 Audrey Olson
Audrey Olson Fuquan Zhang
Fuquan Zhang Hongbao Cao
Hongbao Cao Ancha Baranova
Ancha Baranova Margaret Slavin
Margaret Slavin- 1Department of Nutrition and Food Studies, College of Health and Human Services, George Mason University, Fairfax, VA, United States
- 2School of Systems Biology, College of Science, George Mason University, Manassas, VA, United States
- 3Department of Psychiatry, Nanjing Medical University, Nanjing, China
- 4Department of Psychiatry, Shanxi Medical University, Taiyuan, China
- 5Research Centre for Medical Genetics, Moscow, Russia
Food is the primary human source of choline, an essential precursor to the neurotransmitter acetylcholine, which has a central role in signaling pathways that govern sensorimotor functions. Most Americans do not consume their recommended amount of dietary choline, and populations with neurodevelopmental conditions like autism spectrum disorder (ASD) may be particularly vulnerable to consequences of choline deficiency. This study aimed to identify a relationship between ASD and cholinergic signaling through gene set enrichment analysis and interrogation of existing database evidence to produce a systems biology model. In gene set enrichment analysis, two gene ontologies were identified as overlapping for autism-related and for cholinergic pathways-related functions, both involving ion transport regulation. Subsequent modeling of ion transport intensive cholinergic signaling pathways highlighted the importance of two genes with autism-associated variants: GABBR1, which codes for the gamma aminobutyric acid receptor (GABAB1), and KCNN2, which codes for calcium-activated, potassium ion transporting SK2 channels responsible for membrane repolarization after cholinergic binding/signal transmission events. Cholinergic signal transmission pathways related to these proteins were examined in the Pathway Studio environment. The ion transport ontological associations indicated feasibility of a dietary choline support as a low-risk therapeutic intervention capable of modulating cholinergic sensory signaling in autism. Further research at the intersection of dietary status and sensory function in autism is warranted.
Introduction
Sensory processing dysfunction, which is commonly experienced by persons diagnosed with autism spectrum disorder, extends across auditory, visual, pressure, temperature, pain, vestibulary, and interoceptive processing domains (Craig, 2002; Ashburner et al., 2008; Case-Smith et al., 2015; Boterberg and Warreyn, 2016; Crasta et al., 2016). Some features of sensory processing, including heightened sensitivity to sound (hyperacusis), appear to be prevalent in the majority of the autistic population (Wilson et al., 2017; Williams et al., 2020). All types of sensory processing rely on sensory gating—the ligand-activated, ion-channel mediated pathways built upon cholinergic neurotransmission signals. Acetylcholine, a primary neurotransmitter ligand in these pathways, plays a key role in cognitive function, memory, learning, and sensory processing signal transduction. Acetylcholine is made in an enzymatic reaction between acetyl-CoA and choline, which is facilitated by the choline acetyltransferase (Oda, 1999). Neuronal molecules associated with acetylcholinergic signal transmission, including the nicotinic receptor family, are heavily implicated in propagation of auditory signals through respective transmission pathways (Simmons et al., 2011; Bertrand et al., 2015; Deutsch et al., 2015; Dineley et al., 2015; Sinkus et al., 2015; Gillentine et al., 2017). Research of cholinergic function in autism has led to clinical trials examining efficacy of cholinergic enhancement and/or acetylcholinesterase inhibitors—more commonly used in Alzheimer’s disease—to address social and cognitive aspects of autistic behaviors (Bentley et al., 2003; Hertzman, 2003; Nicolson et al., 2006; Ghaleiha et al., 2014; Karvat and Kimchi, 2014; Rossignol and Frye, 2014; Eissa et al., 2018). However, cholinergic support to sensory processing in autism remains less examined. Recent developments in autism genome-wide association studies (GWAS) have provided estimates of an overlap of autism-related gene sets with those involved in sensory processing and in cholinergic signal transmission.
While humans can make small amounts of choline endogenously, the replenishment of the pool of acetylcholine available in the human body depends heavily upon dietary choline intake (Gibellini and Smith, 2010). In the United States, nearly nine in 10 Americans over 2 years of age do not meet recommended daily consumption levels of dietary choline. Moreover, an estimated 60–93% of children on the autism spectrum are not meeting their adequate intake (AI) level for choline either, even as experts in choline metabolism continue to emphasize its criticality for nutrition across the lifespan, particularly at critical stages of neurodevelopment (Caudill, 2010; Hamlin et al., 2013; Wallace and Fulgoni, 2016; Blusztajn et al., 2017; Ganz et al., 2017; Wallace et al., 2018, 2019; Zeisel, 2019). In rodents, choline-deficient diets are associated with decreased levels of brain acetylcholine (Cohen and Wurtman, 1976). In both rodents and humans, choline deficiency is associated with impaired sensory gating function (Wu et al., 1993; Fendt and Koch, 1999; Stevens et al., 2008; Knott et al., 2014; Swerdlow et al., 2016). Given possible risk of choline underconsumption and/or its deficiency in humans, and the known impact of choline deficiency upon brain acetylcholine and sensory function in rodents, further investigations into the potential impact of choline underconsumption upon sensory processing are warranted, particularly with respect to those on the autism spectrum. Such research may eventually open an avenue for modulation of sensory processing through nutritional interventions.
To explore the potential relationship between autism spectrum disorders (ASDs) and choline intake, we conducted an enrichment analysis of gene sets associated with ASD and cholinergic pathways, and constructed the model reflecting this relationship. Results of our study highlight the potential impact of dietary choline deficiency upon cholinergic signaling within the genetic context of autism.
Materials and Methods
Overview of Workflow
This study has focused on gene ontologies cataloged in the Gene Ontology (GO) database for enrichment analysis of an overlap between two gene sets, one for cholinergic pathways and one for autism-associated genes. Gene ontological enrichment analysis was conducted within the Pathway Studio environment (Elsevier, Inc.)1 between December 2019 and March 2020. The Pathway Studio database contains functional relationships and pathways of mammalian proteins, including human, mouse, and rat genes. It contains over 1.4 million entities of 14 well-defined categories, including cells, proteins, disease, and small molecules, and more than 13.4 million relationships among these entities. The database includes over 24 million PubMed abstracts and 3.5 million Elsevier and third party full-text papers. Using the natural language processing (NLP) functionality of Pathway Studio, intersections of these gene sets’ pathways were rendered graphically, with connections indicating directional relationships being supported by the current scientific literature. This exploratory analysis and figure development which employed in part some of Pathway Studio’s graphical capabilities took place between March 2020 and November 2020. Since many of the autism-associated genes in the GWAS study have been recently identified as autism-associated only within the past 2 years, this gene/pathway modeling effort was supported by secondary analyses, including use of predictive tools in splice site identification.
Identification of Ontologies Associated With Each Gene Set, and Shared Ontologies
To identify GO database pathways shared between cholinergic pathways and autism-associated genes, gene set enrichment analysis was conducted. Molecular Signatures Database v7.1 (MSigDB)—one of the most widely used repositories of thoroughly annotated gene sets, used in research on both neurological conditions and on cholinergic pathways, specifically—was chosen as a precurated resource for gene sets connected with cholinergic activity (Tan et al., 2010; Turcan et al., 2010; Liberzon et al., 2011; Koker et al., 2018; Schijven et al., 2018; Zhang et al., 2019). A key word search for “cholin∗,” identifying any word beginning with cholin- (choline, cholinergic, cholinesterase, etc.), was used to identify cholinergic-relevant gene sets encompassing a total of 345 genes, which constituted the cholinergic activity gene set for this ontological enrichment analysis. An autism gene set containing 47 genes from a recent cross-trait genome-wide meta-analysis (Wu et al., 2020) was used because it identified more risk genes for ASD compared with another GWAS study for ASD, thus providing enhanced statistical power for the present study (Grove et al., 2019).
Gene ontology association analysis was conducted with each respective set of genes within Pathway Studio. The lists of ontologies assigned to each of these gene sets were trimmed at approximately the lowest 100 p-values (p < 0.01). A shift in the magnitude of p-values, nearest the lowest 100 p-values, served as the final determinant of cut-off for each respective gene sets’ ontology lists. The overlaps were identified using the Venn diagram web tool, Venny 2.1 (Oliveros, 2007–2015).
Genes from the autism gene set were then examined for their potential upstream or downstream relationships with acetylcholine, with additional attention paid to potential roles related to the identified shared ontologies, as they were related to sensory processing and cholinergic sensory signal transmission. The identification of upstream and downstream elements and related pathways was conducted using Pathway Studio’s natural language processing tools within its menu architecture, which generate indices of related entities (genes, proteins, physiological conditions, etc.) per tool sorting specifications. The main two tool specifications used in this exploratory analysis were the Pathway Studio options to view first degree connections (known in Pathway Studio as Direct Interactions) and to view the shortest path between entities within the literature (known in Pathway Studio as Shortest Path). Both of these tool specifications were the primary means of building a broader understanding and figure illustration of literature-supported cholinergic networks related to both genes within the autism gene set and to theoretical cholinergic signal disruption related to dietary choline deficiency.
Gene Set Pathway Intersection Analysis of Shared Ontologies
Shared ontologies and associated pathways were rendered graphically using Pathway Studio’s NLP-driven platform. Additionally, certain elements of interest from the ontological overlap and the intersection between pathways involving genes from these sets were inserted.
Graphical models were developed to examine specific pathways or networks with key autism-associated genes, and respective genetic variants which may alter choline and/or acetylcholine levels available for neuronal sensory signal transport. Each instance of a relationship identified by NLP-based word triplet identification was graded by Pathway Studio in terms of confidence levels of 1–3, with a maximum confidence level of 3 being defined as an entity-to-entity relationship supported by at least three peer reviewed publications reflected by the graphical rendering. For this study and related models, the confidence threshold for including elements in graphical representation was set at 3.
Supplemental Analyses for Model Refinement
Splice site identification algorithm Alternative Splice Site Predictor (ASSP) (Wang and Marín, 2006) was used to examine potential changes in the splicing expression which may arise from intronic variants located within autism-associated genes.
Results
Shared Gene Ontologies
Two ontologies were shared between the cholinergic pathway and the autism GWAS gene sets (p < 0.05), namely, those for ion transport regulation and positive ion transport regulation (Figure 1). These ion-transport-related pathways are associated with sensory processing functionality across broad spectrums of the relevant domains, including nociception, tactile response, vestibular reflex, startle response, pupillary light reflex, and sensory gating in general.
