., et al. (1987). Ontogeny of the serotonergic projection to rat neocortex: transient expression of a dense innervation to primary sensory areas. Proc. Natl. Acad. Sci. U.S.A. 84, 4322–4326. doi: 10.1073/pnas.84.12.4322
Day, F. R., Forouhi, N. G., Ong, K. K., and Perry, J. R. (2015). Season of birth is associated with birth weight, pubertal timing, adult body size and educational attainment: a UK Biobank study. Heliyon 1:e00031. doi: 10.1016/j.heliyon.2015.e00031
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215.
Del’Guidice, T., Lemay, F., Lemasson, M., Levasseur-Moreau, J., Manta, S., Etievant, A., et al. (2014). Stimulation of 5-HT2C receptors improves cognitive deficits induced by human tryptophan hydroxylase 2 loss of function mutation. Neuropsychopharmacology 39, 1125–1134. doi: 10.1038/npp.2013.313
Désir, D., Van Cauter, E., Golstein, J., Fang, V. S., Leclercq, R., Refetoff, S., et al. (1980). Circadian and ultradian variations of ACTH and cortisol secretion. Horm. Res. 13, 302–316. doi: 10.1159/000179297
Devlin, B., Cook, E. H. Jr., Coon, H., Dawson, G., Grigorenko, E. L., Mcmahon, W., et al. (2005). Autism and the serotonin transporter: the long and short of it. Mol. Psychiatry 10, 1110–1116. doi: 10.1038/sj.mp.4001724
Dijk, D. J., Duffy, J. F., Silva, E. J., Shanahan, T. L., Boivin, D. B., and Czeisler, C. A. (2012). Amplitude reduction and phase shifts of melatonin, cortisol and other circadian rhythms after a gradual advance of sleep and light exposure in humans. PLoS One 7:e30037. doi: 10.1371/journal.pone.0030037
Doblhammer, G., and Vaupel, J. W. (2001). Lifespan depends on month of birth. Proc. Natl. Acad. Sci. U.S.A 98, 2934–2939. doi: 10.1073/pnas.041431898
Doenyas, C., Mutluer, T., Genç, E., and Balcı, F. (2019). Error monitoring in decision-making and timing is disrupted in autism spectrum disorder. Autism Res. 12, 239–248. doi: 10.1002/aur.2041
Duncan, M. J. (2020). Interacting influences of aging and Alzheimer’s disease on circadian rhythms. Eur. J. Neurosci. 51, 310–325. doi: 10.1111/ejn.14358
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Ebert-Zavos, E., Horvat-Gordon, M., Taylor, A., and Bartell, P. A. (2013). Biological clocks in the duodenum and the diurnal regulation of duodenal and plasma serotonin. PLoS One 8:e58477. doi: 10.1371/journal.pone.0058477
El Helou, J., Bélanger-Nelson, E., Freyburger, M., Dorsaz, S., Curie, T., La Spada, F., et al. (2013). Neuroligin-1 links neuronal activity to sleep-wake regulation. Proc. Natl. Acad. Sci. U.S.A. 110, 9974–9979. doi: 10.1073/pnas.1221381110
Etchegaray, J. P., Machida, K. K., Noton, E., Constance, C. M., Dallmann, R., Di Napoli, M. N., et al. (2009). Casein kinase 1 delta regulates the pace of the mammalian circadian clock. Mol. Cell Biol. 29, 3853–3866. doi: 10.1128/mcb.00338-09
Evans, J. A., Leise, T. L., Castanon-Cervantes, O., and Davidson, A. J. (2013). Dynamic interactions mediated by nonredundant signaling mechanisms couple circadian clock neurons. Neuron 80, 973–983. doi: 10.1016/j.neuron.2013.08.022
Facciolá, G., Hidestrand, M., Von Bahr, C., and Tybring, G. (2001). Cytochrome P450 isoforms involved in melatonin metabolism in human liver microsomes. Eur. J. Clin. Pharmacol. 56, 881–888. doi: 10.1007/s002280000245
Farrelly, L. A., Thompson, R. E., Zhao, S., Lepack, A. E., Lyu, Y., Bhanu, N. V., et al. (2019). Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567, 535–539. doi: 10.1038/s41586-019-1024-7
Franken, P., Dudley, C. A., Estill, S. J., Barakat, M., Thomason, R., O’hara, B. F., et al. (2006). NPAS2 as a transcriptional regulator of non-rapid eye movement sleep: genotype and sex interactions. Proc. Natl. Acad. Sci. U.S.A. 103, 7118–7123. doi: 10.1073/pnas.0602006103
Franklin, T. B., Russig, H., Weiss, I. C., Gräff, J., Linder, N., Michalon, A., et al. (2010). Epigenetic transmission of the impact of early stress across generations. Biol. Psychiatry 68, 408–415. doi: 10.1016/j.biopsych.2010.05.036
Gabriele, S., Sacco, R., and Persico, A. M. (2014). Blood serotonin levels in autism spectrum disorder: a systematic review and meta-analysis. Eur. Neuropsychopharmacol. 24, 919–929. doi: 10.1016/j.euroneuro.2014.02.004
Garcia, J. A., Zhang, D., Estill, S. J., Michnoff, C., Rutter, J., Reick, M., et al. (2000). Impaired cued and contextual memory in NPAS2-deficient mice. Science 288, 2226–2230. doi: 10.1126/science.288.5474.2226
Geoffray, M. M., Nicolas, A., Speranza, M., and Georgieff, N. (2016). Are circadian rhythms new pathways to understand autism spectrum disorder? J. Physiol. Paris 110, 434–438. doi: 10.1016/j.jphysparis.2017.06.002
Gery, S., Komatsu, N., Baldjyan, L., Yu, A., Koo, D., and Koeffler, H. P. (2006). The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell 22, 375–382. doi: 10.1016/j.molcel.2006.03.038
Giannotti, F., Cortesi, F., Cerquiglini, A., and Bernabei, P. (2006). An open-label study of controlled-release melatonin in treatment of sleep disorders in children with autism. J. Autism Dev. Disord. 36, 741–752. doi: 10.1007/s10803-006-0116-z
Gillberg, C. (1990). Do children with autism have March birthdays? Acta Psychiatr. Scand. 82, 152–156. doi: 10.1111/j.1600-0447.1990.tb01373.x
Gillberg, C., Steffenburg, S., and Schaumann, H. (1991). Is autism more common now than ten years ago? Br. J. Psychiatry 158, 403–409. doi: 10.1192/bjp.158.3.403
Girgis, R. R., Slifstein, M., Xu, X., Frankle, W. G., Anagnostou, E., Wasserman, S., et al. (2011). The 5-HT(2A) receptor and serotonin transporter in Asperger’s disorder: a PET study with [11C]MDL 100907 and [11C]DASB. Psychiatry Res. 194, 230–234.
