 Yanming Chen1†
Yanming Chen1† Xuchen Dong2†
Xuchen Dong2† Ye Wang3
Ye Wang3 Haijun Lv4
Haijun Lv4 Nan Chen5Zhongyong Wang1Si Chen5Ping Chen5
Nan Chen5Zhongyong Wang1Si Chen5Ping Chen5 Sheng Xiao6Jizong Zhao1,7,8
Sheng Xiao6Jizong Zhao1,7,8 Jun Dong1*
Jun Dong1*- 1Department of Neurosurgery, Second Affiliated Hospital of Soochow University, Suzhou, China
- 2Department of Neurosurgery, Huashan Hospital, Fudan University, Shanghai, China
- 3Health Management Center, Second Affiliated Hospital of Soochow University, Suzhou, China
- 4Department of Pathology, Second Affiliated Hospital of Soochow University, Suzhou, China
- 5Suzhou Sano Precision Medicine Ltd., Suzhou, China
- 6Department of Pathology, Brigham and Women’s Hospital, Boston, MA, United States
- 7Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
- 8China National Clinical Research Center for Neurological Diseases, Beijing, China
Cerebral cavernous malformations (CCMs) are common vascular anomaly diseases in the central nervous system associated with seizures, cerebral microbleeds, or asymptomatic mostly. CCMs can be classified as sporadic or familial, with familial cerebral cavernous malformations (fCCMs) being the autosomal dominant manner with incomplete penetrance. Germline mutations of KRIT1, CCM2, and PDCD10 are associated with the pathogenesis of fCCMs. Till now, little is known about the fCCMs mutation spectrum in the Han Chinese population. In this study, we enrolled a large, aggregated family, 11/26 of the family members were diagnosed with CCMs by pathological or neuroradiological examination, with a high percentage (5/9) of focal spinal cord involvement. Genomic DNA sequencing verified a novel duplication mutation (c.1119dupT, p.L374Sfs*9) in exon 9 of the Krev interaction trapped 1 (KRIT1) gene. The mutation causes a frameshift and is predicted to generate a truncated KRIT1/CCM1 protein of 381 amino acids. All our findings confirm that c.1119dupT mutation of KRIT1 is associated with fCCMs, which enriched the CCM genes’ mutational spectrum in the Chinese population and will be beneficial for deep insight into the pathogenesis of Chinese fCCMs. Additionally, with a retrospective study, we analyzed the molecular genetic features of Chinese fCCMs, most of the Chinese fCCMs variants are in the KRIT1 gene, and all these variants result in the functional deletion or insufficiency of the C-terminal FERM domain of the KRIT1 protein.
Introduction
Cerebral cavernous malformations (CCMs) are vascular anomalies histologically characterized by tightly packed and abnormal capillary cavities collections, mainly located in the central nervous system (CNS) (Zafar et al., 2019). The clinical presentation of CCMs may be various, including seizures, recurrence headaches, focal neurological deficits, and hemorrhage (Ricci et al., 2021). However, many patients with CCMs are asymptomatic; hence, the real-world pre-valence of CCMs may be hard to accurately evaluate (Moore et al., 2014). CCMs most often occur sporadically or in familial form. Compared to sporadic CCMs usually present with CNS single lesions, fCCMs are characterized by CNS multiple lesions and autosomal dominant inheritance with incomplete penetrance (Riolo et al., 2021).
The pathogenic alterations of fCCMs have been mapped into three loci: KRIT1/CCM1 (OMIM 604214) (Dubovsky et al., 1995; Gunel et al., 1995), CCM2 (OMIM 607929) (Craig et al., 1998; Liquori et al., 2003), and PDCD10/CCM3 (OMIM 609118) (Bergametti et al., 2005), and are located on chromosomes 7q, 7p, and 3q, respectively. The incidence of fCCMs accounts for about 10−15% of all CCM cases (Zhao et al., 2011), and its incidence rate displays obvious geographical characteristics, with fCCMs incidence rates as high as 50% in Hispanic Americans and 10−40% in Caucasian populations (Rigamonti et al., 1988; Labauge et al., 2007). Epidemiologically, the incidence of fCCMs in Han Chinese has not been reported, systematic studies on Han Chinese fCCMs families are extremely rare, and clinically definite diagnosis of Han Chinese fCCMs is also rare (Yang et al., 2017; Liu et al., 2022). However, some Chinese-specific CCMs gene mutation loci have been found sporadically (Chen et al., 2002; Yang et al., 2017, 2018; Jih et al., 2018; Yang L. et al., 2020). Therefore, it is meaningful to uncover the genetic phenotype of Han Chinese fCCMs by continuously enriching the fCCMs’ genomic alteration spectrum in the Chinese population.
In this study, we report the clinical presentations, neuroradiological features, and genomic defects of a large Chinese fCCMs family. It’s beneficial for us to further characterize the clinical features of CCM patients with KRIT1 mutations and expand the mutational spectrum of this gene. Furthermore, a thorough retrospective study of molecular genetic features in Chinese fCCMs was conducted to gain a deeper insight into the etiology and pathogenic characteristics of Chinese fCCMs.
Materials and methods
Subjects
This study was approved by the ethics committee of the Second Affiliated Hospital of Soochow University. The family includes 26 members, 11/26 family members were diagnosed with the CNS CCMs, and two of them have departed. The clinical features of the 11 fCCMs patients have been shown in Table 1. The index patient (proband; II-1) is a 69-year-old male patient, suffering from progressive bilateral lower limbs dystonia. Over the next 6 years, the members of III-2, III-3, III-5, and IV-6 sequentially received surgeries and were confirmed to suffer the CNS CCMs. A retrospective study revealed that the family member of II-11 received surgery to remove an intracranial lesion due to seizures when he was young. The post-operative pathological diagnosis was “vascular malformation.” Unfortunately, no pathological or imaging sources were available, and without offspring. The family members of II-3, III-4, III-6, and IV-3 were all neuroradiological confirmations of CNS CCMs and were clinically silent.
