 Iliya Lefterov
Iliya Lefterov Nicholas F. Fitz
Nicholas F. Fitz Yi Lu
Yi Lu Radosveta Koldamova*†
Radosveta Koldamova*†- Department of Environmental and Occupational Health, School of Public Health, University of Pittsburgh, Pittsburgh, PA, United States
The inheritance of Apolipoprotein E4 (APOEε4) brings the highest genetic risk of Alzheimer’s disease (AD), arguably the highest genetic risk in human pathology. Since the discovery of the association, APOE protein isoforms have been at the center of tens of thousands of studies and reports. While, without a doubt, our knowledge about the normal physiological function of APOE isoforms in the brain has increased tremendously, the questions of how the inheritance of the APOEε4 allele translates into a risk of AD, and the risk is materialized, remain unanswered. Moreover, the knowledge about the risk associated with APOEε4 has not helped design a meaningful preventative or therapeutic strategy. Animal models with targeted replacement of Apoe have been generated and, thanks to the recent NIH/NIA/Alzheimer’s disease Association initiative, are now freely available to AD researchers. While helpful in many aspects, none of the available models recapitulates normal physiological transcriptional regulation of the human APOE gene cluster. Changes in epigenetic regulation of APOE alleles in animal models in response to external insults have rarely been if ever, addressed. However, these animal models provide a useful tool to handle questions and investigate protein–protein interactions with proteins expressed by other recently discovered genes and gene variants considered genetic risk factors of AD, like Triggering Receptor expressed on Myeloid cells 2 (TREM2). In this review, we discuss genetic and epigenetic regulatory mechanisms controlling and influencing APOE expression and focus on interactions of APOE and TREM2 in the context of microglia and astrocytes’ role in AD-like pathology in animal models.
Introduction
Alzheimer’s Disease (AD) is the sixth leading cause of death in the United States. There are two forms of AD: early-onset or familial AD (EOAD), which develops before age 65, and late-onset AD (LOAD). EOAD is caused by autosomal dominant mutations in 3 genes – Amyloid Precursor Protein (APP), Presenilin 1 (PS1), and Presenilin 2 (PS2). The LOAD develops later in life, and in some individuals above 85, with no causative gene mutations known. LOAD cases account for more than 95% of all AD cases. An estimated 6.5 million Americans aged 65 and older were living with Alzheimer’s in 2022. Seventy-three percent are age 75 or older. The cost of Alzheimer’s and other dementias (ADOD) to the nation in 2022 was calculated at $321 billion, and by 2050, these costs could reach nearly $1 trillion. More than 11 million Americans provide unpaid care for people with Alzheimer’s or other dementias. In 2021, these caregivers provided more than 16 billion hours of care valued at nearly $272 billion. The financial burden on American society from ADOD is enormous.
In addition to the cognitive decline, there are two morphological hallmarks of AD: extracellular deposits of β-amyloid (Aβ) peptide, called amyloid plaques and intracellular neurofibrillary tangles of tau protein (Jack et al., 2018; Chen and Holtzman, 2022). While it has been established for more than 30 years now that the highest risk for LOAD is associated with a specific allele of APOE gene – APOEε4, other common gene variants have been added to a long and ever-increasing list of genetic risk factors of various significance (Pimenova et al., 2017; Sims et al., 2017; Ando et al., 2022; Holstege et al., 2022). Half of those are associated with immune response (Wes et al., 2016). Among those, rare variants of TREM2, expressed in microglia, are associated with a risk close to a risk associated with the inheritance of a single APOEε4 allele (Wolfe et al., 2018b). Environmental exposures, lifestyle, diet, traumatic brain injury, and an array of comorbidities have been implicated in LOAD risk, early pathogenesis, and progression, as well (Rao et al., 2023). Unfortunately, none of the knowledge regarding the risk factors of LOAD has translated into early diagnosis of the disease or meaningful direction toward successful therapeutic strategies. In this review, we discuss genetic and epigenetic regulatory mechanisms controlling and influencing APOE expression and focus on interactions of APOE and TREM2 in the context of microglia and astrocytes’ role in AD-like pathology in animal models.
APOE genotype and the risk of Alzheimer’s disease is the strongest genetic association in human pathology
APOE an extremely important and indispensable protein expressed in multiple tissues and organs. It is important to underline that no pathological condition or disease presents with the lack of APOE due to genomic deletion. Functionally, at a biochemical level, APOE provides a scaffold for and is an integral structural part of lipoproteins. In brain, APOE is secreted primarily by astrocytes and, unlike in the periphery, is the major apolipoprotein of High Density Lipoprotein (HDL)-like discoidal particles. These brain HDL-like particles do not contain APOA-I. Thus, the transport of cholesterol and phospholipids in the interstitial fluid and between neural cells highly depends on APOE. The transport of cholesterol and phospholipids is the major function of APOE. Therefore, in case of presumptive dysfunctional APOE, it is reasonable to expect, as a consequence, multiple disturbed molecular and cellular processes. The term dysfunctional APOE is poorly defined, however (Fazio et al., 1994). In AD nonmutated APOE, regardless of the isoform, retains its normal biochemical function.
In an excellent review published 23 years ago, R. Mahley and S. Rall concluded that “APOE plays a part in many processes beyond its traditional role in cholesterol and lipoprotein metabolism” (Mahley and Rall, 2000). A limitation of understanding APOE as a risk of AD is that it is impossible to disentangle the “traditional”/biochemical role of APOE in cholesterol metabolism and its role in AD risk and pathology. Human APOE and its ε2/ε3/ε4 alleles have been associated with many diseases and pathological conditions, including nonpathological aging, and AD (Corder et al., 1993, 1994; Roses et al., 1994; Davies et al., 2014; Table 1). This supports the statement that the genetic risk conferred by APOE is related to a perturbed primary function of APOE isoforms: cholesterol and phospholipid transport and metabolism. This, inevitably, includes AD. Of course, it would have been naïve to explain all of the above phenotypes, particularly AD, only by the disturbed major biological effect of APOE – cholesterol and phospholipid transport.
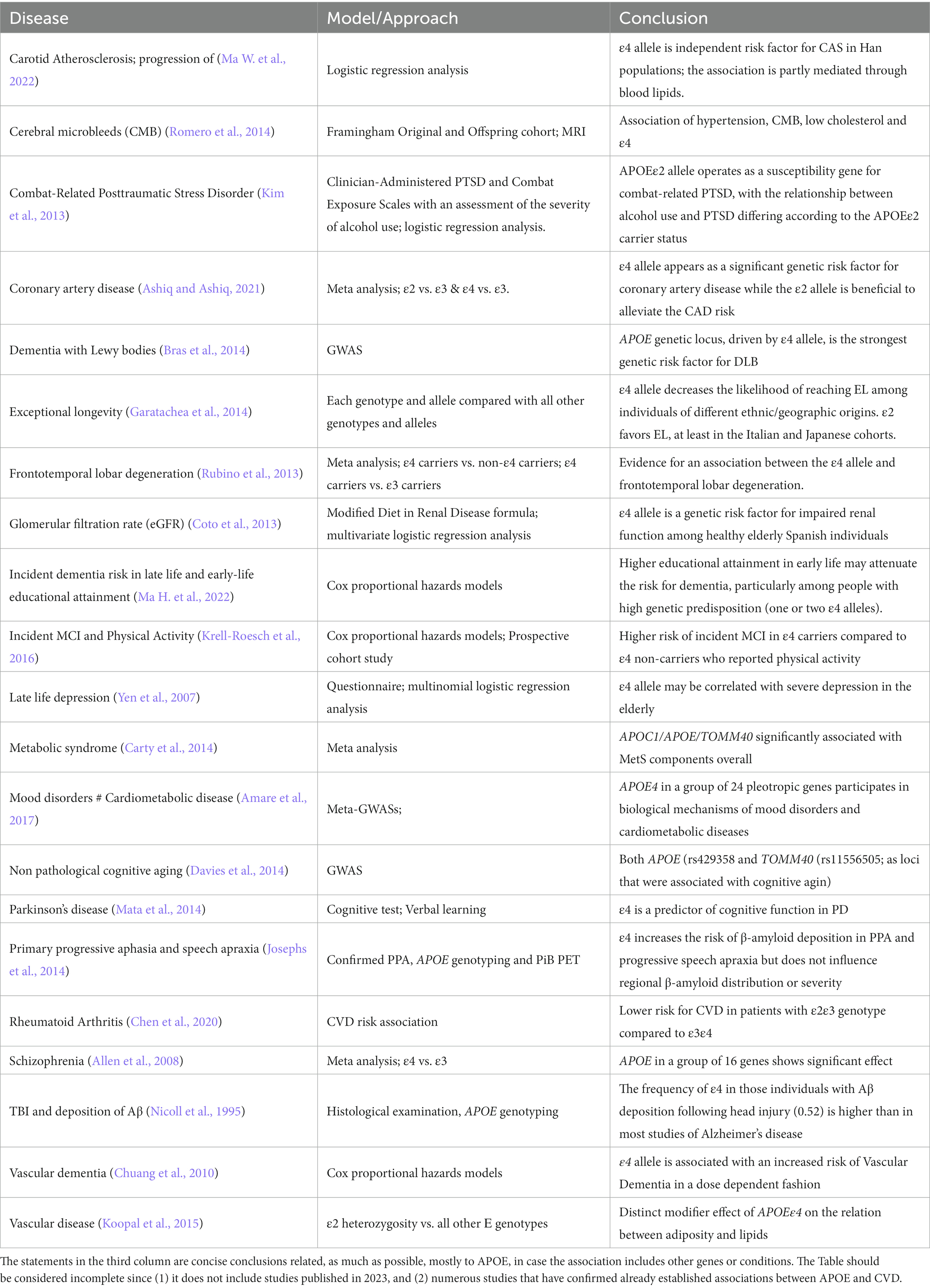
Table 1. List of diseases where an association with APOE variants has been identified using different models and approaches.
