 Manon Dobrigna1
Manon Dobrigna1 Sandrine Poëa-Guyon1Véronique Rousseau1Aline Vincent2Annick Toutain3
Sandrine Poëa-Guyon1Véronique Rousseau1Aline Vincent2Annick Toutain3 Jean-Vianney Barnier1*
Jean-Vianney Barnier1*- 1Institut des Neurosciences Paris-Saclay, UMR 9197, CNRS, Université Paris-Saclay, Saclay, France
- 2Department of Genetics, EA7450 BioTARGen, University Hospital of Caen, Caen, France
- 3Department of Genetics, University Hospital of Tours, UMR 1253, iBrain, Université de Tours, INSERM, Tours, France
Although the identification of numerous genes involved in neurodevelopmental disorders (NDDs) has reshaped our understanding of their etiology, there are still major obstacles in the way of developing therapeutic solutions for intellectual disability (ID) and other NDDs. These include extensive clinical and genetic heterogeneity, rarity of recurrent pathogenic variants, and comorbidity with other psychiatric traits. Moreover, a large intragenic mutational landscape is at play in some NDDs, leading to a broad range of clinical symptoms. Such diversity of symptoms is due to the different effects DNA variations have on protein functions and their impacts on downstream biological processes. The type of functional alterations, such as loss or gain of function, and interference with signaling pathways, has yet to be correlated with clinical symptoms for most genes. This review aims at discussing our current understanding of how the molecular changes of group I p21-activated kinases (PAK1, 2 and 3), which are essential actors of brain development and function; contribute to a broad clinical spectrum of NDDs. Identifying differences in PAK structure, regulation and spatio-temporal expression may help understanding the specific functions of each group I PAK. Deciphering how each variation type affects these parameters will help uncover the mechanisms underlying mutation pathogenicity. This is a prerequisite for the development of personalized therapeutic approaches.
Introduction
The 5th edition of the Diagnostic and Statistical Manual of mental disorders (Crocq and Guelfi, 2015) defines neurodevelopmental disorders (NDDs) as a broad class of brain diseases characterized by a spectrum of early clinical manifestations with developmental delay and cognitive/social impairments representing the most recurrent phenotypes. A non-exhaustive list of NDDs includes intellectual disability (ID), Autism Spectrum Disorders (ASD), Attention-Deficit/Hyperactivity Disorder (ADHD), developmental epilepsies and motor disorders such as cerebral palsy. Schizophrenia (SCZ) and Bipolar disorders (BP) could also be considered as NDDs (Crocq and Guelfi, 2015; Michetti et al., 2022). These psychiatric and neurological disorders participate in a network of neuropsychiatric diseases that share etiology and clinical commonalities related to their complex nosology (i.e., classification of diseases) (Vissers et al., 2016; Owen and O’Donovan, 2017; Glasson et al., 2020; Morris-Rosendahl and Crocq, 2020; Leblond et al., 2021; Vilela et al., 2022). Next-generation sequencing (NGS) has helped identify pathogenic variants in many genes for a large proportion of patients presenting ID and highlights the genetic complexity in NDDs (de Ligt et al., 2012). For instance, more than 1,000 genes have been associated with ID risk and more than 400 with ASD (Willsey et al., 2017; OMIM, 2022). The large number of genes involved makes the identification of pathogenic variants by clinicians and geneticists extremely complex (Piton et al., 2013; Wright et al., 2015; Chiurazzi and Pirozzi, 2016). Hence, distinguishing deleterious variants from neutral polymorphisms is still a challenge for many single nucleotide variations (SNVs), even if they are located in coding sequences. Moreover, different mutations in the same gene can lead to a broad spectrum of clinical symptoms, which makes the etiological characterization of NDDs difficult. An ongoing challenge is to understand how various genetic changes can cause different NDDs. The many approaches developed based on robust algorithms and systematically updated databases open the possibility of elaborating frameworks of analysis for the relationship between genetics and clinics (Gussow et al., 2016; Geisheker et al., 2017; Sanders et al., 2019).
The genes involved in NDDs regulate central signaling pathways that often share a limited number of functions such as chromatin regulation, proliferation and differentiation of neural stem cells, and synaptic plasticity (Ernst, 2016; Forrest et al., 2018; Moretto et al., 2018; Liaci et al., 2021). This convergence of signaling pathways in NDDs can help identify common therapeutic targets. Thus, uncovering the way signaling pathways are disrupted remains a priority to better understand the pathology and define new therapeutic approaches. The RAC1/CDC42 pathway is central to brain development and function since it regulates actin cytoskeleton dynamics, which is essential to cell migration, synaptic plasticity, axon guidance and neurogenesis (Nadif Kasri and Van Aelst, 2008; van Bokhoven, 2011; Penzes and Cahill, 2012; Gentile et al., 2022). The switch between cytosolic, GDP-loaded inactive-GTPases and membrane-bound, GTP-loaded active forms usually regulates this pathway. Two large families control these states: the Guanine Exchange Factor (GEF) and the GTPase-Activating Proteins (GAP). Variations in RAC1, CDC42 as well as those in several GEF such as the PIX guanine exchange factors, and in several GAP such as Oligophrenin, and other genes connected to this RHO GTPase pathway are responsible for NDDs, epilepsy and are often associated with brain structural anomalies (Figure 1A; Frangiskakis et al., 1996; Billuart et al., 1998; Kutsche et al., 2000; Reijnders et al., 2017; Martinelli et al., 2018; Tastet et al., 2019; Barbosa et al., 2020; Halder et al., 2022). Genetic data also helped identify pathogenic variations in Lim Kinases and cofilin involved in neuropsychiatric conditions, confirming the role of the RAC/CDC42/PAKs/LimK/Cofilin pathway in NDDs (Frangiskakis et al., 1996; Tastet et al., 2019; Halder et al., 2022). Cofilin activity is regulated by LIM kinase-dependent phosphorylation, Slingshot-dependent dephosphorylation, and other interactions with protein partners. Cofilin controls dendritic spine dynamics and synaptic expression of glutamate receptors in the post-synaptic compartment, as well as vesicle exocytosis and neurotransmitter release in the presynaptic compartment (Ben Zablah et al., 2020).
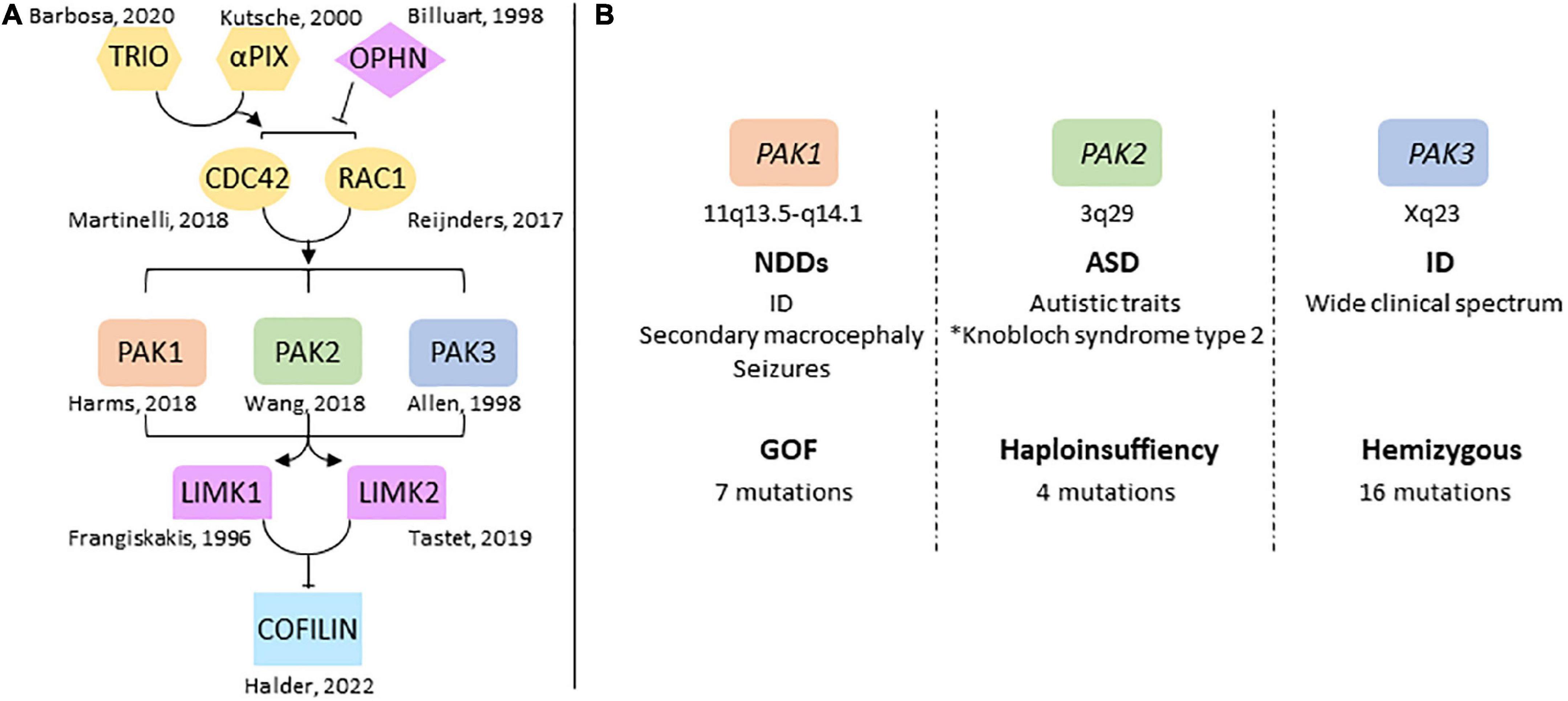
Figure 1. Overview of group 1-PAKs in NDDs (A) genes of the RAC1/CDC42 pathway are involved in NDDs. The identification of pathogenic variations in several genes highlights the central role the dysregulation of this pathway plays in NDDs, notably via its control of actin cytoskeleton dynamics through cofilin. (B) Chromosomal location of PAK genes and conditions generally associated with PAK variations. PAKs are associated with very different mutational and clinical landscapes. *Antonarakis et al. (2021).
p21-activated kinases (PAKs) mediate several functions downstream of RAC1 and CDC42. They regulate actin cytoskeleton dynamics via Lim kinase activation and phosphorylation-dependent cofilin inhibition (Bokoch, 2003; Boda et al., 2010; Ben Zablah et al., 2020; Zhang K. et al., 2022). PAKs are divided in two groups according to their sequence identity, their structural properties, and regulatory mechanisms (Eswaran et al., 2008). Group I PAKs includes PAK1, 2 and 3; group II includes PAK4, 5 and 6 which have not been associated with NDDs yet (Jaffer and Chernoff, 2002; Bokoch, 2003). Several reviews describe the biological functions of PAKs in cell signaling and neuronal pathophysiology (Bokoch, 2003; Kreis and Barnier, 2009; Koth et al., 2014; Kumar and Li, 2016; Civiero and Greggio, 2018; Wang and Guo, 2022; Zhang K. et al., 2022). Functions of group I PAKs are similar in a broad way because of their high sequence identity, their common modes of regulation as well as the substrates they share. Recent data from human genetics highlight their implication in NDDs. Pathogenic variations of PAK1, PAK2 and PAK3 are responsible for ID and/or psychiatric disorders, and are often associated with brain anomalies (Allen et al., 1998; Harms et al., 2018; Wang et al., 2018). However, the clinical spectrum associated with mutations in each PAK genes is different (Figure 1B). This review aims to provide an overview of the different clinical cases associated with PAKs thus far, to reflect on the effects of mutations on PAK functions, and how PAK expression may be involved in the symptomatology of PAK-associated disorders.
History of case reports and clinical data
PAK3: From non-syndromic XLID to a complex clinical pattern
We briefly describe here the history of the identification of PAK genetic variants and the implementation of concepts that have accompanied these discoveries over the past 25 years (Table 1). The autosomal location of the two PAK1 and PAK2 genes explains the delayed timing of the discovery of their genetic involvement in NDDs, compared to the X-linked PAK3 gene. The first evidence of a genetic factor in ID comes from studies of its transmission to men in large families and the analysis of the segregation of chromosomal markers. Because of the male-to-female ratio of patients affected with ID, the X chromosome has been linked to this pathology early on (Lehrke, 1972; Chelly and Mandel, 2001; Gécz et al., 2009; Lubs et al., 2012). Variant identification was carried out by analyzing karyotypes and segregation of markers, thus requiring cross-referencing of data from several patients. Then, the advent of NGS has made it possible to identify variations in the DNA of isolated patients. The first pathogenic mutation in the PAK3 gene was discovered in a family (MRX30) described in 1996. The region involved was mapped between Xq21.3 and Xq24 (Donnelly et al., 1996) and the variant was later identified as p.(Arg419*), and corresponds to a nonsense mutation in the sequence encoding the kinase domain of the PAK3 gene (Allen et al., 1998). The second variant identified, p.(Arg67Cys), was the first missense mutation found within the PAK3 gene (des Portes et al., 1997; Bienvenu et al., 2000). The MRX47 family bearing this mutation was previously described in 1997 (des Portes et al., 1997). This mutation localized in the regulatory domain modifies PAK3 affinity for GTPases (Kreis et al., 2007). The p.(Ala365Glu) substitution within a highly conserved region of the kinase domain was detected in 19 males in a family spread over five generations. Patients bearing this PAK3 variant were diagnosed with mild non-syndromic X-linked ID with a few individuals also presenting neuropsychiatric problems such as aggressiveness, antisocial behavior, psychosis, depression or epilepsy (Gedeon et al., 2003). In 2007, a p.(Trp446Ser) mutation, located in the kinase domain and affecting 12 patients from a four-generation family was published (Peippo et al., 2007). It was the first description of learning difficulties in four carrier females that displayed borderline to mild ID (Peippo et al., 2007). Two males had drooling, otherwise all four males presented inarticulate speech, short attention span, anxiety, restlessness and aggressiveness and one of them had a small head. These data indicate that PAK3 variations can be associated with psychiatric traits and brain anomalies, thus being responsible for a more complex clinical description than non-syndromic ID (Figure 2). Indeed, microcephaly is now often associated with PAK3 variations. Notably, in 2008, two brothers bearing the first frameshift variant due to a four-bases insertion at the end of the sixth exon p.(Gly92Valfs*35), located after the second coding exon were described with ID, aggressiveness and microcephaly (Figure 3; Rejeb et al., 2008). This clinical report, combined with the previous literature, prompted these authors to suggest that PAK3-associated disorders should be considered as a distinguishable ID syndrome. In 2014, the p.(Lys389Asn) mutation associated with severe ID was described in two boys that displayed microcephaly, corpus callosum agenesis (CCA) and cerebellar hypoplasia and one fetus that displayed CCA and septum pellucidum agenesis (Magini et al., 2014). The two patients later died from epileptic seizures at age 6 and 8. The in vitro and in vivo analysis of the functional consequences of this variant indicated a large effect on cell signaling and on craniofacial structures. A less severe case was described soon after, the p.(Arg493Cys), responsible for borderline ID, epilepsy and hemiplegic cerebral palsy (McMichael et al., 2015). During the same period, the p.(Val294Met) was identified in patients displaying moderate ID, ADHD and microcephaly (Muthusamy et al., 2017). Soon after, a missense mutation, p.(Tyr427His), described in monozygotic twins, was associated with macrocephaly in addition to developmental delay, ID and autistic features, such as sensory processing issues (Hertecant et al., 2017). It was the first time macrocephaly was associated with PAK3 mutations. More recently, a patient bearing a p.(Ser527Gly) mutation, located in the kinase domain, was described with severe ID, epilepsy, and self-injurious behavior (Horvath et al., 2018). Since 2018, almost only severe ID was described, such as the p.(Val326Leu) variation in a boy, the p.(Trp428Arg) variation in two siblings or the p.(Gly424Arg) variation in a familial case (Iida et al., 2019; Duarte et al., 2020; Nagy et al., 2020). The latter case displayed global developmental delay, stereotypies, and was later diagnosed with secondary microcephaly in addition to ID and corpus callosum dysgenesis. The same pathogenic variant was found retrospectively in the fetus from a previous pregnancy terminated after detection of CCA and hydrocephaly (Duarte et al., 2020). It was not until 2021 that female cases were reported as probands. Indeed, some pathogenic variations are now described in girls. The first variation responsible for such a case is the p.(Lys389Thr), which was also the second described substitution affecting the Lys389 residue. The patient bearing this mutation displayed ID, secondary microcephaly and ADHD (Pascolini et al., 2021). The published p.(Pro229Ser) variation indexed in PAK3cb, which corresponds to a p.(Pro193Ser) variation in the PAK3a isoform (Figure 3B), is associated with ID, microcephaly and combined immunodeficiency syndrome in the proband girl (Almutairi et al., 2021). To summarize, the clinical picture associated with PAK3 mutations was initially characterized as non-syndromic ID but the identification of additional PAK3 mutations has unveiled the comorbidity of ID with many psychiatric traits as well as morphological abnormalities of the brain. PAK3 mutations can lead to ID of varying severity and may be associated with autistic traits, ADHD, paranoid psychosis, aggressiveness, self-harm, and other neurological signs such as epilepsy and motor disorders.
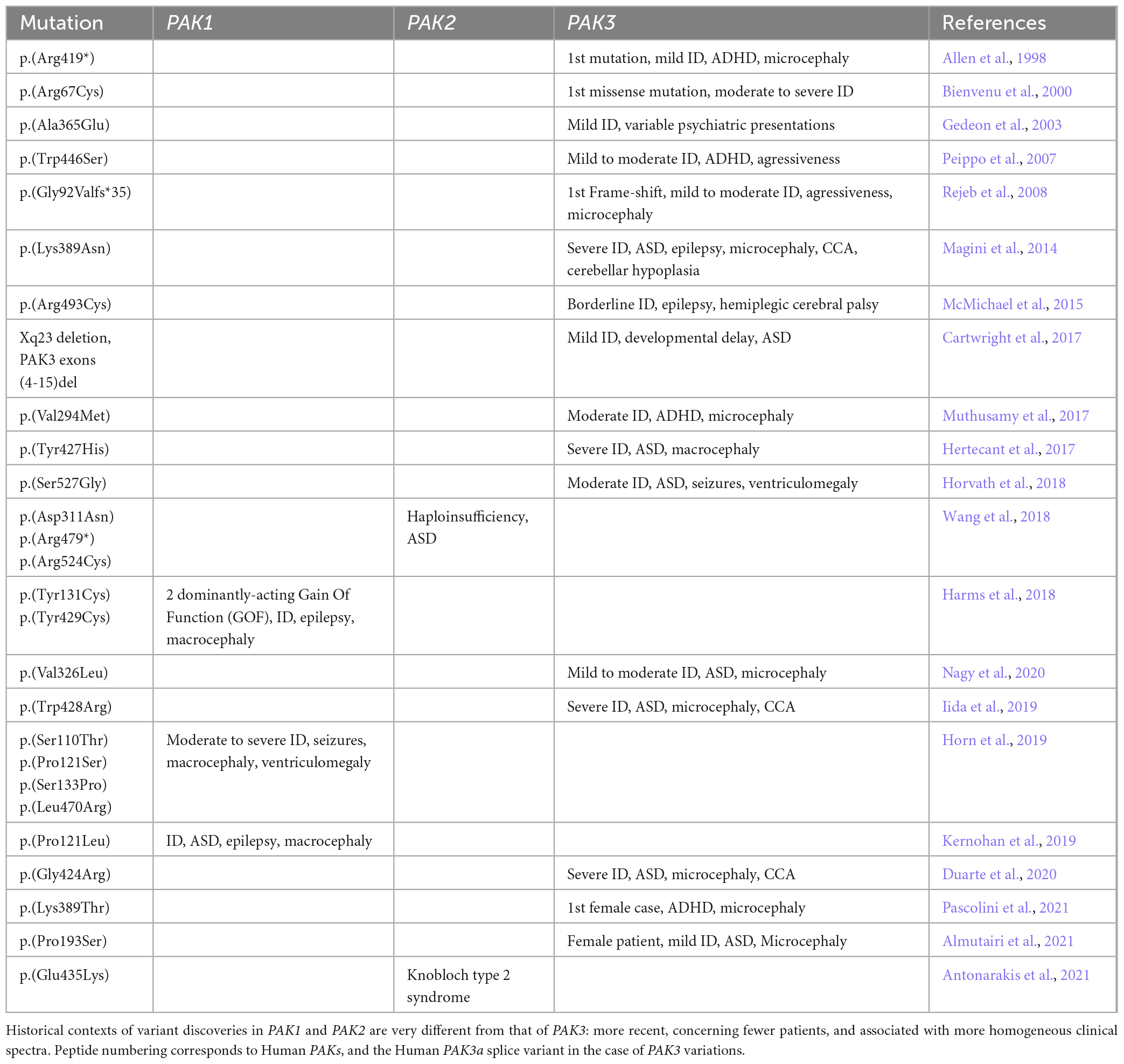
Table 1. Historical discoveries of PAK variations in NDDs.
