 Vanlalrinchhani Varte
Vanlalrinchhani Varte Jeremy W. Munkelwitz
Jeremy W. Munkelwitz Diego E. Rincon-Limas
Diego E. Rincon-Limas- 1Department of Neurology, McKnight Brain Institute, Norman Fixel Institute for Neurological Diseases, University of Florida, Gainesville, FL, United States
- 2Department of Neuroscience, University of Florida, Gainesville, FL, United States
- 3Genetics Institute, University of Florida, Gainesville, FL, United States
Alzheimer’s disease (AD) is the most prevalent neurodegenerative dementia in older adults worldwide. Sadly, there are no disease-modifying therapies available for treatment due to the multifactorial complexity of the disease. AD is pathologically characterized by extracellular deposition of amyloid beta (Aβ) and intracellular neurofibrillary tangles composed of hyperphosphorylated tau. Increasing evidence suggest that Aβ also accumulates intracellularly, which may contribute to the pathological mitochondrial dysfunction observed in AD. According with the mitochondrial cascade hypothesis, mitochondrial dysfunction precedes clinical decline and thus targeting mitochondria may result in new therapeutic strategies. Unfortunately, the precise mechanisms connecting mitochondrial dysfunction with AD are largely unknown. In this review, we will discuss how the fruit fly Drosophila melanogaster is contributing to answer mechanistic questions in the field, from mitochondrial oxidative stress and calcium dysregulation to mitophagy and mitochondrial fusion and fission. In particular, we will highlight specific mitochondrial insults caused by Aβ and tau in transgenic flies and will also discuss a variety of genetic tools and sensors available to study mitochondrial biology in this flexible organism. Areas of opportunity and future directions will be also considered.
Introduction
Drosophila has been used in genetic research for over 100 years (Morgan, 1910). The conservation of genes between Drosophila and human (Rubin et al., 2000), the simplicity of genes with lesser isoform/redundancy in flies, and the smaller size of the Drosophila genome compared to the human counterpart, make Drosophila an excellent model organism. Drosophila also has the advantage of having a short life cycle, around 10 days from embryo to adulthood, is easy to maintain in a small space, and is very economical. Approximately 75% of human disease-causing genes are present in the fly (Rubin et al., 2000; Reiter et al., 2001; Ugur et al., 2016) and its genome has been fully sequenced and extensively annotated (Adams et al., 2000). Fly genes can be over-expressed or knocked-down through RNAi, mutated by targeted knock-out or deletion of the gene, or replaced by knock-in of a gene. Another technical advantage in flies is the presence of the Gal4-UAS system, where the yeast transcription factor Gal4 can drive the expression of any gene of interest at any developmental stage in a tissue-specific manner (Brand and Perrimon, 1993). Thousands of Gal4 drivers and other valuable fly strains are available at stock repositories including the Bloomington Drosophila Stock Center (BDSC), the Harvard Transgenic RNAi Project (TRiP), the Vienna Drosophila Resource Center (VDRC), the Japan National Institute of Genetics (NIG) Stock Center, the Kyoto Drosophila Stock Center (DGGR), and the Zurich ORFeome Project (FlyORF) (Dietzl et al., 2007; Cook et al., 2010; Ni et al., 2011; Yeh et al., 2018). This is highly advantageous as researchers can easily procure essential stocks for experiments in a matter of days. Also, FlyBase is a regularly updated website that provides valuable information about any fly gene, which facilitates rapid progress in the field. All these Drosophila resources and tools make the fly system an ideal platform to approach the pathological complexity underlying neuronal dysfunction and degeneration.
Drosophila as a model to study neurodegenerative diseases
Drosophila melanogaster has been used to study mechanistic aspects of human neurodegenerative diseases for over two decades (Nayak and Mishra, 2022). This is facilitated by the functional conservation of many genes and pathways between humans and flies and their remarkable similarities in neuronal functions. The nervous system of Drosophila is quite complex and includes functionally distinct neurons in the eyes, olfactory, gustatory and auditory organs, ventral nerve cord, brain, as well as peripheral sensory neurons (Hirth, 2010). Multiple assays have been developed to assess neurodegeneration in many of these tissues. For instance, the fly eyes contain photoreceptor neurons and are made up of 800 ommatidia that are uniformly arranged with bristles. Human disease-causing genes can be overexpressed in the eye and the effects of these genes can be determined based on the degree of alterations in eye morphology, including the size of the eye, pigmentation, ommatidial organization, and cell death. All these phenotypic outcomes are very easy to score under a dissecting microscope, making the Drosophila eye the preferred screening platform for modifiers of neurodegenerative processes. Apart from the eye, neurodegeneration can be also assessed based on vacuolization of the brain, locomotor performance such as climbing and flight assays, analysis of neuromuscular junction morphology, life span analysis, as well as assessment of learning and memory decline (McGurk et al., 2015; Gevedon et al., 2019). Interestingly, the accumulation of amyloid aggregates and pathological tau, the main neuropathological hallmarks in AD, can be also assessed in the fly brain by staining with Thioflavin and with antibodies against phosphorylated and conformational tau (Greeve et al., 2004). Thus, neurodegeneration-related genes and their pathways can be easily studied at molecular and pathological level in Drosophila to understand their impact on disease progression. In the next sections, we will discuss the contributions of Drosophila as experimental platform to study mitochondrial dysfunction in Alzheimer’s disease.
Alzheimer’s disease, amyloid beta, and tau
Alzheimer’s disease (AD) is an incurable neurodegenerative brain disorder that leads to cognitive impairment and memory deficits in affected individuals (Knopman et al., 2021) and displays a reduction in the hippocampal and temporal lobe of the brain (Ramos Bernardes da Silva Filho et al., 2017). It accounts for 50–60% of dementia (Blennow et al., 2006), affecting mostly people above 65 years of age. The disease is named after the German neurologist Alois Alzheimer who examined a 51 years old patient, Auguste Deter, suffering from memory loss, hallucinations, disorientation, and language problems. Her autopsy showed an accumulation of amyloid plaques and tangles in the cerebral cortex. The etiology of the disease is not well defined. However, the amyloid cascade theory is the predominant hypothesis where cognitive deficits are due to the deposition of amyloid beta peptides forming extracellular plaques (Morishima-Kawashima and Ihara, 2002; Cummings, 2004; de Vrij et al., 2004) and subsequent hyperphosphorylation of the microtubule associated protein tau, resulting in formation of neurofibrillary tangles (NFT) (Cummings, 2004; Blennow et al., 2006; Braak et al., 2011).
A variety of amyloid beta fragments (37–43 amino acid residues) are released into the extracellular space through proteolytic processing of APP by γ-secretase (Haass et al., 2012). Among these, Abeta 42 (hereafter referred to as Aβ) is the most toxic peptide as it is highly insoluble and more prone to aggregation (Moore et al., 2018). Monomeric Aβ aggregates into oligomers and protofibrils, while insoluble amyloid beta-fibril aggregates form amyloid plaques that interfere with signaling at the synapse (Chen and Yan, 2010; Crews and Masliah, 2010). Studies have shown that Aβ aggregation causes neuronal death by altering calcium homeostasis, elevating mitochondrial oxidative stress, and reducing energy metabolism. In addition, Aβ triggers microglial priming by interacting with the microglia, making it more prone to secondary inflammations. Aβ stimulates microglia to release pro-inflammatory cytokines, and these interfere with anti-inflammatory cytokines and transforming growth factor-beta1 (TGF-β1), which can induce neuroinflammation and neurodegeneration (Torrisi et al., 2019; Merlini et al., 2021). Aβ also causes a neuroinflammatory response by activating astrocytes to release various pro-inflammatory molecules (cytokines, interleukins, complement components, nitric oxide, and other cytotoxic compounds) (Brosseron et al., 2014; van Eldik et al., 2016; Arranz and DeStrooper, 2019). Polymerization of Aβ fibrils leads its aggregation into plaques and to the activation of Glycogen Synthase Kinase 3 (GSK3), causing hyperphosphorylation of microtubule-associated Tau and subsequent formation of neurofibrillary tangles (NFT) (Eftekharzadeh et al., 2018; Tiwari et al., 2019).
Tau is a microtubule-associated protein (MAP) that plays a role in the stabilization of neuronal microtubules and regulates axonal growth. Tau exists in a set of six isoform proteins (3R0N, 3R1N, 3R2N, 4R0N, 4R1N, and 4R2N) and is expressed in neurons via alternate splicing of MAPT. Depending on the exclusion or inclusion of exon 10, the expression of tau isoforms will contain three (3R) or four (4R) microtubule-binding repeats, whereas isoforms with 0, 1, or 2 N-terminal inserts are determined by the inclusion of exons 2 and 3 (Goedert et al., 1989). The ratio of 3R to 4R is 1 in the healthy human brain (Kosik et al., 1989; Goedert and Jakes, 1990), but this ratio is altered in tauopathies (D’Souza and Schellenberg, 2005; Goedert and Jakes, 2005; Sergeant et al., 2005). Tau exists normally as soluble and unfolded protein (Mandelkow and Mandelkow, 2012) and interacts with tubulin, promoting its assembly into microtubules and helping stabilize the structure (Weingarten et al., 1975). It has the potential for multiple phosphorylation at Serine (S), Threonine (T), and Proline (P) residues within its Proline-rich region (PRG) or C-terminal region (CTR), and only 2–3 residues are phosphorylated in the healthy brain. However, there are around five to nine moles of phosphate per mole of tau in AD and other tauopathies (Grundke-Iqbal et al., 1986; Kopke et al., 1993; Holper et al., 2022). Hyperphosphorylation of Tau is associated with an aggregation of multimers and fibers (Despres et al., 2017) that seem to mediate cognitive defects. Microtubule-associated kinases such as Cyclin-dependent Kinase 5 (CDK5), Glycogen Synthase Kinase 3 (GSK3β), casein kinase II (CKII), Src-family tyrosine kinase Fyn, c-Abl tyrosine kinase (c-Abl), lemur tyrosine kinase 2 (LTK), dual specificity tyrosine-phosphorylation-regulated kinase 1A (Dyrk1A), and thousand-and-one amino acid kinases (TAOKs), casein kinase 1 (CK1), c-Jun amino-terminal kinase (JNK),extracellular signal-regulated kinases 1 and 2 (Erk1 and Erk2), adenosine-monophosphate activated protein kinase (AMPK), cyclic AMP (cAMP)-dependent protein kinase (PKA), protein kinase N1, tau-tubulin kinases 1 and 2 (TTBK1 and TTBK2), Ca2+/calmodulin-dependent protein kinase II (CaMKII) and microtubule-affinity regulating kinases (MARKs) are known enzymes to be involved in tau phosphorylation (Limorenko and Lashuel, 2022). In the AD brain, approximately 85 serine, threonine, and tyrosine residues have been found phosphorylated (Tavares et al., 2013; Limorenko and Lashuel, 2022). Therefore, deposition of extracellular Aβ plaques and intracellular accumulation of NFT constitute the main pathological hallmarks in AD (Knopman et al., 2021).
Mitochondrial dysfunction in Alzheimer’s disease
The mitochondrion is a membrane-bound cytoplasmic organelle that carries out essential functions like ATP production through oxidative phosphorylation. It is critical for multiple cellular processes and interacts with several organelles to regulate energy metabolism (Rossmann et al., 2021; Collier et al., 2023). It is worth noting that the brain utilizes 25% of body glucose and 20% oxygen consumption, but it constitutes only 2% of body weight. As a result, the brain is very susceptible to changes in energy metabolism and, therefore, disturbances of mitochondrial functions are associated with neurodegenerative disorders (Lin and Beal, 2006; Wang et al., 2020; Zhang X. et al., 2021), including Alzheimer’s disease (Wang et al., 2014; Swerdlow, 2018). Several studies have shown that mitochondrial abnormalities are early events in AD (Reddy et al., 2012; Swerdlow, 2018). Cell lines expressing mutant APP or treated with Aβ (Schmidt et al., 2007; Diana et al., 2008; Wang et al., 2008b,2009; Calkins and Reddy, 2011; Manczak et al., 2011), AD mouse models (Smith et al., 1997; Li et al., 2004; Reddy et al., 2004; Caspersen et al., 2005; Manczak et al., 2006; Yao et al., 2009), and AD post-mortem brains (Parker et al., 1990; Gibson et al., 1998; Maurer et al., 2000; Butterfield et al., 2001, Dragicevic et al., 2010) are reported to have mitochondrial dysfunction. Brains from AD patients show decreased production of ATP, increased production of free radicals, lipid peroxidation, oxidative damage of DNA and protein, and cellular damage compared to control specimens (Parker et al., 1990; Gibson et al., 1998; Maurer et al., 2000; Wang et al., 2005; Devi et al., 2006; Dragicevic et al., 2010), suggesting that mitochondrial dysfunction is a main pathological feature of AD.
The mitochondrial cascade hypothesis was first reported in 2004 (Swerdlow and Khan, 2004). According to this hypothesis, mitochondrial dysfunction disrupts multiple pathways connected to AD, resulting in a variety of clinical phenotypes including cognitive decline. It also states that mitochondrial dysfunction alters Aβ homeostasis triggering its overproduction and accumulation, and that an overall bioenergetic dysfunction might be the main culprit in AD [reviewed in Swerdlow (2023)]. Accordingly, the mitochondrial ROS production, calcium homeostasis, mitochondrial morphology and number, transport along the neuronal axon, neurotransmitters levels, mitophagy, and mtDNA mutation and oxidation, are all clearly compromised in AD (Johnson and Blum, 1970; Hirai et al., 2001; Hauptmann et al., 2009; Wang et al., 2009; Calkins and Reddy, 2011; Butterfield and Halliwell, 2019; Wong et al., 2020; Figure 1). Next, we will examine specific mechanisms mediating mitochondrial dysfunction and abnormalities in the context of AD.
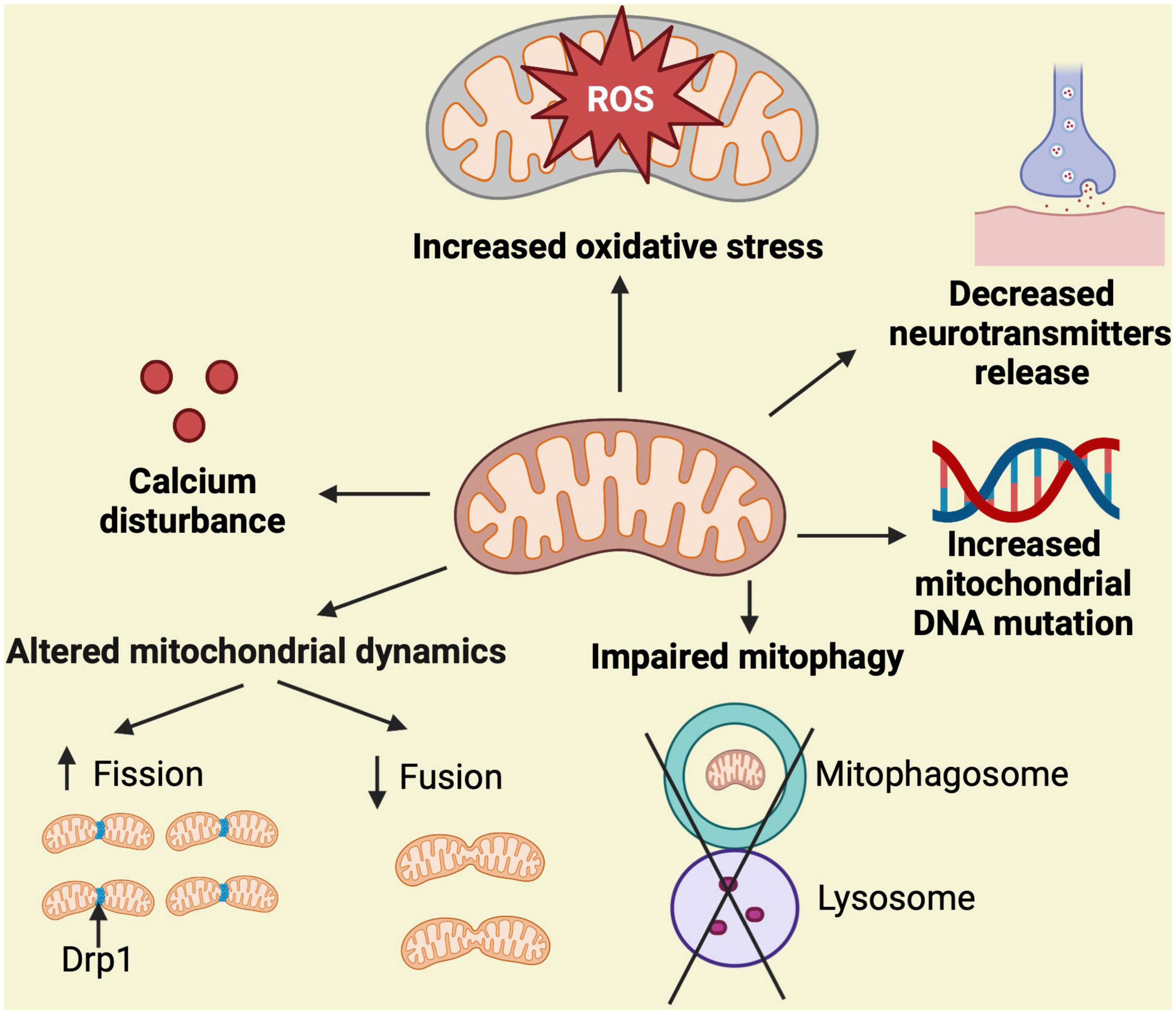
Figure 1. Simplified overview of mechanisms associated with mitochondrial dysfunction. Mitochondrial homeostasis is regulated by mitochondrial dynamics (fission and fusion), proper handling of calcium, control of ROS production, neurotransmitters function, maintenance of mtDNA replication, and elimination of damaged mitochondria through mitophagy. Stressful and persistent insults to the mitochondria, such as the presence of neurotoxic proteins, alter these processes resulting in the indicated abnormalities, which leads to mitochondrial dysfunction and potential cell death. Fission eliminates abnormal mitochondria from healthy ones and this process is mediated by Drp1.
