 Melissa Talita Wiprich1,2
Melissa Talita Wiprich1,2 Carla Denise Bonan1,2,3*
Carla Denise Bonan1,2,3*- 1Programa de Pós-Graduação em Medicina e Ciências da Saúde, Escola de Medicina, Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, Brazil
- 2Laboratório de Neuroquímica e Psicofarmacologia, Escola de Ciências da Saúde e da Vida, Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, Brazil
- 3Instituto Nacional de Ciência e Tecnologia em Doenças Cerebrais, Excitotoxicidade e Neuroproteção, Porto Alegre, Brazil
Huntington’s disease (HD) is a devastating, progressive, and fatal neurodegenerative disorder inherited in an autosomal dominant manner. This condition is characterized by motor dysfunction (chorea in the early stage, followed by bradykinesia, dystonia, and motor incoordination in the late stage), psychiatric disturbance, and cognitive decline. The neuropathological hallmark of HD is the pronounced neuronal loss in the striatum (caudate nucleus and putamen). The striatum is related to the movement control, flexibility, motivation, and learning and the purinergic signaling has an important role in the control of these events. Purinergic signaling involves the actions of purine nucleotides and nucleosides through the activation of P2 and P1 receptors, respectively. Extracellular nucleotide and nucleoside-metabolizing enzymes control the levels of these messengers, modulating the purinergic signaling. The striatum has a high expression of adenosine A2A receptors, which are involved in the neurodegeneration observed in HD. The P2X7 and P2Y2 receptors may also play a role in the pathophysiology of HD. Interestingly, nucleotide and nucleoside levels may be altered in HD animal models and humans with HD. This review presents several studies describing the relationship between purinergic signaling and HD, as well as the use of purinoceptors as pharmacological targets and biomarkers for this neurodegenerative disorder.
Introduction
Huntington’s disease (HD) is a devastating, progressive, and fatal neurodegenerative disorder inherited in an autosomal dominant manner (Smith-Dijak et al., 2019; Blumenstock and Dudanova, 2020). It is triggered by an expansion of a cytosine-adenine-guanine (CAG) triplet repeat in exon 1 of the huntingtin (HTT) gene, located on chromosome 4 (The Huntington’s Disease Collaborative Research Group, 1993; Capiluppi et al., 2020). This change leads to an expanded polyglutamine (polyQ) region in the encoded HTT protein (Bailus et al., 2017; Rai et al., 2019). As a result, the expressed HTT protein is a mutant (mHTT; Cybulska et al., 2020). Individuals with up to 35 CAG repeats are usually considered healthy, while people with 36 to 39 CAG repeats may or may not develop the signs and symptoms of HD (Shoulson and Young, 2011; Capiluppi et al., 2020). More than 50 CAG repeats always cause the disease (Capiluppi et al., 2020). There is an inverse correlation between the number of CAG repeats, age at onset, and the severity of HD symptoms (Bates et al., 2015; Petersén and Weydt, 2019).
It is estimated that the mean HD prevalence is 5 in 100,000 people (Baig et al., 2016; Illarioshkin et al., 2018). HD is characterized by a neurobehavioral progressive triad with motor dysfunction, psychiatric disturbance, and cognitive decline (Stahl and Feigin, 2020). The motor dysfunction is subdivided into two stages: In the early stage, there are abnormal involuntary movements, known as chorea, while in the late stage, the voluntary movements are impaired, causing bradykinesia, dystonia, and motor incoordination. The observed neuropsychiatric symptoms include depression, apathy, irritability, anxiety, and psychosis. The cognitive impairment often precedes the motor abnormalities. The cognitive alterations include impaired attention and visuospatial functions and slow planning processing speed. The cognitive decline progresses to dementia (Stahl and Feigin, 2020), and death becomes imminent 15–20 years after disease onset (Blumenstock and Dudanova, 2020). These dysfunctions can be attributed to multiple brain regions that exhibit neurodegeneration, including the cerebral cortex, thalamus, subthalamic nucleus, globus pallidus, substantia nigra, and hypothalamus. However, the hallmark of the disease is the pronounced neuronal loss in the striatum (caudate nucleus and putamen; Rubinsztein, 2002; Ramaswamy et al., 2007; Coppen and Roos, 2017). Furthermore, HD patients may develop metabolic symptoms including weight loss and cardiac and musculoskeletal dysfunction, among others (Blum et al., 2018; Croce and Yamamoto, 2019; Dufour and McBride, 2019).
The striatum is a region responsible for the control of many behaviors, such as movement, flexibility behavior, motivation, and learning (Koch and Raymond, 2019). Two different striatal pathways express distinct neurotransmitters and neuropeptides (Graybiel, 2000). The indirect pathway contains cholinergic interneurons that express dopamine D2 receptors (D2R), adenosine A2A receptors (A2AR), and enkephalin; it projects to the globus pallidus external (Figure 1; GPe; Albin et al., 1989). This pathway acts by inhibiting voluntary movements; because the neurons are degenerated in the early stage of HD, there is a decrease in D2R and A2AR and thus uncontrolled voluntary movements, coinciding with chorea symptoms (Figure 1; Albin et al., 1989; Graybiel, 2000; Koch and Raymond, 2019). The direct pathway expresses spiny projection neurons (SPNs) that contain gamma-aminobutyric acid (GABA) coexisting with neuropeptide P and dynorphin. Besides, dopamine D1 receptors (D1R) project into the substantia nigra pars reticulate (SNpr) and globus pallidus internal (GPi), initiating voluntary movements (Figure 1; Albin et al., 1989). In the late stage of HD, besides the damaged indirect pathway, there is the degeneration of direct pathway neurons, a phenomenon that decreases D1R and cortex stimulation. This phenomenon leads to the hypokinetic symptoms, which are typical of this stage (Figure 1; Albin et al., 1989; Graybiel, 2000; Koch and Raymond, 2019).
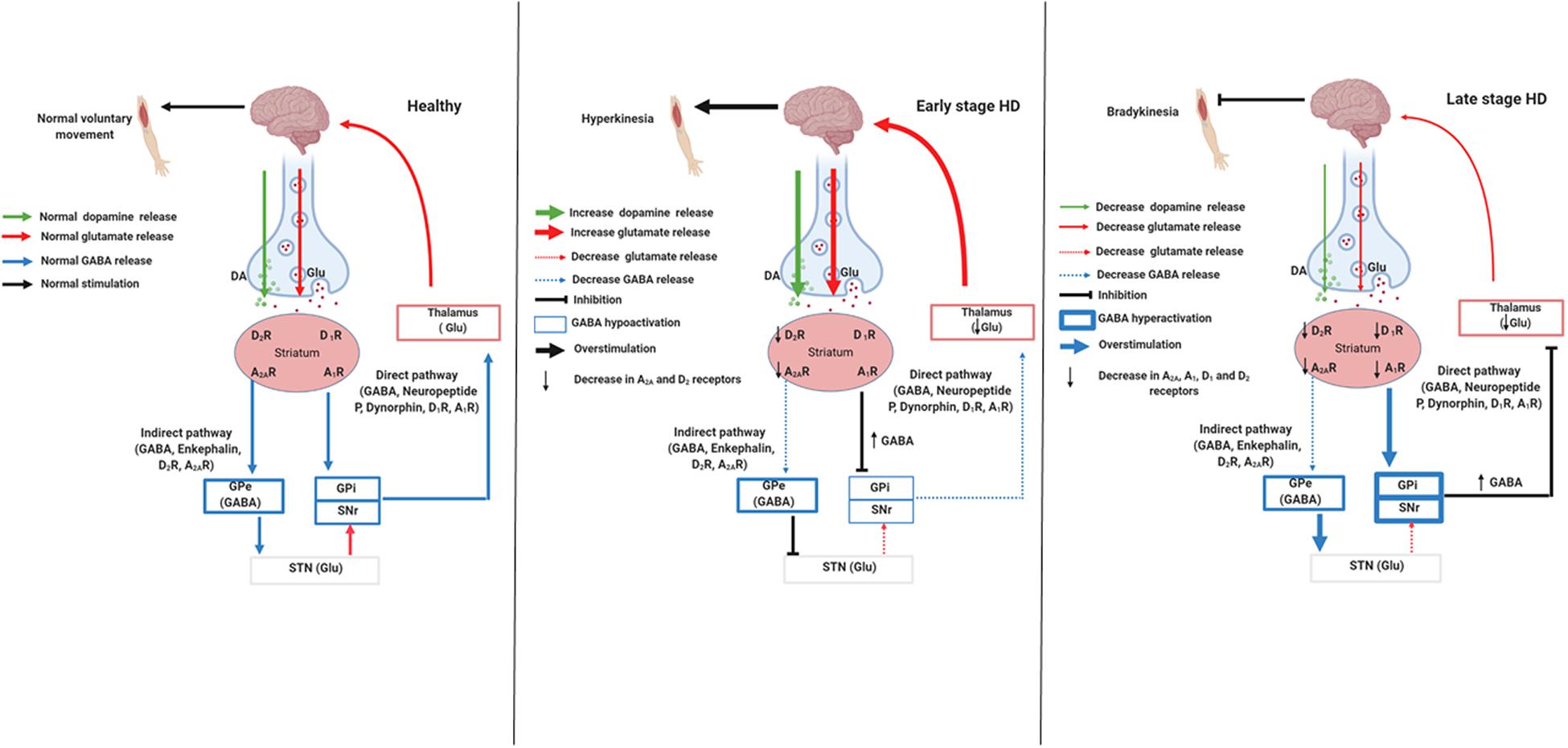
Figure 1. Signaling pathways involved in stages of Huntington’s disease. Created with Biorender.com.