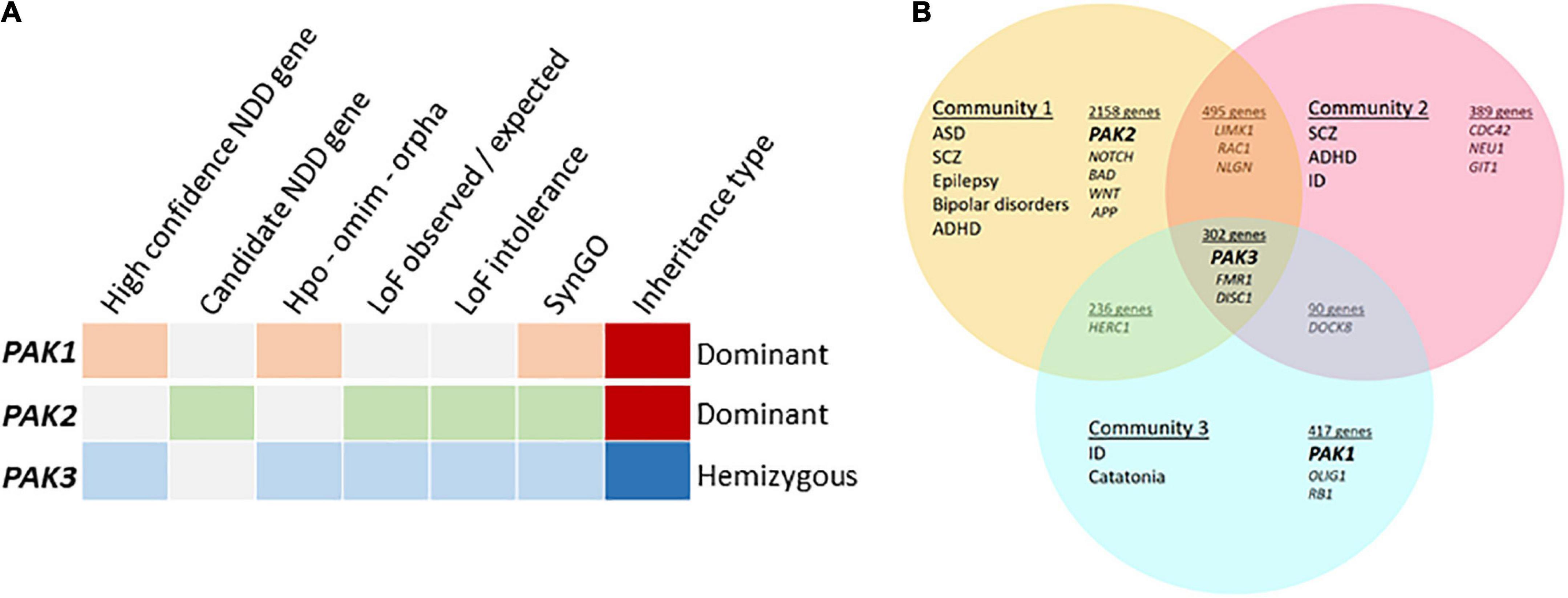
Figure 2. Meta-analyses of group I PAK-associated NDDs (A) PAKs in different databases, categories highlighted when PAK1 (orange), PAK2 (green) or PAK3 (blue) are detected. The high confidence NDD gene category was created by GeneTrek (Leblond et al., 2021) using data-mining from SFARI, SPARK, SyslD, DDG2P, and DBD databases. The candidate NDD gene category includes candidate risk genes from the same databases (except SPARK). HPO-OMIM-Orpha: other databases. Loss of function (LOF) observed or expected: LOF can be expected from genes in this category. LOF intolerance corresponds to genes for which LOF leads to pathogenic effects. Genes from SYNGO database encode synaptic proteins. (B) PAK genes are associated with different community of diseases identified by Vilela et al. (2022).
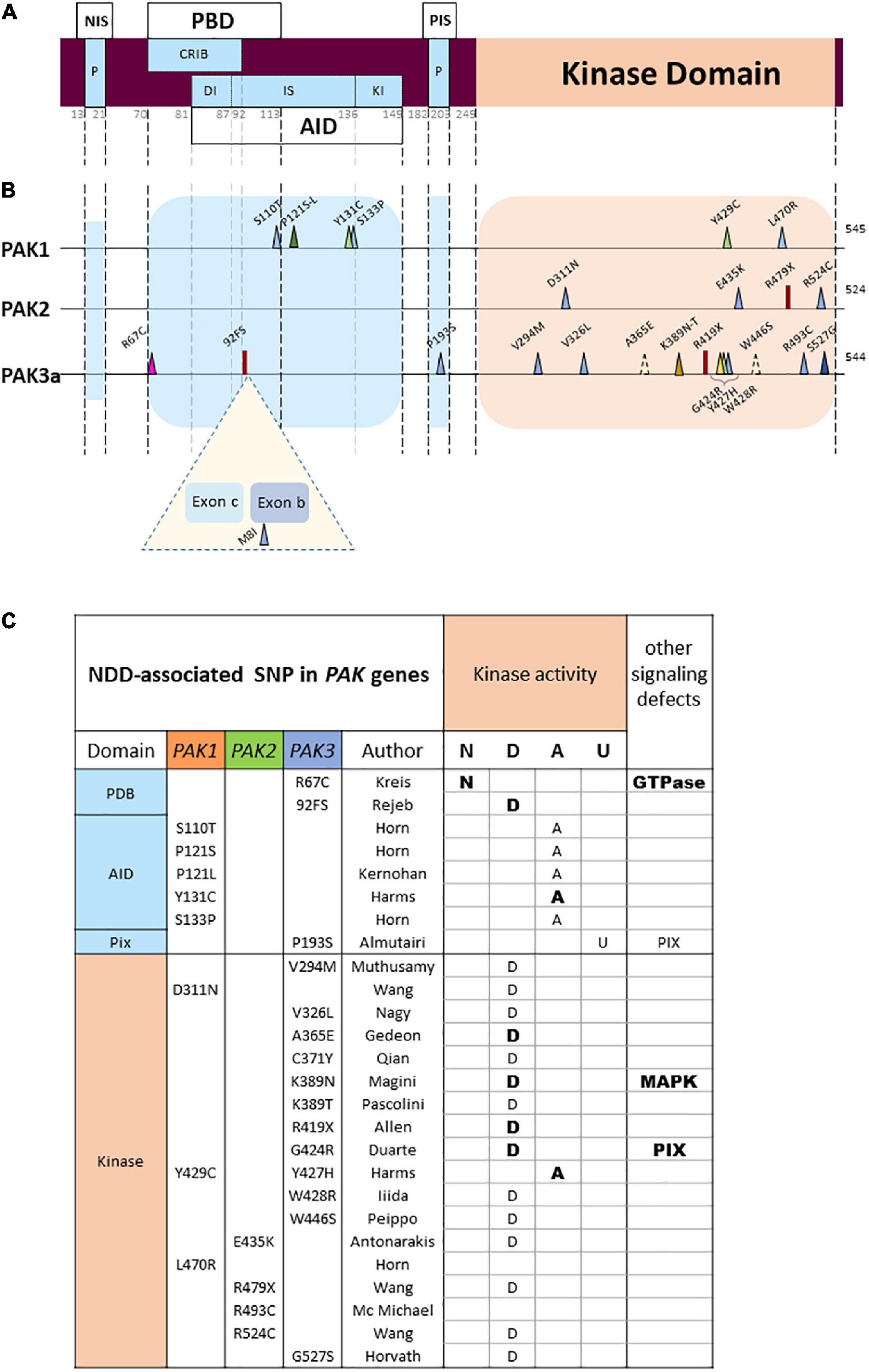
Figure 3. PAK general structure and localization of variants (A) Domain structure of PAKs. NIS, Nck interacting sequences; P, praline-rich sequences; PBD, p21 binding domain; AID, auto-inhibitory domain; DI, Dimerization segment; IS, Inhibitory switch domain; Kl, kinase inhibitory segment; PIS, PIX interacting sequences. Numbering of the general structure is based on PAK 1. (B) Location of missense and LGD (likely gene disruptive) variations. Darker colors represent reccurrences. Both PAK3 alternative exons are represented (Exon b: 15 amino-acids; exon c: 21 amino-acids). (C) Validated or hypothetical functional alterations of PAK variants. N: normal kinase activity; D: defective; A: activated; U: unknown. Bold: experimentally validated; normal: hypothetical.
PAK1 in developmental delay and macrocephaly
The identification of deleterious variations in the two other PAK genes is recent. The first mutation in the PAK1 gene, p.(Arg500His), was reported in a meta-analysis of more than 2,000 patients with ID, but PAK1 was not considered as a gene responsible for ID at that time (Lelieveld et al., 2016). In 2018, two p.(Tyr131Cys) and a p.(Tyr429Cys) mutations were identified in two unrelated patients displaying macrocephaly, ID, seizures, speech impairment and attention deficits (Harms et al., 2018). In 2019, other PAK1 variants including p.(Ser133Pro), p.(Ser110Thr), p.(Pro121Ser) and p.(Leu470Arg) were reported in subjects with ID, macrocephaly and seizures in three boys and one girl, respectively, (Horn et al., 2019; Ohori et al., 2020). They are localized either in the regulatory domain or in the kinase domain such that their translated variants are predicted to suppress the auto-inhibitory mechanism, thereby directly activating or indirectly leading to PAK1 activation. In 2019, a second occurrence affecting the Pro121 residue was reported. This p.(Pro121Leu) mutation is responsible for an autosomal dominant disorder with severe regressive ASD, ID and epilepsy (Kernohan et al., 2019).
PAK2 variants are associated with ASD
The PAK2 gene is localized at the end of the long arm of chromosome 3 and belongs to the 3q29 locus that is relatively frequently rearranged in patients with NDDs. The 3q29 duplication syndrome is associated with ID, microcephaly, cerebral palsy, and epilepsy. The minimal duplicated interval involves 22 annotated genes, including PAK2 and DLG1 (Lisi et al., 2008; Fernández-Jaén et al., 2014; Wen et al., 2022). Likewise, deletion of the 3q29 locus is associated with ASD (Sanders et al., 2015). Clinical studies reported ASD in patients bearing PAK2 missense mutations or with PAK2 deletions, causing PAK2 haploinsufficiency (Wang et al., 2018). One missense p.(Glu435Lys) variation was recently identified by exome sequencing in two siblings displaying a Knobloch-like syndrome designated as KNO2 with ID, autistic behavior, retinal degeneration, and encephalocele (Antonarakis et al., 2021).
Despite the descriptions above, non-cognitive recurring symptoms have also been identified for several patients bearing PAK1 or PAK3 mutations. Some traits such as general hypotonia, drooling, nystagmus and strabismus, speech difficulties, and tremor, that may be due to neurological and/or neuromuscular defects, are described for patients bearing PAK1 or PAK3 variations. There are also some non-neurological symptoms associated with PAK variants. Interestingly, skin anomalies, such as café-au-lait spots, were described for the majority of patients bearing PAK1 mutations and associated with some PAK3 variations (Magini et al., 2014; Hertecant et al., 2017; Harms et al., 2018; Pascolini et al., 2021). These anomalies may be due to defects in the activation of the Ras-MAP kinase pathway, leading to RASopathy (Magini et al., 2014). Facial dysmorphic features are also commonly associated with PAK1 and PAK3 variations (deeply set eyes, large ears, hair alopecia, bushy eyebrows, nose shape, palate, etc.). A gross analysis suggests that the more severe the cognitive defects, the more non-neurological symptoms are present. This is probably due to the more damaging PAK defects in these cases. It should be noted that there is still no thorough clinical description of patients bearing PAK2 variants, except for the KNO2 syndrome.
Clinical overview of PAK variants
The description above indicates that variations are associated with different conditions depending on each PAK gene: PAK1 pathogenic variants are associated with developmental delay, ID, ASD and macrocephaly, PAK2 variants with ASD only and PAK3 variants with ID, often along with ASD, ADHD, epilepsy, and, in numerous cases, microcephaly. The pathophysiological landscape associated with each PAK may also be observed from genetic database mining or from meta-analysis data in web sites such as GeneTrek, which surveys the association of human genes with NDDs (Figure 2A; Leblond et al., 2021). This could also be illustrated by the analysis of disease communities based on comorbidities and clinical characteristics in a disease network approach to explore the shared genetics between ASD, ID, ADHD, SCZ, BP, and epilepsy (Figure 2B; Vilela et al., 2022). As expected, the three PAK genes segregate with different disease communities. PAK1 is found within the community mostly defined by ID, PAK2 is found in the community mostly characterized by ASD, though the comorbidities associated with ASD make this community highly heterogeneous. Indeed, around 70% of individuals with ASD display at least one co-occurring NDD or form of epilepsy (Rosen et al., 2018). Furthermore, even though PAK2 variations are mostly associated with ASD, the number of cases described at present is not sufficient to get a precise idea of the extent of clinical consequences. For example, the PAK2 mutation associated with KNO2 syndrome (Antonarakis et al., 2021) already differs from previous descriptions of PAK2 clinical cases that were exclusively associated with ASD. For PAK3, there is a large clinical variability so that PAK3 is found in all three communities (Figure 2B). However, the exhaustive list of clinical traits associated with PAK3 pathogenic variations is difficult to establish because of heterogeneous clinical descriptions of the patients, such as the relative importance given to certain traits compared to others or the context of medical practices. The absence of mutation hot spots precludes the description of a precise PAK3-associated syndrome and prevents ruling out effects other mutations or the environment might have on PAK3-associated NDDs (Lelieveld et al., 2016).
The meta-analyses previously mentioned (Leblond et al., 2021; Vilela et al., 2022) highlight the complex relationship between PAK variations and the extent of neuropsychiatric symptoms. Several reasons might explain the differences observed in the clinical symptoms associated with each PAK: the paucity of described variations, especially for PAK1 (7) and PAK2 (4), and the different functions of each PAK isoform. Furthermore, the scarcity of biochemical characterization of PAK variants limits our understanding of these differences. To summarize, the study of PAK-associated disorders is currently incomplete due to several hindrances. Yet, the improvement of variant detection methods, the homogenization of clinical analyses and the collaborative approaches between clinicians and researchers will support a more thorough approach.
PAK genetic context governs the expression of patient conditions
An important difference between PAKs is that the variations in PAK1 and PAK2 genes are transmitted according to an autosomal and dominant inheritance pattern, while those in PAK3 are X-linked and hemizygous. Both PAK1 and PAK2 are autosomal genes, respectively, located on chromosome 11 and 3. Pathogenic variations in PAK1 are monoallelic de novo mutations displaying dominant functional effects (Harms et al., 2018; Horn et al., 2019; Kernohan et al., 2019). The missense PAK2 variations are de novo or inherited (Zhang K. et al., 2022). The newly described PAK2 mutation, responsible for the KNO2 syndrome, is monoallelic and detected in both siblings, indicating a likely germ-line mosaicism in one of the parents (Antonarakis et al., 2021). Whether this variant is responsible for haploinsufficiency is still unknown. For PAK3, most variants are inherited and only two of them, p.(Lys389Thr) and p.(Ser527Gly) occurred de novo (Horvath et al., 2018; Pascolini et al., 2021).
Variants in genes localized on the X chromosome form a special class because of the unique mode of allele expression: in males, the mutated allele is expressed in all cells whereas there is a mosaic expression of only one of the two alleles in female cells. In this instance the genetic approaches for patient cohorts with X-linked disorders is particular (Piton et al., 2013; Leitão et al., 2022). Until 2014, PAK3 mutations were identified in large families over several generations and were found to induce less severe symptoms than in most recent cases, whom mutations are responsible for severe ID associated with other psychiatric traits. This change in case reports is probably due to a bias induced by the evolution of diagnostic methods, such as NGS, and to the fact that PAK3 is now more investigated because it is recognized as a gene involved in NDDs. The women bearing PAK3 mutations were either unaffected female carriers or mothers presenting borderline to moderate ID (Peippo et al., 2007; Rejeb et al., 2008). Although skewed X-inactivation has been observed in several women, this could not be correlated with clinical characteristics. Unfortunately, there are no further data on more recently identified female probands (Almutairi et al., 2021; Pascolini et al., 2021). Despite the difference between men and women in PAK-associated disease transmission, no evidence suggests that differences in PAK functions exist between the two sexes.
Another observation is that each PAK displays a unique mutational pattern (Figure 3B). The landscape of genetic abnormalities of the three PAK genes is also particular insofar as current published data are mainly made up of missense mutations. For the three genes combined, there are twenty-three missense mutations, two nonsense mutations and one splice site mutation inducing a frameshift, as well as one multi-exonic deletion (Cartwright et al., 2017). There are only few recurrences. One double occurrence is on the PAK1 proline 121 residue (Horn et al., 2019; Kernohan et al., 2019). For PAK3, two identical mutations p.(Ser527Gly) have been published as well as two variants affecting the same lysine 389 residue (Magini et al., 2014; Tzschach et al., 2015; Horvath et al., 2018; Pascolini et al., 2021). Interestingly, two mutations affecting homologous residues in the PAK1 and PAK3 paralogs, the p.(Tyr429Cys) and p.(Tyr427His), respectively, lead to similar clinical phenotypes associating ID, ASD and macrocephaly (Hertecant et al., 2017; Harms et al., 2018). Of note, the low number of described cases may explain the low number of recurrences and the absence of mutation hotspots.
PAK mutations affect kinase functions, mechanisms of regulation and partner interactions
Nonsense mutations, monogenic deletion, and splice site mutations inducing a frameshift are qualified as Likely Gene Disrupting variations (LGD) that are responsible for loss of function or loss of protein (Figure 3B). These mutations could lead to truncated forms lacking kinase activity and exerting dominant-negative effects or signaling interference, but this possibility has not been thoroughly explored yet. However, mRNAs bearing premature termination codon can be degraded by the translation control system, named Nonsense-Mediated mRNA decay (Supek et al., 2021). This would lead to the total absence of defective proteins, responsible for a complete loss of function. PAK3 LGD mutations are responsible for relatively mild to moderate severity of the disease. The PAK2 gene is considered intolerant to loss-of-function (Figure 2A), and the PAK2 nonsense mutation is also associated with haploinsufficiency. Furthermore, heterozygous LGD mutations in ASD-related genes are frequently predicted to cause haploinsufficiency (Parenti et al., 2020).
Other mutations are missense mutations and their variations could be associated with Loss Of Function (LOF) or Gain Of Function (GOF). In the case of kinases, these denominations correspond to complete loss of kinase activity or high constitutive kinase activity, respectively. This classification as LOF and GOF may be used to qualify the functional defects of the PAK1, PAK2 and some PAK3 variants. However, it does not cover the full range of functional alterations observed for other PAK3 variants. Indeed, PAKs are multifunctional proteins, with a kinase activity but also scaffolding functions, thus leading to kinase-dependent and kinase-independent roles. The best described of these kinase-independent functions is the recruitment of PIXs, which makes it possible to assemble a GEF/GTPase/effector complex, thus ensuring specificity and efficiency of the GEF/GTPase/PAK module (Obermeier et al., 1998). In other words, interactions with certain partners such as α/βPIX, Nck1/2 and Grb2 promote the formation of signaling complexes associated with membrane recruitments, and mutations in these interaction domains could significantly affect PAK signaling. Finally, pathogenic variations have only been identified in the coding sequences of group I PAKs. Given the importance of non-coding region variations in the etiology of NDDs, it would be advisable to look more closely for these kinds of changes (Takata, 2019).