Mitochondrial ROS and oxidative stress
The mitochondrion contributes with approximately 90% of cellular reactive oxygen species (ROS), a by-product of electron transport of aerobic respiration in mitochondria (Balaban et al., 2005). When cellular homeostasis is disrupted, mitochondria produce less ATP and more ROS, resulting in oxidative stress (Gibson et al., 1998). AD patients are reported to have increased oxidative damage of proteins, nucleic acids, sugars, and lipids (Butterfield and Halliwell, 2019). For instance, markers of protein oxidation, such as protein carbonyl content, are significantly increased in the parietal lobe, superior, and middle temporal gyrus, and hippocampus of AD brains (Hensley et al., 1995; Lyras et al., 1997; Aksenov et al., 2001). Similarly, 3-nitrotyrosine, another protein oxidative modification, was significantly increased in various brain regions and cerebrospinal fluid from AD cases (Good et al., 1996; Smith et al., 1997; Tohgi et al., 1999; Castegna et al., 2003; Reed et al., 2009; Butterfield and Halliwell, 2019). Oxidative damage of DNA/RNA in AD brains causes double-strand breaks of DNA, crosslinking of DNA/DNA or DNA/protein, and extensive modification of DNA bases. Consequently, levels of DNA breaks in the AD hippocampus and cerebral cortex are found to be high (Mullaart et al., 1990; Anderson et al., 1996). Both mitochondrial DNA and nuclear DNA from AD brains have a significant increase of 8-hydroxydeoxyguanosine (8-OHdG) and 8-hydroxyguanosine (8-OHG), markers of DNA oxidation (Mecocci et al., 1994; Good et al., 1996; Lyras et al., 1997). Similarly, the levels of oxidized rRNA or mRNAs are significantly elevated in AD cases (Shan et al., 2003; Ding et al., 2005; Honda et al., 2005). Moreover, lipid peroxidation products such as 4-hydroxynonal, malondialdehyde (MDA), and 2-propenal (acrolein) are increased in different regions of AD brains (Wang et al., 2014). In contrasts, the level of antioxidant factors and enzymes is decreased in AD brains compared to control specimens (Marcus et al., 1998; Doré, 2002; Kim et al., 2006; Venkateshappa et al., 2012). Altogether, these observations suggest that mitochondrial dysfunction and oxidative stress are closely linked and, thus, are considered primary triggers of neuronal death.
Calcium dyshomeostasis
The mitochondria play an essential role in the maintenance of calcium homeostasis in neurons which is essential for their survival (Jouaville et al., 1999). The level of mitochondrial calcium affects the activity of mitochondria and the supply of ATP. The mitochondrial calcium uniporter (MCU) protein complex conducts calcium transport into the matrix through MICU1 and MICU2/3 proteins (MICU gatekeepers) that sense calcium levels (Perocchi et al., 2010). AD patients have reduced expression of cytosolic calcium-binding proteins calmodulin, calbindin D28K, and parvalbumin which might cause activation of MCU after free calcium binds to MICU1 and MICU2/3 (McLachlan et al., 1987; Riascos et al., 2011; Ahmadian et al., 2015; Ali et al., 2019). Therefore, the level of calcium in mitochondria is elevated in AD. Consistently, exposure of cortical neurons to Aβ increases calcium concentration and promotes neurodegeneration, which is rescued by blocking MCU proteins (Hedskog et al., 2013). Aβ oligomers are also reported to form a calcium-permeable pore in the mitochondrial membrane that regulates calcium uptake and may disrupt the homeostasis of calcium in the mitochondria (Lashuel et al., 2002; Shirwany et al., 2007). In addition, Aβ oligomers promote the release of calcium stored in the ER to increase mitochondrial calcium levels, leading to mitochondrial dysfunction (Ferreira et al., 2015). Indeed, the expression of Ryanodine receptors (RyR), which regulate the release of intracellular Ca2 + in the ER, varies during AD progression and the RyR fly homologue has been found as modifier of Abeta and tau toxicity in transgenic flies (Casas-Tinto et al., 2011; Feuillette et al., 2020). Calcium homeostasis also depends on the sarcoplasmic/endoplasmic reticulum Ca2 + ATPase (SERCA), an essential ER protein that pumps Ca2 + into the ER, and modulation of SERCA expression modifies Aβ levels (Green et al., 2008). However, the direct role of RyR and SERCA in the AD-related mitochondrial malfunction remains to be elucidated. On the other side, it has been demonstrated that dyshomeostasis of mitochondrial calcium can be restored through calcium efflux pathways, mainly through the sodium-calcium exchanger (NCLX) (Boyman et al., 2013). Interestingly, AD pathology in mouse models is associated with the loss of mitochondrial NCLX expression and function, and rescue of NCLX in these mice restores both cognitive decline and cellular pathology (Jadiya et al., 2019). Additional studies in AD mouse models demonstrated that an elevated mitochondrial calcium level is associated with induction of apoptosis, highlighting the importance of mitochondrial calcium concentration in this process (Sanz-Blasco et al., 2008; Calvo-Rodriguez et al., 2020). Of note, a recent study found an oscillation of cytosolic Ca2 + in primary neurons after incubation with tau K18, a protein fragment carrying the four repeat (4R) domain of the protein. As a result, both mitochondrial and cytosolic Ca2 + are increased, indicating that Tau also impairs Ca2 + homeostasis (Britti et al., 2020).
Mitochondrial fusion and fission
Mitochondria undergo fission and fusion in the cytoplasm to maintain proper distribution (Zhu et al., 2013), and disruption of either of these processes leads to neurological disorders (Mishra and Chan, 2014). The fission of mitochondria is regulated by the GTPase-related dynamin-related-protein1 (Drp1), also known as Dynamin-1-like protein (DLP1) in humans. It triggers the fragmentation of mitochondria and acts as a mitochondrial fission factor; however, its downregulation promotes mitochondrial fusion (Wenger et al., 2013). Of note, the structural damage in mitochondria was documented in AD brains over two decades ago (Hirai et al., 2001). Subsequent studies demonstrated that AD brains have a reduction of mitochondrial size and number (Wang et al., 2008b,2009), which could cause a shortage of mitochondrial bioenergetics either by enhancing ROS generation (Yu et al., 2006) or by having a negative effect on electron transport chain (ETC) function (Liu et al., 2011; Zhou et al., 2017). AD patients are reported to have increased mitochondrial fission (Manczak et al., 2011; Kandimalla et al., 2016). Interestingly, the pathology of AD in the brain not only includes an interaction between oligomeric Aβ and DLP1, but also an interaction between hyperphosphorylated tau and DLP1 (Manczak et al., 2011). This suggests that these abnormal interactions induce mitochondrial fragmentation, which is supported by in vivo and in vitro models of AD. For instance, transgenic flies expressing Aβ display abnormal dynamics and distribution of mitochondria (Iijima-Ando et al., 2009; Zhao et al., 2010). In neuronal cell culture, where Aβ or APP is overexpressed, mitochondria undergo fragmentation with altered distribution (Wang et al., 2008b,2009; Manczak et al., 2011). In M17 neuroblastoma cells and primary neurons, wild-type or mutant APP overexpression also induces fragmentation of mitochondria (Wang et al., 2008b,2009). Interestingly, APP- or Aβ-induced mitochondrial deficit in neurons is rescued by blocking mitochondrial fission, confirming the role of this process in AD pathogenesis (Wang et al., 2008b,2009). On the other hand, mitochondria fusion is regulated by dynamin-related GTPase proteins mitofusin-1 (Mfn1), mitofusin-2 (Mfn2), and the optic atrophy type 1 (OPA1) protein where Mfn1 and Mfn2 bind to the outer membrane of mitochondria and OPA1 binds to the inner mitochondrial membrane and mediates the fusion of the inner mitochondrial membrane (Koshiba et al., 2004; Song et al., 2009). In mice cortexes and the hippocampus, knockout of Mfn2 caused structural and functional damage to mitochondria as well as neuroinflammation, oxidative stress and neuronal death, illuminating the role of mitochondrial fragmentation in AD pathology (Jiang et al., 2018; Han et al., 2020). Confocal and electron microscopy studies from another group confirmed the structural damage and fragmentation of mitochondria in the brain of CRND8 APP transgenic mice at 3 months of age, much before visible amyloid deposition, suggesting that abnormal mitochondrial dynamics is an early event in AD pathogenesis (Wang et al., 2017). It is worth noting that overexpression of Tau also causes abnormal mitochondrial fusion (Li et al., 2016; Kandimalla et al., 2018). A study showed that overexpression of human wild-type tau alters mitochondrial dynamics and results in mitochondrial elongation by increasing the fusion proteins OPA1, Mfn1, and Mfn2, which reduces neuronal viability (Li et al., 2016; Szabo et al., 2020). In addition, the knockdown of mfn2 reduced the human tau-enhanced mitochondrial fusion and restored mitochondrial function, suggesting that Mitofusin-associated mitochondrial fusion might contribute to tau toxicity (Szabo et al., 2020). Another group has shown that expression of caspase-cleaved tau in cortical neurons from tau-/- knockout mice, as well as in immortalized cortical neurons, led to mitochondrial fragmentation with a decline in OPA1 levels (Pérez et al., 2017; Szabo et al., 2020), indicating the impact of tau on mitochondrial fusion.
Axonal trafficking deficits and abnormal mitochondrial distribution
In addition to defects in mitochondrial structure, reduced expression of mitochondrial fission and fusion proteins in AD can lead to the absence of mitochondria in axons or dendritic segments (Wang et al., 2009; Pickett et al., 2018). Mitochondria undergo anterograde and retrograde transport that helps maintain healthy mitochondria by inhabiting axons with fresh mitochondria and recycling the damaged ones (Sheng and Cai, 2012; Lin et al., 2017). Alteration of either of these processes causes a reduction of healthy mitochondria, which leads to impaired mitochondrial function. Dysregulation of axonal mitochondrial transport contributes to AD as this transport is crucial for the maintenance of neurons and their synaptic function (Hollenbeck and Saxton, 2005). In mammals, Milton (OIP106 and GRIF1) and Miro (Miro1 and Miro2) regulate the attachment of mitochondria to microtubules via kinesin heavy chains (Fransson et al., 2006). In Drosophila, Milton and Miro proteins perform similar functions (Guo et al., 2005) and, thus, mitochondria are absent in synaptic terminals and axons when there is no Milton or Miro (Stowers et al., 2002). Interestingly, it has been reported that the number of mitochondria is reduced in the axons of hippocampal neurons upon Aβ treatment (Du et al., 2010), which is consistent with a lower number of mitochondria in axons from AD brains (Stokin et al., 2005). Mislocalization of mitochondria and its decrease in dendrites, axons, and soma (Iijima-Ando et al., 2009) was observed as consequence of Aβ expression in flies. Similar results were reported in neuronal cell cultures exposed to Aβ oligomers, which led to reduced mitochondrial trafficking/motility in axons (Du et al., 2010; Wang et al., 2010; Calkins and Reddy, 2011; Rui and Zheng, 2016). On the other hand, tau has been also associated with abnormal mitochondrial trafficking. For instance, earlier investigations showed that over-expression of tau in neuroblastoma cells alters the kinase-dependent anterograde axonal transport of mitochondria by enhancing the binding of microtubules, resulting in neurites almost devoid of these organelle (Ebneth et al., 1998). In vivo studies in transgenic mice confirmed the reduced mitochondrial movement in axons upon overexpression of human tau (Stoothoff et al., 2009; Vossel et al., 2015). Intriguingly, not only the expression of tau but also its phosphorylation impairs mitochondrial transport. For instance, in neuronal PC12 cells and mouse cortical neurons, phosphorylation of tau at AT8 sites (Ser 199, Ser202, and Thr 205) inhibits mitochondrial movement (Shahpasand et al., 2012). This is consistent with abnormal mitochondrial distribution in neurons positively stained with the anti-phospho tau Alz50 antibody in AD brains (Kopeikina et al., 2011), suggesting that tau plays a key role in the mitochondrial loss observed in AD (Wee et al., 2018).
Neurotransmitters and mitochondrial dysfunction
Mitochondrial dysfunction affects a number of neurotransmitters in AD. For instance, memory and learning deficits result from insufficient cholinergic transmission. In this case, the hyperpermeability of the mitochondrial membrane leads to degeneration of cholinergic neurons and deficiency of acetylcholine (ACh) (Wong et al., 2020). Dysfunctional mitochondria also alter the activity of acetylcholine esterase (AChE) and recycling of choline from the synapse is hampered by mitochondrial-induced oxidative stress via nitrosative stress, which results in ACh deficiency (Wong et al., 2020). Serotoninergic, dopaminergic, norepinephrinergic, and histaminergic systems comprise the diverse monoaminergic neurotransmission network. Through membrane permeabilization and altered serotoninergic metabolism, mitochondrial dysfunction in AD causes serotoninergic inefficiency (Yamamoto and Hirano, 1985; Lai et al., 2011). Mitochondrial dysfunction also causes the loss of serotoninergic neurons via caspase-dependent apoptosis resulting in the reduction of 5-hydroxytryptamine (5-HT) or Serotonin neurotransmission (Wong et al., 2020). In AD, an excessive 5-HT breakdown is the result of mitochondrial dysfunction, which leads to a 5-HT deficit. The inadequate serotoninergic transmission also contributes to AD progression by causing ROS accumulation and further mitochondrial dysfunction (Wong et al., 2020).
Impaired mitophagy
Mitophagy is a mechanism that eliminates damaged mitochondria by activating PINK1 at the outer mitochondrial membrane (Nguyen et al., 2016). Studies have shown that Parkin, an E3-ubiquitin ligase, is drawn to the mitochondria by PINK1 and phosphorylated there to initiate the mitophagy pathway for protein ubiquitination and degradation (Geisler et al., 2010; Matsuda et al., 2010; Narendra et al., 2010; Vives-Bauza et al., 2010). Accumulation of damaged mitochondria is due to insufficient mitophagy to remove them (Ye et al., 2015) or to defects in lysosomal degradation (Martín-Maestro et al., 2016, 2017; Kerr et al., 2017; Sorrentino et al., 2017). Nonetheless, it has been suggested that AD is mainly associated with mitophagy impairment (Nixon and Yang, 2011). For instance, post-mortem hippocampal brain samples from AD patients showed a reduction of mitophagy by 30–50% compared to control patients (Fang et al., 2019). Proteins believed to be involved in autophagy and mitophagy processes, such as Optineurin (OPTN), ATG5, ATG12, Beclin-1 (Bcl-1), PI3K class III, ULK1, AMBRA1, BNIP3, BNIP3L, FUNDC1, VDAC1, and VCP/P97 were decreased in AD brains (Martín-Maestro et al., 2017). The mitochondrial structure and function in the brains of AD patients and mouse models change into a swollen round shape with deformed cristae, low ATP production, reduced LC3 recruitment to the mitochondria, dysfunctional AMP-activated protein kinase (AMPK), and inhibition of its targets ULK1 and TBK1, which collectively impairs the mitophagy process (Hirai et al., 2001; Martín-Maestro et al., 2016; Wang et al., 2017; Fang et al., 2019). Parkin’s ability to translocate to damaged mitochondria is also affected by abnormal contacts between the projection domain of tau protein and Parkin, which prevents mitophagy (Cummins et al., 2019). Moreover, addressing mitophagy with urolithin A or actinonin improves memory in APP/PS1 mice and C. elegans expressing Aβ or tau, emphasizing the role of mitophagy in AD pathogenesis (Fang et al., 2019).
Mitochondrial genome abnormalities
Mitochondria have their own DNA called mtDNA that codes for the 13 mitochondrial core proteins of the electron transport chain complexes, two rRNAs, and 22 tRNAs (Taanman, 1999; D’Souza and Minczuk, 2018). Although mitochondrial DNA plays a crucial role in mitochondrial function, it is prone to mutations due to the lack of histone proteins necessary to protect DNA and to mediate DNA repair mechanisms (Yana et al., 2013; Boczonadi et al., 2018). MtDNA mutations, via inheritance or gradual somatic mutation, affect mitochondrial function, which leads to cell death and disease (Swerdlow, 2018). Like AD patients, individuals with mtDNA mutations are reported to have similar cognitive deficits.