Furthermore, neurotransmitters, such as dopamine, acetylcholine, glutamate, and GABA, are involved in motor coordination and alterations in their levels induce motor deficits. Evidence has demonstrated alterations in these neurotransmitter levels in early- and late-stage HD (Spokes, 1980; Kish et al., 1987; Jamwal et al., 2015; Jamwal and Kumar, 2019). These changes in neurotransmitter levels might cause important intracellular biochemical changes, such as a decrease in mitochondrial complex II, III, and IV activity and adenosine triphosphate (ATP) levels, calcium (Ca2+) overload, excitotoxicity, oxidative stress, and mitochondrial dysfunction (Johri et al., 2013; Carmo et al., 2018; Jodeiri Farshbaf and Kiani-Esfahani, 2018), triggering cell death (Liot et al., 2017). Thus, there is an imbalance in the activity between the direct and indirect pathways, resulting in an inadequate functioning of different neurotransmitter systems in HD. One of the neurotransmitter systems involved in the pathophysiology of HD is the purinergic signaling (Burnstock, 2015), mediated by the action of nucleotides and nucleosides in the P2 and P1 receptors, respectively. Both ATP and adenosine are the most important messengers in the purinergic system, which participates in the control of several behaviors (Burnstock, 2015). Adenosine acts as a neuromodulator; specifically, it modulates dopaminergic and glutamatergic neurotransmission systems (Ferré et al., 2007; Fuxe et al., 2007; Ciruela et al., 2015). Changes in ATP and adenosine levels have been observed in HD (Seong et al., 2005; Kao et al., 2017). Studies have focused on the impact of purinergic signaling on HD as well as the development of pharmacological strategies related to the purinergic system as therapies for HD (Blum et al., 2002; Chou et al., 2005; Simonin et al., 2013; Villar-Menéndez et al., 2013; Kao et al., 2017). Therefore, this review will discuss the role of purinergic signaling in HD as well as the involvement of purinoceptors in the disease progression and their relevance for application as pharmacological targets and biomarkers for HD.
Purinergic Signaling
Adenosine triphosphate and adenosine are recognized as the most powerful purinergic signaling messengers (Burnstock, 1972). Purinergic receptors are classified into P1 and P2 according to their biochemical and pharmacological properties (Burnstock, 2018; Cheffer et al., 2018). P2 receptors are activated by purines [ATP, adenosine diphosphate (ADP)] and pyrimidines (uridine triphosphate, uridine diphosphate) and classified as P2X and P2Y receptors (Abbracchio and Burnstock, 1994; Burnstock, 2011). P2X receptors are ATP-gated ion channels permeable to sodium (Na+) and Ca2+ influx and potassium (K+) efflux, which leads to depolarization of the cell membrane. Seven subunits of these receptors (P2X1–7) are expressed by different cells (Burnstock, 2008). P2Y receptors are metabotropic, activated by purines and pyrimidines, and subdivided into eight receptor subtypes (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, and P2Y14; Burnstock, 2008; Puchałowicz et al., 2014). P1 receptors are metabotropic, selective for adenosine, and exert physiological actions through four subtypes named A1 (A1R), A2A (A2AR), A2B (A2BR), and A3 (A3R) (Fredholm et al., 2001; Burnstock, 2018; Ciruela, 2020). Low adenosine levels activate A1R and A3R receptors, whereas high adenosine levels activate A2AR and A2BR receptors (Borea et al., 2018). A1R and A3R activate Gi/o protein, inhibiting the production of cyclic adenosine monophosphate (cAMP), adenylate cyclase (AC), protein kinase A (PKA), and, consequently, GABA uptake. On the other hand, A2AR is activated through Gs protein that stimulates cAMP production, activating AC and PKA, increasing GABA uptake. In addition, A2BR also act through Gs protein (Sheth et al., 2014; Borea et al., 2018).
Nucleotide levels are regulated by ectonucleotidases, a group of enzymes constituted by nucleotide pyrophosphatases/phosphodiesterases (NPPs), nucleoside triphosphate diphosphohydrolases (NTPDases; CD39), alkaline phosphatase, and ecto-5′-nucleotidase (5′-NT, CD73; Bonan, 2012; Zimmermann, 2021). Ectonucleotidases promote the extracellular hydrolysis of ATP, producing ADP, adenosine monophosphate (AMP), and adenosine controlling their extracellular concentrations (Burnstock, 1980; Bonan, 2012; Cheffer et al., 2018; Cieslak et al., 2019).
Adenosine, through the action of adenosine deaminase (ADA), can be subsequently deaminated to inosine (Latini and Pedata, 2001). Inosine is phosphorylated by purine nucleoside phosphorylase (PNP) into hypoxanthine and then degraded to the stable end product uric acid (Yegutkin, 2008; Ribeiro et al., 2016). Adenosine levels are also regulated by unidirectional and bidirectional transporters, which allow nucleosides to move between the intracellular and extracellular compartments (Fredholm et al., 2001; Ribeiro et al., 2016; Stockwell et al., 2017). Finally, the action of nucleosides is then limited either through their conversion to other products of purine catabolism or through re-synthesis into nucleotides (Ribeiro et al., 2016).
The Role of P1 Receptors in Huntington’s Disease
As mentioned above, adenosine mediates its effect through four adenosine receptors (A1, A2A, A2B, and A3). A1R and A2AR are mainly involved in the central effects of adenosine (Ciruela et al., 2015). A1R is the most abundant receptor in the brain–widely expressed in the hippocampus, cerebellum, thalamus, brain stem, and spinal cord–whereas A2AR is concentrated abundantly in the striatum (Burnstock, 2008; Borea et al., 2018).
It is known that A1R can interact with D1R to form heterodimers. Pre-synaptic A1R activation causes depression of excitatory transmission through Ca2+ channel inhibition and neuronal hyperpolarization by regulation of potassium channel. This action leads to a reduction in the release of many neurotransmitters, including acetylcholine, glutamate, dopamine, noradrenaline, and serotonin; this reduction may be beneficial in some central nervous system diseases (Yoon and Rothman, 1991; Von Lubitz et al., 1994; Gundlfinger et al., 2007).
A2A receptors can co-localize with D2R, resulting in A2AR/D2R heteromers, which have a crucial role in the modulation of motor function. Thus, A2AR activation decreases the affinity and function of D2R for agonist or antagonist drugs (Borea et al., 2018). In addition, A2AR plays an important role in facilitating glutamate release, potentiating their effects via N-methyl-D-aspartate (NMDA) receptors as well as other neurotransmitters, such as GABA, glycine, acetylcholine, noradrenaline, and serotonin (Cunha, 2005; León-Navarro et al., 2018). In addition, there is functional cooperation between A2AR and A1R (heteromers), leading to antagonist actions on dopamine release–that is, when A1R is stimulated it inhibits dopamine release, which would oppose the stimulating effects of A2AR through action on striatal D2R (León-Navarro et al., 2018). A2AR can also form heterodimers with glutamate receptors (metabotropic 5 subtype [mGlu5]). The A2AR/mGlu5 heterodimers exert a synergistic inhibitory effect on dopamine binding to D2R (León-Navarro et al., 2018). Together, these findings lead to the hypothesis that dysfunction in adenosine receptors may cause an imbalance between dopaminergic, glutamatergic, and GABAergic neurotransmission systems, which would explain pathological processes underlying HD. This hypothesis is supported by several studies that focus on adenosine receptors, mainly A2AR, and to a lesser extent A1R. Here we described the involvement of A1R and A2AR in the progression of HD.
A1R in Huntington’s Disease
The first study investigating the involvement of P1 receptors in HD was focused on the analysis of the A1R subtype in the striatum and parietal–frontal culture of post-mortem cerebral samples from HD subjects. Using a selective A1R agonist–[3N] (N6-cyclohexyl)-adenosine–the authors demonstrated a 60% decrease in A1R with an increase in the affinity of the binding drug for the receptor in the striatum (Whitehouse et al., 1985). Later, Matusch et al. (2014) quantified, through position emission tomography (PET) imaging, the cerebral binding of A1R in HD subjects subdivided into four groups: pre-manifest individuals far from (pre-HD-A) or pre-manifest individuals near (pre-HD-B) the predicted symptom onset, manifest HD patients, and controls. The results demonstrated a decrease in caudate and putaminal volumes from pre-HD and HD patients compared with control, more A1R in the thalamus of pre-HD-A individuals compared with control, and less A1R in caudate and amygdala in all stages of the disease. There was also a strong direct correlation between A1R with the years since disease onset–that is, the more advanced the disease, the larger the loss of A1R (Matusch et al., 2014). This finding indicates that A1R is involved in the pathogenesis of HD and might be a biomarker in specific brain areas for HD progression.
To shed further light on the possible role of A1R in HD, pharmacological and genetic mouse models have been used to understand the functionality of these receptors. In a pharmacological model of HD induced by 3-nitropropionic acid (3-NPA)–the main toxin used to induce an HD-like phenotype in an animal model through mitochondrial inhibition–the acute (two or three injections) and chronic (8 days) treatment with A1R agonist adenosine amine congener (ADAC) provoked different behavioral and neurochemical responses in mice (Blum et al., 2002). In the study, animals that received two injections of ADAC were called ADAC4/5, whereas animals that received three injections were called ADAC3/5. ADAC4/5 and ADAC3/5 treatment attenuated dystonia of hindlimbs caused by 3-NPA, while the ADAC4/5 group showed increased striatal succinate dehydrogenase (SDH) activity inhibited by 3-NPA and a reduced striatal lesion volume (Blum et al., 2002). Chronic ADAC treatment did not alter the motor symptoms or striatal lesion size, and there was no increase in the SDH activity (Blum et al., 2002). Interestingly, chronic treatment dramatically decreased the A1R density in the striatum and cortex, whereas acute treatment did not modify the A1R density (Blum et al., 2002).