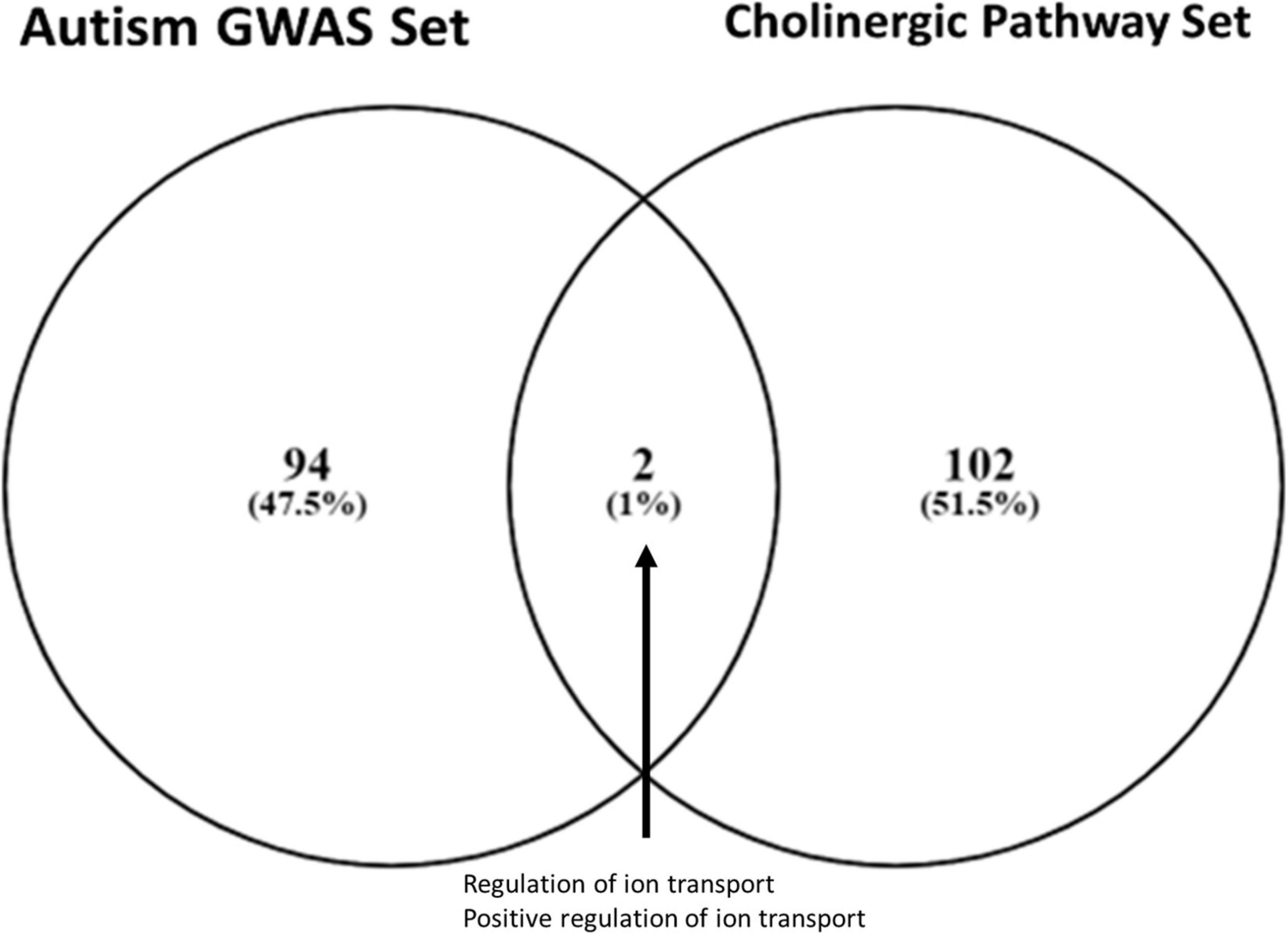
Figure 1. Gene ontology overlap between autism and cholinergic metabolism gene sets. Overlap between an autism-associated genome-wide associated studies gene set (Wu et al., 2020) and cholinergic pathways gene set (MSigDB v7.1) was identified using Venny 2.1. The two shared ontologies were (1) regulation of ion transport and (2) positive regulation of ion transport, identifying ion transport functionality as a critical area of enrichment for further analysis.
Within these two ion transport regulation ontologies, two common genes were identified as implicated in neuronal signal transmission: the gene GABBR1, which encodes the gamma aminobutyric acid membrane receptor GABAB1, also known as “GABA receptor” elsewhere in the literature, and the gene KCNN2, which encodes potassium intermediate/small conductance calcium-activated channels of SK2 type. The latter channels are often co-located with cholinergic receptors and are capable of modulating cholinergic signaling through membrane repolarization.
Analysis of the Intersection of Autism-Related Cholinergic Gene Sets
Analysis with Pathway Studio’s graphical pathway rendering platform identified regulatory and functional relationships between acetylcholine, ion transport/ion levels, GABBR1 and KCNN2 activity, and aspects of sensory processing. All depicted relationships are supported by experimental evidence extracted from the scientific literature (Figure 2).
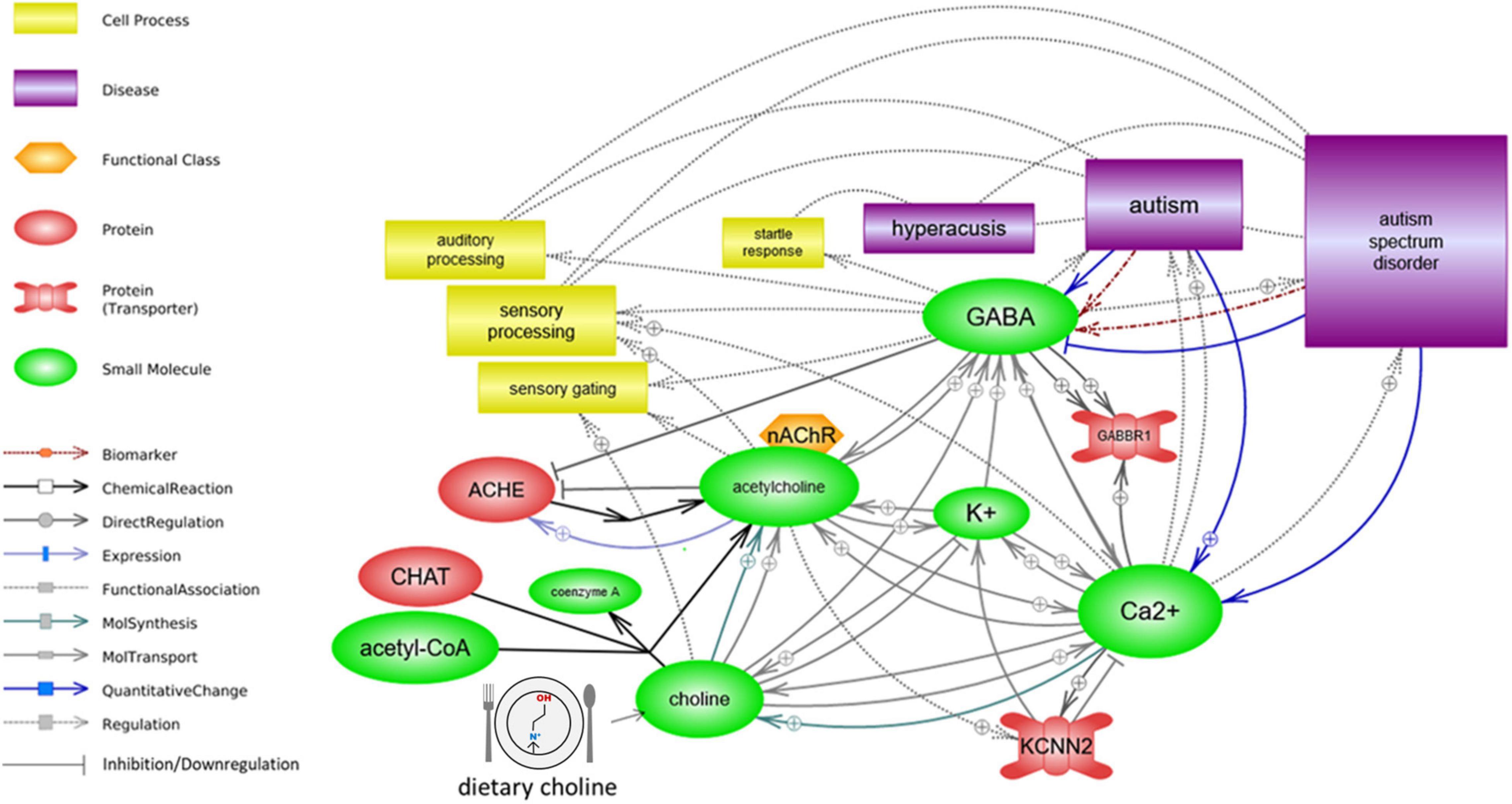
Figure 2. Sensory processing is associated with autism functionally, in the context of ion transport and cholinergic signaling. Acetylcholine regulates both the ion transport and the GABA signaling. A deficiency in its precursor, dietary choline, may impact ion transport, GABA⇒GABBR1 signaling, and KCNN2/SK2 channel function, either or all of which may result in altered sensory processing function.
Choline and acetylcholine were linked to ion transport function across several pathways, with the most direct implication being the neuronal influx of membrane depolarizing calcium resulting from the binding of acetylcholine to cholinergic receptors, such as the alpha-7 nicotinic receptor. This transition from chemical signal transduction (release of acetylcholine into the synapse which binds with the nicotinic receptor on the dendrite of the signal recipient neuron) to ionic signal transduction down the axon is a core aspect of sensory signal pathway functionality (Figure 3).
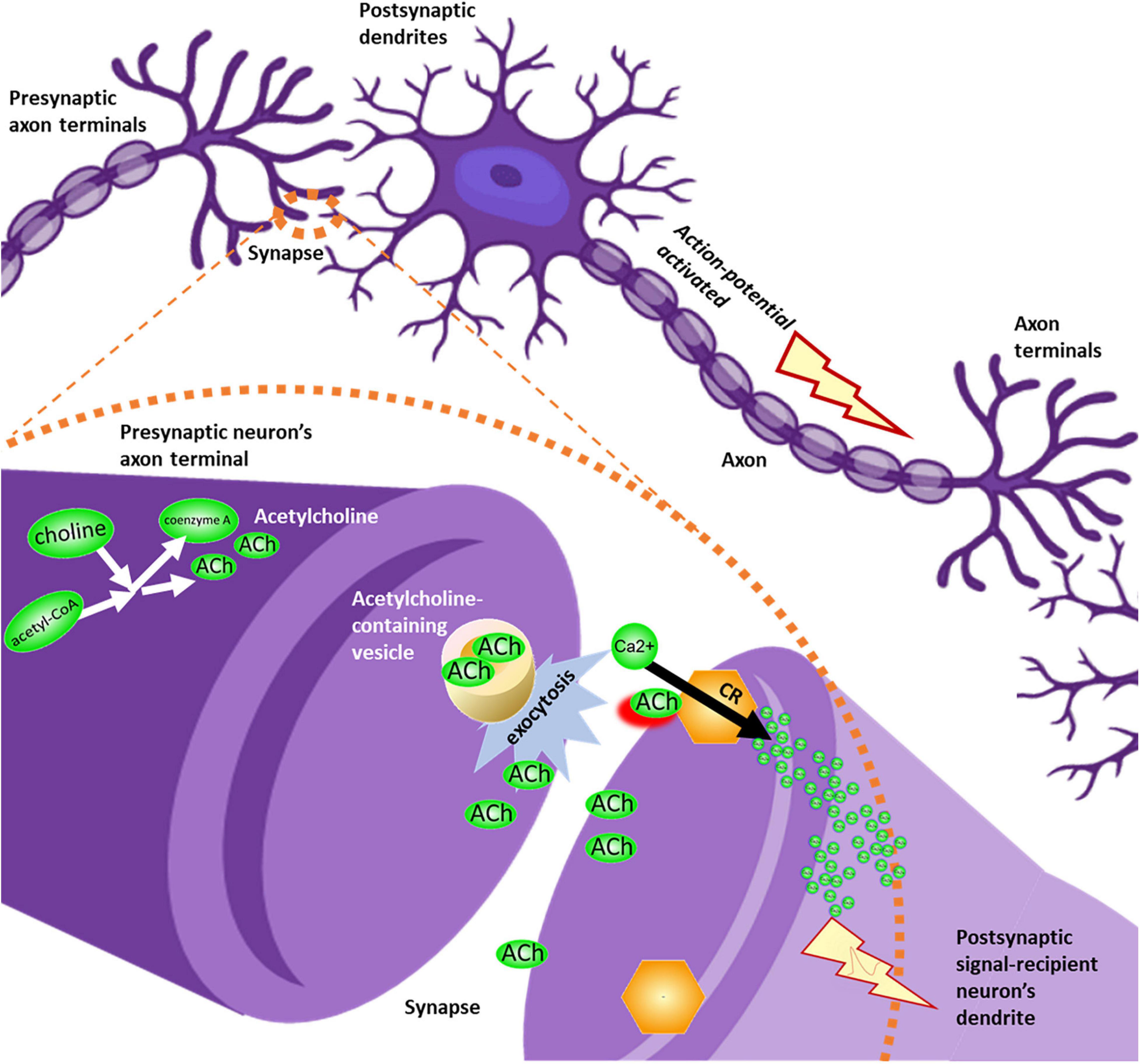
Figure 3. Overview of acetylcholinergic signaling. Acetylcholine (ACh), sourced from precursors choline and acetyl-CoA, leaves the presynaptic neuron’s axon terminal through vesicle-mediated exocytosis. Acetylcholine molecules diffuse through the synapse, until binding with cholinergic receptors (CR) in the membrane of the postsynaptic signal-recipient neuron’s dendrite. This binding event permits influx of calcium ions into the dendrite. Ionic membrane depolarization activates an action potential, causing an electrical signal to be propagated down the length of the axon in a series of ion transport events, until the signal reaches the axon terminal and is translated again into neurotransmitter signals destined to reach the next neuron’s dendrite.