PAK1 to 3 share an overall structure composed of a carboxy-terminal catalytic domain and an amino-terminal regulatory region (Figure 3A; Bokoch, 2003). Notwithstanding, PAK amino-acid sequences diverge in specific areas and those primary sequence differences, that are conserved among vertebrates, may support specific functions. The regulatory region consists of two overlapping domains, one is the p21-binding domain (PBD), and the other one is an auto-inhibitory domain (AID). The kinase domain comprises two lobes: the small lobe in N-ter is essentially composed of β-sheets while the larger lobe in C-ter is mainly constituted of α-helixes, in which several residues can interact with the auto-inhibitory domain (Lei et al., 2000; Parrini et al., 2002). In resting conditions, when PAKs are inactive, they form dimer comprised of two molecules in an asymmetric, antiparallel, manner, one monomer adopting an active conformation, and the other one an inactive (Wang et al., 2011). This trans-inhibited dimer is activated by several mechanisms that modify its conformation, induce a multi-step dissociation, and trigger several events of cis- and trans-phosphorylation. It is, however, possible that there was a bias in some crystallographic analyses, since the kinase-dead variant of PAK1 retains a very low, but sufficient, residual activity that allows its autophosphorylation (Wang et al., 2011). The crystallization carried out in the presence of phosphatases suppress this residual phosphorylation and generates inactive proteins in a monomeric conformation. Furthermore, the first step in the activation process corresponds to the transient formation of dimers (Sorrell et al., 2019).
Almost all pathogenic mutations currently identified are located in two domains corresponding to the kinase domain and the PBD/AID regulatory domain (Figure 3B), a fact that is probably due to the strong structural constraint observed in the catalytic and regulatory domains of kinases. In contrast, non-pathogenic single nucleotide polymorphisms (SNPs) extracted from databases are mainly distributed in sequences coding for non-functional, more flexible, zones. Note also the seemingly total penetrance of PAK1 and PAK2 mutations, as well as PAK3 mutations in boys.
In addition to the slight structural differences between the three group I PAKs, the PAK3 gene also shows specificity as it can encode four splice variants (Rousseau et al., 2003; Kreis et al., 2007). PAK3a is devoid of any insertion between the position 92 and 93 of the amino acid sequence. PAK3b contains an insertion of 15 amino acids at this position. PAK3c displays an insertion of 21 amino acids, located between the coding exon 2 and exon b. Finally, PAK3cb (also called PAK3d) exhibits both b and c alternative exons (Figure 3B). To date, a gross characterization of the expression of PAK3 splice isoforms has been conducted using semi quantitative RT-PCR, since the splice variant-specific antibodies available at present cannot work for immunofluorescent detection. All spliced isoforms have been detected in the mouse adult brain and observed as early as E15. Western blot analysis of the 544aa-PAK3a and 559aa-PAK3b proves that these isoforms are equally expressed in the adult mouse brain. However, there is a significantly lower expression of the two 565aa-PAK3c and 580aa-PAK3cb. These alternative exons change drastically the biochemical properties of PAK3. For example: the PAK3b AID cannot inhibit PAK3a (Rousseau et al., 2003). Furthermore, PAK3b, c and cb display high constitutive kinase activity, and thus do not require GTPase activation. The difference in regulatory mechanisms and expression levels between PAK3a, PAK3b, PAK3c, and PAK3cb should be taken into account when estimating mutation pathogenicity. For example, one PAK3 mutation was identified in the alternatively spliced exon b (Figure 3B) in a patient reported in a cohort of cerebral palsy (McMichael et al., 2015), even though new mining from a polymorphism database suggests that this mutation actually corresponds to a neutral polymorphism.
Variations in functional domains
The PBD directly interacts with active GTP-loaded RAC1 and CDC42 (Lei et al., 2000). At first glance, the classical mechanism of activation by GTPases appears to be similar for the three PAKs, considering the similarity of the protein structures involved. The CDC42 and RAC1 interactive binding domain (CRIB) is a consensus sequence found in downstream effectors of RAC1 and CDC42 that constitutes the core of the PBD and is necessary for PAK activation and its subcellular location. Other PBD residues are also heavily involved in PAK selective interactions with GTPases (Knaus et al., 1998; Sun et al., 2019). Only one mutation affects this domain: the missense PAK3 p.(Arg67Cys) mutation that is located at the limit of the PBD. PAK3 normally displays a higher affinity for CDC42 than for RAC1 according to in vitro assays, but the ID-associated p.(Arg67Cys) variant has more affinity for RAC1 than for CDC42, which changes its dynamics of activation and explains its property to induce immature dendritic spines in hippocampal cells (Kreis et al., 2007). GTPases of the Rac/Cdc42 family, that also interact with PAKs and may be involved in the pathophysiology of PAK variants, are still insufficiently explored in the context of PAK-associated disorders. Especially when taking into consideration that pathogenic variations in RAC3 were recently identified in patients with developmental delay, brain anomalies, and facial dysmorphic features (Scala et al., 2022). Like RAC1, RAC3 is ubiquitously expressed in the brain and evidence suggests that RAC1 and RAC3 share functions in neuron survival (Katayama et al., 2023). In double Rac1/3-KO mice, which display severe neuronal loss in the postnatal cerebral cortex, PAK is downregulated and the over-expression of constitutively active PAK1 rescues neuron survival and differentiation. All other GTPases of the Rac/Cdc42 family interact with PAKs but with no indication of their involvement in NDDs, as currently indicated by Decipher or ClinVar databases.
The AID is made of three highly conserved subdomains: a dimerization interface (DI) that contributes to the stabilization of PAK dimers, the inhibitory switch (IS) that partially overlaps the GTPase binding region, and a kinase inhibitor domain (KI) carboxy-terminal to the IS. PAK1 crystallographic structure highlights its ability to form dimers (Lei et al., 2000; Parrini et al., 2002). However, PAK3 forms preferentially heterodimers with PAK1 rather than homodimers, in vitro and in vivo (Combeau et al., 2012). Dimers are maintained inactive via the DI and the IS (Figure 4). The IS is folded against the catalytic region which favors the positioning of the KI domain into the catalytic cleft. The KI domain of each PAK within the dimer interacts with the catalytic site of the other PAK, thus inducing a trans-inhibition (Lei et al., 2000; Parrini et al., 2002). In their dimeric form, the CRIB domain in the PBD of each PAK is exposed at the surface, allowing PAK binding to an active small G protein.
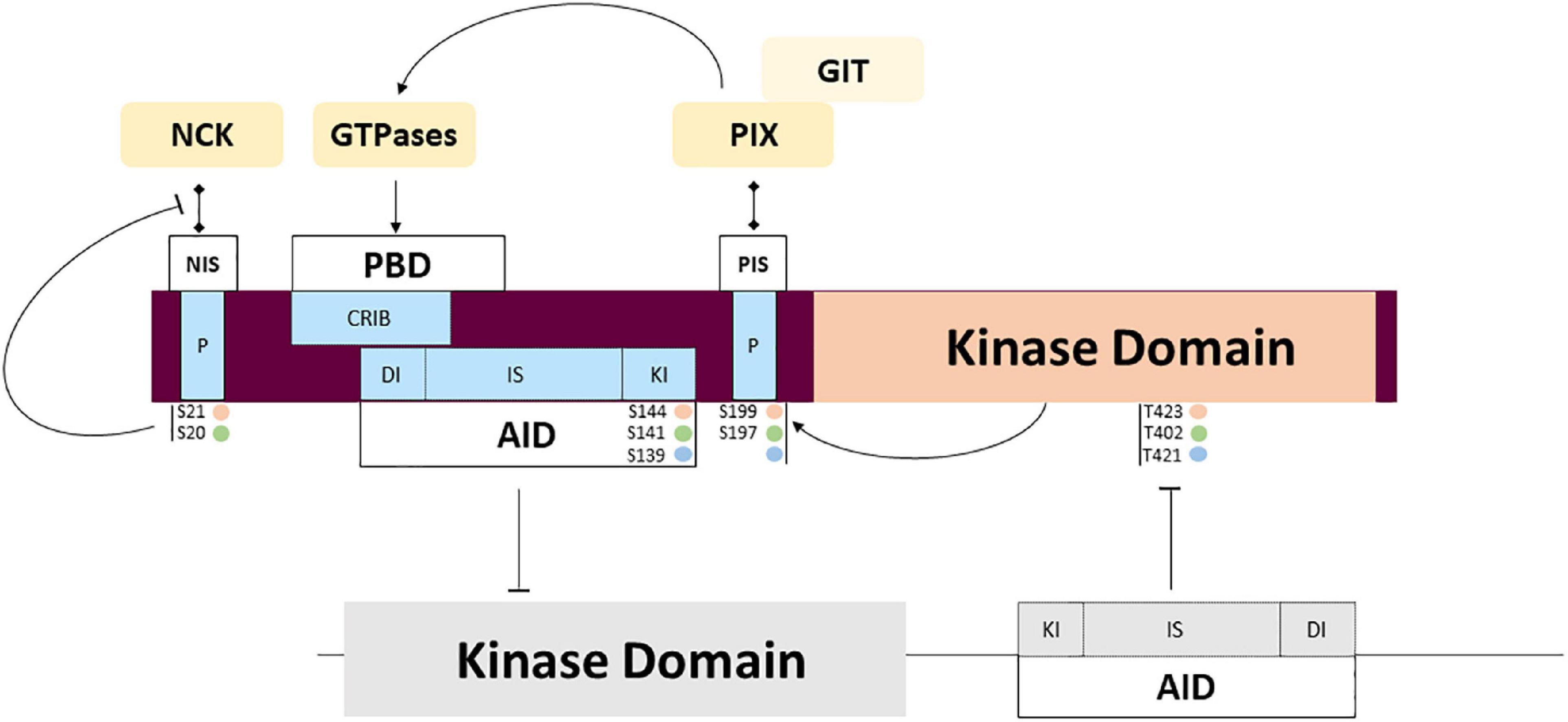
Figure 4. PAK regulation through partner interactions and intramolecular signaling. Complex mechanisms of regulation are triggered by interaction with activators and adaptors that may regulate activity and subcellular location. Several auto phosphorylation sites also participate in the regulation of interaction with the partners mentioned above, and the transitory location and activation of PAKs. Phosphorylation sites corresponding to PAKl (orange), PAK2 (green) and PAK3 (blue).
PAK1 mutations are mainly located in the regulatory domain (5/7). It is expected that mutations affecting the AID impair inhibitory interactions and induce constitutive activation (Tu and Wigler, 1999). To date, five PAK1 mutations affecting the Ser110, Pro121, Tyr131 and Ser133 residues were thus identified in the AID and associated with ASD, ID and macrocephaly (Harms et al., 2018; Horn et al., 2019; Kernohan et al., 2019). Indirect experimental data strongly suggest that these PAK1 variants display constitutive or higher basal kinase activity (Harms et al., 2018). Moreover, the inhibitory domain of a given isoform acts on all three paralogs, and heterodimer formation facilitates crosstalk between PAK signaling. Consequently, mutations in the AID domain should lead to kinase activation of the variant and possibly heterodimerization defects, thus altering the kinase activity of other isoforms (Combeau et al., 2012).
In contrast, a greater proportion of missense mutations are located in the kinase domain of PAK2 (3/3) and PAK3 (11/14). Several PAK mutations are located in the kinase domain: eleven are inactivating mutations and four are activating. Indeed, mutations in the kinase domain, such as the ones mostly found in PAK2 and PAK3, usually disrupt kinase activity. This was particularly well described in the biochemical characterization of several PAK3 mutations in vitro (Allen et al., 1998; Kreis et al., 2007; Magini et al., 2014; Duarte et al., 2020). PAK2 mutations, also located in the kinase domain, are probably associated with loss of function. Structural data, computational analysis, and mutation compilations already established in the field of cancer may help analyzing the effect that kinase variants have on kinase activity, but biochemical characterization is often necessary (Dixit and Verkhivker, 2014).
Proline-rich sequences are involved in regulated interactions with important partners. Among the other conserved features, several highly conserved proline-rich regions found in the amino-terminal tail serve as binding sites for SH3 domains. Two sites are involved in binding adaptor proteins NCK1/2 and GRB2 which is involved in axonal guidance and synaptic transmission through PAK recruitment at the membrane. Isoform particularity is that the autophosphorylation of the PAK1 Serine 21 residue negatively regulates PAK/NCK interactions (Zhao et al., 2000). Interestingly, PAK3 was shown to preferentially bind NCK2/GRB4 compared to NCK1 and this interaction is not regulated by autophosphorylation (Thévenot et al., 2011). The absence of mutations identified in the NCK-binding domain is probably due to the current low sampling of mutations. The second proline-rich sequence is not completely conserved between the different PAK proteins and GRB2 interaction was only demonstrated for PAK1 (Puto et al., 2003). This pathway is probably impacted by mutation of the non-conventional kinesin KIF26A that inhibits GRB2, as observed in patients with severe brain malformations (Qian et al., 2022).
The central proline-rich sequence located between the AID and the kinase domain corresponds to an atypical SH3 binding domain that strongly binds the PAK interacting exchange factor (PIX/COOL) (Bagrodia et al., 1998; Manser et al., 1998). There are two PIX isoforms in mammals: the ubiquitously expressed ɑPIX, encoded by the ARHGEF6 gene (Manser et al., 1998) and βPIX encoded by ARHGEF7 and more specific to the brain (Koh et al., 2001). The numerous splice variants of PIX form different transient complexes with PAKs. These PIX-PAK complexes are regulated by phosphorylation. PIXs are also important in the recruitment of PAKs to the membrane and their activation. This complex located at the membrane enables the activation of GTPases and selectively favors PAK activation. The particularities of the different PAK isoforms in these molecular mechanisms are not known (Zhang, 2005; Zhou et al., 2016). ɑ/βPIX and GIT1/2 form larger complexes that interact with numerous proteins such as Paxillin and Shank, to coordinate their activity in cells and at synapses (Hashimoto et al., 2001; Parker et al., 2013; López-Colomé et al., 2017; Zhu et al., 2020). Disruption of the ARHGEF6 gene leads to severe ID, dysmorphic features and sensorineural hearing loss (Kutsche et al., 2000). The mutation located near the PIX-binding sequence in PAK3 was recently described in a girl with ID, microcephaly and immunological disease but without the biochemical analysis of the variant (Almutairi et al., 2021). The other proline-rich domains identified are not yet associated with partners and functions: indeed, PAK1 contains five proline-rich domains, while PAK2 has two and PAK3 has four. Whether all these proline-rich sequences bind the same partners with all three PAK isoforms is uncertain and has yet to be experimentally demonstrated.
The differences in peptide sequences also influence phosphosites. At the membrane, Rho-GTPase interactions induce a conformational change via the first event of trans-phosphorylation (threonine 423 for PAK1, 402 for PAK2 and 421 for PAK3). This restores some activity at the catalytic domain, which triggers cis-phosphorylation of sites, thus reinforcing kinase activity and completing the dissociation of dimers as well as maintaining monomers in an active conformation (Lei et al., 2000; Parrini et al., 2002; Pirruccello et al., 2006). No mutation affecting phosphosites has been described so far. Whether some PAK mutations can alter autophosphorylation or phosphorylation via other regulatory kinases has yet to be demonstrated. The caspase cleavage site, specific to PAK2, plays an important role in signaling but is not yet associated with NDDs (Jakobi et al., 2003).
PAK interactions with lipids, such as membrane lipids like PIP2, also participate in the opening of the kinase and its activation (Parrini et al., 2009; Malecka et al., 2013). PAKs are also enriched in dendritic spines (Zhang, 2005; Hayashi et al., 2007). As early as 2004, it was shown that PAKs copurify with synaptic markers (Hayashi et al., 2004). This analysis highlighted the colocalization of activated PAK with phospho-Cofilin and PSD95 upon neuronal activation in cortical neuron cultures and in organotypic hippocampal slices (Hayashi et al., 2004; Chen et al., 2007). Biochemical fractionation followed by Western blot analysis revealed the presence of active PAKs in the post-synaptic density (PSD) fractions of mouse brains, whereas inactive PAKs copurified mainly with presynaptic markers such as synaptophysin (Hayashi et al., 2004). However, fractionation experiments using adult mouse cortex suggest that PAK1 strongly localizes in the presynaptic fraction while PAK3 (especially PAK3a and PAK3b) co-purifies more with post-synaptic densities (Combeau et al., 2012). This suggests they may play different roles depending on their location and association with distinct synaptic elements.
PAK expression patterns and mutation pathogenicity
Group I PAKs share many structural and regulatory similarities, but their temporal and spatial expressions diverge. Globally, PAK1 expression is high in muscles, in the spleen, and the brain except in the dentate gyrus (BrainSpan, 2022 Atlas of the Developing Human Brain) while PAK2 expression is ubiquitous (Manser et al., 1995; Teo et al., 1995; Whale et al., 2011). PAK3 is mostly expressed in the brain, with a particularly strong expression in the hippocampus. At the cellular level in the brain, PAK1 is found only in neural lineage cells, PAK2 is expressed in every cell types and PAK3 can be found in neurons as well as oligodendrocyte progenitor cells (Figure 5A; Zhang et al., 2014; Maglorius Renkilaraj et al., 2017; Magne et al., 2021; Wang and Guo, 2022). However, some differences were noticed in PAK expression between mouse and human brain (Figure 5A; Civiero and Greggio, 2018; Zhang K. et al., 2022). Such differences should be taken into account when experiments are conducted on animal models and for extrapolation to humans.