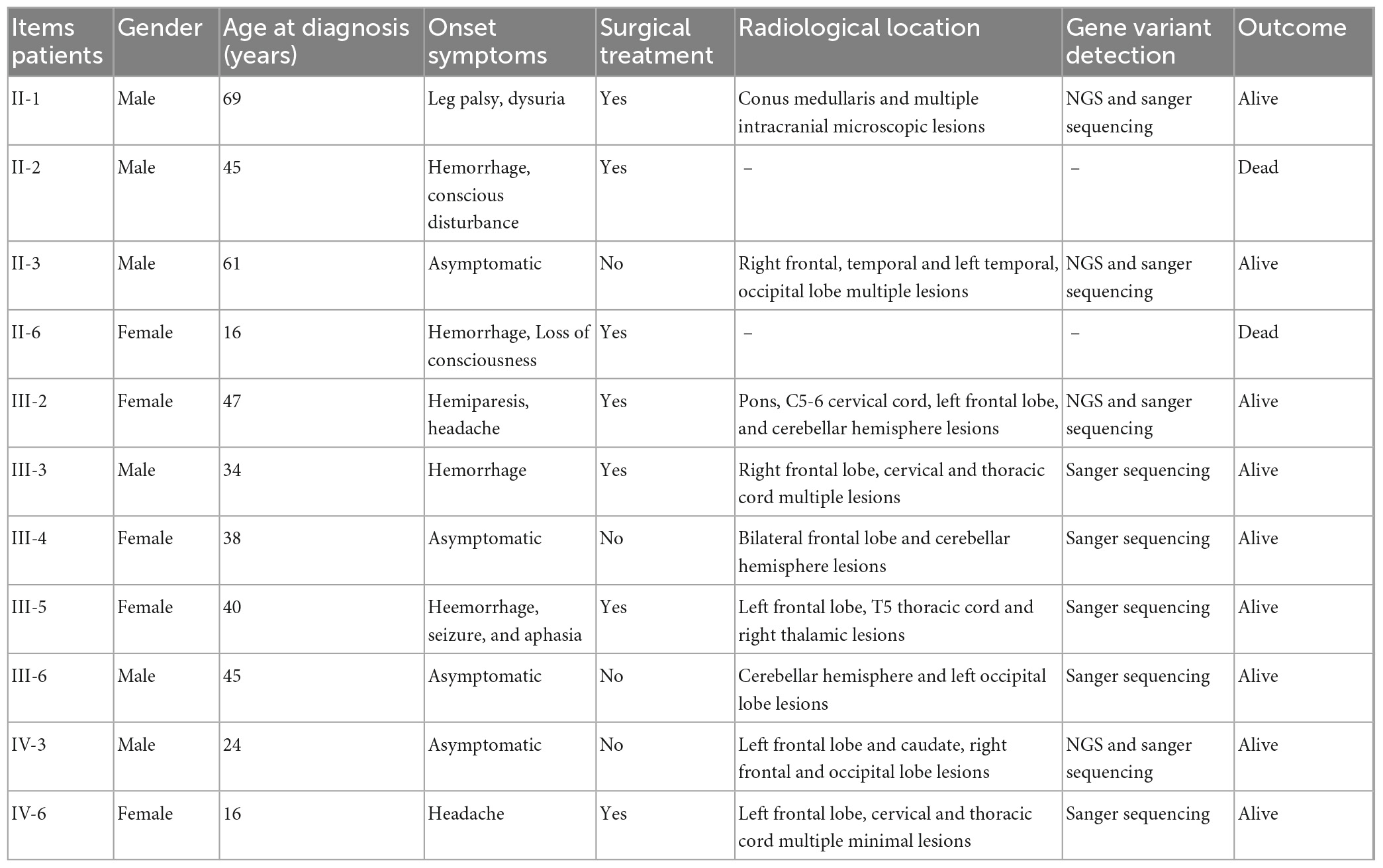
Table 1. Clinical features and genetic data of the fCCMs members.
Whole-exome sequencing and causative variants filtering
We utilized whole-exome sequencing (WES) to detect potential pathogenic germline mutations within the fCCMs family members. Four fCCMs patient members (II-1, III-2, III-5, and IV-3) and two fCCMs health members (III-1 and III-7) were included in the WES study. The genomic DNA was extracted from each family member’s peripheral blood. DNA libraries were constructed and sequenced to a target depth of 100 × on the Illumina X-TEN platform, as previously reported (Han et al., 2020). Reads were aligned to the reference genome (hg19) using BWA-MEM. Sequencing results for single-nucleotide variations (SNVs) and insertion/deletion (Indels) were analyzed by GATK HaplotypeCaller.
The filtering process of candidate pathogenic variants: (1) Remove the variants with minor allele frequency (MAF) higher than 1% in the normal population database (gnomAD and 1000 Genomes); (2) The variants that occur in healthy Chinese and unaffected members of the family were removed; (3) Preference was given to variants that occur in both affected individuals and that are associated with cavernous malformations in the human genetic variation database ClinVar. Finally, manual identification of possible pathogenic variants, especially in the pathogenic genes KRIT1, CCM2, and PDCD10.
Polymerase chain reaction (PCR) and sanger sequencing
PCR was performed with primers specific to KRIT1 (KRIT1-F: 5′-CCACTGAACTGTACGCCTAAA-3′, KRIT1-R: 5′-AAGTTGAGGCCACTCGCATAT-3′). 10 ng gDNA were used, and the PCR conditions were 95°C 5 min for 1 cycle followed by 35 cycles of 95°C 30 s, 60°C 30 s, and 72°C 1 min. A 424 bp PCR product was generated, which contained the candidate pathogenic mutation of KRIT1. The PCR product was analyzed by gel electrophoresis and directly Sanger sequenced.
Three-dimensional structure analysis of KRIT1 protein
Currently, the most commonly used method for unknown protein structure prediction is based on the known protein structure. However, this method largely relies on homologous templates, which leads to the inability to predict protein structures without homologous templates. In this study, the Alphafold2 algorithm is used, which is based on a deep learning algorithm and can predict protein structures with atomic precision. It can still be predicted in the absence of homologous templates. As previously reported (Liang et al., 2022), using the default parameters of Alphafold2 to predict the structure of KRIT1 wild-type and mutant proteins, the optimal model was selected and obtained the protein data bank (PDB) files of the three-dimensional structure of KRIT1 protein. Meanwhile, to visualize the mutant and wild-type KRIT1 protein structures, the predicted PDB files were imported into Swiss-PDB Viewer (Version 4.0.4) for protein structure drawing.
Results
Clinical features
The pedigree was established with the help of family members (Figure 1). The index patient (proband; II-1) is a 69-year-old male patient. Spinal magnetic resonance imaging (MRI) of the T1-weighted imaging revealed a 1.0 cm diameter inhomogeneous hyperintense lesion in the conus medullaris (Figure 2A), and multiple intracranial mixed signal lesions of variable sizes were observed in the T2-weighted brain MR imaging (Figure 2A). Of the 11 fCCMs patient members, seven individuals were symptomatic, and four individuals were asymptomatic. The clinical presentation included paralysis (n = 2), headache (n = 2), loss of consciousness (n = 2), seizure (n = 1), and hemorrhage (n = 4) (Table 2). Seven patients underwent surgical treatment, and post-operative pathology verified the diagnosis of CCMs (Figures 2B-E). Four individuals neuroradiologically exhibited the CNS CCMs but were clinically asymptomatic (Figure 2F). Multiple lesions were found in 9/9 affected familial individuals (Figure 2). In this family of patients with CCMs, a high percentage (5/9) of patients had lesions involving the spinal cord (Table 2).