Consistently, in R6/2 mice–the most widely used transgenic model of HD–A1R was significantly reduced in the cortex and striatum compared with the wild type (WT) group (Ferrante et al., 2014). Moreover, corticostriatal slices of R6/2 treated with the selective A1R agonist cyclopentyladenosine (CPA) showed a marked reduction in synaptic transmission, with consequent inhibition of glutamate release in the pre-synaptic terminal (Ferrante et al., 2014). On the contrary, in a pharmacological model of HD induced by malonate, Alfinito et al. (2003) observed that intraperitoneal and intrastriatal A1R blockade with the A1R antagonist CPX potentiated the effect of malonate, reducing striatal dopamine levels, tyrosine hydroxylase (TH) content, and GABA levels as well as GABAergic and dopaminergic neuronal loss (Alfinito et al., 2003). Together, these findings lead to the hypothesis that the pharmacological therapy with A1R agonists may be a beneficial protective treatment in the early stage of HD (Figure 2). However, A1R antagonist drugs did not seem to be an alternative for the treatment of HD due to the crosstalk with GABAergic neurons, which enhanced the susceptibility to toxic insults, reducing A1R in these two populations of neurons.
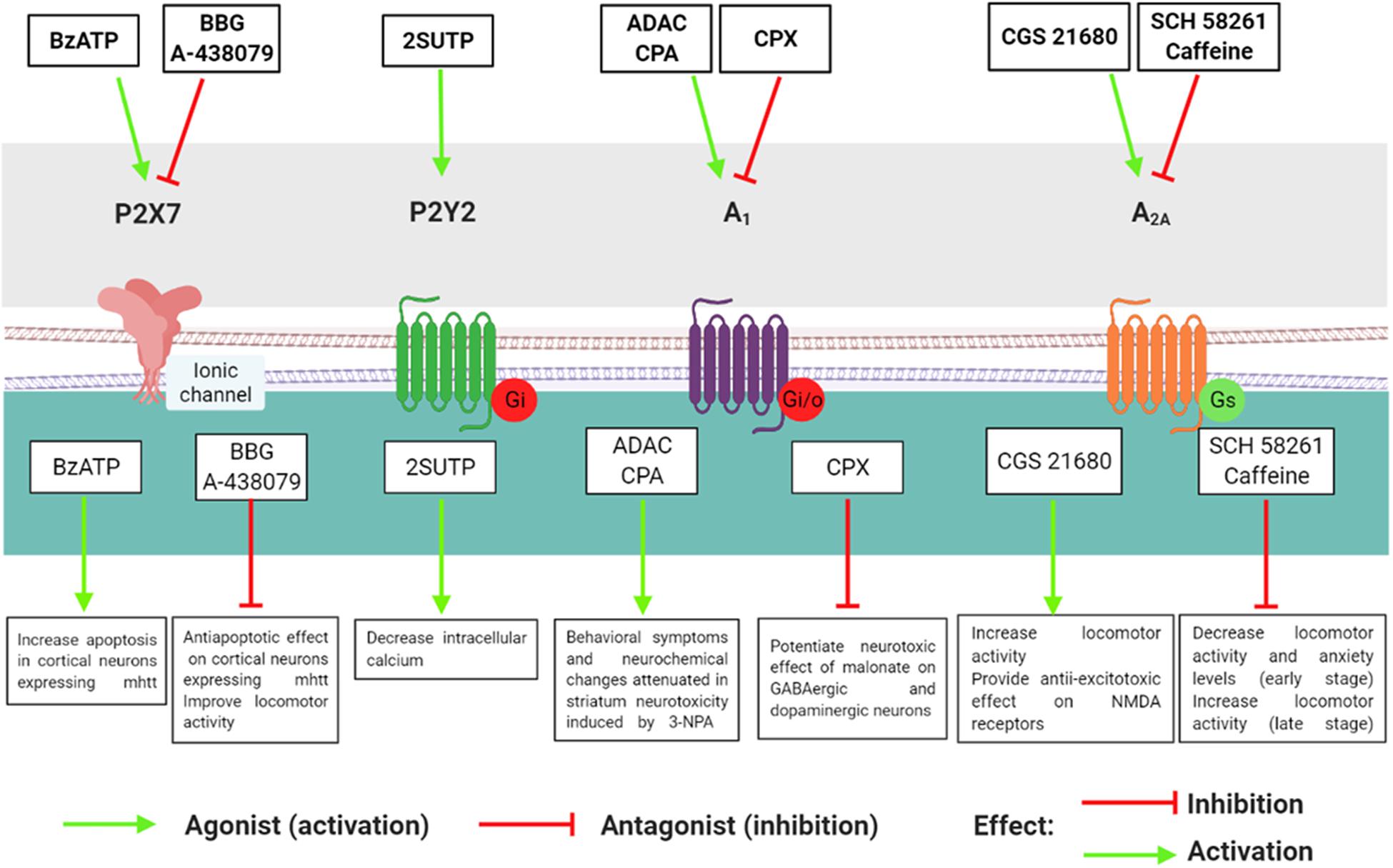
Figure 2. Effects of P2 and P1 agonists and antagonists in models of Huntington’s disease.
A2AR Alterations in Huntington’s Disease Progression
A2AR: Focus on Human Studies
Contrary to A1R, the role of A2AR in HD has been more widely investigated in human and animal models. The first evidence for the involvement of A2AR in HD was provided by autoradiographic mapping using the selective A2AR agonist ligand CGS 21680 in brain sections from HD patients and controls without the pathology. The results showed a dramatic decrease in A2AR in the caudate nucleus, putamen, nucleus accumbens, olfactory tubercle, and globus pallidus lateral in HD compared with control samples (Martinez-Mir et al., 1991). In a subsequent study, analyses of post-mortem neuronal tissue in HD subjects in the early, intermediate, and late stages of the disease showed A2AR bound the selective A2AR agonist ligand CGS 21680 within the caudate nucleus and putamen of the control brain. In the early and intermediate stages of HD, there was a dramatic loss of A2AR binding, whereas in brain tissue with the late stage of the disease there was no detectable A2AR binding (Glass et al., 2000).
On the other hand, higher A2AR levels are detected in peripheral blood cells, such as platelets, lymphocytes, and neutrophils at pre-symptomatic, early, and late stages of HD patients (Varani et al., 2003; Maglione et al., 2005). In addition, the higher A2AR levels in platelets correlated with CAG expansion and with anticipation in years since the onset of symptoms (Maglione et al., 2005, 2006). Thus, these findings indicate that there are A2AR alterations in the peripheral and central nervous systems in HD patients, and A2AR in blood cells might be an easy biomarker to detect and monitor HD progression.
Genetic studies support the relationship between A2AR and HD pathology. Two similar cohort studies performed in France and Germany with HD subjects showed gene polymorphisms in the A2AR gene (ADORA2A), mainly the rs5751876 variant, which was associated with a variation in the age at onset on the disease (Dhaenens et al., 2009; Taherzadeh-Fard et al., 2010). Compelling evidence showed increased methylcytosine and decreased 5-methylhydroxylation (enzymes responsible by DNA methylation) in the 5′ untranslated region (UTR) of ADORA2A in the putamen of HD patients compared with their respective controls (Villar-Menéndez et al., 2013). These data suggest that a dysfunction in A2AR gene expression might also contribute to the pathophysiology of HD.
Some habitually consumed drinks and foods–such as coffee, tea, chocolate, and soda–contain caffeine, a non-selective A1R and A2AR antagonist (Chen and Chern, 2011). Supported by the evidence that adenosine receptor antagonists could exert beneficial effects in neurodegenerative diseases, Simonin et al. (2013) raised the hypothesis that caffeine may be a life-style modifier in HD. For this reason, a retrospective study evaluated a possible relationship between caffeine consumption and age at onset in 80 subjects with HD. The data showed >190 mg/day caffeine consumption was significantly associated with an earlier age at onset of the disease (Simonin et al., 2013). This finding leads to the hypothesis that this association may not be a relationship with food habits but might be related to genetic determinants, such as ADORA2A or CYP1A genes, that may cause an earlier age at onset of the disease, influence a higher caffeine intake, and modulate behavioral effects.
A2AR: Evidence in Animal Models
Changes in A2AR gene expression and density have been found in transgenic animal models of HD. R6/2, HD100, and tgHD mice at 4 weeks to 24 months of age had a decrease in A2AR levels and density (Cha et al., 1999; Chan et al., 2002; Bauer et al., 2005; Tarditi et al., 2006); R6/2 animals less than 3 weeks of age had an increase in A2AR levels and density (Tarditi et al., 2006). In contrast, Benn et al. (2007) did not find alterations in A2AR gene expression in the YAC128 genetic model of HD (Benn et al., 2007). These results suggest that alterations in gene expression A2AR could have an association with HTT length and age of disease.
