 Ulrich Schweizer
Ulrich Schweizer Simon Bohleber
Simon Bohleber Wenchao Zhao
Wenchao Zhao Noelia Fradejas-Villar
Noelia Fradejas-Villar- Institut für Biochemie und Molekularbiologie, Medizinische Fakultät, Rheinische Friedrich-Wilhelms-Universität Bonn, Bonn, Germany
Eighteen years ago, unexpected epileptic seizures in Selenop-knockout mice pointed to a potentially novel, possibly underestimated, and previously difficult to study role of selenium (Se) in the mammalian brain. This mouse model was the key to open the field of molecular mechanisms, i.e., to delineate the roles of selenium and individual selenoproteins in the brain, and answer specific questions like: how does Se enter the brain; which processes and which cell types are dependent on selenoproteins; and, what are the individual roles of selenoproteins in the brain? Many of these questions have been answered and much progress is being made to fill remaining gaps. Mouse and human genetics have together boosted the field tremendously, in addition to traditional biochemistry and cell biology. As always, new questions have become apparent or more pressing with solving older questions. We will briefly summarize what we know about selenoproteins in the human brain, glance over to the mouse as a useful model, and then discuss new questions and directions the field might take in the next 18 years.
A Brief History of the Field
When selenoprotein P (Selenop)-knockout mice were made independently in two laboratories, it was not expected that their most dramatic phenotype was to reveal the essential requirement of selenium (Se) in the brain (Hill et al., 2003; Schomburg et al., 2003). However, the model was tricky: the neurological phenotype depended exquisitely on the level of dietary Se supply and, more precisely, on the timing of Se deficiency during the ontogeny of the animal. The neurological phenotype varied between none at all, epileptic seizures, movement phenotype with ataxia and/or dystonia, overt neurodegeneration with premature death, or death before weaning (Hill et al., 2004; Schweizer et al., 2004b, 2005; Valentine et al., 2005; Schweizer, 2016). Since Se levels in commercial diets may not always precisely reflect the printed specifications in every lot of mouse chow, and because the Selenop-knockout mice reflected so sensitively the dietary Se supply, working with the model was difficult, to say the least. At the time, antibodies against selenoproteins were not widely available, and enzymatic assays for glutathione peroxidase (GPX) and thioredoxin reductase (TXNRD), Se measurements, and metabolic labeling with 75Se were the methods of choice. At least, these methods clearly demonstrated, that inactivation of Selenop, a gene mostly expressed in liver and secreted into the plasma, reduced Se levels and selenoenzyme activities in the brain to a degree impossible to achieve with dietary Se restriction alone (Hill et al., 2003; Schomburg et al., 2003). New selenoproteins were still being discovered, until a landmark genomic study fixed the number at the final 25 in humans and 24 in mice (Kryukov et al., 2003). In an early review (Schweizer et al., 2004c), the key questions about the neurobiology of Se were: Which selenoproteins are expressed in the brain, in which regions and in which cell types? What are their functions? How does Se enter the brain and is it distributed in a hierarchical manner? Is there a causal connection to neurological disorders?
To cut the answers short: most selenoproteins are expressed in the brain, mainly in neurons, in all major brain regions (Zhang et al., 2008). Gene targeting in mice for all or single selenoproteins has revealed that GABAergic interneurons are particularly vulnerable, but also basal ganglia, cerebellum, cortex, and brain stem (Valentine et al., 2005; Wirth et al., 2010, 2014; Pitts et al., 2012; Seeher et al., 2014). At first it remained an open question whether myelination defects were caused by neuronal or oligodendroglial selenoprotein dysfunction (Valentine et al., 2008). Several selenoproteins are essential for the brain development and function, in particular glutathione peroxidase 4 (GPX4), but also thioredoxin reductase 1 (TXNRD1), and SELENOT (Castex et al., 2015). Currently the function of GPX4 as a major regulator of ferroptosis in development and disease is receiving a great deal of attention (Stockwell et al., 2017; Friedmann Angeli et al., 2019). It will be seen whether GPX4 is an indispensable protein for neurons in its own right or whether there is a causal connection to neurodegeneration in neurological disorders. What is now clear, is that Se enters the brain either as non-physiological selenite salt or, more physiologically, in the form of SELENOP, loaded in the liver with several atoms of Se (Schweizer et al., 2005; Renko et al., 2008; Hill et al., 2012), and taken up at the blood-brain-barrier (BBB) and by individual cells via endocytosis using receptors of the LRP family (Burk and Hill, 2015). The most important receptor is APOER2 (Olson et al., 2007), but also LRP2/MEGALIN (Olson et al., 2008; Chiu-Ugalde et al., 2010) and, most likely, LRP1 contribute to SELENOP internalization. Among these receptors, APOER2 clearly is the most important, providing preferential Se supply at the BBB and in the testis. MEGALIN appears more involved in Se supply through the blood-cerebrospinal fluid barrier and in the kidney (Burk and Hill, 2015). SELENOP is also expressed in the brain and may contribute to local Se storage and recycling (Scharpf et al., 2007; Renko et al., 2008). SELENOP and its receptors contribute to a “hierarchy” of selenoprotein expression in organs in the sense that some organs, like the brain, can be preferentially supplied with Se at the expense of others, like the liver. There is, however, a second “hierarchy” of selenoprotein expression that depends on the relative sensitivity of individual selenoproteins to Se availability. Thus, GPX1 is more sensitive to cellular Se levels than, e.g., GPX4. The reasons behind this observation are multifarious and beyond the scope of this review. Both hierarchies may work hand-in-hand as GPX1 mRNA is highly expressed in liver and contains a highly efficient SECIS element. Depending on dietary Se supply, the amount of GPX1 protein covers orders of magnitude, and likely provides a safe Se storage device, until the Se is needed for distribution via SELENOP to preferred target tissues. The amount of Se in a tissue does not necessarily inform about its physiological importance, e.g., brain Se levels are much lower than liver Se levels, yet selenoproteins are essential in brain, but not liver (Schweizer et al., 2005; Wirth et al., 2010).
Genetic Deficiency of Single Selenoproteins
At that time, only one human disorder was genetically linked to deficiency of a selenoprotein-encoding gene, now known as selenoprotein N (SELENON)-related myopathies (Moghadaszadeh et al., 2001; Castets et al., 2012), but the situation was soon to change with exome sequencing entering clinical practice. SELENON is an ER-resident membrane protein of unknown function. Mutations in SELENON that disrupt the gene or prevent selenocysteine (Sec) insertion into the protein lead to myopathy (Villar-Quiles et al., 2020). Mouse and zebrafish models of SELENON-deficiency reflect aspects of the muscular phenotype, in particular the preferential affection of axial muscles (Rederstorff et al., 2011). This example suggests that the mouse may represent an acceptable model for humans regarding selenoprotein deficiency. It should be noted that dietary deficiency for Se in livestock was recognized early as a cause for white muscle disease (Muth et al., 1958). So far there is no evidence to link SELENON mutations to impaired neuronal or neurological function.