(Inczedy-Farkas et al., 2014), suggesting the potential role of mtDNA in cognition. In the case of AD, affected individuals have an increase in mtDNA mutations, possibly due to higher oxidative damage (Swerdlow, 2018). Indeed, AD patients have 10-fold higher levels of oxidized bases in mtDNA than nuclear DNA compared to healthy controls (Mecocci et al., 1994; Wang et al., 2005). This is consistent with higher oxidized nucleic acid in mtDNA in MCI patients and preclinical AD (Lovell et al., 2011). All these observations illustrate the role of mitochondrial genome abnormalities in AD pathogenesis.
Assays for studying mitochondrial biology
Many assays have been developed and improved over the years to analyze the structure and function of the mitochondria. Flow cytometry is a standard assay to record mitochondrial mass (Doherty and Perl, 2017). However, it was only in recent decades that this procedure could be performed independently of mitochondrial potential due to the production of the MitoTracker family of dyes in the mid-1990s. Specifically, MitoTracker probes-green (MTG) is used for its ability to accumulate inside the mitochondria despite the measured membrane potential, unlike the positively charged red dye (Doherty and Perl, 2017; Rana et al., 2017). MTG collects inside the mitochondrial matrix and binds to free thiol groups in cysteine residues located on the mitochondrial membrane proteins; the amount of accumulated dye can then be quantified via fluorescence relative to the mitochondrial size (Presley et al., 2003; Doherty and Perl, 2017).
Along with the size, it is also important to measure the mitochondrial membrane potential (MMP) to assess the global mitochondrial function, as this parameter directly correlates with the ability of the mitochondria to generate ATP for the cells. The MMP is created from the electrochemical gradient formed through a series of coupled redox reactions in the electron transport chain steps; decreases in the MMP can be detected using lipophilic cationic fluorescent dyes (Sakamuru et al., 2012). Other membrane-potential dependent fluorescent dyes that can be used to measure MMP include tetramethylrhodamine, methyl ester (TMRM), and tetramethylrhodamine, ethyl ester (TMRE). The more polarized the mitochondria, the brighter the signal when measured due to higher accumulation of dye (Perry et al., 2011). Other dyes have been developed to analyze and measure oxygen consumption, which is directly related to oxidative phosphorylation (OXPHOS) activity (Yarosh et al., 2008). Spectrophotometric assays have also been developed to measure the activity of mitochondrial enzymes or the concentrations of cellular metabolites. For instance, by measuring enzyme activity through spectrophotometry, it was found that the activity of the mitochondrial complex I is slightly reduced in the brain during aging, but highly reduced in the context of neurodegeneration (Pollard et al., 2016).
In many cell types, the mitochondria create a complex, reticular network to support its critical functions, such as energy production. These networks undergo constant dynamic changes, which require the complementary processes of fission and fusion to occur (Hoppins et al., 2020). Several in vitro and in vivo studies were performed to assess mitochondrial fission and fusion and earlier assays provided evidence that mitochondrial fission involves multiple constriction steps that are marked by the endoplasmic reticulum (ER) and dynamin-related proteins (DRPs) (Friedman et al., 2011; Anand et al., 2014). Recent advances in light and electron microscopy demonstrated that the inner and outer membrane dynamics may be uncoupled during fission and fusion, which suggests that both processes are uniquely required to uphold the morphology of the mitochondria during its dynamic equilibrium; problems in this relationship can cause a diverse array of diseases as mitochondrial fission and fission are required for mitochondrial replication, a process critical to maintain energy levels and overall cell health (Cho and Sun, 2020; Hoppins et al., 2020). To correctly visualize mitochondrial fusion, stable cell lines that express fluorescent mitochondrial matrices via targeted proteins must be generated; once this is done, the mitochondria can be isolated, and the fusion assay can be performed (Hoppins et al., 2020). The fusion efficiency of the mitochondria can be quantified by dividing the number of fused mitochondria by the total number of mitochondria seen in the microscope field. It is anticipated that about 15–20% of total mitochondria are fused in wild-type control specimens (Hoppins et al., 2020).
Mitochondrial fission is required for growing and dividing cells and is mediated by the cytosolic dynamin family member, Drp1, in Drosophila and mammals (Youle and van der Bliek, 2012). Assays to measure fission in vitro involve quantification of the GTPase cycle kinetics and biochemical activity of Drp1 (Ingerman et al., 2005). Electron microscopy has also been included to study mitochondrial fission through Drp1-mediated liposome tubulation and constriction assays. Once images are obtained, the ultrastructural changes can be analyzed and measured using the ImageJ software (Schneider et al., 2012).
Mitochondrial biology in Drosophila
Drosophila is a prominent model for studying mitochondrial diseases because researchers can manipulate the mitochondrial genome to express the characteristics of many human mitochondrial disorders (Chen et al., 2019). By utilizing certain restriction enzymes on Drosophila mitochondrial DNA, it has been observed that many mitochondrial mutations are heritable by isolation of specific disease-causing genes; when these methods are used with genetic drivers, such as GMR-GAL4, heteroplasmic flies with different mitochondrial genomes can be produced in order to study the molecular and phenotypic effects of a specific disease gene (Xu et al., 2008; Chen et al., 2015).
From a genomics perspective, human and Drosophila genomic mtDNA are very similar despite the human genome being about 3 kb shorter (Lewis et al., 1995). The 16,559 kb human mtDNA genome encodes for 13 proteins, 22 tRNAs, and two rRNA; all thirteen of these mitochondrial proteins make up components of the four complexes found in the electron transport chain, and almost all the DNA sequences in each specific protein have small introns that must be spliced out before translation can occur. Drosophila mtDNA encodes the same transcripts as its human counterpart but has a slightly different genomic order, predominantly due to its expanded “A + T-rich” regions (Sen and Cox, 2017). Crucial molecular functions of the mitochondria, including transport, oxidative phosphorylation (OXPHOS), and nucleotide biosynthesis, have highly conserved nuclear-encoded genes across both species, making Drosophila an excellent model for studying metabolic issues (Chen et al., 2019). Each of the 13 mitochondrial proteins is translated in the mitochondrial matrix using mtDNA-encoded tRNAs and most of the mRNA sequences for the mitochondrial proteins are separated by at least one mtDNA-encoded tRNA. It is critical that each mtDNA-encoded tRNA is properly and systematically excised since both human and Drosophila mtDNA are transcribed in a polycistronic manner. Without proper excision of each previous transcript, normal translation and processing of the proteins cannot occur which can lead to illnesses or even the death of the organism (Sen and Cox, 2017).
Drosophila research can also be used to define phenotypes associated with defective mitochondria. It has become clear that the number of mitochondria present in different organisms can fluctuate and the structure of the organelle dynamically changes depending on which type of cell they reside in Zhang et al. (2016). Neuronal cells are in a constant state of high energy demand so they are considered to be especially vulnerable to dysfunction in the mitochondrial equilibrium as this can lead to abnormally low levels of ATP; specifically, there is an abundance of mutations in the MFN2 gene that have been commonly linked to a peripheral neuropathy caused by Charcot-Marie-Tooth disease Type 2A (CMT2A) (Kyriakoudi et al., 2021). Severe symptoms of this disease, most likely caused by mutations in the functional domains of the gene, include distal limb atrophy leading to loss of leg function as well as distal sensory loss in the limbs (Züchner et al., 2004). Other mutations in the MFN2 gene can be linked to other CMT diseases, which can damage the optic nerve and other crucial neurons in different sensory pathways (Zhou et al., 2019). In addition to genetic research, behavioral assays such as flight and climbing tests can be performed in flies as behavioral defects correlate with key features observed in patients with certain mitochondrial disorders (Jacobs et al., 2004).
Tools for studying mitochondrial biology in flies
Genetically encoded sensors have been generated in Drosophila to monitor mitochondrial structure, function, and metabolites. These sensors are placed under the control of the UAS promoter and have been used to perform mitochondrial assessments in different fly tissues using tissue-specific Gal4 drivers such as C155-Gal4 (pan-neuronal), MB-GS Gal4 (mushroom body), MB296B-Gal4 (dopaminergic neurons), TH-Gal4 (dopaminergic neurons), vGlutOK371 Gal4 (glutamatergic neurons), esgts-Gal4 (intestinal stem cell), apt-Gal4 (perineurial glial cells), and vGlutVGN6341 Gal4 (glutamatergic interneurons) (Hwang et al., 2014; Arce-Molina et al., 2020; Morris et al., 2020; Sharma and Hasan, 2020; Cho et al., 2021; Hartwig et al., 2021; Wong et al., 2021; Houlihan et al., 2022). Some sensors are substrate-dependent, while some are light-dependent. On the other hand, UAS-mitoGFP is used as a mitochondrial marker to visualize mitochondria in any tissue because, in this case, the mitochondrial import sequence is fused to GFP (Lutas et al., 2012; DeVorkin et al., 2014; Morris et al., 2020; Hartwig et al., 2021; Figure 2A).
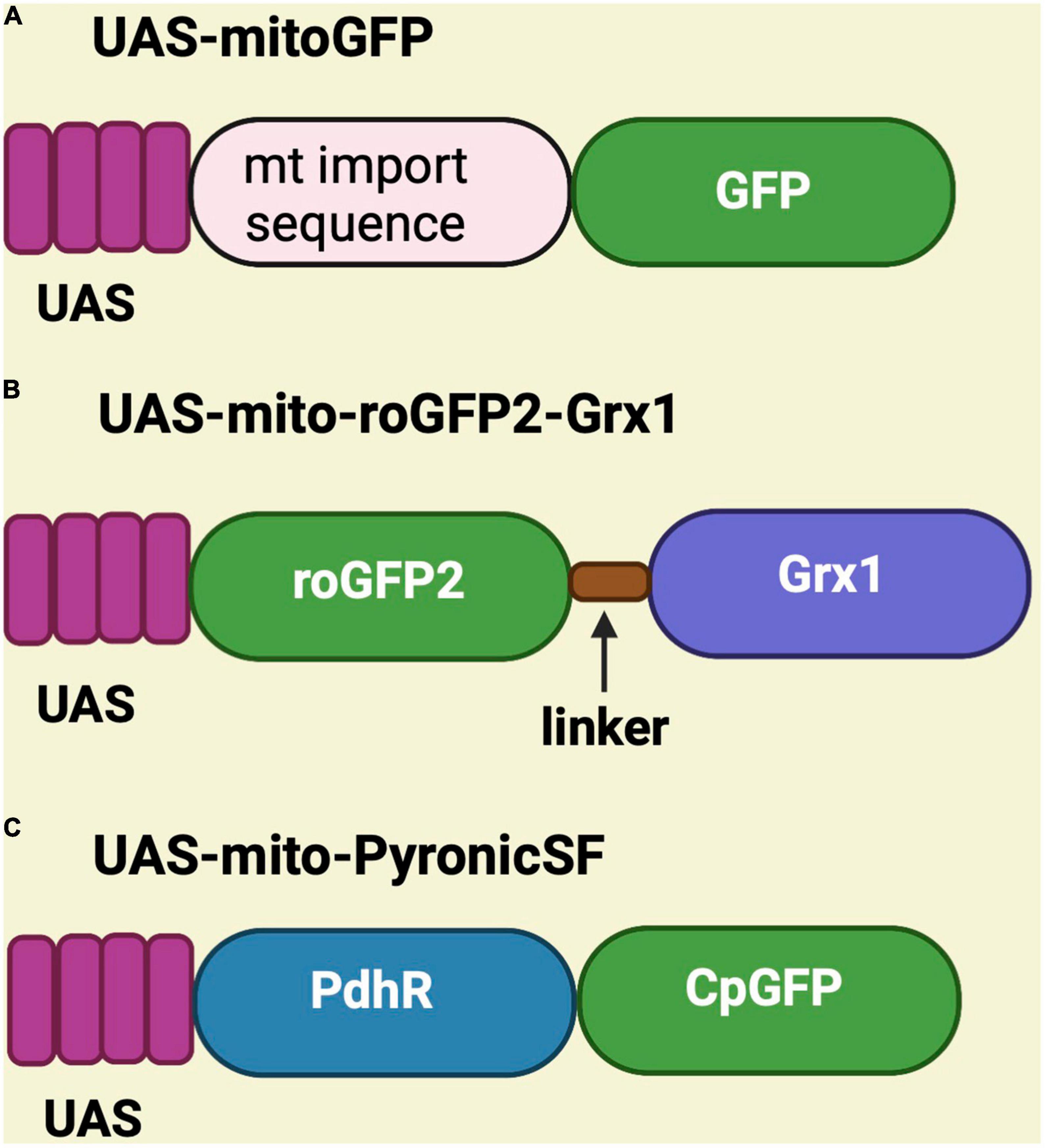
Figure 2. Schematic representation of UAS-mitoGFP, UAS-mito-roGFP2-Grx1 and UAS-mito-PyronicSF constructs. (A) UAS-mitoGFP contains a mitochondrial import sequence fused to GFP and serves as mitochondrial marker. (B) UAS-mito-roGFP2-Grx1 is a redox sensor transgene in which glutaredoxin-1 (Grx1) is fused to redox-sensitive GFP. (C) UAS-mito-PyronicSF is a pyruvate sensor containing the bacterial pyruvate-sensitive transcription factor PdhR linked to circulated permuted GFP (CpGFP), which allows real-time assessment of mitochondrial pyruvate transport.
Substrate-dependent sensors
Various substrate-dependent sensors were created to detect substrates like hydrogen peroxide, glutathione, pyruvate, ATP, NAD/NADH, and calcium. To see redox changes in live mitochondria, the UAS-mito-roGFP2-Grx1 is available, which encodes a redox-sensitive GFP cassette fused to glutaredoxin-1 (Grx1) (Albrecht et al., 2014; Krzystek et al., 2021; Houlihan et al., 2022; Figure 2B). The sensor for pyruvate is known as UAS-mito-PyronicSF and consist of a circularly permuted GFP fused to the bacterial pyruvate-sensitive transcription factor PdhR. It binds to pyruvate and induces conformational changes that increase the readout of FRET signal (Arce-Molina et al., 2020). Therefore, this sensor can be used to study mitochondrial metabolism (Figure 2C).
To monitor calcium, the UAS-mito-GCaMP3 was designed to express a circularly permutated EGFP M13/Calmodulin fusion protein under control of the UAS promoter. Upon binding to calcium, this fusion protein undergoes conformational changes that elicit GFP signal (Figure 3A; Lutas et al., 2012; Morris et al., 2020; Sharma and Hasan, 2020). UAS-AT1.03NL is an ATP sensor that consists of two fluorescent proteins (mVenus and mseCFP) linked to the ε subunit from bacterial F0F1-type ATP synthase. The ε subunit has two C-terminal helices and an N-terminal barrel domain. Low FRET efficiency results from the loose and flexible subunit separating two fluorescent proteins in ATP-free mode. When ATP is bound, the ε subunit configuration switches from open to close, bringing the two fluorescent proteins closer together and increasing FRET efficiency (Imamura et al., 2009; Tsuyama et al., 2013; Dong and Zhao, 2016; Cho et al., 2021; Figure 3B). UAS-PercevalHR is also used as ATP/ADP sensor and contains the ATP-binding protein GlnK1 from Methanocaldococcus jannaschii and circularly permutated monomeric Venus (CpmVenus) connected by a peptide linker. Upon binding to ATP, the T-loop of GlnK1 undergoes a dramatic conformational change from a loose, disordered structure to a tight, ordered loop, which integrates CpmVenus into the T-loop for sensing ATP (Berg et al., 2009). It has an excitation peak at 405 nm for ATP binding and 488 nm for ADP binding (Figure 3C; Broyles et al., 2018; Morris et al., 2020; Wong et al., 2021). Lastly, the UAS-SoNaR transgene is used as NADH/NAD + sensor. It has the NADH-binding domain of Rex protein from Thermus aquaticus (T-Rex) connected to a circularly permuted yellow fluorescent protein (cpYFP) (Figure 3D). It has excitation at 420 or 485 nm with emission at 528 nm to determine NADH/NAD + ratio (Zhao et al., 2015; Bonnay et al., 2020; Morris et al., 2020).