Binding of acetylcholinergic receptors is modulated through multiple means, both preemptive and ex post facto. The enzyme acetylcholinesterase functions by cleaving acetylcholine in the synapse, including either acetylcholine recently made available for signal transduction, or acetylcholine released from membrane-bound acetylcholinergic receptors in the signal recipient neuron (Figure 4).
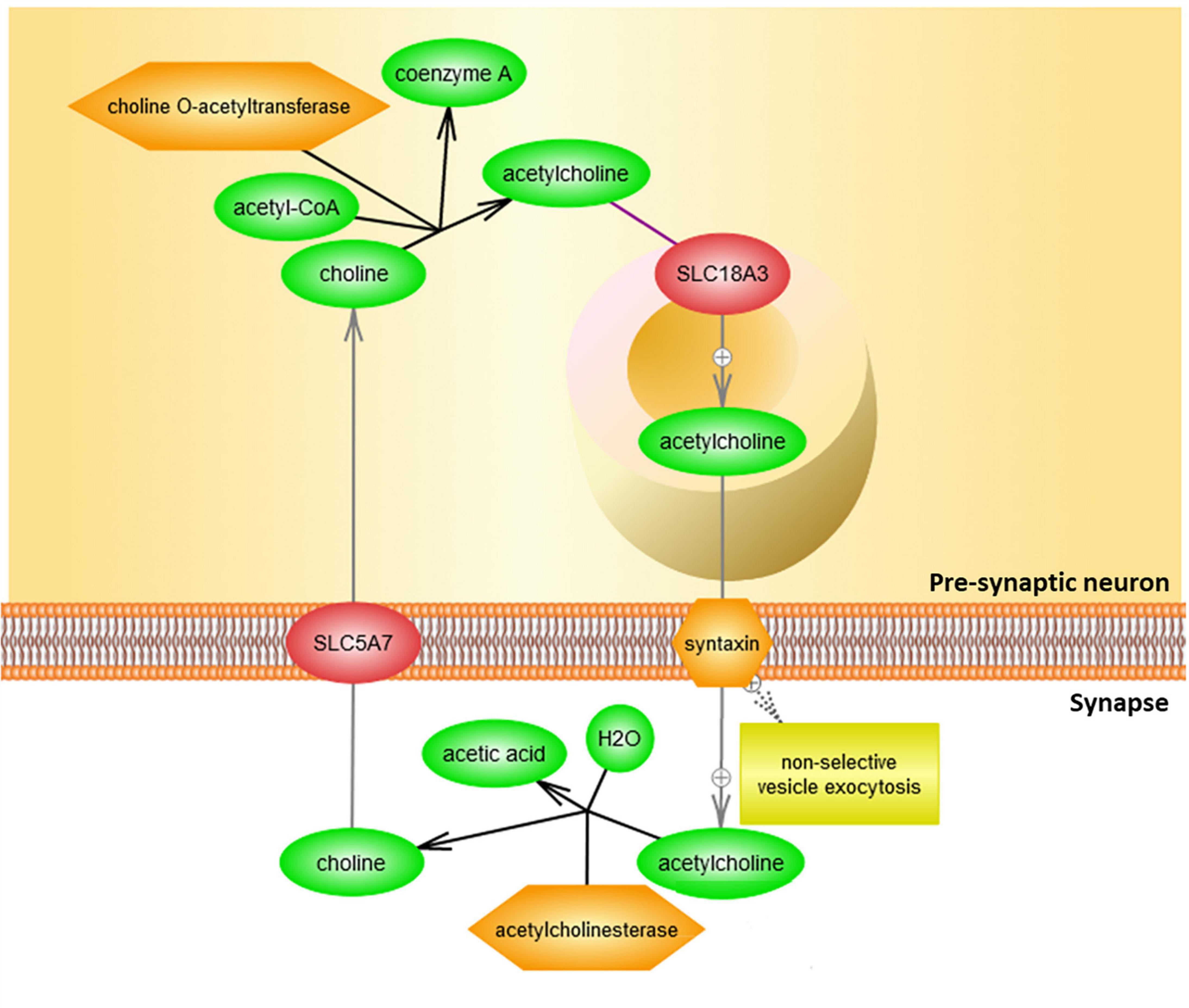
Figure 4. Clearance of acetylcholine from the synapse by acetylcholinesterase. Acetylcholine (ACh) is delivered to the synapse as a result of vesicle exocytosis from pre-synaptic neuronal signaling and a release of the recycled acetylcholine from the postsynaptic neuron’s cholinergic receptors. In the synaptic cleft, acetylcholinesterase (ACHE) then degrades acetylcholine. Resulting choline is then taken back by the presynaptic neuron to be recycled in order to make more acetylcholine for future signaling. This figure was extracted from Pathway Studio’s curated pathways and was edited according to the needs of this study.
The regulatory role of GABA-ergic binding in acetylcholinergic activity is nuanced. Typically, GABA is released from the signal-transmitting neuron synchronously with acetylcholine through separate GABA-specific vesicles, which serve as an immediate signal modulator. The subsequent cascade that arises from GABA binding to the GABAB1 receptor suppresses further upregulation of acetylcholinesterase (ACHE) and preserves acetylcholine in the synaptic cleft from destruction. The signal propagated through the GABAB1 receptor is, therefore, hypothesized to modulate an availability of acetylcholinesterase within the cleft. In other words, GABA-ergic suppression of ACHE upregulation may lead to decrease of synaptic acetylcholine degradation, leaving more acetylcholine available for cholinergic signal transmission. This cascade’s impact upon synaptic acetylcholine levels is depicted in Figure 5. A hypothetical impact of genetically determined differences in GABA–GABAB1 binding activity upon cholinergic sensory signal modulation is depicted in Figure 6A.
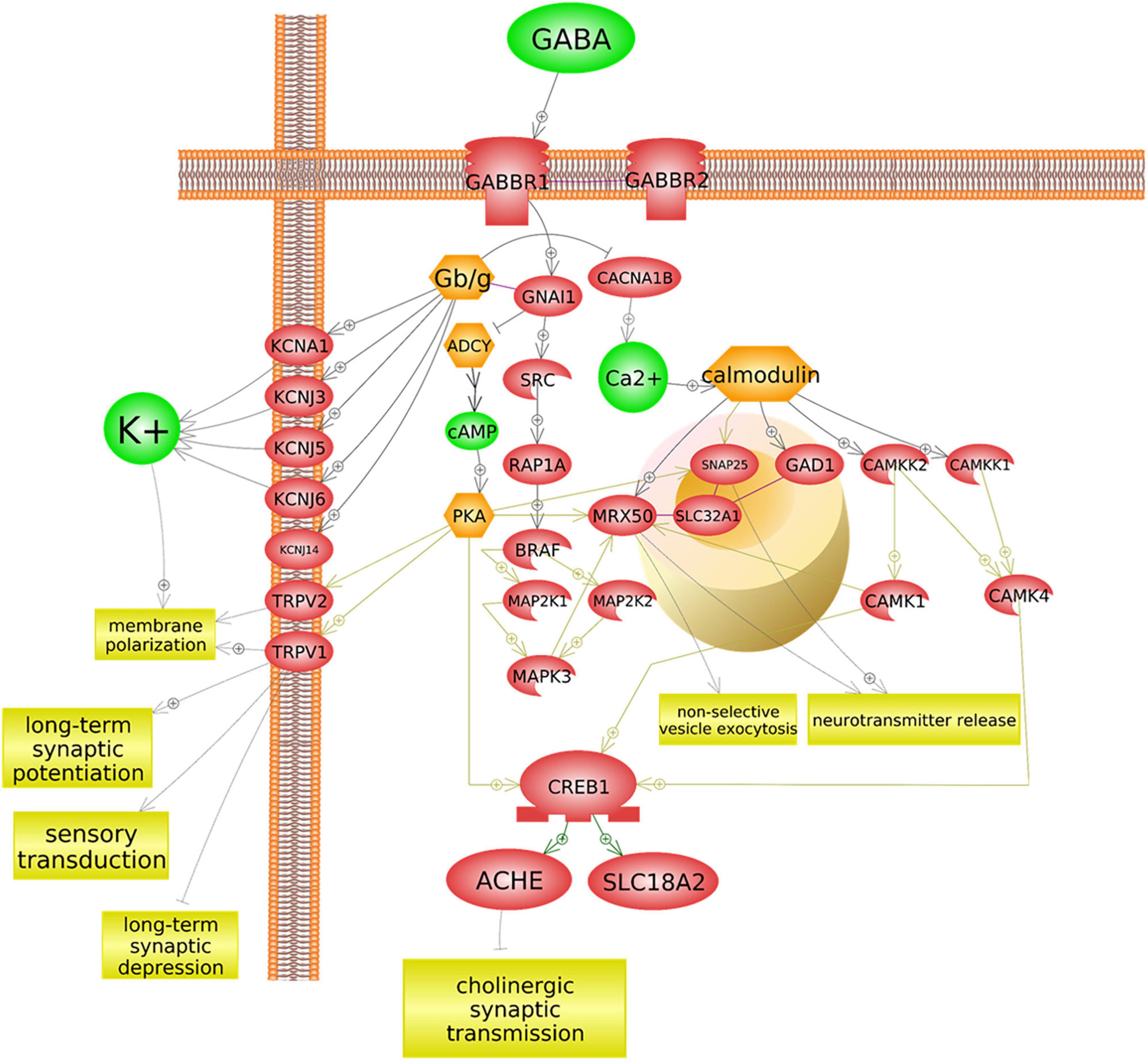
Figure 5. GABBR1 may influence acetylcholinesterase regulation. GABBR1 is an autism-associated gene that codes for a membrane-bound protein (GABAB1) (Wu et al., 2020). The binding of the neurotransmitter GABA to GABAB1 has several implications for cholinergic signaling, sensory signal transduction, and ion transport, across multiple cascades. As depicted here, a typical GABA/GABBR1 binding cascade inhibits adenylyl cyclases (ADCY), thereby preventing positive upregulation of acetylcholinesterase (ACHE). This figure was extracted from Pathway Studio’s curated pathways and was edited according to the needs of this study.