Several studies with controversial results have evaluated whether A2AR genetic and pharmacological activation or blockade affects behavioral symptoms and/or striatal degeneration in genetic or pharmacological models of HD. A2AR knockout between 12 and 21 weeks of age in the N171-82-Q transgenic model of HD had a deleterious impact on survival, motor coordination, body weight, striatal volume, and enkephalin and neuropeptide messenger RNA (mRNA) levels (Mievis et al., 2011). On the other hand, A2AR knockout in two R6/2 transgenic lines of HD (CAG120 and CAG240) did not cause working memory deficits and locomotor impairment compared with HD CAG120 and HD CAG240 WT mice (Li et al., 2015). Moreover, pharmacological blockade of A2AR with 1 mg/kg of the antagonist KW60002 in R6/2 (CAG240) mice at 3 months of age also reversed the working memory deficits compared with R6/2 (CAG240) mice that did not receive KW60002 (Li et al., 2015). Consistent with that finding, Fink et al. (2004) reported that A2AR genetic inactivation is beneficial to the HD animal model. The authors used two HD mouse models on different genetic backgrounds, namely C57BL/6 and 129-Steel; these models had A2AR knockout (KO; C57BL/6-A2A-KO and 129-Steel-A2A-KO). C57BL/6-A2A-KO mice and their respective WT mice at 6 months of age were administered 3-NPA twice daily for 5 days. C57BL/6-WT mice developed bradykinesia with dystonic movements and bilateral striatal lesion compared with C57BL/6-A2A-KO mice (Fink et al., 2004). Furthermore, in the 129-Steel line, 3-NPA administration also resulted in bilateral striatal lesions in WT compared with A2AR KO (Fink et al., 2004).
More recently, Domenici et al. (2018) investigated in vivo and in vitro the effect of 3-NPA in a transgenic rat strain overexpressing A2AR. Surprisingly, there was a reduction in striatal lesion volume induced by 3-NPA in transgenic rats overexpressing A2AR compared with WT animals. There was greater striatal cell viability in transgenic rats overexpressing A2AR than WT rats after exposing corticostriatal slices to 10 mM 3-NPA. In addition, 3-NPA treatment depressed synaptic transmission in corticostriatal slices of WT animals compared with rats overexpressing A2AR (Domenici et al., 2018). These data lead to the idea that A2AR is an important target involved in the pathophysiology of HD and A2AR modulation might be beneficial to attenuate the neurodegenerative progression of the disease.
A2A receptors modulation has demonstrated beneficial effects against neurodegeneration and HD-like behavioral symptoms induced by quinolinic acid and 3-NPA. The behavioral symptoms like hyperkinesia (increased locomotor activity) and the increase in anxiety levels persistent in early stage HD were decreased by blocking A2AR using two A2AR antagonists (SCH 58261 and caffeine; Popoli et al., 2002; Scattoni et al., 2007; Mishra and Kumar, 2014). In addition, the bradykinesia (decrease in locomotor activity) present in late stage of the disease was reverted with the use of SCH 58261 and caffeine (Mishra and Kumar, 2014; Bortolatto et al., 2017). Blocking A2AR using SCH 58261 and caffeine protected against the elevated glutamate extracellular levels, excessive stimulation of NMDA receptors, increased reactive oxygen species, oxidative stress, and mitochondrial dysfunction (Popoli et al., 2002; Mishra and Kumar, 2014; Bortolatto et al., 2017). Furthermore, Fink et al. (2004) showed that striatal neurotoxicity induced by 3-NPA in C57BL/6 mice resulted in bilateral striatal lesions, while mice pre-treated with the A2AR antagonist caffeine before 3-NPA injections did not show striatal lesions.
A2A receptors pharmacological blockade also attenuated the levels of cyclooxygenase (COX-2), prostaglandin E2, and brain-derived neurotrophic factor, a member of the neurotrophin family that participates in synaptic transmission and regulates neuronal proliferation and survival (Minghetti et al., 2007; Potenza et al., 2007). In R6/2 mice, A2AR blockade prevented alterations in anxious responses and abolished the increase in NMDA-receptor-induced striatal toxicity (Domenici et al., 2007). However, A2AR antagonism had a potentially detrimental effect on neurotoxicity induced by quinolinic acid in an animal model of HD; specifically, there was increased striatal glutamate outflow (Gianfriddo et al., 2003).
Considering that a pronounced loss of A2AR occurs in HD, researchers have tested whether A2A agonists could ameliorate HD symptoms. The selective A2AR agonist CGS 21680 reverted the hypolocomotor profile and the increase in the ventricle/brain ratio in R6/2 mice (Chou et al., 2005). CGS 21680 also attenuated the NMDA toxic effects in corticostriatal slices (Martire et al., 2007) and reduced the expression of NMDA receptor subunits NR1 in the striatum of R6/2 and WT mice. In addition, there was a decrease in the NR2A/NR2B ratio in the striatum of WT mice, inducing a pro-excitotoxic effect, whereas the NR2A/NR2B ratio was increased in HD mice (R6/2), and thus the NMDA receptors provided an anti-excitotoxic effect (Ferrante et al., 2010). Finally, in the liver of R6/2 mice, CGS 21680 treatment also had a protective effect, suppressing mHTT aggregates and decreasing the levels of crucial transcription factor and protein chaperones (Hsp27 and Hsp70; Chiang et al., 2009). Taken together, it is possible to suggest that A2AR alterations in HD occur in the central nervous system as well as other tissues. Thus, pharmacological modulation of A2AR through agonist or antagonist administration might exert a neuroprotective effect against HD progression (Figure 2), considering the stage of the disease, drug dosage, and period of pharmacological administration.
P2 Receptor Involvement in Huntington’s Disease
Contrary to P1 receptors, there are few studies about P2 receptors in HD and their roles in this neurodegenerative disease remain elusive. Díaz-Hernández et al. (2009) investigated P2X7 receptor involvement in HD using two genetic mouse models of HD (Tet/HD94 and R6/1). First, cortical and striatal neurons showed an increase in P2X7 receptor protein and mRNA levels in Tet/HD94 and R6/1 compared with WT mice (Díaz-Hernández et al., 2009). Moreover, transgenic HD mice had higher P2X7 receptor levels in synaptosomes and were more sensitive to the P2X7 agonist BzATP than WT mice. This increase in sensitivity induced by BzATP led to apoptosis of cultured cortical neurons expressing mHTT, which was prevented by the P2X7 antagonist brilliant blue (BBG) (Díaz-Hernández et al., 2009). Due to the protective effect of BBG in neuronal culture, the authors investigated the efficacy of P2X7 antagonists BBG and A-438079 in R6/1 mice and their WT on body weight and locomotor parameters. BBG administration fully prevented body weight loss and significantly improved motor coordination and locomotor performance in R6/1 compared with WT mice. The antagonist A-438079 also prevented body weight loss and improved locomotor parameters in R6/1 mice, but the effect was only moderate (Díaz-Hernández et al., 2009). These data support the hypothesis that changes in P2X7 receptor levels and function lead to an increase in Ca2+ permeability; this eventuality could induce excitotoxicity and oxidative stress in neurons, changes that are related to HD pathogenesis (Figure 2). In the future, studies using P2X7 receptor antagonists should be performed because these compounds may have therapeutic potential for HD treatment.
Martire et al. (2021) investigated the expression and the functioning of P2X7R in two genetic models of HD: ST14A rat striatal cells, expressing full-length wild-type (WT, Q15) or mutant (Q120) htt and R6/2 mice, which resembles to juvenile forms of HD. In the presence of HD mutation, there is an altered P2X7R expression and a larger P2X7R response to the agonist BzATP, inducing cell death and reducing synaptic transmission. BzATP effect observed in the electrophysiology experimental setting are dependent of A1R activation. These findings may permit a better understanding of the P2X7R mechanisms and evaluate the therapeutic potential of P2X7 antagonists in HD. Certainly, this topic requires a deepen investigation since recent studies demonstrated changes in P2X7R in the brain of HD subjects (Ollà et al., 2020). Ollà et al. (2020) observed that the protein levels of the full-length form of P2X7R, also called P2X7R-A, as well as the exclusively human naturally occurring variant lacking the C-terminus region, named P2X7R-B, are upregulated. These augmented protein levels can be explained by elevated P2X7R mRNA levels. In addition, P2X7R introns 10 and 11 are more retained in HD subjects when compared with controls patients (Ollà et al., 2020). Therefore, further studies are required to evaluate the implications of P2X7R in HD and if this receptor may be a target for the development of new pharmacological therapies for this pathology.
Regarding to other P2 receptors, Glaser et al. (2020) analyzed whether P2Y2 receptors might be involved in the pathogenesis of HD. They investigated the role of the P2Y2 receptor and spontaneous Ca2+ concentrations in embryonic stem cells in in vitro HD models and controls. In basal state HD, there were higher intracellular Ca2+ levels than controls. Besides, there was elevated P2X7 and P2Y2 receptor levels in the HD cell line compared with the controls (Glaser et al., 2020). Moreover, the cells from controls (WT cells) were responsive to the P2X7 receptor agonist BzATP and the P2Y2 receptor agonist 2SUTP, while HD cells (expressing mHTT) showed a decrease in the 2SUTP response, with impaired P2Y2 receptor activation (Glaser et al., 2020). The administration of ATP or 2SUTP in HD cells decreased the concentration of intracellular Ca2+ transients (Glaser et al., 2020). Therefore, these results indicate that P2 receptors contribute to the pathogenesis of HD (Figure 2) and provided the first evidence for the involvement of the P2Y2 receptor in this neurodegenerative disease.
Together, the above-mentioned studies shed new light on the mechanism underlying HD, leading to the idea that pharmacological therapies based on P2X7 and P2Y2 receptors should be better investigated for HD treatment.