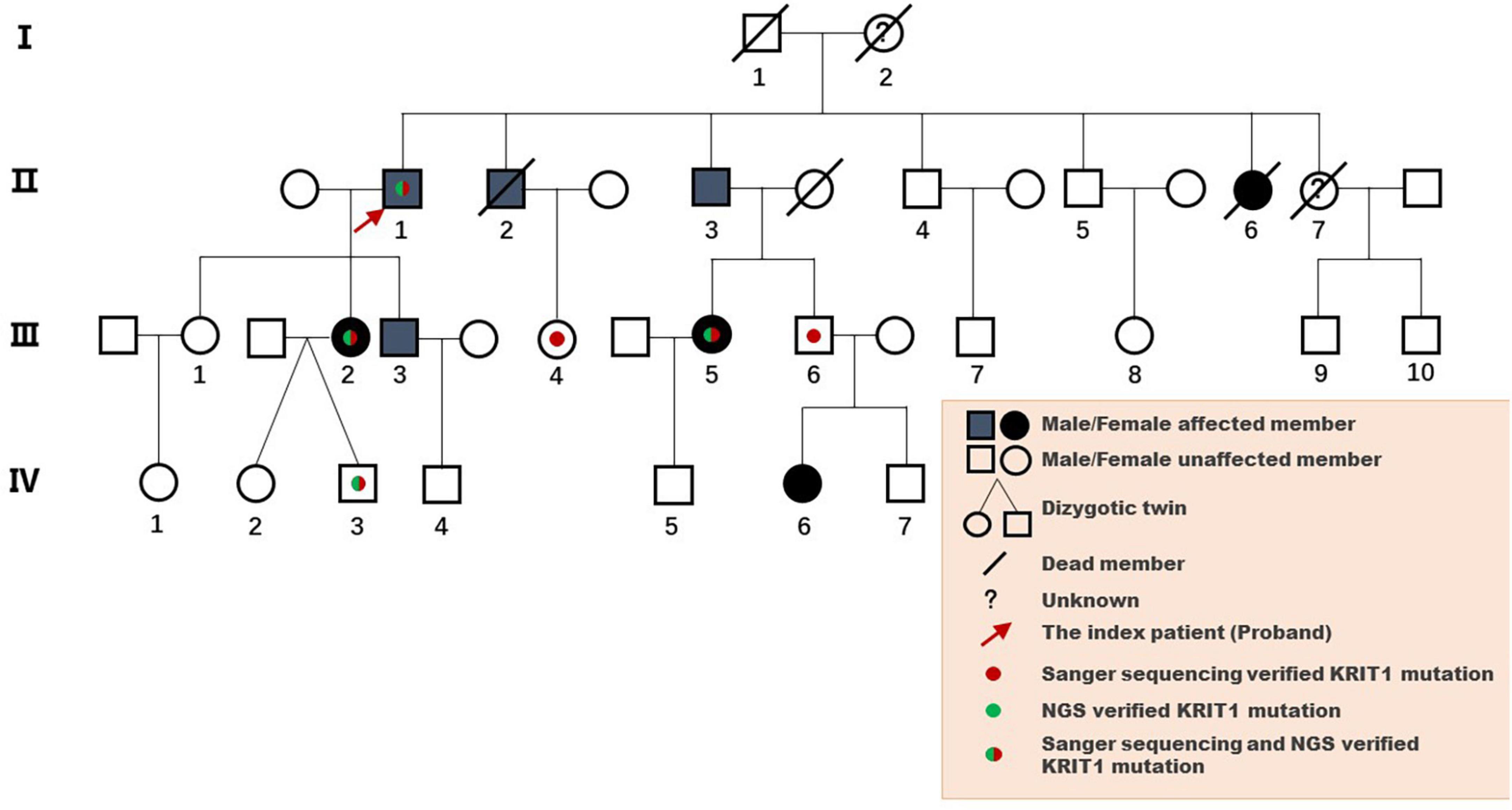
Figure 1. Pedigree diagram of the Chinese family with fCCMs. The affected family members were diagnosed with pathology or T2-weighted MRI of the CNS.
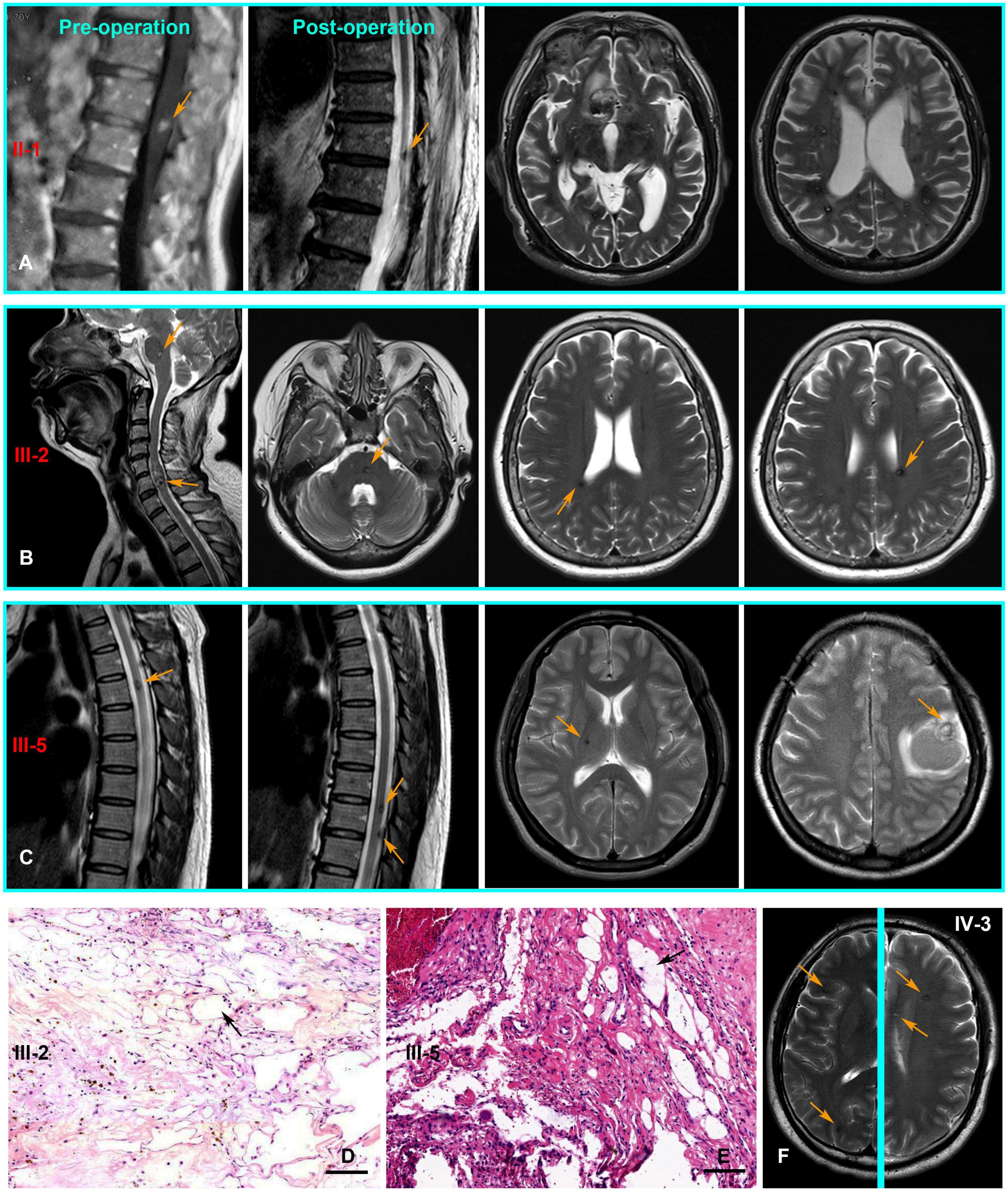
Figure 2. The CNS MRI and histopathology findings of patients with the CCM1 variant. (A) T1-weighted MR imaging of the proband (II-1) detected a lesion in the conus medullaris. Post-operative MR imaging revealed residual microfoci in the spinal cord, and the brain of T2-weighted MR imaging show multiple lesions of variable sizes in hemispheres. (B,C) Cerebral and spinal MR imaging showing there are multiple lesions in the CNS of III-3 and III-5, respectively. (D,E) Pathologically, hematoxylin and eosin (H and E, Scale bar = 100 μm) staining revealed vascular malformation associated with dilated vessels and thin-walled capillaries (black arrows), III-3 and III-5, respectively. (F) Multiple lesions were observed in an asymptomatic familial member (IV-3) with the KRIT1/CCM1 gene variant from T2-weighted MR imaging of different levels (Yellow arrows point to the lesions).
