 Maged M. Harraz1,2*
Maged M. Harraz1,2*- 1Department of Psychiatry, University of Maryland School of Medicine, Baltimore, MD, United States
- 2Department of Pharmacology, University of Maryland School of Medicine, Baltimore, MD, United States
One of the hallmarks of Parkinson’s disease (PD) is the progressive loss of dopaminergic neurons and associated dopamine depletion. Several mechanisms, previously considered in isolation, have been proposed to contribute to the pathophysiology of dopaminergic degeneration: dopamine oxidation-mediated neurotoxicity, high dopamine transporter (DAT) expression density per neuron, and autophagy-lysosome pathway (ALP) dysfunction. However, the interrelationships among these mechanisms remained unclear. Our recent research bridges this gap, recognizing autophagy as a novel dopamine homeostasis regulator, unifying these concepts. I propose that autophagy modulates dopamine reuptake by selectively degrading DAT. In PD, ALP dysfunction could increase DAT density per neuron, and enhance dopamine reuptake, oxidation, and neurotoxicity, potentially contributing to the progressive loss of dopaminergic neurons. This integrated understanding may provide a more comprehensive view of aspects of PD pathophysiology and opens new avenues for therapeutic interventions.
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative movement disorder. While mainly characterized by motor syndrome, PD is associated with non-motor symptoms (NMS) in almost all patients. More than 10 million primarily older adults worldwide suffer from PD. The most common symptom associated with PD is tremor, which typically begins on one side before eventually affecting both sides over time. Other symptoms may include muscle rigidity (stiffness), gait disturbances (difficulty walking), slowness/decreased mobility, impaired speech/swallowing problems, depression/anxiety disorders, hyposmia, constipation, urinary dysfunction, orthostatic hypotension, pain, sleep disturbances, and cognitive decline, including dementia or psychosis later in life if left untreated for too long. While there is no cure for PD, symptomatic dopaminergic therapy helps improve the quality of life for those with the disease. PD is characterized by the presence of certain hallmarks in its pathology. These include progressive selective loss of dopaminergic neurons in the midbrain substantia nigra pars compacta (SNpc); Lewy bodies containing aggregates of α-synuclein protein; gliosis or inflammation around affected areas; and a dramatic decrease in dopamine, a neurotransmitter associated with movement and reward. While the motor syndrome of PD is related to nigral degeneration and dopamine depletion, NMS may be linked to the degeneration of other neural types, including the peripheral autonomic nervous system (Girault and Greengard, 2004; Tolosa et al., 2021; Haider et al., 2023). PD is a multifactorial disease with genetic and environmental factors that converge on common pathophysiological processes (Simon et al., 2020). Dopaminergic therapy can improve motor symptoms but does not alter the underlying progressive neurodegeneration. As such, there is a need to develop disease-modifying therapeutic strategies that target underlying pathophysiological mechanisms to slow or arrest the progression of PD (Murakami et al., 2023).
The molecular pathogenesis of PD involves a complex interplay of several factors, such as impaired mitochondria, protein homeostasis, autophagy-lysosome pathway (ALP) dysfunction at the synapse, trafficking of the dopamine transporter (DAT), DA toxicity, oxidative stress, disruptions in synaptic vesicle endocytosis, autonomous pacemaking, calcium homeostasis imbalance, iron-content, extensive axonal arborization size, prion-like α-synuclein transmission, and neuroinflammation. The reader is referred to several review articles that discuss these pathophysiological mechanisms in detail (Lohr et al., 2017; Giguère et al., 2018; Nguyen et al., 2019; Lu et al., 2020; Bu et al., 2021). Acknowledging that neuronal loss in PD cannot be explained by one pathway, I focus on uniting three of the above-mentioned pathophysiological mechanisms: (1) Dysfunction of the ALP. (2) High expression density of DAT per neuron, which strongly correlates with the extent of dopaminergic neuron loss. (3) Dopamine oxidation mediated neurotoxicity. Several groups characterized each of these mechanisms for their potential role in the pathophysiology of PD. Still, there was no apparent connection to link them together (Figure 1A). This minireview unites these concepts and clarifies their interrelationships considering our recent discovery of autophagy as a novel mechanism for controlling dopamine homeostasis (Figure 1B). Here, I discuss how dysregulated autophagic degradation of DAT could be considered a contributing element in mediating selective dopaminergic degeneration in PD through DA toxicity.