Numerous proposed hypotheses explain how APOE isoform-specific differences might increase the risk of AD, ranging from neurotoxic effect based on domain interaction, binding to, deposition and clearance of Aβ, differential lipidation of isoforms, and neurotoxic and neuroprotection effects. More than 30 years since the association of APOEε4 and the risk of AD has been established (Corder et al., 1993), we still do not know how exactly the role of APOE coded by APOEε4 allele (APOE4) in cholesterol and lipoprotein metabolism – normal or disturbed, translates into an increased risk of AD. Nevertheless, in the last 2 years, several reviews and research papers have been published pointing to aspects of APOE biology that might be worth considering in terms of deciphering the role of APOE as a risk factor of AD and even designing new therapeutic strategies (Parhizkar et al., 2019; Chen and Holtzman, 2022; Li et al., 2022; Lindner et al., 2022; Martens et al., 2022; Raulin et al., 2022). In an in-vitro experimental system, Lindner et al. (2022) explored isoform-specific lipidation and revealed different lipidation pathways. While ATP Binding Cassette transporter A1 (ABCA1)-regulated APOE lipidation is isoform independent and cholesterol-rich HDL-like lipid particles are secreted by astrocytes, in stress-associated conditions, assembling and secretion of triacylglycerol-rich lipoproteins is boosted by the APOE4 isoform. The authors showed that APOE4 was a strong triacylglycerol binder and thus had a reduced capacity to clear toxic fatty acids from the extracellular milieu. Since 2003 (Koldamova et al., 2003), the importance of Nuclear Receptor’s Liver X Receptors/Retinoid X receptors/ABCA1-APOA-I/APOE (LXR/RXR-ABCA1-APOA-I/APOE) regulatory axis for normal/physiological function of APOE, and its relevance to AD pathogenesis in particular, has been demonstrated in tens of studies [for comprehensive reviews see Fitz et al. (2019), Koldamova and Lefterov (2007), Pahnke et al. (2021), Wolfe et al. (2018a), and Kim et al. (2009)]. The regulatory axis does not simply imply transcriptional regulation. While ABCA1 is a primary LXR/RXR target gene, APOE and APOA-I are part of the axis because transcriptional upregulation of ABCA1 and ABCA1-mediated lipidation of APOA-I and APOE are prerequisites for their stability, avoiding fast apolipoprotein degradation.
Perhaps the most comprehensive review of disturbed molecular, cellular, and pathophysiological processes in AD that might be associated with APOE4 has been published by researchers previously or currently affiliated with Mayo Clinic and Washington University at St Louis (Martens et al., 2022). Alike to Amyloid cascade hypothesis proposed in the late 90s [see J. Hardy for a comprehensive review and critical reappraisal (Hardy, 2009)], the authors coined the term “APOE cascade hypothesis” in the pathogenesis of AD and related dementias. Besides the excellent description of disturbed molecular and cellular processes during AD progression, Martens et al. propose a targeted engagement type of therapeutic strategy based on APOE biology and the physical properties of multimolecular complexes where APOE is an integral part. To be successful, these strategies should nevertheless consider that APOE is not a cause of AD as well as the role of APOE isoforms in development and normal CNS physiology.
Expression of the Apolipoprotein E/C-I/C-IV/C-II gene cluster is highly regulated and differs in humans and mice
Molecular clues to understand the regulated expression of Apolipoprotein E/Apolipoprotein C-I/Apolipoprotein C-IV/Apolipoprotein C-II (E/C-I/C-IV/C-II) gene cluster came from a series of reports based on studies conducted in J. Taylor’s and D. Mangelsdorf’s laboratories in the late 90s and the beginning of this century (Simonet et al., 1993; Allan et al., 1995, 1997; Shih et al., 2000; Grehan et al., 2001; Laffitte et al., 2001, 2003; Mak et al., 2002). The characterization of transgenic mice overexpressing individual genes of the cluster revealed diverse functions. However, the expression of all members of this apolipoprotein gene cluster is reported to be coordinately regulated by distal enhancer regions (Simonet et al., 1993; Allan et al., 1995). It was demonstrated that the regulatory sequences in the MultiEnhancers ME1 and ME2 downstream from the Apolipoprotein C-I (APOC-I) gene are transcription factors Liver X Receptor/Retinoid X receptor (LXR/RXR) Nuclear Receptor dimers canonical response elements (Mak et al., 2002; Figure 1). The regulatory sequences in the promoter regions of the human APOE gene cluster represent binding sites/response elements of additional transcription factors – APP-2, SP1, Estrogen Receptor, as well as transcriptional elements A, B, B1, and B2 (Smith et al., 1988; Jo et al., 1995; Paik et al., 1995; Bullido and Valdivieso, 2000). Furthermore, polymorphisms in the proximal promoter and the first intron of the APOE gene cluster (−1,019 to +407) affecting APOE expression were identified in the late 90s and early 2000s (Mui et al., 1996; Lambert et al., 1997, 1998a,b, 2000; Bullido and Valdivieso, 2000; Lumsden et al., 2020). Importantly, these polymorphic sites have been associated with a differential risk of AD (Artiga et al., 1998; Sims et al., 2017). However, the association of those polymorphic sites, sequence variability in the proximal promoter and ME1 and ME2, and the level of APOE protein in AD are not clearly understood. The data on the expression level of APOE RNA and the correlation with APOE protein level differ across the studies, and the question why remains unanswered. Importantly, in human postmortem brain, a CGI island that overlaps with exon four and downstream is highly methylated, and the methylation is altered in AD frontal lobe (Lee et al., 2020). However, the methylation level of APOE CGI correlates to the expression level of four known APOE transcripts, of which only one is full-length. Surprisingly, circRNAs, miRNAs, and truncated APOE transcripts constitute a significant part of the total APOE mRNA with higher expression in the AD frontal lobe. While the precise clinical significance of these variations in the amounts of RNA and methylation level of CGI in the APOE 3′-exon are not fully understood, the results of an increasing number of studies point to the regulatory role of epigenomic signatures and changes in epigenome associated with risk or clinical presentation of a variety of neurological disorders (Lee et al., 2020).
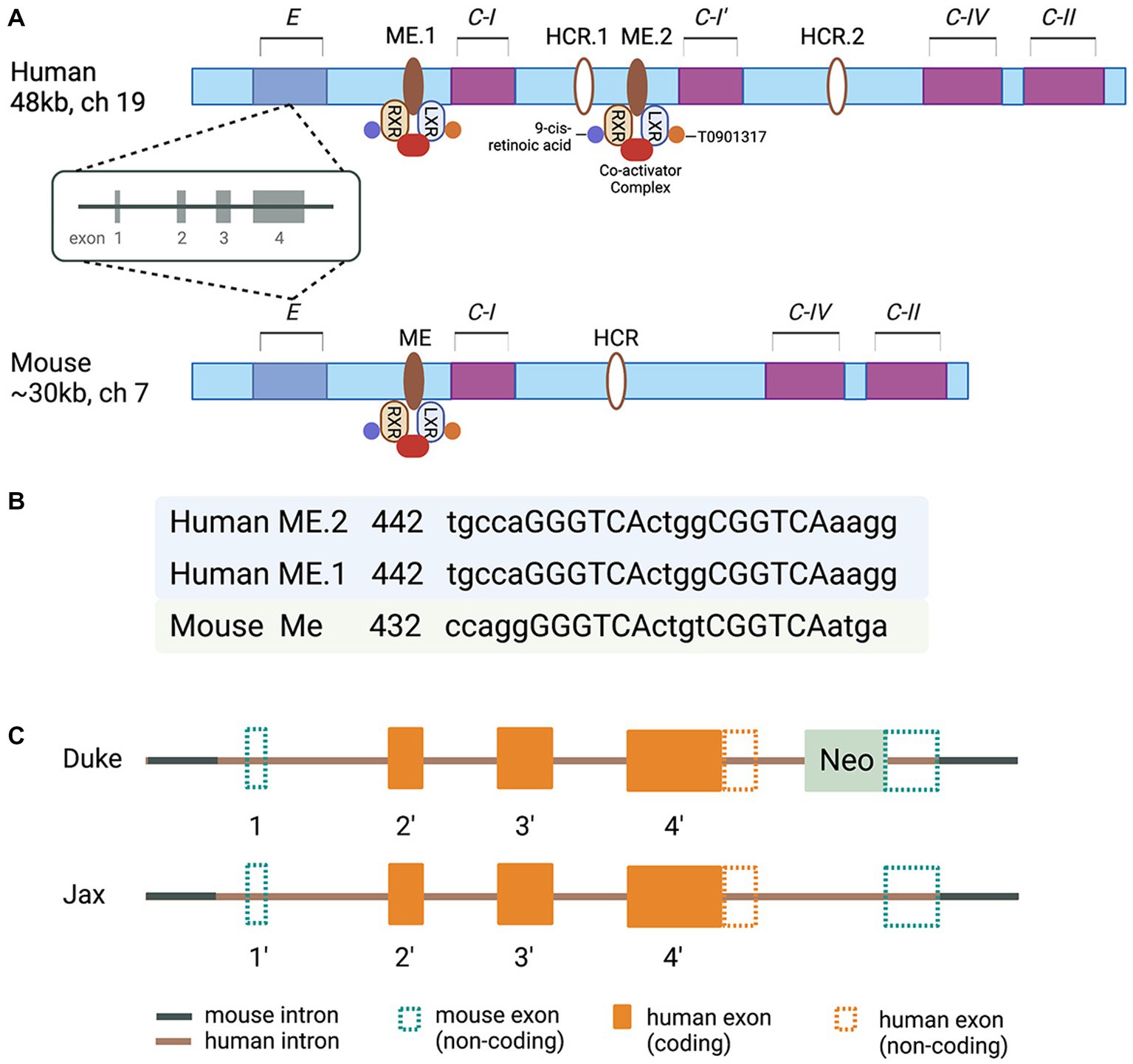
Figure 1. (A) Schematic of the human and murine APOE/C-I/C-IV/C-II gene clusters. Human cluster contains the APOC-I pseudogene. In the multienhancer regions downstream from human and mouse APOE/Apoe genes are located multienhancer elements ME1 in human and ME in mouse clusters. Human and mouse Hepatic Control Regions HCR.1 and HCR are located downstream of C-I. Human APOC-I pseudogene is indicated as C-I′; the pseudogene is a result of APOC-I duplication and does not exist in the mouse cluster. ME2 is a duplicated ME1 and exists only in humans. (B) The nucleotide sequences corresponding to putative LXR response elements with the 4-bp spacer within the human and murine MEs are shown as capital letters. The direct repeats are separated by 4-bp spacers. The locations of these sequences relative to the 5′ end of each ME are given. (C) A diagram of chimeric genes in TR APOE model mice generated at Duke (Sullivan et al., 1997) and Jax Labs (Foley et al., 2022). Detailed explanations are provided in the text.