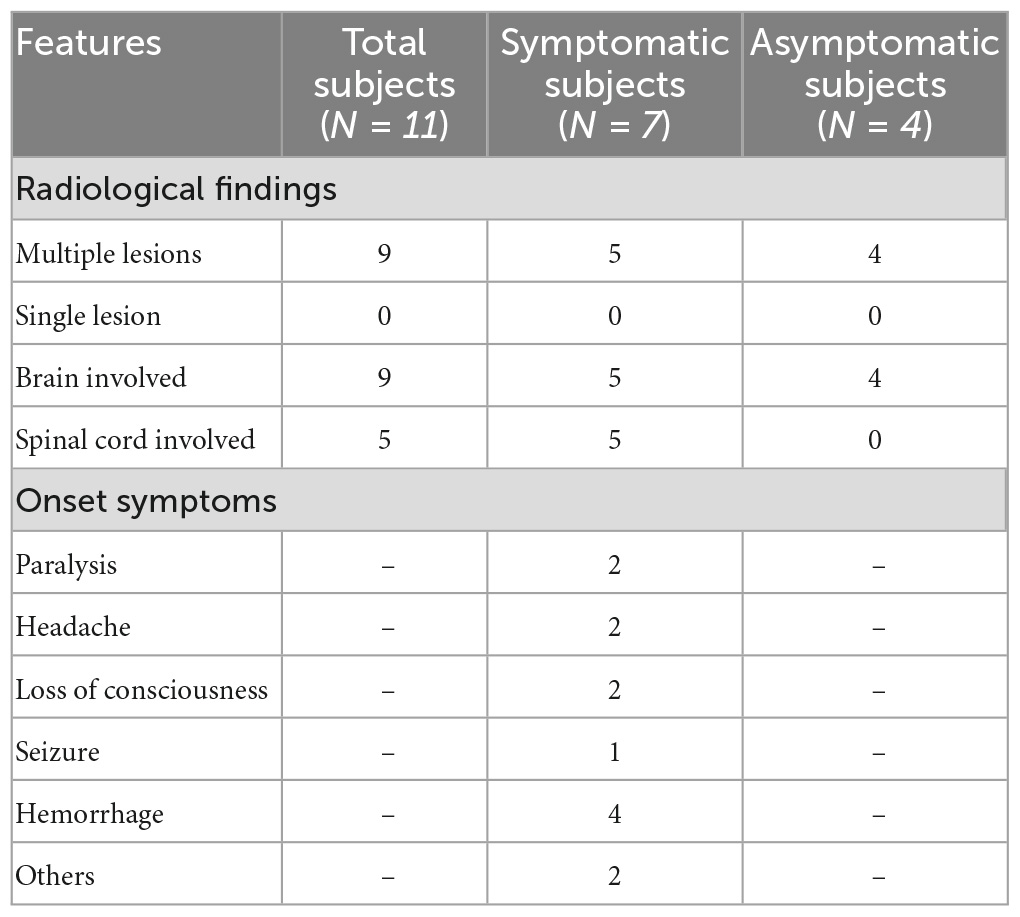
Table 2. Clinical features and radiological findings in the fCCMs patients.
Molecular studies
Analysis of the WES data found that four fCCMs individuals shared a common KRIT1/CCM1 (NM_004912) frameshift mutation (c.1119dupT, p.L374Sfs*9), while the candidate variation was exclusive in the two healthy familial individuals. The subsequent Sanger sequencing analysis of 16 familial individuals further confirmed that the candidate mutation of KRIT1/CCM1 was present in all members of the CCMs patients, while this mutation was unobserved in any of the ten healthy family members (Figure 3). This heterozygous duplication mutation (c.1119dupT) in exon 9 leads to a frameshift and is predicted to cause a premature termination codon to generate a truncated CCM1 protein of 381 amino acids. This mutation (c.1119dupT, p.L374Sfs*9) has never been reported in the following publicly available databases: CCM mutation database,1 NCBI single-nucleotide polymorphism (SNP),2 and the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server.3
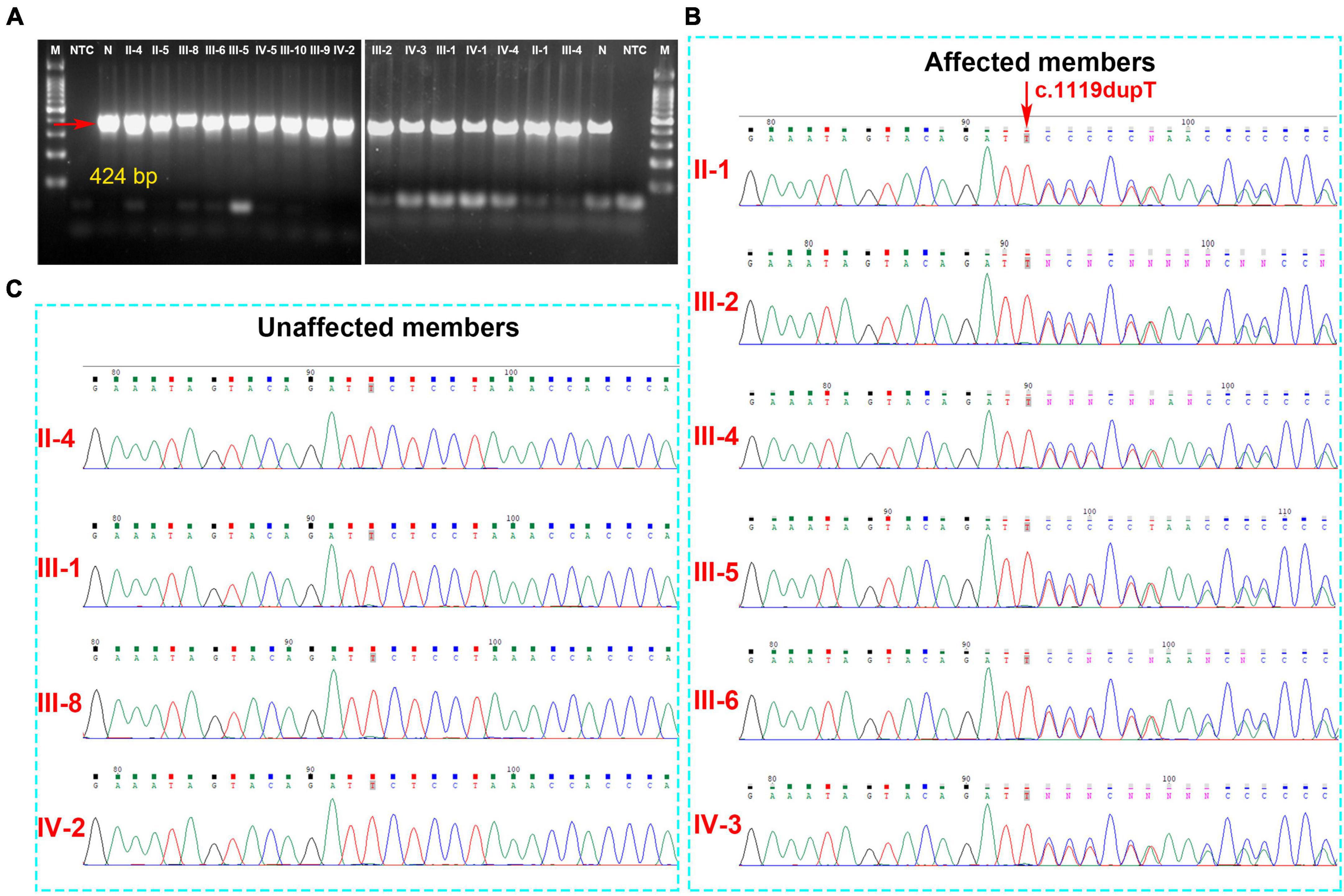
Figure 3. (A) The gel electrophoresis of PCR products shows KRIT1/CCM1 DNA segment containing the candidate mutant sites, and the PCR products were sequenced by the Sanger sequencing method. (B) A novel heterozygous duplication mutation (c.1119dupT, p.L374Sfs*9) of the KRIT1/CCM1 gene was detected by DNA sequencing in the CCM-affected members. The red arrow indicates a duplication of thymine at that site. (C) Representative sequencing results from the healthy family members indicate the absence of the KRIT1/CCM1 gene variant.