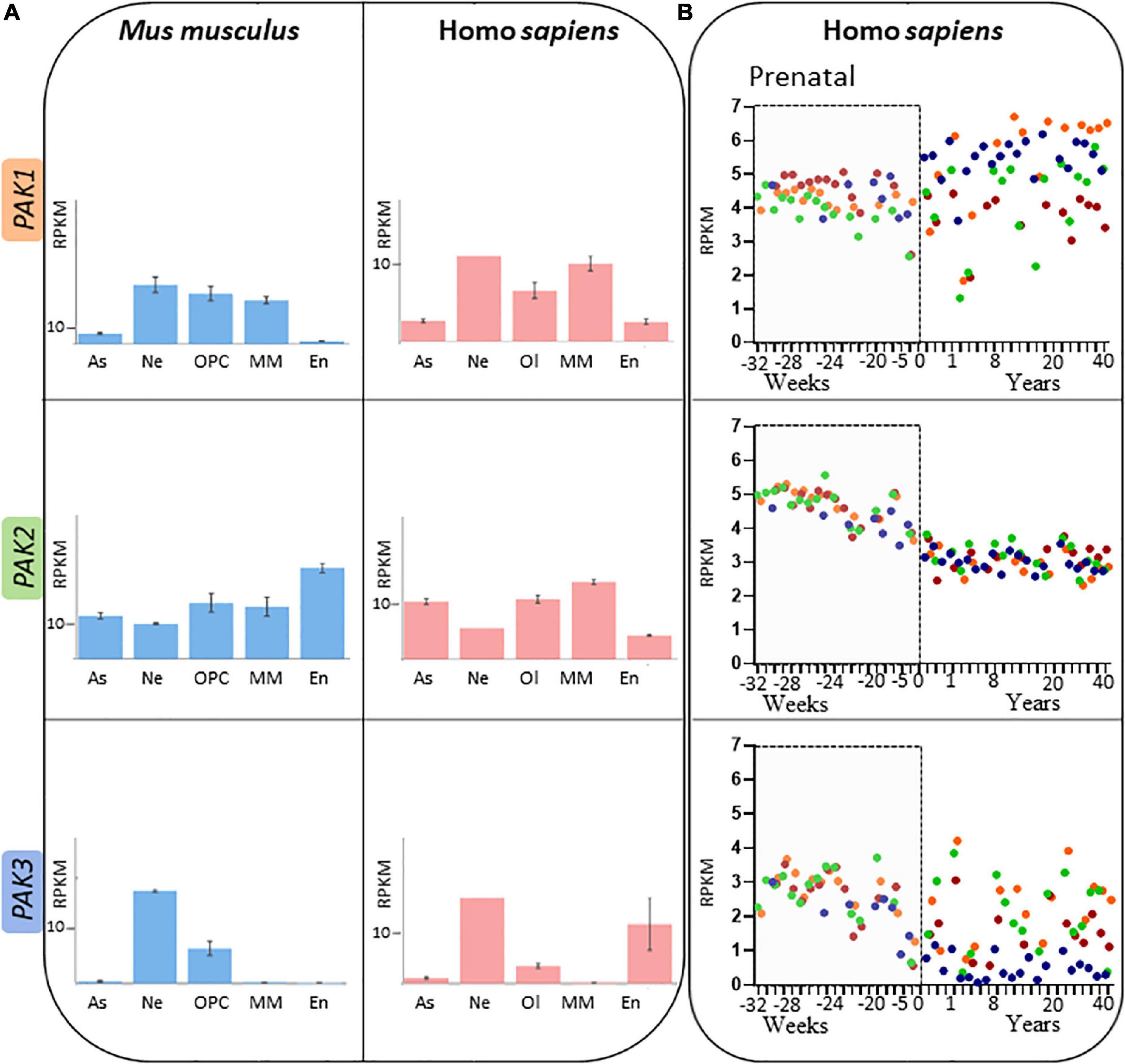
Figure 5. Spatio-temporal expression patterns of PAK isoforms (A) PAK expression depending on brain cell types, expressed in RPKM (reads per kilobase of exon per million reads mapped) in adult mouse (blue) and human (pink). Astrocytes (As), neurons (Ne), Oligodendrocyte precursor cells (OPC) or Oligodendrocyte (Ol), microglia/macrophages (MM) and endothelial cells (En). Data mining from brainrnaseq.org. (B) PAK expression throughout human lifespan in neurons of the cerebellar cortex (blue), hippocampus (green), striatum (brown), and frontal cortex (orange). Data mining from brainspan.org.
PAK expression also differs along human lifespan (Figure 5B; BrainSpan, 2022: Atlas of the Developing Human Brain; Miller et al., 2014). The differences in pattern of expression may indicate that the different PAKs do not have the same role over the course of human brain development. PAK expression also presents variations depending on the brain region, which might reflect region-dependent specificities in addition to their concomitant functions. PAK1 expression increases after birth in several regions such as the cerebellar and frontal cortices, but remains at a stable level in the striatum. PAK2 expression profile appears to be similar in all brain regions (Figure 5B). Steadily decreasing after birth (Iossifov et al., 2014; Wang et al., 2018), PAK3 expression is consistent throughout human brain development in the hippocampus, striatum and the frontal cortex but decreases after birth in the cerebellar cortex.
PAK expression is altered in several NDDs, suggesting that the decrease in PAK may be partly involved in cognitive or developmental symptoms. Therefore, the post-mortem meta-analysis of differentially expressed genes in the brain of ASD patients shows that neural PAK1 expression is reduced in this pathology (Rahman et al., 2020). Hearing loss is correlated with ASD in children and this symptom may be due to the dysregulation of PAK1 expression. Indeed, PAK1 is highly expressed in the postnatal mouse cochlea, but the study of the Pak1-knockout mouse model showed that PAK1 deficiency downregulates cofilin phosphorylation and the expression of βII-spectrin, decreases the hair synapse density in the cochlea, and finally leads to hair cell apoptosis and severe hearing loss (Cheng et al., 2021).
Another example is PAK3 downregulation in a model displaying iron deficiency-dependent cognitive alterations (Schachtschneider et al., 2016). PAK3 is also one of the differentially expressed genes in BP and its expression is also regulated by valproate, a histone deacetylase inhibitor used to treat BP (Sinha et al., 2021). PAK1 and PAK3 mRNA levels are significantly reduced in the post-mortem brain of subjects affected with depression (Fuchsova et al., 2016). Transcriptomic analysis and genome-wide association in schizophrenia and bipolar disorder identify PAK1 and PAK3 as the differentially expressed genes along with several other genes in the RAC1/CDC42 pathway, highlighting the role of this pathway in these psychiatric disorders (Zhao et al., 2015).
Thus, it is important to consider PAK regulation of expression and the way it might change in the context of NDDs. Transcriptional and post-transcriptional regulations of each PAK present differences that may also change the way mutations affect brain development and functions. PAK1 expression is activated via FOXO and E2F2 transcription factors and is inhibited by Rb (de la Torre-Ubieta et al., 2010; Sosa-García et al., 2015). PAK2 transcriptional regulation is still an understudied subject, however, PAK2 was found as differentially methylated in children with ASD (Jasoliya et al., 2022). PAK3 expression is inhibited by Notch, ZEB1, SP8/9 and DLX1/2 transcription factors, highlighting its role in the proneural pathway (Souopgui, 2002; Cobos et al., 2007; Liu et al., 2019; Shen et al., 2019; Wei et al., 2019). PAK3 transcription is also activated via Neurogenin and cJUN/AP-1 (Parker et al., 2013; Piccand et al., 2014). Several microRNAs (miRNAs) were identified in the regulation of PAK expression. miRNAs are gene regulators frequently involved in neurological disorders (Kim and Pak, 2020; Yao et al., 2020). At least seven PAK1-targeting miRNAs regulate PAK1 transcription: miR-7, 34b, 96, 140-5p, 145, 485-5p, and 494 (Yue et al., 2016; Yao et al., 2020; Zhao et al., 2022). More than 19 microRNAs target directly PAK2 expression, such as miR4779 (Koo and Kwon, 2018), miR-7-5p (Li et al., 2019) and miR455-3p (Hu et al., 2019). Several miRNA such as miR-134-5p, miR-1252, miR-125b-5p, miR193b-3p, miRNA-495, miRNA542 and miRNA133 were shown to be involved in PAK3 downregulation, mainly in cancer (Chiurazzi and Pirozzi, 2016; Zhang et al., 2020; Tan et al., 2021). Interestingly, expression of the miR-495 is also involved in schizophrenia (Santarelli et al., 2019). Alternative polyadenylation (APA) are post-transcriptional events that regulate gene expression and that play a key role in cell proliferation and differentiation, and in neuronal functions. APA events may produce different protein isoforms. PAK3 exhibits bimodality of distal APA usage in a type of GABAergic interneurons, the chandelier cells. These interneurons have specific spatial and temporal origins, target the axon initial segment of pyramidal neurons of the hippocampus and are implicated in brain disorders, including schizophrenia, epilepsy, and ASD (Yang et al., 2022).
To our knowledge, no data exists concerning the role of PAKs in other brain cells, except for the oligodendrocyte lineage (Maglorius Renkilaraj et al., 2017). Interestingly, MRI analysis of white matter reveals hypomyelination in patients bearing PAK1 pathogenic variations whereas partial agenesis of corpus callosum and thin fiber tracts have been observed in some patients with PAK3 mutations (6/16). In vitro experiments demonstrated that PAK3 deletion impairs the differentiation of oligodendrocyte precursors, supporting a developmental and cell-autonomous role for PAK3 during the first steps of myelination. These fragmentary data suggest that defects induced by PAK variations in oligodendrocytes or oligodendrocyte precursors may also account for some phenotypical traits observed in affected patients.
To summarize, these different PAK isoforms may be involved in one of the several crucial cellular mechanism that shape the brain, through proliferation and apoptosis, cell type specification, migration, arborization, and synaptic genesis. Nonetheless, experimental evidence attributing unique and specific functions to one isoform is currently lacking, making it difficult to link spatiotemporal expression characteristics to clinical phenotypes.
The specific roles of PAKs isoforms
Among group I PAK isoforms, PAK1 is the most studied and is considered as representative of group I PAKs in general. Thus, there are currently more than sixty identified substrates of PAK1. These are involved in different major cellular processes such as proliferation, survival, motility and regulation of the actin cytoskeleton (Kanumuri et al., 2020; Liu et al., 2021). Such exhaustive list of specific substrates remains partial and cannot currently be compiled for the other isoforms (Ye and Field, 2012; Kumar and Li, 2016). Among the PAK-activated pathways, mentions should be made of the MAP kinase ERK pathways, the alteration of which leads to RASopathies, the Akt-PI3Kinase-mTor and Wnt pathways involved in ASD, the NF-κB pathway involved in apoptosis, among others (Yao et al., 2020; Caracci et al., 2021; Sharma and Mehan, 2021; Tartaglia et al., 2022). Many functions of the three PAK isoforms probably derive from their role in the regulation of actin cytoskeleton dynamics. It remains to be seen whether some of these functions are supported by only one of the three isoforms, which has not always been demonstrated experimentally. We can cite, for example, the following data. PAK1 is involved in the proliferation and migration of neural progenitors in young mice (Pan et al., 2015) and dendritic maturation in the hippocampus (Dagliyan et al., 2017), which is essential to the regulation of postnatal cortical development. The abnormal increase in actin polymerization described in a mouse model of functional demyelination was caused by an over activation of PAK1 (Hu et al., 2016). PAK2 probably plays an essential role during the first developmental stages as it regulates neuronal migration in the fetal brain and is, as such, essential for brain development. Interestingly, among PAKs, only PAK2 suppression leads to the death of mouse embryos (Meng et al., 2005; Asrar et al., 2009; Wang et al., 2018). PAK3 does not play the same role in cell cycle and cell division as PAK1 and PAK2. While PAK1 and PAK2 have been characterized as oncogenes, PAK3 is instead a probable tumor suppressor (Magne et al., 2021). In xenopus laevis, PAK3 induces cell cycle arrest of precursors and their differentiation into neuroblasts during primitive neurogenesis (Souopgui, 2002). Furthermore, PAK3 is also involved in axonal and dendritic arborization of immature interneurons and favors their tangential migration toward the cortex in mice (Cobos et al., 2007; Dai et al., 2014).
However, several studies have deciphered and attributed isoform-specific functions independent of actin. For example, PAK1 takes on actin-independent functions such as positive regulation of tonic GABA transmission via the downregulation of tonic endocannabinoïd (eCBs) secretion (Xia et al., 2018). The phosphorylation of CtBP1 by PAK1 is also a key step in synaptic vesicle retrieval in cultured hippocampal slices (Ivanova et al., 2020). Indeed, recordings of synaptic currents from CA1 neurons in Pak1-KO mice showed a decreased GABA signaling. This was also the case with acute PAK1 inhibition in hippocampal slices. Interestingly, the maturation of GABA function plays a major role in postnatal brain development (Deidda et al., 2014; Peerboom and Wierenga, 2021). PAK2 displays actin-independent pro-apoptotic functions, since PAK2, but not PAK1 nor PAK3, is cleaved by caspase 3, 8 and 10 in response to Fas signaling, which leads to PAK2-dependent apoptosis (Rudel and Bokoch, 1997; Fischer et al., 2006). PAK2 activated by RAC and CDC42 promotes cell survival while the caspase/PAK2 pathway is indeed pro-apoptotic (Marlin et al., 2011). Finally, one of PAK3 specific functions is the controls of AMPA receptor (AMPAR) trafficking via GluA1 subunit phosphorylation, upregulating the number of AMPAR at the membrane (Hussain et al., 2015).
Several data illustrate the link between PAK signaling and NDDs. PAKs were shown to regulate several proteins involved in NDDs, such as LIMK and cofilin (Figure 1). PAKs family members also interact with other downstream proteins that are dysregulated in NDDs or whose variations are causative factors in NDDs. For example, it was demonstrated that PAK1 binding to ERK2 facilitates ERK2 signaling, or activates their upstream activators RAF1 and MEK, which are involved in RASopathies (Sundberg-Smith et al., 2005; Motta et al., 2020; Liu et al., 2021). PAK1 is also required for the activation of NFκB pathway (Frost et al., 2000) via its modulation of the PPARy/NFκB signaling (Dammann et al., 2015). The expression of NFκB pro-inflammatory transcription factor is dysregulated in the brain of ASD patients and animal models of ASD. This dysregulation could be a pathogenic mechanism of neuro-inflammation involved in ASD (Young et al., 2011; Honarmand Tamizkar et al., 2021). Furthermore, PAK1, but not PAK2 nor PAK3, interacts with FRX family proteins involved in Fragile X syndrome, notably by phosphorylating FRX1 at Ser-420 (Say et al., 2010; Majumder et al., 2020). PAK2 modulates Wnt and Hedgehog signaling (Sementino et al., 2022). These proteins are essential to brain development and are involved in ASD and brain structural anomalies (Mulligan and Cheyette, 2017; Memi et al., 2018). Variations in the GluA1 subunit of AMPA receptors, one of PAK3-specific targets, are associated with moderate to severe ID and sometimes epilepsy, ASD, ADHD and movement disorders (Ismail et al., 2022).
PAK dysregulation in neurodegenerative diseases: Cause or consequence?
Group I PAK dysfunctions and dysregulations, that are the root causes in several NDDs, are also involved in neurodegenerative diseases (Hayashi et al., 2004; Kreis et al., 2008; Ma et al., 2012). Numerous observations describe dysfunctions and abnormal decreases in the expression or activity of PAK during the evolution of neurodegenerative diseases such as Alzheimer’s, Parkinson’s and Huntington’s diseases, as well as some ataxias and prion diseases. These dysfunctions are often concomitant with dendritic spine abnormalities, emphasizing the question of whether PAK alteration is a cause or a consequence of synaptic deterioration. We can summarize the data as follows. In post-mortem brains of Alzheimer’s disease (AD) patients, PAK proteins are markedly reduced and their phosphorylated forms present abnormal localization, which is also correlated with abnormal cofilin activation and aggregates in neurites (Ma et al., 2008; Arsenault et al., 2013; Bories et al., 2017; Lauterborn et al., 2020). Concordant results were observed in the Tg2576 mouse model of AD which display peptide Aβ-containing plaques, but not apoptosis or microfibrillary tangles (Zhao et al., 2006). Interestingly, PAK inhibition in the brains of older mice triggers neuronal damage and cognitive deficits similar to those of the Tg2576 mice, clearly indicating a role of PAK in the Aβ pathology. It was shown in primary neuron cultures that interaction of PAK3 dominant-negative variant with the amyloid precursor protein (APP) blocks APP-mediated neuronal apoptosis in familial AD (McPhie et al., 2003). The C-terminal peptide cleaved from the APP at Asp664 may be responsible for these abnormal PAK activation (Nguyen et al., 2008). The inhibition of cofilin dephosphorylation in the Familial Autosomal dominant (FAD) mouse model of AD that express several pathogenic mutations in APP and Presinilin1 genes, decreases Aβ plaques, cellular defects and improves cognitive function, confirming the link between Aβ pathologies and actin cytoskeleton (Deng et al., 2016).
PAK1-mediated oligomerization of Huntingtin (HTT), which is independent of PAK1 kinase activity, is associated with Huntington’s disease (Luo et al., 2008). Interestingly, αPIX also enhances HTT aggregation via PAK1 (Eriguchi et al., 2010). Furthermore, HTT negatively regulates PAK2 apoptotic functions by preventing PAK2 cleavage via caspases: this protective function is retained by mutated HTT. These studies suggest that PAK1 and PAK2 have completely different associations with Huntington disease, PAK1 increases the toxicity of mutated HTT while PAK2 possibly has protective effects (Luo and Rubinsztein, 2009). PAK1 also regulates the expression of Ataxin-1 that is involved in spinocerebellar ataxia type I, suggesting that PAK1 inhibition may be a therapeutic approach in this orphan neurodegenerative disease (Bondar et al., 2018).
Synaptic activity requires a significant energy supply, provided mainly by the mitochondria. The routing of presynaptic mitochondria is under the control of a recently identified signaling pathway, involving AMP-activated protein kinase, which is mediated by PAKs via their kinase activity (Kong et al., 2016). This pathway regulates the recruitment of mitochondria and their anchoring to the vicinity of presynaptic terminals via myosin VI phosphorylation (Li et al., 2020). Defects in energy metabolism are also features of neurodegenerative diseases and could involve this AMPK/PAK/MYOVI pathway. It is interesting to note that the hippocampus, a key brain region for memory formation, is one of the cerebral structures most sensitive to hypoxia. However, the involvement of PAKs in this respect has not been documented (Schmidt-Kastner, 2015). PAK1 and PAK3 are also down-expressed in the post-mortem brains of Parkinson patients. In conclusion, alterations in PAK expression and activity are also involved in neurodegenerative pathologies, associated with excessive apoptosis, energy deficit, and dendritic spine shrinkage.
In summary, these numerous observations describe PAK down-expression, dysfunction, mislocation, and dysregulation throughout the progression of neurodegenerative diseases. PAKs are at the center of three signaling pathways involved in neurodegeneration, namely the regulation of dendritic spine dynamics, apoptosis and neuronal energy homeostasis. However, signaling pathways that link together synaptic defects, energy balance dysregulations, and abnormal neuronal death are complex and the role of PAK defects in these diseases remains unclear. A seminal experiment in mice showed that PAK inhibition causes cofilin anomalies and memory impairments similar to AD defects, strongly suggesting a causal role of PAK dysfunctions in AD-associated cognitive deficits (Zhao et al., 2006). Are PAK dysfunctions a cause in the emergence of neurodegenerative diseases in the patients with NDDs? At the moment, the absence of published prospective clinical follow up of patients with PAK-associated conditions prevents us from knowing the incidence of neurogenerative diseases on these patients. This has not been addressed by experimental approaches either. While the role of PAKs is well documented in worsening actin-dependent synaptic dysfunctions in AD, their role in apoptosis and energy failure during neurodegenerative disease progression is still elusive. Moreover, although several isoform specificities have been observed, the role of each PAK in these pathologies is still unclear. Nevertheless, recent data suggest that the activation of the acetylcholine muscarinic receptor improves aversive memory in striatal and accumbens nuclei via a Protein kinase C/Rac1/PAK pathway and may thus be a new therapeutic approach, at least for this type of behavioral symptoms (Yamahashi et al., 2022).
Experiments in PAKs studies: Finding the right model
These recent and scarce data open the way toward a new understanding in the etiology of many cerebral pathologies. Yet, there is still much to uncover in the field of PAK-associated disorders. A deeper exploration of each PAK specificity, a more thorough investigation of the molecular mechanisms underlying PAK mutation pathogenicity and adapted therapeutic strategies are needed to improve diagnosis and treatment in PAK-related disorders. After the discovery of PAKs in 1994 (Manser et al., 1995) and after establishing their role in the regulation of cytoskeleton dynamics (Sells et al., 1997, 1999), studies conducted in cell and neuronal cultures helped characterizing PAKs regulation (Manser et al., 1998; Banerjee et al., 2002; Brown et al., 2002) and functions in migration (Causeret et al., 2009), neurite outgrowth (Daniels et al., 1998), spine morphogenesis, synapse formation and plasticity (Boda et al., 2004; Hayashi et al., 2007; Kreis et al., 2008; Dubos et al., 2012). Other studies have been conducted in animal models, widely used to understand mechanistic aspects of neurodevelopmental pathogenesis. Such models can be of translational significance or bring insight into fundamental mechanisms involved in NDDs (Fallah and Eubanks, 2020; Mariano et al., 2020; Rotaru et al., 2020). Even though some PAK studies have been conducted in non-mammalian models (Santiago-Medina et al., 2013; Magini et al., 2014), most are carried out in mouse models. Thus, Pak1-knockout mice have been generated and display MAPK signaling defects (Arias-Romero and Chernoff, 2008) and, accordingly, present slight immune deficiencies (Allen et al., 2009). LTP deficits and a decrease in NMDA activity-induced cofilin phosphorylation have also been identified in Pak1-KO mice (Asrar et al., 2009). The fact that the phenotype of Pak1 and Pak3 KO mice are relatively mild, while the double Pak1/3 KO generates substantial cognitive loss, brain morphological abnormalities, as well as functional and structural synaptic defects suggests that these two isoforms have redundant functions allowing functional complementation (Meng et al., 2005; Asrar et al., 2009; Huang et al., 2011). Nevertheless, all PAK1 variations generate GOF that cannot be compensated by other isoforms.