The human APOE targeted replacement and other humanized mouse models
For the last 25 years or so, the availability of a mouse model with a targeted replacement of mouse Apoe gene (Sullivan et al., 1997) provided an opportunity to address a myriad of questions ranging from the role of APOE isoforms in AD pathogenesis to how successful variety of therapeutic strategies have been so far. The model has been used in multiple studies aiming to reveal the role of APOE isoforms in the response to traumatic brain injury, as well (Crawford et al., 2002, 2009; Castranio et al., 2017, 2018). Considering the differences between mouse and human APOE gene clusters, how complex the transcriptional control of human APOE is, the structure of the targeting construct (s), and the strategy to replace mouse Apoe in the TR APOE models (Sullivan et al., 1997, 2009) become important factors. An NIH/NIA initiative recently established the MODEL-AD (Model Organism Development and Evaluation for Late-onset AD) consortium to provide the scientific community with AD animal models of LOAD that better mimic human disease (www.model-ad.org). The consortium has created knock-in humanized coding and non-coding LOAD risk variants expressed at endogenous levels, including mice expressing all APOE isoforms (Oblak et al., 2020; Kotredes et al., 2021; Foley et al., 2022). The targeting constructs used to generate APOE TR at Jax (Foley et al., 2022) differ from those originally used at Duke (Sullivan et al., 1997). In the Duke model the 3′ homology arm of the targeting construct, manipulated according to the classic genetic engineering technology, is upstream of the mouse Apoe exon 4 and 3′ noncoding sequences. That means no human regulatory sequences downstream of the human APOE gene exist in the chimeric gene. The targeting construct used in the Jax APOE TR mouse model was generated using a technology called recombineering (Sharan et al., 2009), which allows engineering of large constructs (>100 kb) and recombination events without leaving any ‘footprints’ behind homologous recombination. There are two differences between the targeting constructs/chimeric genes used in the Duke and Jax models: (1) in the Duke model the distal part of human APOE 1st intron, followed by the entire downstream sequences and part of 3′ sequences downstream of exon 4, followed by a Neo cassette with a stop codon are inserted between Sac I recognition site downstream of mouse 1st exon/intron junction, and Pvu I recognition site within mouse Apoe exon 4 (Sullivan et al., 1997, 2009). (2) in the Jax model, the chimeric gene retains part of human regulatory sequences upstream of the noncoding exon 1and the entire genomic sequence of APOE. There is no Neo cassette and downstream of human APOE 3′ noncoding sequences the chimeric gene retains a small part of mouse distal noncoding exon 4 (Foley et al., 2022; Figure 1). Neither of the chimeric genes has human regulatory sequences downstream of APOE. While this is hard to predict with certainty, most probably the expression levels and response to external stimuli/insults in TR mice will not differ, regardless of the model. The availability of Jax APOE TR mice to the AD research community and the opportunity to generate, examine and compare APOEε3/ε4 heterozygous mice to APOEε3/ε3 and APOEε4/ε4 mice is what makes the models hugely different. So far, the patent restrictions have not allowed the generation of heterozygous TR mice. We can speculate, however, that the expression of APOE isoforms, in the brains of Jax TR mouse models, does not recapitulate the transcriptional control and the regulation of the expression of APOE isoforms in the human brain. While this poses some questions about how good the models are at studying the risk of AD, we can assume that in a mouse model without overexpression of human APP, different protein–protein interactions, receptor-ligand interactions, and downstream intracellular and extracellular effects, replicate true physiological or pathophysiological conditions responsive to regulatory mechanisms at various stages of a neurodegenerative disorder, including AD. We believe these are the principal arguments and a justification to conduct in vivo experiments using animal models expressing different APOE isoforms where mouse Trem2, for example, is physiologically expressed, globally deleted by genetic engineering, or expressed as a mutant form shown to be associated with AD. We expect that animal models allowing the analysis of APOE in the context of gene–gene and gene–environment interactions will appear soon.
In APP transgenic mice expressing human APOE isoforms, APOE affects Aβ clearance and deposition in an isoform-dependent manner (Holtzman et al., 2000; Bales et al., 2009; Castellano et al., 2011; Kim et al., 2011), and APOE lipidation level is of significance (Fitz et al., 2012; Liao et al., 2018). Data from ABCA1 deficient mice (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004) and Tangier disease patients with non-functional ABCA1 demonstrated that in the absence of cholesterol efflux plasma APOA-I is virtually missing and APOE in plasma and brain is significantly reduced [reviewed in Oram and Vaughan (2000) and Koldamova et al. (2014)]. It has been shown independently by three groups that in APP transgenic mice, global deletion of Abca1 decreases APOE lipidation and significantly increases amyloid deposition (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005; Wahrle et al., 2005). Recently, Fitz et al., using preclinical AD mouse models, demonstrated that APOE3 lipoproteins, compared to APOE4, prompted faster microglial migration toward injected Aβ, facilitated Aβ uptake, and abrogated damaging effects of Aβ oligomers on cognition (Fitz et al., 2021). Applying in vivo two-photon imaging, they showed that the APOE3 lipoproteins caused microglia to move faster toward Aβ and surround it, thus decreasing the spread of Aβ (Fitz et al., 2021). In tau mouse models of AD, expression of APOE4 has been shown to exacerbate tau-mediated neurodegeneration compared to mice with APOE3 expression, while the selective removal of astrocytic APOE4 is protective (Wang et al., 2021).
TREM2: expression, function, and effects
TREM2 – gene and protein structure
TREM2, a member of the TREM family, is a cell surface transmembrane receptor with an extracellular Ig-like domain, a cytoplasmic tail, and a transmembrane domain [reviewed in Colonna (2023) and Wolfe et al. (2018b)] (see Figure 2). TREM2 is expressed in cells of the myeloid lineage, such as microglia, osteoclasts, tissue macrophages, monocytes, and dendritic cells (Colonna, 2023). Most of the TREMs are evolutionarily conserved in mice and humans. Human TREM2 is located on chromosome 6p21.1 in the TREM gene cluster near other TREM and TREM-like (TREML) genes: TREML1, TREM2, TREML2, TREML3, TREML4, and TREM1 (Klesney-Tait et al., 2006). Mouse Trem2 is located on mouse chromosome 17C in a cluster including Trem1, Treml1, Trem2, Treml2, Trem3, Trem4, Treml4, Trem5, Treml6 (Colonna, 2023).
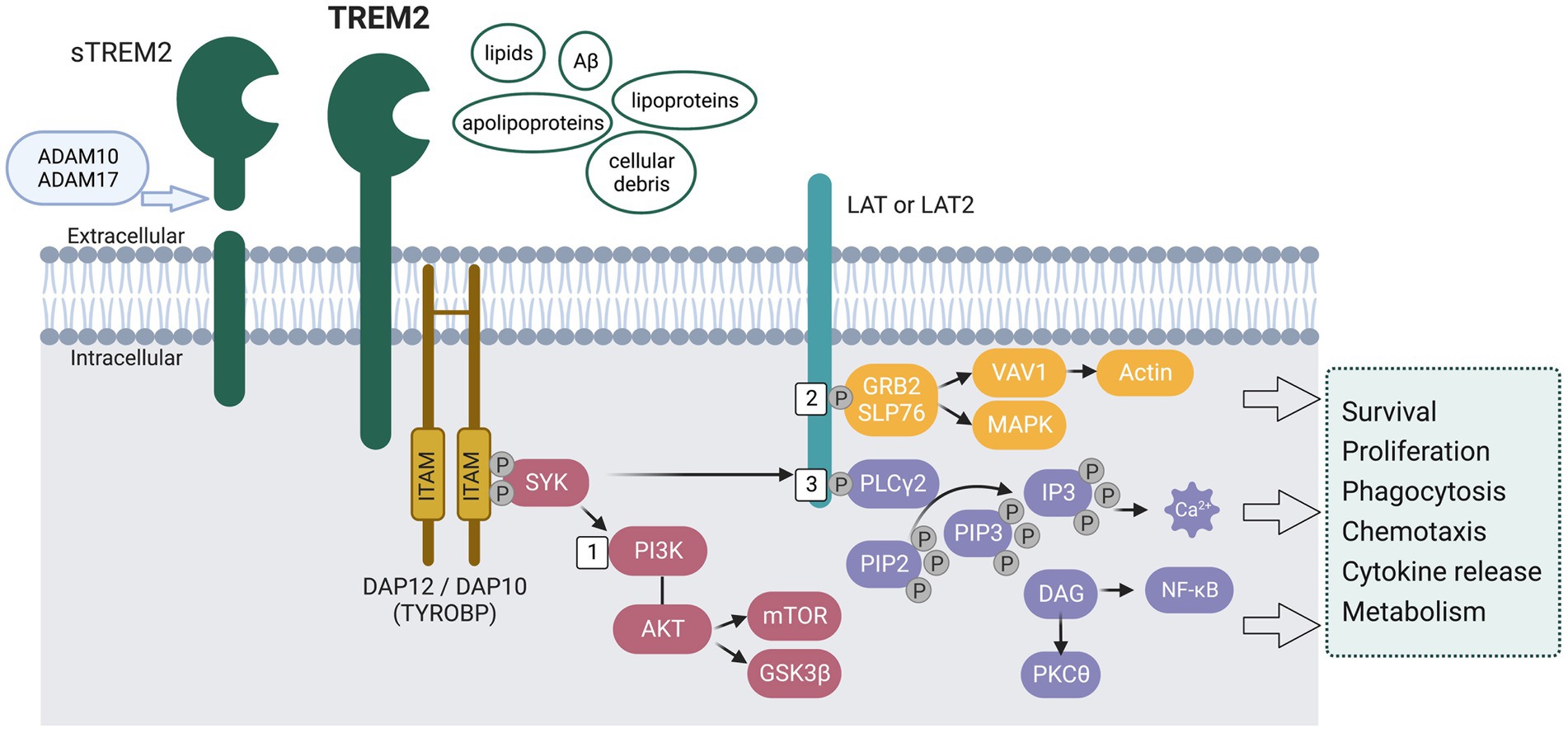
Figure 2. Schematic representation of the signaling pathways activated by the transmembrane receptor TREM2. The extracellular region of TREM2 is suggested to recognize and bind lipids, Aβ, lipoproteins, apolipoproteins, and cellular debris. Upon ligand binding, the cytoplasmic immunoreceptor tyrosine-based activation motifs (ITAMs) on DNAX-activating protein of 12 kDa (DAP12) recruit tyrosine protein kinase SKY, leading to activation of the phosphoinositide 3-kinase (PI3K) - AKT pathway and subsequent phosphorylation of linker for activation of T-cells family member 1 (LAT) and/or LAT2. Specifically, (1) The activation of PI3K leads to the activation of AKT, which in turn leads to the activation of mechanistic target of rapamycin (mTOR) signaling and the phosphorylation of glycogen synthase kinase 3β (GSK3β), resulting in GSK3β inactivation, stabilization of β-catenin, and cell cycling. (2) Upon phosphorylation of LAT/LAT2, adaptor molecules GRB2 and SLP76 are recruited, thereby activating the mitogen-activated protein kinase (MAPK) pathway and facilitating the recruitment of guanine exchange factors of the VAV family. This leads to the promotion of actin cytoskeleton rearrangement. (3) LAT/LAT2 also activates phospholipase Cγ2 (PLCγ2), which degrades phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). This results in Ca2+ mobilization and NF-κB activation, respectively. These pathways have been found to affect microglia survival and functions, including phagocytosis, chemotaxis, cytokine release, and energy metabolism. Full-length TREM2 can be cleaved by a disintegrin and metalloproteinase domain-containing protein (ADAM10 and ADAM17), releasing soluble TREM2 (sTREM2).