Gkogkas, C. G., Khoutorsky, A., Ran, I., Rampakakis, E., Nevarko, T., Weatherill, D. B., et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. doi: 10.1038/nature11628
Glass, J. D., Dinardo, L. A., and Ehlen, J. C. (2000). Dorsal raphe nuclear stimulation of SCN serotonin release and circadian phase-resetting. Brain Res. 859, 224–232. doi: 10.1016/s0006-8993(00)01963-6
Glass, J. D., Grossman, G. H., Farnbauch, L., and Dinardo, L. (2003). Midbrain raphe modulation of nonphotic circadian clock resetting and 5-HT release in the mammalian suprachiasmatic nucleus. J. Neurosci. 23, 7451–7460. doi: 10.1523/jneurosci.23-20-07451.2003
Goto, M., Mizuno, M., Matsumoto, A., Yang, Z., Jimbo, E. F., Tabata, H., et al. (2017). Role of a circadian-relevant gene NR1D1 in brain development: possible involvement in the pathophysiology of autism spectrum disorders. Sci. Rep. 7:43945.
Graf, E. R., Zhang, X., Jin, S.-X., Linhoff, M. W., and Craig, A. M. (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026. doi: 10.1016/j.cell.2004.11.035
Grant, W. B., and Soles, C. M. (2009). Epidemiologic evidence supporting the role of maternal vitamin D deficiency as a risk factor for the development of infantile autism. Dermatoendocrinol 1, 223–228. doi: 10.4161/derm.1.4.9500
Gray, T. S., Carney, M. E., and Magnuson, D. J. (1989). Direct projections from the central amygdaloid nucleus to the hypothalamic paraventricular nucleus: possible role in stress-induced adrenocorticotropin release. Neuroendocrinology 50, 433–446. doi: 10.1159/000125260
Green, N. H., Jackson, C. R., Iwamoto, H., Tackenberg, M. C., and Mcmahon, D. G. (2015). Photoperiod programs dorsal raphe serotonergic neurons and affective behaviors. Curr. Biol. 25, 1389–1394. doi: 10.1016/j.cub.2015.03.050
Gurney, J. G., Mcpheeters, M. L., and Davis, M. M. (2006). Parental report of health conditions and health care use among children with and without autism: national Survey of Children’s Health. Arch. Pediatr. Adolesc. Med. 160, 825–830. doi: 10.1001/archpedi.160.8.825
Hannibal, J. (2002). Pituitary adenylate cyclase-activating peptide in the rat central nervous system: an immunohistochemical and in situ hybridization study. J. Comp. Neurol. 453, 389–417. doi: 10.1002/cne.10418
Happé, F., and Frith, U. (2006). The weak coherence account: detail-focused cognitive style in autism spectrum disorders. J. Autism Dev. Disord. 36, 5–25. doi: 10.1007/s10803-005-0039-0
Harmar, A. J., Marston, H. M., Shen, S., Spratt, C., West, K. M., Sheward, W. J., et al. (2002). The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell 109, 497–508. doi: 10.1016/s0092-8674(02)00736-5
Hastings, M. H., Maywood, E. S., and Brancaccio, M. (2018). Generation of circadian rhythms in the suprachiasmatic nucleus. Nat. Rev. Neurosci. 19, 453–469.
Hazlett, H. C., Poe, M., Gerig, G., Smith, R. G., Provenzale, J., Ross, A., et al. (2005). Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Arch. Gen. Psychiatry 62, 1366–1376. doi: 10.1001/archpsyc.62.12.1366
Hery, F., Chouvet, G., Kan, J. P., Pujol, J. F., and Glowinski, J. (1977). Daily variations of various parameters of serotonin metabolism in the rat brain. II. Circadian variations in serum and cerebral tryptophan levels: lack of correlation with 5-HT turnover. Brain Res. 123, 137–145. doi: 10.1016/0006-8993(77)90648-5
Hermelin, B. (1972). Locating events in space and time: experiments with autistic, blind, anddeaf children. J. Autism Child Schizophr. 2, 288–298. doi: 10.1007/BF01537619
Herzog, E. D., Takahashi, J. S., and Block, G. D. (1998). Clock controls circadian period in isolated suprachiasmatic nucleus neurons. Nat. Neurosci. 1, 708–713. doi: 10.1038/3708
Hill, S. D., Wagner, E. A., Shedlarski, J. G. Jr., and Sears, S. P. (1977). Diurnal cortisol and temperature variation of normal and autistic children. Dev. Psychobiol. 10, 579–583. doi: 10.1002/dev.420100612
Hirano, A., Yumimoto, K., Tsunematsu, R., Matsumoto, M., Oyama, M., Kozuka-Hata, H., et al. (2013). FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell 152, 1106–1118. doi: 10.1016/j.cell.2013.01.054
Hoshino, Y., Yamamoto, T., Kaneko, M., Tachibana, R., Watanabe, M., Ono, Y., et al. (1984). Blood serotonin and free tryptophan concentration in autistic children. Neuropsychobiology 11, 22–27. doi: 10.1159/000118045
Hoshino, Y., Yokoyama, F., Watanabe, M., Murata, S., Kaneko, M., and Kumashiro, H. (1987). The diurnal variation and response to dexamethasone suppression test of saliva cortisol level in autistic children. Jpn. J. Psychiatry Neurol. 41, 227–235. doi: 10.1111/j.1440-1819.1987.tb00406.x
Ingiosi, A. M., Schoch, H., Wintler, T., Singletary, K. G., Righelli, D., Roser, L. G., et al. (2019). Shank3 modulates sleep and expression of circadian transcription factors. eLife 8:e42819.
Iossifov, I., O’roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221.
Jacinto, E., Facchinetti, V., Liu, D., Soto, N., Wei, S., Jung, S. Y., et al. (2006). SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137. doi: 10.1016/j.cell.2006.08.033
Jamain, S., Quach, H., Betancur, C., Råstam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi: 10.1038/ng1136
James, F. O., Cermakian, N., and Boivin, D. B. (2007). Circadian rhythms of melatonin, cortisol, and clock gene expression during simulated night shift work. Sleep 30, 1427–1436. doi: 10.1093/sleep/30.11.1427
Jensen, J. B., Realmuto, G. M., and Garfinkel, B. D. (1985). The dexamethasone suppression test in infantile autism. J. Am. Acad. Child Psychiatry 24, 263–265. doi: 10.1016/s0002-7138(09)61085-2
Jin, X., Von Gall, C., Pieschl, R. L., Gribkoff, V. K., Stehle, J. H., Reppert, S. M., et al. (2003). Targeted disruption of the mouse Mel(1b) melatonin receptor. Mol. Cell Biol. 23, 1054–1060. doi: 10.1128/mcb.23.3.1054-1060.2003
Johansson, C., Willeit, M., Smedh, C., Ekholm, J., Paunio, T., Kieseppä, T., et al. (2003). Circadian clock-related polymorphisms in seasonal affective disorder and their relevance to diurnal preference. Neuropsychopharmacology 28, 734–739. doi: 10.1038/sj.npp.1300121
Johnson, K. P., and Zarrinnegar, P. (2021). Autism spectrum disorder and sleep. Child Adolesc. Psychiatr. Clin. N. Am. 30, 195–208.