Focusing on the Relationship Between Nucleotide and Nucleoside Metabolism and Huntington’s Disease
Reduced mitochondrial ATP levels and ATP/ADP ratios have been found in striatal cells containing mHTT (Seong et al., 2005). It is known that ATP depletion occurs in HD, so researchers have investigated nucleotide and nucleoside metabolism in this pathological condition. Researchers have demonstrated reduced mitochondrial ATP levels in cortical cells from an animal model of HD induced by 3-NPA, in cardiac tissue from two genetic models of HD (R6/2 and HdhQ150), and in a HEK293T cell line containing mHTT (Riepe et al., 1994; Toczek et al., 2016a, 2018). There was a decrease in the ATP/ADP ratio in cardiac tissue from R6/2 and HdhQ150 mice (Toczek et al., 2016a). There was also a reduction in ATP and AMP degradation in the HEK293T cell line containing mHTT (Toczek et al., 2018). Furthermore, studies in cardiac and cerebral tissue have shown no alteration in ADP levels in R6/2 and HdhQ150 mice. However, AMP levels were increased in R6/2 mice (Toczek et al., 2016a; Kao et al., 2017).
A genetic study of HD using different lines, including Tg51, zQ175, and R6/2, and a pharmacological model of HD induced by quinolinic acid and 3-NPA demonstrated a decrease in adenosine levels in the striatum of Tg51, zQ175, R6/2 lines, and HD animals induced by quinolinic acid and 3-NPA (Gianfriddo et al., 2003; Guitart et al., 2016; Jamwal and Kumar, 2016; Kao et al., 2017). Importantly, researchers have also found a decrease in adenosine levels in the cerebrospinal fluid of patients with HD compared with controls (Kao et al., 2017). Kao et al. (2017) observed that ATP was indirectly correlated with the number of CAG repeats and had a direct correlation with the age at onset of the disease while the adenosine/ATP ratio was negatively correlated with the disease duration of patients with HD (Kao et al., 2017).
Consistently, there was a decrease in ecto-5′-nucleotidase (an enzyme that converts AMP to adenosine) activity in HEK 293T cell line containing mHTT and cardiac tissue from R6/2 mice (Toczek et al., 2016b, 2018), while there was an increase in ADA activity in cardiac tissue from R6/2 mice (Toczek et al., 2016b). Furthermore, in the striatum of rats treated with 3-NPA, there was a decrease in inosine and hypoxanthine levels compared with the control group (Jamwal and Kumar, 2016). On the other hand, Toczek et al. (2016a) found an increase in inosine, hypoxanthine, xanthine, uric acid, and uridine levels in cardiac tissue in R6/2 and HdhQ150 mice (Toczek et al., 2016a).
Genes involved in purine metabolism (Entpd2, Ampd3, Pnp, and Xdh), adenosine metabolism (Ada), conversion of adenine nucleotides (adenylate kinase 1 [Ak1] and inosine monophosphatase dehydrogenase 2 [Impdh2]), and equilibrative nucleoside transporter-like ENT1 and ENT2 were altered in cardiac tissue, striatum, and skeletal muscle of R6/2 mice of HD (Toczek et al., 2016a; Kao et al., 2017; Mielcarek et al., 2017). Moreover, in HD patients at the early stage of the disease, there was an upregulation of the gene that encodes ENT1 (Guitart et al., 2016). Finally, nitrobenzylthioinosine (NBTI), a selective inhibitor of ENT1, and dipyridamole (DPR), a non-specific inhibitor of ENT1 and ENT2, were intrastriatally perfused in R6/2 mice. Administration of NBTI alone increased adenosine levels 1-h post-treatment, whereas both NBTI and DPR administration increased adenosine levels 1-h post-treatment, sustaining the effects up to 5 h post-treatment (Kao et al., 2017).
Regarding to studies in HD patients, it has been observed differences in the metabolism of nucleoside and its derivates. In plasma samples from HD patients, there was a significant increase in hypoxanthine and uridine levels, but there were not changes in uric acid levels compared with healthy subjects (Toczek et al., 2016a). Interestingly, hypoxanthine and uridine levels were directly correlated with HD duration and indirectly correlated with motor scores and chorea intensity (Toczek et al., 2016a). In contrast, Corey-Bloom et al. (2020) recently investigated uric acid levels in plasma and saliva samples from HD subjects and normal controls, as well as in post-mortem prefrontal cortical samples from HD subjects and controls. The authors surprisingly revealed that plasma and salivary uric acid levels were significantly lower in female pre-manifest HD and manifest HD subjects compared with normal controls, whereas salivary levels of uric acid were also significantly lower in male manifest HD subjects than controls. In male HD patients, plasma and salivary uric acid levels were negatively correlated with total functional capacity, while there were direct correlations with the total motor score. Female HD patients showed a direct correlation between plasma uric acid levels and total functional capacity, while salivary uric acid levels were significantly correlated with disease burden. Finally, in post-mortem prefrontal cortical samples from HD subjects, there was a decrease in uric acid levels (Corey-Bloom et al., 2020).
Overall, these findings indicate that purinergic signaling plays an important role in HD by inhibiting the cascade of ATP hydrolysis, modulating the concentration of adenine nucleotides and nucleosides.
Conclusion
This review sheds light on the important regulatory role of purinergic signaling in HD pathophysiology. Notably, ATP, adenosine, and A2AR are the main actors in HD. Although some results in animal models and HD patients are controversial–depending on the model tested, type of pharmacological treatment, and period of drug administration–pharmacological modulation of A2AR, through agonist and antagonist drugs, has shown a neuroprotective effect by attenuating the behavioral symptoms and improving neurochemical parameters during HD progression. Besides, A2AR in the central and peripheral nervous system might be considered a powerful biomarker for HD progression and ought to be used in clinical practice. A2AR heterodimerizes with several other G-protein coupled receptors involved in striatal dysfunction and degeneration in HD; thus, A2AR could be considered a target for the development of pharmacological therapies for HD patients.
Several small molecules acting as A2AR antagonists have already been developed and tested in patients several neurological diseases, such as Parkinson’s Disease. Although the efficacy of these agents in Parkinson’s disease was not proved, the use of antagonists targeting A2AR in cancer immunotherapy has been also investigated. The treatment with small molecules or mAbs aiming to block adenosine signaling, either by limiting its production or its binding to adenosine receptors, has yielded important tumor control in pre-clinical studies. Moreover, simultaneous blockade of adenosine production and receptor binding, achieved by an anti-CD73 mAb co-administered with an A2AR antagonist, for example, have demonstrated synergy. Therefore, it is important to deepen the investigation of A2AR as a target for the development of existing or new agents targeting this axis, along with further testing of combinatorial strategies, which may be relevant in the search for pharmacological therapies for HD patients. Moreover, the role of A1R and P2 receptors in HD pathogenesis needs to be reconsidered; there should be more specific investigation on these receptors, because they could provide a powerful contribution to understanding the mechanism underlying HD. In addition, nucleoside metabolism and the control of hypoxanthine, xanthine, and uric acid levels should also be deeply investigated, because these nucleosides might be important candidates for future drugs or biomarkers for HD. In summary, purinergic signaling represents a promising research area, and the main players, such as ATP, adenosine, and A2AR, as well as the respective coadjutants, such as A1R, P2 receptors, and other components of nucleotide and nucleoside metabolism, should be considered possible targets for drug development for HD treatment.
Author Contributions
Both authors contributed for the conceptualization, performed the literature review, read and approved the submitted version. MW wrote the original draft of the manuscript. CB wrote and review and editing the final version of the manuscript.
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–Brasil (CAPES)–Finance Code 001, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; 420695/2018-4 and 304450/2019-7), Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS; 17/2551-0000977-0), and Instituto Nacional de Ciência e Tecnologia para Doenças Cerebrais, Excitotoxicidade e Neuroproteção.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbracchio, M. P., and Burnstock, G. (1994). Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacol. Ther. 64, 445–475. doi: 10.1016/0163-7258(94)00048-4
Albin, R. L., Young, A. B., and Penney, J. B. (1989). The functional anatomy of basal ganglia disorders. Trends. Neurosci. 12, 366–375.