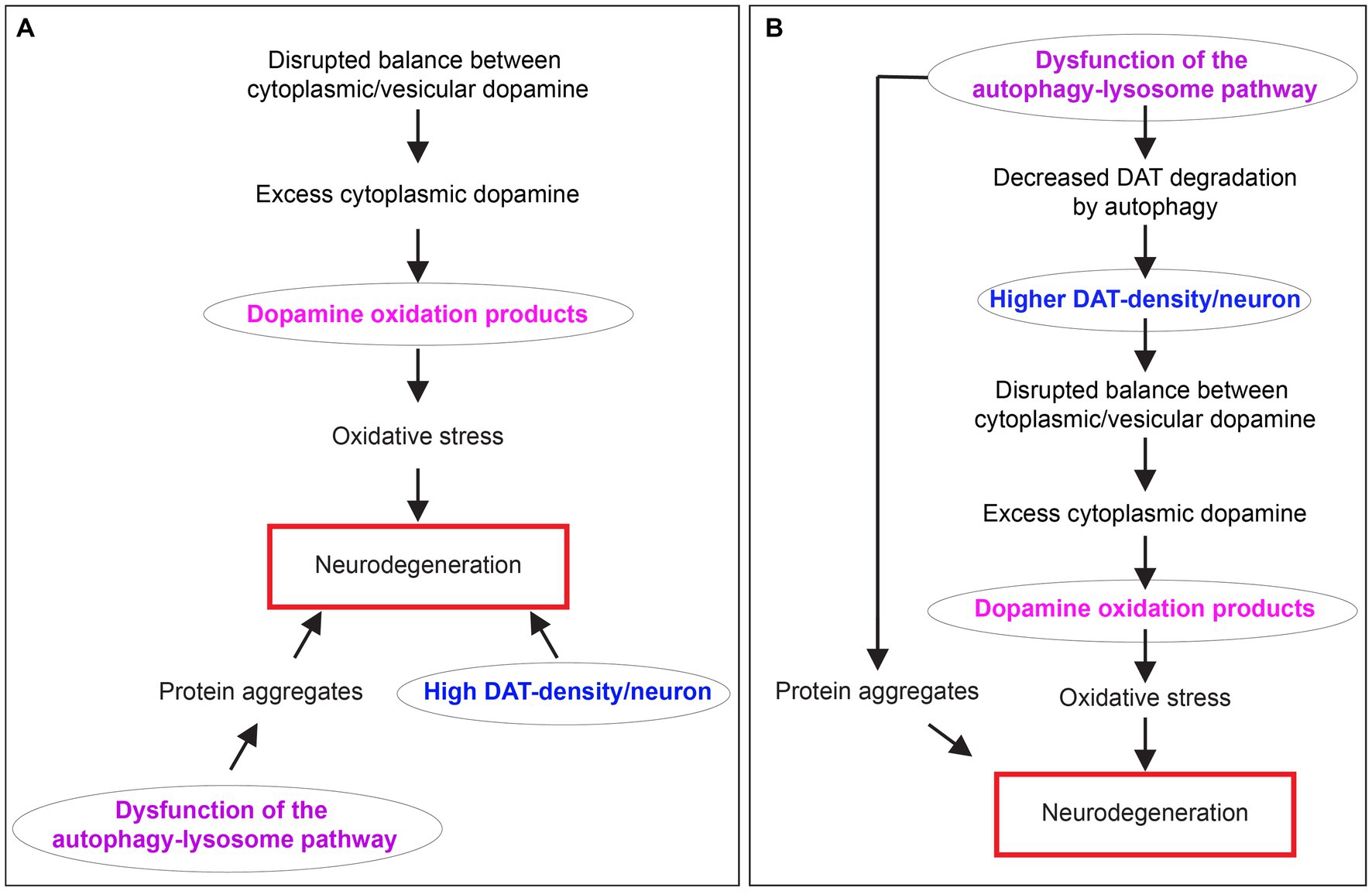
Figure 1. The relationship between dopamine oxidation, DAT expression density, and autophagy-lysosome pathway dysfunction in neurodegeneration in PD. (A) A diagram showing three separate mechanisms are proposed to contribute to neurodegeneration in PD. (B) A diagram depicting the interrelationship between dopamine oxidation, DAT expression density, and autophagy-lysosome pathway dysfunction in PD neurodegeneration unified by DAT autophagy.