An in silico prediction of the three-dimensional structures of wild-type and mutated KRIT1 proteins were performed based on Alphafold2. A structural comparison of the wild-type and mutated KRIT1 proteins indicated that ankyrin (ANK) repeats domains in the C-terminal were partially disrupted (Figure 4).
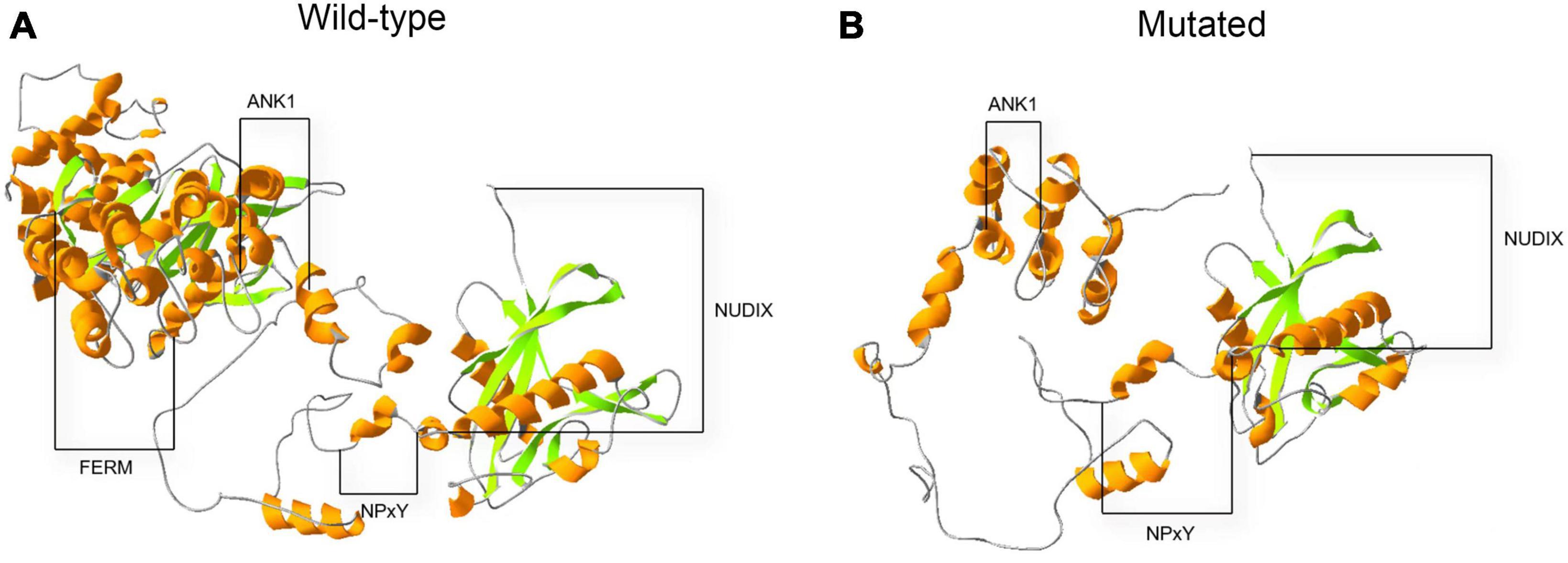
Figure 4. Three-dimensional structure prediction of wild-type and mutated KRIT1 proteins. The image compares native (A) and mutated (B) KRIT1 tertiary protein structures. As indicated, the main structural modifications affect ANK repeats domains at the C-terminal with consequent loss of the FERM domain.
Retrospective study
Till now, little is known referring to genetic alterations associated with fCCMs in the Chinese population. A retrieved result from Web of Science, PubMed, and Wanfang Data Knowledge Service Platform databases since 2000, with the terms familial cerebral cavernous malformations, mutation, and Chinese, articles published in English or Chinese with essential information about the mutations were included. We collected 24 pathogenetic alterations of fCCMs first identified in the Chinese population (Chen et al., 2002; Xu et al., 2003; Mao et al., 2005, 2016; Ji et al., 2006; Zhao et al., 2011; Wang et al., 2013, 2017, 2018; Zhu et al., 2014; Yang et al., 2017; Du et al., 2019; Han et al., 2020; Jiang et al., 2020; Yang L. et al., 2020; Yang Q. et al., 2020; Zhang et al., 2020; Liu et al., 2022). As exhibited in Table 3, the majority (18/24) of Chinese fCCMs pathogenetic alterations are in KRIT1/CCM1 gene. The incidence of CCM2 mutations followed, with only one Chinese fCCM carrying the PDCD10/CCM3 mutation (Figure 5A). Frameshift and Nonsense were the most common mutation types (Figure 5B), which always lead to a premature termination codon and cause to generate a truncated protein. In Chinese fCCMs carrying KRIT1/CCM1 variants, all these variants result in the functional deletion or insufficiency of the C-terminal FERM (a band four-point-one, ezrin, radixin, moesin) domain of the KRIT1 protein (Figure 5C). KRIT1 binds to microtubules, and when activated RAP1 binds to the FERM domain, KRIT1 translocates to adherens junctions and binds β-catenin (Francalanci et al., 2009; Nardella et al., 2018). However, as previously reported (Liu et al., 2011), the depletion of KRIT1-RAP1 interaction caused a deleterious decreased endothelial barrier function. CCM2 can interact with CCM1 via conserved NPxY motifs, and act as a bridge-like role in the CCM complex (Zawistowski et al., 2005; Draheim et al., 2015). Unlike Hispanic-American or Italian fCCMs (Ricci et al., 2021), no variants located at the three NPxY motifs were identified in this cohort of KRIT1/CCM1 variants of Chinese fCCMs.