Pak2-knockout mice are not viable (Arias-Romero and Chernoff, 2008). However, Pak2+/– mice have been studied and display ASD-related behaviors and reduced spine density, defective LTP and impaired actin polymerization (Wang et al., 2018), which is concordant with the effects of Pak2 haploinsufficiency in humans. The high PAK2 expression during embryonic development, the lethality of its gene invalidation and its role in the establishment of cerebral vascularization, are in agreement with the fact that bi-allelic loss have not been identified in patients. However, it is surprising that no variant with gain of function has not been identified yet.
The deficits in late-LTP and CREB activity in the brain of Pak3-KO mice indicate a more important role of PAK3 in the actin-independent regulation of plasticity-associated genes (Meng et al., 2005). Interestingly, double Pak1/Pak3-knockout mice display hyperactivity, anxiety, learning and memory deficits, secondary microcephaly, reduced synaptic density but increased size of individual synapses and increased basal synaptic transmission, disrupted LTP and LTD, decreased cofilin regulation and reduced CREB activity (Huang et al., 2011). PAK1 displays a higher expression during development than in adulthood, in contrast to PAK3, suggesting that PAK3 has more developmental functions.
Thus, the global inhibition of PAKs in post-mitotic neurons of mice forebrain via the expression of the PAK kinase activity-inhibiting AID (dnPak) cause an increase in dendritic spine size, enhanced synaptic transmission and LTP as well as LTD deficits in the cortex (Hayashi et al., 2004). Consequently, mice expressing dnPAK displayed memory deficits. One can also wonder whether Pak3-AID only blocks kinase-activity-dependent functions, leaving PAK signaling at least partially functional. The AID of PAK3 was also used to post-natally block PAK activity in regions associated with social functions, which impaired social memory retrieval (Leung et al., 2018; Zhou and Jia, 2021). These models shed light on the roles of PAKs and how PAK disruption might affect brain functions. However, they only represent the loss of PAK proteins (in the case of Pak-knockouts) or loss of PAK kinase function (in the case of PAK inhibition via dnPak) while PAK1 mutations are usually gain of functions. There are also differences in PAK3 nonsense mutations, that might lead to a complete loss of protein, and PAK3 missense mutations that would have more variable effects.
New models, such as the knock-in mouse line which expresses the Pak3-Arg67Cys mutation, responsible for a moderate to severe ID in humans are expected to mimic more closely the precise biochemical alterations caused by PAK mutations. Thus the effects of a PAK3 variant displaying functional kinase activity, but with changes in partner interactions in vitro, has been studied and furthers our understanding pathogenic mechanisms underlying PAK3 pathogenicity in vivo. The PAK3-Arg67Cys variant impacts newly generated neurons circuit integration in the adult mouse hippocampus, thus disturbing complex memory tasks performance such as pattern separation (Castillon et al., 2020). Through the study of animal models bearing Pak mutations, directly linking behavioral symptoms and brain dysfunctions to molecular mechanisms is now achievable. However, as shown in Figure 5, gene expression might not be the same between animal models and humans. Thus, patient-derived cell models are important to complement animal studies. PAK1 variants have been proposed to display high kinase activity in patient fibroblasts (Harms et al., 2018). A similar study was conducted in keratinocytes, which have a higher level of PAK3 expression than fibroblasts (Magini et al., 2014). Obviously, the development of iPSCs from patient fibroblasts and models of embryonic bodies would help put together a more elaborated analysis of PAK mutation-dependent cell abnormalities. For example, the first data obtained for Down syndrome, the most frequent cause of ID, confirms the hyperactivation of the DsCam/PAK1 module in patient-derived iPSCs and highlight the possibility to target PAKs in this disease (Tang et al., 2021).
PAKs as therapeutic targets in brain diseases
Researchers are confronted with the contradiction of wanting, on one hand, to target a molecule in a strategy applicable to many psychiatric and neurodevelopmental disorders, and on the other hand, to develop approaches applicable to specific cases, with the perspective of personalized medicine. Cofilin which is a focal point of the Rho GTPase pathway, is a therapeutic target that has already been validated to correct behavioral and synaptic plasticity defects in several NDD models (Shaw and Bamburg, 2017). PAKs are also points of convergence and therefore potential therapeutic targets. Indeed, since the phenotypes associated with some NDDs appear to be due to exacerbations of the Rac/Cdc42/PAK pathway activity, PAK inhibition may be an interesting therapeutic approach. Several PAK inhibitors were developed and display high affinity and selectivity against group I-PAKs (Semenova and Chernoff, 2017). First evidence of this concept is that the expression of an autoinhibitory domain of PAK3, which targets all three group I PAKs improves the cellular and behavioral phenotype of Fmr1 KO mice, a model of Fragile X syndrome (Hayashi et al., 2007). Interestingly the bioavailable and brain–permeant catalytic inhibitor of group I PAK, FRAX486, rescues the spine defects, seizures, stereotypical behavior, and hyperactivity displayed by Frm1-KO mice (Dolan et al., 2013). FRAX486 also rescues neurobehavioral alterations in a mouse model of CDKL5 disorders, probably via the regulation of the complex between the main CDKL5 substrate ARHGEF2 and PAK1 (Zenke et al., 2004; Fuchs et al., 2022). PAK over-activation was also observed in neurons cultured from Disc1-KO mice, a mouse model of schizophrenia. DISC1 downregulates Kalirin7, a potent RAC1-activating GEF, upon NMDAR activation (Hayashi-Takagi et al., 2014). PAK inhibition using FRAX486 prevents DISC1 RNAi-induced spine deterioration (prophylactic effect) and reverses already existing spine deterioration triggered by DISC1 RNAi (treatment effect) in neuronal cultures. It also partially recues spine morphology in adult mice (Hayashi-Takagi et al., 2014). In the case of neurofibromatosis type 1 (NF1) there is also cognitive and learning disabilities that are associated to MAP Kinase activation and dendritic spine regulation. PAK1 gene invalidation or pharmacological inhibition of PAK by intracerebral injection of the IPA3 inhibitor restores social interaction and memory deficits (Molosh et al., 2014). PAK1 activity is increased in a mouse model of hereditary demyelinating neuropathy of demyelination, in which disease progression is associated with an increase of actin polymerization at the nerve myelin junction (Hu et al., 2016). Pharmacological inhibition of PAK using PF3758309 normalized levels of F-actin, completely prevented the progression of the myelin junction disruption, and restored nerve conduction.
Several PAK inhibitors target the ATP binding site of the catalytic domain via a competitive mechanism (FRAX compounds, PF3758309), but the high identity of peptide sequences within the catalytic domain prevents poor PAK-isoform specificity of inhibitors toward the three PAK isoforms. Another inhibitor, the IPA-3 compound, is a brain-permeant allosteric inhibitor that binds to the PDB/AID, impairs GTPase binding, and thus prevents group I-PAK activation but has no effect on already activated kinases (Deacon et al., 2008). However, recently, a compound (NVS-PAK1-1) that limits the PAK access to ATP was characterized as the first PAK1 specific inhibitor, especially compared to PAK2 (Semenova and Chernoff, 2017). It would be interesting to test to what extent these PAK inhibitors could constitute therapeutic approaches in cases where mutations confer kinase activity over-activation (6/23). However, for the majority of variants (15/23), mutations in the PAK genes lead to the loss of kinase activity (Figure 3C). In these situations, it would probably be more appropriate to target a downstream molecule, such as cofilin.
Conclusion and perspectives
PAK mutations are rare events but their genetic traceability as monogenic defects, their total penetrance, the expanding collection of human mutations and curated phenotypes, make the study of these genes ideal to discover the biological mechanisms that go awry in common human NDDs. The exploration of fundamental and novel biological processes can greatly benefit from the study of rare genetic disorders (Lee et al., 2020). Group I PAKs occupy a central place in neuron physiology by being RAC1/CDC42 downstream effectors as well as the convergence point of many other pathways that control cell fate, motility, cell proliferation and apoptosis. Furthermore, an increasing number of experiments also indicate that PAKs are heavily involved in synaptic transmission and plasticity. Nevertheless, clinical data and fundamental experiments in controlled paradigms strongly suggest that these kinases present both redundant and isoform-specific functions essential for the development and processes of the brain and other organs (Figure 6).
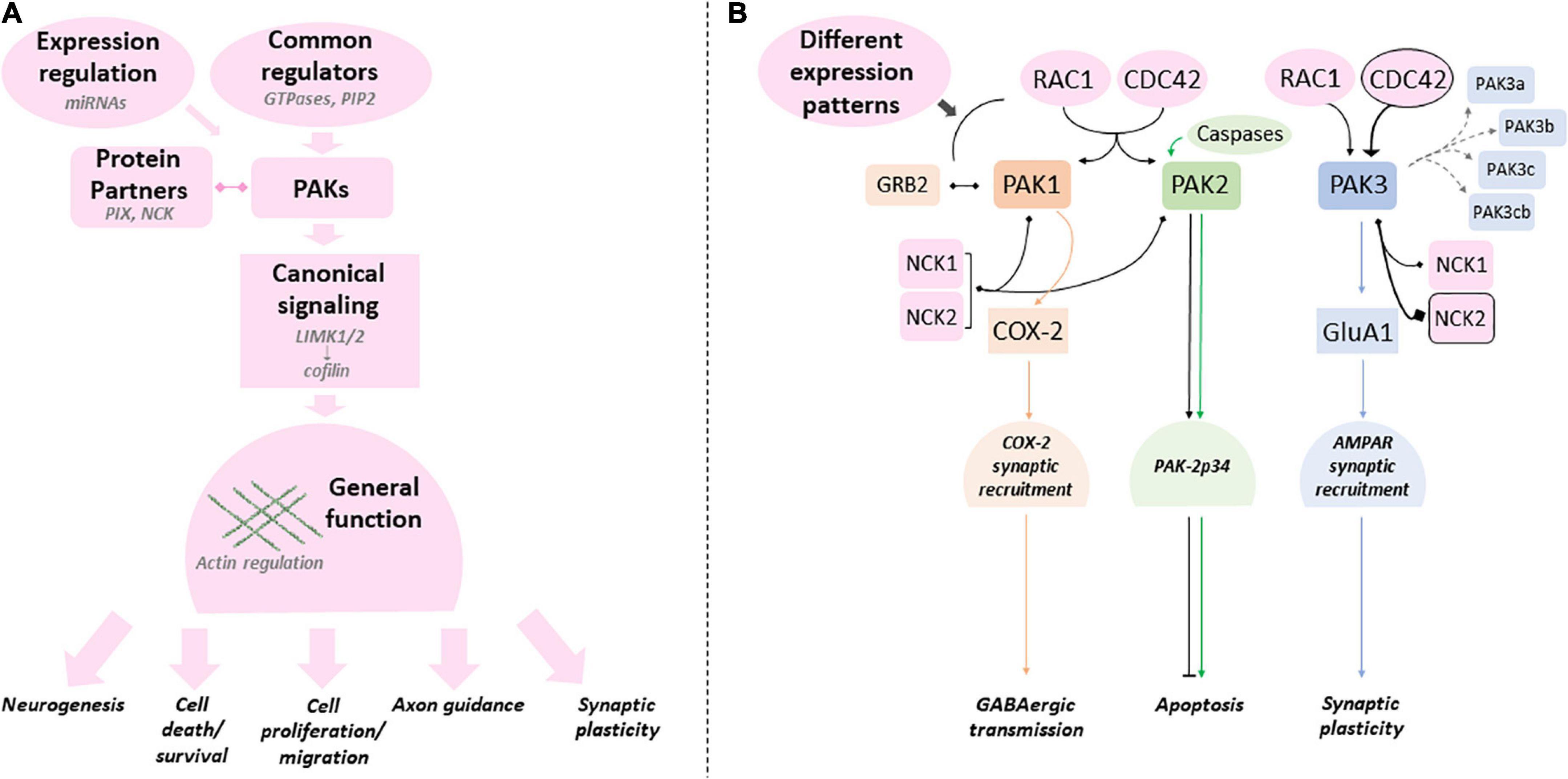
Figure 6. Canonical and specific PAK-dependent processes (A) Group I PAKs share several regulatory mechanisms, protein partners and substrates (canonical processes in pink - examples in light gray). PAK mutations can affect any of these processes. (B) Several studies emphasize the differences between PAKs (canonical processes in pink can still present quantitative differences). PAKl (orange), PAK2 (green) and PAK3 (blue) were shown to have their own specificities (colors matching the corresponding PAK). Protein partners and regulators that are favored by PAK are emphasized with a black frame. Mutations can have different effects on these specific processes and explain the variations found between PAKs and intragenic variability.
In summary, PAK1 mutations are dominant gain of function, PAK2 mutations generate haploinsufficiency, whereas PAK3 pathogenic variations have various functional consequences. The genotype/phenotype correlation is relatively easy to comprehend for the first two genes, associated with developmental delay and macrocephaly or with ASD, respectively. However, this relation remains to be established for PAK3. The main goal is to identify the molecular and cellular processes at the foundation of the differences between each isoform genotype/phenotype correlation. Why PAK1 mutations are GOF and PAK2 mutations are LOF is elusive, and there is still a gap preventing the full comprehension of the link between isoform-specific functions and the phenotypes associated with mutations in each PAK gene. More specifically, the biochemical alterations caused by different PAK mutations are still understudied. It is necessary to obtain a deeper understanding of the expression, localization, regulation, substrates and partners of each PAK isoform in the more global context of PAK interactome in order to gauge the impact of mutations on these processes. As listed above, several publications already demonstrate the role of these kinases in several neurological disorders such as Fragile X, neurofibromatosis, Down syndrome and several rare diseases caused by synaptic gene mutations, but also in neurodegenerative diseases such as Alzheimer’s, Huntington’s, Parkinson’s and some ataxias. With very few exceptions, it does not currently seem possible to decipher whether the three PAKs underlie the same dysfunctions in some or all of these neurological diseases. Understanding this is a challenge for future translational studies. It is perhaps also interesting to note that PAKs are associated with COVID pathologies, in particular PAK1 in the virus infection process and PAK3 in certain immunological signatures of patients (Yu et al., 2021; Zhang K. et al., 2022). This could be linked to the neurological damage associated with the long forms of COVID-19 (Monje and Iwasaki, 2022). Many experimental approaches already brought a proof of the concept that group I PAKs are interesting therapeutic targets in NDDs and neuropsychiatric disorders. The joint effort in the fields of cancerology, neurodegenerative disorders, rare and neurodevelopmental diseases will greatly advance our knowledge on PAK functions, dysfunctions and adapted therapeutic strategies in the view of personalized treatments. One of the remaining weaknesses in the study of the genotype/phenotype relationship in PAK-associated diseases is the rarity of reported cases. However, the efforts of clinicians and geneticists to implement clinical databases provide access to more genetic and clinical data, thus allowing the genotype/phenotype correlation to be established more precisely. Clinician and researcher collaborations, improved experimental models, precise biochemical characterization of variants and conceptual frameworks in the context of convergent neurosciences could be the missing stone to fully comprehend the biological mechanisms underlying mutation pathogenicity and to successfully translate research findings into clinical practices (Willsey et al., 2017; Srivastava et al., 2018).
Author contributions
MD and J-VB conceptualized the review and wrote it. AV and AT supervised the clinical data. SP-G and VR followed the successive steps of writing with their advice and corrections. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Centre National de la Recherche Scientifique (CNRS) and the Université Paris-Saclay and grants from Agence National de la Recherche (ANR) ANR-21-CE17-0053 ID-GePhe-PAK and from the Fondation Jérôme Lejeune 2022-#2160.