The autophagy lysosome pathway in PD
The ALP plays a crucial role in the pathogenesis of PD. Autophagy, or self-eating, is a process by which cells degrade and recycle their cellular components, such as organelles, proteins, lipids, and nucleic acids, for use in metabolic processes. The autophagic process is regulated by several molecular pathways involving the lysosomes and other organelles, including the endoplasmic reticulum and mitochondria (Fleming et al., 2022). The lysosomes are the destination for autophagic cargo for degradation. There are three types of autophagy:
1. Macroautophagy: characterized by the formation of a double membrane organelle, the autophagosome, which forms around its cargo and transports it to the lysosome. There are two types of macroautophagy in terms of cargo recognition: selective and non-selective. Relevant examples of selective macroautophagy include mitophagy (mitochondria) and aggrephagy (protein aggregates), among many others (Galluzzi et al., 2017).
2. Microautophagy: characterized by the lysosome’s direct internalization of small cytoplasmic cargo. Microautophagy can also be selective and non-selective (Wang et al., 2023).
3. Chaperone-mediated autophagy (CMA): characterized by selective recognition of cytoplasmic proteins harboring a KFERQ or KFERQ-like motif by chaperones that escort them to the lysosome where they undergo translocation through the lysosomal membrane to its lumen for degradation (Kaushik and Cuervo, 2012).
In PD, the ALP pathway has been found to be dysregulated in both familial (Beilina et al., 2014) and sporadic (Kabuta and Wada, 2008; Wu et al., 2008, 2011; Lonskaya et al., 2013; Beilina et al., 2014; Moors et al., 2019) forms of the disease, leading to the accumulation of toxic protein aggregates within neurons resulting in neuronal death or dysfunction.
Several genes related to ALP have been implicated in PD due to their mutations. These genes have roles in various aspects of the pathway, from regulating autophagy initiation to the proper functioning of lysosomes. Here are a few examples of some key genes:
• Leucine-rich repeat kinase 2 (LRRK2) mutations are one of the most common genetic causes of autosomal dominant PD. The LRRK2 protein is involved in various cellular processes, including autophagy and endolysosomal functions. Mutations can lead to impaired autophagy and increased susceptibility to PD (Gómez-Suaga and Hilfiker, 2012; Orenstein et al., 2013; Ysselstein et al., 2019).
• GBA mutations are the most frequent genetic risk factor for PD. GBA encodes the lysosomal enzyme glucocerebrosidase, which is involved in the breakdown of glucosylceramide. Reduced glucocerebrosidase activity is associated with impaired lysosomal function and α-synuclein accumulation (Pang et al., 2022; Pradas and Martinez-Vicente, 2023).
• Vacuolar protein sorting ortholog 35 (VPS35) is a retromer complex component. VPS35 mutations have been linked to autosomal dominant PD. VPS35/retromer complex is essential for the retrieving specific membrane proteins from endosomes to the trans-Golgi network, thereby maintaining lysosomal function (Zavodszky et al., 2014; Cui et al., 2021).
• ATPase 13A2 (ATP13A2), also known as PARK9, is a lysosomal P-type ATPase. Mutations in this gene are associated with a rare form of early-onset parkinsonism (Kufor-Rakeb syndrome). ATP13A2 is involved in maintaining lysosomal pH and manganese homeostasis, and its dysfunction contributes to impaired autophagy (Dhanushkodi et al., 2023).