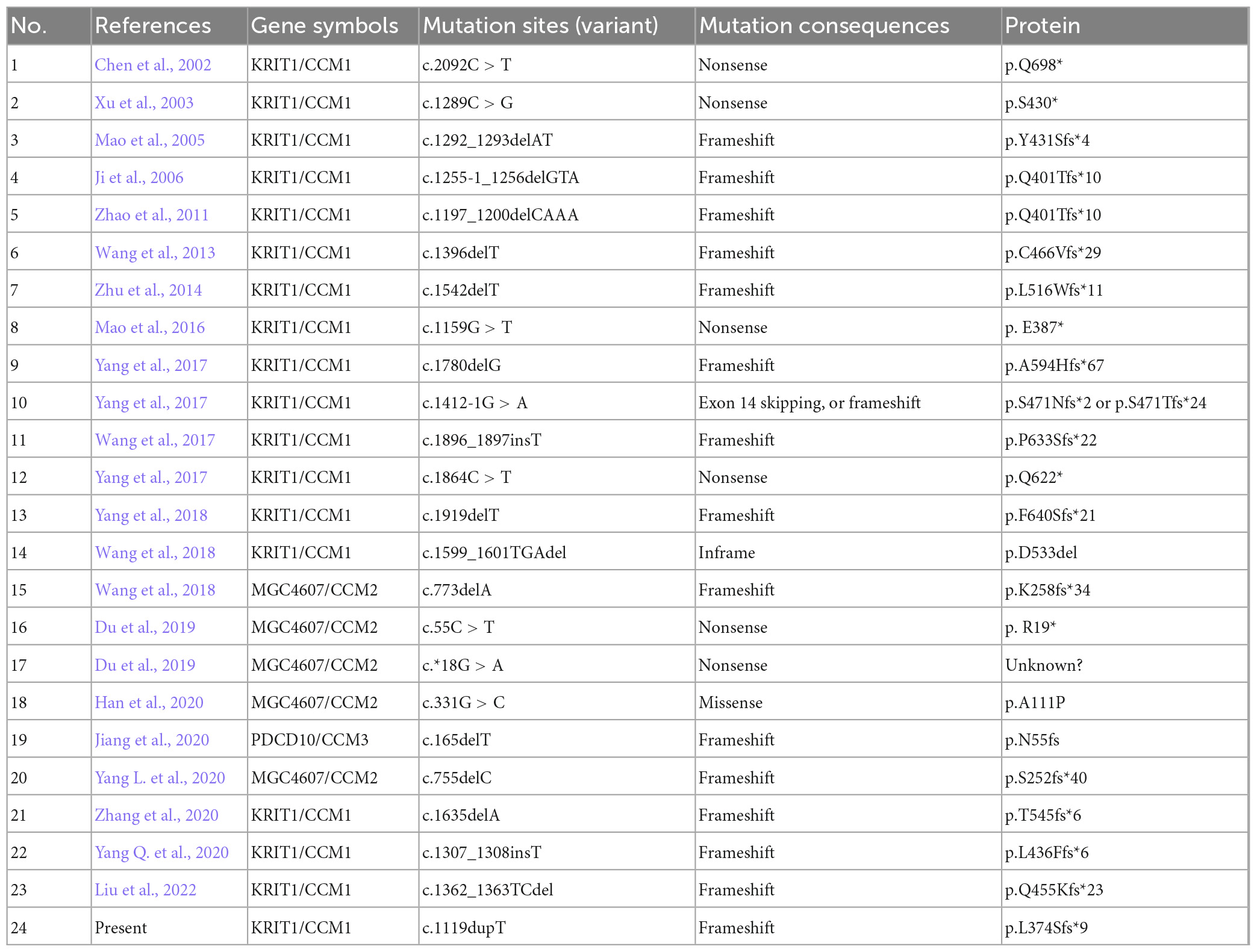
Table 3. Summary of identified pathogenetic alterations of CCMs in the Chinese population.
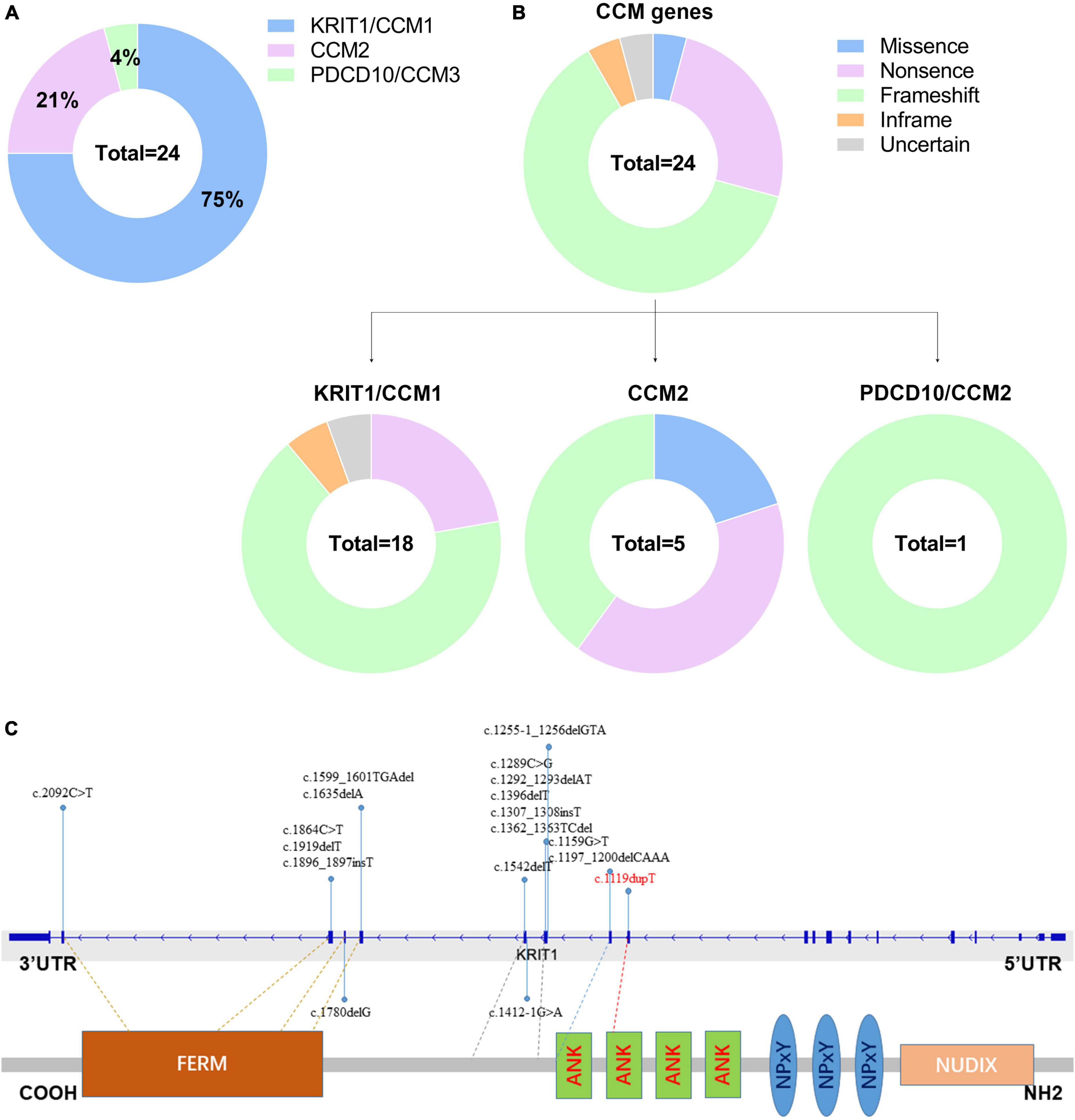
Figure 5. (A) Mutation spectrum of CCM genes in this cohort of Chinese fCCMs patients. (B) Distribution of different types of variants identified by each CCM gene, respectively. (C) Schematic representation of the gene structure and protein domains of KRIT1/CCM1 in the Chinese population. The upper panel represents the genetic structure, and the lower panel represents a corresponding protein product and its specific domain. Each reported KRIT1/CCM1 gene variant is marked in the corresponding genetic and amino acid position, respectively.