TREM2 signaling
As shown in Figure 2, upon ligand binding, intracellular signals are conveyed through DNAX-activating protein DAP12 (12 kDA disulphide-bonded protein homodimer containing an immunoreceptor tyrosine-based activation motif), also known as TYROBP (tyrosine kinase-binding protein). The cytosolic immunoreceptor tyrosine-based activation motifs (ITAMs) on DAP12 will recruit the tyrosine protein kinase SKY, which leads to the activation of phosphoinositide 3-kinase (PI3K) - AKT pathway (Figure 2 #1) and phosphorylation of linker for activation of T-cells family member 1 (LAT) and/or LAT2, which recruit other signaling adaptors such as phospholipase Cγ2 (PLCγ2) (Figure 2 #3), and guanine exchange factor proto-oncogene VAV1 [reviewed in Colonna (2023)]. PLCγ2 further degrades phosphatidylinositol-3,4,5-trisphosphate (PIP3) to inositol trisphosphate (IP3) and diacylglycerol (DAG), thus mobilizing Ca2+. VAV1 induces actin remodeling that controls migration and adhesion (Figure 2 #2). All these signaling pathways affect the survival and proliferation of microglia (mTOR signaling), phagocytosis, and release of cytokines and chemokines (Wang et al., 2015; Wu et al., 2015; Raha et al., 2017; Ulland et al., 2017; Zhong et al., 2017; Zheng et al., 2018). Full-length TREM2 can be cleaved by disintegrin and metalloproteinase domain-containing proteins (ADAM10 and ADAM17, see Figure 2), thus releasing soluble TREM2 (sTREM2) (Wunderlich et al., 2013; Thornton et al., 2017). The exact biological and pathological role of sTREM2 is not clear, with some proposing it acts as a decoy receptor against TREM2 (Zhong et al., 2017) and others suggesting that sTREM2 plays an important role in promoting microglia survival and regulating inflammatory responses (Wu et al., 2015; Zhong et al., 2017). Overall, TREM2 expression and TREM2-induced signaling are essential in regulating microglia survival and proliferation.
TREM2 ligands
The specific ligands that bind and activate TREM2 remain unclear. The extracellular region of TREM2 with an immunoglobulin domain binds many Gram-negative and Gram-positive bacteria and microbial products, such as LPS (Daws et al., 2003) and lipids [reviewed in Colonna (2023) and Kober et al. (2016)]. It has been shown that TREM2 binds phospholipids such as the phosphatidylserine on the surface of apoptotic cells (Poliani et al., 2015; Wang et al., 2015; Krasemann et al., 2017; Shirotani et al., 2019), lipoproteins including HDL and low-density lipoproteins (LDL) (Song et al., 2017). TREM2 also binds apolipoproteins, such as APOE (Atagi et al., 2015; Bailey et al., 2015; Yeh et al., 2016; Jendresen et al., 2017; Kober et al., 2020). TREM2 binds myelin lipids and participates in debris clearance and remyelination (Cantoni et al., 2015; Poliani et al., 2015; Nugent et al., 2020). It has also been reported that in vitro soluble TREM2 directly binds to Aβ oligomers (Lessard et al., 2018) and, at least in vitro, can facilitate Aβ degradation (Zhao et al., 2018).
Regulation of Trem2 expression
Expression of TREM2 is affected by inflammation; for example, pro-inflammatory molecules such as lipopolysaccharide (LPS) downregulate TREM2 expression, and in-vitro anti-inflammatory molecules upregulated TREM2 expression (Bhattacharjee et al., 2016; Liu et al., 2016; Zheng et al., 2016). TREM2 expression is increased with the progression of neurodegeneration in AD patients (Perez et al., 2017) and mouse models of AD (Keren-Shaul et al., 2017; Krasemann et al., 2017) – traumatic brain injury (Castranio et al., 2017; Saber et al., 2017), Amyotrophic lateral sclerosis (Jerico et al., 2023), Parkinson’s disease (Liu et al., 2016), and post-stroke remodeling (Song et al., 2022). In terms of transcription factor regulation, a study by Daniel et al. (2014) demonstrated that the Retinoid X receptor (RXR) ligand LG268 increased RXR binding at a site upstream of the TREM2 locus, which regulates murine Trem2 expression [also reviewed in Jay et al. (2017b)]. Treatment with another RXR agonist – bexarotene, enhanced the expression of TREM2 mRNA in the cortex of AD mice (Lefterov et al., 2015). A recent study demonstrated that HX600, a synthetic agonist for RXR-Nuss1 heterodimers, increased TREM2 immunoreactivity in an ischemic mouse model (Loppi et al., 2018).
TREM2, amyloid deposition, and tau
The effects of Trem2 deficiency on amyloid pathology have been studied in APP transgenic mice with different results based on the mouse model used and the stage of amyloid pathology. Wang et al. (Wang et al., 2015) were the first to report that Trem2 deficiency significantly decreased the number of plaque-associated microglia in the 5XFAD AD mouse model, indicating that the proliferation of local microglia around the plaques is impaired, which other groups later confirmed (Yuan et al., 2016; Fitz et al., 2020). In their study, Wang et al. reported a significant increase of amyloid load in the hippocampus but not in the cortex at 8.5 months and no difference in amyloid load at an earlier age of 5XFAD mice (Wang et al., 2016).
Similarly, in APPPS1-21 model mice, there was no change in the amyloid pathology in the cortex but a significant decrease in the hippocampus of Trem2−/− mice at 4 months (Jay et al., 2015). Interestingly, using the same AD mouse model at 8 months of age, Jay et al. showed an increase in 6e10 staining in the cortex and no changes in the hippocampus of Trem2 deficient mice when compared to controls (Jay et al., 2017a) and concluded that in the early stages of amyloid deposition (2-month cortex, 4-month hippocampus) Trem2 deficiency reduces both plaque number and size and at later stages of the disease Trem2 deficiency increases plaque size and area. Using three different AD mouse models and employing high-resolution STORM imaging, Yuan et al. showed that Trem2 deficiency increased the diffuse amyloid plaques associated with increased neuronal dystrophy (Yuan et al., 2016). What looks to be an undisputed feature of TREM2 is its role in microglia barrier around the amyloid plaques and in amyloid compaction (Wang et al., 2015; Yuan et al., 2016; Fitz et al., 2020; Meilandt et al., 2020; Wood et al., 2022). The conclusion from these studies is that the lack of microglia barrier in Trem2 deficient mice impedes amyloid from forming dense plaques, thus allowing the spreading of more toxic Aβ oligomers.
The effect of Trem2 deficiency on tau pathology using tau mouse models is less clear and more complex. In P301S tau mice expressing mouse Apoe the loss of Trem2 (Sayed et al., 2018; Leyns et al., 2019) or the expression of loss-of-function R47H variant (Gratuze et al., 2020) decreased brain atrophy and neurodegeneration as well as microgliosis compared to control mice expressing wild type Trem2. Surprisingly, the deletion of Trem2 in the same P301S-tau model expressing human APOE4 isoform exacerbated tau-mediated brain atrophy (Gratuze et al., 2023). The most probable explanation for the observed discrepancies is that the mutated tau interacts differentially with human and mouse APOE. It is difficult to say the reason for these discrepancies without more experiments, including the expression of APOE2 and E3 isoforms and comparing their effect on tau pathology.
In 2017 using different models of neurodegeneration, several groups have identified and characterized the phenotype and transcriptomics of novel microglia type associated with neurodegenerative diseases called either Disease Associate microglia (DAM) (Keren-Shaul et al., 2017) or microglial neurodegenerative phenotype (MGnD) (Krasemann et al., 2017). DAM signature represents a unique set of genes that overlapped in different studies and includes the upregulation of genes such as Apoe, Trem2, Clec7a, Axl, Lpl, Spp1, and Mpeg1 and downregulation of another set of genes termed “homeostatic” microglia (for example Tmem119, P2ry12) (Keren-Shaul et al., 2017; Krasemann et al., 2017; Sala Frigerio et al., 2019; Fitz et al., 2020). Keren-Shaul et al. determined that Trem2 is required for the transition from one disease state to another, but the shift from homeostatic to DAM is Trem2 independent (Keren-Shaul et al., 2017). The finding that Trem2 deficiency suppresses the DAM program, including the expression of Apoe, was confirmed by other studies (Krasemann et al., 2017; Sala Frigerio et al., 2019; Fitz et al., 2020, 2021).
APOE, TREM2, and AD
APOE and TREM2 are part of a large group of GWAS-identified genes associated with AD risk with glial-specific expression (microglia and astrocytes) and are related to immune response (Villegas-Llerena et al., 2016). As noted above, at least in vitro TREM2 binds to APOE (Atagi et al., 2015; Bailey et al., 2015; Yeh et al., 2016; Lessard et al., 2018; Kober et al., 2020). In some studies, the R47H variant of TREM2 has markedly reduced binding between APOE and TREM2 (Atagi et al., 2015; Bailey et al., 2015; Yeh et al., 2016) and in others with no effect on their interaction (Lessard et al., 2018). In terms of the affinity of TREM2 to bind different APOE isoforms, two studies showed that APOE4 demonstrates a slightly higher affinity to bind TREM2 than the other two isoforms (Jendresen et al., 2017; Kober et al., 2020) but other – no significant difference between the isoforms (Atagi et al., 2015; Bailey et al., 2015; Yeh et al., 2017; Lessard et al., 2018). Regarding APOE lipidation, TREM2 was shown to bind lipidated and non-lipidated APOE. However, it is an open question if APOE lipidation affects TREM2 binding. Some studies found that their interaction is enhanced by lipidation (Yeh et al., 2016) and others to be slightly decreased by it (Kober et al., 2020). As noted above, bearing in mind the data from Tangier patients and Abca1 knockout mice, it is possible that in vivo APOE does not exist in lipid-free form [reviewed in Oram and Vaughan (2000) and Koldamova et al. (2014)]. Thus, whether APOE lipidation has a role in TREM2 binding is more of a theoretical than practical significance.