• PTEN-induced putative kinase 1 (PINK1) mutations are associated with autosomal recessive early-onset PD. PINK1 is a mitochondrial kinase that plays a key role in mitophagy, a selective form of autophagy responsible for removing damaged mitochondria (Jones, 2010).
• Parkin RBR E3 Ubiquitin Protein Ligase (PARKIN) mutations are also linked to autosomal recessive early-onset PD. PARKIN functions as an E3 ubiquitin ligase and works with PINK1 in the mitophagy pathway, promoting the clearance of damaged mitochondria (Herman and Moussa, 2011).
• Protein deglycase DJ-1, also known as Parkinson’s disease protein 7 (DJ-1), mutations are implicated in autosomal recessive early-onset PD. DJ-1 has multiple roles in the cell, including acting as a redox-sensitive chaperone and a sensor for oxidative stress. DJ-1 is also involved in regulating autophagy and mitophagy (Wang et al., 2016).
• Auxilin (DNAJC6/PARK19) is a cofactor for uncoating clathrin-coated vesicles for the heat shock cognate protein-70 (Hsc70) (Nguyen et al., 2019). Its loss-of-function mutations cause early-onset Parkinson’s disease (PD) (Olgiati et al., 2016; Ng et al., 2020), and its mutations also occur in late-onset PD (Gialluisi et al., 2021).
Most PD cases are sporadic and not linked to known genetic mutations. Sporadic PD is thought to result from a combination of genetic and environmental factors, with aging being the most significant risk factor. Yet, recent studies implicate a significant role of heredity in PD that was not previously appreciated, suggesting that 27 to 60% is due to genetic factors (Hamza and Payami, 2010; Do et al., 2011; Keller et al., 2012). Many genes and genetic loci are now linked to the risk for PD using modern genetic methods. Many studies suggest that ALP might be involved in sporadic PD (Orenstein et al., 2013; Zavodszky et al., 2014; Ysselstein et al., 2019; Cui et al., 2021; Pang et al., 2022; Shrivastava et al., 2022; Pradas and Martinez-Vicente, 2023). A recent meta-analysis of genome-wide association studies (GWAS), interrogating data from >13,000 PD patients and > 95,000 controls, identified 26 genetic loci across the genome associated with PD. Intriguingly, 16 out of these 26 genetic loci have 18 genes nearby that modulate ALP, namely SYT11 (Bento et al., 2016), RAB7L1 (Kuwahara et al., 2016; Pradhan et al., 2018; Shrivastava et al., 2022), NUCKS1 (Zhao et al., 2020), GBA (Murphy and Halliday, 2014; Pang et al., 2022; Pradas and Martinez-Vicente, 2023), SIPA1L2 (Andres-Alonso et al., 2019), TMEM163 (Cuajungco et al., 2014; Cuajungco and Kiselyov, 2017), STK39 (Gallolu Kankanamalage et al., 2016, 2017; He et al., 2022), LAMP3 (Tanaka et al., 2022), TMEM175 (Jinn et al., 2017; Hu et al., 2022), GAK (Miyazaki et al., 2021; Munson et al., 2021), SCARB2 (Velayati et al., 2011), GPNMB (Robinet et al., 2021; Zhu et al., 2022), VPS13C (Cai et al., 2022; Hancock-Cerutti et al., 2022), RIT2 (Obergasteiger et al., 2023), DDRGK1 (Cao et al., 2021), SNCA (Sampaio-Marques et al., 2012; Song et al., 2014), LRRK2 (Gómez-Suaga and Hilfiker, 2012; Orenstein et al., 2013; Ysselstein et al., 2019), and MAPT (Zhu et al., 2017; Feng et al., 2020).
Dopaminergic degeneration in PD, potential role of the dopamine transporter
DAT is a membrane-spanning protein that plays a principal role in regulating dopaminergic neurotransmission mainly through modulating the availability and duration of action of dopamine. It is responsible for the reuptake of released dopamine back into presynaptic neurons (Bu et al., 2021). Multiple lines of evidence support the theory that DAT is involved in selective dopaminergic neurotoxicity and degeneration in PD (Storch et al., 2004). Studies have demonstrated a correlation between DAT expression levels and the extent of neurodegeneration, as well as an association between genetic polymorphisms of DAT and PD risk. Additionally, animal models have shown increased vulnerability to toxins with higher concentrations of DAT in striatal regions affected by PD pathology. These findings suggest that dysregulation or malfunctioning of DAT may contribute to neuronal death associated with PD pathogenesis.