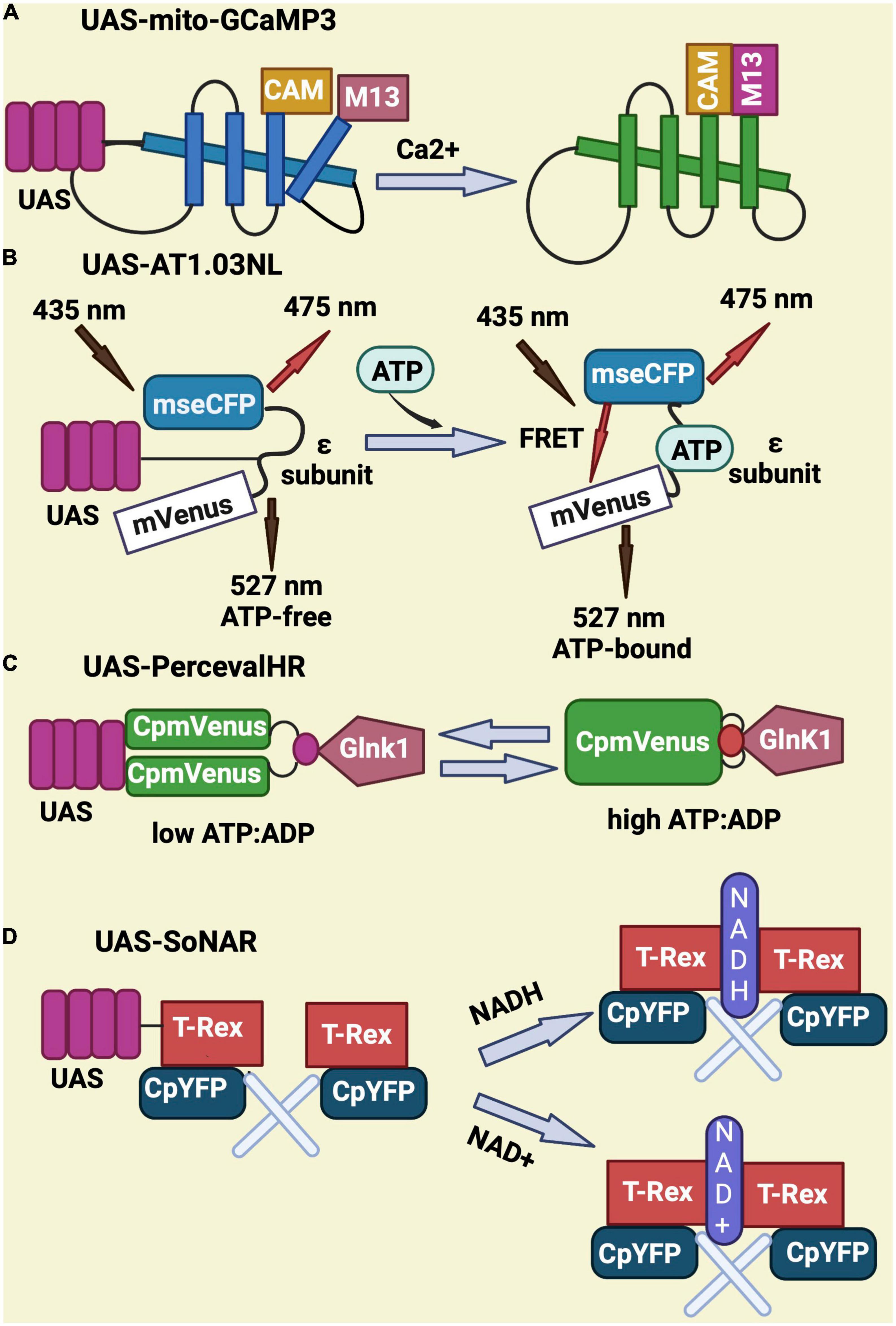
Figure 3. Schematic representation of genetically-encoded sensors for Calcium, ATP, ATP/ADP, NADH, and NAD. (A) UAS-mito-GCaMP3 encodes for a circularly permutated EGFP M13 and calmodulin fusion protein that gives GFP signal upon binding to calcium. (B) UAS-AT1.03NL consist of two fluorescent proteins (mVenus and mseCFP) and an ATP binding sequence (ε subunit) that elicits a FRET signal upon binding to ATP. (C) UAS-PercevalHR contains the ATP-binding protein GlnK1 from Methanocaldococcus jannaschii linked to circularly permutated mVenus and allows ATP sensing through conformational changes in Glnk1. (D) UAS-SoNar serves as NADH/NAD + sensor and encodes for a fusion of the NADH-binding domain of T-Rex (Rex protein from Thermus aquaticus) with a circularly permuted yellow fluorescent protein (cpYFP).
Light-dependent sensors
UAS-Dendra2.mito is a light-dependent sensor where the photoconvertible protein Dendra2 (green) from the filamentous fungus A. nidulans is tagged to the mitochondrial matrix. The green fluorescence of this tagged protein can be localized and examined prior to irradiation. The region of interest within a cell or the entire cell is then exposed to UV (405 nm) or blue light (488 nm) lasers. Dendra2 is immediately photo-converted from green to red fluorescence by UV or blue light (Figure 4). It is used to monitor mitochondria over time (Hwang et al., 2014; Perez-de-Nanclares-Arregi and Etxebeste, 2014; Bertolin et al., 2018). This sensor allows easy tracking of green signal before photoconversion and the corresponding shift to red fluorescence upon UV or blue light irradiation. Importantly, with excitation and emission occurring at 553 and 573 nm, respectively, the activated red Dendra2 signal exhibits high photo-stability (Perez-de-Nanclares-Arregi and Etxebeste, 2014).

Figure 4. Schematic representation of the photo-convertible process associated with UAS-Dendra2.mito. Green Dendra2 is photo-converted to the red form upon UV or blue light exposure.
Mitochondrial dysfunction in Aβ-expressing flies
Wang and Davis (2021) recently assessed mitochondrial function and dynamics in flies expressing Aβ in the mushroom body neurons through super-resolution microscopy, calcium imaging and behavioral assays. They found that Aβ induces mitochondrial fragmentation and dysfunction at a very early age, consistent with detectable apoptosis. Interestingly, learning was impaired much later than the initial mitochondrial abnormalities, confirming the proximal role of mitochondria in AD pathogenesis. In terms of structural changes, Aβ was found to induce formation of aberrant mitochondria with a build-up of vacuoles or damaged cristae in the pre-synapse of the fly dorsal longitudinal flight muscle (DLM). In this case, Aβ decreased the age-dependent anterograde and retrograde axonal trafficking of mitochondria (Zhao et al., 2010). To determine if manipulation of mitochondrial fission could modify Aβ-induced phenotypes, the fission regulator Drp1 was pan-neuronally co-expressed with Aβ. This work demonstrated that overexpression of Drp1 improves survival, climbing capacity, neuronal degeneration and ATP levels in Aβ flies (Lv et al., 2017). On the other hand, over-expression of Aβ in all neurons decreases drp1 and marf mRNA levels in older flies (Abtahi et al., 2020).
Another protein essential for mitochondrial function and transport is Milton. It connects Miro to kinesin, (Stowers et al., 2002) and enables them to move in axons and dendrites (Glater et al., 2006). Knock-down of Milton enhanced Aβ-induced locomotion defects. Consistently, heterozygous miro mutants led to an enhancement of Aβ-induced locomotor impairment associated with mitochondrial mislocalization (Iijima-Ando et al., 2009), whereas over-expression of Miro improved the eye phenotype, climbing performance and ATP levels in Aβ flies (Panchal and Tiwari, 2020).
To understand the effects of Aβ on mitochondrial distribution, Aβ was expressed in glutamatergic motor neurons of the fly leg. Aβ significantly reduces the number of mitochondria in the motor neurons and shortens the fly lifespan when overexpressed in glutamatergic neurons (Fernius et al., 2017). This suggests that Aβ affects mitochondrial distribution in the neuron, possibly contributing to the reduction in life span.
Mitochondria and endoplasmic reticulum connect to create mitochondria-ER contact sites (MERCs), which facilitates the exchange of lipids and calcium ions. However, MERCs dysfunction or miscommunication affects ATP generation and mitochondrial division by disturbing calcium shuttling and possibly through interaction with Drp1 and mitochondrial fission factor (MFF), although the molecular mechanism is not well understood (Rowland and Voeltz, 2012; Wilson and Metzakopian, 2021). This is relevant because alterations of the mitochondria–endoplasmic reticulum contacts have been reported in AD (Schon and Area-Gomez, 2013). In an effort to improve contacts between mitochondria and ER, synthetic linkers have been designed to enhance the proximity between both organelles. For instance, over-expression of a synthetic linker carrying mitochondrial and ER targeting sequences extended lifespan and suppressed climbing deficits in Aβ flies, which suggests that improving the interaction between mitochondria and ER could alleviate AD pathologies associated with mitochondrial dysfunction (Garrido-Maraver et al., 2020). However, a different team recently demonstrated that the knockdown of pdzd8, a putative Drosophila homolog of the mammalian MERC tethering protein, decreases contacts between the ER and mitochondria and restores locomotor deficits in Aβ flies (Hewitt et al., 2022). Thus, further research is needed to clarify these contradictory findings.
Calcium homeostasis is very important for proper mitochondrial function and must be preserved. Flies expressing human Aβ in the mushroom body have significantly reduced calcium import compared to control flies, suggesting an Aβ-mediated impairment of mitochondrial function in mushroom body neurons (Wang and Davis, 2021). In addition, pan-neuronal expression of Aβ reduced the levels of phosphoproteins predicted to be substrates of PKA, indicating that Aβ42 might modulate cAMP/PKA signaling (Iijima-Ando et al., 2009). This cAMP/PKA signaling is known to be affected by mitochondrial dysfunction which, in turn, reduces synaptic strength and prevents synaptic vesicle movement in the presynaptic terminal (Verstreken et al., 2005).
NDUFS3, a core component of the mitochondrial complex, is involved in the electron transport chain. Its expression is down-regulated in Aβ-expressing flies, which causes a decrease in the generation of ATP (Lin et al., 2021). Given that vitamin K serves as a mitochondrial electron transporter during oxidative respiration (Vos et al., 2012), there is an interest in investigating its therapeutic potential in fly models of AD. For instance, Lin et al. (2021) treated Aβ flies with vitamin K, which led to an improvement of mitochondrial function, reduction of Aβ neurotoxicity, and autophagy activation with concomitant increase in NDUFS3 expression and ATP levels.
Taken together, all these findings demonstrate that Aβ over-expression in flies triggers abnormal structural and functional changes in mitochondria, disrupts their dynamics and transport, and impairs learning and memory in late stages of the disease, all of which is relevant to understand AD pathogenesis (Figure 5).
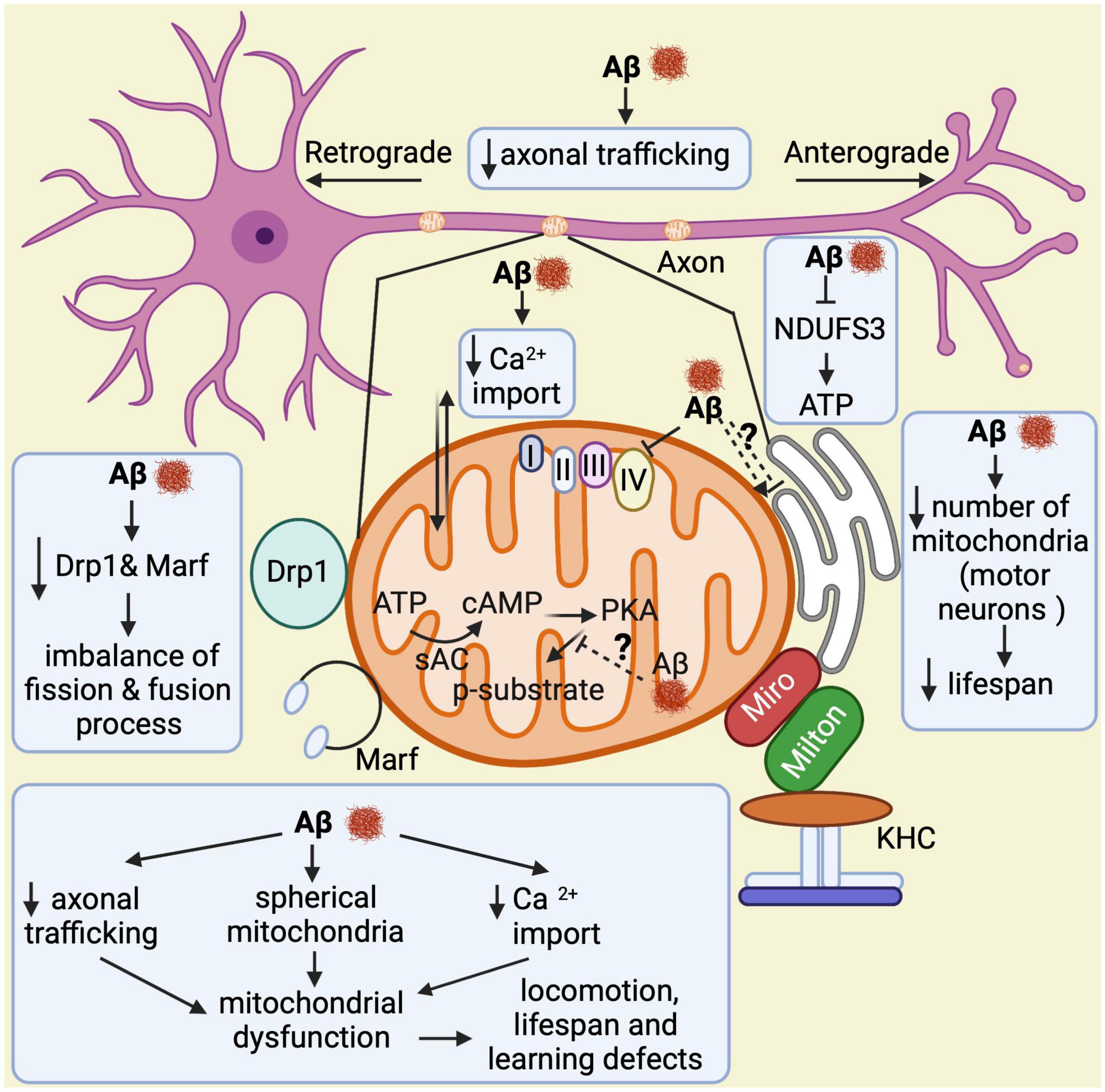
Figure 5. Overview of mitochondrial abnormalities found in Aβ-expressing flies. Aβ induces formation of spherical mitochondria, which disrupts their function. It also decreases calcium import into the mitochondria and affects their axonal trafficking, which impacts learning, lifespan, and locomotor behaviors. Aβ down-regulates NDUFS3, a crucial factor for the electron transport chain, resulting in low ATP production. In motor neurons, it reduces the number of mitochondria leading to shortened lifespan. Furthermore, Aβ decreases Drp1 and Marf expression, which causes an imbalance in the fission and fusion processes. Lastly, it seems to alter the mitochondria–endoplasmic reticulum contacts, although contradictory results have been found in this regard. Thus, this is highlighted with a question mark.
Mitochondrial dysfunction in tau-expressing flies
Tau is a protein involved in the polymerization and stabilization of microtubules and is also associated with the axonal transport of sub-cellular organelles (Gendron and Petrucelli, 2009; Dolan and Johnson, 2010). Phosphorylation of tau reduces its binding affinity for the tubulin subunits of microtubules, which enhances the self-aggregation and fibrillization of phosphorylated tau (Cohen et al., 2011; Cisek et al., 2014; Singh et al., 2015) and leads to defects in axonal transport of mitochondria (Ittner and Götz, 2011; Mondragón-Rodríguez et al., 2013; Mietelska-Porowska et al., 2014). Over-expression of human wild-type and mutant tau R406W in flies induces elongation of mitochondria, resulting in mitochondrial dysfunction and apoptotic neurodegeneration with cell cycle activation (Wittmann et al., 2001; Khurana et al., 2006). Tau-dependent activation of the cell cycle requires tau phosphorylation and it has been shown that TOR signaling is also involved in the cell-cycle activation that mediates tau-induced neurodegeneration (Khurana et al., 2006). Interestingly, stimulation of mitochondrial fission by concomitantly increasing the expression of Drp1 and reducing Marf levels reversed mitochondrial elongation and alleviated tau neurotoxicity in flies, suggesting that restoring the proper balance of mitochondrial fission and fusion is necessary to alleviate mitochondrial dysfunction and cell cycle-mediated cell death (DuBoff et al., 2012). In contrast, increasing fusion by upregulation of Marf and downregulation of Drp1 further increased the mitochondrial length in tau flies, resulting in more aggressive neurodegeneration (DuBoff et al., 2012).
Dynamin-related-protein1 is a cytoplasmic protein that translocates to the mitochondrial outer membrane to drive mitochondrial fission; however, in the context of mutant tau overexpression, Drp1 staining does not colocalize with mitoGFP and stays primarily in the cytosol, as evidenced by its distribution in cytoplasmic and mitochondrial fractions from fly heads (Frank et al., 2001). This tau-related blockage of Drp1 translocation is thought to be mediated by stabilization of actin because reversing actin stabilization rescues tau-induced mitochondrial defects (DuBoff et al., 2012). Another study demonstrated that over-expression of leucine-rich repeat kinase 2 (LRRK2) also increases tau neurotoxicity through excessive actin stabilization and subsequent mislocalization of Drp1 (Bardai et al., 2018). This seems to be a relevant pathway as pharmacological suppression of actin polymerization was found to reverse neurodegeneration and mitochondrial impairments in tau transgenic flies (Bardai et al., 2018). On the other hand, given that the balance between fusion and fission is disturbed in AD (Wang et al., 2008a), Abtahi and coworkers looked at how tau affects the expression of Marf and Drp1, which are essential for mitochondrial fusion and fission, respectively, (Abtahi et al., 2020). The authors found that pan-neural expression of both wild-type and mutant tau R406W in flies decreased the expression of Marf mRNA in older flies, suggesting that the decline in the mitochondrial fusion process takes place at a later stages in AD. The expression of Drp1 is modulated differently by wild-type and mutant tau; wild-type tau up-regulates Drp1 mRNA, whereas mutant tau down-regulates it. Despite the fact that mutant and wild type tau express Marf and Drp1 differently, the increased ratio of Drp1/Marf suggests that both wild type and mutant tau display more mitochondrial fission (Abtahi et al., 2020), uncovering an imbalance between fusion and fission in tau flies.