Jülich, K., and Sahin, M. (2014). Mechanism-based treatment in tuberous sclerosis complex. Pediatr. Neurol. 50, 290–296. doi: 10.1016/j.pediatrneurol.2013.12.002
Jure, R., Pogonza, R., and Rapin, I. (2016). Autism spectrum disorders (ASD) in blind children: very high prevalence, potentially better outlook. J. Autism Dev. Disord., 46, 749–759. doi: 10.1007/s10803-015-2612-5
Kalsbeek, A., Palm, I., La Fleur, S., Scheer, F., Perreau-Lenz, S., Ruiter, M., et al. (2006). SCN outputs and the hypothalamic balance of life. J. Biol. Rhythms 21, 458–469. doi: 10.1177/0748730406293854
Karthikeyan, R., Cardinali, D. P., Shakunthala, V., Spence, D. W., Brown, G. M., and Pandi-Perumal, S. R. (2020). Understanding the role of sleep and its disturbances in Autism spectrum disorder. Int. J. Neurosci. 130, 1033–1046. doi: 10.1080/00207454.2019.1711377
Kidd, S. A., Corbett, B. A., Granger, D. A., Boyce, W. T., Anders, T. F., and Tager, I. B. (2012). Daytime secretion of salivary cortisol and alpha-amylase in preschool-aged children with autism and typically developing children. J. Autism Dev. disorders 42, 2648–2658. doi: 10.1007/s10803-012-1522-z
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475.
Konopka, R. J., and Benzer, S. (1971). Clock mutants of Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 68, 2112–2116. doi: 10.1073/pnas.68.9.2112
Konopka, R. J., Kyriacou, C. P., and Hall, J. C. (1996). Mosaic analysis in the Drosophila CNS of circadian and courtship-song rhythms affected by a period clock mutation. J. Neurogenet. 11, 117–139. doi: 10.3109/01677069609107066
Konstantareas, M. M., Hauser, P., Lennox, C., and Homatidis, S. (1986). Season of birth in infantile autism. Child Psychiatry Hum. Dev. 17, 53–65. doi: 10.1007/bf00707913
Korczak, A., Martynhak, B., Pedrazzoli, M., Brito, A., and Louzada, F. (2008). Influence of chronotype and social zeitgebers on sleep/wake patterns. Braz. J. Med. Biol.Res. 41, 914–919. doi: 10.1590/s0100-879x2008005000047
Kulman, G., Lissoni, P., Rovelli, F., Roselli, M. G., Brivio, F., and Sequeri, P. (2000). Evidence of pineal endocrine hypofunction in autistic children. Neuro Endocrinol. Lett. 21, 31–34.
Kwon, O., Yu, J. H., Jeong, E., Yoo, H. J., and Kim, M. S. (2018). Meal-related oscillations in the serum serotonin levels in healthy young men. Clin. Endocrinol. 88, 549–555. doi: 10.1111/cen.13545
Lamia, K. A., Sachdeva, U. M., DiTacchio, L., Williams, E. C., Alvarez, J. G., Egan, D. F., et al. (2009). AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 326, 437–440. doi: 10.1126/science.1172156
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Leboyer, M., Philippe, A., Bouvard, M., Guilloud-Bataille, M., Bondoux, D., Tabuteau, F., et al. (1999). Whole blood serotonin and plasma beta-endorphin in autistic probands and their first-degree relatives. Biol. Psychiatry 45, 158–163. doi: 10.1016/s0006-3223(97)00532-5
Lee, B., Li, A., Hansen, K. F., Cao, R., Yoon, J. H., and Obrietan, K. (2010). CREB influences timing and entrainment of the SCN circadian clock. J. Biol. Rhythms 25, 410–420. doi: 10.1177/0748730410381229
Lee, B. K., Magnusson, C., Gardner, R. M., Blomström, Å, Newschaffer, C. J., Burstyn, I., et al. (2015). Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav. Immun. 44, 100–105. doi: 10.1016/j.bbi.2014.09.001
Lee, C., Etchegaray, J. P., Cagampang, F. R., Loudon, A. S., and Reppert, S. M. (2001). Posttranslational mechanisms regulate the mammalian circadian clock. Cell 107, 855–867. doi: 10.1016/s0092-8674(01)00610-9
Lee, H. M., Chen, R., Kim, H., Etchegaray, J. P., Weaver, D. R., and Lee, C. (2011). The period of the circadian oscillator is primarily determined by the balance between casein kinase 1 and protein phosphatase 1. Proc. Natl. Acad. Sci. U.S.A. 108, 16451–16456. doi: 10.1073/pnas.1107178108
Lee, Y., Kim, H., Kim, J.-E., Park, J.-Y., Choi, J., Lee, J.-E., et al. (2018). Excessive D1 dopamine receptor activation in the dorsal striatum promotes autistic-like behaviors. Mol. Neurobiol. 55, 5658–5671. doi: 10.1007/s12035-017-0770-5
Li, S., and Kirouac, G. J. (2012). Sources of inputs to the anterior and posterior aspects of the paraventricular nucleus of the thalamus. Brain Struct. Funct. 217, 257–273. doi: 10.1007/s00429-011-0360-7
Lipton, J. O., Yuan, E. D., Boyle, L. M., Ebrahimi-Fakhari, D., Kwiatkowski, E., Nathan, A., et al. (2015). The circadian protein BMAL1 regulates translation in response to s6k1-mediated phosphorylation. Cell 161, 1138–1151. doi: 10.1016/j.cell.2015.04.002
Liu, C., Weaver, D. R., Jin, X., Shearman, L. P., Pieschl, R. L., Gribkoff, V. K., et al. (1997). Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron 19, 91–102. doi: 10.1016/s0896-6273(00)80350-5
Liu, D., Stowie, A., De Zavalia, N., Leise, T., Pathak, S. S., Drewes, L. R., et al. (2018). mTOR signaling in VIP neurons regulates circadian clock synchrony and olfaction. Proc. Natl. Acad. Sci. U.S.A. 115, E3296–E3304.