Discussion
Since the three pathogenic gene loci (CCM1-3) were mapped, KRIT1/CCM1 mutations were disclosed in more than 40% of familial cases (Yang L. et al., 2020). As the most common pathogenetic mutational gene, more than 250 KRIT1/CCM1 mutations have been identified with a low degree of redundancy (Stenson et al., 2020). Noteworthily, all these mutations lead to premature stop codons that result in truncated proteins. However, pathogenic mutations of fCCMs exhibit remarkable genetic heterogeneity among different races and ethnicities. As a large ethnic population, the incidence of fCCMs is relatively lower among the Chinese population, and novel confirmed pathogenic mutations from the Chinese population are extremely rare. To date, little is known about the genomic alterations associated with fCCMs in the Chinese population. Hence, the identification of this novel CCM1 mutation expanded the mutational spectrum of CCM1 underlining familial CCMs.
In this study, we performed DNA sequencing to analyze a large Chinese family, with years of suffering from CNS cavernous malformations. We identified a novel pathogenic mutation of KRIT1/CCM1 in this family. This novel frameshift mutation of KRIT1 in exon 9 is predicted to cause a premature termination codon to generate a truncated KRIT1 protein of 381 amino acids. The truncated KRIT1 protein is speculated to suffer a severe structural and functional defect, which causes dysregulation of angiogenesis. Intriguingly, a high percentage (5/9) of lesions involving the spinal cord was observed in this family, which was rarely reported in previous studies.
Additionally, to deep insight into the molecular genomic alteration features of the Chinese fCCMs, we compiled the most comprehensive mutational spectrum of Chinese fCCMs to date. Our retrospective study indicated that KRIT1/CCM1 gene mutations were still the most common (18/24) pathogenic variants in the Chinese fCCMs, most of which result in the functional deletion or insufficiency of the C-terminal FERM domain of the KRIT1 protein. PDCD10/CCM3 mutations were extremely rare in this cohort of Chinese fCCMs. Consistent with previous reports, frameshift, and nonsense were the most common mutation types, which always lead to a premature termination codon and cause to generate a truncated protein. However, unlike Hispanic-American or Italian fCCMs, no variants were identified at the three NPxY motifs of KRIT1 protein in this cohort of Chinese fCCMs.
Emerging evidence indicated that fCCMs might exhibit great genetic heterogeneity among different geographic ethnicities. Till now, little is known regarding germline mutations leading to fCCMs in the Chinese population. In this study, we described a novel mutation in KRIT1/CCM1 gene, and retrospectively analyzed the scarcity and sporadic reports of germline mutations of fCCMs in the Chinese population. The population-specific mutational analysis of fCCMs is of a warrant to deep insight into the mutational spectrum of the Chinese population.
Conclusion
We identified a novel germline mutation (c.1119dupT) of KRIT1/CCM1 from a Chinese fCCMs family, which enriches the fCCMs pathogenic mutational spectrum in the Chinese population. The family member carrying this novel CCM1 variant suffered approximately 100% neuroradiological penetrance, which will be beneficial for prenatal guidance and genetic counseling. Moreover, a thorough retrospective study of molecular genetic features in Chinese fCCMs facilitates us to gain a deeper insight into the etiology and pathogenic characteristics of Chinese fCCMs.
Data availability statement
The original contributions presented in this study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/sra/PRJNA958149.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Second Affiliated Hospital of Soochow University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YC designed this study. XD and HL were in charge of pathological diagnosis. YW, ZW, and PC supported and analyzed clinical data. NC and SC supported the genomic analysis. SX, JZ, and JD guided this work and reviewed the manuscript. All authors approved the final version of the submitted manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (81602183), the Youth Medical Talent Foundation of Jiangsu (QNRC2016870), and the Project of Suzhou Health Talents (2020090).
Conflict of interest
NC, SC, and PC were employed by Suzhou Sano Precision Medicine Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
- ^ http://www.angiomaalliance.org
- ^ http://www.ncbi.nlm.nih.gov/snp
- ^ http://evs.gs.washington.edu/EVS/
References
Bergametti, F., Denier, C., Labauge, P., Arnoult, M., Boetto, S., Clanet, M., et al. (2005). Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 76, 42–51.
Chen, D., Lipe, H., Qin, Z., and Bird, T. (2002). Cerebral cavernous malformation: novel mutation in a Chinese family and evidence for heterogeneity. J. Neurol. Sci. 196, 91–96. doi: 10.1016/s0022-510x(02)00031-x
Craig, H., Gunel, M., Cepeda, O., Johnson, E., Ptacek, L., Steinberg, G., et al. (1998). Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum. Mol. Genet. 7, 1851–1858. doi: 10.1093/hmg/7.12.1851
Draheim, K., Li, X., Zhang, R., Fisher, O., Villari, G., Boggon, T., et al. (2015). CCM2-CCM3 interaction stabilizes their protein expression and permits endothelial network formation. J. Cell Biol. 208, 987–1001. doi: 10.1083/jcb.201407129
Du, Q., Shi, Z., Chen, H., Zhang, Y., Wang, J., and Zhou, H. (2019). Two novel CCM2 heterozygous mutations associated with cerebral cavernous malformation in a Chinese family. J. Mol. Neurosci. 67, 467–471. doi: 10.1007/s12031-018-1254-4
Dubovsky, J., Zabramski, J., Kurth, J., Spetzler, R., Rich, S., Orr, H., et al. (1995). A gene responsible for cavernous malformations of the brain maps to chromosome 7q. Hum. Mol. Genet. 4, 453–458. doi: 10.1093/hmg/4.3.453
Francalanci, F., Avolio, M., De Luca, E., Longo, D., Menchise, V., Guazzi, P., et al. (2009). Structural and functional differences between KRIT1A and KRIT1B isoforms: a framework for understanding CCM pathogenesis. Exp. Cell Res. 315, 285–303. doi: 10.1016/j.yexcr.2008.10.006
Gunel, M., Awad, I., Anson, J., and Lifton, R. (1995). Mapping a gene causing cerebral cavernous malformation to 7q11.2-q21. Proc. Natl. Acad. Sci. U.S.A. 92, 6620–6624.
Han, G., Ma, L., Qiao, H., Han, L., Wu, Q., and Li, Q. (2020). A novel CCM2 missense variant caused cerebral cavernous malformations in a Chinese family. Front. Neurosci. 14:604350. doi: 10.3389/fnins.2020.604350
Ji, B., Qin, W., Sun, T., Feng, G., He, L., and Wang, Y. J. (2006). A novel deletion mutation in CCM1 gene (krit1) is detected in a Chinese family with cerebral cavernous malformations. Yi Chuan Xue Bao. 33, 105–110. doi: 10.1016/S0379-4172(06)60028-0
Jiang, X., Zhang, Y., Yin, X., Nan, D., Wang, X., Feng, J., et al. (2020). A novel CCM3 mutation associated with cerebral cavernous malformation in a Chinese family. Ther. Adv. Neurol. Dis. 13:1756286420902664. doi: 10.1177/1756286420902664
Jih, K., Chung, C., Chang, Y., Hung, P., Soong, B., Liao, Y., et al. (2018). Mutational analysis of CCM1, CCM2 and CCM3 in a han Chinese cohort with multiple cerebral cavernous malformations in Taiwan. Clin. Genet. 94, 389–390. doi: 10.1111/cge.13377
Labauge, P., Denier, C., Bergametti, F., and Tournier-Lasserve, E. (2007). Genetics of cavernous angiomas. Lancet Neurol. 6, 237–244.