In Drosophila larval motor neurons, overexpression of human tau (0N3R) disrupts axonal transport as well as the morphology and function of neuromuscular junctions (Chee et al., 2005). This is caused by a marked decrease in the amount of detectable mitochondria in the pre-synaptic terminal, which causes synaptic dysfunction accompanied by a lower number of functional mitochondria (Chee et al., 2005). To understand the effects of tau on mitochondrial distribution, both UAS-tau0N4R and UAS-tau0N4R–E14 were expressed in the fly leg neurons. The results showed that “clump-like” aggregation of mitochondria is seen in the motor neurons projecting into the muscles while the distribution of mitochondria is even in control flies, indicating that tau affects mitochondrial distribution (Fernius et al., 2017). Accordingly, wild-type UAS-tau0N4R and UAS-tau0N4R–E14 expression in glutamatergic neurons significantly shorten the fly lifespan (Fernius et al., 2017).
A recent genome-wide RNAi screen in flies expressing human mutant tau pan-neuronally led to the identification of several modifiers involved in the mitochondrial pathway, such as biotinidase, NDUFS4, ALDH6A1, and TFB1M (Lohr et al., 2020). The authors found that the knock-down of biotinidase in tau flies disrupts the structure and function of mitochondria, the function of carboxylase enzymes, and leads to a more aggressive neurodegeneration. Interestingly, administration of biotin through feeding rescues toxicity of both wild-type and mutant tau in transgenic flies (Lohr et al., 2020). This is relevant because the authors also found reduced carboxylase biotinylation in the brain of some AD patients. However, the extent to which biotin levels contribute to AD pathogenesis is largely unknown. On the other hand, to understand the possible interaction between toxicity of tau and axonal mitochondria, the adaptor proteins essential for axonal mitochondrial transport, Milton and Miro, were knocked-down in flies expressing wild type tau. Knock-down of either Milton or Miro enhances tau-induced neurodegeneration. Moreover, Milton knock-down accelerates the accumulation of autophagic bodies and vacuole formation in presynaptic vesicles and axons (Iijima-Ando et al., 2012). Additionally, it increases tau phosphorylation at Ser262 via the partitioning defective-1 (PAR-1) protein, which decreases tau ability to bind microtubules. This suggests that both tau phosphorylation at Ser262 and PAR-1 are essential for enhancing tau-induced axon degeneration in Milton knockdown. Whereas pan-neuronal knockdown of Milton or Miro results in age-dependent neurodegeneration in the fly brain, knock-down of PAR-1 or endogenous fly tau suppresses Milton knockdown-induced neurodegeneration, demonstrating that both PAR-1 and tau participate in Milton knockdown-mediated neuropathology (Iijima-Ando et al., 2012).
Lastly, another study showed that pan-neuronal expression of wild-type and phosphomimetic mutant tau (tauE14) disrupts the circadian rhythm (Zhang et al., 2022). While wild type tau expression in clock neurons reduces the levels of neuropeptide pigment dispersing factor (PDF), a neurotransmitter essential for circadian function in Drosophila, expression of tauE14 in clock neurons disrupts the circadian rhythm and reduces PDF distribution in the dorsal axonal projections. Interestingly, tauE14 also induces a complete loss of mitochondria in dorsal projections indicating that tauE14 impairs axonal transport of neuropeptides and mitochondria in circadian pacemaker neurons, affecting circadian rhythm (Zhang et al., 2022). Further studies will be required to better understand the association between circadian rhythm, mitochondrial biology and Alzheimer’s disease. In summary, all these observations highlight multiple ways in which abnormal tau contributes to mitochondrial malfunction (Figure 6).
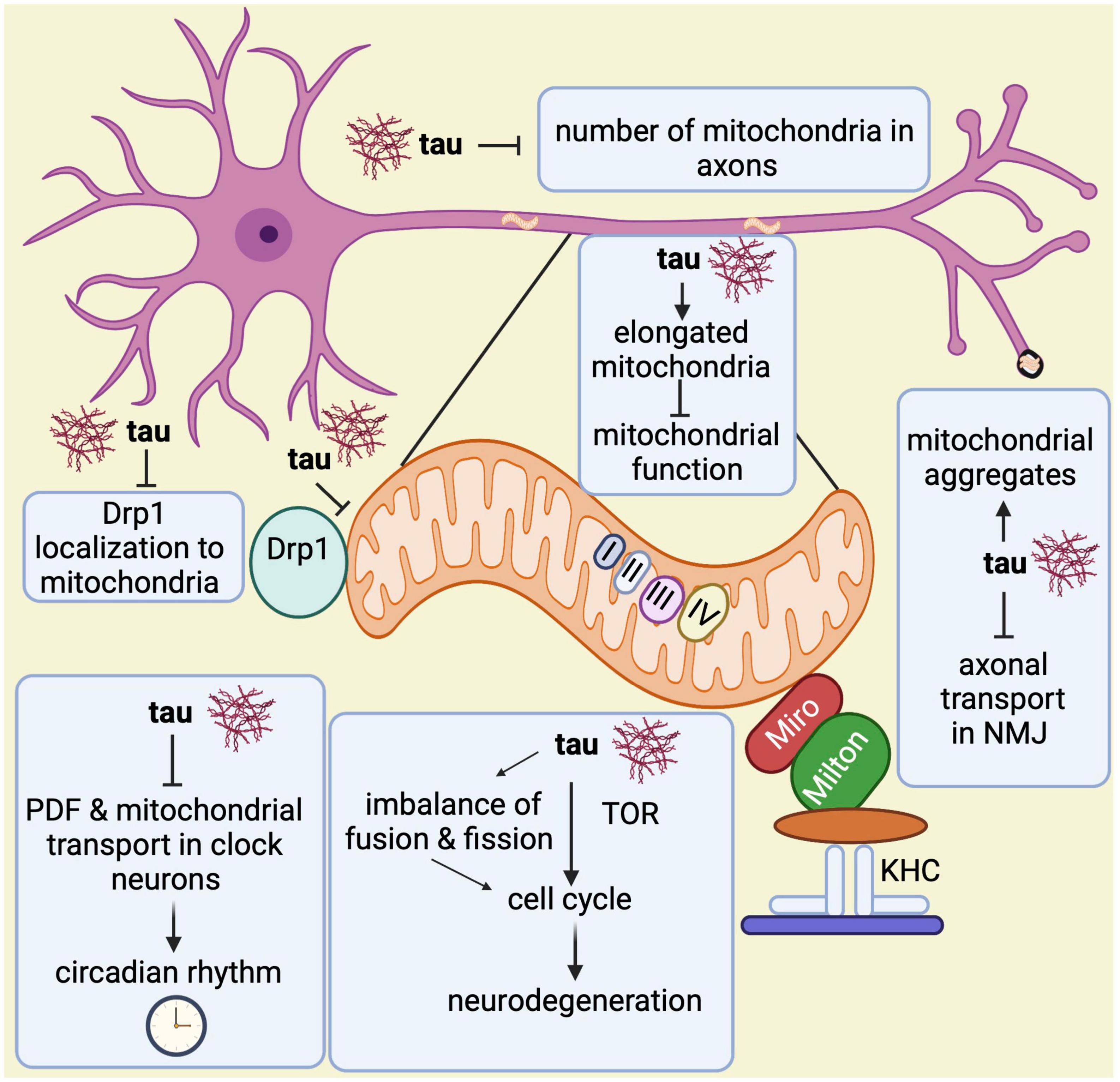
Figure 6. Overview of mitochondrial abnormalities found in tau-expressing flies. Tau induces elongated mitochondria, which causes mitochondrial dysfunction and apoptotic neurodegeneration with cell cycle activation. The cell cycle activation brings neurodegeneration via TOR and through an imbalance in the fusion and fission processes. Tau disrupts the circadian rhythm by inhibiting the neuropeptide pigment dispersing factor (PDF) and reducing mitochondrial transport in axons of clock neurons. Additionally, it prevents Drp1 from localizing to the outer mitochondrial membrane, which affects the fission process. Tau also disrupts axonal transport in the neuromuscular junction (NMJ) leading to mitochondrial aggregation. Lastly, it has been found that tau also reduces mitochondrial numbers in axons.
Concluding remarks and future directions
Alzheimer’s disease is a devastating neurodegenerative brain disorder characterized by extracellular Aβ plaques and intracellular aggregates of hyperphosphorylated tau, along with progressive cognitive decline. Despite decades of research and impressive efforts from multiple groups, there is still no treatment available for this dreadful disorder. Recent evidence suggested that AD and other neurodegenerative diseases are impacted by mitochondrial dysfunction, an area that may provide new targets for future therapeutic strategies.
In this review, we emphasized how Drosophila closely resembles molecular and pathological features of Aβ- and tau-related mitochondrial disfunction observed in AD. We compiled relevant studies in flies showing how Aβ42 or tau affect the structure of mitochondria, mitochondrial dynamics, calcium homeostasis, axonal transport of mitochondria, cAMP/PKA signaling, mitochondria-ER contact sites as well as the expression of several mitochondrial factors. We also discussed available assays and tools for examining mitochondrial function and dynamics in Drosophila. Considering the flexibility and power of Drosophila genetics, it is clear that this model organism will continue improving our understanding of the critical role of mitochondria in AD pathogenesis and its crosstalk with other pathomechanisms.
It is worth noting that there are still many opportunities for Drosophila-based research in this field. For instance a recent study found that the glutathione S-transferase (GST) Gfzf prevents mitochondrial hyperfusion in axons and regulates mitochondrial dynamics (Smith et al., 2019). This is important because several GST polymorphisms in humans have been associated with the development of AD (Allen et al., 2012). Thus, future manipulation of Gfzf in fly models of AD will help understand the potential contribution of GST activity to this devastating disorder.
One limitation of the studies discussed here is that they were performed in either Aβ42- or tau-expressing flies. Since Aβ42 and tau display synergistic interactions (Zhang H. et al., 2021), it will be imperative to study mitochondrial dynamics and function in flies co-expressing Aβ42 and tau to provide a more physiological context. This is because a recent longitudinal positron emission tomography (PET) study found that Aβ accelerated tau deposition in the inferior temporal cortex of older people with cognitively normal function over a 7-year follow-up period and that the rate of such accumulation was linked to the degree of cognitive decline (Hanseeuw et al., 2019). This finding is consistent with another PET study in cognitively healthy individuals showing that Aβ-tau interactions (rather than Aβ or tau alone) accelerated cognitive decline (Sperling et al., 2019). Moreover, individuals with primary age-related tauopathy, who exhibited equivalent tau burdens but negligible amounts of Aβ, had lesser high-molecular-weight (HMW) tau levels than patients with AD, who showed typical Aβ-plaque and tau-tangle burden (Bennett et al., 2017). Taken together, these studies confirm the synergistic interaction between Aβ and tau in AD pathology and emphasize the need of using fly models with concurrent Aβ and tau pathologies. On the other hand, TDP-43, an RNA/DNA binding protein linked to frontotemporal lobar degeneration and amyotrophic lateral sclerosis, was recently found to mediate prominent structural and functional damage to mitochondria along with activation of the mitochondrial unfolded protein response (UPRmt) (Wang et al., 2019). This is also relevant because more than 50% of AD cases display TDP-43 pathology in the brain (Meneses et al., 2021). It is, therefore, critical to concurrently manipulate Aβ42, tau and TDP-43 in transgenic flies to decipher their overall mitochondrial insults in AD cases with TDP-43 pathology. Approaching these and other unknown aspects of mitochondrial dysfunction with Drosophila will provide a more comprehensive portrait of molecular abnormalities and, thus, may lead to the identification of multiple and promising therapeutic targets in the years to come.
Author contributions
VV and DER-L conceived the original idea and revised the manuscript. VV and JM designed the outlines of the study, performed the literature review, wrote the first draft, and prepared the figures. All authors read and approved the final manuscript.
Funding
This work was supported by the National Institutes of Health grants R01AG059871, R01AG077534, and R21AG069050 to DER-L.
Acknowledgments
We thank all members of the DER-L’s laboratory for critical comments and Deepak Chhangani for suggestions in the preparation of figures. All figures in this review article were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abtahi, S. L., Masoudi, R., and Haddadi, M. (2020). The distinctive role of tau and amyloid beta in mitochondrial dysfunction through alteration in Mfn2 and Drp1 mRNA levels: A comparative study in Drosophila melanogaster. Gene 754:144854. doi: 10.1016/j.gene.2020.144854
Adams, M. D., Celniker, S. E., Holt, R. A., Evans, C. A., Gocayne, J. D., Amanatides, P. G., et al. (2000). The genome sequence of Drosophila melanogaster. Science 287, 2185–2195. doi: 10.1126/science.287.5461.2185
Ahmadian, S. S., Rezvanian, A., Peterson, M., Weintraub, S., Bigio, E. H., Mesulam, M. M., et al. (2015). Loss of calbindin-D28K is associated with the full range of tangle pathology within basal forebrain cholinergic neurons in Alzheimer’s disease. Neurobiol. Aging 36, 3163–3170. doi: 10.1016/j.neurobiolaging.2015.09.001
Aksenov, M. Y., Aksenova, M. V., Butterfield, D. A., Geddes, J. W., and Markesbery, W. R. (2001). Protein oxidation in the brain in Alzheimer’s disease. Neuroscience 103, 373–383. doi: 10.1016/s0306-4522(00)00580-7
Albrecht, S. C., Sobotta, M. C., Bausewein, D., Aller, I., Hell, R., Dick, T. P., et al. (2014). Redesign of genetically encoded biosensors for monitoring mitochondrial redox status in a broad range of model eukaryotes. J. Biomol. Screen. 19, 379–386. doi: 10.1177/1087057113499634
Ali, F., Baringer, S. L., Neal, A., Cho, E. Y., and Kwan, A. C. (2019). Parvalbumin-positive neuron loss and amyloid-β deposits in the frontal cortex of Alzheimer’s disease-related mice. J. Alzheimers Dis. 72, 1323–1339. doi: 10.3233/JAD-181190
Allen, M., Zou, F., Chai, H. S., Younkin, C. S., Miles, R., Nair, A. A., et al. (2012). Glutathione S-transferase omega genes in Alzheimer and Parkinson disease risk, age-at- diagnosis and brain gene expression: An association study with mechanistic implications. Mol. Neurodegener. 7:13. doi: 10.1186/1750-1326-7-13
Anand, R., Wai, T., Baker, M. J., Kladt, N., Schauss, A. C., Rugarli, E., et al. (2014). The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 204, 919–929. doi: 10.1083/jcb.201308006
Anderson, A. J., Su, J. H., and Cotman, C. W. (1996). DNA damage and apoptosis in Alzheimer’s disease: Colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J. Neurosci. 16, 1710–1719. doi: 10.1523/JNEUROSCI.16-05-01710.1996
Arce-Molina, R., Cortes-Molina, F., Sandoval, P. Y., Galaz, A., Alegria, K., Schirmeier, S., et al. (2020). A highly responsive pyruvate sensor reveals pathway-regulatory role of the mitochondrial pyruvate carrier MPC. eLife 9:e53917. doi: 10.7554/eLife.53917
Arranz, A. M., and DeStrooper, B. (2019). The role of astroglia in Alzheimer’s disease: Patho- physiology and clinical implications. Lancet Neurol. 18, 406–414. doi: 10.1016/S1474-4422(18)30490-3
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. doi: 10.1016/j.cell.2005.02.001
Bardai, F. H., Ordonez, D. G., Bailey, R. M., Hamm, M., Lewis, J., and Feany, M. B. (2018). Lrrk promotes tau neurotoxicity through dysregulation of actin and mitochondrial dynamics. PLoS Biol. 20:e2006265. doi: 10.1371/journal.pbio.2006265
Bennett, R. E., DeVos, S. L., Dujardin, S., Corjuc, B., Gor, R., Gonzalez, J., et al. (2017). Enhanced tau aggregation in the presence of Amyloid β. Am. J. Pathol. 187, 1601–1612. doi: 10.1016/j.ajpath.2017.03.011
Berg, J., Hung, Y. P., and Yellen, G. (2009). A genetically encoded fluorescent reporter of ATP:ADP ratio. Nat. Methods 6, 161–166. doi: 10.1038/nmeth.1288
Bertolin, G., Bulteau, A. L., Alves-Guerra, M. C., Burel, A., Lavault, M. T., Gavard, O., et al. (2018). Aurora kinase a localises to mitochondria to control organelle dynamics and energy production. eLife 7, 1–28. doi: 10.7554/eLife.38111
Blennow, K., de Leon, M. J., and Zetterberg, H. (2006). Alzheimer’s disease. Lancet 368, 387–403. doi: 10.1016/S0140-6736(06)69113-7
Boczonadi, V., Ricci, G., and Horvath, R. (2018). Mitochondrial DNA transcription and translation: Clinical syndromes. Essay Biochem. 62, 321–340. doi: 10.1042/EBC20170103
Bonnay, F., Veloso, A., Steinmann, V., Koöcher, T., Abdusselamoglu, M. D., Bajaj, S., et al. (2020). Oxidative metabolism drives immortalization of neural stem cells during tumorigenesis. Cell 182, 1490–1507.e19. doi: 10.1016/j.cell.2020.07.039
Boyman, L., Williams, G. S. B., Khananshvili, D., Sekler, I., and Lederer, W. J. (2013). NCLX: The mitochondrial sodium calcium exchanger. J. Mol. Cell Cardiol. 59, 205–213. doi: 10.1016/j.yjmcc.2013.03.012
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. Neuropathol. Exp. Neurol. 7, 960–969. doi: 10.1097/NEN.0b013e318232a379
Brand, A. H., and Perrimon, N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. doi: 10.1242/dev.118.2.401
Britti, E., Ros, J., Esteras, N., and Abramov, A. Y. (2020). Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 86:102150. doi: 10.1016/j.ceca.2019.102150
Brosseron, F., Krauthausen, M., Kummer, M., and Heneka, M. T. (2014). Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 50, 534–544. doi: 10.1007/s12035-014-8657-1
Broyles, C. N., Robinson, P., and Daniels, M. J. (2018). Fluorescent, bioluminescent, and optogenetic approaches to study excitable physiology in the single cardiomyocyte. Cells 7:51. doi: 10.3390/cells7060051
Butterfield, D. A., and Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160. doi: 10.1038/s41583-019-0132-6
Butterfield, D. A., Drake, J., Pocernich, C., and Castegna, A. (2001). Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol. Med. 7, 548–554. doi: 10.1016/s1471-4914(01)02173-6
Calkins, M. J., and Reddy, P. H. (2011). Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta 1812, 507–513. doi: 10.1016/j.bbadis.2011.01.007
Calvo-Rodriguez, M., Hou, S. H., Synder, A. C., Kharitonova, E. K., Russ, A. N., Das, S., et al. (2020). Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 11, 1–17. doi: 10.1038/s41467-020-16074-2
Casas-Tinto, S., Zhang, Y., Sanchez-Garcia, J., Gomez-Velazquez, M., Rincon-Limas, D. E., and Fernandez-Funez, P. (2011). The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 20, 2144–2160. doi: 10.1093/hmg/ddr100
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., et al. (2005). Mitochondrial Aβ: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 19, 2040–2041. doi: 10.1096/fj.05-3735fje
Castegna, A., Thongboonkerd, V., Klein, J. B., Lynn, B., Markesbery, W. R., and Butterfield, D. A. (2003). Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J. Neurochem. 85, 1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x
Chee, F. C., Mudher, A., Cuttle, M. F., Newman, T. A., Mackay, D., Lovestone, S., et al. (2005). Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol. Dis. 20, 918–928. doi: 10.1016/j.nbd.2005.05.029
Chen, J. X., and Yan, S. S. (2010). Role of mitochondrial amyloid-β in Alzheimer’s disease. Alzheimers Dis. 20(Suppl. 2), S569–S578. doi: 10.3233/JAD-2010-100357