Only a few reports examine the interaction between TREM2 deficiency and APOE isoforms in vivo either in APP transgenic models (Fitz et al., 2020) or in AD patients (Nguyen et al., 2020). Fitz et al. demonstrated that in APP mice expressing human APOE3 or APOE4 (APP/E3 and APP/E4), the lack of Trem2 impaired microglia barrier in both isoforms but did not change steady-state plaque load (Fitz et al., 2020). However, APOE mRNA expression measured in plaque-associated microglia was significantly reduced by Trem2 deficiency only in APP/E4 and not in APP/E3 mice, suggesting that APOE4 microglia respond differently to the absence of Trem2 (Fitz et al., 2020). Since another DAM gene – Clec7a, was also significantly decreased in plaque microenvironment only in APP/E4 without Trem2, this might imply that DAM response to a challenge (in this case, amyloid deposition and Trem2 absence) is less effective on an APOE4 background, thus preventing microglia around plaques to form a complete barrier (Fitz et al., 2020, 2021). A recent study by Nguyen et al. used single-nucleus RNA sequencing of postmortem human brain expressing APOE and TREM2 variants and identified four distinct microglia clusters. One was the homeostatic microglia; the rest were “active” clusters designated based on different pathology and differentially expressed genes. Nguyen et al. established that one of the active clusters includes a subpopulation of CD163-positive microglia, which they named “amyloid-responsive microglia.” They determined that this cluster is enriched in APOE3/3 AD patients and is relatively depleted in cases with APOE4 carriers and TREM2 risk variants. Nguyen et al. proposed an amyloid-responsive microglia subpopulation primed to elicit an activated immune response and note the reduced response in APOE4 carriers. In a recent study, Fitz et al. showed that in mice injected with Aβ plus native APOE lipoproteins, there was a higher number of differentially expressed genes between WT vs. Trem2ko if the mice were injected with APOE4 compared to APOE3, particularly genes associated with interferon signaling (Ifit2, Ifi27l2a, Ifi207, and Axl) and endocytosis (Cd14, Cxcl16, Fth1, and Ifitm3) (Fitz et al., 2021). Additionally, the lack of TREM2 decreases Aβ phagocytosis only by APOE4-treated microglia, thus suggesting that APOE4 lipoproteins compared to APOE3 are insufficient to resist TREM2 deficiency, particularly in the presence of Aβ (Fitz et al., 2021).
Concluding remarks
It is hard to admit that 30 years after the discovery of APOEε4 as the highest genetic risk of AD, the nature and the molecular and cellular mechanisms that materialize the risk are still poorly understood. Multiple and well-supported hypotheses have been proposed, pointing to various mechanisms explaining the risk conferred by APOE4. Most importantly, however, loss of function – decreased level of APOE4 and fast degradation of poorly lipidated Apolipoprotein E4, as well as gain of function – generation of neurotoxic fragments due to domain interaction and reduced stability of APOE4, seem to work in concert and gradually lead to dysbalanced Aβ clearance, facilitated tau fibrillation and higher order behavioral disturbances. It is possible that these two pathogenic pathways work in concert and should be addressed together. A better understanding of those seemingly distant pathways and the interaction of APOE4 with other signaling and regulatory molecules – ABCA1 and TREM2, for example, conferring an increased risk themselves, are very important and hopefully will point to reasonable and probably successful therapeutic strategies based on APOEε4. Until then, the ideas of replacing/eliminating APOE4, inhibiting its interactions at an ill-defined age, or ignoring the intervention time seem poorly substantiated. In this review, we cautiously, although briefly, emphasized the complexity of transcriptional regulation of the E/C-I/C-IV/C-II gene cluster and differences in regulatory, including epigenetic, mechanisms in humans and mice. These differences become even more important in APOE TR mice. An attempt to formulate a strong hypothesis on how the risk conferred by the inheritance of the APOEε4 allele is materialized, based on the studies that use more or less complex or extremely complex animal models, is doomed to failure. The overwhelming controversies and inconsistencies in conclusions from otherwise perfectly conducted studies indicate the lack of the appropriate mouse model. Perhaps a model that includes APOE/CI part of APOE cluster with critical regulatory sequences is one promising option. The scientific community needs such a model/models to move further from square one – the inheritance of the APOEε4 allele is the highest genetic risk of AD.
Author contributions
IL and RK contributed to the conception of the review. YL wrote the first draft. IL, RK, and NF co-edited the manuscript. All authors contributed to the writing, have read and agreed on the final manuscript.
Funding
This work was funded by the National Institute on Aging – National Institutes of Health, USA: R01AG06619, R01AG077636, R01AG075992, R01AG057565, and R01AG052978.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Allan, C. M., Taylor, S., and Taylor, J. M. (1997). Two hepatic enhancers, HCR.1 and HCR.2, coordinate the liver expression of the entire human apolipoprotein E/C-I/C-IV/C-II gene cluster. J. Biol. Chem. 272, 29113–29119. doi: 10.1074/jbc.272.46.29113
Allan, C. M., Walker, D., Segrest, J. P., and Taylor, J. M. (1995). Identification and characterization of a new human gene (APOC4) in the apolipoprotein E, C-I, and C-II gene locus. Genomics 28, 291–300. doi: 10.1006/geno.1995.1144
Allen, N. C., Bagade, S., McQueen, M. B., Ioannidis, J. P. A., Kavvoura, F. K., Khoury, M. J., et al. (2008). Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat. Genet. 40, 827–834. doi: 10.1038/ng.171
Amare, A. T., Schubert, K. O., Klingler-Hoffmann, M., Cohen-Woods, S., and Baune, B. T. (2017). The genetic overlap between mood disorders and cardiometabolic diseases: a systematic review of genome wide and candidate gene studies. Transl. Psychiatry 7, –e1007. doi: 10.1038/tp.2016.261
Ando, K., Nagaraj, S., Küçükali, F., De Fisenne, M.-A., Kosa, A.-C., Doeraene, E., et al. (2022). PICALM and Alzheimer’s disease: an update and perspectives. Cells 11:3994. doi: 10.3390/cells11243994
Artiga, M. J., Bullido, M. J., Frank, A., Sastre, I., Recuero, M., García, M. A., et al. (1998). Risk for Alzheimer’s disease correlates with transcriptional activity of the APOE gene. Hum. Mol. Genet. 7, 1887–1892. doi: 10.1093/hmg/7.12.1887
Ashiq, S., and Ashiq, K. (2021). The association of apolipoprotein-E (APOE) gene polymorphisms with coronary artery disease: a systematic review and meta-analysis. Egypt. J. Med. Hum. Genet. 22. doi: 10.1186/s43042-021-00135-2
Atagi, Y., Liu, C. C., Painter, M. M., Chen, X. F., Verbeeck, C., Zheng, H., et al. (2015). Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J. Biol. Chem. 290, 26043–26050. doi: 10.1074/jbc.M115.679043
Bailey, C. C., DeVaux, L. B., and Farzan, M. (2015). The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J. Biol. Chem. 290, 26033–26042. doi: 10.1074/jbc.M115.677286
Bales, K. R., Liu, F., Wu, S., Lin, S., Koger, D., DeLong, C., et al. (2009). Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009
Bhattacharjee, S., Zhao, Y., Dua, P., Rogaev, E. I., and Lukiw, W. J. (2016). micro RNA-34a-mediated down-regulation of the microglial-enriched triggering receptor and phagocytosis-sensor TREM2 in age-related macular degeneration. PLoS One 11:e0150211. doi: 10.1371/journal.pone.0150211
Bras, J., Guerreiro, R., Darwent, L., Parkkinen, L., Ansorge, O., Escott-Price, V., et al. (2014). Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum. Mol. Genet. 23, 6139–6146. doi: 10.1093/hmg/ddu334
Bullido, M. A. J., and Valdivieso, F. (2000). Apolipoprotein E gene promoter polymorphisms in Alzheimer’s disease. Microsc. Res. Tech. 50, 261–267. doi: 10.1002/1097-0029(20000815)50:4<261::aid-jemt2>3.0.co;2-b
Cantoni, C., Bollman, B., Licastro, D., Xie, M., Mikesell, R., Schmidt, R., et al. (2015). TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 129, 429–447. doi: 10.1007/s00401-015-1388-1
Carty, C. L., Bhattacharjee, S., Haessler, J., Cheng, I., Hindorff, L. A., Aroda, V., et al. (2014). Analysis of metabolic syndrome components in >15 000 African Americans identifies pleiotropic variants. Circulation 7, 505–513. doi: 10.1161/circgenetics.113.000386
Castellano, J. M., Kim, J., Stewart, F. R., Jiang, H., DeMattos, R. B., Patterson, B. W., et al. (2011). Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 3:89ra57. doi: 10.1126/scitranslmed.3002156
Castranio, E. L., Mounier, A., Wolfe, C. M., Nam, K. N., Fitz, N. F., Letronne, F., et al. (2017). Gene co-expression networks identify Trem2 and Tyrobp as major hubs in human APOE expressing mice following traumatic brain injury. Neurobiol. Dis. 105, 1–14. doi: 10.1016/j.nbd.2017.05.006
Castranio, E. L., Wolfe, C. M., Nam, K. N., Letronne, F., Fitz, N. F., Lefterov, I., et al. (2018). ABCA1 haplodeficiency affects the brain transcriptome following traumatic brain injury in mice expressing human APOE isoforms. Acta Neuropathol. Commun. 6:69. doi: 10.1186/s40478-018-0569-2