Acknowledgments
We thank Glenn Dallerac and Stéphane Jamain for reviewing and providing thoughtful comments on the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Allen, J. D., Jaffer, Z. M., Park, S.-J., Burgin, S., Hofmann, C., Sells, M. A., et al. (2009). P21-activated kinase regulates mast cell degranulation via effects on calcium mobilization and cytoskeletal dynamics. Blood 113, 2695–2705. doi: 10.1182/blood-2008-06-160861
Allen, K. M., Gleeson, J. G., Bagrodia, S., Partington, M. W., MacMillan, J. C., Cerione, R. A., et al. (1998). PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 20, 25–30. doi: 10.1038/1675
Almutairi, O., Almutairi, H. A., and Al Rushood, M. (2021). Protein-activated kinase 3 (PAK3)-related intellectual disability associated with combined immunodeficiency: a case report. Am. J. Case Rep. 22:e930966. doi: 10.12659/AJCR.930966
Antonarakis, S. E., Holoubek, A., Rapti, M., Rademaker, J., Meylan, J., Iwaszkiewicz, J., et al. (2021). Dominant monoallelic variant in the PAK2 gene causes Knobloch syndrome type 2. Hum. Mol. Genet. 31, 1–9. doi: 10.1093/hmg/ddab026
Arias-Romero, L. E., and Chernoff, J. (2008). A tale of two paks. Biol. Cell 100, 97–108. doi: 10.1042/BC20070109
Arsenault, D., Dal-Pan, A., Tremblay, C., Bennett, D. A., Guitton, M. J., De Koninck, Y., et al. (2013). PAK inactivation impairs social recognition in 3xTg-AD Mice without increasing brain deposition of tau and Aβ. J. Neurosci. Offi. J. Soc. Neurosci. 33, 10729–10740. doi: 10.1523/JNEUROSCI.1501-13.2013
Asrar, S., Meng, Y., Zhou, Z., Todorovski, Z., Huang, W. W., and Jia, Z. (2009). Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 56, 73–80. doi: 10.1016/j.neuropharm.2008.06.055
Bagrodia, S., Taylor, S. J., Jordon, K. A., Van Aelst, L., and Cerione, R. A. (1998). A novel regulator of p21-activated kinases. J. Biol. Chem. 273, 23633–23636. doi: 10.1074/jbc.273.37.23633
Banerjee, M., Worth, D., Prowse, D. M., and Nikolic, M. (2002). Pak1 phosphorylation on t212 affects microtubules in cells undergoing mitosis. Curr. Biol. 12, 1233–1239. doi: 10.1016/s0960-9822(02)00956-9
Barbosa, S., Greville-Heygate, S., Bonnet, M., Godwin, A., Fagotto-Kaufmann, C., Kajava, A. V., et al. (2020). Opposite modulation of RAC1 by mutations in TRIO is associated with distinct, domain-specific neurodevelopmental disorders. Am. J. Hum. Genet. 106, 338–355. doi: 10.1016/j.ajhg.2020.01.018
Ben Zablah, Y., Merovitch, N., and Jia, Z. (2020). The role of ADF/cofilin in synaptic physiology and Alzheimer’s disease. Front. Cell Dev. Biol. 8:594998. doi: 10.3389/fcell.2020.594998
Bienvenu, T., des Portes, V., McDonell, N., Carrié, A., Zemni, R., Couvert, P., et al. (2000). Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am. J. Med. Genet. 93, 294–298. doi: 10.1002/1096-8628(20000814)93:4<294::AID-AJMG8<3.0.CO;2-F
Billuart, P., Bienvenu, T., Ronce, N., des Portes, V., Vinet, M. C., Zemni, R., et al. (1998). Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 392, 923–926. doi: 10.1038/31940
Boda, B., Alberi, S., Nikonenko, I., Node-Langlois, R., Jourdain, P., Moosmayer, M., et al. (2004). The mental retardation protein PAK3 contributes to synapse formation and plasticity in hippocampus. J. Neurosci. Offi. J. Soc. Neurosci. 24, 10816–10825. doi: 10.1523/JNEUROSCI.2931-04.2004
Boda, B., Dubos, A., and Muller, D. (2010). Signaling mechanisms regulating synapse formation and function in mental retardation. Curr. Opin. Neurobiol. 20, 519–527. doi: 10.1016/j.conb.2010.03.012
Bokoch, G. M. (2003). Biology of the p21-activated kinases. Ann. Rev. Biochem. 72, 743–781. doi: 10.1146/annurev.biochem.72.121801.161742
Bondar, V. V., Adamski, C. J., Onur, T. S., Tan, Q., Wang, L., Diaz-Garcia, J., et al. (2018). PAK1 regulates ATXN1 levels providing an opportunity to modify its toxicity in spinocerebellar ataxia type 1. Hum. Mol. Genet. 27, 2863–2873. doi: 10.1093/hmg/ddy200
Bories, C., Arsenault, D., Lemire, M., Tremblay, C., De Koninck, Y., and Calon, F. (2017). Transgenic autoinhibition of p21-activated kinase exacerbates synaptic impairments and fronto-dependent behavioral deficits in an animal model of Alzheimer’s disease. Aging 9, 1386–1403. doi: 10.18632/aging.101239
BrainSpan (2022). Developmental transcriptome: brainspan: atlas of the developing human brain. Available online at: https://www.brainspan.org/rnaseq/search/index.html (accessed December 7, 2022)
Brown, M. C., West, K. A., and Turner, C. E. (2002). Paxillin-dependent paxillin kinase linker and p21-activated kinase localization to focal adhesions involves a multistep activation pathway. Mol. Biol. Cell 13, 1550–1565. doi: 10.1091/mbc.02-02-0015
Caracci, M. O., Avila, M. E., Espinoza-Cavieres, F. A., López, H. R., Ugarte, G. D., and De Ferrari, G. V. (2021). Wnt/β-catenin-dependent transcription in autism spectrum disorders. Front. Mol. Neurosci. 14:764756. doi: 10.3389/fnmol.2021.764756
Cartwright, A., Smith, K., and Balasubramanian, M. (2017). Short case report: Xq23 deletion involving PAK3 as a novel cause of developmental delay in a 6-year-old boy. Clin. Dysmorphol. 26, 38–40. doi: 10.1097/MCD.0000000000000154
Castillon, C., Gonzalez, L., Domenichini, F., Guyon, S., Da Silva, K., Durand, C., et al. (2020). The intellectual disability PAK3 R67C mutation impacts cognitive functions and adult hippocampal neurogenesis. Hum. Mol. Genet. 29, 1950–1968. doi: 10.1093/hmg/ddz296
Causeret, F., Terao, M., Jacobs, T., Nishimura, Y. V., Yanagawa, Y., Obata, K., et al. (2009). The p21-activated kinase is required for neuronal migration in the cerebral cortex. Cerebral Cortex 19, 861–875. doi: 10.1093/cercor/bhn133
Chelly, J., and Mandel, J.-L. (2001). Monogenic causes of X-linked mental retardation. Nat. Rev. Genet. 2:9. doi: 10.1038/35088558
Chen, L. Y., Rex, C. S., Casale, M. S., Gall, C. M., and Lynch, G. (2007). Changes in synaptic morphology accompany actin signaling during LTP. J. Neurosci. 27, 5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007
Cheng, C., Hou, Y., Zhang, Z., Wang, Y., Lu, L., Zhang, L., et al. (2021). Disruption of the autism-related gene Pak1 causes stereocilia disorganization, hair cell loss, and deafness in mice. J. Genet. Genom. 48, 324–332. doi: 10.1016/j.jgg.2021.03.010
Chiurazzi, P., and Pirozzi, F. (2016). Advances in understanding—genetic basis of intellectual disability. F1000 Res. 5:599. doi: 10.12688/f1000research.7134.1
Civiero, L., and Greggio, E. (2018). PAKs in the brain: function and dysfunction. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1864, 444–453. doi: 10.1016/j.bbadis.2017.11.005
Cobos, I., Borello, U., and Rubenstein, J. L. R. (2007). Dlx transcription factors promote migration through repression of axon and dendrite growth. Neuron 54, 873–888. doi: 10.1016/j.neuron.2007.05.024
Combeau, G., Kreis, P., Domenichini, F., Amar, M., Fossier, P., Rousseau, V., et al. (2012). The p21-activated kinase PAK3 forms heterodimers with PAK1 in brain implementing trans-regulation of PAK3 activity. J. Biol. Chem. 287, 30084–30096. doi: 10.1074/jbc.M112.355073
Crocq, M.-A., and Guelfi, J.-D. (2015). DSM-5: manuel diagnostique et statistique des troubles mentaux. Amsterdam: Elsevier Masson.
Dagliyan, O., Karginov, A. V., Yagishita, S., Gale, M. E., Wang, H., DerMardirossian, C., et al. (2017). Engineering pak1 allosteric switches. ACS Synth. Biol. 6, 1257–1262. doi: 10.1021/acssynbio.6b00359
Dai, X., Iwasaki, H., Watanabe, M., and Okabe, S. (2014). Dlx1 transcription factor regulates dendritic growth and postsynaptic differentiation through inhibition of neuropilin-2 and PAK3 expression. Eur. J. Neurosci. 39, 531–547. doi: 10.1111/ejn.12413
Dammann, K., Khare, V., Lang, M., Claudel, T., Harpain, F., Granofszky, N., et al. (2015). PAK1 modulates a PPARγ/NF-κB cascade in intestinal inflammation. Biochim. Biophys. Acta 1853, 2349–2360. doi: 10.1016/j.bbamcr.2015.05.031
Daniels, R. H., Hall, P. S., and Bokoch, G. M. (1998). Membrane targeting of p21-activated kinase 1 (PAK1) induces neurite outgrowth from PC12 cells. EMBO J. 17, 754–764. doi: 10.1093/emboj/17.3.754
de la Torre-Ubieta, L., Gaudillière, B., Yang, Y., Ikeuchi, Y., Yamada, T., DiBacco, S., et al. (2010). A FOXO-Pak1 transcriptional pathway controls neuronal polarity. Genes Dev. 24, 799–813. doi: 10.1101/gad.1880510
de Ligt, J., Willemsen, M. H., van Bon, B. W. M., Kleefstra, T., Yntema, H. G., Kroes, T., et al. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929. doi: 10.1056/NEJMoa1206524
Deacon, S. W., Beeser, A., Fukui, J. A., Rennefahrt, U. E. E., Myers, C., Chernoff, J., et al. (2008). An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Biol. 15, 322–331. doi: 10.1016/j.chembiol.2008.03.005
Deidda, G., Bozarth, I. F., and Cancedda, L. (2014). Modulation of GABAergic transmission in development and neurodevelopmental disorders: investigating physiology and pathology to gain therapeutic perspectives. Front. Cell. Neurosci. 8:119. doi: 10.3389/fncel.2014.00119
Deng, Y., Wei, J., Cheng, J., Zhong, P., Xiong, Z., Liu, A., et al. (2016). Partial amelioration of synaptic and cognitive deficits by inhibiting cofilin dephosphorylation in an animal model of Alzheimer’s disease. J. Alzheimer’s Dis. JAD 53, 1419–1432. doi: 10.3233/JAD-160167
des Portes, V., Soufir, N., Carrié, A., Billuart, P., Bienvenu, T., Vinet, M. C., et al. (1997). Gene for nonspecific X-linked mental retardation (MRX 47) is located in Xq22.3-q24. Am. J. Med. Genet. 72, 324–328. doi: 10.1002/(sici)1096-8628(19971031)72:3<324::aid-ajmg14<3.0.co;2-v
Dixit, A., and Verkhivker, G. M. (2014). Structure-functional prediction and analysis of cancer mutation effects in protein kinases. Comput. Mathe. Methods Med. 2014:653487. doi: 10.1155/2014/653487
Dolan, B. M., Duron, S. G., Campbell, D. A., Vollrath, B., Shankaranarayana Rao, B. S., Ko, H.-Y., et al. (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. U.S.A. 110, 5671–5676. doi: 10.1073/pnas.1219383110
Donnelly, A. J., Partington, M. W., Ryan, A. K., and Mulley, J. C. (1996). Regional localization of two non-specific X-linked mental retardation genes (MRX30 and MRX31). Am. J. Med. Genet. 64, 113–120. doi: 10.1002/(SICI)1096-8628(19960712)64:1<113::AID-AJMG19<3.0.CO;2-Q
Duarte, K., Heide, S., Poëa-Guyon, S., Rousseau, V., Depienne, C., Rastetter, A., et al. (2020). PAK3 mutations responsible for severe intellectual disability and callosal agenesis inhibit cell migration. Neurobiol. Dis. 136:104709. doi: 10.1016/j.nbd.2019.104709
Dubos, A., Combeau, G., Bernardinelli, Y., Barnier, J.-V., Hartley, O., Gaertner, H., et al. (2012). Alteration of synaptic network dynamics by the intellectual disability protein PAK3. J. Neurosci. 32, 519–527. doi: 10.1523/JNEUROSCI.3252-11.2012
Eriguchi, M., Mizuta, H., Luo, S., Kuroda, Y., Hara, H., and Rubinsztein, D. C. (2010). Alpha pix enhances mutant huntingtin aggregation. J. Neurol. Sci. 290, 80–85. doi: 10.1016/j.jns.2009.11.003
Ernst, C. (2016). Proliferation and differentiation deficits are a major convergence point for neurodevelopmental disorders. Trends Neurosci. 39, 290–299. doi: 10.1016/j.tins.2016.03.001
Eswaran, J., Soundararajan, M., Kumar, R., and Knapp, S. (2008). UnPAKing the class differences among p21-activated kinases. Trends Biochem. Sci. 33, 394–403. doi: 10.1016/j.tibs.2008.06.002
Fallah, M. S., and Eubanks, J. H. (2020). Seizures in mouse models of rare neurodevelopmental disorders. Neuroscience 445, 50–68. doi: 10.1016/j.neuroscience.2020.01.041
Fernández-Jaén, A., Castellanos, M., Fernández-Perrone, A. L., Fernández-Mayoralas, D. M., de la Vega, A. G., Calleja-Pérez, B., et al. (2014). Cerebral palsy, epilepsy, and severe intellectual disability in a patient with 3q29 microduplication syndrome. Am. J. Med. Genet. 164, 2043–2047. doi: 10.1002/ajmg.a.36559
Fischer, U., Stroh, C., and Schulze-Osthoff, K. (2006). Unique and overlapping substrate specificities of caspase-8 and caspase-10. Oncogene 25:1. doi: 10.1038/sj.onc.1209015
Forrest, M. P., Hill, M. J., Kavanagh, D. H., Tansey, K. E., Waite, A. J., and Blake, D. J. (2018). The psychiatric risk gene transcription factor 4 (TCF4) regulates neurodevelopmental pathways associated with schizophrenia, autism, and intellectual disability. Schizophr. Bull. 44, 1100–1110. doi: 10.1093/schbul/sbx164
Frangiskakis, J. M., Ewart, A. K., Morris, C. A., Mervis, C. B., Bertrand, J., Robinson, B. F., et al. (1996). LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell 86, 59–69. doi: 10.1016/S0092-8674(00)80077-X
Frost, J. A., Swantek, J. L., Stippec, S., Yin, M. J., Gaynor, R., and Cobb, M. H. (2000). Stimulation of NFkappa B activity by multiple signaling pathways requires PAK1. J. Biol. Chem. 275, 19693–19699. doi: 10.1074/jbc.M909860199
Fuchs, C., Cosentino, L., Urbinati, C., Talamo, M. C., Medici, G., Quattrini, M. C., et al. (2022). Treatment with FRAX486 rescues neurobehavioral and metabolic alterations in a female mouse model of CDKL5 deficiency disorder. CNS Neurosci. Ther. 28, 1718–1732. doi: 10.1111/cns.13907
Fuchsova, B., Juliá, A. A., Rizavi, H. S., Frasch, A. C., and Pandey, G. N. (2016). Expression of p21-activated kinases 1 and 3 is altered in the brain of subjects with depression. Neuroscience 333, 331–344. doi: 10.1016/j.neuroscience.2016.07.037
Gécz, J., Shoubridge, C., and Corbett, M. (2009). The genetic landscape of intellectual disability arising from chromosome X. Trends Genetics TIG 25, 308–316. doi: 10.1016/j.tig.2009.05.002
Gedeon, A. K., Nelson, J., Gécz, J., and Mulley, J. C. (2003). X-linked mild non-syndromic mental retardation with neuropsychiatric problems and the missense mutation A365E in PAK3. Am. J. Med. Genet. Part A 120A, 509–517. doi: 10.1002/ajmg.a.20131
Geisheker, M. R., Heymann, G., Wang, T., Coe, B. P., Turner, T. N., Stessman, H. A. F., et al. (2017). Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nat. Neurosci. 20, 1043–1051. doi: 10.1038/nn.4589
Gentile, J. E., Carrizales, M. G., and Koleske, A. J. (2022). Control of synapse structure and function by actin and its regulators. Cells 11:4. doi: 10.3390/cells11040603
Glasson, E. J., Buckley, N., Chen, W., Leonard, H., Epstein, A., Skoss, R., et al. (2020). Systematic review and meta-analysis: mental health in children with neurogenetic disorders associated with intellectual disability. J. Am. Acad. Child Adoles. Psychiatry 59, 1036–1048. doi: 10.1016/j.jaac.2020.01.006
Gussow, A. B., Petrovski, S., Wang, Q., Allen, A. S., and Goldstein, D. B. (2016). The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Geno. Biol. 17:9. doi: 10.1186/s13059-016-0869-4
Halder, S. K., Rafi, M., Shahriar, E. B., Albogami, S., El-Shehawi, A. M., Daullah, S. M., et al. (2022). Identification of the most damaging nsSNPs in the human CFL1 gene and their functional and structural impacts on cofilin-1 protein. Gene 819:146206. doi: 10.1016/j.gene.2022.146206
Harms, F. L., Kloth, K., Bley, A., Denecke, J., Santer, R., Lessel, D., et al. (2018). Activating mutations in PAK1, encoding p21-activated kinase 1, cause a neurodevelopmental disorder. Am. J. Hum. Genet. 103, 579–591. doi: 10.1016/j.ajhg.2018.09.005
Hashimoto, S., Tsubouchi, A., Mazaki, Y., and Sabe, H. (2001). Interaction of paxillin with p21-activated Kinase (PAK). Association of paxillin alpha with the kinase-inactive and the Cdc42-activated forms of PAK3. J. Biol. Chem. 276, 6037–6045. doi: 10.1074/jbc.M005854200
Hayashi, K., Ohshima, T., Hashimoto, M., and Mikoshiba, K. (2007). Pak1 regulates dendritic branching and spine formation. Dev. Neurobiol. 67, 655–669. doi: 10.1002/dneu.20363
Hayashi, M. L., Choi, S.-Y., Rao, B. S. S., Jung, H.-Y., Lee, H.-K., Zhang, D., et al. (2004). Altered cortical synaptic morphology and impaired memory consolidation in forebrain- specific dominant-negative PAK transgenic mice. Neuron 42, 773–787. doi: 10.1016/j.neuron.2004.05.003
Hayashi-Takagi, A., Araki, Y., Nakamura, M., Vollrath, B., Duron, S. G., Yan, Z., et al. (2014). PAKs inhibitors ameliorate schizophrenia-associated dendritic spine deterioration in vitro and in vivo during late adolescence. Proc. Natl. Acad. Sci. U.S.A. 111, 6461–6466. doi: 10.1073/pnas.1321109111
Hertecant, J., Komara, M., Nagi, A., Al-Zaabi, O., Fathallah, W., Cui, H., et al. (2017). A de novo mutation in the X-linked PAK3 gene is the underlying cause of intellectual disability and macrocephaly in monozygotic twins. Eur. J. Med. Genet. 60, 212–216. doi: 10.1016/j.ejmg.2017.01.004
Honarmand Tamizkar, K., Badrlou, E., Aslani, T., Brand, S., Arsang-Jang, S., Ghafouri-Fard, S., et al. (2021). Dysregulation of NF-κB-associated LncRNAs in autism spectrum disorder. Front. Mol. Neurosci. 14:747785. doi: 10.3389/fnmol.2021.747785
Horn, S., Au, M., Basel-Salmon, L., Bayrak-Toydemir, P., Chapin, A., Cohen, L., et al. (2019). De novo variants in PAK1 lead to intellectual disability with macrocephaly and seizures. Brain J. Neurol. 142, 3351–3359. doi: 10.1093/brain/awz264
Horvath, G. A., Tarailo-Graovac, M., Bartel, T., Race, S., Van Allen, M. I., Blydt-Hansen, I., et al. (2018). Improvement of self-injury with dopamine and serotonin replacement therapy in a patient with a hemizygous PAK3 mutation: a new therapeutic strategy for neuropsychiatric features of an intellectual disability syndrome. J. Child Neurol. 33, 106–113. doi: 10.1177/0883073817740443
Hu, B., Arpag, S., Zhang, X., Möbius, W., Werner, H., Sosinsky, G., et al. (2016). Tuning PAK activity to rescue abnormal myelin permeability in HNPP. PLoS Genet. 12:e1006290. doi: 10.1371/journal.pgen.1006290
Hu, S., Zhao, X., Mao, G., Zhang, Z., Wen, X., Zhang, C., et al. (2019). MicroRNA-455-3p promotes TGF-β signaling and inhibits osteoarthritis development by directly targeting PAK2. Exp. Mol. Med. 51:10. doi: 10.1038/s12276-019-0322-3
Huang, W., Zhou, Z., Asrar, S., Henkelman, M., Xie, W., and Jia, Z. (2011). P21-activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Mol. Cell. Biol. 31, 388–403. doi: 10.1128/MCB.00969-10
Hussain, N. K., Thomas, G. M., Luo, J., and Huganir, R. L. (2015). Regulation of AMPA receptor subunit GluA1 surface expression by PAK3 phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 112, E5883–E5890. doi: 10.1073/pnas.1518382112
Iida, A., Takano, K., Takeshita, E., Abe-Hatano, C., Hirabayashi, S., Inaba, Y., et al. (2019). A novel PAK3 pathogenic variant identified in two siblings from a Japanese family with X-linked intellectual disability: case report and review of the literature. Cold Spring Harbor Mol. Case Stud. 5:a003988. doi: 10.1101/mcs.a003988
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Ismail, V., Zachariassen, L. G., Godwin, A., Sahakian, M., Ellard, S., Stals, K. L., et al. (2022). Identification and functional evaluation of GRIA1 missense and truncation variants in individuals with ID: an emerging neurodevelopmental syndrome. Am. J. Hum. Genet. 109, 1217–1241. doi: 10.1016/j.ajhg.2022.05.009
Ivanova, D., Imig, C., Camacho, M., Reinhold, A., Guhathakurta, D., Montenegro-Venegas, C., et al. (2020). CtBP1-mediated membrane fission contributes to effective recycling of synaptic vesicles. Cell Rep. 30, 2444–2459.e7. doi: 10.1016/j.celrep.2020.01.079
Jaffer, Z. M., and Chernoff, J. (2002). p21-activated kinases: three more join the pak. Int. J. Biochem. Cell Biol. 34, 713–717. doi: 10.1016/s1357-2725(01)00158-3
Jakobi, R., McCarthy, C. C., Koeppel, M. A., and Stringer, D. K. (2003). Caspase-activated PAK-2 is regulated by subcellular targeting and proteasomal degradation*. J. Biol. Chem. 278, 38675–38685. doi: 10.1074/jbc.M306494200
Jasoliya, M., Gu, J., AlOlaby, R. R., Durbin-Johnson, B., Chedin, F., and Tassone, F. (2022). Profiling genome-wide DNA methylation in children with autism spectrum disorder and in children with fragile X syndrome. Genes 13:1795. doi: 10.3390/genes13101795
Kanumuri, R., Saravanan, R., Pavithra, V., Sundaram, S., Rayala, S. K., and Venkatraman, G. (2020). Current trends and opportunities in targeting p21 activated kinase-1(PAK1) for therapeutic management of breast cancers. Gene 760:144991. doi: 10.1016/j.gene.2020.144991
Katayama, K.-I., Zheng, Y., and Inoue, N. (2023). Rac is required for the survival of cortical neurons. Exp. Neurol. 361:114316. doi: 10.1016/j.expneurol.2022.114316
Kernohan, K. D., McBride, A., Hartley, T., Rojas, S. K., Care4Rare Canada Consortium, Dyment, D. A., et al. (2019). P21 protein-activated kinase 1 is associated with severe regressive autism, and epilepsy. Clin. Genet. 96, 449–455. doi: 10.1111/cge.13618
Kim, C. K., and Pak, T. R. (2020). MiRNA degradation in the mammalian brain. Am. J. Physiol. Cell Physiol. 319, C624–C629. doi: 10.1152/ajpcell.00303.2020
Knaus, U. G., Wang, Y., Reilly, A. M., Warnock, D., and Jackson, J. H. (1998). Structural requirements for PAK activation by Rac GTPases. J. Biol. Chem. 273, 21512–21518. doi: 10.1074/jbc.273.34.21512
Koh, C. G., Manser, E., Zhao, Z. S., Ng, C. P., and Lim, L. (2001). Beta1PIX, the PAK-interacting exchange factor, requires localization via a coiled-coil region to promote microvillus-like structures and membrane ruffles. J. Cell Sci. 114, 4239–4251. doi: 10.1242/jcs.114.23.4239
Kong, D., Dagon, Y., Campbell, J. N., Guo, Y., Yang, Z., Yi, X., et al. (2016). A postsynaptic AMPK→p21-activated kinase pathway drives fasting-induced synaptic plasticity in AgRP neurons. Neuron 91, 25–33. doi: 10.1016/j.neuron.2016.05.025
Koo, K. H., and Kwon, H. (2018). MicroRNA miR-4779 suppresses tumor growth by inducing apoptosis and cell cycle arrest through direct targeting of PAK2 and CCND3. Cell Death Dis. 9:77. doi: 10.1038/s41419-017-0100-x
Koth, A. P., Oliveira, B. R., Parfitt, G. M., Buonocore, J., de, Q., and Barros, D. M. (2014). Participation of group I p21-activated kinases in neuroplasticity. J. Physiol. Paris 108, 270–277. doi: 10.1016/j.jphysparis.2014.08.007
Kreis, P., and Barnier, J.-V. (2009). PAK signalling in neuronal physiology. Cell. Sign. 21, 384–393. doi: 10.1016/j.cellsig.2008.11.001
Kreis, P., Rousseau, V., Thévenot, E., Combeau, G., and Barnier, J.-V. (2008). The four mammalian splice variants encoded by the p21-activated kinase 3 gene have different biological properties. J. Neurochem. 106, 1184–1197. doi: 10.1111/j.1471-4159.2008.05474.x
Kreis, P., Thévenot, E., Rousseau, V., Boda, B., Muller, D., and Barnier, J.-V. (2007). The p21-activated kinase 3 implicated in mental retardation regulates spine morphogenesis through a Cdc42-dependent pathway. J. Biol. Chem. 282, 21497–21506. doi: 10.1074/jbc.M703298200
Kumar, R., and Li, D.-Q. (2016). PAKs in human cancer progression: from inception to cancer therapeutic to future oncobiology. Adv. Cancer Res. 130, 137–209. doi: 10.1016/bs.acr.2016.01.002
Kutsche, K., Yntema, H., Brandt, A., Jantke, I., Nothwang, H. G., Orth, U., et al. (2000). Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat. Genet. 26, 247–250. doi: 10.1038/80002
Lauterborn, J. C., Cox, C. D., Chan, S. W., Vanderklish, P. W., Lynch, G., and Gall, C. M. (2020). Synaptic actin stabilization protein loss in down syndrome and alzheimer disease. Brain Pathol. 30, 319–331. doi: 10.1111/bpa.12779
Leblond, C. S., Le, T.-L., Malesys, S., Cliquet, F., Tabet, A.-C., Delorme, R., et al. (2021). Operative list of genes associated with autism and neurodevelopmental disorders based on database review. Mol. Cell. Neurosci. 113:103623. doi: 10.1016/j.mcn.2021.103623
Lee, C. E., Singleton, K. S., Wallin, M., and Faundez, V. (2020). Rare genetic diseases: nature’s experiments on human development. IScience 23:101123. doi: 10.1016/j.isci.2020.101123
Lehrke, R. (1972). A theory of X-linkage of major intellectual traits. Am. J. Mental Deficiency 76, 611–619.