Alfinito, P. D., Wang, S. P., Manzino, L., Rijhsinghani, S., Zeevalk, G. D., and Sonsalla, P. K. (2003). Adenosinergic protection of dopaminergic and GABAergic neurons against mitochondrial inhibition through receptors located in the substantia nigra and striatum, respectively. J. Neurosci. 23, 10982–10987. doi: 10.1523/JNEUROSCI.23-34-10982-2003
Baig, S. S., Strong, M., and Quarrell, O. W. (2016). The global prevalence of Huntington’s disease: a systematic review and discussion. Neurodegener. Dis. Manag. 6, 331–343. doi: 10.2217/nmt-2016-0008
Bailus, B., Zhang, N., and Ellerby, L. M. (2017). “Using genome engineering to understand huntington’s disease,” in Genome Editing in Neurosciences, eds R. Jaenisch, F. Zhang, and F. Gage (Brlin: Springer), 87–101. doi: 10.1007/978-3-319-60192-2_9
Bates, G. P., Dorsey, R., Gusella, J. F., Hayden, M. R., Kay, C., Leavitt, B. R., et al. (2015). Huntington disease. Nat. Rev. Dis. Primers. 1:15005. doi: 10.1038/nrdp.2015.5
Bauer, A., Zilles, K., Matusch, A., Holzmann, C., Riess, O., and Von Hörsten, S. (2005). Regional and subtype selective changes of neurotransmitter receptor density in a rat transgenic for the Huntington’s disease mutation. J. Neurochem. 94, 639–650. doi: 10.1111/j.1447.4159.2005.03169.x
Benn, C. L., Slow, E. J., Farrell, L. A., Graham, R., Deng, Y., Hayden, M. R., et al. (2007). Glutamate receptor abnormalities in the YAC128 transgenic mouse model of huntington’s disease. Neuroscience 147, 354–372. doi: 10.1016/j.neuroscience.2007.03.010
Blum, D., Chern, Y., Domenici, M. R., Buée, L., Lin, C. Y., Rea, W., et al. (2018). The role of adenosine tone and adenosine receptors in huntington’s disease. J. Caffeine. Adenosine. Res. 8, 43–58. doi: 10.1089/caff.2018.0006
Blum, D., Gall, D., Galas, M. C., d’Alcantara, P., Bantubungi, K., and Schiffmann, S. N. (2002). The adenosine A1 receptor agonist adenosine amine congener exerts a neuroprotective effect against the development of striatal lesions and motor impairments in the 3-nitropropionic acid model of neurotoxicity. J. Neurosci. 22, 9122–9133. doi: 10.1523/JNEUROSCI.22.20.09122.2002
Blumenstock, S., and Dudanova, I. (2020). Cortical and striatal circuits in huntington’s disease. Front. Neurosci. 14:82. doi: 10.3389/fnins.2020.00082
Bonan, C. D. (2012). Ectonucleotidases and nucleotide/nucleoside transporters as pharmacological targets for neurological disorders. CNS. Neurol. Disord. Drug. Targets. 11, 739–750. doi: 10.2174/187152712803581092
Borea, P. A., Gessi, S., Merighi, S., Vincenzi, F., and Varani, K. (2018). Pharmacology of adenosine receptors: the state of the art. Physiol. Rev. 98, 1591–1625. doi: 10.1152/physrev.00049.2017
Bortolatto, C. F., Reis, A. S., Pinz, M. P., Voss, G. T., Oliveira, R. L., Vogt, A. G., et al. (2017). Selective A2A receptor antagonist SCH 58261 modulates striatal oxidative stress and alleviates toxicity induced by 3-Nitropropionic acid in male Wistar rats. Metab. Brain. Dis. 32, 1919–1927. doi: 10.1007/s11011-017-0086-1
Burnstock, G. (2008). Purinergic signalling and disorders of the central nervous system. Nat. Rev. Drug. Discov. 7, 575–590. doi: 10.1038/nrd2605
Burnstock, G. (2011). Adenosine and ATP receptors in the brain. Curr. Top. Med. Chem. 11, 973–1011. doi: 10.2174/156802611795347627
Burnstock, G. (2015). Purinergic signalling in neuroregeneration. Neural. Regn. Res. 10:1919. doi: 10.4103/1673-5374.165300
Burnstock, G. (2018). Purine and purinergic receptors. Brain. Neurosci. Adv. 2:2398212818817494. doi: 10.1177/2398212818817494
Capiluppi, E., Romano, L., Rebora, P., Nanetti, L., Castaldo, A., Gellera, C., et al. (2020). Late-onset huntington’s disease with 40–42 CAG expansion. Neurol. Sci. 41, 869–876. doi: 10.1007/s10072-019-04177-8
Carmo, C., Naia, L., Lopes, C., and Rego, A. C. (2018). Mitochondrial dysfunction in huntington’s disease. Adv. Exp. Med. Biol. 1049, 59–83. doi: 10.1007/978-3-319-71779-1_3
Cha, J. H. J., Frey, A. S., Alsdorf, S. A., Kerner, J. A., Kosinski, C. M., Mangiarini, L., et al. (1999). Altered neurotransmitter receptor expression in transgenic mouse models of Huntington’s disease. Philos. Trans. R. Soc. B Biol. Sci. 354, 981–989. doi: 10.1098/rstb.1999.0449
Chan, E. Y. W., Luthi-Carter, R., Strand, A., Solano, S. M., Hanson, S. A., DeJohn, M. M., et al. (2002). Increased huntingtin protein length reduces the number of polyglutamine-induced gene expression changes in mouse models of Huntington’s disease. Hum. Mol. Genet. 11, 1939–1951. doi: 10.1093/hmg/11.17.1939
Cheffer, A., Castillo, A. R. G., Corrêa-Velloso, J., Gonçalves, M. C. B., Naaldijk, Y., Nascimento, I. C., et al. (2018). Purinergic system in psychiatric diseases. Mol. Psychiatry. 23, 94–106. doi: 10.1038/mp.2017.188
Chen, J. F., and Chern, Y. (2011). Impacts of methylxanthines and adenosine receptors on neurodegeneration: human and experimental studies. Handb. Exp. Pharmacol. 200, 267–310. doi: 10.1007/978-3-642-13443-2_10
Chiang, M. C., Chen, H. M., Lai, H. L., Chen, H. W., Chou, S. Y., Chen, C. M., et al. (2009). The A2A adenosine receptor rescues the urea cycle deficiency of Huntington’s disease by enhancing the activity of the ubiquitin-proteasome system. Hum. Mol. Genet. 18, 2929–2942. doi: 10.1093/hmg/ddp230
Chou, S. Y., Lee, Y. C., Chen, H. M., Chiang, M. C., Lai, H. L., Chang, H. H., et al. (2005). CGS21680 attenuates symptoms of huntington’s disease in a transgenic mouse model. J. Neurochem. 93, 310–320. doi: 10.1111/j.1471.4159.2005.03029.x
Cieslak, M., Roszek, K., and Wujak, M. (2019). Purinergic implication in amyotrophic lateral sclerosis- from pathological mechanisms to therapeutic perspectives. Purinergic. Signal 15, 1–15. doi: 10.1007/s11302-018-9633-4
Ciruela, F. (2020). Special issue: G protein-coupled adenosine receptors: molecular aspects and beyond. Int. J. Mol. Sci. 21:1997. doi: 10.3390/ijms21061997
Ciruela, F., Fernández-Dueñas, V., and Jacobson, K. A. (2015). Lighting up G protein-coupled purinergic receptors with engineered fluorescent ligands. Neuropharmacology 98, 58–67. doi: 10.1016/j.neuropharm.2015.04.001
Coppen, E. M., and Roos, R. A. C. (2017). Current pharmacological approaches to reduce chorea in huntington’s disease. Drugs. 77, 29–46. doi: 10.1007/s40265-016-0670-4
Corey-Bloom, J., Haque, A., Aboufadel, S., Snell, C., Fischer, R. S., Granger, S. W., et al. (2020). Uric acid as a potential peripheral biomarker for disease features in huntington’s patients. Front. Neurosci. 14:73. doi: 10.3389/fnins.2020.00073
Croce, K. R., and Yamamoto, A. A. (2019). A role for autophagy in huntington’s disease. Neurobiol. Dis. 122, 16–22. doi: 10.1016/j.nbd.2018.08.010
Cunha, R. A. (2005). Neuroprotection by adenosine in the brain: From A(1) receptor activation to A(2A) receptor blockade. Purinergic. Signal 1, 111–134. doi: 10.1007/s11302-005-0649-1
Cybulska, K., Perk, L., Booij, J., Laverman, P., and Rijpkema, M. (2020). Huntington’s disease: a review of the known pet imaging biomarkers and targeting radiotracers. Molecules 25:482.
Dhaenens, C. M., Burnouf, S., Simonin, C., Brussel, E., Van Duhamel, A., Defebvre, L., et al. (2009). A genetic variation in the ADORA2A gene modifies age at onset in huntington’s disease. Neurobiol. Dis. 35, 474–476. doi: 10.1016/j.nbd.2009.06.009
Díaz-Hernández, M., Díez-Zaera, M., Sánchez-Nogueiro, J., Gómez-Villafuertes, R., Canals, J. M., Alberch, J., et al. (2009). Altered P2X7-receptor level and function in mouse models of huntington’s disease and therapeutic efficacy of antagonist administration. FASEB. J. 23, 1893–1906. doi: 10.1096/fj.08.122275
Domenici, M. R., Chiodi, V., Averna, M., Armida, M., Pèzzola, A., Pepponi, R., et al. (2018). Neuronal adenosine A2Areceptor overexpression is neuroprotective towards 3-nitropropionic acid-induced striatal toxicity: a rat model of huntington’s disease. Purinergic. Signal 14, 235–243. doi: 10.1007/s11302.018-0609-4
Domenici, M. R., Scattoni, M. L., Martire, A., Lastoria, G., Potenza, R. L., Borioni, A., et al. (2007). Behavioral and electrophysiological effects of the adenosine A2A receptor antagonist SCH 58261 in R6/2 Huntington’s disease mice. Neurobiol. Dis. 28, 197–205. doi: 10.1016/j.nbd.2007.07.009
Dufour, B. D., and McBride, J. L. (2019). Normalizing glucocorticoid levels attenuates metabolic and neuropathological symptoms in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 121, 214–229. doi: 10.1016/j.nbd.2018.09.025
Ferrante, A., Martire, A., Armida, M., Chiodi, V., Pézzola, A., Potenza, R. L., et al. (2010). Influence of CGS 21680, a selective adenosine A2A receptor agonist, on NMDA receptor function and expression in the brain of Huntington’s disease mice. Brain. Res. 1323, 184–191. doi: 10.1016/jbrainres.2010.01.080
Ferrante, A., Martire, A., Pepponi, R., Varani, K., Vincenzi, F., Ferraro, L., et al. (2014). Expression, pharmacology and functional activity of adenosine A1 receptors in genetic models of Huntington’s disease. Neurobiol. Dis. 71, 193–204. doi: 10.1016/j.nbd.2014.08.013
Ferré, S., Ciruela, F., Quiroz, C., Luján, R., Popoli, P., Cunha, R. A., et al. (2007). Adenosine receptor heteromers and their integrative role in striatal function. Scientific. World. J. 7, 74–85. doi: 10.1100/tsw.2007.211
Fink, J. S., Kalda, A., Ryu, H., Stack, E. C., Schwarzschild, M. A., Chen, J. F., et al. (2004). Genetic and pharmacological inactivation of the adenosine A2A receptor attenuates 3-nitropropionic acid-induced striatal damage. J. Neurochem. 88, 538–544. doi: 10.1046/j.1471.4159.2003.02145.x
Fredholm, B. B., Ijzerman, A. P., Jacobson, K. A., Klotz, K. N., and Linden, J. (2001). International Union of Pharmacology. XXV. nomenclature and classification of adenosine receptors. Pharmacol. Rev. 53, 527–552.