The syndromes associated with selenoprotein deficiency can be broadly divided into two categories: the first category represents mutations in single genes encoding selenoproteins. The second category is represented by mutations in genes involved in selenoprotein biosynthesis. A comprehensive compilation of individual mutations and patient phenotypes can be found in recent summaries of the state of the field (Schweizer and Fradejas-Villar, 2016; Fradejas-Villar, 2018; Schoenmakers and Chatterjee, 2020). Here, we will only briefly summarize these results and rather focus on new developments since then.
Conditional gene inactivation of Gpx4 in mice demonstrated that GPX4 is an essential selenoprotein for several types of neurons as discussed above (Seiler et al., 2008). Conditional inactivation of Gpx4 in forebrain neurons after development lead to cognitive decline and hippocampal neurodegeneration (Hambright et al., 2017). Furthermore, constitutive gene inactivation of Gpx4 lead to embryonic lethality around embryonic day 7 (Yant et al., 2003; Seiler et al., 2008). Thus, it came as a complete surprise to find newborn children affected with Sedaghatian-type spondylometaphyseal dysplasia with inactivating non-sense mutations in the GPX4 gene (Aygun et al., 2012; Smith et al., 2014). These patients show massive brain atrophy and usually die shortly after birth. In stark contrast to mice, where Gpx4-deficiency leads to early embryonic lethality, human fetuses obviously progress much further in their development.
A lesser sensitivity of humans compared to mice regarding the lack of selenoproteins was also observed with respect to TXNRD2. Txnrd2–/– mice died around embryonic day 13 with thinned ventricular walls in the heart and impaired hematopoiesis (Conrad et al., 2004). Conditional ablation of Txnrd2 in the heart lead to a fatal cardiomyopathy (Conrad et al., 2004). Similarly, heterozygous missense mutations were associated with dilated cardiomyopathy in humans (Sibbing et al., 2011) and were reminiscent of Keshan disease, a fatal cardiomyopathy observed in Se-deficient regions in China (Ge et al., 1983; Loscalzo, 2014). It thus came as a complete surprise that a homozygous truncating mutation in human TXNRD2 merely resulted in familial glucocorticoid deficiency without a cardiac phenotype (Prasad et al., 2014).
Similar to inactivation of Txnrd2, constitutive inactivation of Txnrd1 is embryonic lethal in mice (Jakupoglu et al., 2005). Conditional ablation of Txnrd1 in neuronal precursors shows only a mild cerebellar defect (Soerensen et al., 2008), while neuron-specific Txnrd1 ablation leads to neurodegeneration with aging (Schweizer and Schomburg, 2006). Since our last review of this subject, we have found that homozygous mutations in TXNRD1, which reduce enzymatic activity, are associated with genetic generalized epilepsy in human (Kudin et al., 2017). In summary, upon Txnrd mutation the mouse phenotypes appear throughout more severe than the corresponding human phenotypes. This raises the question whether variants of TXNRDs may be able to compensate for the loss of the other TXNRD in humans, but not in mice, or whether, e.g., the glutaredoxin system may be able to partially compensate in some human cell types.
Ethanolamine-phosphotransferase 1 (EPT1/SELENOI) is one of two enzymes catalyzing the same step in phospholipid biosynthesis (Gladyshev et al., 2016). This enzyme is obviously important for myelin biosynthesis in the human brain (Ahmed et al., 2017; Horibata et al., 2018). Inactivation of the gene in mice is embryonic lethal (Avery et al., 2020). Se deficiency of the brains of Selenop-deficient mice impaired the myelin sheath in the brain stem, at least under conditions of low Se diet (Valentine et al., 2005). Thus, it is possible that the myelination defect is a primary phenotype of myelin formation in oligodendrocytes and not a result of retrograde signaling from selenoprotein-deficient neurons, as we initially suspected.
The only other selenoprotein that has been shown to play an essential role in the brain is SELENOT (Castex et al., 2015; Boukhzar et al., 2016). How this relates to its role in protein glycosylation is not clear, yet (Hamieh et al., 2017). It is intriguing, that other selenoproteins have also been implicated in protein glycosylation or protein folding, e.g., SELENOF and SELENOM. The respective knockout mouse models, however, did not show any apparent neurological defects (Kasaikina et al., 2011). SELENOS, another selenoprotein implicated in endoplasmic reticulum associated degradation of proteins (ERAD) (Curran et al., 2005), has not been studied by gene targeting in mice. While selenoproteins have often simply been classified as “anti-oxidant,” it is remarkable that inactivation of a selenoenzyme with a defined reductase activity, methionine-R-sulfoxide-reductase B1 (MSRB1), has not produced a neurological phenotype (Fomenko et al., 2008; Lee et al., 2013).
There is a rich literature on mouse models of neurodegenerative diseases whose phenotypes can be exacerbated by additional deficiency of “antioxidant” selenoproteins (Schweizer et al., 2004a; Zhang et al., 2020). In such models there is always the conceptual question whether the mutation in the selenoprotein specifically abrogates (and thus reveals) a specific protective mechanism or whether the selenoprotein mutation simply tips over a dysbalanced system that is already vulnerable to any other possible stressor. Given the availability of many powerful genome-wide association studies on important neurodegenerative disorders, and their failure to identify mutations in selenoprotein genes, it seems unlikely for us that mutations in selenoproteins are important causes or modifiers of common neurological disorders. Yet, mutations in selenoproteins or their biosynthesis pathways may reveal specific cell biological or developmental functions of selenoproteins.