Chen, Z., Qi, Y., French, S., Zhang, G., Covian, G. R., Balaban, R., et al. (2015). Genetic mosaic analysis of a deleterious mitochondrial DNA mutation in Drosophila reveals novel aspects of mitochondrial regulation and function. Mol. Biol. Cell. 26, 674–684. doi: 10.1091/mbc.E14-11-1513
Chen, Z., Zhang, F., and Xu, H. (2019). Human mitochondrial DNA diseases and Drosophila models. J. Genet. Genomics 46, 201–212. doi: 10.1016/j.jgg.2019.03.009
Cho, H. M., and Sun, W. (2020). Molecular cross talk among the components of the regulatory machinery of mitochondrial structure and quality control. Exp. Mol. Med. 52, 730–737. doi: 10.1038/s12276-020-0434-9
Cho, Y. H., Kim, G. H., and Park, J. J. (2021). Mitochondrial aconitase 1 regulates age-related memory impairment via autophagy/mitophagy-mediated neural plasticity in middle-aged flies. Aging Cell 20:e13520. doi: 10.1111/acel.13520
Cisek, K., Cooper, L. G., Huseby, J. C., and Kuret, J. (2014). Structure and mechanism of action of tau aggregation inhibitors. Curr. Alzheimer Res. 11, 918–927. doi: 10.2174/1567205011666141107150331
Cohen, T. J., Guo, J. L., Hurtado, D. E., Kwong, L. K., Mills, I. P., Trojanowski, J. Q., et al. (2011). The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2:252. doi: 10.1038/ncomms1255
Collier, J. J., Oláhová, M., Mcwilliams, T. G., and Taylor, R. W. (2023). Neurosciences mitochondrial signalling and homeostasis: From cell biology to neurological disease. Trends Neurosci. 46, 137–152. doi: 10.1016/j.tins.2022.12.001
Cook, K. R., Parks, A. L., Jacobus, L. M., Kaufman, T. C., and Matthews, K. A. (2010). New research resources at the Bloomington Drosophila Stock Center. Fly 4, 88–91. doi: 10.4161/fly.4.1.11230
Crews, L., and Masliah, E. (2010). Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 19, 12–20. doi: 10.1093/hmg/ddq160
Cummins, N., Tweedie, A., Zuryn, S., Bertran-Gonzalez, J., and Götz, J. (2019). Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 38:e99360. doi: 10.15252/embj.201899360
D’Souza, I., and Schellenberg, G. D. (2005). Regulation of tau isoform expression and dementia. Biochim. Biophys. Acta 1739, 104–115. doi: 10.1016/j.bbadis.2004.08.009
de Vrij, F. M., Fischer, D. F., van Leeuwen, F. W., and Hol, E. M. (2004). Protein quality control in Alzheimer’s disease by the ubiquitin proteasome system. Prog. Neurobiol. 74, 249–270. doi: 10.1016/j.pneurobio.2004.10.001
Despres, C., Byrne, C., Qi, H., Cantrelle, F. X., Huvent, I., Chambraud, B., et al. (2017). Identification of the tau phosphorylation pattern that drives its aggregation. Proc. Natl. Acad. Sci. U.S.A. 114, 9080–9085. doi: 10.1073/pnas.1708448114
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006
DeVorkin, L., Go, N. E., Hou, Y. C. C., Moradian, A., Morin, G. B., and Gorski, S. M. (2014). The Drosophila effector caspase Dcp-1 regulates mitochondrial dynamics and autophagic flux via SesB. J. Cell Biol. 205, 477–492. doi: 10.1083/jcb.201303144
Diana, A., Simic, G., Sinforiani, E., Orru, N., Pichiri, G., and Bono, G. (2008). Mitochondria morphology and DNA content upon sublethal exposure to beta-amyloid (1-42) peptide. Coll. Antropol. 32(Suppl. 1), 51–58.
Dietzl, G., Chen, D., Schnorrer, F., Su, K. C., Barinova, Y., Fellner, M., et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156. doi: 10.1038/nature05954
Ding, Q., Markesbery, W. R., Chen, Q., Li, F., and Keller, J. N. (2005). Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 25, 9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005
Doherty, E., and Perl, A. (2017). Measurement of mitochondrial mass by flow cytometry during oxidative stress. React. Oxyg. Species 4, 275–283. doi: 10.20455/ros.2017.839
Dolan, P. J., and Johnson, G. V. W. (2010). The role of tau kinases in Alzheimer’s disease. Curr. Opin. Drug Discov. Dev. 13, 595–603.
Dong, J., and Zhao, M. (2016). In-vivo fluorescence imaging of adenosine 5′-triphosphate. Anal. Chem. 80, 190–203.
Doré, S. (2002). Decreased activity of the antioxidant heme oxygenase enzyme: Implications in ischemia and in Alzheimer’s disease. Free Radic. Biol. Med. 32, 1276–1282. doi: 10.1016/s0891-5849(02)00805-5
Dragicevic, N., Mamcarz, M., Zhu, Y., Buzzeo, R., Tan, J., Arendash, G. W., et al. (2010). Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J. Alzheimers Dis. 20(Suppl. 2), S535–S550. doi: 10.3233/JAD-2010-100342
D’Souza, A. R., and Minczuk, M. (2018). Mitochondrial transcription and translation: Overview. Essays Biochem. 62, 309–320. doi: 10.1042/EBC20170102
Du, H., Guo, L., Yan, S., Sosunov, A. A., McKhann, G. M., and Yan, S. S. (2010). Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 107, 18670–18675. doi: 10.1073/pnas.1006586107
DuBoff, B., Götz, J., and Feany, M. B. (2012). Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75, 618–632. doi: 10.1016/j.neuron.2012.06.026
Ebneth, A., Godemann, R., Stamer, K., Illenberger, S., Trinczek, B., and Mandelkow, E. (1998). Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 143, 777–794. doi: 10.1083/jcb.143.3.777
Eftekharzadeh, B., Daigle, J. G., Kapinos, L. E., Coyne, A., Schiantarelli, J., Carlomagno, Y., et al. (2018). Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99, 925–940.e7. doi: 10.1016/j.neuron.2018.07.039
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Fernius, J., Starkenberg, A., and Thor, S. (2017). Bar-coding neurodegeneration: Identifying subcellular effects of human neurodegenerative disease proteins using Drosophila leg neurons. Dis. Model Mech. 10, 1027–1038. doi: 10.1242/dmm.029637
Ferreira, I. L., Ferreiro, E., Schmidt, J., Cardoso, J. M., Pereira, C. M., Carvalho, A. L., et al. (2015). Aβ and NMDAR activation cause mitochondrial dysfunction involving ER calcium release. Neurobiol. Aging. 36, 680–692. doi: 10.1016/j.neurobiolaging.2014.09.006
Feuillette, S., Charbonnier, C., Frebourg, T., Campion, D., and Lecourtois, M. A. (2020). Connected network of interacting proteins is involved in human-tau toxicity in Drosophila. Front. Neurosci. 14:68. doi: 10.3389/fnins.2020.00068
Frank, S., Gaume, B., Bergmann-Leitner, E. S., Leitner, W. W., Robert, E. G., Catez, F., et al. (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell. 1, 515–525. doi: 10.1016/s1534-5807(01)00055-7
Fransson, S., Ruusala, A., and Aspenström, P. (2006). The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 344, 500–510. doi: 10.1016/j.bbrc.2006.03.163
Friedman, J. R., Lackner, L. L., West, M., DiBenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Garrido-Maraver, J., Loh, S. H., and Martins, L. M. (2020). Forcing contacts between mitochondria and the endoplasmic reticulum extends lifespan in a Drosophila model of Alzheimer’s disease. Biol. Open 9:bio047530. doi: 10.1242/bio.047530
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Gendron, T. F., and Petrucelli, L. (2009). The role of tau in neurodegeneration. Mol. Neurodegen. 4:13. doi: 10.1186/1750-1326-4-13
Gevedon, O., Bolus, H., Lye, S. H., Schmitz, K., Fuentes-Gonzalez, J., Hatchell, K., et al. (2019). In vivo forward genetic screen to identify novel neuroprotective genes in Drosophila melanogaster. J. Vis. Exp. 149:e59720. doi: 10.3791/59720
Gibson, G. E., Sheu, K. F. R., and Blass, J. P. (1998). Abnormalities of mitochondrial enzymes in Alzheimer disease. Neural Transm. 105, 855–870. doi: 10.1007/s007020050099
Glater, E. E., Megeath, L. J., Stowers, R. S., and Schwarz, T. L. (2006). Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 173, 545–557. doi: 10.1083/jcb.200601067
Goedert, M., and Jakes, R. (1990). Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9, 4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x
Goedert, M., and Jakes, R. (2005). Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 1739, 240–250. doi: 10.1016/j.bbadis.2004.08.007
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989). Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526. doi: 10.1016/0896-6273(89)90210-9
Good, P. F., Werner, P., Hsu, A., Olanow, C. W., and Perl, D. P. (1996). Evidence for neuronal oxidative damage in Alzheimer’s disease. Am. J. Pathol. 149, 21–28.
Green, K. N., Demuro, A., Akbari, Y., Hitt, B. D., Smith, I. F., Parker, I., et al. (2008). SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 181, 1107–1116. doi: 10.1083/jcb.200706171
Greeve, I., Kretzschmar, D., Tschäpe, J. A., Beyn, A., Brellinger, C., Schweizer, M., et al. (2004). Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J. Neurosci. 24, 3899–3906. doi: 10.1523/JNEUROSCI.0283-04.2004
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917. doi: 10.1073/pnas
Guo, X., Macleod, G. T., Wellington, A., Hu, F., Panchumarthi, S., Schoenfield, M., et al. (2005). The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. doi: 10.1016/j.neuron.2005.06.027
Haass, C., Kaether, C., Thinakaran, G., and Sisodia, S. (2012). Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2, 1–26. doi: 10.1101/cshperspect.a006270
Han, Y., Wang, N., Kang, J., and Fang, Y. (2020). β-Asarone improves learning and memory in Aβ1-42-induced Alzheimer’s disease rats by regulating PINK1-Parkin-mediated mitophagy. Metab. Brain Dis. 35, 1109–1117. doi: 10.1007/s11011-020-00587-2
Hanseeuw, B. J., Betensky, R. A., Jacobs, H. I. L., Schultz, A. P., Sepulcre, J., Becker, J. A., et al. (2019). Association of amyloid and tau with cognition in preclinical Alzheimer disease: A longitudinal study. JAMA Neurol. 76, 915–924. doi: 10.1001/jamaneurol.2019.1424
Hartwig, C., Méndez, G. M., Bhattacharjee, S., Vrailas-Mortimer, A. D., Zlatic, S. A., Freeman, A., et al. (2021). Golgi-dependent copper homeostasis sustains synaptic development and mitochondrial content. J. Neurosci. 41, 215–233. doi: 10.1523/JNEUROSCI.1284-20.2020
Hauptmann, S., Scherping, I., Dröse, S., Brandt, U., Schulz, K. L., Jendrach, M., et al. (2009). Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 30, 1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005
Hedskog, L., Pinho, C. M., Filadi, R., Rönnbäck, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U.S.A. 110, 7916–7921. doi: 10.1073/pnas.1300677110
Hensley, K., Hall, N., Subramaniam, R., Cole, P., Harris, M., Aksenov, M., et al. (1995). Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 65, 2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x
Hewitt, V. L., Miller-fleming, L., Twyning, M. J., Andreazza, S., Mattedi, F., Prudent, J., et al. (2022). Decreasing pdzd8-mediated mito – ER contacts improves organismal fi tness and mitigates Aβ42 toxicity. Life Sci. Alliance 5, 1–15. doi: 10.26508/lsa.202201531
Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., et al. (2001). Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 21, 3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001
Hirth, F. (2010). On the origin and evolution of the tripartite brain. Brain Behav. Evol. 76, 3–10. doi: 10.1159/000320218
Hollenbeck, P. J., and Saxton, W. M. (2005). The axonal transport of mitochondria. J. Cell Sci. 118, 5411–5419. doi: 10.1242/jcs.02745
Holper, S., Watson, R., and Yassi, N. (2022). Tau as a biomarker of neurodegeneration. Int. J. Mol. Sci. 23:7307. doi: 10.3390/ijms23137307
Honda, K., Smithi, M., Zhu, X., Baus, D., Merrick, W. C., Tartakoff, A. M., et al. (2005). Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J. Biol. Chem. 280, 20978–20986. doi: 10.1074/jbc.M500526200
Hoppins, S., Lackner, L. L., Lee, J. E., and Mears, J. A. (2020). In vitro and in vivo assays for mitochondrial fission and fusion. Methods Cell Biol. 155, 491–518. doi: 10.1016/bs.mcb.2019.11.010
Houlihan, K. L., Keoseyan, P. P., Juba, A. N., Margaryan, T., Voss, M. E., Babaoghli, A. M., et al. (2022). Folic acid improves parkin-null Drosophila phenotypes and transiently reduces vulnerable dopaminergic neuron mitochondrial hydrogen peroxide levels and glutathione redox equilibrium. Antioxidants 11:2068. doi: 10.3390/antiox11102068
Hwang, R. D., Wiemerslage, L., LaBreck, C. J., Khan, M., Kannan, K., Wang, X., et al. (2014). The neuroprotective effect of human uncoupling protein 2 (hUCP2) requires cAMP-dependent protein kinase in a toxin model of Parkinson’s disease. Neurobiol. Dis. 69, 180–191. doi: 10.1016/j.nbd.2014.05.032
Iijima-Ando, K., Hearn, S. A., Shenton, C., Gatt, A., Zhao, L., and Iijima, K. (2009). Mitochondrial mislocalization underlies aβ42-induced neuronal dysfunction in a Drosophila model of Alzheimer’s disease. PLoS One 4:e8310. doi: 10.1371/journal.pone.0008310
Iijima-Ando, K., Sekiya, M., Maruko-Otake, A., Ohtake, Y., Suzuki, E., Lu, B., et al. (2012). Loss of axonal mitochondria promotes tau-mediated neurodegeneration and Alzheimer’s disease-related tau phosphorylation via PAR-1. PLoS Genet. 8:e1002918. doi: 10.1371/journal.pgen.1002918
Imamura, H., Huynh Nhat, K. P., Togawa, H., Saito, K., Iino, R., Kato-Yamada, Y., et al. (2009). Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. U.S.A. 106, 15651–15656. doi: 10.1073/pnas.0904764106
Inczedy-Farkas, G., Trampush, J. W., Perczel Forintos, D., Beech, D., Andrejkovics, M., Varga, Z., et al. (2014). Mitochondrial DNA mutations and cognition: A case-series report. Arch. Clin. Neuropsychol. 29, 315–321. doi: 10.1093/arclin/acu016
Ingerman, E., Perkins, E. M., Marino, M., Mears, J. A., McCaffery, J. M., Hinshaw, J. E., et al. (2005). Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 170, 1021–1027. doi: 10.1083/jcb.200506078
Ittner, L. M., and Götz, J. (2011). Amyloid-β and tau–A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 12, 67–72. doi: 10.1038/nrn2967
Jacobs, H. T., Fernández-Ayala, D. J. M., Manjiry, S., Kemppainen, E., Toivonen, J. M., and O’Dell, K. M. (2004). Mitochondrial disease in flies. Biochim. Biophys. Acta 1659, 190–196. doi: 10.1016/j.bbabio.2004.07.004
Jadiya, P., Kolmetzky, D. W., Tomar, D., Di Meco, A., Lombardi, A. A., Lambert, J. P., et al. (2019). Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 10:3885. doi: 10.1038/s41467-019-11813-6
Jiang, S., Nandy, P., Wang, W., Ma, X., Hsia, J., Wang, C., et al. (2018). Mfn2 ablation causes an oxidative stress response and eventual neuronal death in the hippocampus and cortex. Mol. Neurodegen. 13, 1–15. doi: 10.1186/s13024-018-0238-8
Johnson, A. B., and Blum, N. R. (1970). Nucleoside phosphatase activities associated with the tangles and plaques of Alzheimer’s disease: A histochemical study of natural and experimental neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 29, 463–478. doi: 10.1097/00005072-197007000-00009
Jouaville, L. S., Pinton, P., Bastianutto, C., Rutter, G. A., and Rizzuto, R. (1999). Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. U.S.A. 96, 13807–13812. doi: 10.1073/pnas.96.24.13807
Kandimalla, R., Manczak, M., Fry, D., Suneetha, Y., Sesaki, H., and Reddy, P. H. (2016). Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 25, 4881–4897. doi: 10.1093/hmg/ddw312
Kandimalla, R., Manczak, M., Yin, X., Wang, R., and Reddy, P. H. (2018). Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 27, 30–40. doi: 10.1093/hmg/ddx381
Kerr, J. S., Adriaanse, B. A., Greig, N. H., Mattson, M. P., Cader, M. Z., Bohr, V. A., et al. (2017). Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 40, 151–166. doi: 10.1016/j.tins.2017.01.002
Khurana, V., Lu, Y., Steinhilb, M. L., Oldham, S., Shulman, J. M., and Feany, M. B. (2006). Article TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol. 16, 230–241. doi: 10.1016/j.cub.2005.12.042
Kim, T. S., Pae, C. U., Yoon, S. J., Jang, W. Y., Lee, N. J., Kim, J. J., et al. (2006). Decreased plasma antioxidants in patients with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 21, 344–348. doi: 10.1002/gps.1469
Knopman, D. S., Holtzman, D. M., and Hyman, B. T. (2021). Alzheimer disease. Nat. Rev. Dis. Primers 7:33. doi: 10.1038/s41572-021-00269-y
Kopeikina, K. J., Carlson, G. A., Pitstick, R., Ludvigson, A. E., Peters, A., Luebke, J. I., et al. (2011). Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 179, 2071–2082. doi: 10.1016/j.ajpath.2011.07.004
Kopke, E., Tung, Y. C., Shaikh, S., Del Alonso, C. A., Iqbal, K., and Grundke-Iqbal, I. (1993). Microtubule-associated protein tau. Abnormal phosphorylation of a non- paired helical filament pool in Alzheimer disease. J. Biol. Chem. 268, 24374–24384.