Lockley, S. W., Skene, D. J., Tabandeh, H., Bird, A. C., Defrance, R., and Arendt, J. (1997). Relationship between napping and melatonin in the blind. J. Biol. Rhythms 12, 16–25. doi: 10.1177/074873049701200104
Logan, R. W., and McClung, C. A. (2019). Rhythms of life: circadian disruption and brain disorders across the lifespan. Nat. Rev. Neurosci. 20, 49–65. doi: 10.1038/s41583-018-0088-y
Losh, M., Esserman, D., Anckarsäter, H., Sullivan, P. F., and Lichtenstein, P. (2012). Lower birth weight indicates higher risk of autistic traits in discordant twin pairs. Psychol. Med. 42, 1091–1102. doi: 10.1017/s0033291711002339
Lotter, V. (1966). Epidemiology of autistic conditions in young children. Soc. Psychiatry 1, 124–135. doi: 10.1007/bf00584048
Makkonen, I., Riikonen, R., Kokki, H., Airaksinen, M. M., and Kuikka, J. T. (2008). Serotonin and dopamine transporter binding in children with autism determined by SPECT. Dev. Med. Child Neurol. 50, 593–597. doi: 10.1111/j.1469-8749.2008.03027.x
Malow, B., Adkins, K. W., Mcgrew, S. G., Wang, L., Goldman, S. E., Fawkes, D., et al. (2012). Melatonin for sleep in children with autism: a controlled trial examining dose, tolerability, and outcomes. J. Autism Dev. Disord. 42, 1729–1737. author reply 1738, doi: 10.1007/s10803-011-1418-3
Mandell, D. S., Cao, J., Ittenbach, R., and Pinto-Martin, J. (2006). Medicaid expenditures for children with autistic spectrum disorders: 1994 to 1999. J. Autism Dev. Disord. 36, 475–485. doi: 10.1007/s10803-006-0088-z
Marinović-Curin, J., Marinović-Terzić, I., Bujas-Petković, Z., Zekan, L., Skrabić, V., Dogas, Z., et al. (2008). Slower cortisol response during ACTH stimulation test in autistic children. Eur. Child Adolesc. Psychiatry 17, 39–43. doi: 10.1007/s00787-007-0632-1
Maronde, E., Saade, A., Ackermann, K., Goubran-Botros, H., Pagan, C., Bux, R., et al. (2011). Dynamics in enzymatic protein complexes offer a novel principle for the regulation of melatonin synthesis in the human pineal gland. J. Pineal Res. 51, 145–155. doi: 10.1111/j.1600-079x.2011.00880.x
Martin, J. S., Poirier, M., and Bowler, D. M. (2010). Brief report: impaired temporal reproduction performance in adults with autism spectrum disorder. J. Autism Dev. Disord. 40, 640–646. doi: 10.1007/s10803-009-0904-3
Maywood, E. S., Reddy, A. B., Wong, G. K., O’neill, J. S., O’brien, J. A., Mcmahon, D. G., et al. (2006). Synchronization and maintenance of timekeeping in suprachiasmatic circadian clock cells by neuropeptidergic signaling. Curr. Biol. 16, 599–605. doi: 10.1016/j.cub.2006.02.023
Mazurek, M. O., Handen, B. L., Wodka, E. L., Nowinski, L., Butter, E., and Engelhardt, C. R. (2014). Age at first autism spectrum disorder diagnosis: the role of birth cohort, demographic factors, and clinical features. J. Dev. Behav. Pediatr. 35, 561–569. doi: 10.1097/dbp.0000000000000097
Mazurek, M. O., and Petroski, G. F. (2015). Sleep problems in children with autism spectrum disorder: examining the contributions of sensory over-responsivity and anxiety. Sleep Med. 16, 270–279. doi: 10.1016/j.sleep.2014.11.006
McArthur, A. J., Gillette, M. U., and Prosser, R. A. (1991). Melatonin directly resets the rat suprachiasmatic circadian clock in vitro. Brain Res 565, 158–161. doi: 10.1016/0006-8993(91)91748-p
Melke, J., Goubran Botros, H., Chaste, P., Betancur, C., Nygren, G., Anckarsäter, H., et al. (2008). Abnormal melatonin synthesis in autism spectrum disorders. Mol. Psychiatry 13, 90–98.
Meng, Q. J., Logunova, L., Maywood, E. S., Gallego, M., Lebiecki, J., Brown, T. M., et al. (2008). Setting clock speed in mammals: the CK1 epsilon tau mutation in mice accelerates circadian pacemakers by selectively destabilizing PERIOD proteins. Neuron 58, 78–88. doi: 10.1016/j.neuron.2008.01.019
Meyer-Bernstein, E., Blanchard, J., and Morin, L. (1997). The serotonergic projection from the median raphe nucleus to the suprachiasmatic nucleus modulates activity phase onset, but not other circadian rhythm parameters. Brain Res. 755, 112–120. doi: 10.1016/s0006-8993(97)00111-x
Meyer-Bernstein, E. L., and Morin, L. P. (1996). Differential serotonergic innervation of the suprachiasmatic nucleus and the intergeniculate leaflet and its role in circadian rhythm modulation. J. Neurosci. 16, 2097–2111. doi: 10.1523/jneurosci.16-06-02097.1996
Modahl, C., Green, L. A., Fein, D., Morris, M., Waterhouse, L., Feinstein, C., et al. (1998). Plasma oxytocin levels in autistic children. Biol. Psychiatry 43, 270–277. doi: 10.1016/s0006-3223(97)00439-3
Moga, M. M., and Moore, R. Y. (1997). Organization of neural inputs to the suprachiasmatic nucleus in the rat. J. Comp. Neurol. 389, 508–534. doi: 10.1002/(sici)1096-9861(19971222)389:3<508::aid-cne11>3.0.co;2-h
Moore, R. Y., Speh, J. C., and Leak, R. K. (2002). Suprachiasmatic nucleus organization. Cell Tissue Res. 309, 89–98.
Morgan, C. P., and Bale, T. L. (2011). Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J. Neurosci. 31, 11748–11755. doi: 10.1523/jneurosci.1887-11.2011
Mosienko, V., Beis, D., Alenina, N., and Wöhr, M. (2015). Reduced isolation-induced pup ultrasonic communication in mouse pups lacking brain serotonin. Mol. Autism 6:13.
Mughal, S., Faizy, R. M., and Saadabadi, A. (2020). “Autism spectrum disorder,” in StatPearls (Treasure Island, FL: StatPearls Publishing). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK525976
Muller, C. L., Anacker, A. M. J., and Veenstra-Vanderweele, J. (2016). The serotonin system in autism spectrum disorder: from biomarker to animal models. Neuroscience 321, 24–41. doi: 10.1016/j.neuroscience.2015.11.010
Murphy, D. G., Daly, E., Schmitz, N., Toal, F., Murphy, K., Curran, S., et al. (2006). Cortical serotonin 5-HT2A receptor binding and social communication in adults with Asperger’s syndrome: an in vivo SPECT study. Am. J. Psychiatry 163, 934–936. doi: 10.1176/ajp.2006.163.5.934
Musiek, E. S., Xiong, D. D., and Holtzman, D. M. (2015). Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp. Mol. Med. 47:e148. doi: 10.1038/emm.2014.121
Nakai, N., Nagano, M., Saitow, F., Watanabe, Y., Kawamura, Y., Kawamoto, A., et al. (2017). Serotonin rebalances cortical tuning and behavior linked to autism symptoms in 15q11-13 CNV mice. Sci. Adv. 3:e1603001. doi: 10.1126/sciadv.1603001
Nakamura, K., Sekine, Y., Ouchi, Y., Tsujii, M., Yoshikawa, E., Futatsubashi, M., et al. (2010). Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch. Gen. Psychiatry 67, 59–68. doi: 10.1001/archgenpsychiatry.2009.137
Newschaffer, C. J., Croen, L. A., Daniels, J., Giarelli, E., Grether, J. K., Levy, S. E., et al. (2007). The epidemiology of autism spectrum disorders. Annu. Rev. Public Health 28, 235–258.