Selenoprotein Deficiency Resulting From Mutations Impairing Selenoprotein Biosynthesis
A landmark paper on the identification of mutations in the selenoprotein biosynthesis factor SECISBP2 in humans called into question the possibly simple-minded concept of selenoproteins as “anti-oxidants.” The key phenotype that brought the patients to medical attention, was a growth retardation in puberty (Dumitrescu et al., 2005). Abnormal thyroid function tests (TFT), i.e., the constellation of thyroid-stimulating hormone (TSH) and thyroid hormone levels, guided the discovery of a congenital deficiency of selenoprotein biosynthesis. The pubertal growth spurt depends not only on growth hormone, but requires permissive action of thyroid hormone. The TFT suggested deficiency of deiodinase 2 (DIO2) activity in these patients which was confirmed in patient fibroblasts. Deiodinases are selenoenzymes capable of removing iodide from iodothyronines (Köhrle et al., 2005; Mondal et al., 2016). The prohormone thyroxine (T4) requires 5′-deiodination to yield triiodothyronine (T3), which binds the nuclear T3-receptors (Figure 1). 5-deiodination of T4 and T3 yields reverse T3 (rT3) and 3,3′-T2, respectively. Moreover, the two plasma selenoproteins SELENOP and GPX3 were reduced in these patients (Dumitrescu et al., 2005). Thus, the congenital deficiency of selenoprotein biosynthesis revealed itself not in neurodegeneration, epilepsy, heart disease or a muscular disorder, but in altered thyroid hormone levels in the sense of a blunted response to T4! Later, more patients with apparently stronger mutations in SECISBP2 were identified (Di Cosmo et al., 2009; Azevedo et al., 2010; Schoenmakers et al., 2010). Some of these patients exhibited a SELENON-related myopathy, infertility, and an immune phenotype (Schoenmakers and Chatterjee, 2018). The importance of local conversion of T3 is illustrated by the Thr92Ala polymorphism in DIO2. People with the homozygous Ala92 version of this polymorphism have a reduced ability to convert T4 to T3 (McAninch et al., 2015), hence when being treated for hypothyroidism, have improved psychological well-being on combination T4/T3 therapy than on T4 treatment alone (Bianco and Kim, 2018). Remarkably, the first patient with a mutation in the tRNASec gene (TCA-TRU in human, Trsp in mouse) showed the same phenotype of a blunted response to T4 (Schoenmakers et al., 2016). Findings from Dio1- and Dio2-knock-out mouse models are entirely compatible with the above conclusions drawn from SECISBP2-deficiency (Schneider et al., 2001, 2006). Selenoprotein deficiency does not fundamentally impair thyroid gland function (Chiu-Ugalde et al., 2012).
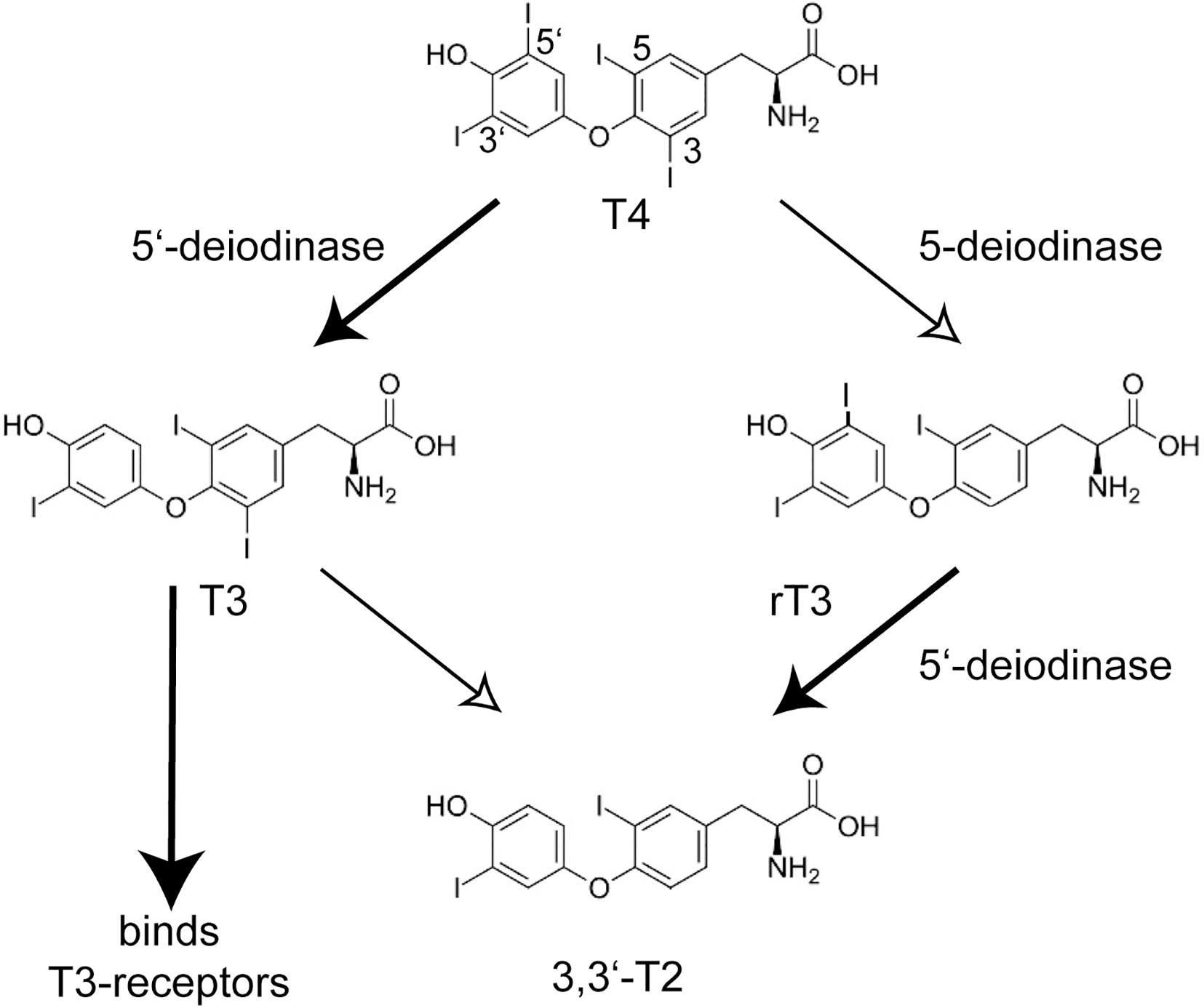
Figure 1. Conversion of iodothyronines by deiodinases and activation of the nuclear T3-receptors. The main product of the thyroid gland is thyroxine (T4, 3,3′,5,5′-tetraiodothyronine). The actions of 5′-deiodinases (DIO1 and DIO2) and 5-deiodinases (DIO1 and DIO3) lead to T3 (3,3′,5-triiodothyronine) and rT3 (3,3′,5′-triiodothyronine), respectively. Only T3 activates the nuclear T3-receptors TRα and TRβ. T2 and rT3 are thus inactive metabolites and a cell can shape its local T3 level through DIO expression. Inactivation of Dio1 as well as SECISBP2-deficiency lead to increased plasma rT3.
This leads us to the obvious question whether other factors involved in selenoprotein biosynthesis have been found mutated in humans. And what kind of phenotypes are presented by affected individuals? The first patients with mutations in the selenocysteine synthase gene (SEPSECS) have been identified in Agamy et al. (2010). The patients presented with “progressive cerebello cerebral atrophy,” now systematically designated pontocerebellar hypoplasia 2D (PCH2D). The names of the syndromes capture quite well the observed phenotypes (Schoenmakers and Chatterjee, 2020). The predominantly neurological condition with neurodegeneration and epilepsy is likely based on dysfunction of GPX4 and other essential selenoproteins, possibly TXNRD1 or 2 (Anttonen et al., 2015). Some patients display milder phenotypes and may grow into adulthood with intellectual disability, but no overt neurodegeneration (Iwama et al., 2016). Interestingly, we are not aware of reports of abnormal TFT in these patients. Likewise, we do not know of a SELENON-related myopathy in one of these patients.