Fuxe, K., Ferré, S., Genedani, S., Franco, R., and Agnati, L. F. (2007). Adenosine receptor-dopamine receptor interactions in the basal ganglia and their relevance for brain function. Physiol. Behav. 92, 210–217. doi: 10.1016/j.physbeh.2007.05.034
Gianfriddo, M., Corsi, C., Melani, A., Pèzzola, A., Reggio, R., Popoli, P., et al. (2003). Adenosine A2A antagonism increases striatal glutamate outflow in the quinolinic acid rat model of Huntington’s disease. Brain. Res. 979, 225–229. doi: 10.1016/s0006-8993(03)02942-1
Glaser, T., Hiromi, S., Ribeiro, D. E., Martins, P. P. L., Beco, R. P., Kosinski, M., et al. (2020). ATP and spontaneous calcium oscillations control neural stem cell fate determination in Huntington’s disease: a novel approach for cell clock research. Mol. Psychiatry. doi: 10.1038/s41380-020-0717-5 [Epub ahead of print].
Glass, M., Dragunow, M., and Faull, R. L. M. (2000). The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience 97, 505–519. doi: 10.1016/s0306-4522(00)00008-7
Graybiel, A. M. (2000). The basal ganglia. Curr. Biol. 10, R509–R511. doi: 10.1016/s0960-9822(00)00593-5
Guitart, X., Bonaventura, J., Rea, W., Orrú, M., Cellai, L., Dettori, I., et al. (2016). Equilibrative nucleoside transporter ENT1 as a biomarker of Huntington disease. Neurobiol Dis. 96, 47–53. doi: 10.1016/j.nbd.2016.08.013
Gundlfinger, A., Bischofberger, J., Johenning, F. W., Torvinen, M., Schmitz, D., and Breustedt, J. (2007). Adenosine modulates transmission at the hippocampal mossy fiber synapse via direct inhibition of presynaptic calcium channels. J. Physiol. 582, 263–277. doi: 10.1113/jphysiol.2007.132618
Illarioshkin, S. N., Klyushnikov, S. A., Vigont, V. A., Seliverston, Y. A., and Kaznacheyeva, E. V. (2018). Molecular pathogenesis in Huntington’s Disease. Biochemistry (Mosc.) 83, 1030–1039. doi: 10.1134/S0006297918090043
Jamwal, S., and Kumar, P. (2016). Spermidine ameliorates 3-nitropropionic acid (3-NP)- induced striatal toxicity: possible role of oxidative stress, neuroinflammation, and neurotransmitters. Physiol. Behav. 155, 180–187. doi: 10.1016/j.physbeh.2015.12.015
Jamwal, S., and Kumar, P. (2019). Insight into the emerging role of striatal neurotransmitters in the pathophysiology of Parkinson’s disease and Huntington’s disease: a review. Curr. Neuropharmacol. 17, 165–175. doi: 10.2174/1570159X6666180302115032
Jamwal, S., Singh, S., Kaur, N., and Kumar, P. (2015). Protective effect of spermidine against excitotoxic neuronal death induced by quinolinic acid in rats: possible neurotransmitters and neuroinflammatory mechanisms. Neurotox. Res. 28, 171–184. doi: 10.1007/s12640-015-9535-y
Jodeiri Farshbaf, M., and Kiani-Esfahani, A. (2018). Succinate dehydrogenase: prospect for neurodegenerative diseases. Mitochondrion 42, 77–83. doi: 10.1016/j.mito.2017.12.002
Johri, A., Chandra, A., and Beal, M. F. (2013). PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free. Radic. Biol. Med. 62, 37–46. doi: 10.1016/j.freeradbiomed.2013.04.016
Kao, Y. H., Lin, M. S., Chen, C. M., Wu, Y. R., Chen, H. M., Lai, H. L., et al. (2017). Targeting ENT1 and adenosine tone for the treatment of Huntington’s disease. Hum. Mol. Genet. 26, 467–478. doi: 10.1093/hmg/ddw402
Kish, S. J., Shannak, K., and Hornykiewicz, O. (1987). Elevated serotonin and reduced dopamine in subregionally divided Huntington’s disease striatum. Ann. Neurol. 22, 386–389. doi: 10.1002/ana.410220318
Koch, E. T., and Raymond, L. A. (2019). Dysfunctional striatal dopamine signaling in Huntington’s disease. J. Neurosci. Res. 97, 1636–1654. doi: 10.1002/jnr.24495
Latini, S., and Pedata, F. (2001). Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J. Neurochem. 79, 463–484. doi: 10.1046/j.1471-4159.2001.00607.x
León-Navarro, D. A., Albasanz, J. L., and Martín, M. (2018). Functional cross-talk between adenosine and metabotropic glutamate receptors. Curr. Neuropharmacol. 17, 422–437. doi: 10.2174/1570159X16666180416093717
Li, W., Silva, H. B., Real, J., Wang, Y. M., Rial, D., Li, P., et al. (2015). Inactivation of adenosine A2A receptors reverses working memory deficits at early stages of Huntington’s disease models. Neurobiol. Dis. 79, 70–80. doi: 10.1016/j.nbd.2015.03.030
Liot, G., Valette, J., Pépin, J., Flament, J., and Brouillet, E. (2017). Energy defects in Huntington’s disease: Why “in vivo” evidence matters. Biochem. Biophys. Res. Commun. 483, 1084–1095. doi: 10.1016/j.bbrc.2016.09.065
Maglione, V., Cannella, M., Martino, T., De Blasi, A., Frati, L., and Squitieri, F. (2006). The platelet maximum number of A2A-receptor binding sites (Bmax) linearly correlates with age at onset and CAG repeat expansion in Huntington’s disease patients with predominant chorea. Neurosci. Lett. 393, 27–30. doi: 10.1016/j.neulet.2005.09.037
Maglione, V., Giallonardo, P., Cannella, M., Martino, T., Frati, L., and Squitieri, F. (2005). Adenosine A2A receptor dysfunction correlates with age at onset anticipation in blood platelets of subjects with Huntington’s disease. Am. J. Med. Genet. Neuropsychiatr. Genet. 139 B, 101–105. doi: 10.1002/ajmg.b.30223
Martinez-Mir, M. I., Probst, A., and Palacios, J. M. (1991). Adenosine A2 receptors: selective localization in the human basal ganglia and alterations with disease. Neuroscience 42, 697–706. doi: 10.1016/0306-4522(91)90038-p
Martire, A., Calamandrei, G., Felici, F., Scattoni, M. L., Lastoria, G., Domenici, M. R., et al. (2007). Opposite effects of the A2A receptor agonist CGS21680 in the striatum of Huntington’s disease versus wild-type mice. Neurosci. Lett. 417, 78–83. doi: 10.1016/j.neulet.2007.02.034
Martire, A., Pepponi, R., Liguori, F., Volonté, C., and Popoli, P. (2021). P2X7 receptor agonist 2’(3’)-O-(4-Benzoylbenzoyl)ATP differently modulates cell viability and coricostriatal synaptic transmission in experimental models of Huntington’s disease. Front. Pharmacol. 19:633861. doi: 10.3389/fphar.2020.633861
Matusch, A., Saft, C., Elmenhorst, D., Kraus, P. H., Gold, R., Hans-Peter, H., et al. (2014). Cross-sectional PET study of cerebral adenosine A1 receptors in premanifest and manifest Huntington’s disease. Eur. J. Nucl. Med. Mol. Imaging. 41, 1210–1220. doi: 10.1007/s00259-014-2724-8
Mielcarek, M., Smolenski, R. T., and Isalan, M. (2017). Transcriptional signature of an altered purine metabolism in the skeletal muscle of a Huntington’s disease mouse model. Front. Physio. 8:127. doi: 10.3389/fphys.2017.00127
Mievis, S., Blum, D., and Ledent, C. (2011). A2A receptor knockout worsens survival and motor behavior in a transgenic mouse model of Huntington’s disease. Neurobiol. Dis. 41, 570–576. doi: 10.1016/j.nbd.2010.09.021
Minghetti, L., Greco, A., Potenza, R. L., Pezzola, A., Blum, D., Bantubungi, K., et al. (2007). Effects of the adenosine A2A receptor antagonist SCH 58621 on cyclooxygenase-2 expression, glial activation, and brain-derived neurotrophic factor availability in a rat model of striatal neurodegeneration. J. Neuropathol. Exp. Neurol. 66, 363–371. doi: 10.1097/nen.0b013e3180517477
Mishra, J., and Kumar, A. (2014). Improvement of mitochondrial NAD+/FAD+-linked state-3 respiration by caffeine attenuates quinolinic acid-induced motor impairment in rats: implications in Huntington’s disease. Pharmacol. Rep. 66, 1148–1155. doi: 10.1016/j.pharep.2014.07.006
Ollà, I., Santos-Galindo, M., Elorza, A., and Lucas, J. J. (2020). P2X7 receptor upregulation in Huntington’s disease brains. Front. Mol. Neurosci. 6:567430. doi: 10.3389/fnmol.2020.567430
Petersén, A., and Weydt, P. (2019). The psychopharmacology of Huntington disease. Handb. Clin. Neurol. 165, 179–189.