Nguyen, A., Rauch, T. A., Pfeifer, G. P., and Hu, V. W. (2010). Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. Faseb J. 24, 3036–3051. doi: 10.1096/fj.10-154484
Nicholas, B., Rudrasingham, V., Nash, S., Kirov, G., Owen, M. J., and Wimpory, D. C. (2007). Association of Per1 and Npas2 with autistic disorder: support for the clock genes/social timing hypothesis. Mol. Psychiatry 12, 581–592. doi: 10.1038/sj.mp.4001953
Nir, I., Meir, D., Zilber, N., Knobler, H., Hadjez, J., and Lerner, Y. (1995). Brief report: circadian melatonin, thyroid-stimulating hormone, prolactin, and cortisol levels in serum of young adults with autism. J. Autism Dev. Disord. 25, 641–654. doi: 10.1007/bf02178193
Obsil, T., Ghirlando, R., Klein, D. C., Ganguly, S., and Dyda, F. (2001). Crystal structure of the 14-3-3zeta:serotonin N-acetyltransferase complex. a role for scaffolding in enzyme regulation. Cell 105, 257–267.
Obrietan, K., Impey, S., and Storm, D. R. (1998). Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat. Neurosci. 1, 693–700. doi: 10.1038/3695
Ornitz, E. M., Tanguay, P. E., Lee, J. C., Ritvo, E. R., Sivertsen, B., and Wilson, C. (1972). The effect of stimulus interval on the auditory evoked response during sleep in autistic children. J. Autism Child Schizophr. 2, 140–150. doi: 10.1007/bf01537567
O’Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. doi: 10.1126/science.1227764
Oster, H., Damerow, S., Kiessling, S., Jakubcakova, V., Abraham, D., Tian, J., et al. (2006). The circadian rhythm of glucocorticoids is regulated by a gating mechanism residing in the adrenal cortical clock. Cell Metab. 4, 163–173. doi: 10.1016/j.cmet.2006.07.002
Owens, J. A., Spirito, A., Mcguinn, M., and Nobile, C. (2000). Sleep habits and sleep disturbance in elementary school-aged children. J. Dev. Behav. Pediatr, 21, 27–36. doi: 10.1097/00004703-200002000-00005
Pagan, C., Goubran-Botros, H., Delorme, R., Benabou, M., Lemière, N., Murray, K., et al. (2017). Disruption of melatonin synthesis is associated with impaired 14-3-3 and miR-451 levels in patients with autism spectrum disorders. Sci. Rep. 7:2096.
Patke, A., Young, M. W., and Axelrod, S. (2020). Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 21, 67–84. doi: 10.1038/s41580-019-0179-2
Patton, A. P., Chesham, J. E., and Hastings, M. H. (2016). Combined pharmacological and genetic manipulations unlock unprecedented temporal elasticity and reveal phase-specific modulation of the molecular circadian clock of the mouse suprachiasmatic nucleus. J. Neurosci. 36, 9326–9341. doi: 10.1523/jneurosci.0958-16.2016
Paulus, E. V., and Mintz, E. M. (2012). Developmental disruption of the serotonin system alters circadian rhythms. Physiol. Behav. 105, 257–263. doi: 10.1016/j.physbeh.2011.08.032
Pearce, L. R., Huang, X., Boudeau, J., Pawłowski, R., Wullschleger, S., Deak, M., et al. (2007). Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 405, 513–522. doi: 10.1042/bj20070540
Peirson, S., and Foster, R. G. (2006). Melanopsin: another way of signaling light. Neuron 49, 331–339. doi: 10.1016/j.neuron.2006.01.006
Pezük, P., Mohawk, J. A., Wang, L. A., and Menaker, M. (2012). Glucocorticoids as entraining signals for peripheral circadian oscillators. Endocrinology 153, 4775–4783. doi: 10.1210/en.2012-1486
Pilcher, J. J., and Huffcutt, A. I. (1996). Effects of sleep deprivation on performance: a meta-analysis. Sleep 19, 318–326. doi: 10.1093/sleep/19.4.318
Pittendrigh, C. S. (1993). Temporal organization: reflections of a Darwinian clock-watcher. Annu. Rev. Physiol. 55, 16–54.
Polimeni, M., Richdale, A., and Francis, A. (2005). A survey of sleep problems in autism. Asperger’s disorder and typically developing children. J. Intell. Disabil. Res. 49, 260–268. doi: 10.1111/j.1365-2788.2005.00642.x
Porcu, A., Riddle, M., Dulcis, D., and Welsh, D. K. (2018). Photoperiod-Induced neuroplasticity in the circadian system. Neural Plast 2018:5147585.
Preitner, N., Damiola, F., Zakany, J., Duboule, D., Albrecht, U., and Schibler, U. (2002). The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110, 251–260.
Provencio, I., Rollag, M. D., and Castrucci, A. M. (2002). Photoreceptive net in the mammalian retina. Nature 415, 493–493.
Purcell, A., Jeon, O., Zimmerman, A., Blue, M. E., and Pevsner, J. (2001). Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 57, 1618–1628.
Putnam, S. K., Lopata, C., Thomeer, M. L., Volker, M. A., and Rodgers, J. D. (2015). Salivary cortisol levels and diurnal patterns in children with autism spectrum disorder. J. Dev. Phys. Disabil. 27, 453–465.
Ramanathan, C., Kathale, N. D., Liu, D., Lee, C., Freeman, D. A., Hogenesch, J. B., et al. (2018). mTOR signaling regulates central and peripheral circadian clock function. PLoS Genet. 14:e1007369. doi: 10.1371/journal.pgen.1007369
Ramanathan, C., Nunez, A. A., Martinez, G. S., Schwartz, M. D., and Smale, L. (2006). Temporal and spatial distribution of immunoreactive PER1 and PER2 proteins in the suprachiasmatic nucleus and peri-suprachiasmatic region of the diurnal grass rat (Arvicanthis niloticus). Brain Res. 107, 348–358.
Reppert, S. M., and Weaver, D. R. (2002). Coordination of circadian timing in mammals. Nature 418, 935–941.
Richdale, A. L. (1999). Sleep problems in autism: prevalence, cause, and intervention. Dev. Med. Child Neurol. 41, 60–66.
Richdale, A. L., and Prior, M. R. (1992). Urinary cortisol circadian rhythm in a group of high-functioning children with autism. J. Autism Dev. Disord. 22, 433–447. doi: 10.1007/BF01048245
Richdale, A. L., and Prior, M. R. (1995). The sleep/wake rhythm in children with autism. Eur. Child Adolesc. Psychiatry 4, 175–186.
Rijo-Ferreira, F., and Takahashi, J. S. (2019). Genomics of circadian rhythms in health and disease. Genome Med. 11:82.
Ritvo, E. R., Ritvo, R., Yuwiler, A., Brothers, A., Freeman, B., and Plotkin, S. (1993). Elevated daytime melatonin concentrations in autism: a pilot study. Eur. Child Adolesc. Psychiatry 2, 75–78.
Rojas, D. C. (2014). The role of glutamate and its receptors in autism and the use of glutamate receptor antagonists in treatment. J. Neural Transmiss. 121, 891–905.