Individuals with mutations in EEFSEC, SEPHS2, and PSTK have not yet been found (Figure 2). Selenoprotein expression in knockout mouse models for these genes have not been described. Conditional inactivation of the suggested biosynthesis factor SECP43, encoded by the Trnau1ap gene, in liver and in neurons did not support a role for this gene in selenoprotein expression (Mahdi et al., 2015). Mouse models for hypomorphic mutations in the tRNASec have been generated. One model has a mutation in the promoter and, as a simple transgene, is inserted somewhere in the genome (Carlson et al., 2009). This mouse displays a neurological phenotype that resembles in several aspects of Selenop- and neuron-specific Trsp-knockout mutants (Schomburg et al., 2003; Wirth et al., 2010, 2014). Point mutations have been made in Trsp affecting the anticodon loop of tRNASec. Overexpression of the A37G-Trsp mutant (made as a simple transgene) resulted in neurological defects, in particular when fed a high Se diet (Kasaikina et al., 2013). TFTs have not been determined for the Trsp mutant mouse models.
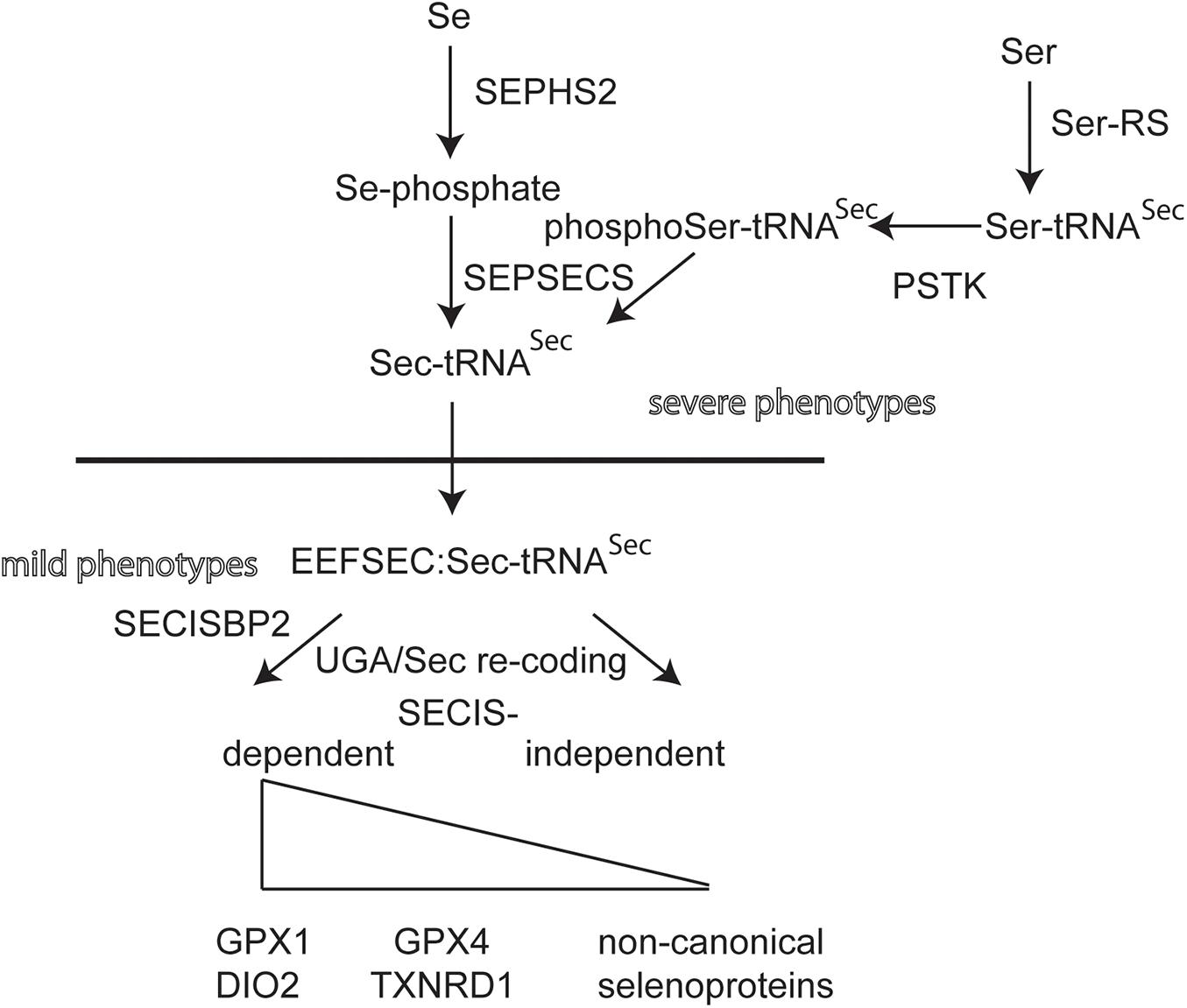
Figure 2. Biosynthetic pathway of selenoprotein translation. Transfer-RNASec is charged with Ser by Seryl-tRNA synthase (Ser-RS), hence the more accurate designation tRNA[Ser]Sec. The kinase PSTK phosphorylates Ser-tRNASec. Selenophosphate synthase (SEPHS2) provides selenophosphate which is used by selenocysteine synthase (SEPSECS) to convert phosphoSer-tRNASec into Sec-tRNASec. EEFSEC is a translation elongation factor specific for Sec-tRNASec. Canonical selenoproteins carry a selenocysteine insertion sequence (SECIS) in their mRNA in order to re-code the UGA codon as Sec codon. The dependence of UGA/Sec re-coding varies among canonical selenoproteins. Several non-canonical selenoprotein genes have been described that do not contain a SECIS element (Guo et al., 2018).
Looking at these results, there seems to exist a strange dichotomy of phenotypes being related either to neurobiology or endocrinology, when selenoprotein biosynthesis is impaired. This observation holds for both mice and humans. A naïve epistatic model of selenoprotein biosynthesis should predict more or less the same phenotypes, if selenoprotein translation is globally impaired (Figure 2). Clearly, we have not yet analyzed all possible mutants and not all available mutants have been systematically analyzed side by side. Yet, in humans, mutations most often come as missense, splicing or other mutations that may not completely abrogate 100% of gene/protein expression/activity. We have recently shown that the effect of a missense mutation in vitro and in vivo may differ, in particular stability of a mutated protein may depend on the cell type (Zhao et al., 2019). For the SECISBP2R543Q mutant, we demonstrated that the protein is a complete NULL in mouse liver, but partially functioning and supporting selenoprotein expression in neurons (Zhao et al., 2019).