Popoli, P., Pintor, A., Domenici, M. R., Frank, C., Tebano, M. T., Pezzola, A., et al. (2002). Blockade of striatal adenosine A2A receptor reduces, through a presynaptic mechanism, quinolinic acid-induced excitotoxicity: possible relevance to neuroprotective interventions in neurodegenerative diseases of the striatum. J. Neurosci. 22, 1967–1975. doi: 10.1523/JNEUROOSCI.22-05-01967.2002
Potenza, R. L., Tebano, M. T., Martire, A., Domenici, M. R., Pepponi, R., Armida, M., et al. (2007). Adenosine A2A receptors modulate BDNF both in normal conditions and in experimental models of Huntington’s disease. Purinergic. Signal. 3, 333–338. doi: 10.1007/s11302-007-9066-y
Puchałowicz, K., Tarnowski, M., Baranowska-Bosiacka, I., Chlubek, D., and Dziedziejko, V. (2014). P2X and P2Y receptors—role in the pathophysiology of the nervous system. Int. J. Mol. Sci. 15, 23672–23704. doi: 10.3390/ijms151223672
Rai, S. N., Singh, B. K., Rathore, A. S., Zahra, W., Keswani, C., Birla, H., et al. (2019). Quality control in Huntington’s disease: a therapeutic target. Neurotox. Res. 36, 612–626. doi: 10.1007/s12640-019-00087-x
Ramaswamy, S., Mcbride, J. L., and Kordower, J. H. (2007). Animal models of Huntington’s Disease. ILAR. J. 48, 356–373. doi: 10.1093/ilar.48.4.356
Ribeiro, F. F., Xapelli, S., Miranda-Lourenço, C., Tanqueiro, S. R., Fonseca-Gomes, J., Diógenes, M. J., et al. (2016). Purine nucleosides in neuroregeneration and neuroprotection. Neuropharmacology 104, 226–242. doi: 10.1016/neuropharm.2015.11.006
Riepe, M., Ludolph, A., Seelig, M., Spencer, P. S., and Ludolph, A. C. (1994). Increase of ATP levels by glutamate antagonists is unrelated to neuroprotection. Neuroreport 5, 2130–2132. doi: 10.1097/00001756-199410270-00035
Rubinsztein, D. C. (2002). Lessons from animal models of Huntington’s disease. Trends. Genet. 18, 202–209. doi: 10.1016/s0168-9525(01)02625-7
Scattoni, M. L., Valanzano, A., Pezzola, A., March, Z., De Fusco, F. R., Popoli, P., et al. (2007). Adenosine A2A receptor blockade before striatal excitotoxic lesions prevents long term behavioral disturbances in the quinolinic rat model of Huntington’s disease. Behav. Brain. Res. 176, 216–221. doi: 10.1016/j.bbr.2006.10.004
Seong, I. S., Ivanova, E., Lee, J. M., Choo, Y. S., Fossale, E., Anderson, M., et al. (2005). HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 14, 2871–2880. doi: 10.1093/hmg/ddi319
Sheth, S., Brito, R., Mukherjea, D., Rybak, L. P., and Ramkumar, V. (2014). Adenosine receptors: expression, function and regulation. Int. J. Mol. Sci. 15, 2024–2052. doi: 10.3390/ijms15022024
Shoulson, I., and Young, A. B. (2011). Milestones in huntington disease. Mov. Disord. 26, 1127–1133. doi: 10.1002/mds.23685
Simonin, C., Duru, C., Salleron, J., Hincker, P., Charles, P., Delval, A., et al. (2013). Association between caffeine intake and age at onset in Huntington’s disease. Neurobiol. Dis. 58, 179–182. doi: 10.1016/j.nbd.2013.05.013
Smith-Dijak, A. I., Sepers, M. D., and Raymond, L. A. (2019). Alterations in synaptic functions and plasticity in Huntington disease. J. Neurochem. 150, 346–365. doi: 10.1111/jnc.14723
Spokes, E. G. S. (1980). Neurochemical alterations in Huntington’s chorea: a study of post-mortem brain tissue. Brain 103, 179–210. doi: 10.1093/brain/103.1.179
Stahl, C. M., and Feigin, A. (2020). Medical, surgical, and genetic treatment of Huntington disease. Neurol. Clin. 38, 367–378. doi: 10.1016/j.ncl.2020.01.010
Stockwell, J., Jakova, E., and Cayabyab, F. S. (2017). Adenosine A1 and A2A receptors in the brain: current research and their role in neurodegeneration. Molecules 22, 1–18. doi: 10.3390/molecules22040676
Taherzadeh-Fard, E., Saft, C., Wieczorek, S., Epplen, J. T., and Arning, L. (2010). Age at onset in Huntington’s disease: replication study on the associations of ADORA2A, HAP1 and OGG1. Neurogenetics 11, 435–439. doi: 10.1007/s10048-010-0248-3
Tarditi, A., Camurri, A., Varani, K., Borea, P. A., Woodman, B., Bates, G., et al. (2006). Early and transient alteration of adenosine A2A receptor signaling in a mouse model of Huntington disease. Neurobiol. Dis. 23, 44–53. doi: 10.1016/j.nbd.2006.01.014
The Huntington’s Disease Collaborative Research Group (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-e
Toczek, M., Kutryb-Zajac, B., Zukowska, P., Slominska, E. M., Isalan, M., Mielcarek, M., et al. (2016b). Changes in cardiac nucleotide metabolism in Huntington’s disease. Nucleosides. Nucleotides. Nucleic. Acids. 35, 707–712. doi: 10.1080/1527770.2016.1154969
Toczek, M., Pierzynowska, K., Kutryb-Zajac, B., Gaffke, L., Slominska, E. M., Wegrzyn, G., et al. (2018). Characterization of adenine nucleotide metabolism in the cellular model of Huntington’s disease. Nucleosides. Nucleotides. Nucleic. Acids. 37, 630–638. doi: 10.1080/15257770.2018.1481508
Toczek, M., Zielonka, M., Zukowska, P., Marcnkow ski, J. T., Slominska, E., Isalan, M., et al. (2016a). An impaired metabolism of nucleotides underpins a novel mechanism of cardiac remodeling leading to Huntington’s disease related cardiomyopathy. Biochim. Biophys. Acta 1862, 2147–2157. doi: 10.1016/j.bbadis.2016.08.019
Varani, K., Abbracchio, M. P., Cannella, M., Cislaghi, G., Giallonardo, P., Mariotti, C., et al. (2003). Aberrant A2A receptor function in peripheral blood cells in Huntington’s disease. FASEB. J. 17, 2148–2150. doi: 10.1002/ajmg.b.30223
Villar-Menéndez, I., Blanch, M., Tyebji, S., Pereira-Veiga, T., Albasanz, J. L., Martín, M., et al. (2013). Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington’s disease. NeuroMolecular. Med. 15, 295–309. doi: 10.1007/s12017-013-8219-0
Von Lubitz, D. K., Paul, I. A., Ji, X. D., Carter, M., and Jacobson, K. A. (1994). Chronic adenosine A1 receptor agonist and antagonist: effect on receptor density and N-methyl-D-aspartate induced seizures in mice. Eur. J. Pharmacol. 253, 95–99. doi: 10.1016/0014-2999(94)90762-5
Whitehouse, P. J., Trifiletti, R. R., Jones, B. E., Folstein, S., Price, D. L., Snyder, S. H., et al. (1985). Neurotransmitter receptor alterations in Huntington’s disease: Autoradiographic and homogenate studies with special reference to benzodiazepine receptor complexes. Ann. Neurol. 18, 202–210. doi: 10.1002/ana.410180207
Yegutkin, G. G. (2008). Nucleotide- and nucleoside- converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochem. Biophys. Acta. 1783, 673–694. doi: 10.1016/j.bbamcr.2008.01.024
Yoon, K. W., and Rothman, S. M. (1991). Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus. J. Neurosci. 11, 1375–1380. doi: 10.1523/JNEUROSCI.11-05-01375-1991
Keywords: Huntington’s disease, motor dysfunction, A2A receptors, adenosine, ATP, nucleotide metabolism
Citation: Wiprich MT and Bonan CD (2021) Purinergic Signaling in the Pathophysiology and Treatment of Huntington’s Disease. Front. Neurosci. 15:657338. doi: 10.3389/fnins.2021.657338
Received: 22 January 2021; Accepted: 04 June 2021;
Published: 01 July 2021.
Edited by:
Francisney Pinto Nascimento, Universidade Federal da Integração Latino-Americana, BrazilReviewed by:
Peter McCormick, Queen Mary University of London, United KingdomHyung Nam, Louisiana State University Health Shreveport, United States
Adair Roberto Soares Santos, Federal University of Santa Catarina, Brazil
Copyright © 2021 Wiprich and Bonan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carla Denise Bonan, Y2JvbmFuQHB1Y3JzLmJy; Y2FybGFkYm9uYW5AZ21haWwuY29t