Rosbash, M. (2009). The implications of multiple circadian clock origins. PLoS Biol. 7:e62. doi: 10.1371/journal.pbio.1000062
Ross, A. W., Barrett, P., Mercer, J. G., and Morgan, P. J. (1996). Melatonin suppresses the induction of AP-1 transcription factor components in the pars tuberalis of the pituitary. Mol. Cell Endocrinol. 123, 71–80. doi: 10.1016/0303-7207(96)03897-X
Rudolf, G., Lesca, G., Mehrjouy, M. M., Labalme, A., Salmi, M., Bache, I., et al. (2016). Loss of function of the retinoid-related nuclear receptor (RORB) gene and epilepsy. Eur. J. Hum. Genet. 24, 1761–1770. doi: 10.1038/ejhg.2016.80
Rusak, B., Meijer, J. H., and Harrington, M. E. (1989). Hamster circadian rhythms are phase-shifted by electrical stimulation of the geniculo-hypothalamic tract. Brain Res. 493, 283–291. doi: 10.1016/0006-8993(89)91163-3
Russell, G., and Lightman, S. (2019). The human stress response. Nat. Rev. Endocrinol. 15, 525–534. doi: 10.1038/s41574-019-0228-0
Russo, A. J. (2013). Correlation between hepatocyte growth factor (HGF) and gamma-aminobutyric acid (GABA) plasma levels in autistic children. Biomarker Insights 8, 69–75. doi: 10.4137/BMI.S11448
Santini, E., Huynh, T. N., Macaskill, A. F., Carter, A. G., Pierre, P., Ruggero, D., et al. (2013). Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493, 411–415. doi: 10.1038/nature11782
Santini, E., and Klann, E. (2014). Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci. Signal 7:re10. doi: 10.1126/scisignal.2005832
Sarai, K., and Kayano, M. (1968). The level and diurnal rhythm of serum serotonin in manic-depressive patients. Folia Psychiatr. Neurol. Jpn. 22, 271–281. doi: 10.1111/j.1440-1819.1968.tb01425.x
Saudou, F., Amara, D. A., Dierich, A., Lemeur, M., Ramboz, S., Segu, L., et al. (1994). Enhanced aggressive behavior in mice lacking 5-HT1B receptor. Science 265, 1875–1878. doi: 10.1126/science.8091214
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth. Metab. Disease Cell 169, 361–371. doi: 10.1016/j.cell.2017.03.035
Sayad, A., Noroozi, R., Omrani, M. D., Taheri, M., and Ghafouri-Fard, S. (2017). Retinoic acid-related orphan receptor alpha (RORA) variants are associated with autism spectrum disorder. Metab. Brain Dis, 32, 1595–1601. doi: 10.1007/s11011-017-0049-6
Scammell, T. E., Arrigoni, E., and Lipton, J. O. (2017). Neural circuitry of wakefulness and sleep. Neuron 93, 747–765. doi: 10.1016/j.neuron.2017.01.014
Schauder, K. B., Muller, C. L., Veenstra-Vanderweele, J., and Cascio, C. J. (2015). Genetic variation in serotonin transporter modulates tactile hyperresponsiveness in ASD. Res. Autism Spectr. Disord. 10, 93–100. doi: 10.1016/j.rasd.2014.11.008
Seok, B. S., Cao, F., Bélanger-Nelson, E., Provost, C., Gibbs, S., Jia, Z., et al. (2018). The effect of Neuroligin-2 absence on sleep architecture and electroencephalographic activity in mice. Mol. Brain 11:52. doi: 10.1186/s13041-018-0394-3
Shalev, H., Solt, I., and Chodick, G. (2017). Month of birth and risk of autism spectrum disorder: a retrospective cohort of male children born in Israel. BMJ Open 7:e014606. doi: 10.1136/bmjopen-2016-014606
Shearman, L. P., Sriram, S., Weaver, D. R., Maywood, E. S., Chaves, I., Zheng, B., et al. (2000). Interacting molecular loops in the mammalian circadian clock. Science 288, 1013–1019. doi: 10.1126/science.288.5468.1013
Shinohe, A., Hashimoto, K., Nakamura, K., Tsujii, M., Iwata, Y., Tsuchiya, K. J., et al. (2006). Increased serum levels of glutamate in adult patients with autism. Prog. Neuro Psychopharmacol. Biol. Psychiatry 30, 1472–1477.
Shu, T., Wu, T., Pang, M., Liu, C., Wang, X., Wang, J., et al. (2016). Effects and mechanisms of melatonin on neural differentiation of induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 474, 566–571. doi: 10.1016/j.bbrc.2016.04.108
Siemann, J. K., Muller, C. L., Forsberg, C. G., Blakely, R. D., Veenstra-Vanderweele, J., and Wallace, M. T. (2017). An autism-associated serotonin transporter variant disrupts multisensory processing. Transl. Psychiatry 7:e1067. doi: 10.1038/tp.2017.17
Singh, R., Turner, R. C., Nguyen, L., Motwani, K., Swatek, M., and Lucke-Wold, B. P. (2016). Pediatric traumatic brain injury and autism: elucidating shared mechanisms. Behav. Neurol. 2016:8781725. doi: 10.1155/2016/8781725
Smith, R., Reichenberg, A., Kember, R., Buxbaum, J., Schalkwyk, L., Fernandes, C., et al. (2013). Advanced paternal age is associated with altered DNA methylation at brain-expressed imprinted loci in inbred mice: implications for neuropsychiatric disease. Mol. Psychiatry 18, 635–636. doi: 10.1038/mp.2012.88
Smit-Rigter, L. A., Wadman, W. J., and Van Hooft, J. A. (2010). Impaired social behavior in 5-HT(3A) receptor knockout mice. Front. Behav. Neurosci. 4:169.