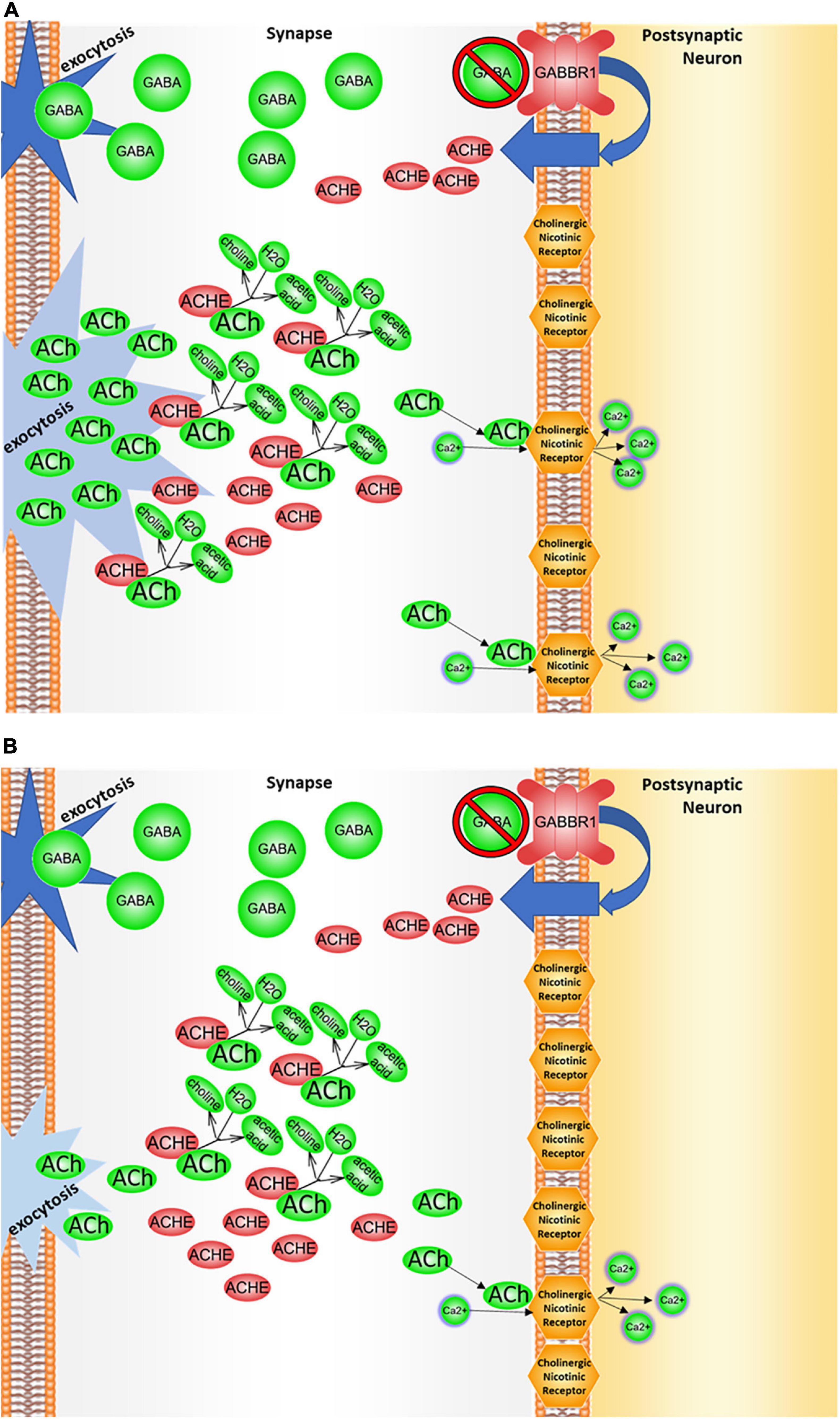
Figure 6. GABBR1/ACHE regulation. (A) In the presence of GABBR1 variant. The autism-associated GABBR1 variant may affect levels of acetylcholinesterase (ACHE) in the synaptic cleft by decreasing either total levels of GABBR1 expression and/or function of its gene product, the GABAB1 receptor. Under this condition, the cascade that normally downregulates acetylcholinesterase expression may be suppressed. Because of that, larger amounts of the enzyme are produced, and the acetylcholine degrades at an elevated rate, resulting in less acetylcholine in the synaptic cleft and less signaling through the cholinergic receptor. (B) In the presence of GABBR1 variant and a dietary choline deficiency. When choline levels are deficient, less acetylcholine (ACh) is available in the neurons for signaling, and a decrease in cholinergic binding/signaling attributed to GABBR1 variant would be exacerbated.
Other modulators of acetylcholinergic signal transmission include SK2 ion channels, which counter the membrane depolarization in the signal recipient neuron (Figure 7A). These types of membrane repolarizing ion channels prepare the signal recipient neuron to receive other action potential-activated sensory signals immediately after processing the preceding signal.
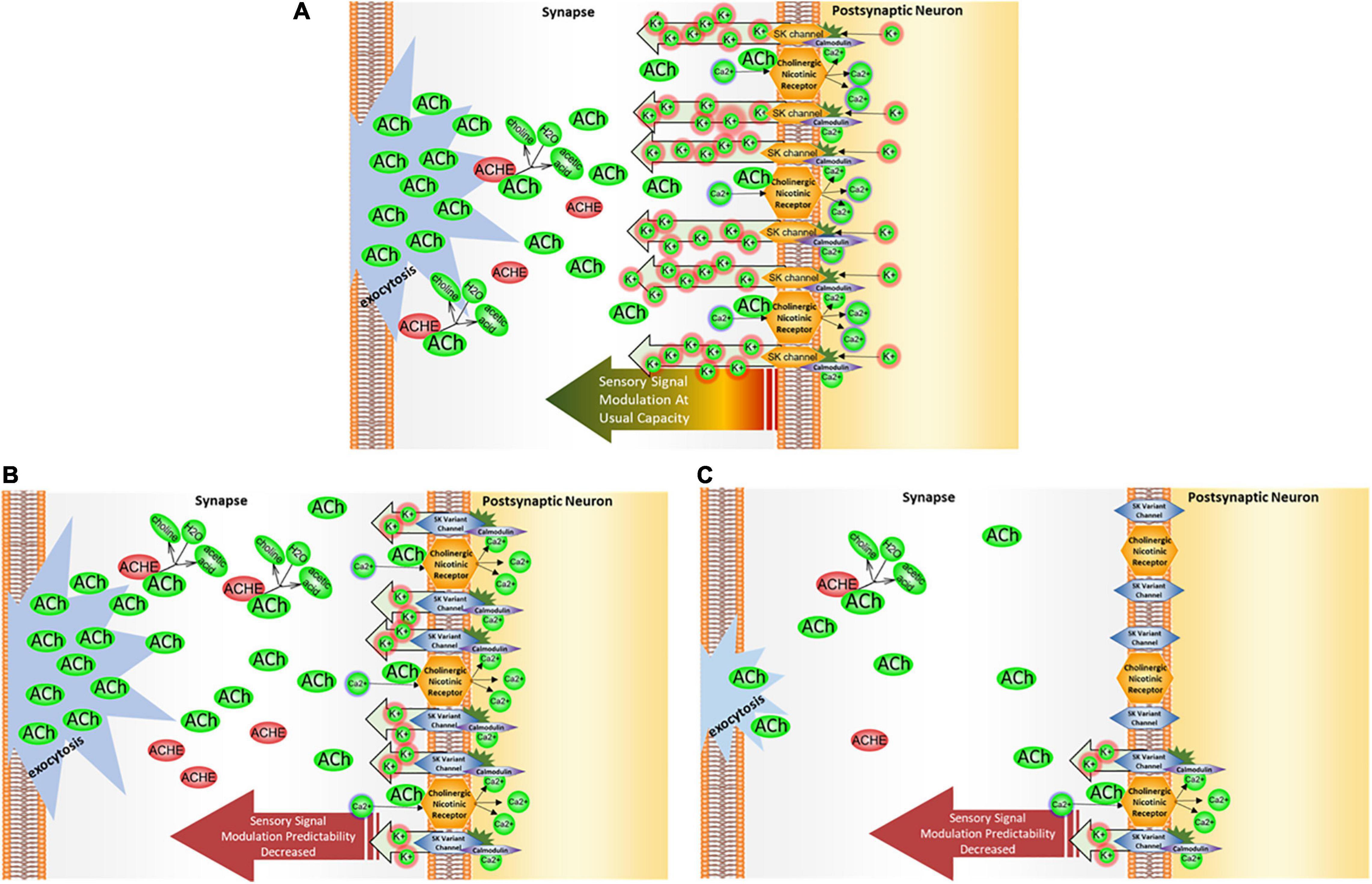
Figure 7. KCNN2/SK2 impact on cholinergic signal modulation. (A) SK2 channel operation. SK2 channels operate in tandem with a propagation of cholinergic signal as they counter the membrane depolarization brought about by calcium influx through cholinergic receptors. The calcium influx activates the calmodulin domains on SK2 channels, permitting passage of potassium back into the synapse to restore membrane hyperpolarization. Thus, SK2 channels serve a critical role in helping a neuron to recover and re-hyperpolarize before receiving the next cholinergic signal. (B) In the presence of KCNN2/SK2 variant. When SK2 channel binding activity and/or functionality is altered, the sensory signaling modulation could either be too efficient—that is, the membrane repolarizes too soon due to overflow of K + ions back into the synapse, or not efficient enough, with membrane taking too long to repolarize, leaving the neuron less ready to process any subsequent signals transmitted from the presynaptic neuron. In either of these two conditions, the neuron’s capacity to modulate sensory signaling through membrane repolarization may be diminished. (C) In the presence of KCNN2/SK2 variant and a dietary choline deficiency. When choline levels are deficient, less acetylcholine (ACh) is available in the neurons for signaling, and a post-signal repolarization may be less amenable to modulation by SK2 channels. When these channels are working in a suboptimal regimen due to the presence of KCNN2 variant, modulation leeway may be curtailed even further.
In part, the magnitude of the sensory signal depends upon SK2 channels’ ability to effectively counter membrane depolarization in the wake of a cholinergic signal, thus, serving as an ex post facto modulator of cholinergic activity. The autism-associated KCNN2 variant may either change the level of the baseline SK2 channel expression or lead to the alteration of the splicing, and the shift of the open reading frame. In either case, sensory signal stands to be impacted. These hypothetical impacts of a KCNN2 genetic variant upon cholinergic sensory signal can be seen in Figure 7B.
The final modeling stage sought to evaluate and depict these hypothesized sensory signaling outcomes in light of a deficiency sufficient to reduce available acetylcholine. The intersection of dietary choline deficiency with GABBR1-related pathways can be seen in Figure 6B, and the intersection of dietary choline deficiency with KCNN2-related pathways can be seen in Figure 7C.
Supportive Evidence for KCNN2 Variant as a Modifier of Splicing
To examine a possible change in splicing patterns attributable to autism-associated variants in GABBR1 and KCNN2, both the wildtype loci and the variant loci representing each of these two genes were compared using the Alternative Splice Site Predictor (ASSP) tool. While the (rs740883) SNP-related differences in predicted splicing patterns of GABBR1 were minimal, the KCNN2 variant rs13188074 yielded a high confidence prediction of a lowered strength within the splice site between the seventh and eighth exons (Table 1), indicating the possibility of a lower rate of splicing than may occur in the wildtype version of this site.