Chen, Y.-M., Chen, P.-K., Chang, C.-K., Lin, C.-C., Chen, H.-H., Lan, J.-L., et al. (2020). Association of Apolipoprotein E Polymorphism with Adipokines and cardiovascular disease risk in rheumatoid arthritis patients. Life 10:330. doi: 10.3390/life10120330
Chen, X., and Holtzman, D. M. (2022). Emerging roles of innate and adaptive immunity in Alzheimer’s disease. Immunity 55, 2236–2254. doi: 10.1016/j.immuni.2022.10.016
Chuang, Y.-F., Hayden, K. M., Norton, M. C., Tschanz, J., Breitner, J. C. S., Welsh-Bohmer, K. A., et al. (2010). Association between APOE ε4 allele and vascular dementia: the cache county study. Dement. Geriatr. Cogn. Disord. 29, 248–253. doi: 10.1159/000285166
Colonna, M. (2023). The biology of TREM receptors. Nat. Rev. Immunol., 7, 1–15. doi: 10.1038/s41577-023-00837-1
Corder, E. H., Saunders, A. M., Risch, N. J., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C. Jr., et al. (1994). Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 7, 180–184. doi: 10.1038/ng0694-180
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Coto, E., Gómez, J., Tavira, B., Tranche, S., Ortega, F., Rodríguez, M. I., et al. (2013). A common APOE polymorphism is an independent risk factor for reduced glomerular filtration rate in the Spanish RENASTUR cohort. Cardiorenal Med. 3, 113–119. doi: 10.1159/000351158
Crawford, F. C., Vanderploeg, R. D., Freeman, M. J., Singh, S., Waisman, M., Michaels, L., et al. (2002). APOE genotype influences acquisition and recall following traumatic brain injury. Neurology 58, 1115–1118. doi: 10.1212/wnl.58.7.1115
Crawford, F., Wood, M., Ferguson, S., Mathura, V., Gupta, P., Humphrey, J., et al. (2009). Apolipoprotein E-genotype dependent hippocampal and cortical responses to traumatic brain injury. Neuroscience 159, 1349–1362. doi: 10.1016/j.neuroscience.2009.01.033
Daniel, B., Nagy, G., Hah, N., Horvath, A., Czimmerer, Z., Poliska, S., et al. (2014). The active enhancer network operated by liganded RXR supports angiogenic activity in macrophages. Genes Dev. 28, 1562–1577. doi: 10.1101/gad.242685.114
Davies, G., Harris, S. E., Reynolds, C. A., Payton, A., Knight, H. M., Liewald, D. C., et al. (2014). A genome-wide association study implicates the APOE locus in nonpathological cognitive ageing. Mol. Psychiatry 19, 76–87. doi: 10.1038/mp.2012.159
Daws, M. R., Sullam, P. M., Niemi, E. C., Chen, T. T., Tchao, N. K., and Seaman, W. E. (2003). Pattern recognition by TREM-2: binding of anionic ligands. J. Immunol. 171, 594–599. doi: 10.4049/jimmunol.171.2.594
Fazio, S., Sanan, D. A., Lee, Y. L., Ji, Z. S., Mahley, R. W., and Rall, S. C. (1994). Susceptibility to diet-induced atherosclerosis in transgenic mice expressing a dysfunctional human apolipoprotein E(Arg 112,Cys 142). Arterioscler. Thromb. 14, 1873–1879. doi: 10.1161/01.atv.14.11.1873
Fitz, N. F., Cronican, A. A., Saleem, M., Fauq, A. H., Chapman, R., Lefterov, I., et al. (2012). Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J. Neurosci. 32, 13125–13136. doi: 10.1523/JNEUROSCI.1937-12.2012
Fitz, N. F., Nam, K. N., Koldamova, R., and Lefterov, I. (2019). Therapeutic targeting of nuclear receptors, liver X and retinoid X receptors, for Alzheimer’s disease. Br. J. Pharmacol. 176, 3599–3610. doi: 10.1111/bph.14668
Fitz, N. F., Nam, K. N., Wolfe, C. M., Letronne, F., Playso, B. E., Iordanova, B. E., et al. (2021). Phospholipids of APOE lipoproteins activate microglia in an isoform-specific manner in preclinical models of Alzheimer’s disease. Nat. Commun. 12:3416. doi: 10.1038/s41467-021-23762-0
Fitz, N. F., Wolfe, C. M., Playso, B. E., Biedrzycki, R. J., Lu, Y., Nam, K. N., et al. (2020). Trem2 deficiency differentially affects phenotype and transcriptome of human APOE3 and APOE4 mice. Mol. Neurodegener. 15:41. doi: 10.1186/s13024-020-00394-4
Foley, K. E., Hewes, A. A., Garceau, D. T., Kotredes, K. P., Carter, G. W., Sasner, M., et al. (2022). The APOE (ε3/ε4) genotype drives distinct gene signatures in the cortex of young mice. Front. Aging Neurosci. 14:838436. doi: 10.3389/fnagi.2022.838436
Garatachea, N., Emanuele, E., Calero, M., Fuku, N., Arai, Y., Abe, Y., et al. (2014). ApoE gene and exceptional longevity: insights from three independent cohorts. Exp. Gerontol. 53, 16–23. doi: 10.1016/j.exger.2014.02.004
Gratuze, M., Leyns, C. E., Sauerbeck, A. D., St-Pierre, M. K., Xiong, M., Kim, N., et al. (2020). Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J. Clin. Invest. 130, 4954–4968. doi: 10.1172/JCI138179
Gratuze, M., Schlachetzki, J. C. M., D’Oliveira Albanus, R., Jain, N., Novotny, B., Brase, L., et al. (2023). TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4. Neuron 111, 202–219 e7. doi: 10.1016/j.neuron.2022.10.022
Grehan, S., Tse, E., and Taylor, J. M. (2001). Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. J. Neurosci. 21, 812–822. doi: 10.1523/jneurosci.21-03-00812.2001
Hardy, J. (2009). The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J. Neurochem. 110, 1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x
Hirsch-Reinshagen, V., Maia, L. F., Burgess, B. L., Blain, J. F., Naus, K. E., McIsaac, S. A., et al. (2005). The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J. Biol. Chem. 280, 43243–43256. doi: 10.1074/jbc.M508781200
Hirsch-Reinshagen, V., Zhou, S., Burgess, B. L., Bernier, L., McIsaac, S. A., Chan, J. Y., et al. (2004). Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 279, 41197–41207. doi: 10.1074/jbc.M407962200
Holstege, H., Hulsman, M., Charbonnier, C., Grenier-Boley, B., Quenez, O., Grozeva, D., et al. (2022). Exome sequencing identifies rare damaging variants in ATP8B4 and ABCA1 as risk factors for Alzheimer’s disease. Nat. Genet. 29113–29119. doi: 10.1038/s41588-022-01208-7
Holtzman, D. M., Bales, K. R., Tenkova, T., Fagan, A. M., Parsadanian, M., Sartorius, L. J., et al. (2000). Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 97, 2892–2897. doi: 10.1073/pnas.050004797
Jack, C. R., Bennett, D. A., Blennow, K., Carrillo, M. C., Dunn, B., Haeberlein, S. B., et al. (2018). NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 14, 535–562. doi: 10.1016/j.jalz.2018.02.018
Jay, T. R., Hirsch, A. M., Broihier, M. L., Miller, C. M., Neilson, L. E., Ransohoff, R. M., et al. (2017a). Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 37, 637–647. doi: 10.1523/JNEUROSCI.2110-16.2016
Jay, T. R., Miller, C. M., Cheng, P. J., Graham, L. C., Bemiller, S., Broihier, M. L., et al. (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 212, 287–295. doi: 10.1084/jem.20142322
Jay, T. R., von Saucken, V. E., and Landreth, G. E. (2017b). TREM2 in neurodegenerative diseases. Mol. Neurodegener. 12:56. doi: 10.1186/s13024-017-0197-5
Jendresen, C., Arskog, V., Daws, M. R., and Nilsson, L. N. (2017). The Alzheimer’s disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J. Neuroinflammation 14:59. doi: 10.1186/s12974-017-0835-4
Jerico, I., Vicuna-Urriza, J., Blanco-Luquin, I., Macias, M., Martinez-Merino, L., Roldan, M., et al. (2023). Profiling TREM2 expression in amyotrophic lateral sclerosis. Brain Behav. Immun. 109, 117–126. doi: 10.1016/j.bbi.2023.01.013
Jo, D.-W., Leren, T. P., Yang, Z.-Y., Chung, Y.-H., Taylor, J. M., and Paik, Y.-K. (1995). Characterization of an upstream regulatory element of the human apolipoprotein E gene, and purification of its binding protein from the human Placenta1. J. Biochem. 117, 915–922. doi: 10.1093/oxfordjournals.jbchem.a124796
Josephs, K. A., Duffy, J. R., Strand, E. A., Machulda, M. M., Senjem, M. L., Lowe, V. J., et al. (2014). APOE ε4 influences β-amyloid deposition in primary progressive aphasia and speech apraxia. Alzheimer’s Dement. 10, 630–636. doi: 10.1016/j.jalz.2014.03.004
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cells 169, 1276–1290 e17. doi: 10.1016/j.cell.2017.05.018
Kim, J., Basak, J. M., and Holtzman, D. M. (2009). The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303. doi: 10.1016/j.neuron.2009.06.026
Kim, T. Y., Chung, H. G., Shin, H.-S., Kim, S. J., Choi, J. H., Chung, M. Y., et al. (2013). Apolipoprotein E gene polymorphism, alcohol use, and their interactions in combat-related posttraumatic stress disorder. Depress. Anxiety 30, 1194–1201. doi: 10.1002/da.22138
Kim, J., Jiang, H., Park, S., Eltorai, A. E., Stewart, F. R., Yoon, H., et al. (2011). Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J. Neurosci. 31, 18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011
Klesney-Tait, J., Turnbull, I. R., and Colonna, M. (2006). The TREM receptor family and signal integration. Nat. Immunol. 7, 1266–1273. doi: 10.1038/ni1411
Kober, D. L., Alexander-Brett, J. M., Karch, C. M., Cruchaga, C., Colonna, M., Holtzman, M. J., et al. (2016). Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. elife 5, e20391. doi: 10.7554/eLife.20391
Kober, D. L., Stuchell-Brereton, M. D., Kluender, C. E., Dean, H. B., Strickland, M. R., Steinberg, D. F., et al. (2020). Functional insights from biophysical study of TREM2 interactions with apoE and Abeta (1-42). Alzheimers Dement. 17, 475–488. doi: 10.1002/alz.12194
Koldamova, R., Fitz, N. F., and Lefterov, I. (2014). ATP-binding cassette transporter A1: from metabolism to neurodegeneration. Neurobiol. Dis. 72, 13–21. doi: 10.1016/j.nbd.2014.05.007