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., McCaffery, J. M., and Chan, D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862. doi: 10.1126/science.1099793
Kosik, K. S., Orecchio, L. D., Bakalis, S., and Neve, R. L. (1989). Developmentally regulated expression of specific tau sequences. Neuron 2, 1389–1397. doi: 10.1016/0896-6273(89)90077-9
Krzystek, T. J., Banerjee, R., Thurston, L., Huang, J. Q., Swinter, K., Rahman, S. N., et al. (2021). Differential mitochondrial roles for α-synuclein in DRP1-dependent fission and PINK1/Parkin-mediated oxidation. Cell Death Dis. 12, 1–16. doi: 10.1038/s41419-021-04046-3
Kyriakoudi, S., Drousiotou, A., and Petrou, P. P. (2021). When the balance tips: Dysregulation of mitochondrial dynamics as a culprit in disease. Int. J. Mol. Sci. 22:4617. doi: 10.3390/ijms22094617
Lai, M. K., Tsang, S. W., Esiri, M. M., Francis, P. T., Wong, P. T., and Chen, C. P. (2011). Differential involvement of hippocampal serotonin1A receptors and re-uptake sites in non-cognitive behaviors of Alzheimer’s disease. Psychopharmacology 213, 431–439. doi: 10.1007/s00213-010-1936-2
Lashuel, H. A., Hartley, D., Petre, B. M., Walz, T., and Lansbury, P. T. (2002). Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 418:291. doi: 10.1038/418291a
Lewis, O. L., Farr, C. L., and Kaguni, L. S. (1995). Drosophila melanogaster mitochondrial DNA: Completion of the nucleotide sequence and evolutionary comparisons. Insect Mol. Biol. 4, 263–278. doi: 10.1111/j.1365-2583.1995.tb00032.x
Li, F., Calingasan, N. Y., Yu, F., Mauck, W. M., Toidze, M., Almeida, C. G., et al. (2004). Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J. Neurochem. 89, 1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x
Li, X. C., Hu, Y., Wang, Z. H., Luo, Y., Zhang, Y., Liu, X. P., et al. (2016). Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 6:24756. doi: 10.1038/srep24756
Limorenko, G., and Lashuel, H. A. (2022). Revisiting the grammar of Tau aggregation and pathology formation: How new insights from brain pathology are shaping how we study and target tauopathies. Chem. Soc. Rev. 51, 513–565. doi: 10.1039/d1cs00127b
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Lin, M. Y., Chenga, X. T., Xiea, Y., Caib, Q., and Shenga, Z. H. (2017). Removing dysfunctional mitochondria from axons independent of mitophagy under pathophysiological conditions. Autophagy 13, 1792–1794. doi: 10.1080/15548627.2017.1356552
Lin, X., Wen, X., Wei, Z., Guo, K., Shi, F., Huang, T., et al. (2021). Vitamin K2protects against Aβ42-induced neurotoxicity by activating autophagy and improving mitochondrial function in Drosophila. Neuroreport 32, 431–437. doi: 10.1097/WNR.0000000000001599
Liu, W., Acin-Perez, R., Geghman, K. D., Manfredi, G., Lu, B., and Li, C. (2011). Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. U.S.A. 108, 12920–12924. doi: 10.1073/pnas.1107332108
Lohr, K. M., Frost, B., Scherzer, C., and Feany, M. B. (2020). Biotin rescues mitochondrial dysfunction and neurotoxicity in a tauopathy model. Proc. Natl. Acad. Sci. U.S.A. 117, 33608–33618. doi: 10.1073/pnas.192239211
Lovell, M. A., Soman, S., and Bradley, M. A. (2011). Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech. Ageing Dev. 132, 443–448. doi: 10.1016/j.mad.2011.08.003
Lutas, A., Wahlmark, C. J., Acharjee, S., and Kawasaki, F. (2012). Genetic analysis in Drosophila reveals a role for the mitochondrial protein P32 in synaptic transmission. G3 2, 59–69. doi: 10.1534/g3.111.001586
Lv, F., Yang, X., Cui, C., and Su, C. (2017). Exogenous expression of Drp1 plays neuroprotective roles in the Alzheimer’s disease in the Aβ42 transgenic Drosophila model. PLoS One 12:e0176183. doi: 10.1371/journal.pone.0176183
Lyras, L., Cairns, N. J., Jenner, A., Jenner, P., and Halliwell, B. (1997). An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. J. Neurochem. 68, 2061–2069. doi: 10.1046/j.1471-4159.1998.71010302.x
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449. doi: 10.1093/hmg/ddl066
Manczak, M., Calkins, M. J., and Reddy, P. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 20, 2495–2509. doi: 10.1093/hmg/ddr139
Mandelkow, E. M., and Mandelkow, E. (2012). Biochemistry and cell biology of Tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2:a006247. doi: 10.1101/cshperspect.a006247
Marcus, D. L., Thomas, C., Rodriguez, C., Simberkoff, K., Tsai, J. S., Strafaci, J. A., et al. (1998). Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp. Neurol. 150, 40–44. doi: 10.1006/exnr.1997.6750
Martín-Maestro, P., Gargini, R., García, E., Perry, G., Avila, J., and García-Escudero, V. (2017). Slower dynamics and aged mitochondria in sporadic Alzheimer’s disease. Oxid. Med. Cell. Longev. 9302761:14. doi: 10.1155/2017/9302761
Martín-Maestro, P., Gargini, R., Perry, G., Avila, J., and García-Escudero, V. (2016). PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 25, 792–806. doi: 10.1093/hmg/ddv616
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189, 211–221. doi: 10.1083/jcb.200910140
Maurer, I., Zierz, S., and Moöller, H. J. (2000). A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 21, 455–462. doi: 10.1016/S0197-4580(00)00112-3
McGurk, L., Berson, A., and Bonini, N. M. (2015). Drosophila as an in vivo model for human neurodegenerative disease. Genetics 201, 377–402. doi: 10.1534/genetics.115.179457
McLachlan, D. R., Wong, L., Bergeron, C., and Baimbridge, K. G. (1987). Calmodulin and calbindin D28K in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1, 171–179. doi: 10.1097/00002093-198701030-00009
Mecocci, P., MacGarvey, U., and Beal, M. F. (1994). Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 36, 747–750. doi: 10.1002/ana.410360510
Meneses, A., Koga, S., Leary, J. O., Dickson, D. W., Bu, G., and Zhao, N. (2021). TDP-43 pathology in Alzheimer’s disease. Mol. Neurodegener. 16:84. doi: 10.1186/s13024-021-00503-x
Merlini, M., Rafalski, V. A., Ma, K., Kim, K. Y., Bushong, E. A., Rios Coronado, P. E., et al. (2021). Microglial Gi-dependent dynamics regulate brain network hyperexcitability. Nat. Neurosci. 24, 19–23. doi: 10.1038/s41593-020-00756-7
Mietelska-Porowska, A., Wasik, U., Goras, M., Filipek, A., and Niewiadomska, G. (2014). Tau protein modifications and interactions: Their role in function and dysfunction. Int. J. Mol. Sci. 15, 4671–4713. doi: 10.3390/ijms15034671
Mishra, P., and Chan, D. C. (2014). Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 15, 634–646. doi: 10.1038/nrm3877
Mondragón-Rodríguez, S., Perry, G., Zhu, X., Moreira, P. I., Acevedo-Aquino, M. C., and Williams, S. (2013). Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: Implications for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013:940603. doi: 10.1155/2013/940603
Moore, B. D., Martin, J., de Mena, L., Sanchez, J., Cruz, P. E., Ceballos-Diaz, C., et al. (2018). Short Aβ peptides attenuate Aβ42 toxicity in vivo. J. Exp. Med. 215, 283–301. doi: 10.1084/jem.20170600
Morgan, T. H. (1910). Sex limited inheritance in Drosophila. Science 32, 120–122. doi: 10.1126/science.32.812.120
Morishima-Kawashima, M., and Ihara, Y. (2002). Alzheimer’s disease: β-amyloid protein and tau. J. Neurosci. Res. 70, 392–401. doi: 10.1002/jnr.10355
Morris, O., Deng, H., Tam, C., and Jasper, H. (2020). Warburg-like metabolic reprogramming in aging intestinal stem cells contributes to tissue hyperplasia. Cell Rep. 33:108423. doi: 10.1016/j.celrep.2020.108423
Mullaart, E., Boerrigter, M. E. T. I., Ravid, R., Swaab, D. F., and Vijg, J. (1990). Increased levels of DNA breaks in cerebral cortex of Alzheimer’s disease patients. Neurobiol. Aging 11, 169–173. doi: 10.1016/0197-4580(90)90542-8
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Nayak, N., and Mishra, M. (2022). Drosophila melanogaster as a model to understand the mechanisms of infection mediated neuroinflammation in neurodegenerative diseases. J. Integr. Neurosci. 21:66. doi: 10.31083/j.jin2102066
Nguyen, T. N., Padman, B. S., and Lazarou, M. (2016). Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 26, 733–744. doi: 10.1016/j.tcb.2016.05.008
Ni, J. Q., Zhou, R., Czech, B., Liu, L. P., Holderbaum, L., Yang-Zhou, D., et al. (2011). A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 8, 405–407. doi: 10.1038/nmeth.1592
Nixon, R. A., and Yang, D. S. (2011). Autophagy failure in Alzheimer’s disease–locating the primary defect. Neurobiol. Dis. 43, 38–45. doi: 10.1016/j.nbd.2011.01.021
Panchal, K., and Tiwari, A. K. (2020). Miro, a Rho GTPase genetically interacts with Alzheimer’s disease-associated genes (Tau, Aβ42 and Appl) in Drosophila melanogaster. Biol. Open 9:bio049569. doi: 10.1242/bio.049569
Parker, W. D. J., Filley, C. M., and Parks, J. K. (1990). Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 8, 1302–1303. doi: 10.1212/wnl.40.8.1302
Pérez, M. J., Vergara-Pulgar, K., Jara, C., Cabezas-Opazo, F., and Quintanilla, R. A. (2017). Caspase-cleaved tau impairs mitochondrial dynamics in Alzheimer’s disease. Mol. Neurobiol. 55, 1004–1018. doi: 10.1007/s12035-017-0385-x
Perez-de-Nanclares-Arregi, E., and Etxebeste, O. (2014). Photo-convertible tagging for localization and dynamic analyses of low-expression proteins in filamentous fungi. Fungal Genet. Biol. 70, 33–41. doi: 10.1016/j.fgb.2014.06.006
Perocchi, F., Gohil, V. M., Girgis, H. S., Bao, X. R., McCombs, J. E., Palmer, A. E., et al. (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296. doi: 10.1038/nature09358
Perry, S. W., Norman, J. P., Barbieri, J., Brown, E. B., and Gelbard, H. A. (2011). Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Biotechniques 50, 98–115. doi: 10.2144/000113610
Pickett, E. K., Rose, J., McCrory, C., McKenzie, C. A., King, D., Smith, C., et al. (2018). Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. 136, 747–757. doi: 10.1007/s00401-018-1903-2
Pollard, A. K., Craig, E. L., and Chakrabarti, L. (2016). Mitochondrial complex 1 activity measured by spectrophotometry is reduced across all brain regions in ageing and more specifically in neurodegeneration. PLoS One 11:e0157405. doi: 10.1371/journal.pone.0157405
Presley, A. D., Fuller, K. M., and Arriaga, E. A. (2003). MitoTracker Green labeling of mitochondrial proteins and their subsequent analysis by capillary electrophoresis with laser-induced fluorescence detection. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 793, 141–150. doi: 10.1016/s1570-0232(03)00371-4
Ramos Bernardes da Silva Filho, S., Oliveira Barbosa, J. H., Rondinoni, C., dos Santos, A. C., Garrido Salmon, C. E., da Costa Lima, N. K., et al. (2017). Neuro-degeneration profile of Alzheimer’s patients: A brain morphometry study. Neuroimage 15, 15–24. doi: 10.1016/j.nicl.2017.04.001
Rana, A., Oliveira, M. P., Khamoui, A. V., Aparicio, R., Rera, M., Rossiter, H. B., et al. (2017). Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 8:448. doi: 10.1038/s41467-017-00525-4
Reddy, P. H., McWeeney, S., Park, B. S., Manczak, M., Gutala, R. V., Partovi, D., et al. (2004). Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Mol. Genet. 13, 1225–1240. doi: 10.1093/hmg/ddh140
Reddy, P. H., Tripathi, R., Troung, Q., Tirumala, K., Reddy, T. P., Anekonda, V., et al. (2012). Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 1822, 639–649. doi: 10.1016/j.bbadis.2011.10.011
Reed, T. T., Pierce, W. M. J., Turner, D. M., Markesbery, W. R., and Butterfield, D. A. (2009). Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. J. Cell Mol. Med. 13, 2019–2029. doi: 10.1111/j.1582-4934.2008.00478.x
Reiter, L. T., Potocki, L., Chien, S., Gribskov, M., and Bier, E. (2001). A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 11, 1114–1125. doi: 10.1101/gr.169101
Riascos, D., De Leon, D., Baker-Nigh, A., Nicholas, A., Yukhananov, R., Bu, J., et al. (2011). Age-related loss of calcium buffering and selective neuronal vulnerability in Alzheimer’s disease. Acta Neuropathol. 122, 565–576. doi: 10.1007/s00401-011-0865-4
Rossmann, M. P., Dubois, S. M., Agarwal, S., and Zon, L. I. (2021). Mitochondrial function in development and disease. Dis. Model Mech. 14:dmm048912. doi: 10.1242/dmm.048912
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625. doi: 10.1038/nrm3440
Rubin, G. M., Yandell, M. D., Wortman, J. R., Gabor, M. G. L., Nelson, C. R., Hariharan, I. K., et al. (2000). Comparative genomics of the eukaryotes. Science 287, 2204–2215. doi: 10.1126/science.287.5461.2204
Rui, Y., and Zheng, J. Q. (2016). Amyloid β oligomers elicit mitochondrial transport defects and fragmentation in a time-dependent and pathway-specific manner. Mol. Brain 9:79. doi: 10.1186/s13041-016-0261-z
Sakamuru, S., Li, X., Attene-Ramos, M. S., Huang, R., Lu, J., Shou, L., et al. (2012). Application of a homogenous membrane potential assay to assess mitochondrial function. Physiol. Genomics 44, 495–503. doi: 10.1152/physiolgenomics.00161.2011
Sanz-Blasco, S., Valero, R. A., Rodríguez-Crespo, I., Villalobos, C., and Núñez, L. (2008). Mitochondrial Ca2+ overload underlies Aβ oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 3:e2718. doi: 10.1371/journal.pone.0002718
Schmidt, V., Sporbert, A., Rohe, M., Reimer, T., Rehm, A., Andersen, O. M., et al. (2007). SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J. Biol. Chem. 282, 32956–32964. doi: 10.1074/jbc.M705073200
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089
Schon, E. A., and Area-Gomez, E. (2013). Mitochondria-associated ER membranes in Alzheimer disease. Mol. Cell Neurosci. 55, 26–36. doi: 10.1016/j.mcn.2012.07.011
Sen, A., and Cox, R. T. (2017). “Chapter one–Fly models of human diseases: Drosophila as a model for understanding human mitochondrial mutations and disease,” in Fly models of human diseases, ed. L. Pick (Cambridge, MA: Academic Press), 1–27. doi: 10.1016/bs.ctdb.2016.07.001
Sergeant, N., Delacourte, A., and Buée, L. (2005). Tau protein as a differential biomarker of tauopathies. Biochim. Biophys. Acta 1739, 179–197. doi: 10.1016/j.bbadis.2004.06.020
Shahpasand, K., Uemura, I., Saito, T., Asano, T., Hata, K., Shibata, K., et al. (2012). Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J. Neurosci. 32, 2430–2441. doi: 10.1523/JNEUROSCI.5927-11.2012
Shan, X., Tashiro, H., and Lin, C. G. (2003). The identification and characterization of oxidized RNAs in Alzheimer’s disease. J. Neurosci. 23, 4913–4921. doi: 10.1523/JNEUROSCI.23-12-04913.2003
Sharma, A., and Hasan, G. (2020). Modulation of flight and feeding behaviours requires presynaptic IP3 RS in dopaminergic neurons. eLife 9:e62297. doi: 10.7554/eLife.62297
Sheng, Z. H., and Cai, Q. (2012). Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Shirwany, N. A., Payette, D., Xie, J., and Guo, Q. (2007). The amyloid beta ion channel hypothesis of Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 3, 597–612.