Future Directions
In order to understand the neurobiology of Se, we need both, the precise biochemical or cell biological function of each selenoprotein and the full understanding of the phenotypes under conditions of its absence in an entire mammalian organism. This goal has only been achieved for a small subset of selenoproteins. For some of the others, we may have a biochemical reaction and a phenotype of cells grown in a dish, but we are convinced that nobody would have been able to predict the complex phenotype of patients with mutations in SECISBP2 based on the finding of reduced selenoprotein expression in SECISBP2-deficient cells in culture. If we just focus on the brain with its many neuronal and glial cell types, we are confronted with perplexing complexity (Zhang et al., 2008). All of these cell types are involved in mechanisms of development, exert a function in the mature organism, and may play are role in neurodegeneration. Thus, it is obvious how wide this field still is and how much expertise is required to address this question.
Another question related to the discussion above, is to what extent mice are valid models for humans with regard to understanding the functions of selenoproteins. The answer will again rely on the comparison of genetic models in mice and patients with congenital defects in selenoprotein genes or genes encoding selenoprotein biosynthesis factors. We can expect that exome-sequencing approaches that are now broadly available will help us identify patients with such mutations. A recent thought-provoking paper has looked at the same question from just the opposite perspective: Instead of searching for mutations in the genes of patients with clinical phenotypes, Santesmasses et al. (2020) searched human genome data for inactivating mutations in selenoprotein genes. They found that humans can carry homozygous inactivating mutations in SELENOO without apparently presenting with a phenotype. SELENOO is a novel mitochondrial protein Ser/Thr-AMP transferase that has not yet been inactivated in mice (Sreelatha et al., 2018).
The question whether the selenoproteome is completely known seemed to have been solved through the landmark paper by Kryukov et al. (2003) who identified genes encoding selenoproteins based in part on the presence of the SECIS element. A recent proteome paper now suggested there are additional Sec-containing proteins with UGA/Sec codons, but lacking recognizable SECIS elements (Guo et al., 2018). This provocative finding is, interestingly, in line with the demonstration of selenoprotein translation in the absence of functional SECISBP2 (Fradejas-Villar et al., 2017; Zhao et al., 2019). If mutations in SEPSECS, unlike mutations in SECISBP2, would also affect selenoproteins that do not depend on a SECIS for biosynthesis, the dichotomy of phenotypes could be explained and some of the non-canonical selenoproteins would likely be important for the brain.
The arguably most dynamic field of selenoprotein research, again related to the neurobiology of Se, is the wider context of the function of GPX4. The whole field of ferroptosis is blossoming. This type of cell death emerges as an important cell biological process on which much hope is placed in the context of cancer treatment and prevention of neurodegenerative disease. Can ferroptosis be modulated pharmacologically to the benefit of patients? Do other pathways related to selenoproteins play a role in these processes? What is the role of lipid peroxidation in this context? In mitochondria? This reminds one of us (US) of a lab rotation in organic chemistry long ago: during his undergraduate study he separated lipid-hydroperoxides and their respective alcohols on a chiral gas-chromatographic column and observed that the peroxides and alcohols were chiral. An enzymatic process seemed the most likely explanation, while biologists argued that spontaneous lipid peroxidation was most likely overinterpreted… Keeping this in mind, who knows what exciting findings there are just around the corner revealing a part of themselves seemingly as oddities or artifacts?
Author Contributions
US wrote the initial draft of the manuscript. All authors refined and rewrote parts of the manuscript and contributed to research associated with the thoughts presented here.
Funding
Recent research on selenoproteins in the authors’ laboratory was supported by the Deutsche Forschungsgemeinschaft DFG (Schw914/2, Schw914/5, and Schw914/6), German Israeli Fund, and Medizinische Fakultät, Rheinische Friedrich-Wilhelms-Universität Bonn.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agamy, O., Ben Zeev, B., Lev, D., Marcus, B., Fine, D., and Su, D. (2010). Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am. J. Hum. Genet. 87, 538–544. doi: 10.1016/j.ajhg.2010.09.007
Ahmed, M. Y., Al-Khayat, A., Al-Murshedi, F., Al-Futaisi, A., Chioza, B. A., and Pedro Fernandez-Murray, J. (2017). A mutation of EPT1 (SELENOI) underlies a new disorder of kennedy pathway phospholipid biosynthesis. Brain 140, 547–554.
Anttonen, A. K., Hilander, T., Linnankivi, T., Isohanni, P., French, R. L., and Liu, Y. (2015). Selenoprotein biosynthesis defect causes progressive encephalopathy with elevated lactate. Neurology 85, 306–315. doi: 10.1212/wnl.0000000000001787
Avery, J. C., Yamazaki, Y., Hoffmann, F. W., Folgelgren, B., and Hoffmann, P. R. (2020). Selenoprotein i is essential for murine embryogenesis. Arch. Biochem. Biophys. 689:108444. doi: 10.1016/j.abb.2020.108444
Aygun, C., Celik, F. C., Nural, M. S., Azak, E., Kucukoduk, S., Ogur, G., et al. (2012). Simplified gyral pattern with cerebellar hypoplasia in Sedaghatian type spondylometaphyseal dysplasia: a clinical report and review of the literature. Am. J. Med. Genet. A 158A, 1400–1405. doi: 10.1002/ajmg.a.35306
Azevedo, M. F., Barra, G. B., Naves, L. A., Ribeiro Velasco, L. F., Godoy Garcia, C. P., and de Castro, L. C. (2010). Rocha, selenoprotein-related disease in a young girl caused by nonsense mutations in the SBP2 gene. J. Clin. Endocrinol. Metab. 95, 4066–4071. doi: 10.1210/jc.2009-2611