Socaciu, A. I., Ionuţ, R., Socaciu, M. A., Ungur, A. P., Bârsan, M., Chiorean, A., et al. (2020). Melatonin, an ubiquitous metabolic regulator: functions, mechanisms and effects on circadian disruption and degenerative diseases. Rev. Endocr. Metab. Disord. 21, 465–478. doi: 10.1007/s11154-020-09570-9
Souders, M. C., Mason, T. B., Valladares, O., Bucan, M., Levy, S. E., Mandell, D. S., et al. (2009). Sleep behaviors and sleep quality in children with autism spectrum disorders. Sleep 32, 1566–1578. doi: 10.1093/sleep/32.12.1566
Souders, M. C., Zavodny, S., Eriksen, W., Sinko, R., Connell, J., Kerns, C., et al. (2017). Sleep in children with autism spectrum disorder. Curr. Psychiatry Rep. 19:34. doi: 10.1007/s11920-017-0782-x
Spratt, E. G., Nicholas, J. S., Brady, K. T., Carpenter, L. A., Hatcher, C. R., Meekins, K. A., et al. (2012). Enhanced cortisol response to stress in children in autism. J. Autism Dev. Disord. 42, 75–81. doi: 10.1007/s10803-011-1214-0
Su, Y., Cailotto, C., Foppen, E., Jansen, R., Zhang, Z., Buijs, R., et al. (2016). The role of feeding rhythm, adrenal hormones and neuronal inputs in synchronizing daily clock gene rhythms in the liver. Mol. Cell. Endocrinol. 422, 125–131. doi: 10.1016/j.mce.2015.12.011
Sumová, A., Sládek, M., Jác, M., and Illnerová, H. (2002). The circadian rhythm of Per1 gene product in the rat suprachiasmatic nucleus and its modulation by seasonal changes in daylength. Brain Res. 947, 260–270. doi: 10.1016/S0006-8993(02)02933-5
Sumová, A., Trávnícková, Z., Peters, R., Schwartz, W. J., and Illnerová, H. (1995). The rat suprachiasmatic nucleus is a clock for all seasons. Proc. Natl. Acad. Sci. U.S.A. 92, 7754–7758. doi: 10.1073/pnas.92.17.7754
Sun, W., Poschmann, J., Del Rosario, R. C.-H., Parikshak, N. N., Hajan, H. S., Kumar, V., et al. (2016). Histone acetylome-wide association study of autism spectrum disorder. Cell 167, 1385.e11–1397.e11. doi: 10.1016/j.cell.2016.10.031
Sundström, E., Kölare, S., Souverbie, F., Samuelsson, E. B., Pschera, H., Lunell, N. O., et al. (1993). Neurochemical differentiation of human bulbospinal monoaminergic neurons during the first trimester. Brain Res. Dev. Brain Res. 75, 1–12. doi: 10.1016/0165-3806(93)90059-J
Sutcliffe, J. S., Delahanty, R. J., Prasad, H. C., Mccauley, J. L., Han, Q., Jiang, L., et al. (2005). Allelic heterogeneity at the serotonin transporter locus (SLC6A4) confers susceptibility to autism and rigid-compulsive behaviors. Am. J. Hum. Genet. 77, 265–279. doi: 10.1086/432648
Szatmari, P., Paterson, A. D., Zwaigenbaum, L., Roberts, W., Brian, J., Liu, X.-Q., et al. (2007). Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 39, 319–328. doi: 10.1038/ng1985
Szymanski, C. A., Brice, P. J., Lam, K. H., and Hotto, S. A. (2012). Deaf children with autism spectrum disorders. J. Autism Dev. Disord. 42, 2027–2037. doi: 10.1007/s10803-012-1452-9
Takahashi, J. S., Hong, H.-K., Ko, C. H., and Mcdearmon, E. L. (2008). The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat. Rev. Genet. 9, 764–775. doi: 10.1038/nrg2430
Takata, A., Miyake, N., Tsurusaki, Y., Fukai, R., Miyatake, S., Koshimizu, E., et al. (2018). Integrative analyses of de novo mutations provide deeper biological insights into autism spectrum disorder. Cell Rep. 22, 734–747. doi: 10.1016/j.celrep.2017.12.074
Takumi, T., Tamada, K., Hatanaka, F., Nakai, N., and Bolton, P. F. (2020). Behavioral neuroscience of autism. Neurosci. Biobehav. Rev. 110, 60–76. doi: 10.1016/j.neubiorev.2019.04.012
Thirumalai, S. S., Shubin, R. A., and Robinson, R. (2002). Rapid eye movement sleep behavior disorder in children with autism. J. Child Neurol. 17, 173–178. doi: 10.1177/088307380201700304
Tomarken, A. J., Han, G. T., and Corbett, B. A. (2015). Temporal patterns, heterogeneity, and stability of diurnal cortisol rhythms in children with autism spectrum disorder. Psychoneuroendocrinology 62, 217–226. doi: 10.1016/j.psyneuen.2015.08.016
Tong, H., Li, Q., Zhang, Z. C., Li, Y., and Han, J. (2016). Neurexin regulates nighttime sleep by modulating synaptic transmission. Sci. Rep. 6:38246. doi: 10.1038/srep38246
Tordjman, S., Anderson, G. M., Mcbride, P. A., Hertzig, M. E., Snow, M. E., Hall, L. M., et al. (1997). Plasma beta-endorphin, adrenocorticotropin hormone, and cortisol in autism. J. Child Psychol. Psychiatry 38, 705–715. doi: 10.1111/j.1469-7610.1997.tb01697.x
Tordjman, S., Anderson, G. M., Pichard, N., Charbuy, H., and Touitou, Y. (2005). Nocturnal excretion of 6-sulphatoxymelatonin in children and adolescents with autistic disorder. Biol. Psychiatry 57, 134–138. doi: 10.1016/j.biopsych.2004.11.003
Tordjman, S., Gutknecht, L., Carlier, M., Spitz, E., Antoine, C., Slama, F., et al. (2001). Role of the serotonin transporter gene in the behavioral expression of autism. Mol. Psychiatry 6, 434–439. doi: 10.1038/sj.mp.4000873
Tsai, P. T., Hull, C., Chu, Y., Greene-Colozzi, E., Sadowski, A. R., Leech, J. M., et al. (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. doi: 10.1038/nature11310
Uchiwa, T., Takai, Y., Tashiro, A., Furuse, M., and Yasuo, S. (2016). Exposure of C57BL/6J mice to long photoperiod during early life stages increases body weight and alters plasma metabolomic profiles in adulthood. Physiol. Rep. 4:e12974. doi: 10.14814/phy2.12974
Ueda, H. R., Hayashi, S., Chen, W., Sano, M., Machida, M., Shigeyoshi, Y., et al. (2005). System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat. Genet. 37, 187–192. doi: 10.1038/ng1504
Van Cauter, E., Leproult, R., and Kupfer, D. J. (1996). Effects of gender and age on the levels and circadian rhythmicity of plasma cortisol. J. Clin. Endocrinol. Metab. 81, 2468–2473. doi: 10.1210/jc.81.7.2468
Vargas, I., and Lopez-Duran, N. (2020). The cortisol awakening response after sleep deprivation: is the cortisol awakening response a “response” to awakening or a circadian process? J. Health Psychol. 25, 900–912. doi: 10.1177/1359105317738323
Varoqueaux, F., Jamain, S., and Brose, N. (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell Biol. 83, 449–456. doi: 10.1078/0171-9335-00410
Veenstra-VanderWeele, J., Muller, C. L., Iwamoto, H., Sauer, J. E., Owens, W. A., Shah, C. R., et al. (2012). Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc. Natl. Acad. Sci. U.S.A. 109, 5469–5474. doi: 10.1073/pnas.1112345109
Volk, H. E., Lurmann, F., Penfold, B., Hertz-Picciotto, I., and Mcconnell, R. (2013). Traffic-related air pollution, particulate matter, and autism. JAMA Psychiatry 70, 71–77.