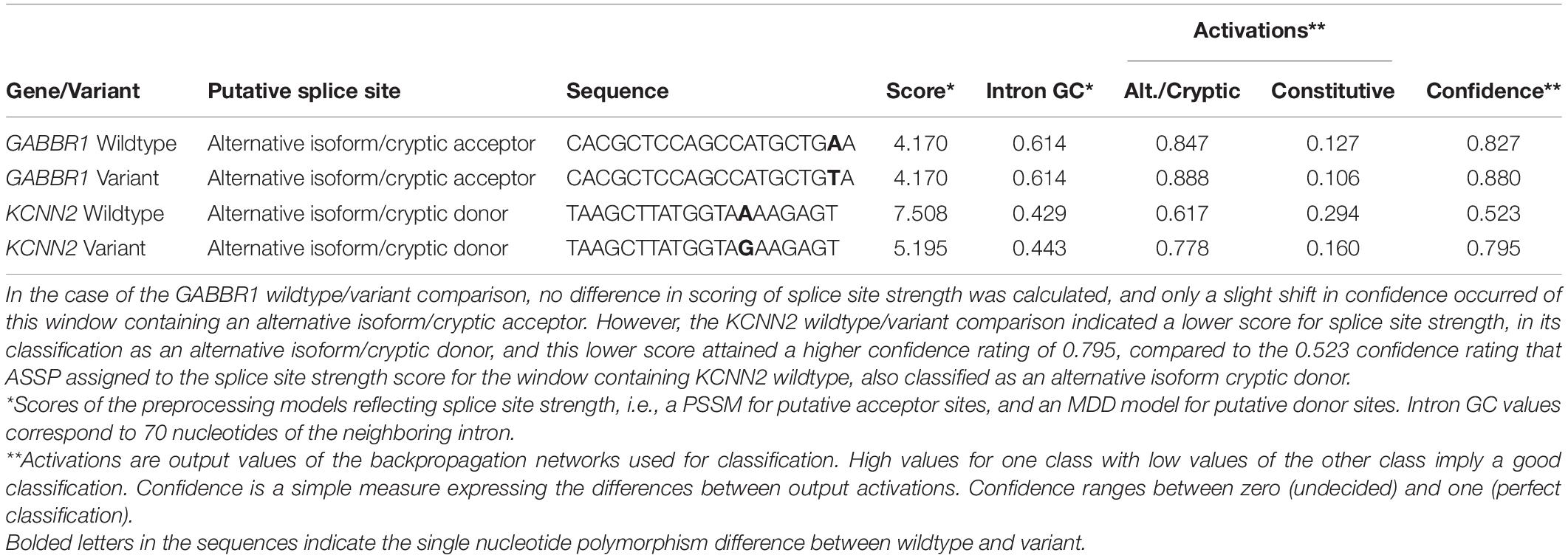
Table 1. The Alternative Splice Site Predictor (ASSP) tool (Wang and Marín, 2006) determined short windows containing the loci for each of the GABBR1 and KCNN2 autism-associated genes.
Discussion
This paper presents the results of a gene set enrichment analysis focusing on shared gene ontologies between an autism-associated gene set obtained through meta-analysis of GWAS and a gene set centering on cholinergic function, which has been collated using a combination of curated gene sets in the Molecular Signatures Database (MSigDB) (Liberzon et al., 2011; Liu et al., 2020). Our study identified two shared gene ontologies, an ion transport regulation and a positive ion transport regulation. Within these ontologies, two genes were further identified as being involved with cholinergic neuronal signal transmission, GABBR1 and KCNN2. The role of ion transport in neurological signaling is complex, as it features a series of ion transport enablers, which boost ion transport and depolarize membranes, as well as counter-transporters, which work to maintain membrane hyperpolarization. These ion transport players in cholinergic and GABA-ergic pathways amplify or inhibit transmission of synaptic signals, respectively. As a result of these interactions, a carefully tuned balance emerges. Potential implications of the presence of GABBR1 and KCNN2 variants in the context of autism and sensory processing are discussed below. Further consideration is given to how operation of these pathways may be altered during acetylcholine deficiency resulting from an inadequate supply of dietary choline.
GABBR1/GABAB1 Implications
The GABBR1 gene encodes for the membrane protein known as GABAB1 (or sometimes GABAB). This membrane protein, which constitutes the metabotropic class of GABA-binding membrane-bound receptors, functions at a slow, steady level, in partnership with another membrane-bound GABA-binding protein, GABAB2. Through activation of potassium ion channels that release potassium (K+) out of the neuron and inhibition of calcium ion (Ca2+) channels which permit passage of Ca2+ ions into the neuron, this set of proteins brings about membrane hyperpolarization. In this circuit, GABA–GABAB1 binding activity serves an inhibitory function, which counters the possibility of excitation of membrane depolarized neurons. This functionality helps to modulate a magnitude of incoming stimulation by inhibiting sensory signals transmitted by cholinergic membrane depolarization. Thus, activation of neuronal action potentials and a subsequent, consistent state of neuronal overexcitation are prevented (Figure 5).
Additionally, GABAB1 receptor binding initiates a cascade which eventually results in the inhibition of acetylcholinesterase—the enzyme that degrades acetylcholine in the synapse. This indicates a possibility that GABAB1 receptor binding activity may at least in part regulate the size of the acetylcholine pool available for cholinergic signaling.
The GABBR1 variant associated with autism—variant rs740883—is an intronic variant in which a thymine replaces an adenine (Liu et al., 2020). This single-nucleotide polymorphism (SNP) occurs in approximately 9–10% of individual human genomes, and is located within a 3 prime untranslated region, commonly associated with a stability of a transcript. GABBR1-encoded GABAB1 receptors are expressed at lower levels in autistic brains, in both the superior frontal and parietal cortices, and in the cerebellum (Fatemi et al., 2009, 2010). Levels of related GABA-ergic biomarkers also differ in the brains of people with an autism diagnosis, when compared to levels of GABA-ergic biomarkers in control brains (Blatt and Fatemi, 2011). While the autism-associated GABBR1 intron variant discussed here is discovered too recently to be analyzed for its relation to these epigenetic differences, examining the possibility that this variant may explain the observed autism-specific differences in GABA-ergic transcriptomic regulation described in the scientific literature is warranted.
Impact of GABA/GABAB1 Binding on the Synaptic Supply of Acetylcholine
GABA is a common neuromediator which often functions in tandem with acetylcholine, in either inhibitory or excitatory roles that span learning, memory, sensory signal transmission, neuromuscular function, and cardiac function. In the hippocampus/somatosensory cortex, the mammalian central nervous system, and elsewhere in the body, GABA and acetylcholine may be co-released, in a relationship which is not yet fully understood (Bianchi et al., 1982; Kusunoki et al., 1984; Granger et al., 2016; Takács et al., 2018). Both of these mediators rely upon vesicle-based exocytosis to be released into the synapse, with GABA and acetylcholine transporting vesicles within the same presynaptic vesicle pools being filled independently of each other (Takács et al., 2018).
Notably, the binding of GABAB1 to its ligand impacts available interneuronal acetylcholine supply (Figures 5, 6A). More specifically, GABA–GABAB1 binding inhibits a cascade in which the transcription factor CREB1 boosts expression of acetylcholinesterase (ACHE), an enzyme that degrades acetylcholine in the interneuronal junction, thereby lessening the magnitude of cholinergic signal. If GABA–GABAB1 binding activity were to decrease as a result of a presence of a genetic variant in a GABBR1 gene—for example, rs740883, which is strongly associated with autism—an increase in acetylcholinesterase production would ensue after being prompted by an increase in the binding of CREB1 ACHE promoter. In this scenario, the increased supply of acetylcholinesterase would degrade acetylcholine in the interneuronal junction faster, thus suppressing cholinergic signaling to a greater extent than in the brain with wildtype GABBR1. Figure 6A systematically depicts hypothesized impact of GABBR1’s autism-associated intronic variant on GABA-ergic and cholinergic signaling.
Impact of Acetylcholine Deficiency on Cholinergic Output, and Hypothesized Interplay With GABBR1 Genetics
Both our modeling and limited experimental evidence suggest that insufficient dietary intake of a choline may contribute to a reduced acetylcholine supply in the brain (Cohen and Wurtman, 1976). The combination of a dietary choline deficiency with a GABBR1 variant may result in cholinergic signaling being lowered further, or even markedly impaired. Moreover, given that GABA sometimes co-transmits with acetylcholine, reduced acetylcholinergic signaling may also be tied to the temporal restriction on the GABA-driven neuronal inhibition as a secondary event. Sensory processing depends upon function of both of these neurotransmitters, so it is reasonable to extrapolate that the combination of dysregulated ion transport due to the presence of GABBR1/GABAB1 variants and dietary-driven acetylcholine deficiency may result in altered sensory processing for the individual in question.
KCNN2/SK2 Channel Implications
The gene KCNN2 encodes for the membrane-bound SK2 calcium-activated potassium ion channel, which modulates the excitability of neurons and neuromuscular activity. After being activated through co-located acetylcholine nicotinic receptors, SK2 channel modulates cholinergic signals through neuronal membrane repolarization (Figure 7A). SK2 channels are abundant throughout the body, including the brain and the cardiorespiratory system (Gu et al., 2018). The SK2 channel encoded by KCNN2 is associated with sensory processing in a variety of contexts, with much research focusing on auditory processing with respect to cholinergic nicotinic receptors in the cochlear hair (Elgoyhen et al., 2001, 2009; Kong et al., 2008; Murthy et al., 2009),vestibular awareness (Wangemann, 2002), and nociception (Bahia et al., 2005; Mongan et al., 2005; Pagadala et al., 2013; Hipólito et al., 2015; Thompson et al., 2015).
Impact of Acetylcholine Supply on the Function of KCNN2/SK2 Channel
As discussed above in relation to the GABAB1 receptor, dietary choline deficiency may markedly reduce the pool of acetylcholine in the brain (Cohen and Wurtman, 1976). It follows that diminished binding of the acetylcholine to cholinergic receptors may reduce activation of co-located SK2 channels. In turn, reduced acetylcholine supply in the brain may require increased sensory input, which, in practical terms, translates into sensory hyposensitivity, or sensory underresponsiveness. Reduced signaling may alter the closely-tied counterfeedback of SK2 ion transport signal modulation.
Impact of Acetylcholine Deficiency on Cholinergic Output, and Its Possible Interplay With KCNN2 Genetics
Similar to the autism-associated epigenetic regulation of GABBR1 observed in the literature, an autism-specific epigenetic regulation of SK2 ion transport activity was noted. In the brains of people with an autism diagnosis, cholinergic nicotinic receptors are expressed at lower levels when compared to that in neurotypical brains (Lee et al., 2002; Andersen et al., 2013). A lower level of available cholinergic receptors may translate to fewer opportunities for acetylcholine binding, and, as collateral, the normally colocalized SK2 channels may also fire less frequently, cumulatively altering the degree of membrane repolarization, and therefore, the timing of subsequent cholinergic sensory signaling.
Because SK2 signaling depends upon the signals from co-located cholinergic nicotinic receptors, diminished or altered SK2 activity arising from the presence of autism-associated KCNN2 coding variant(s) may be compounded by the synaptic acetylcholine deficiency. Thus, dietary choline deficiency may ultimately exacerbate genetic weakness in the SK2 activity, resulting in altered sensory processing, possibly resulting in sensory over-responsiveness, under-responsiveness, or a combination of both, for a given autistic person.
rs13188074 Variant May Influence KCNN2 Splicing Pattern
Currently, there is a little evidence of direct impact of intronic variants in GABBR1 and KCNN2 on the function of the respective proteins. ASSP analysis hints that, in case of KCNN2, the change in a strength of a splice site may influence an amount of functional mRNA encoding for full-size KCNN2. Experimental gauging of the impact of KCNN2 variant upon sensory processing is warranted.