Bianco, A. C., and Kim, B. S. (2018). Pathophysiological relevance of deiodinase polymorphism. Curr. Opin. Endocrinol. Diabetes Obes. 25, 341–346. doi: 10.1097/med.0000000000000428
Boukhzar, L., Hamieh, A., Cartier, D., Tanguy, Y., Alsharif, I., and Castex, M. (2016). Selenoprotein t exerts an essential oxidoreductase activity that protects dopaminergic neurons in mouse models of parkinson’s disease. Antioxid. Redox Signal. 24, 557–574. doi: 10.1089/ars.2015.6478
Burk, R. F., and Hill, K. E. (2015). Regulation of selenium metabolism and transport. Annu. Rev. Nutr. 35, 109–134. doi: 10.1146/annurev-nutr-071714-034250
Carlson, B. A., Schweizer, U., Perella, C., Shrimali, R. K., Feigenbaum, L., and Shen, L. (2009). The selenocysteine tRNA STAF-binding region is essential for adequate selenocysteine tRNA status, selenoprotein expression and early age survival of mice. Biochem. J. 418, 61–71. doi: 10.1042/bj20081304
Castets, P., Lescure, A., Guicheney, P., and Allamand, V. (2012). Selenoprotein N in skeletal muscle: from diseases to function. J. Mol. Med. 90, 1095–1107. doi: 10.1007/s00109-012-0896-x
Castex, M. T., Arabo, A., Benard, M., Roy, V., Le Joncour, V., and Prevost, G. (2015). Selenoprotein T deficiency leads to neurodevelopmental abnormalities and hyperactive behavior in mice. Mol. Neurobiol. 53, 5818–5832. doi: 10.1007/s12035-015-9505-7
Chiu-Ugalde, J., Theilig, F., Behrends, T., Drebes, J., Sieland, C., and Subbarayal, P. (2010). Mutation of megalin leads to urinary loss of selenoprotein P and selenium deficiency in serum, liver, kidneys and brain. Biochem. J. 431, 103–111. doi: 10.1042/bj20100779
Chiu-Ugalde, J., Wirth, E. K., Klein, M. O., Sapin, R., Fradejas-Villar, N., and Renko, K. (2012). Thyroid function is maintained despite increased oxidative stress in mice lacking selenoprotein biosynthesis in thyroid epithelial cells. Antioxid. Redox Signal. 17, 902–913. doi: 10.1089/ars.2011.4055
Conrad, M., Jakupoglu, C., Moreno, S. G., Lippl, S., Banjac, A., and Schneider, M. (2004). Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol. Cell. Biol. 24, 9414–9423. doi: 10.1128/mcb.24.21.9414-9423.2004
Curran, J. E., Jowett, J. B., Elliott, K. S., Gao, Y., Gluschenko, K., and Wang, J. (2005). Genetic variation in selenoprotein S influences inflammatory response. Nat. Genet. 37, 1234–1241. doi: 10.1038/ng1655
Di Cosmo, C., McLellan, N., Liao, X. H., Khanna, K. K., Weiss, R. E., Papp, L., et al. (2009). Clinical and molecular characterization of a novel selenocysteine insertion sequence-binding protein 2 (SBP2) gene mutation (R128X). J. Clin. Endocrinol. Metab. 94, 4003–4009. doi: 10.1210/jc.2009-0686
Dumitrescu, A. M., Liao, X. H., Abdullah, M. S., Lado-Abeal, J., Majed, F. A., and Moeller, L. C. (2005). Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat. Genet. 37, 1247–1252. doi: 10.1038/ng1654
Fomenko, D. E., Novoselov, S. V., Natarajan, S. K., Lee, B. C., Koc, A., and Carlson, B. A. (2008). Methionine-R-sulfoxide reductase 1 (MsrB1) knockout mice: roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J. Biol. Chem. 284, 5986–5993. doi: 10.1074/jbc.m805770200
Fradejas-Villar, N. (2018). Consequences of mutations and inborn errors of selenoprotein biosynthesis and functions. Free Radic. Biol. Med. 127, 206–214. doi: 10.1016/j.freeradbiomed.2018.04.572
Fradejas-Villar, N., Seeher, S., Anderson, C. B., Doengi, M., Carlson, B. A., and Hatfield, D. L. (2017). The RNA-binding protein Secisbp2 differentially modulates UGA codon reassignment and RNA decay. Nucleic Acids Res. 45, 4094–4107. doi: 10.1093/nar/gkw1255
Friedmann Angeli, J. P., Krysko, D. V., and Conrad, M. (2019). Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 19, 405–414. doi: 10.1038/s41568-019-0149-1
Ge, K., Xue, A., Bai, J., and Wang, S. (1983). Keshan disease-an endemic cardiomyopathy in China. Virchows Arch A Pathol. Anat. Histopathol. 401, 1–15. doi: 10.1007/bf00644785
Gladyshev, V. N., Arner, E. S., Berry, M. J., Brigelius-Flohe, R., Bruford, E. A., and Burk, R. F. (2016). Selenoprotein gene nomenclature. J. Biol. Chem. 291, 24036–24040.
Guo, L., Yang, W., Huang, Q., Qiang, J., Hart, J. R., and Wang, W. (2018). Selenocysteine-specific mass spectrometry reveals tissue-distinct selenoproteomes and candidate selenoproteins. Cell Chem. Biol. 25, 1380–1388.e4.
Hambright, W. S., Fonseca, R. S., Chen, L., Na, R., and Ran, Q. (2017). Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 12, 8–17. doi: 10.1016/j.redox.2017.01.021
Hamieh, A., Cartier, D., Abid, H., Calas, A., Burel, C., and Bucharles, C. (2017). Selenoprotein T is a novel OST subunit that regulates UPR signaling and hormone secretion. EMBO Rep. 18, 1935–1946. doi: 10.15252/embr.201643504
Hill, K. E., Wu, S., Motley, A. K., Stevenson, T. D., Winfrey, V. P., and Capecchi, M. R. (2012). Production of selenoprotein P (Sepp1) by hepatocytes is central to selenium homeostasis. J. Biol. Chem. 287, 40414–40424. doi: 10.1074/jbc.m112.421404
Hill, K. E., Zhou, J., McMahan, W. J., Motley, A. K., and Burk, R. F. (2004). Neurological dysfunction occurs in mice with targeted deletion of the selenoprotein p gene. J. Nutr. 134, 157–161. doi: 10.1093/jn/134.1.157
Hill, K. E., Zhou, J., McMahan, W. J., Motley, A. K., Atkins, J. F., and Gesteland, R. F. (2003). Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 278, 13640–13646. doi: 10.1074/jbc.m300755200
Horibata, Y., Elpeleg, O., Eran, A., Hirabayashi, Y., Savitzki, D., and Tal, G. (2018). EPT1 (selenoprotein I) is critical for the neural development and maintenance of plasmalogen in humans. J. Lipid Res. 59, 1015–1026. doi: 10.1194/jlr.p081620
Iwama, K., Sasaki, M., Hirabayashi, S., Ohba, C., Iwabuchi, E., and Miyatake, S. (2016). Milder progressive cerebellar atrophy caused by biallelic SEPSECS mutations. J. Hum. Genet. 61, 527–531. doi: 10.1038/jhg.2016.9
Jakupoglu, C., Przemeck, G. K., Schneider, M., Moreno, S. G., Mayr, N., and Hatzopoulos, A. (2005). Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol. Cell. Biol. 25, 1980–1988. doi: 10.1128/mcb.25.5.1980-1988.2005