Koldamova, R., and Lefterov, I. (2007). Role of LXR and ABCA1 in the pathogenesis of Alzheimer’s disease - implications for a new therapeutic approach. Curr. Alzheimer Res. 4, 171–178. doi: 10.2174/156720507780362227
Koldamova, R. P., Lefterov, I. M., Ikonomovic, M. D., Skoko, J., Lefterov, P. I., Isanski, B. A., et al. (2003). 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid β secretion. J. Biol. Chem. 278, 13244–13256. doi: 10.1074/jbc.m300044200
Koldamova, R., Staufenbiel, M., and Lefterov, I. (2005). Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 280, 43224–43235. doi: 10.1074/jbc.M504513200
Koopal, C., Van Der Graaf, Y., Asselbergs, F. W., Westerink, J., and Visseren, F. L. J. (2015). Influence of APOE-2 genotype on the relation between adiposity and plasma lipid levels in patients with vascular disease. Int. J. Obes. 39, 265–269. doi: 10.1038/ijo.2014.105
Kotredes, K. P., Oblak, A., Pandey, R. S., Lin, P. B., Garceau, D., Williams, H., et al. (2021). Uncovering disease mechanisms in a novel mouse model expressing humanized APOEepsilon4 and Trem2*R47H. Front. Aging Neurosci. 13:735524. doi: 10.3389/fnagi.2021.735524
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581 e9. doi: 10.1016/j.immuni.2017.08.008
Krell-Roesch, J., Pink, A., Roberts, R. O., Stokin, G. B., Mielke, M. M., Spangehl, K. A., et al. (2016). Timing of physical activity, apolipoprotein E ε4 genotype, and risk of incident mild cognitive impairment. J. Am. Geriatr. Soc. 64, 2479–2486. doi: 10.1111/jgs.14402
Laffitte, B. A., Joseph, S. B., Chen, M., Castrillo, A., Repa, J., Wilpitz, D., et al. (2003). The phospholipid transfer protein gene is a liver X receptor target expressed by macrophages in atherosclerotic lesions. Mol. Cell. Biol. 23, 2182–2191. doi: 10.1128/mcb.23.6.2182-2191.2003
Laffitte, B. A., Repa, J. J., Joseph, S. B., Wilpitz, D. C., Kast, H. R., Mangelsdorf, D. J., et al. (2001). LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc. Natl. Acad. Sci. U. S. A. 98, 507–512. doi: 10.1073/pnas.98.2.507
Lambert, J. C., Berr, C., Pasquier, F., Delacourte, A., Frigard, B., Cottel, D., et al. (1998a). Pronounced impact of Th1/E47cs mutation compared with-491 AT mutation on neural APOE gene expression and risk of developing Alzheimer’s disease. Hum. Mol. Genet. 7, 1511–1516. doi: 10.1093/hmg/7.9.1511
Lambert, J. C., Brousseau, T., Defosse, V., Evans, A., Arveiler, D., Ruidavets, J. B., et al. (2000). Independent association of an APOE gene promoter polymorphism with increased risk of myocardial infarction and decreased APOE plasma concentrations-the ECTIM study. Hum. Mol. Genet. 9, 57–61. doi: 10.1093/hmg/9.1.57
Lambert, J. C., Pasquier, F., Cottel, D., Frigard, B., Amouyel, P., and Chartier-Harlin, M. C. (1998b). A new polymorphism in the APOE promoter associated with risk of developing Alzheimer’s disease. Hum. Mol. Genet. 7, 533–540. doi: 10.1093/hmg/7.3.533
Lambert, J. C., Perez-Tur, J., Dupire, M. J., Galasko, D., Mann, D., Amouyel, P., et al. (1997). Distortion of allelic expression of apolipoprotein E in Alzheimer’s disease. Hum. Mol. Genet. 6, 2151–2154. doi: 10.1093/hmg/6.12.2151
Lee, E.-G., Tulloch, J., Chen, S., Leong, L., Saxton, A. D., Kraemer, B., et al. (2020). Redefining transcriptional regulation of the APOE gene and its association with Alzheimer’s disease. PLoS One 15:e0227667. doi: 10.1371/journal.pone.0227667
Lefterov, I., Schug, J., Mounier, A., Nam, K. N., Fitz, N. F., and Koldamova, R. (2015). RNA-sequencing reveals transcriptional up-regulation of Trem2 in response to bexarotene treatment. Neurobiol. Dis. 82, 132–140. doi: 10.1016/j.nbd.2015.05.019
Lessard, C. B., Malnik, S. L., Zhou, Y., Ladd, T. B., Cruz, P. E., Ran, Y., et al. (2018). High-affinity interactions and signal transduction between Abeta oligomers and TREM2. EMBO Mol. Med. 10, e9027. doi: 10.15252/emmm.201809027
Leyns, C. E. G., Gratuze, M., Narasimhan, S., Jain, N., Koscal, L. J., Jiang, H., et al. (2019). TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 22, 1217–1222. doi: 10.1038/s41593-019-0433-0
Li, Y., Macyczko, J. R., Liu, C.-C., and Bu, G. (2022). ApoE4 reduction: An emerging and promising therapeutic strategy for Alzheimer’s disease. Neurobiol. Aging 115, 20–28. doi: 10.1016/j.neurobiolaging.2022.03.011
Liao, F., Li, A., Xiong, M., Bien-Ly, N., Jiang, H., Zhang, Y., et al. (2018). Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Invest. 128, 2144–2155. doi: 10.1172/JCI96429
Lindner, K., Beckenbauer, K., Van Ek, L. C., Titeca, K., De Leeuw, S. M., Awwad, K., et al. (2022). Isoform-and cell-state-specific lipidation of ApoE in astrocytes. Cell Rep. 38:110435. doi: 10.1016/j.celrep.2022.110435
Liu, G., Liu, Y., Jiang, Q., Jiang, Y., Feng, R., Zhang, L., et al. (2016). Convergent genetic and expression datasets highlight TREM2 in Parkinson’s disease susceptibility. Mol. Neurobiol. 53, 4931–4938. doi: 10.1007/s12035-015-9416-7
Loppi, S., Kolosowska, N., Karkkainen, O., Korhonen, P., Huuskonen, M., Grubman, A., et al. (2018). HX600, a synthetic agonist for RXR-Nurr1 heterodimer complex, prevents ischemia-induced neuronal damage. Brain Behav. Immun. 73, 670–681. doi: 10.1016/j.bbi.2018.07.021
Lumsden, A. L., Mulugeta, A., Zhou, A., and Hypponen, E. (2020). Apolipoprotein E (APOE) genotype-associated disease risks: a phenome-wide, registry-based, case-control study utilising the UK biobank. EBioMedicine 59:102954. doi: 10.1016/j.ebiom.2020.102954
Ma, W., Zhang, L., Luo, L., Zhang, S., Yang, S., Yao, H., et al. (2022). Effect of apolipoprotein E ε4 allele on the progression of carotid atherosclerosis through apolipoprotein levels. Pharmgenomics Pers. Med. 15, 653–661. doi: 10.2147/pgpm.s367471
Ma, H., Zhou, T., Li, X., Maraganore, D., Heianza, Y., and Qi, L. (2022). Early-life educational attainment, APOE ε4 alleles, and incident dementia risk in late life. Gero Sci. 44, 1479–1488. doi: 10.1007/s11357-022-00545-z
Mahley, R. W., and Rall, S. C. (2000). Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 1, 507–537. doi: 10.1146/annurev.genom.1.1.507
Mak, P. A., Laffitte, B. A., Desrumaux, C., Joseph, S. B., Curtiss, L. K., Mangelsdorf, D. J., et al. (2002). Regulated expression of the apolipoprotein E/C-I/C-IV/C-II gene cluster in murine and human macrophages. J. Biol. Chem. 277, 31900–31908. doi: 10.1074/jbc.m202993200
Martens, Y. A., Zhao, N., Liu, C.-C., Kanekiyo, T., Yang, A. J., Goate, A. M., et al. (2022). ApoE cascade hypothesis in the pathogenesis of Alzheimer’s disease and related dementias. Neuron 110, 1304–1317. doi: 10.1016/j.neuron.2022.03.004
Mata, I. F., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Hurtig, H. I., Van Deerlin, V. M., et al. (2014). APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 71:1405. doi: 10.1001/jamaneurol.2014.1455
Meilandt, W. J., Ngu, H., Gogineni, A., Lalehzadeh, G., Lee, S. H., Srinivasan, K., et al. (2020). Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Abeta42:Abeta40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J. Neurosci. 40, 1956–1974. doi: 10.1523/JNEUROSCI.1871-19.2019
Mui, S., Briggs, M., Chung, H., Wallace, R. B., Gomez-Isla, T., Rebeck, G. W., et al. (1996). A newly identified polymorphism in the apolipoprotein E enhancer gene region is associated with Alzheimer’s disease and strongly with the epsilon 4 allele. Neurology 47, 196–201. doi: 10.1212/wnl.47.1.196
Nguyen, A. T., Wang, K., Hu, G., Wang, X., Miao, Z., Azevedo, J. A., et al. (2020). APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 140, 477–493. doi: 10.1007/s00401-020-02200-3
Nicoll, J. A. R., Roberts, G. W., and Graham, D. I. (1995). Apolipoprotein E ε4 allele is associated with deposition of amyloid β-protein following head injury. Nat. Med. 1, 135–137. doi: 10.1038/nm0295-135
Nugent, A. A., Lin, K., van Lengerich, B., Lianoglou, S., Przybyla, L., Davis, S. S., et al. (2020). TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837–854 e9. doi: 10.1016/j.neuron.2019.12.007
Oblak, A. L., Forner, S., Territo, P. R., Sasner, M., Carter, G. W., Howell, G. R., et al. (2020). Model organism development and evaluation for late-onset Alzheimer’s disease: MODEL-AD. Alzheimers Dement 6:e12110. doi: 10.1002/trc2.12110
Oram, J. F., and Vaughan, A. M. (2000). ABCA1-mediated transport of cellular cholesterol and phospholipids to HDL apolipoproteins. Curr. Opin. Lipidol. 11, 253–260. doi: 10.1097/00041433-200006000-00005
Pahnke, J., Bascunana, P., Brackhan, M., Stefan, K., Namasivayam, V., Koldamova, R., et al. (2021). Strategies to gain novel Alzheimer’s disease diagnostics and therapeutics using modulators of ABCA transporters. Free Neuropathol. 2, 33. doi: 10.17879/freeneuropathology-2021-3528
Paik, Y.-K., Reardon, C. A., Taylor, J. M., and Choi, B.-K. (1995). Characterization of an upstream regulatory sequence and its binding protein in the mouse apolipoprotein E gene. Biochim. Biophys. Acta 1262, 124–132. doi: 10.1016/0167-4781(95)00048-L
Parhizkar, S., Arzberger, T., Brendel, M., Kleinberger, G., Deussing, M., Focke, C., et al. (2019). Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 22, 191–204. doi: 10.1038/s41593-018-0296-9
Perez, S. E., Nadeem, M., He, B., Miguel, J. C., Malek-Ahmadi, M. H., Chen, K., et al. (2017). Neocortical and hippocampal TREM2 protein levels during the progression of Alzheimer’s disease. Neurobiol. Aging 54, 133–143. doi: 10.1016/j.neurobiolaging.2017.02.012