Targeting Cholinergic and GABA-ergic Signaling Pathways May Modulate Sensory Processing in Autism
The models indicated in Figures 2, 3 are consistent with extant literature examining the possibility of cholinergic pharmacotherapy of autism, as increasing the pool of available interneuronal acetylcholine may improve function in several domains related to cholinergic signaling activity and related ion transport dynamics. For example, acetylcholinesterase inhibitors galantamine and memantine, which are typically prescribed for Alzheimer’s disease patients, are also actively explored in autism (Maire and Wurtman, 1984; Hertzman, 2003; Nicolson et al., 2006; Ghaleiha et al., 2014; Rossignol and Frye, 2014; Rahman et al., 2018). A systematic review examining the impact of these drugs in individuals with autism identified an improvement across several domains, including communication and social interaction, and a decrease in disruptive behavior, hyperactivity, inattention, and irritability (Rossignol and Frye, 2014). Many of these domains are linked to sensory processing difficulties (Ashburner et al., 2008; Sanz-Cervera et al., 2015). In the present study, we posit that variant function of GABBR1 may lead to a smaller pool of available acetylcholine (Figure 5). For patients with GABBR1 variants, including individuals with autism, prescription of acetylcholinesterase inhibitors such as galantamine or memantine may counter the disturbance in GABA/GABBR1/acetylcholinesterase cascade, by boosting the synaptic acetylcholine levels to match these seen in neurotypical brains.
In the context of addiction, the literature supports a relationship between acetylcholinergic transmission and genetic variation of GABBR1. Individuals with genetics variants of the GABBR1 locus are more likely to develop dependency upon a major cholinergic nicotinic receptor agonist, nicotine. This observation highlights the complexity of the influence of genetic GABBR1 variation upon acetylcholinergic function (Li et al., 2009; Li, 2018). Notably, nicotinic receptors have been of particular interest as a therapeutic target in autism, as their stimulation affects both working memory and executive functions. In a placebo-controlled trial, nicotine patches have been investigated as a mean to improve sleep and to address aggressive behavior in autistic adults (Deutsch et al., 2015; Olincy et al., 2016; Lewis et al., 2018; Deutsch and Burket, 2020).
The evidence for pharmacological intervention on GABAB1 upon autism-related sensory domains is equivocal. GABAB1 receptor agonists such as arbaclofen have been investigated through evaluation of their efficiency across measurable features of irritability, lethargy, and social responsiveness (Veenstra-VanderWeele et al., 2017). When examining a specific sensory subdomain—auditory processing—a subset of teenagers with autism taking arboclofen were found to exhibit improved magnetoencephalographic (MEG) measures of their auditory response (Roberts et al., 2019).
It is of certain importance that in addition to pharmacological means, cholinergic transmission may also be influenced by change in the dietary intake of the choline, one of the common nutrients abundant in milk, liver, eggs, and peanuts. As discussed, many Americans do not consume the daily AI level of choline, and indeed, recent surveys indicate that a majority of a sample of autistic children (60–93%) do not meet their choline AI level recommended respective of age (Hamlin et al., 2013; Wallace and Fulgoni, 2016). A number of autism-associated traits such as preference for routine, aversion to particular foods, as well as sensory aversions to particular smells, textures, or temperatures are known to profoundly shape the diets of people on the autism spectrum (Dunn, 1997; Baranek et al., 2006; Cermak et al., 2010; Nadon et al., 2011; Kral et al., 2013; Hubbard et al., 2014; Sanz-Cervera et al., 2015; Bogdashina and Casanova, 2016; Boterberg and Warreyn, 2016; Crasta et al., 2016; Bitsika and Sharpley, 2018; Weeden, 2019). Because of that, the availability of an adequate pool of acetylcholine for cholinergic signaling may be a legitimate concern, particularly in light of the potentially exacerbated need for proper function of sensory signaling pathways discussed above.
Dietary Choline: A Low-Risk Intervention to Support Acetylcholine Supply, Cholinergic Signaling, and Sensory Processing in Autism
The recommended dietary consumption levels for choline are set by the Food and Nutrition Board of the National Academies as assessed by measuring serum alanine aminotransferase levels reflecting overall liver function. Whether or not the choline at AI level would be sufficient to modulate sensory processing symptoms is not known. It is notable, though, that most Americans do not achieve even this level of the choline intake (Wallace and Fulgoni, 2016). For autistic individuals, who experience particular difficulty in meeting dietary guidelines for choline through diet alone, choline supplementation may provide an alternate effective means of ameliorating daily choline intake levels. As opposed to pharmacological interventions, using the dietary modifications or choline supplements is considered low risk, because neither dietary nor supplementary forms of choline are known to interact with medications. Additionally, overconsuming choline through diet or supplementation to the point of excess—which is defined by the tolerable upper limit of 3,500 mg/day—would require Americans to consume more than seven times their recently estimated mean intake levels of choline, which is currently at less than 500 mg/day for all age groups (Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline, 1988; Wallace and Fulgoni, 2016; Wallace et al., 2018).
To call dietary intervention for choline status “low risk” does not negate the challenges specific to supporting the increased consumption of choline in autistic children and adults. The research and development which is required to determine targeted, effective ways for elevating choline consumption and identifying personalized intake baselines sufficient for fulfilling physiological need for choline in this population is considerable. As with any autism research, investigations should purposefully include representatives and researchers with an autism diagnosis who can help to shape such interventions optimally for autistic children and adults (Chown et al., 2017; Hoekstra et al., 2018; Lebenhagen, 2019; Cascio et al., 2020; Hogan et al., 2020).
Future Research Directions
The autism-associated GABBR1 and KCNN2 genetic variants have been highlighted as such only recently, and therefore there is an opportunity to collect more definitive experimental data, which can inform the modeling of sensory processing dynamics as they relate to these variants. Studies of GABBR1 and KCNN2 in autism should examine possibilities such as structure/function differences in the autism-associated variant version of these membrane proteins, as well as measurement of GABBR1 and KCNN2 expression levels in those presenting with the respective autism-associated variants.
Because of the relative likelihood that autism-associated intron variants such as those found in GABBR1 and KCNN2 would result in transcriptomic changes, transcriptomics should be a primary focus of future research on these genetic variants’ impact. Because of the measured difference in GABAB1 protein expression in autism, examining a potential link between the autism-associated GABBR1 variant and these expression differences should be a priority. Similarly, SK2 channel expression levels in autism should be examined with respect to this SK2 channel variant. Any transcriptomic differences which would be found to be associated with these genetic variants would set the foundation for more refined examination of impact upon cholinergic signal transmission and modulation.
In terms of further examination of potential alterations to autism-related variant versions of GABAB1 and SK2 channel structure and/or function, there are several tools and methods available, which vary in scope and performance. For example, if it can be determined that a reading frame shift has occurred as a consequence of an altered splice site, basic computational homology modeling may be helpful. Without the context of a frame shift, if structural differences are still suspected for the variant version of either of these proteins, validated structural assessment through means of electron microscopy, NMR spectroscopy, and X-ray crystallography remains as resource-intensive possibilities. Additionally, new structure prediction AI presented by AlphaFold, exhibiting unprecedented accuracy in structural prediction, could be considered as a potential future tool (DeepMind, 2020). Additionally, if structural differences were uncovered, molecular dynamics modeling may provide insights into the relative binding frequencies and strength in wildtype vs. variant versions of GABAB1 receptors and SK2 channels. If these models provide actionable information, they may merit further development in terms of broadening molecular dynamics model scope to include various elements from the cholinergic sensory signal pathways and cascades discussed in this study. Computational modeling and pathway exploration may continue to provide support and context as researchers continue to explore potential impact of these genetic variants upon sensory processing in autism.
In terms of clinical interventions, while available evidence on dietary interventions for autism remains limited, this work provides mechanistic support for further exploration of exogenous choline—through diet or supplementation—as a potential low-risk sensory processing support intervention. Future clinical interventions in humans will de facto require the input of multidisciplinary teams of researchers and stakeholders—including those with autism diagnoses—to effectively develop and test autism-tailored clinical interventions that carefully measure inputs through diligent nutritional assessment, and outcomes such as standardized sensory processing scores, and choline activity measured through functional magnetic resonance imaging (fMRI). Such teams may include researchers from a broad range of fields including nutrition, neuroscience, occupational therapy, bioinformatics, and computational biology. Additionally, choline-related biomarkers such as serum alanine aminotransferase may provide useful outcome measurements, particularly in cases where nutritional assessment indicates risk of deficiency.
Conclusion
In connecting dietary choline intake as a mediator of acetylcholinergic pathways, especially for sensory signal ion transport in the context of two autism-associated genes, GABBR1 and KCNN2, this study is consistent with the growing evidence base concerning the role of choline in pre- and postnatal nutrition and neurodevelopment. In our opinion, clinical evaluation of choline intake interventions with respect to validated sensory processing scores is warranted in future studies whose age ranges include children, teens, and adults.
Because of the physically, mentally, and logistically demanding requirements involved in collection of experimental data through clinical trials, particularly from participant individuals with autism and their families, it is incumbent upon researchers to ensure that clinical trials examining dietary status, genetic variants, and sensory processing are effective, efficient, and respectful of their participants. Advance planning and implementation for these clinical trials is therefore at a premium. Accordingly, in preparation for clinical trials, research employing computational biology, bioinformatics, and predictive modeling should also be considered as essential precursor elements in examining the intersection of dietary choline status with sensory processing in the context of autism-associated genes.
Data Availability Statement
The autism GWAS dataset is available as a supplementary file for the original article (Wu et al., 2020).
Author Contributions
AO, AB, and MS were responsible for the conception of the project and editing the final manuscript. AO carried out analyses and interpretation with input from all other authors and drafted the manuscript with feedback from all other authors. All authors contributed to the design.
Funding
Funding to conduct this research was received from the American Egg Board/Egg Nutrition Center. The funder was not involved in the conducting of research or generation of the manuscript. Article processing fees were partially covered by George Mason University’s Open Access Publishing Fund (OAPF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Andersen, N., Corradi, J., Sine, S. M., and Bouzat, C. (2013). Stoichiometry for activation of neuronal α7 nicotinic receptors. Proc. Natl. Acad. Sci. U.S.A. 110, 20819–20824. doi: 10.1073/pnas.1315775110
Ashburner, J., Ziviani, J., and Rodger, S. (2008). Sensory processing and classroom emotional, behavioral, and educational outcomes in children with autism spectrum disorder. Am. J. Occup. Ther. Bethesda 62, 564–573.
Bahia, P. K., Suzuki, R., Benton, D. C. H., Jowett, A. J., Chen, M. X., Trezise, D. J., et al. (2005). A functional role for small-conductance calcium-activated potassium channels in sensory pathways including nociceptive processes. J. Neurosci. 25, 3489–3498. doi: 10.1523/JNEUROSCI.0597-05.2005
Baranek, G. T., David, F. J., Poe, M. D., Stone, W. L., and Watson, L. R. (2006). Sensory experiences questionnaire: discriminating sensory features in young children with autism, developmental delays, and typical development. J. Child Psychol. Psychiatry 47, 591–601. doi: 10.1111/j.1469-7610.2005.01546.x
Bentley, P., Vuilleumier, P., Thiel, C. M., Driver, J., and Dolan, R. J. (2003). Cholinergic enhancement modulates neural correlates of selective attention and emotional processing. Neuroimage 20, 58–70. doi: 10.1016/S1053-8119(03)00302-1
Bertrand, D., Lee, C.-H. L., Flood, D., Marger, F., and Donnelly-Roberts, D. (2015). Therapeutic potential of α7 nicotinic acetylcholine receptors. Pharmacol. Rev. 67, 1025–1073. doi: 10.1124/pr.113.008581
Bianchi, C., Tanganelli, S., Marzola, G., and Beani, L. (1982). GABA induced changes in acetylcholine release from slices of guinea-pig brain. Naunyn. Schmiedebergs Arch. Pharmacol. 318, 253–258. doi: 10.1007/BF00501162
Bitsika, V., and Sharpley, C. F. (2018). Specific aspects of repetitive and restricted behaviours are of greater significance than sensory processing difficulties in eating disturbances in high-functioning young girls with ASD. J. Dev. Phys. Disabil. 30, 259–267. doi: 10.1007/s10882-017-9583-8
Blatt, G. J., and Fatemi, S. H. (2011). Alterations in GABAergic biomarkers in the autism brain: research findings and clinical implications. Anat. Rec. 294, 1646–1652. doi: 10.1002/ar.21252
Blusztajn, J. K., Slack, B. E., and Mellott, T. J. (2017). Neuroprotective actions of dietary choline. Nutr. Basel 9:815. doi: 10.3390/nu9080815
Bogdashina, O., and Casanova, M. (2016). Sensory Perceptual Issues in Autism and Asperger Syndrome, Second Edition: Different Sensory Experiences - Different Perceptual Worlds. London: Jessica Kingsley Publishers.