Kasaikina, M. V., Fomenko, D. E., Labunskyy, V. M., Lachke, S. A., Qiu, W., and Moncaster, J. A. (2011). Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J. Biol. Chem. 286, 33203–33212. doi: 10.1074/jbc.m111.259218
Kasaikina, M. V., Turanov, A. A., Avanesov, A., Schweizer, U., Seeher, S., and Bronson, R. T. (2013). Contrasting roles of dietary selenium and selenoproteins in chemically induced hepatocarcinogenesis. Carcinogenesis 34, 1089–1095. doi: 10.1093/carcin/bgt011
Köhrle, J., Jakob, F., Contempre, B., and Dumont, J. E. (2005). Selenium, the thyroid, and the endocrine system. Endocr. Rev. 26, 944–984. doi: 10.1210/er.2001-0034
Kryukov, G. V., Castellano, S., Novoselov, S. V., Lobanov, A. V., Zehtab, O., Guigo, R., et al. (2003). Characterization of mammalian selenoproteomes. Science 300, 1439–1443. doi: 10.1126/science.1083516
Kudin, A. P., Baron, G., Zsurka, G., Hampel, K. G., Elger, C. E., and Grote, A. (2017). Homozygous mutation in TXNRD1 is associated with genetic generalized epilepsy. Free Radic. Biol. Med. 106, 270–277. doi: 10.1016/j.freeradbiomed.2017.02.040
Lee, B. C., Peterfi, Z., Hoffmann, F. W., Moore, R. E., Kaya, A., and Avanesov, A. (2013). MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol. Cell. 51, 397–404. doi: 10.1016/j.molcel.2013.06.019
Loscalzo, J. (2014). Keshan disease, selenium deficiency, and the selenoproteome. N. Engl. J. Med. 370, 1756–1760. doi: 10.1056/nejmcibr1402199
Mahdi, Y., Xu, X. M., Carlson, B. A., Fradejas, N., Gunter, P., and Braun, D. (2015). Expression of selenoproteins is maintained in mice carrying mutations in SECp43, the tRNA selenocysteine 1 associated protein (Trnau1ap). PLoS One 10:e0127349. doi: 10.1371/journal.pone.0127349
McAninch, E. A., Jo, S., Preite, N. Z., Farkas, E., Mohacsik, P., and Fekete, C. (2015). Prevalent polymorphism in thyroid hormone-activating enzyme leaves a genetic fingerprint that underlies associated clinical syndromes. J. Clin. Endocrinol. Metab. 100, 920–933. doi: 10.1210/jc.2014-4092
Moghadaszadeh, B., Petit, N., Jaillard, C., Brockington, M., Roy, S. Q., and Merlini, L. (2001). Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat. Genet. 29, 17–18. doi: 10.1038/ng713
Mondal, S., Raja, K., Schweizer, U., and Mugesh, G. (2016). Chemistry and biology in the biosynthesis and action of thyroid hormones. Angew. Chem. Int. Ed. Engl. 55, 7606–7630. doi: 10.1002/anie.201601116
Muth, O. H., Oldfield, J. E., Remmert, L. F., and Schubert, J. R. (1958). Effects of selenium and vitamin E on white muscle disease. Science 128:1090. doi: 10.1126/science.128.3331.1090
Olson, G. E., Winfrey, V. P., Hill, K. E., and Burk, R. F. (2008). Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J. Biol. Chem. 283, 6854–6860. doi: 10.1074/jbc.m709945200
Olson, G. E., Winfrey, V. P., Nagdas, S. K., Hill, K. E., and Burk, R. F. (2007). Apolipoprotein E receptor-2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. J. Biol. Chem. 282, 12290–12297. doi: 10.1074/jbc.m611403200
Pitts, M. W., Raman, A. V., Hashimoto, A. C., Todorovic, C., Nichols, R. A., and Berry, M. J. (2012). Deletion of selenoprotein P results in impaired function of parvalbumin interneurons and alterations in fear learning and sensorimotor gating. Neuroscience 208, 58–68. doi: 10.1016/j.neuroscience.2012.02.017
Prasad, R., Chan, L. F., Hughes, C. R., Kaski, J. P., Kowalczyk, J. C., and Savage, M. O. (2014). Thioredoxin Reductase 2 (TXNRD2) mutation associated with familial glucocorticoid deficiency (FGD). J. Clin. Endocrinol. Metab. 99, E1556–E1563.
Rederstorff, M., Castets, P., Arbogast, S., Laine, J., Vassilopoulos, S., and Beuvin, M. (2011). Increased muscle stress-sensitivity induced by selenoprotein N inactivation in mouse: a mammalian model for SEPN1-related myopathy. PLoS One 6:e23094. doi: 10.1371/journal.pone.0023094
Renko, K., Werner, M., Renner-Muller, I., Cooper, T. G., Yeung, C. H., and Hollenbach, B. (2008). Hepatic selenoprotein P (SePP) expression restores selenium transport and prevents infertility and motor-incoordination in sepp-knockout mice. Biochem. J. 409, 741–749. doi: 10.1042/bj20071172
Santesmasses, D., Mariotti, M., and Gladyshev, V. N. (2020). Tolerance to selenoprotein loss differs between human and mouse. Mol. Biol. Evol. 37, 341–354. doi: 10.1093/molbev/msz218
Scharpf, M., Schweizer, U., Arzberger, T., Roggendorf, W., Schomburg, L., and Köhrle, J. (2007). Neuronal and ependymal expression of selenoprotein P in the human brain. J. Neural. Transm. 114, 877–884. doi: 10.1007/s00702-006-0617-0
Schneider, M. J., Fiering, S. N., Pallud, S. E., Parlow, A. F., St Germain, D. L., and Galton, V. A. (2001). Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol. Endocrinol. 15, 2137–2148. doi: 10.1210/mend.15.12.0740
Schneider, M. J., Fiering, S. N., Thai, B., Wu, S. Y., St Germain, E., and Parlow, A. F. (2006). Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology 147, 580–589. doi: 10.1210/en.2005-0739
Schoenmakers, E., Agostini, M., Mitchell, C., Schoenmakers, N., Papp, L., and Rajanayagam, O. (2010). Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J. Clin. Invest. 120, 4220–4235. doi: 10.1172/jci43653
Schoenmakers, E., and Chatterjee, K. (2018). Identification of genetic disorders causing disruption of selenoprotein biosynthesis. Methods Mol. Biol. 1661, 325–335. doi: 10.1007/978-1-4939-7258-6_23
Schoenmakers, E., and Chatterjee, K. (2020). Human disorders affecting the selenocysteine incorporation pathway cause systemic selenoprotein deficiency. Antioxid. Redox Signal. 33, 481–497. doi: 10.1089/ars.2020.8097
Schoenmakers, E., Carlson, B., Agostini, M., Moran, C., Rajanayagam, O., and Bochukova, E. (2016). Mutation in human selenocysteine transfer RNA selectively disrupts selenoprotein synthesis. J. Clin. Invest. 126, 992–996. doi: 10.1172/jci84747
Schomburg, L., Schweizer, U., Holtmann, B., Flohé, L., Sendtner, M., and Köhrle, J. (2003). Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 370, 397–402. doi: 10.1042/bj20021853
Schweizer, U. (2016). “Selenoproteins in nervous system development, function, and degeneration,” in Selenium. its Molecular Biology and Role in Human Health, eds D. L. S. Hatfield, P. A. Tsuji, and V. N. Gladyshev (Berlin: Springer).