Pimenova, A. A., Marcora, E., and Goate, A. M. (2017). A tale of two genes: microglial Apoe and Trem2. Immunity 47, 398–400. doi: 10.1016/j.immuni.2017.08.015
Poliani, P. L., Wang, Y., Fontana, E., Robinette, M. L., Yamanishi, Y., Gilfillan, S., et al. (2015). TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest. 125, 2161–2170. doi: 10.1172/JCI77983
Raha, A. A., Henderson, J. W., Stott, S. R., Vuono, R., Foscarin, S., Friedland, R. P., et al. (2017). Neuroprotective effect of TREM-2 in aging and Alzheimer’s disease model. J. Alzheimers Dis. 55, 199–217. doi: 10.3233/JAD-160663
Rao, R. V., Subramaniam, K. G., Gregory, J., Bredesen, A. L., Coward, C., Okada, S., et al. (2023). Rationale for a multi-factorial approach for the reversal of cognitive decline in Alzheimer’s disease and MCI: a review. Int. J. Mol. Sci. 24:1659. doi: 10.3390/ijms24021659
Raulin, A. C., Doss, S. V., Trottier, Z. A., Ikezu, T. C., Bu, G., and Liu, C. C. (2022). ApoE in Alzheimer’s disease: pathophysiology and therapeutic strategies. Mol. Neurodegener. 17:72. doi: 10.1186/s13024-022-00574-4
Romero, J. R., Preis, S. R., Beiser, A., Decarli, C., Viswanathan, A., Martinez-Ramirez, S., et al. (2014). Risk factors, stroke prevention treatments, and prevalence of cerebral microbleeds in the Framingham heart study. Stroke 45, 1492–1494. doi: 10.1161/strokeaha.114.004130
Roses, A. D., Strittmatter, W. J., Pericak-Vance, M. A., Corder, E. H., Saunders, A. M., and Schmechel, D. E. (1994). Clinical application of apolipoprotein E genotyping to Alzheimer’s disease. Lancet 343, 1564–1565. doi: 10.1016/s0140-6736(94)92960-2
Rubino, E., Vacca, A., Govone, F., De Martino, P., Pinessi, L., and Rainero, I. (2013). Apolipoprotein E polymorphisms in frontotemporal lobar degeneration: a meta-analysis. Alzheimer’s Dement. 9, 706–713. doi: 10.1016/j.jalz.2012.10.013
Saber, M., Kokiko-Cochran, O., Puntambekar, S. S., Lathia, J. D., and Lamb, B. T. (2017). Triggering receptor expressed on myeloid cells 2 deficiency alters acute macrophage distribution and improves recovery after traumatic brain injury. J. Neurotrauma 34, 423–435. doi: 10.1089/neu.2016.4401
Sala Frigerio, C., Wolfs, L., Fattorelli, N., Thrupp, N., Voytyuk, I., Schmidt, I., et al. (2019). The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Abeta plaques. Cell Rep. 27, 1293–1306.e6. doi: 10.1016/j.celrep.2019.03.099
Sayed, F. A., Telpoukhovskaia, M., Kodama, L., Li, Y., Zhou, Y., Le, D., et al. (2018). Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc. Natl. Acad. Sci. U. S. A. 115, 10172–10177. doi: 10.1073/pnas.1811411115
Sharan, S. K., Thomason, L. C., Kuznetsov, S. G., and Court, D. L. (2009). Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 4, 206–223. doi: 10.1038/nprot.2008.227
Shih, S.-J., Allan, C., Grehan, S., Tse, E., Moran, C., and Taylor, J. M. (2000). Duplicated downstream enhancers control expression of the human apolipoprotein E gene in macrophages and adipose tissue. J. Biol. Chem. 275, 31567–31572. doi: 10.1074/jbc.m005468200
Shirotani, K., Hori, Y., Yoshizaki, R., Higuchi, E., Colonna, M., Saito, T., et al. (2019). Aminophospholipids are signal-transducing TREM2 ligands on apoptotic cells. Sci. Rep. 9:7508. doi: 10.1038/s41598-019-43535-6
Simonet, W. S., Bucay, N., Lauer, S. J., and Taylor, J. M. (1993). A far-downstream hepatocyte-specific control region directs expression of the linked human apolipoprotein E and C-I genes in transgenic mice. J. Biol. Chem. 268, 8221–8229.
Sims, R., van der Lee, S. J., Naj, A. C., Bellenguez, C., Badarinarayan, N., Jakobsdottir, J., et al. (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49, 1373–1384. doi: 10.1038/ng.3916
Smith, J. D., Melian, A., Leff, T., and Breslow, J. L. (1988). Expression of the human apolipoprotein E gene is regulated by multiple positive and negative elements. J. Biol. Chem. 263, 8300–8308.
Song, W., Hooli, B., Mullin, K., Jin, S. C., Cella, M., Ulland, T. K., et al. (2017). Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 13, 381–387. doi: 10.1016/j.jalz.2016.07.004
Song, S., Yu, L., Hasan, M. N., Paruchuri, S. S., Mullett, S. J., Sullivan, M. L. G., et al. (2022). Elevated microglial oxidative phosphorylation and phagocytosis stimulate post-stroke brain remodeling and cognitive function recovery in mice. Commun. Biol. 5:35. doi: 10.1038/s42003-021-02984-4
Sullivan, P. M., Han, B., Liu, F., Mace, B. E., Ervin, J. F., Wu, S., et al. (2009). Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol. Aging 32, 791–801. doi: 10.1016/j.neurobiolaging.2009.05.011
Sullivan, P. M., Mezdour, H., Aratani, Y., Knouff, C., Najib, J., Reddick, R. L., et al. (1997). Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 272, 17972–17980. doi: 10.1074/jbc.272.29.17972
Thornton, P., Sevalle, J., Deery, M. J., Fraser, G., Zhou, Y., Stahl, S., et al. (2017). TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol. Med. 9, 1366–1378. doi: 10.15252/emmm.201707673
Ulland, T. K., Song, W. M., Huang, S. C., Ulrich, J. D., Sergushichev, A., Beatty, W. L., et al. (2017). TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cells 170, 649–663 e13. doi: 10.1016/j.cell.2017.07.023
Villegas-Llerena, C., Phillips, A., Garcia-Reitboeck, P., Hardy, J., and Pocock, J. M. (2016). Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 36, 74–81. doi: 10.1016/j.conb.2015.10.004
Wahrle, S. E., Jiang, H., Parsadanian, M., Hartman, R. E., Bales, K. R., Paul, S. M., et al. (2005). Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 280, 43236–43242. doi: 10.1074/jbc.M508780200
Wahrle, S. E., Jiang, H., Parsadanian, M., Legleiter, J., Han, X., Fryer, J. D., et al. (2004). ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 279, 40987–40993. doi: 10.1074/jbc.m407963200
Wang, Y., Cella, M., Mallinson, K., Ulrich, J. D., Young, K. L., Robinette, M. L., et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cells 160, 1061–1071. doi: 10.1016/j.cell.2015.01.049
Wang, Y., Ulland, T. K., Ulrich, J. D., Song, W., Tzaferis, J. A., Hole, J. T., et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675. doi: 10.1084/jem.20151948
Wang, C., Xiong, M., Gratuze, M., Bao, X., Shi, Y., Andhey, P. S., et al. (2021). Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 109, 1657–1674.e7. doi: 10.1016/j.neuron.2021.03.024
Wes, P. D., Sayed, F. A., Bard, F., and Gan, L. (2016). Targeting microglia for the treatment of Alzheimer’s disease. Glia 64, 1710–1732. doi: 10.1002/glia.22988
Wolfe, C., Fitz, N., Nam, K., Lefterov, I., and Koldamova, R. (2018). The role of APOE and TREM2 in Alzheimer′s disease—current understanding and perspectives. Int. J. Mol. Sci. 20:81. doi: 10.3390/ijms20010081
Wood, J. I., Wong, E., Joghee, R., Balbaa, A., Vitanova, K. S., Stringer, K. M., et al. (2022). Plaque contact and unimpaired Trem2 is required for the microglial response to amyloid pathology. Cell Rep. 41:111686. doi: 10.1016/j.celrep.2022.111686
Wu, K., Byers, D. E., Jin, X., Agapov, E., Alexander-Brett, J., Patel, A. C., et al. (2015). TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J. Exp. Med. 212, 681–697. doi: 10.1084/jem.20141732
Wunderlich, P., Glebov, K., Kemmerling, N., Tien, N. T., Neumann, H., and Walter, J. (2013). Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J. Biol. Chem. 288, 33027–33036. doi: 10.1074/jbc.M113.517540
Yeh, F. L., Hansen, D. V., and Sheng, M. (2017). TREM2, microglia, and neurodegenerative diseases. Trends Mol. Med. 23, 512–533. doi: 10.1016/j.molmed.2017.03.008
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yen, Y.-C., Rebok, G. W., Gallo, J. J., Yang, M.-J., Lung, F.-W., and Shih, C.-H. (2007). ApoE4 allele is associated with late-life depression: a population-based study. Am. J. Geriatr. Psychiatr. 15, 858–868. doi: 10.1097/jgp.0b013e3180f63373
Yuan, P., Condello, C., Keene, C. D., Wang, Y., Bird, T. D., Paul, S. M., et al. (2016). TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739. doi: 10.1016/j.neuron.2016.05.003
Zhao, Y., Wu, X., Li, X., Jiang, L. L., Gui, X., Liu, Y., et al. (2018). TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron 97, 1023–1031.e7. doi: 10.1016/j.neuron.2018.01.031
Zheng, H., Cheng, B., Li, Y., Li, X., Chen, X., and Zhang, Y. W. (2018). TREM2 in Alzheimer’s disease: microglial survival and energy metabolism. Front. Aging Neurosci. 10:395. doi: 10.3389/fnagi.2018.00395
Zheng, H., Liu, C. C., Atagi, Y., Chen, X. F., Jia, L., Yang, L., et al. (2016). Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol. Aging 42, 132–141. doi: 10.1016/j.neurobiolaging.2016.03.004
Keywords: Alzheimer’s disease, risk, APOE, TREM2, transcriptional control, animal model
Citation: Lefterov I, Fitz NF, Lu Y and Koldamova R (2023) APOEε4 and risk of Alzheimer’s disease – time to move forward. Front. Neurosci. 17:1195724. doi: 10.3389/fnins.2023.1195724
Edited by:
Mark P. Burns, Georgetown University, United StatesReviewed by:
Efthimios M. C. Skoulakis, Alexander Fleming Biomedical Sciences Research Center, GreeceYonghe Li, Mayo Clinic Florida, United States
Copyright © 2023 Lefterov, Fitz, Lu and Koldamova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iliya Lefterov, aWxpeWFsQHBpdHQuZWR1; Radosveta Koldamova, cmFkYWtAcGl0dC5lZHU=
†These authors have contributed equally to this work and share senior authorship