Boterberg, S., and Warreyn, P. (2016). Making sense of it all: the impact of sensory processing sensitivity on daily functioning of children. Pers. Individ. Differ. 92, 80–86. doi: 10.1016/j.paid.2015.12.022
Cascio, M. A., Weiss, J. A., and Racine, E. (2020). Empowerment in decision-making for autistic people in research. Disabil. Soc. 36, 1–45. doi: 10.1080/09687599.2020.1712189
Case-Smith, J., Weaver, L. L., and Fristad, M. A. (2015). A systematic review of sensory processing interventions for children with autism spectrum disorders. Autism 19, 133–148. doi: 10.1177/1362361313517762
Caudill, M. A. (2010). Pre- and postnatal health: evidence of increased choline needs. J. Am. Diet Assoc. 110, 1198–1206. doi: 10.1016/j.jada.2010.05.009
Cermak, S. A., Curtin, C., and Bandini, L. G. (2010). Food selectivity and sensory sensitivity in children with autism spectrum disorders. J. Am. Diet Assoc. 110, 238–246. doi: 10.1016/j.jada.2009.10.032
Chown, N., Robinson, J., Beardon, L., Downing, J., Liz, H., Julia, L., et al. (2017). Improving research about us, with us: a draft framework for inclusive autism research. Disabil. Soc. 32, 720–734. doi: 10.1080/09687599.2017.1320273
Cohen, E. L., and Wurtman, R. J. (1976). Brain acetylcholine: control by dietary choline. Science 191, 561–562.
Craig, A. D. (2002). How do you feel? Interoception: the sense of the physiological condition of the body. Nat. Rev. Neurosci. 3, 655–666. doi: 10.1038/nrn894
Crasta, J., LaGasse, B., Gavin, W. J., and Davies, P. (2016). Sensory gating and sensory processing in children with high-functioning autism spectrum disorders. Am. J. Occup. Ther. 70:70115050941. doi: 10.5014/ajot.2016.70S1-RP401A
DeepMind (2020). AlphaFold: a Solution to a 50-Year-Old Grand Challenge in Biology. Deepmind. Available online at: https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology (accessed December 30, 2020).
Deutsch, S. I., and Burket, J. A. (2020). An evolving therapeutic rationale for targeting the α7 nicotinic acetylcholine receptor in autism spectrum disorder. Curr. Top Behav. Neurosci. 45, 167–208. doi: 10.1007/7854_2020_136
Deutsch, S. I., Burket, J. A., Urbano, M. R., and Benson, A. D. (2015). The α7 nicotinic acetylcholine receptor: a mediator of pathogenesis and therapeutic target in autism spectrum disorders and down syndrome. Biochem. Pharmacol. 97, 363–377. doi: 10.1016/j.bcp.2015.06.005
Dineley, K. T., Pandya, A. A., and Yakel, J. L. (2015). Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 36, 96–108. doi: 10.1016/j.tips.2014.12.002
Dunn, W. (1997). The impact of sensory processing abilities on the daily lives of young children and their families: a conceptual model. Infants Young Child 9, 23–35. doi: 10.1097/00001163-199704000-00005
Eissa, N., Al-Houqani, M., Sadeq, A., Ojha, S. K., Sasse, A., and Sadek, B. (2018). Current enlightenment about etiology and pharmacological treatment of autism spectrum disorder. Front. Neurosci. 12:304. doi: 10.3389/fnins.2018.00304
Elgoyhen, A. B., Katz, E., and Fuchs, P. A. (2009). The nicotinic receptor of cochlear hair cells: a possible pharmacotherapeutic target? Biochem. Pharmacol. 78, 712–719. doi: 10.1016/j.bcp.2009.05.023
Elgoyhen, A. B., Vetter, D. E., Katz, E., Rothlin, C. V., Heinemann, S. F., and Boulter, J. (2001). α10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc. Natl. Acad. Sci. U.S.A. 98, 3501–3506. doi: 10.1073/pnas.051622798
Fatemi, S. H., Folsom, T. D., Reutiman, T. J., and Thuras, P. D. (2009). Expression of GABAB receptors is altered in brains of subjects with autism. Cereb. Lond. Engl. 8, 64–69. doi: 10.1007/s12311-008-0075-3
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., Rooney, R. J., Patel, D. H., and Thuras, P. D. (2010). mRNA and protein levels for GABAAα4, α5, β1 and GABABR1 receptors are altered in brains from subjects with autism. J. Autism Dev. Disord. 40, 743–750. doi: 10.1007/s10803-009-0924-z
Fendt, M., and Koch, M. (1999). Cholinergic modulation of the acoustic startle response in the caudal pontine reticular nucleus of the rat. Eur. J. Pharmacol. 370, 101–107. doi: 10.1016/S0014-2999(99)00156-9
Ganz, A. B., Klatt, K. C., and Caudill, M. A. (2017). Common genetic variants alter metabolism and influence dietary choline requirements. Nutrients 9:837. doi: 10.3390/nu9080837
Ghaleiha, A., Ghyasvand, M., Mohammadi, M.-R., Farokhnia, M., Yadegari, N., Tabrizi, M., et al. (2014). Galantamine efficacy and tolerability as an augmentative therapy in autistic children: a randomized, double-blind, placebo-controlled trial. J. Psychopharmacol. 28, 677–685. doi: 10.1177/0269881113508830
Gibellini, F., and Smith, T. K. (2010). The Kennedy pathway—De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 62, 414–428. doi: 10.1002/iub.337
Gillentine, M. A., Berry, L. N., Goin-Kochel, R. P., Ali, M. A., Ge, J., Guffey, D., et al. (2017). The cognitive and behavioral phenotypes of individuals with CHRNA7 duplications. J. Autism Dev. Disord. 47, 549–562. doi: 10.1007/s10803-016-2961-8
Granger, A. J., Mulder, N., Saunders, A., and Sabatini, B. L. (2016). Cotransmission of Acetylcholine and GABA. Neuropharmacology 100, 40–46. doi: 10.1016/j.neuropharm.2015.07.031
Grove, J., Ripke, S., Als, T. D., Mattheisen, M., Walters, R. K., Won, H., et al. (2019). Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444. doi: 10.1038/s41588-019-0344-8
Gu, M., Zhu, Y., Yin, X., and Zhang, D.-M. (2018). Small-conductance Ca 2+ -activated K + channels: insights into their roles in cardiovascular disease. Exp. Mol. Med. 50, 1–7. doi: 10.1038/s12276-018-0043-z
Hamlin, J. C., Pauly, M., Melnyk, S., Yang, S., Li, M., Zhang, H., et al. (2013). Dietary intake and plasma levels of choline and betaine in children with autism spectrum disorders. Autism Res. Treat. 2013:578429. doi: 10.1155/2013/578429
Hertzman, M. (2003). Galantamine in the treatment of adult autism: a report of three clinical cases. Int. J. Psychiatry Med. 33, 395–398. doi: 10.2190/JE5Q-1NFT-FL40-7PMW
Hipólito, L., Fakira, A. K., Cabañero, D., Blandón, R., Carlton, S. M., Morón, J. A., et al. (2015). In vivo activation of the SK channel in the spinal cord reduces the NMDA receptor antagonist dose needed to produce antinociception in an inflammatory pain model. Pain 156, 849–858. doi: 10.1097/j.pain.0000000000000124
Hoekstra, R. A., Girma, F., Tekola, B., and Yenus, Z. (2018). Nothing about us without us: the importance of local collaboration and engagement in the global study of autism. Br. J. Psychol. Int. 15, 40–43. doi: 10.1192/bji.2017.26
Hogan, A., Jain, N. R., Peiris-John, R., and Ameratunga, S. (2020). Disabled people say ‘Nothing about us without us’. Clin. Teach. 17, 70–75. doi: 10.1111/tct.13022
Hubbard, K. L., Anderson, S. E., Curtin, C., Must, A., and Bandini, L. G. A. (2014). Comparison of food refusal related to characteristics of food in children with autism spectrum disorder and typically developing children. J. Acad. Nutr. Diet 114, 1981–1987. doi: 10.1016/j.jand.2014.04.017
Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline (1988). Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: National Academies Press, doi: 10.17226/6015
Karvat, G., and Kimchi, T. (2014). Acetylcholine elevation relieves cognitive rigidity and social deficiency in a mouse model of autism. Neuropsychopharmacol. N.Y. 39, 831–840. doi: 10.1038/npp.2013.274
Knott, V., Smith, D., de la Salle, S., Impey, D., Choueiry, J., Beaudry, E., et al. (2014). CDP-choline: effects of the procholine supplement on sensory gating and executive function in healthy volunteers stratified for low, medium and high P50 suppression. J. Psychopharmacol. 28, 1095–1108. doi: 10.1177/0269881114553254
Koker, S. C., Jahja, E., Shehwana, H., Keskus, A. G., and Konu, O. (2018). Cholinergic receptor nicotinic Alpha 5 (CHRNA5) RNAi is associated with cell cycle inhibition, apoptosis, DNA damage response and drug sensitivity in breast cancer. PLoS One 13:e0208982. doi: 10.1371/journal.pone.0208982
Kong, J.-H., Adelman, J. P., and Fuchs, P. A. (2008). Expression of the SK2 calcium-activated potassium channel is required for cholinergic function in mouse cochlear hair cells. J. Physiol. 586, 5471–5485. doi: 10.1113/jphysiol.2008.160077
Kral, T. V. E., Eriksen, W. T., Souders, M. C., and Pinto-Martin, J. A. (2013). Eating behaviors, diet quality, and gastrointestinal symptoms in children with autism spectrum disorders: a brief review. J. Pediatr. Nurs. 28, 548–556. doi: 10.1016/j.pedn.2013.01.008
Kusunoki, M., Taniyama, K., and Tanaka, C. (1984). Neuronal GABA release and GABA inhibition of ACh release in guinea pig urinary bladder. Am. J. Physiol. 246(4 Pt 2), R502–R509. doi: 10.1152/ajpregu.1984.246.4.R502
Lebenhagen, C. (2019). Including speaking and nonspeaking autistic voice in research. Autism Adulthood 2, 128–131. doi: 10.1089/aut.2019.0002
Lee, M., Martin-Ruiz, C., Graham, A., Court, J., Jaros, E., Perry, R., et al. (2002). Nicotinic receptor abnormalities in the cerebellar cortex in autism. Brain 125, 1483–1495. doi: 10.1093/brain/awf160
Lewis, A. S., van Schalkwyk, G. I., Lopez, M. O., Volkmar, F. R., Picciotto, M. R., and Sukhodolsky, D. G. (2018). An exploratory trial of transdermal nicotine for aggression and irritability in adults with autism spectrum disorder. J. Autism Dev. Disord. 48, 2748–2757. doi: 10.1007/s10803-018-3536-7
Li, M. D. (2018). “Genetic contribution of variants in GABAergic signaling to nicotine dependence,” in Tobacco Smoking Addiction: Epidemiology, Genetics, Mechanisms, and Treatment, ed. M. D. Li (Cham: Springer), 95–105. doi: 10.1007/978-981-10-7530-8_7
Li, M. D., Mangold, J. E., Seneviratne, C., Chen, G.-B., Ma, J. Z., Lou, X.-Y., et al. (2009). Association and interaction analyses of GABBR1 and GABBR2 with nicotine dependence in European- and African-american populations. PLoS One 4:e7055. doi: 10.1371/journal.pone.0007055