Schweizer, U., and Fradejas-Villar, N. (2016). Why 21? The significance of selenoproteins for human health revealed by inborn errors of metabolism. FASEB J. 30, 3669–3681. doi: 10.1096/fj.201600424
Schweizer, U., and Schomburg, L. (2006). “Selenium, selenoproteins, and brain function,” in Selenium: its Molecular Biology and Role in Human Health, eds D. L. Hatfield, M. J. Berry, and V. N. Gladyshev (New York: Springer), 233–248. doi: 10.1007/0-387-33827-6_21
Schweizer, U., Bräuer, A. U., Köhrle, J., Nitsch, R., and Savaskan, N. E. (2004a). Selenium and brain function: a poorly recognized liaison. Brain Res. Brain Res. Rev. 45, 164–178. doi: 10.1016/j.brainresrev.2004.03.004
Schweizer, U., Michaelis, M., Köhrle, J., and Schomburg, L. (2004b). Efficient selenium transfer from mother to offspring in selenoprotein-P-deficient mice enables dose-dependent rescue of phenotypes associated with selenium deficiency. Biochem. J. 378, 21–26. doi: 10.1042/bj20031795
Schweizer, U., Schomburg, L., and Savaskan, N. E. (2004c). The neurobiology of selenium: lessons from transgenic mice. J. Nutr. 134, 707–710. doi: 10.1093/jn/134.4.707
Schweizer, U., Streckfuss, F., Pelt, P., Carlson, B. A., Hatfield, D. L., Köhrle, J., et al. (2005). Hepatically derived selenoprotein P is a key factor for kidney but not for brain selenium supply. Biochem. J. 386, 221–226. doi: 10.1042/bj20041973
Seeher, S., Carlson, B. A., Miniard, A. C., Wirth, E. K., Mahdi, Y., Hatfield, D. L., et al. (2014). Impaired selenoprotein expression in brain triggers striatal neuronal loss leading to co-ordination defects in mice. Biochem. J. 462, 67–75. doi: 10.1042/bj20140423
Seiler, A., Schneider, M., Forster, H., Roth, S., Wirth, E. K., and Culmsee, C. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8, 237–248. doi: 10.1016/j.cmet.2008.07.005
Sibbing, D., Pfeufer, A., Perisic, T., Mannes, A. M., Fritz-Wolf, K., and Unwin, S. (2011). Mutations in the mitochondrial thioredoxin reductase gene TXNRD2 cause dilated cardiomyopathy. Eur. Heart J. 32, 1121–1133. doi: 10.1093/eurheartj/ehq507
Smith, A. C., Mears, A. J., Bunker, R., Ahmed, A., MacKenzie, M., and Schwartzentruber, J. A. (2014). Mutations in the enzyme glutathione peroxidase 4 cause sedaghatian-type spondylometaphyseal dysplasia. J. Med. Genet. 51, 470–474. doi: 10.1136/jmedgenet-2013-102218
Soerensen, J., Jakupoglu, C., Beck, H., Forster, H., Schmidt, J., and Schmahl, W. (2008). The role of thioredoxin reductases in brain development. PLoS One 3:e1813. doi: 10.1371/journal.pone.0001813
Sreelatha, A., Yee, S. S., Lopez, V. A., Park, B. C., Kinch, L. N., and Pilch, S. (2018). Protein AMPylation by an evolutionarily conserved pseudokinase. Cell 175, 809–821.e19.
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Valentine, W. M., Abel, T. W., Hill, K. E., Austin, L. M., and Burk, R. F. (2008). Neurodegeneration in mice resulting from loss of functional selenoprotein P or its receptor apolipoprotein E receptor 2. J. Neuropathol. Exp. Neurol. 67, 68–77. doi: 10.1097/nen.0b013e318160f347
Valentine, W. M., Hill, K. E., Austin, L. M., Valentine, H. L., Goldowitz, D., and Burk, R. F. (2005). Brainstem axonal degeneration in mice with deletion of selenoprotein p. Toxicol. Pathol. 33, 570–576. doi: 10.1080/01926230500243045
Villar-Quiles, R. N., von, der Hagen, M., Metay, C., Gonzalez, V., Donkervoort, S., et al. (2020). The clinical, histologic, and genotypic spectrum of SEPN1-related myopathy: a case series. Neurology 95, e1512–e1527.
Wirth, E. K., Bharathi, B. S., Hatfield, D., Conrad, M., Brielmeier, M., and Schweizer, U. (2014). Cerebellar hypoplasia in mice lacking selenoprotein biosynthesis in neurons. Biol. Trace Elem. Res. 158, 203–210. doi: 10.1007/s12011-014-9920-z
Wirth, E. K., Conrad, M., Winterer, J., Wozny, C., Carlson, B. A., and Roth, S. (2010). Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J. 24, 844–852. doi: 10.1096/fj.09-143974
Yant, L. J., Ran, Q., Rao, L., Van Remmen, H., Shibatani, T., and Belter, J. G. (2003). The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 34, 496–502. doi: 10.1016/s0891-5849(02)01360-6
Zhang, Y., Zhou, Y., Schweizer, U., Savaskan, N. E., Hua, D., Kipnis, J., et al. (2008). Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J. Biol. Chem. 283, 2427–2438. doi: 10.1074/jbc.m707951200
Zhang, Z. H., Chen, C., Jia, S. Z., Cao, X. C., Liu, M., and Tian, J. (2020). Selenium restores synaptic deficits by modulating NMDA receptors and selenoprotein K in an Alzheimer’s disease model. Antioxid. Redox Signal. doi: 10.1089/ars.2019.7990 Online ahead of print.
Keywords: genetics, neurodegeneration, GPX4, ferroptosis, epilepsy
Citation: Schweizer U, Bohleber S, Zhao W and Fradejas-Villar N (2021) The Neurobiology of Selenium: Looking Back and to the Future. Front. Neurosci. 15:652099. doi: 10.3389/fnins.2021.652099
Received: 11 January 2021; Accepted: 08 February 2021;
Published: 25 February 2021.
Edited by:
Peter R. Hoffmann, University of Hawai’i, United StatesReviewed by:
Arturo Hernandez, Maine Medical Center, United StatesMargaret Rayman, University of Surrey, United Kingdom
Copyright © 2021 Schweizer, Bohleber, Zhao and Fradejas-Villar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ulrich Schweizer, VWxyaWNoLnNjaHdlaXplckB1bmktYm9ubi5kZQ==; dXNjaHdlaXpAdW5pLWJvbm4uZGU=