Singh, S. K., Srivastav, S., Yadav, A. K., Srikrishna, S., and Perry, G. (2015). Overview of 900 Alzheimer’s disease and some therapeutic approaches targeting Aβ by using several synthetic 901 and herbal compounds. Oxid. Med. Cell Longev. 2016:7361613. doi: 10.1155/2016/7361613
Smith, G. A., Lin, T. H., Sheehan, A. E., Van der Goes van Naters, W., Neukomm, L. J., Graves, H. K., et al. (2019). Glutathione S-transferase regulates mitochondrial populations in axons through increased glutathione article glutathione S-transferase regulates mitochondrial populations in axons through increased glutathione oxidation. Neuron 103, 52–65.e6. doi: 10.1016/j.neuron.2019.04.017
Smith, M. A., Richey Harris, P. L., Sayre, L. M., Beckman, J. S., and Perry, G. (1997). Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J. Neurosci. 17, 2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997
Song, Z., Ghochani, M., McCaffery, J. M., Frey, T. G., and Chan, D. C. (2009). Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell. 20, 3525–3532. doi: 10.1091/mbc.e09-03-0252
Sorrentino, V., Romani, M., Mouchiroud, L., Beck, J. S., Zhang, H., D’Amico, D., et al. (2017). Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552, 187–193. doi: 10.1038/nature25143
Sperling, R. A., Mormino, E. C., Schultz, A. P., Betensky, R. A., Papp, K. V., Amariglio, R. E., et al. (2019). The impact of amyloid-beta and tau on prospective cognitive decline in older individuals. Ann. Neurol. 85, 181–193. doi: 10.1002/ana.25395
Stokin, G. B., Lillo, C., Falzone, T. L., Brusch, R. G., Rockenstein, E., Mount, S. L., et al. (2005). Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307, 1282–1288. doi: 10.1126/science.1105681
Stoothoff, W., Jones, P. B., Spires-Jones, T. L., Joyner, D., Chhabra, E., Bercury, K., et al. (2009). Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J. Neurochem. 111, 417–427. doi: 10.1111/j.1471-4159.2009.06316.x
Stowers, R. S., Megeath, L. J., Gorska-Andrzejak, J., Meinertzhagen, I. A., and Schwarz, T. L. (2002). Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36, 1063–1077. doi: 10.1016/s0896-6273(02)01094-2
Swerdlow, R. H. (2018). Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimers Dis. 62, 1403–1416. doi: 10.3233/JAD-170585
Swerdlow, R. H. (2023). The Alzheimer’s disease mitochondrial cascade hypothesis: A current overview. J. Alzheimers Dis. 92, 751–768. doi: 10.3233/JAD-221286
Swerdlow, R. H., and Khan, S. M. (2004). A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 63, 8–20. doi: 10.1016/j.mehy.2003.12.045
Szabo, L., Eckert, A., and Grimm, A. (2020). Insights into disease-associated tau impact on mitochondria. Int. J. Mol. Sci. 21:6344. doi: 10.3390/ijms21176344
Taanman, J. W. (1999). The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1410, 103–123. doi: 10.1016/s0005-2728(98)00161-3
Tavares, I. A., Touma, D., Lynham, S., Troakes, C., Schober, M., Causevic, M., et al. (2013). Prostate-derived sterile 20-like kinases (PSKs/TAOKs) phosphorylate tau protein and are activated in tangle-bearing neurons in Alzheimer disease. J. Biol. Chem. 288, 15418–15429. doi: 10.1074/jbc.M112.448183
Tiwari, S., Atluri, V., Kaushik, A., Yndart, A., and Nair, M. (2019). Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 14, 5541–5554. doi: 10.2147/IJN.S200490
Tohgi, H., Abe, T., Yamazaki, K., Murata, T., Ishizaki, E., and Isobe, C. (1999). Alterations of 3-nitrotyrosine concentration in the cerebrospinal fluid during aging and in patients with Alzheimer’s disease. Neurosci. Lett. 269, 52–54. doi: 10.1016/s0304-3940(99)00406-1
Torrisi, S. A., Geraci, F., Tropea, M. R., Grasso, M., Caruso, G., Fidilio, A., et al. (2019). Fluoxetine and vortioxetine reverse depressive-like phenotype and memory deficits induced by Aβ1-42 oligomers in mice: A key role of transforming growth factor-β1. Front. Pharmacol. 10:693. doi: 10.3389/fphar.2019.00693
Tsuyama, T., Kishikawa, J., Han, Y. W., Harada, Y., Tsubouchi, A., Noji, H., et al. (2013). In vivo fluorescent adenosine 5’-triphosphate (ATP) imaging of Drosophila melanogaster and Caenorhabditis elegans by using a genetically encoded fluorescent ATP biosensor optimized for low temperatures. Anal. Chem. 85, 7889–7896. doi: 10.1021/ac4015325
Ugur, B., Chen, K., and Bellen, H. J. (2016). Drosophila tools and assays for the study of human diseases. Dis. Model Mech. 9, 235–244. doi: 10.1242/dmm.023762
van Eldik, L. J., Carrillo, M. C., Cole, P. E., Feuerbach, D., Greenberg, B. D., Hendrix, J. A., et al. (2016). The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimers Dement. 2, 99–109. doi: 10.1016/j.trci.2016.05.001
Venkateshappa, C., Harish, G., Mahadevan, A., Srinivas Bharath, M. M., and Shankar, S. K. (2012). Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: Implications for neurodegeneration in Alzheimer’s disease. Neurochem. Res. 37, 1601–1614. doi: 10.1007/s11064-012-0755-8
Verstreken, P., Ly, C. V., Venken, K. J., Koh, T. W., Zhou, Y., and Bellen, H. J. (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365–378. doi: 10.1016/j.neuron.2005.06.018
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., de, V. R., Kim, J., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383. doi: 10.1073/pnas.0911187107
Vos, M., Esposito, G., Edirisinghe, J. N., Vilain, S., Haddad, D. M., Slabbaert, J. R., et al. (2012). Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science 336, 1306–1310. doi: 10.1126/science.1218632
Vossel, K. A., Xu, J. C., Fomenko, V., Miyamoto, T., Suberbielle, E., Knox, J. A., et al. (2015). Tau reduction prevents Aβ-induced axonal transport deficits by blocking activation of GSK3β. J. Cell Biol. 209, 419–433. doi: 10.1083/jcb.201407065
Wang, J., Xiong, S., Xie, C., Markesbery, W. R., and Lovell, M. A. (2005). Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 93, 953–962. doi: 10.1111/j.1471-4159.2005.03053.x
Wang, P., Deng, J., Dong, J., Liu, J., Bigio, E. H., Mesulam, M., et al. (2019). TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 15:e1007947. doi: 10.1371/journal.pgen.1007947
Wang, W., Yin, J., Ma, X., Zhao, F., Siedlak, S. L., Wang, Z., et al. (2017). Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 26, 4118–4131. doi: 10.1093/hmg/ddx299
Wang, W., Zhao, F., Ma, X., Perry, G., and Zhu, X. (2020). Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegen. 15:30. doi: 10.1186/s13024-020-00376-6
Wang, X., and Davis, R. L. (2021). Early mitochondrial fragmentation and dysfunction in a Drosophila model for Alzheimer’s disease. Mol. Neurobiol. 58, 143–155. doi: 10.1007/s12035-020-02107-w
Wang, X., Perry, G., Smith, M. A., and Zhu, X. (2010). Amyloid-β-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegen. Dis. 7, 56–59. doi: 10.1159/000283484
Wang, X., Su, B., Fujioka, H., and Zhu, X. (2008a). Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 173, 470–482. doi: 10.2353/ajpath.2008.071208
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009
Wang, X., Su, B., Siedlak, S. L., Moreira, P. I., Fujioka, H., Wang, Y., et al. (2008b). Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 19318–19323. doi: 10.1073/pnas.0804871105
Wang, X., Wang, W., Li, L., Perry, G., Lee, H. G., and Zhu, X. (2014). Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 1842, 1240–1247. doi: 10.1016/j.bbadis.2013.10.015
Wee, M., Chegini, F., Power, J. H. T., and Majd, S. (2018). Tau positive neurons show marked mitochondrial loss and nuclear degradation in Alzheimer’s disease. Curr. Alzheimer Res. 15, 928–937. doi: 10.2174/1567205015666180613115644
Weingarten, M. D., Lockwood, A. H., Hwo, S. Y., and Kirschner, M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Wenger, J., Klinglmayr, E., Fröhlich, C., Eibl, C., Gimeno, A., Hessenberger, M., et al. (2013). Functional mapping of human dynamin-1-like GTPase domain based on x-ray structure analyses. PLoS One 8:e71835. doi: 10.1371/journal.pone.0071835
Wilson, E. L., and Metzakopian, E. (2021). ER-mitochondria contact sites in neurodegeneration: Genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ. 28, 1804–1821. doi: 10.1038/s41418-020-00705-8
Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J., Hutton, M., et al. (2001). Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 293, 711–714. doi: 10.1126/science.1062382
Wong, C. O., Karagas, N. E., Jung, J., Wang, Q., Rousseau, M. A., Chao, Y., et al. (2021). Regulation of longevity by depolarization-induced activation of PLC- β-IP3R signaling in neurons. Proc. Natl. Acad. Sci. U.S.A. 118:e2004253118. doi: 10.1073/pnas.2004253118
Wong, K. Y., Roy, J., Fung, M. L., Heng, B. C., Zhang, C., and Lim, L. W. (2020). Relationships between mitochondrial dysfunction and neurotransmission failure in Alzheimer’s disease. Aging Dis. 11, 1291–1316. doi: 10.14336/AD.2019.1125
Xu, H., DeLuca, S. Z., and O’Farrell, P. H. (2008). Manipulating the metazoan mitochondrial genome with targeted restriction enzymes. Science 321, 575–577. doi: 10.1126/science.1160226
Yamamoto, T., and Hirano, A. (1985). Nucleus raphe dorsalis in Alzheimer’s disease: Neurofibrillary tangles and loss of large neurons. Ann Neurol. 17, 573–577. doi: 10.1002/ana.410170608
Yana, M. H., Wang, X., and Zhu, X. (2013). Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 62, 90–101. doi: 10.1016/j.freeradbiomed.2012.11.014
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 106, 14670–14675. doi: 10.1073/pnas.0903563106
Yarosh, W., Monserrate, J., Tong, J. J., Tse, S., Le, P. K., Nguyen, K., et al. (2008). The molecular mechanisms of OPA1-mediated optic atrophy in Drosophila model and prospects for antioxidant treatment. PLoS Genet. 4:e6. doi: 10.1371/journal.pgen.0040006
Ye, X., Sun, X., Starovoytov, V., and Cai, Q. (2015). Parkin mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 24, 2938–2951. doi: 10.1093/hmg/ddv056
Yeh, P. A., Liu, Y. H., Chu, W. C., Liu, J. Y., and Sun, Y. H. (2018). Glial expression of disease-associated poly-glutamine proteins impairs the blood-brain barrier in Drosophila. Hum. Mol. Genet. 27, 2546–2562. doi: 10.1093/hmg/ddy160
Youle, R. J., and van der Bliek, A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. doi: 10.1126/science.1219855
Yu, T., Robotham, J. L., and Yoon, Y. (2006). Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. 103, 2653–2658. doi: 10.1073/pnas.0511154103
Zhang, H., Wei, W., Zhao, M., Ma, L., Jiang, X., Pei, H., et al. (2021). Interaction between A β and tau in the pathogenesis of Alzheimer’s disease. Int. J. Biol. Sci. 17, 2181–2192. doi: 10.7150/ijbs.57078
Zhang, J., Wang, Q., Wang, M., Jiang, M., Wang, Y., Sun, Y., et al. (2016). GASZ and mitofusin-mediated mitochondrial functions are crucial for spermatogenesis. EMBO Rep. 17, 220–234. doi: 10.15252/embr.201540846
Zhang, M. Y., Lear, B. C., and Allada, R. (2022). The microtubule-associated protein tau suppresses the axonal distribution of PDF neuropeptide and mitochondria in circadian clock neurons. Hum. Mol. Genet. 31, 1141–1150. doi: 10.1093/hmg/ddab303
Zhang, X., Alshakhshir, N., Zhao, L., Wood, L., and Kaplin, A. I. (2021). Glycolytic metabolism, brain resilience, and Alzheimer’s disease. Front. Neurosci. 15:662242. doi: 10.3389/fnins.2021.662242
Zhao, X. L., Wang, W. A., Tan, J. X., Huang, J. K., Zhang, X., Zhang, B. Z., et al. (2010). Expression of β-amyloid induced age-dependent presynaptic and axonal changes in Drosophila. J. Neurosci. 30, 1512–1522. doi: 10.1523/JNEUROSCI.3699-09.2010
Zhao, Y., Hu, Q., Cheng, F., Su, N., Wang, A., Zou, Y., et al. (2015). SoNar, a highly responsive NAD+/NADH sensor, allows high- throughput metabolic screening of anti-tumor agents. Cell Metab. 21, 777–789. doi: 10.1016/j.cmet.2015.04.009
Zhou, L., Wang, W., Hoppel, C., Liu, J., and Zhu, X. (2017). Parkinson’s disease associated pathogenic VPS35 mutation causes complex I deficits. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 2791–2795. doi: 10.1016/j.bbadis.2017.07.032
Zhou, Y., Carmona, S., Muhammad, A. K. M. G., Bell, S., Landeros, J., Vazquez, M., et al. (2019). Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Invest. 129, 1756–1771. doi: 10.1172/JCI124194
Zhu, X., Perry, G., Smith, M. A., and Wang, X. (2013). Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis. 33(Suppl. 1), S253–S262. doi: 10.3233/JAD-2012-129005
Keywords: Alzheimer’s disease, amyloid beta, Drosophila, mitochondria, neurodegeneration, tau, mitophagy, Drp1
Citation: Varte V, Munkelwitz JW and Rincon-Limas DE (2023) Insights from Drosophila on Aβ- and tau-induced mitochondrial dysfunction: mechanisms and tools. Front. Neurosci. 17:1184080. doi: 10.3389/fnins.2023.1184080
Received: 10 March 2023; Accepted: 31 March 2023;
Published: 17 April 2023.
Edited by:
Udai Pandey, University of Pittsburgh Medical Center, United StatesReviewed by:
Meghana Tare, Birla Institute of Technology and Science, IndiaMadhuri Kango-Singh, University of Dayton, United States
Copyright © 2023 Varte, Munkelwitz and Rincon-Limas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diego E. Rincon-Limas, ZGllZ28ucmluY29uQG5ldXJvbG9neS51ZmwuZWR1