DAT density per neuron correlates with neurodegeneration vulnerability in PD
Consistent with a role for DAT in the pathophysiology of PD, the positive correlation between specific dopaminergic neuron subtypes’ vulnerability with the relative DAT expression levels. Hence, hypothalamic dopaminergic neurons are spared in PD, while the ventral tegmental area (VTA) dopaminergic neurons are moderately affected. The dopaminergic neurons in SNpc are most affected in PD. In contrast, the dopaminergic neurons spared in PD patients’ postmortem tissues show lower DAT expression per neuron than controls. For example, Uhl et al. reported that DAT mRNA per neuron is higher in SNpc compared to the adjacent nucleus paranigralis of the ventral tegmental area (VTA) in neurologically normal study subjects. Surviving SNpc neurons in age-matched PD patients is almost half that in controls (Uhl et al., 1994). The same finding was reproduced by other groups (Uhl et al., 1994; Joyce et al., 1997). These findings suggest the selective vulnerability of neurons expressing high levels of DAT in PD. An alternative explanation could be a compensatory downregulation of DAT mRNA levels in the low-expressing neurons in PD. However, this alternative scenario is less likely since the same differential expression of DAT mRNA was documented in controls across species (Sanghera et al., 1994; Haber et al., 1995; Bannon and Whitty, 1997; Afonso-Oramas et al., 2009). Along the same lines, a recent meta-analysis study suggests that the SLC6A3 10R variant, associated with relatively lower expression activity of DAT, may be a protective factor in susceptibility to PD (Zeng et al., 2021).
Consistent with the observations mentioned above, preclinical evidence demonstrates that ectopic or elevated DAT expression is sufficient to induce neurodegeneration. Chen et al. generated a Tet-inducible forebrain DAT-transgenic mouse model under the calcium/calmodulin-dependent protein kinase type II alpha (CaMKIIα) promoter. CamDAT mice spontaneously develop motor dysfunctions and progressive neurodegeneration in the striatum, which l-DOPA accelerates (Chen et al., 2008). Masoud et al. showed that overexpression of DAT in dopaminergic neurons (DAT-transgenic mice) shows impromptu loss of midbrain dopaminergic neurons and increased oxidative stress. DAT protein levels are elevated in DAT-tg mice only in dopaminergic neurons. DAT-tg mice show a < 50% increase in dopamine uptake rate, increased metabolite/dopamine ratios, and diminished striatal VMAT2 protein expression. Interestingly, DAT-tg mice show l-DOPA reversible motor deficits (Salahpour et al., 2008; Masoud et al., 2015). These data show that a 30% DAT activity increase in dopaminergic neurons is sufficient to induce oxidative stress and spontaneous dopaminergic neurodegeneration in vivo.
Evidence showing increased DAT levels in DA neurons in PD or PD models
One of the hallmarks of PD progression is the reduction of DAT levels in the brain, which is the direct result of the loss of DA terminals. However, several studies observed increased DAT levels without or preceding dopaminergic neurodegeneration. Here I list examples of these studies.
Clinical evidence
A cross-sectional analysis in the subset of non-manifesting carriers of LRRK2 and GBA mutations enrolled into the Parkinson’s Progression Markers Initiative (PPMI) focused on 123-I Ioflupane DAT imaging reported that non-manifesting carriers of GBA mutation had increased DAT binding in all striatal regions compared with healthy controls and non-manifesting carriers of the LRRK2 mutation (Simuni et al., 2020). Interestingly, many lines of evidence show that glucocerebrosidase (the enzyme encoded by the GBA gene) regulates autophagy (Bento et al., 2016; Lu et al., 2020; Kuo et al., 2022; Navarro-Romero et al., 2022). The biological mechanism driving increased DAT binding in non-manifesting carriers of GBA mutations remains unclear. Since PPMI is a longitudinal study, it will be important to determine whether the observed up-regulation of DAT in GBA non-manifesting carriers is associated with an increased risk of PD.