Lei, M., Lu, W., Meng, W., Parrini, M. C., Eck, M. J., Mayer, B. J., et al. (2000). Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell 102, 387–397. doi: 10.1016/s0092-8674(00)00043-x
Leitão, E., Schröder, C., Parenti, I., Dalle, C., Rastetter, A., Kühnel, T., et al. (2022). Systematic analysis and prediction of genes associated with monogenic disorders on human chromosome X. Nat. Commun. 13:4264. doi: 10.1038/s41467-022-34264-y
Lelieveld, S. H., Reijnders, M. R. F., Pfundt, R., Yntema, H. G., Kamsteeg, E.-J., de Vries, P., et al. (2016). Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 19, 1194–1196. doi: 10.1038/nn.4352
Leung, C., Cao, F., Nguyen, R., Joshi, K., Aqrabawi, A. J., Xia, S., et al. (2018). Activation of entorhinal cortical projections to the dentate gyrus underlies social memory retrieval. Cell Rep. 23, 2379–2391. doi: 10.1016/j.celrep.2018.04.073
Li, Q., Wu, X., Guo, L., Shi, J., and Li, J. (2019). MicroRNA-7-5p induces cell growth inhibition, cell cycle arrest and apoptosis by targeting PAK2 in non-small cell lung cancer. FEBS Open Bio. 9, 1983–1993. doi: 10.1002/2211-5463.12738
Li, S., Xiong, G.-J., Huang, N., and Sheng, Z.-H. (2020). The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2:10. doi: 10.1038/s42255-020-00289-0
Liaci, C., Camera, M., Caslini, G., Rando, S., Contino, S., Romano, V., et al. (2021). Neuronal cytoskeleton in intellectual disability: from systems biology and modeling to therapeutic opportunities. Int. J. Mol. Sci. 22:6167. doi: 10.3390/ijms22116167
Lisi, E. C., Hamosh, A., Doheny, K. F., Squibb, E., Jackson, B., Galczynski, R., et al. (2008). 3q29 interstitial microduplication: a new syndrome in a three-generation family. Am. J. Med. Genet. Part A 146A, 601–609. doi: 10.1002/ajmg.a.32190
Liu, C., Zhang, L., Cui, W., Du, J., Li, Z., Pang, Y., et al. (2019). Epigenetically upregulated GEFT-derived invasion and metastasis of rhabdomyosarcoma via epithelial mesenchymal transition promoted by the Rac1/Cdc42-PAK signalling pathway. EBioMedicine 50, 122–134. doi: 10.1016/j.ebiom.2019.10.060
Liu, H., Liu, K., and Dong, Z. (2021). The role of p21-activated kinases in cancer and beyond: where are we heading? Front. Cell Dev. Biol. 9:641381. doi: 10.3389/fcell.2021.641381
López-Colomé, A. M., Lee-Rivera, I., Benavides-Hidalgo, R., and López, E. (2017). Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 10:50. doi: 10.1186/s13045-017-0418-y
Lubs, H. A., Stevenson, R. E., and Schwartz, C. E. (2012). Fragile X and X-Linked intellectual disability: four decades of discovery. Am. J. Hum. Genet. 90, 579–590. doi: 10.1016/j.ajhg.2012.02.018
Luo, S., Mizuta, H., and Rubinsztein, D. C. (2008). P21-activated kinase 1 promotes soluble mutant huntingtin self-interaction and enhances toxicity. Hum. Mol. Genet. 17, 895–905. doi: 10.1093/hmg/ddm362
Luo, S., and Rubinsztein, D. C. (2009). Huntingtin promotes cell survival by preventing Pak2 cleavage. J. Cell Sci. 122, 875–885. doi: 10.1242/jcs.050013
Ma, Q.-L., Yang, F., Calon, F., Ubeda, O. J., Hansen, J. E., Weisbart, R. H., et al. (2008). P21-activated kinase-aberrant activation and translocation in Alzheimer disease pathogenesis. J. Biol. Chem. 283, 14132–14143. doi: 10.1074/jbc.M708034200
Ma, Q.-L., Yang, F., Frautschy, S. A., and Cole, G. M. (2012). PAK in alzheimer disease. Huntington disease and X-linked mental retardation. Cell. Logist. 2, 117–125. doi: 10.4161/cl.21602
Magini, P., Pippucci, T., Tsai, I.-C., Coppola, S., Stellacci, E., Bartoletti-Stella, A., et al. (2014). A mutation in PAK3 with a dual molecular effect deregulates the RAS/MAPK pathway and drives an X-linked syndromic phenotype. Hum. Mol. Genet. 23, 3607–3617. doi: 10.1093/hmg/ddu070
Maglorius Renkilaraj, M. R. L., Baudouin, L., Wells, C. M., Doulazmi, M., Wehrlé, R., Cannaya, V., et al. (2017). The intellectual disability protein PAK3 regulates oligodendrocyte precursor cell differentiation. Neurobiol. Dis. 98, 137–148. doi: 10.1016/j.nbd.2016.12.004
Magne, N., Rousseau, V., Duarte, K., Poëa-Guyon, S., Gleize, V., Mutel, A., et al. (2021). PAK3 is a key signature gene of the glioma proneural subtype and affects its proliferation, differentiation and growth. Cell. Oncol. 44, 1257–1271. doi: 10.1007/s13402-021-00635-8
Majumder, M., Johnson, R. H., and Palanisamy, V. (2020). Fragile X-related protein family: a double-edged sword in neurodevelopmental disorders and cancer. Crit. Rev. Biochem. Mol. Biol. 55, 409–424. doi: 10.1080/10409238.2020.1810621
Malecka, K. A., Szentpetery, Z., and Peterson, J. R. (2013). Synergistic activation of p21-activated kinase 1 by phosphatidylinositol 4,5-bisphosphate and rho GTPases*. J. Biol. Chem. 288, 8887–8897. doi: 10.1074/jbc.M112.428904
Manser, E., Chong, C., Zhao, Z. S., Leung, T., Michael, G., Hall, C., et al. (1995). Molecular cloning of a new member of the p21-Cdc42/Rac-activated kinase (PAK) family. J. Biol. Chem. 270, 25070–25078. doi: 10.1074/jbc.270.42.25070
Manser, E., Loo, T. H., Koh, C. G., Zhao, Z. S., Chen, X. Q., Tan, L., et al. (1998). PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell 1, 183–192. doi: 10.1016/s1097-2765(00)80019-2
Mariano, V., Achsel, T., Bagni, C., and Kanellopoulos, A. K. (2020). Modelling learning and memory in drosophila to understand intellectual disabilities. Neuroscience 445, 12–30. doi: 10.1016/j.neuroscience.2020.07.034
Marlin, J. W., Chang, Y.-W. E., Ober, M., Handy, A., Xu, W., and Jakobi, R. (2011). Functional PAK-2 knockout and replacement with a caspase cleavage-deficient mutant in mice reveals differential requirements of full-length PAK-2 and caspase-activated PAK-2p34. Mammalian Geno. Offi. J. Int. Mammalian Geno. Soc. 22, 306–317. doi: 10.1007/s00335-011-9326-6
Martinelli, S., Krumbach, O. H. F., Pantaleoni, F., Coppola, S., Amin, E., Pannone, L., et al. (2018). Functional dysregulation of CDC42 causes diverse developmental phenotypes. Am. J. Hum. Genet. 102, 309–320. doi: 10.1016/j.ajhg.2017.12.015
McMichael, G., Bainbridge, M. N., Haan, E., Corbett, M., Gardner, A., Thompson, S., et al. (2015). Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol. Psychiatry 20, 176–182. doi: 10.1038/mp.2014.189
McPhie, D. L., Coopersmith, R., Hines-Peralta, A., Chen, Y., Ivins, K. J., Manly, S. P., et al. (2003). DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J. Neurosci. Offi. J. Soc. Neurosci. 23, 6914–6927. doi: 10.1523/JNEUROSCI.23-17-06914.2003
Memi, F., Zecevic, N., and Radonjiæ, N. (2018). Multiple roles of sonic hedgehog in the developing human cortex are suggested by its widespread distribution. Brain Struct. Funct. 223, 2361–2375. doi: 10.1007/s00429-018-1621-5
Meng, J., Meng, Y., Hanna, A., Janus, C., and Jia, Z. (2005). Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J. Neurosci. Offi. J. Soc. Neurosci. 25, 6641–6650. doi: 10.1523/JNEUROSCI.0028-05.2005
Michetti, C., Falace, A., Benfenati, F., and Fassio, A. (2022). Synaptic genes and neurodevelopmental disorders: from molecular mechanisms to developmental strategies of behavioral testing. Neurobiol. Dis. 173:105856. doi: 10.1016/j.nbd.2022.105856
Miller, J. A., Ding, S.-L., Sunkin, S. M., Smith, K. A., Ng, L., Szafer, A., et al. (2014). Transcriptional landscape of the prenatal human brain. Nature 508, 199–206. doi: 10.1038/nature13185
Molosh, A. I., Johnson, P. L., Spence, J. P., Arendt, D., Federici, L. M., Bernabe, C., et al. (2014). Social learning and amygdala disruptions in Nf1 mice are rescued by blocking p21-activated kinase. Nat. Neurosci. 17, 1583–1590. doi: 10.1038/nn.3822
Monje, M., and Iwasaki, A. (2022). The neurobiology of long COVID. Neuron 110, 3484–3496. doi: 10.1016/j.neuron.2022.10.006
Moretto, E., Murru, L., Martano, G., Sassone, J., and Passafaro, M. (2018). Glutamatergic synapses in neurodevelopmental disorders. Prog. Neuro Psychopharmacol. Biol. Psychiatry 84, 328–342. doi: 10.1016/j.pnpbp.2017.09.014
Morris-Rosendahl, D. J., and Crocq, M.-A. (2020). Neurodevelopmental disorders-the history and future of a diagnostic concept. Dial. Clin. Neurosci. 22, 65–72. doi: 10.31887/DCNS.2020.22.1/macrocq
Motta, M., Pannone, L., Pantaleoni, F., Bocchinfuso, G., Radio, F. C., Cecchetti, S., et al. (2020). Enhanced MAPK1 function causes a neurodevelopmental disorder within the RASopathy clinical spectrum. Am. J. Hum. Genet. 107, 499–513. doi: 10.1016/j.ajhg.2020.06.018
Mulligan, K. A., and Cheyette, B. N. R. (2017). Neurodevelopmental perspectives on wnt signaling in psychiatry. Mol. Neuropsychiatry 2, 219–246. doi: 10.1159/000453266
Muthusamy, B., Selvan, L. D. N., Nguyen, T. T., Manoj, J., Stawiski, E. W., Jaiswal, B. S., et al. (2017). Next-generation sequencing reveals novel mutations in X-linked intellectual disability. Omics J. Int. Biol. 21, 295–303. doi: 10.1089/omi.2017.0009
Nadif Kasri, N., and Van Aelst, L. (2008). Rho-linked genes and neurological disorders. Pflügers Archiv Eur. J. Physiol. 455, 787–797. doi: 10.1007/s00424-007-0385-1
Nagy, D., Farkas, K., Armengol, L., Segura, M., Esi Zodanu, G. K., Csányi, B., et al. (2020). Further delineation of the phenotype of PAK3-associated x-linked intellectual disability: identification of a novel missense mutation and review of literature. Eur. J. Med. Genet. 63:103800. doi: 10.1016/j.ejmg.2019.103800
Nguyen, T.-V. V., Galvan, V., Huang, W., Banwait, S., Tang, H., Zhang, J., et al. (2008). Signal transduction in alzheimer disease: P21-activated kinase signaling requires C-terminal cleavage of APP at Asp664. J. Neurochem. 104, 1065–1080. doi: 10.1111/j.1471-4159.2007.05031.x
Obermeier, A., Ahmed, S., Manser, E., Yen, S. C., Hall, C., and Lim, L. (1998). PAK promotes morphological changes by acting upstream of Rac. EMBO J. 17, 4328–4339. doi: 10.1093/emboj/17.15.4328
Ohori, S., Mitsuhashi, S., Ben-Haim, R., Heyman, E., Sengoku, T., Ogata, K., et al. (2020). A novel PAK1 variant causative of neurodevelopmental disorder with postnatal macrocephaly. J. Hum. Genet. 65, 481–485. doi: 10.1038/s10038-020-0728-8
OMIM (2022). Available online at: https://www.omim.org/ (accessed December 6, 2022)
Owen, M. J., and O’Donovan, M. C. (2017). Schizophrenia and the neurodevelopmental continuum: evidence from genomics. World Psychiatry 16, 227–235. doi: 10.1002/wps.20440
Pan, X., Chang, X., Leung, C., Zhou, Z., Cao, F., Xie, W., et al. (2015). PAK1 regulates cortical development via promoting neuronal migration and progenitor cell proliferation. Mol. Brain 8:36. doi: 10.1186/s13041-015-0124-z
Parenti, I., Rabaneda, L. G., Schoen, H., and Novarino, G. (2020). Neurodevelopmental disorders: from genetics to functional pathways. Trends Neurosci. 43, 608–621. doi: 10.1016/j.tins.2020.05.004
Parker, N. H., Donninger, H., Birrer, M. J., and Leaner, V. D. (2013). P21-activated kinase 3 (PAK3) is an AP-1 regulated gene contributing to actin organisation and migration of transformed fibroblasts. PLoS One 8:e66892. doi: 10.1371/journal.pone.0066892
Parrini, M. C., Camonis, J., Matsuda, M., and de Gunzburg, J. (2009). Dissecting activation of the PAK1 kinase at protrusions in living cells*. J. Biol. Chem. 284, 24133–24143. doi: 10.1074/jbc.M109.015271
Parrini, M. C., Lei, M., Harrison, S. C., and Mayer, B. J. (2002). Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol. Cell 9, 73–83. doi: 10.1016/s1097-2765(01)00428-2
Pascolini, G., Gaudioso, F., Passarelli, C., Novelli, A., Di Giosaffatte, N., Majore, S., et al. (2021). Clinical and molecular aspects of the neurodevelopmental disorder associated with PAK3 perturbation. J. Mol. Neurosci. 71, 2474–2481. doi: 10.1007/s12031-021-01868-w
Peerboom, C., and Wierenga, C. J. (2021). The postnatal GABA shift: a developmental perspective. Neurosci. Biobehav. Rev. 124, 179–192. doi: 10.1016/j.neubiorev.2021.01.024
Peippo, M., Koivisto, A. M., Särkämö, T., Sipponen, M., von Koskull, H., Ylisaukko-oja, T., et al. (2007). PAK3 related mental disability: further characterization of the phenotype. Am. J. Med. Genet. Part A 143A, 2406–2416. doi: 10.1002/ajmg.a.31956
Penzes, P., and Cahill, M. E. (2012). Deconstructing signal transduction pathways that regulate the actin cytoskeleton in dendritic spines. Cytoskeleton 69, 426–441. doi: 10.1002/cm.21015
Piccand, J., Meunier, A., Merle, C., Jia, Z., Barnier, J.-V., and Gradwohl, G. (2014). Pak3 promotes cell cycle exit and differentiation of β-cells in the embryonic pancreas and is necessary to maintain glucose homeostasis in adult mice. Diabetes 63, 203–215. doi: 10.2337/db13-0384
Pirruccello, M., Sondermann, H., Pelton, J. G., Pellicena, P., Hoelz, A., Chernoff, J., et al. (2006). A dimeric kinase assembly underlying autophosphorylation in the p21 activated kinases. J. Mol. Biol. 361, 312–326. doi: 10.1016/j.jmb.2006.06.017
Piton, A., Redin, C., and Mandel, J.-L. (2013). XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am. J. Hum. Genet. 93, 368–383. doi: 10.1016/j.ajhg.2013.06.013
Puto, L. A., Pestonjamasp, K., King, C. C., and Bokoch, G. M. (2003). P21-activated kinase 1 (PAK1) interacts with the Grb2 adapter protein to couple to growth factor signaling. J. Biol. Chem. 278, 9388–9393. doi: 10.1074/jbc.M208414200
Qian, X., DeGennaro, E. M., Talukdar, M., Akula, S. K., Lai, A., Shao, D. D., et al. (2022). Loss of non-motor kinesin KIF26A causes congenital brain malformations via dysregulated neuronal migration and axonal growth as well as apoptosis. Dev. Cell 57, 2381–2396e13. doi: 10.1016/j.devcel.2022.09.011
Rahman, M. R., Petralia, M. C., Ciurleo, R., Bramanti, A., Fagone, P., Shahjaman, M., et al. (2020). Comprehensive analysis of RNA-seq gene expression profiling of brain transcriptomes reveals novel genes, regulators, and pathways in autism spectrum disorder. Brain Sci. 10:747. doi: 10.3390/brainsci10100747
Reijnders, M. R. F., Ansor, N. M., Kousi, M., Yue, W. W., Tan, P. L., Clarkson, K., et al. (2017). RAC1 missense mutations in developmental disorders with diverse phenotypes. Am. J. Hum. Genet. 101, 466–477. doi: 10.1016/j.ajhg.2017.08.007
Rejeb, I., Saillour, Y., Castelnau, L., Julien, C., Bienvenu, T., Taga, P., et al. (2008). A novel splice mutation in PAK3 gene underlying mental retardation with neuropsychiatric features. Eur. J. Hum. Genet. 16, 1358–1363. doi: 10.1038/ejhg.2008.103
Rosen, T. E., Mazefsky, C. A., Vasa, R. A., and Lerner, M. D. (2018). Co-occurring psychiatric conditions in autism spectrum disorder. Int. Rev. Psychiatry 30, 40–61. doi: 10.1080/09540261.2018.1450229
Rotaru, D. C., Mientjes, E. J., and Elgersma, Y. (2020). Angelman syndrome: from mouse models to therapy. Neuroscience 445, 172–189. doi: 10.1016/j.neuroscience.2020.02.017