Liang, X., Zhang, H., Wang, Z., Zhang, X., Dai, Z., Zhang, J., et al. (2022). JMJD8 Is an M2 macrophage biomarker, and it associates with DNA damage repair to facilitate stemness maintenance, chemoresistance, and immunosuppression in pan-cancer. Front. Immunol. 13:875786. doi: 10.3389/fimmu.2022.875786
Liquori, C., Berg, M., Siegel, A., Huang, E., Zawistowski, J., Stoffer, T., et al. (2003). Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am. J. Hum. Genet. 73, 1459–1464. doi: 10.1086/380314
Liu, J., Stockton, R., Gingras, A., Ablooglu, A., Han, J., Bobkov, A., et al. (2011). A mechanism of Rap1-induced stabilization of endothelial cell–cell junctions. Mol. Biol. Cell. 22, 2509–2519. doi: 10.1091/mbc.E11-02-0157
Liu, W., Liu, M., Lu, D., Wang, J., Cao, Z., Liu, X., et al. (2022). A Chinese Family with cerebral cavernous malformation caused by a frameshift mutation of the CCM1 gene: a case report and review of the literature. Front. Neurol. 13:795514. doi: 10.3389/fneur.2022.795514
Mao, C., Yang, J., Zhang, S., Luo, H., Song, B., Liu, Y., et al. (2016). Exome capture sequencing identifies a novel CCM1 mutation in a Chinese family with multiple cerebral cavernous malformations. Int. J. Neurosci. 126, 1071–1076. doi: 10.3109/00207454.2015.1118628
Mao, Y., Zhao, Y., Zhou, L., Huang, C., Shou, X., Gong, J., et al. (2005). A novel gene mutation (1292 deletion) in a Chinese family with cerebral cavernous malformations. Neurosurgery 56, 1149–1153.
Moore, S., Brown, R. Jr., Christianson, T., and Flemming, K. (2014). Long-term natural history of incidentally discovered cavernous malformations in a single-center cohort. J. Neurosurg. 120, 1188–1192. doi: 10.3171/2014.1.JNS131619
Nardella, G., Visci, G., Guarnieri, V., Castellana, S., Biagini, T., Bisceglia, L., et al. (2018). A single-center study on 140 patients with cerebral cavernous malformations: 28 new pathogenic variants and functional characterization of a PDCD10 large deletion. Hum. Mutat. 39, 1885–1900. doi: 10.1002/humu.23629
Ricci, C., Cerase, A., Riolo, G., Manasse, G., and Battistini, S. (2021). KRIT1 gene in patients with cerebral cavernous malformations: clinical features and molecular characterization of novel variants. J. Mol. Neurosci. 71, 1876–1883. doi: 10.1007/s12031-021-01814-w
Rigamonti, D., Hadley, M., Drayer, B., Johnson, P., Hoenig-Rigamonti, K., Knight, J., et al. (1988). Cerebral cavernous malformations. Incidence and familial occurrence. N. Engl. J. Med. 319, 343–347.
Riolo, G., Ricci, C., and Battistini, S. (2021). Molecular genetic features of cerebral cavernous malformations (CCM) patients: an overall view from genes to endothelial cells. Cells 10:3. doi: 10.3390/cells10030704
Stenson, P., Mort, M., Ball, E., Chapman, M., Evans, K., Azevedo, L., et al. (2020). The Human gene mutation database (HGMD((R))): optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 139, 1197–1207.
Wang, H., Pan, Y., Zhang, Z., Li, X., Xu, Z., Suo, Y., et al. (2017). A novel KRIT1/CCM1 gene insertion mutation associated with cerebral cavernous malformations in a Chinese family. J. Mol. Neurosci. 61, 221–226. doi: 10.1007/s12031-017-0881-5
Wang, K., Wu, D., Zhang, B., and Zhao, G. (2018). Novel KRIT1/CCM1 and MGC4607/CCM2 gene variants in Chinese families with cerebral cavernous malformations. Front. Neurol. 9:1128. doi: 10.3389/fneur.2018.01128
Wang, X., Liu, X., Lee, N., Liu, Q., Li, W., Han, T., et al. (2013). Features of a Chinese family with cerebral cavernous malformation induced by a novel CCM1 gene mutation. Chin. Med. J. 126, 3427–3432.
Xu, Y., Zhao, J., Wu, B., Zhong, H., Wang, S., and Heng, W. (2003). [A novel Krit-1 mutation in han family with cerebral cavernous malformation]. Zhonghua Bing Li Xue Za Zhi 32, 220–225.
Yang, C., Wu, B., Zhong, H., Li, Y., Zheng, X., and Xu, Y. (2018). A novel CCM1/KRIT1 heterozygous deletion mutation (c.1919delT) in a Chinese family with familial cerebral cavernous malformation. Clin. Neurol. Neurosurg. 164, 44–46. doi: 10.1016/j.clineuro.2017.11.005
Yang, C., Zhao, J., Wu, B., Zhong, H., Li, Y., and Xu, Y. (2017). Identification of a novel deletion mutation (c.1780delG) and a novel splice-site mutation (c.1412-1G>A) in the CCM1/KRIT1 gene associated with familial cerebral cavernous malformation in the chinese population. J. Mol. Neurosci. 61, 8–15. doi: 10.1007/s12031-016-0836-2
Yang, L., Wu, J., and Zhang, J. (2020). A novel CCM2 gene mutation associated with cerebral cavernous malformation. Front. Neurol. 11:70. doi: 10.3389/fneur.2020.00070
Yang, Q., Liu, H., Guo, J., Liu, Q., and Dou, W. (2020). Case report of familial cerebral cavernous hemangioma with new mutation site and literature review. Neural Injury Funct. Reconstruct. 15, 497–500.
Zafar, A., Quadri, S., Farooqui, M., Ikram, A., Robinson, M., Hart, B., et al. (2019). Familial cerebral cavernous malformations. Stroke 50, 1294–1301.
Zawistowski, J., Stalheim, L., Uhlik, M., Abell, A., Ancrile, B., Johnson, G., et al. (2005). CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 14, 2521–2531.
Zhang, F., Xue, Y., Zhang, F., Wei, X., Zhou, Z., Ma, Z., et al. (2020). Identification of a novel CCM1 frameshift mutation in a Chinese han family with multiple cerebral cavernous malformations. Front. Neurosci. 14:525986. doi: 10.3389/fnins.2020.525986
Zhao, Y., Xie, L., Li, P., Song, J., Qu, T., Fan, W., et al. (2011). A novel CCM1 gene mutation causes cerebral cavernous malformation in a Chinese family. J. Clin. Neurosci. 18, 61–65. doi: 10.1016/j.jocn.2010.04.051
Keywords: cerebral cavernous malformations (CCMs), Krev interaction trapped 1 (KRIT1), DNA sequencing, duplication mutation, frameshift
Citation: Chen Y, Dong X, Wang Y, Lv H, Chen N, Wang Z, Chen S, Chen P, Xiao S, Zhao J and Dong J (2023) Molecular genetic features and clinical manifestations in Chinese familial cerebral cavernous malformation: from a novel KRIT1/CCM1 mutation (c.1119dupT) to an overall view. Front. Neurosci. 17:1184333. doi: 10.3389/fnins.2023.1184333
Received: 11 March 2023; Accepted: 17 April 2023;
Published: 05 May 2023.
Edited by:
Jun Zhang, Texas Tech University Health Sciences Center, United StatesReviewed by:
Xingliang Dai, The First Affiliated Hospital of Anhui Medical University, ChinaGuodong Liu, Chongqing Medical University, China
Copyright © 2023 Chen, Dong, Wang, Lv, Chen, Wang, Chen, Chen, Xiao, Zhao and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Dong, ZG9uZ2p1bkBzdWRhLmVkdS5jbg==
†These authors have contributed equally to this work