Liberzon, A., Subramanian, A., Pinchback, R., Thorvaldsdottir, H., Tamayo, P., and Mesirov, J. P. (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. doi: 10.1093/bioinformatics/btr260
Liu, S., Rao, S., Xu, Y., Li, J., Huang, H., Zhang, X., et al. (2020). Identifying common genome-wide risk genes for major psychiatric traits. Hum. Genet. 139, 185–198. doi: 10.1007/s00439-019-02096-4
Maire, J.-C. E., and Wurtman, R. J. (1984). Choline production from choline-containing phospholipids: a hypothetical role in Alzheimer’s disease and aging. Prog. Neuropsychopharmacol. Biol. Psychiatry 8, 637–642. doi: 10.1016/0278-5846(84)90027-7
Mongan, L. C., Hill, M. J., Chen, M. X., Tate, S. N., Collins, S. D., Buckby, L., et al. (2005). The distribution of small and intermediate conductance calcium-activated potassium channels in the rat sensory nervous system. Neuroscience 131, 161–175. doi: 10.1016/j.neuroscience.2004.09.062
Murthy, V., Maison, S. F., Taranda, J., Haque, N., Bond, C. T., Elgoyhen, A. B., et al. (2009). SK2 channels are required for function and long-term survival of efferent synapses on mammalian outer hair cells. Mol. Cell Neurosci. 40, 39–49. doi: 10.1016/j.mcn.2008.08.011
Nadon, G., Feldman, D. E., Dunn, W., and Gisel, E. (2011). Association of sensory processing and eating problems in children with autism spectrum disorders. Autism Res. Treat. 2011:541926. doi: 10.1155/2011/541926
Nicolson, R., Craven-Thuss, B., and Smith, J. (2006). AProspective, open-label trial of galantamine in autistic disorder. J. Child Adolesc. Psychopharmacol. New Rochelle 16, 621–629. doi: 10.1089/cap.2006.16.621
Oda, Y. (1999). Choline acetyltransferase: the structure, distribution and pathologic changes in the central nervous system. Pathol. Int. 49, 921–937. doi: 10.1046/j.1440-1827.1999.00977.x
Olincy, A., Blakeley-Smith, A., Johnson, L., Kem, W. R., and Freedman, R. (2016). Brief report: initial trial of Alpha7-nicotinic receptor stimulation in two adult patients with autism spectrum disorder. J. Autism Dev. Disord. 46, 3812–3817. doi: 10.1007/s10803-016-2890-6
Oliveros, J. C. (2007–2015). Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online at: https://bioinfogp.cnb.csic.es/tools/venny/index.html
Pagadala, P., Park, C.-K., Bang, S., Xu, Z.-Z., Xie, R.-G., Liu, T., et al. (2013). Loss of NR1 Subunit of NMDARs in primary sensory neurons leads to hyperexcitability and pain hypersensitivity: involvement of Ca2+-activated small conductance potassium channels. J. Neurosci. 33, 13425–13430. doi: 10.1523/JNEUROSCI.0454-13.2013
Rahman, A., Freedman, R., and Hollander, E. (2018). S59: alpha-7 nicotinic acetylcholine receptor positive allosteric modulator galantamine in autism spectrum disorder. Biol. Psychiatry 83, S369–S370. doi: 10.1016/j.biopsych.2018.02.950
Roberts, T. P. L., Bloy, L., Blaskey, L., Kuschner, E., Gaetz, L., Anwar, A., et al. (2019). A MEG study of acute Arbaclofen (STX-209) administration. Front. Integr. Neurosci. 13:69. doi: 10.3389/fnint.2019.00069
Rossignol, D. A., and Frye, R. E. (2014). The use of medications approved for Alzheimer’s disease in autism spectrum disorder: a systematic review. Front. Pediatr. 2:87. doi: 10.3389/fped.2014.00087
Sanz-Cervera, P., Pastor-Cerezuela, G., Fernández-Andrés, M.-I., and Tárraga-Mínguez, R. (2015). Sensory processing in children with autism spectrum disorder: relationship with non-verbal IQ, autism severity and attention deficit/hyperactivity disorder symptomatology. Res. Dev. Disabil. 4, 188–201. doi: 10.1016/j.ridd.2015.07.031
Schijven, D., Kofink, D., Tragante, V., Verkerke, M., Pulit, S. L., Kahn, R. S., et al. (2018). Comprehensive pathway analyses of schizophrenia risk loci point to dysfunctional postsynaptic signaling. Schizophr. Res. 199, 195–202. doi: 10.1016/j.schres.2018.03.032
Simmons, D., Duncan, J., de Caprona, D. C., and Fritzsch, B. (2011). “Development of the inner ear efferent system,” in Auditory and Vestibular Efferents, eds D. K. Ryugo and R. R. Fay (Berlin: Springer), 187–216. doi: 10.1007/978-1-4419-7070-1_7
Sinkus, M. L., Graw, S., Freedman, R., Ross, R. G., Lester, H. A., and Leonard, S. (2015). The human CHRNA7 and CHRFAM7A Genes: a review of the genetics, regulation, and function. Neuropharmacology 96, 274–288. doi: 10.1016/j.neuropharm.2015.02.006
Stevens, K. E., Adams, C. E., Mellott, T. J., Robbins, E., and Kisley, M. A. (2008). Perinatal choline deficiency produces abnormal sensory inhibition in Sprague–Dawley rats. Brain Res. 1237, 84–90. doi: 10.1016/j.brainres.2008.08.047
Swerdlow, N. R., Braff, D. L., and Geyer, M. A. (2016). Sensorimotor gating of the startle reflex: what we said 25 years ago, what has happened since then, and what comes next. J. Psychopharmacol. Oxf. Engl. 30, 1072–1081. doi: 10.1177/0269881116661075
Takács, V. T., Cserép, C., Schlingloff, D., Trezise, D. J., Blandón, R., Carlton, S. M., et al. (2018). Co-transmission of acetylcholine and GABA regulates hippocampal states. Nat. Commun. 9:2848. doi: 10.1038/s41467-018-05136-1
Tan, M. G., Chua, W.-T., Esiri, M. M., Smith, A. D., Vinters, H. V., and Lai, M. K. (2010). Genome wide profiling of altered gene expression in the neocortex of Alzheimer’s disease. J. Neurosci. Res. 88, 1157–1169. doi: 10.1002/jnr.22290
Thompson, J. M., Ji, G., and Neugebauer, V. (2015). Small-conductance calcium-activated potassium (SK) channels in the amygdala mediate pain-inhibiting effects of clinically available riluzole in a Rat Model of Arthritis Pain. Mol. Pain 11:s12990-015-0055-9. doi: 10.1186/s12990-015-0055-9
Turcan, S., Slonim, D. K., and Vetter, D. E. (2010). Lack of nAChR activity depresses cochlear maturation and up-regulates GABA system components: temporal profiling of gene expression in α9 null Mice. PLoS One 5:e9058. doi: 10.1371/journal.pone.0009058
Veenstra-VanderWeele, J., Cook, E. H., King, B. H., Zarevics, P., Cherubini, M., Walton-Bowen, K., et al. (2017). Arbaclofen in children and adolescents with autism spectrum disorder: a randomized, controlled, phase 2 Trial. Neuropsychopharmacology 42, 1390–1398. doi: 10.1038/npp.2016.237
Wallace, T. C., Blusztajn, J. K., Caudill, M. A., Klatt, K. C., Natker, E., Zeisel, S. H., et al. (2018). Choline: the underconsumed and underappreciated essential nutrient. Nutr. Today 53, 240–253. doi: 10.1097/NT.0000000000000302
Wallace, T. C., Blusztajn, J. K., Caudill, M. A., Klatt, K. C., and Zeisel, S. H. (2019). Choline: the neurocognitive essential nutrient of interest to obstetricians and gynecologists. J. Diet Suppl. 17, 1–20. doi: 10.1080/19390211.2019.1639875
Wallace, T. C., and Fulgoni, V. L. (2016). Assessment of total choline intakes in the United States. J. Am. Coll. Nutr. 35, 108–112. doi: 10.1080/07315724.2015.1080127
Wang, M., and Marín, A. (2006). Characterization and prediction of alternative splice sites. Gene 366, 219–227. doi: 10.1016/j.gene.2005.07.015
Wangemann, P. K. (2002). K+ cycling and the endocochlear potential. Hear. Res. 165, 1–9. doi: 10.1016/S0378-5955(02)00279-4
Weeden, A. M. (2019). “Dietetics/Nutrition,” in Handbook of Interdisciplinary Treatments for Autism Spectrum Disorder. Autism and Child Psychopathology Series, ed. R. D. Rieske (Cham: Springer), 279–296. doi: 10.1007/978-3-030-13027-5_15
Williams, Z. J., Suzman, E., and Woynaroski, T. G. (2021). Prevalence of decreased sound tolerance (Hyperacusis) in individuals with autism spectrum disorder: a meta-analysis. Ear Hear. doi: 10.1097/AUD.0000000000001005 [Epub ahead of print].
Wilson, U. S., Sadler, K. M., Hancock, K. E., Guinan, J. J., and Lichtenhan, J. T. (2017). Efferent inhibition strength is a physiological correlate of hyperacusis in children with autism spectrum disorder. J. Neurophysiol. 118, 1164–1172. doi: 10.1152/jn.00142.2017
Wu, M. F., Jenden, D. J., Fairchild, M. D., and Siegel, J. M. (1993). Cholinergic mechanisms in startle and prepulse inhibition: effects of the false cholinergic precursor N-aminodeanol. Behav. Neurosci. 107, 306–316.
Wu, Y., Cao, H., Baranova, A., Huang, H., Li, S., Cai, L., et al. (2020). Multi-trait analysis for genome-wide association study of five psychiatric disorders. Transl. Psychiatry 10:209. doi: 10.1038/s41398-020-00902-6
Zeisel, S. H. (2019). A conceptual framework for studying and investing in precision nutrition. Front. Genet. 10:200. doi: 10.3389/fgene.2019.00200
Zhang, K., Fan, Z., Wang, Y., Faraone, S. V., Yang, L., and Chang, S. (2019). Genetic analysis for cognitive flexibility in the trail-making test in attention deficit hyperactivity disorder patients from single nucleotide polymorphism, gene to pathway level. World J. Biol. Psychiatry 20, 476–485. doi: 10.1080/15622975.2017.1386324
Keywords: acetylcholine, dietary choline, autism, sensory processing, cholinergic, gene set enrichment analysis
Citation: Olson A, Zhang F, Cao H, Baranova A and Slavin M (2021) In silico Gene Set and Pathway Enrichment Analyses Highlight Involvement of Ion Transport in Cholinergic Pathways in Autism: Rationale for Nutritional Intervention. Front. Neurosci. 15:648410. doi: 10.3389/fnins.2021.648410
Received: 31 December 2020; Accepted: 22 March 2021;
Published: 20 April 2021.
Edited by:
Joana M. Gaspar, Federal University of Santa Catarina, BrazilReviewed by:
Karan Kapoor, University of Illinois at Urbana-Champaign, United StatesMengting Liu, University of Southern California, United States
Lydia Manor, American Informatics Consultant LLC, United States
Copyright © 2021 Olson, Zhang, Cao, Baranova and Slavin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Audrey Olson, YW9sc29uMkBtYXNvbmxpdmUuZ211LmVkdQ==; Margaret Slavin, bXNsYXZpbkBnbXUuZWR1