Rousseau, V., Goupille, O., Morin, N., and Barnier, J.-V. (2003). A new constitutively active brain PAK3 isoform displays modified specificities toward Rac and Cdc42 GTPases. J. Biol. Chem. 278, 3912–3920. doi: 10.1074/jbc.M207251200
Rudel, T., and Bokoch, G. M. (1997). Membrane and Morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science 276, 1571–1574. doi: 10.1126/science.276.5318.1571
Sanders, S. J., He, X., Willsey, A. J., Ercan-Sencicek, A. G., Samocha, K. E., Cicek, A. E., et al. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233. doi: 10.1016/j.neuron.2015.09.016
Sanders, S. J., Sahin, M., Hostyk, J., Thurm, A., Jacquemont, S., Avillach, P., et al. (2019). A framework for the investigation of rare genetic disorders in neuropsychiatry. Nat. Med. 25, 1477–1487. doi: 10.1038/s41591-019-0581-5
Santarelli, D. M., Carroll, A. P., Cairns, H. M., Tooney, P. A., and Cairns, M. J. (2019). Schizophrenia-associated MicroRNA-gene interactions in the dorsolateral prefrontal cortex. Genom. Proteom. Bioinform. 17, 623–634. doi: 10.1016/j.gpb.2019.10.003
Santiago-Medina, M., Gregus, K. A., and Gomez, T. M. (2013). PAK–PIX interactions regulate adhesion dynamics and membrane protrusion to control neurite outgrowth. J. Cell Sci. 126, 1122–1133. doi: 10.1242/jcs.112607
Say, E., Tay, H.-G., Zhao, Z., Baskaran, Y., Li, R., Lim, L., et al. (2010). A functional requirement for PAK1 binding to the KH(2) domain of the fragile X protein-related FXR1. Mol. Cell 38, 236–249. doi: 10.1016/j.molcel.2010.04.004
Scala, M., Nishikawa, M., Ito, H., Tabata, H., Khan, T., Accogli, A., et al. (2022). Variant-specific changes in RAC3 function disrupt corticogenesis in neurodevelopmental phenotypes. Brain J. Neurol. 145, 3308–3327. doi: 10.1093/brain/awac106
Schachtschneider, K. M., Liu, Y., Rund, L. A., Madsen, O., Johnson, R. W., Groenen, M. A. M., et al. (2016). Impact of neonatal iron deficiency on hippocampal DNA methylation and gene transcription in a porcine biomedical model of cognitive development. BMC Genom. 17:856. doi: 10.1186/s12864-016-3216-y
Schmidt-Kastner, R. (2015). Genomic approach to selective vulnerability of the hippocampus in brain ischemia-hypoxia. Neuroscience 309, 259–279. doi: 10.1016/j.neuroscience.2015.08.034
Sells, M. A., Boyd, J. T., and Chernoff, J. (1999). P21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 145, 837–849. doi: 10.1083/jcb.145.4.837
Sells, M. A., Knaus, U. G., Bagrodia, S., Ambrose, D. M., Bokoch, G. M., and Chernoff, J. (1997). Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. CB 7, 202–210. doi: 10.1016/s0960-9822(97)70091-5
Semenova, G., and Chernoff, J. (2017). Targeting PAK1. Biochem. Soc. Trans. 45, 79–88. doi: 10.1042/BST20160134
Sementino, E., Kadariya, Y., Cheung, M., Menges, C. W., Tan, Y., Kukuyan, A.-M., et al. (2022). Inactivation of p21-activated kinase 2 (Pak2) inhibits the development of Nf2-deficient tumors by restricting downstream hedgehog and Wnt signaling. Mol. Cancer Res. 20, 699–711. doi: 10.1158/1541-7786.MCR-21-0837
Sharma, A., and Mehan, S. (2021). Targeting PI3K-AKT/mTOR signaling in the prevention of autism. Neurochem. Int. 147:105067. doi: 10.1016/j.neuint.2021.105067
Shaw, A. E., and Bamburg, J. R. (2017). Peptide regulation of cofilin activity in the CNS: a novel therapeutic approach for treatment of multiple neurological disorders. Pharmacol. Ther. 175, 17–27. doi: 10.1016/j.pharmthera.2017.02.031
Shen, W., Ba, R., Su, Y., Ni, Y., Chen, D., Xie, W., et al. (2019). Foxg1 regulates the postnatal development of cortical interneurons. Cerebral Cortex 29, 1547–1560. doi: 10.1093/cercor/bhy051
Sinha, P., Cree, S. L., Miller, A. L., Pearson, J. F., and Kennedy, M. A. (2021). Transcriptional analysis of sodium valproate in a serotonergic cell line reveals gene regulation through both HDAC inhibition-dependent and independent mechanisms. Pharmacogen. J. 21, 359–375. doi: 10.1038/s41397-021-00215-x
Sorrell, F. J., Kilian, L. M., and Elkins, J. M. (2019). Solution structures and biophysical analysis of full-length group a PAKs reveal they are monomeric and auto-inhibited in cis. Biochem. J. 476, 1037–1051. doi: 10.1042/BCJ20180867
Sosa-García, B., Vázquez-Rivera, V., González-Flores, J. N., Engel, B. E., Cress, W. D., and Santiago-Cardona, P. G. (2015). The retinoblastoma tumor suppressor transcriptionally represses pak1 in osteoblasts. PLoS One 10:e0142406. doi: 10.1371/journal.pone.0142406
Souopgui, J. (2002). XPak3 promotes cell cycle withdrawal during primary neurogenesis in Xenopus laevis. EMBO J. 21, 6429–6439. doi: 10.1093/emboj/cdf644
Srivastava, P. K., van Eyll, J., Godard, P., Mazzuferi, M., Delahaye-Duriez, A., Van Steenwinckel, J., et al. (2018). A systems-level framework for drug discovery identifies Csf1R as an anti-epileptic drug target. Nat. Commun. 9:6008. doi: 10.1038/s41467-018-06008-4
Sun, X., Su, V. L., and Calderwood, D. A. (2019). The subcellular localization of type I p21-activated kinases is controlled by the disordered variable region and polybasic sequences. J. Biol. Chem. 294, 14319–14332. doi: 10.1074/jbc.RA119.007692
Sundberg-Smith, L. J., Doherty, J. T., Mack, C. P., and Taylor, J. M. (2005). Adhesion stimulates direct PAK1/ERK2 association and leads to ERK-dependent PAK1 Thr212 phosphorylation. J. Biol. Chem. 280, 2055–2064. doi: 10.1074/jbc.M406013200
Supek, F., Lehner, B., and Lindeboom, R. G. H. (2021). To NMD or Not To NMD: nonsense-mediated mRNA decay in cancer and other genetic diseases. Trends Genet. 37, 657–668. doi: 10.1016/j.tig.2020.11.002
Takata, A. (2019). Estimating contribution of rare non-coding variants to neuropsychiatric disorders. Psychiatry Clin. Neurosci. 73, 2–10. doi: 10.1111/pcn.12774
Tan, X., Tong, L., Li, L., Xu, J., Xie, S., Ji, L., et al. (2021). Loss of Smad4 promotes aggressive lung cancer metastasis by de-repression of PAK3 via miRNA regulation. Nat. Commun. 12:4853. doi: 10.1038/s41467-021-24898-9
Tang, X.-Y., Xu, L., Wang, J., Hong, Y., Wang, Y., Zhu, Q., et al. (2021). DSCAM/PAK1 pathway suppression reverses neurogenesis deficits in iPSC-derived cerebral organoids from patients with down syndrome. J. Clin. Invest. 131:e135763. doi: 10.1172/JCI135763
Tartaglia, M., Aoki, Y., and Gelb, B. D. (2022). The molecular genetics of RASopathies: an update on novel disease genes and new disorders. Am. J. Med. Genet. Part C Sem. Med. Genet. 190, 425–439. doi: 10.1002/ajmg.c.32012
Tastet, J., Cuberos, H., Vallée, B., Toutain, A., Raynaud, M., Marouillat, S., et al. (2019). LIMK2-1 is a hominidae-specific isoform of LIMK2 expressed in central nervous system and associated with intellectual disability. Neuroscience 399, 199–210. doi: 10.1016/j.neuroscience.2018.12.017
Teo, M., Manser, E., and Lim, L. (1995). Identification and molecular cloning of a p21cdc42/rac1-activated serine/threonine kinase that is rapidly activated by thrombin in platelets. J. Biol. Chem. 270, 26690–26697. doi: 10.1074/jbc.270.44.26690
Thévenot, E., Moreau, A. W., Rousseau, V., Combeau, G., Domenichini, F., Jacquet, C., et al. (2011). P21-activated kinase 3 (PAK3) protein regulates synaptic transmission through its interaction with the Nck2/Grb4 protein adaptor. J. Biol. Chem. 286, 40044–40059. doi: 10.1074/jbc.M111.262246
Tu, H., and Wigler, M. (1999). Genetic evidence for pak1 autoinhibition and its release by Cdc42. Mol. Cell. Biol. 19, 602–611. doi: 10.1128/MCB.19.1.602
Tzschach, A., Grasshoff, U., Beck-Woedl, S., Dufke, C., Bauer, C., Kehrer, M., et al. (2015). Next-generation sequencing in X-linked intellectual disability. Eur. J. Hum. Genet. 23, 1513–1518. doi: 10.1038/ejhg.2015.5
van Bokhoven, H. (2011). Genetic and epigenetic networks in intellectual disabilities. Ann. Rev. Genet. 45, 81–104. doi: 10.1146/annurev-genet-110410-132512
Vilela, J., Martiniano, H., Marques, A. R., Santos, J. X., Rasga, C., Oliveira, G., et al. (2022). Disease similarity network analysis of autism spectrum disorder and comorbid brain disorders. Front. Mol. Neurosci. 15:932305. doi: 10.3389/fnmol.2022.932305
Vissers, L. E., Gilissen, C., and Veltman, J. A. (2016). Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 17, 9–18. doi: 10.1038/nrg3999
Wang, J., Wu, J.-W., and Wang, Z.-X. (2011). Structural insights into the autoactivation mechanism of p21-activated protein kinase. Structure 19, 1752–1761. doi: 10.1016/j.str.2011.10.013
Wang, Y., and Guo, F. (2022). Group I PAKs in myelin formation and repair of the central nervous system: what, when, and how. Biol. Rev. 97, 615–639. doi: 10.1111/brv.12815
Wang, Y., Zeng, C., Li, J., Zhou, Z., Ju, X., Xia, S., et al. (2018). PAK2 haploinsufficiency results in synaptic cytoskeleton impairment and autism-related behavior. Cell Rep. 24, 2029–2041. doi: 10.1016/j.celrep.2018.07.061
Wei, S., Du, H., Li, Z., Tao, G., Xu, Z., Song, X., et al. (2019). Transcription factors Sp8 and Sp9 regulate the development of caudal ganglionic eminence-derived cortical interneurons. J. Comparat. Neurol. 527, 2860–2874. doi: 10.1002/cne.24712
Wen, X., Zhu, J., Cai, L., Tang, G., Zeng, W., Luo, Y., et al. (2022). A familial 3q28q29 duplication induced mild intellectual disability: case presentation and literature review. Am. J. Trans. Res. 14, 1663–1671.
Whale, A., Hashim, F. N., Fram, S., Jones, G. E., and Wells, C. M. (2011). Signalling to cancer cell invasion through PAK family kinases. Front. Biosci. 16:849–864. doi: 10.2741/3724
Willsey, A. J., Fernandez, T. V., Yu, D., King, R. A., Dietrich, A., Xing, J., et al. (2017). De novo coding variants are strongly associated with tourette disorder. Neuron 94, 486–499e9. doi: 10.1016/j.neuron.2017.04.024
Wright, C. F., Fitzgerald, T. W., Jones, W. D., Clayton, S., McRae, J. F., van Kogelenberg, M., et al. (2015). Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 385, 1305–1314. doi: 10.1016/S0140-6736(14)61705-0
Xia, S., Zhou, Z., and Jia, Z. (2018). PAK1 regulates inhibitory synaptic function via a novel mechanism mediated by endocannabinoids. Small GTPases 9, 322–326. doi: 10.1080/21541248.2016.1228793
Yamahashi, Y., Lin, Y.-H., Mouri, A., Iwanaga, S., Kawashima, K., Tokumoto, Y., et al. (2022). Phosphoproteomic of the acetylcholine pathway enables discovery of the PKC-β-PIX-Rac1-PAK cascade as a stimulatory signal for aversive learning. Mol. Psychiatry 27:8. doi: 10.1038/s41380-022-01643-2
Yang, J., Yang, X., and Tang, K. (2022). Interneuron development and dysfunction. FEBS J. 289, 2318–2336. doi: 10.1111/febs.15872
Yao, D., Li, C., Rajoka, M. S. R., He, Z., Huang, J., Wang, J., et al. (2020). P21-activated kinase 1: emerging biological functions and potential therapeutic targets in cancer. Theranostics 10, 9741–9766. doi: 10.7150/thno.46913
Ye, D. Z., and Field, J. (2012). PAK signaling in cancer. Cell. Logist. 2, 105–116. doi: 10.4161/cl.21882
Young, A. M. H., Campbell, E., Lynch, S., Suckling, J., and Powis, S. J. (2011). Aberrant NF-KappaB expression in autism spectrum condition: a mechanism for neuroinflammation. Front. Psychiatry 2:27. doi: 10.3389/fpsyt.2011.00027
Yu, S., Di, C., Chen, S., Guo, M., Yan, J., Zhu, Z., et al. (2021). Distinct immune signatures discriminate between asymptomatic and presymptomatic SARS-CoV-2pos subjects. Cell Res. 31:11. doi: 10.1038/s41422-021-00562-1
Yue, K., Wang, X., Wu, Y., Zhou, X., He, Q., and Duan, Y. (2016). MicroRNA-7 regulates cell growth, migration and invasion via direct targeting of PAK1 in thyroid cancer. Mol. Med. Rep. 14, 2127–2134. doi: 10.3892/mmr.2016.5477
Zenke, F. T., Krendel, M., DerMardirossian, C., King, C. C., Bohl, B. P., and Bokoch, G. M. (2004). P21-activated kinase 1 phosphorylates and regulates 14-3-3 binding to GEF-H1, a microtubule-localized Rho exchange factor. J. Biol. Chem. 279, 18392–18400. doi: 10.1074/jbc.M400084200
Zhang, H. (2005). A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J. Neurosci. 25, 3379–3388. doi: 10.1523/JNEUROSCI.3553-04.2005
Zhang, K., Wang, Y., Fan, T., Zeng, C., and Sun, Z. S. (2022). The p21-activated kinases in neural cytoskeletal remodeling and related neurological disorders. Protein Cell 13, 6–25. doi: 10.1007/s13238-020-00812-9
Zhang, X., Xiao, H., Zhang, X., Gong, X., Li, T., Han, Y., et al. (2020). Decreased microRNA-125b-5p disrupts follicle steroidogenesis through targeting PAK3/ERK1/2 signalling in mouse preantral follicles. Metabolism 107:154241. doi: 10.1016/j.metabol.2020.154241
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O’Keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014
Zhao, L., Ma, Q.-L., Calon, F., Harris-White, M. E., Yang, F., Lim, G. P., et al. (2006). Role of p21-activated kinase pathway defects in the cognitive deficits of alzheimer disease. Nat. Neurosci. 9, 234–242. doi: 10.1038/nn1630
Zhao, T., Ren, L., Li, C., Liu, L., Zou, Y., Yan, H., et al. (2022). MiR-7 regulates pathogen-induced immune response via PAK1 in the sea cucumber apostichopus japonicus. Front. Immunol. 13:927796. doi: 10.3389/fimmu.2022.927796
Zhao, Z., Xu, J., Chen, J., Kim, S., Reimers, M., Bacanu, S.-A., et al. (2015). Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder. Mol. Psychiatry 20, 563–572. doi: 10.1038/mp.2014.82
Zhao, Z. S., Manser, E., and Lim, L. (2000). Interaction between PAK and nck: a template for Nck targets and role of PAK autophosphorylation. Mol. Cell. Biol. 20, 3906–3917. doi: 10.1128/MCB.20.11.3906-3917.2000
Zhou, S., and Jia, Z. (2021). Disruption of PAK3 signaling in social interaction induced CFOS positive cells impairs social recognition memory. Cells 10:3010. doi: 10.3390/cells10113010
Zhou, W., Li, X., and Premont, R. T. (2016). Expanding functions of GIT Arf GTPase-activating proteins, PIX Rho guanine nucleotide exchange factors and GIT-PIX complexes. J. Cell Sci. 129, 1963–1974. doi: 10.1242/jcs.179465
Keywords: intellectual disability, autism spectrum disorder (ASD), neurodevelopmental disorders, genotype/phenotype correlation, p21-activated kinase, mutations
Citation: Dobrigna M, Poëa-Guyon S, Rousseau V, Vincent A, Toutain A and Barnier J-V (2023) The molecular basis of p21-activated kinase-associated neurodevelopmental disorders: From genotype to phenotype. Front. Neurosci. 17:1123784. doi: 10.3389/fnins.2023.1123784
Received: 14 December 2022; Accepted: 13 February 2023;
Published: 02 March 2023.
Edited by:
Barbara Bardoni, UMR 7275 Institut de Pharmacologie Moléculaire et Cellulaire (IPMC), FranceReviewed by:
Maria Giuseppina Miano, Institute of Genetics and Biophysics Adriano Buzzati-Traverso (CNR), ItalySangeeta Nath, Manipal Academy of Higher Education, India
Curtis Benson, Yale University, United States
Copyright © 2023 Dobrigna, Poëa-Guyon, Rousseau, Vincent, Toutain and Barnier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Vianney Barnier, amVhbi12aWFubmV5LmJhcm5pZXJAdW5pdmVyc2l0ZS1wYXJpcy1zYWNsYXkuZnI=