von Gall, C., Garabette, M. L., Kell, C. A., Frenzel, S., Dehghani, F., Schumm-Draeger, P. M., et al. (2002). Rhythmic gene expression in pituitary depends on heterologous sensitization by the neurohormone melatonin. Nat. Neurosci. 5, 234–238. doi: 10.1038/nn806
Walder, D. J., Laplante, D. P., Sousa-Pires, A., Veru, F., Brunet, A., and King, S. (2014). Prenatal maternal stress predicts autism traits in 6 year-old children: project Ice Storm. Psychiatry Res. 219, 353–360. doi: 10.1016/j.psychres.2014.04.034
Walther, D. J., Peter, J. U., Winter, S., Höltje, M., Paulmann, N., Grohmann, M., et al. (2003). Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 115, 851–862. doi: 10.1016/S0092-8674(03)01014-6
Wehr, T. A. (1998). Effect of seasonal changes in daylength on human neuroendocrine function. Horm. Res. 49, 118–124. doi: 10.1159/000023157
Wehr, T. A., Duncan, W. C., Sher, L., Aeschbach, D., Schwartz, P. J., Turner, E. H., et al. (2001). A circadian signal of change of season in patients with seasonal affective disorder. Arch. Gen. Psychiatry 58, 1108–1114. doi: 10.1001/archpsyc.58.12.1108
Welsh, D. K., Logothetis, D. E., Meister, M., and Reppert, S. M. (1995). Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron 14, 697–706. doi: 10.1016/0896-6273(95)90214-7
Whyte, A., Jessen, T., Varney, S., and Carneiro, A. M. (2014). Serotonin transporter and integrin beta 3 genes interact to modulate serotonin uptake in mouse brain. Neurochem. Int. 73, 122–126. doi: 10.1016/j.neuint.2013.09.014
Wiggs, L., and Stores, G. (2004). Sleep patterns and sleep disorders in children with autistic spectrum disorders: insights using parent report and actigraphy. Dev. Med. Child Neurol. 46, 372–380. doi: 10.1017/S0012162204000611
Wimpory, D., Nicholas, B., and Nash, S. (2002). Social timing, clock genes and autism: a new hypothesis. J. Intellect. Disabil. Res. 46, 352–358. doi: 10.1046/j.1365-2788.2002.00423.x
Winden, K. D., Ebrahimi-Fakhari, D., and Sahin, M. (2018). Abnormal mTOR Activation in Autism. Annu. Rev. Neurosci. 41, 1–23. doi: 10.1146/annurev-neuro-080317-061747
Wirz-Justice, A., Lichtsteiner, M., and Feer, H. (1977). Diurnal and seasonal variations in human platelet serotonin in man. J. Neural. Transm. 41, 7–15. doi: 10.1007/BF01252961
Wolff, G., and Esser, K. A. (2012). Scheduled exercise phase shifts the circadian clock in skeletal muscle. Med. Sci. Sports Exerc. 44, 1663–1670. doi: 10.1249/MSS.0b013e318255cf4c
World Health Organization (2017). Autism Spectrum Disorders. Avaliable at: https://www.who.int/news-room/fact-sheets/detail/autism-spectrum-disorders#:~:text=Autism%20spectrum%20disorder%20(ASD)%20refers,individual%20and%20carried%20out%20repetitively (accessed September 20, 2020).
Wright, K. P. Jr., Drake, A. L., Frey, D. J., Fleshner, M., Desouza, C. A., Gronfier, C., et al. (2015). Influence of sleep deprivation and circadian misalignment on cortisol, inflammatory markers, and cytokine balance. Brain Behav. Immun. 47, 24–34. doi: 10.1016/j.bbi.2015.01.004
Wulff, K., Dijk, D.-J., Middleton, B., Foster, R. G., and Joyce, E. M. (2012). Sleep and circadian rhythm disruption in schizophrenia. Br. J. Psychiatry 200, 308–316. doi: 10.1192/bjp.bp.111.096321
Wullschleger, S., Loewith, R., and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell 124, 471–484. doi: 10.1016/j.cell.2006.01.016
Xiong, J., Chen, S., Pang, N., Deng, X., Yang, L., He, F., et al. (2019). Neurological diseases with autism spectrum disorder: role of ASD risk genes. Front. Neurosci. 13:349. doi: 10.3389/fnins.2019.00349
Yamazaki, K., Saito, Y., Okada, F., Fujieda, T., and Yamashita, I. (1975). An application of neuroendocrinological studies in autistic children and Heller’s syndrome. J. Autism Child Schizophr. 5, 323–332. doi: 10.1007/BF01540679
Yan, L., Smale, L., and Nunez, A. A. (2020). Circadian and photic modulation of daily rhythms in diurnal mammals. Eur. J. Neurosci. 51, 551–566. doi: 10.1111/ejn.14172
Yang, C. J., Tan, H. P., Yang, F. Y., Wang, H. P., Liu, C. L., He, H. Z., et al. (2015). The cortisol, serotonin and oxytocin are associated with repetitive behavior in autism spectrum disorder. Res. Autism Spectr. Disord. 18, 12–20.
Yang, Z., Matsumoto, A., Nakayama, K., Jimbo, E. F., Kojima, K., Nagata, K., et al. (2016). Circadian-relevant genes are highly polymorphic in autism spectrum disorder patients. Brain Dev. 38, 91–99.
Ye, R., Selby, C. P., Chiou, Y. Y., Ozkan-Dagliyan, I., Gaddameedhi, S., and Sancar, A. (2014). Dual modes of CLOCK:BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes Dev. 28, 1989–1998. doi: 10.1101/gad.249417.114
Yoo, S. H., Mohawk, J. A., Siepka, S. M., Shan, Y., Huh, S. K., Hong, H. K., et al. (2013). Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell 152, 1091–1105. doi: 10.1016/j.cell.2013.01.055
Yuen, R. K., Merico, D., Bookman, M., Howe, J. L., Thiruvahindrapuram, B., Patel, R. V., et al. (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611. doi: 10.1038/nn.4524
Zhang, L., Hirano, A., Hsu, P. K., Jones, C. R., Sakai, N., Okuro, M., et al. (2016). A PERIOD3 variant causes a circadian phenotype and is associated with a seasonal mood trait. Proc. Natl. Acad. Sci. U.S.A. 113, E1536–E1544. doi: 10.1073/pnas.1600039113
Zhao, D., Yu, Y., Shen, Y., Liu, Q., Zhao, Z., Sharma, R., et al. (2019). Melatonin synthesis and function: evolutionary history in animals and plants. Front. Endocrinol. 10:249. doi: 10.3389/fendo.2019.00249
Keywords: circadian rhythms, autism spectrum disorders, melatonin, cortisol, serotonin, clock genes, mTOR, sleep
Citation: Lorsung E, Karthikeyan R and Cao R (2021) Biological Timing and Neurodevelopmental Disorders: A Role for Circadian Dysfunction in Autism Spectrum Disorders. Front. Neurosci. 15:642745. doi: 10.3389/fnins.2021.642745
Received: 16 December 2020; Accepted: 03 February 2021;
Published: 12 March 2021.
Edited by:
Guang-Zhong Wang, Shanghai Institute of Nutrition and Health, Shanghai Institutes for Biological Sciences (CAS), ChinaReviewed by:
Megan Mahoney, University of Illinois at Urbana–Champaign, United StatesGenevieve Konopka, University of Texas Southwestern Medical Center, United States
Copyright © 2021 Lorsung, Karthikeyan and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruifeng Cao, cmNhb0B1bW4uZWR1
†These authors have contributed equally to this work
‡Present address: Ramanujam Karthikeyan, Department of Biological Sciences, Vanderbilt University, Nashville, TN, United States