In a smaller clinical study, a significant increase in striatal DAT was observed in non-manifesting carriers of the 22q11.2 mutation, which is associated with an age-related increased risk of PD, suggesting that a hyperdopaminergic mechanism may be linked to manifestations of PD and its pathogenesis (Butcher et al., 2017).
Preclinical evidence
Data from non-human primates show a robust increase in DAT expression in monkeys treated with α-synuclein delivered via Adeno-associated virus (AAV) as assessed by 123I-PE2I single-photon emission computerized tomography scans and confirmed by post-mortem immunohistochemical analyses. This non-human primate model induces Parkinson’s disease-like pathology, including α-synuclein aggregation with pathological properties such as amyloid-like Lewy body pathology in PD brains and nigral neuronal degeneration (Chu et al., 2019). Similarly, AAV-mediated delivery of human A53T α-synuclein to the rat SN leads to behavioral deficits and PD-like nigro-striatal degeneration in a dose-and time-dependent manner. Human A53T α-synuclein expression in the SN significantly increased striatal DAT and DA turnover 3 weeks post-AAV injection before overt pathology. By 6 weeks post-AAV injection, SN DA neurons, striatal DA, TH, and DAT were reduced, and a sustained behavioral deficit was observed (Koprich et al., 2011). Furthermore, α-synuclein overexpressing (SNCA-OVX) mice show increased DA uptake and upregulation of membrane DAT levels as assessed by in situ autoradiography and DAT-immunostaining, correlating with α-synuclein levels (Threlfell et al., 2021). Further, Bellucci et al. reported a significant increase in DAT levels in the brain of the truncated α-synuclein (SYN120) mice (Bellucci et al., 2011).
LRRK2 G2019S mutant knock-in (GKI) mice show a significant increase in striatal DAT protein as assessed by western blot (Longo et al., 2017; Volta et al., 2017; Domenicale et al., 2022) and dopamine uptake (Longo et al., 2017; Domenicale et al., 2022). These changes to DAT levels are observed in LRRK2 G2019S mutants but not LRRK2 kinase-dead or knock-out mice (Domenicale et al., 2022).
In DJ-1 KO mice, multiple groups found no difference in DAT mRNA or total DAT protein levels in striatal lysates (Chen et al., 2005; Goldberg et al., 2005; Manning-Boğ et al., 2007). However, the same studies indicate an increase in surface DAT, which is the functional pool of the protein. Young adult DJ-1 KO mice (4 months) show faster DA uptake (Vmax) in the dorsal but not ventral caudate-putamen (Chen et al., 2005), indicating a significant increase in surface DAT since the velocity of DA uptake reflects the functional number of DAT molecules on the cell surface (Daniels and Amara, 1999; Melikian and Buckley, 1999; Sorkina et al., 2005). Similarly, Goldberg et al. reported increased DA reuptake in the dorsal striatum of 3 months old DJ-1 KO mice (Goldberg et al., 2005). Manning-Boğ et al. also reported increased DAT protein levels in the synaptosomal fraction of DJ-1 young adult mice striata (3–4 months). This increase in cell surface DAT was confirmed by an increased active DAT as assessed by [125I]-RTI-121 binding and increased [3H]-DA uptake in striatal synaptosomes (Manning-Boğ et al., 2007).
Auxilin KO mice recapitulate all the critical hallmarks of PD; age-dependent α-synuclein pathology, selective dopaminergic degeneration, astrogliosis, and microgliosis leading to motor deficits that are reversed by L-DOPA treatment. Vidyadhara et al. demonstrate that the lack of auxilin leads to dopaminergic degeneration likely through three processes in the nigral nerve terminals: (Girault and Greengard, 2004) a dramatic upregulation of surface DAT in young mice before dopaminergic degeneration (3-month-old mice) (Haider et al., 2023), toxic accumulation of cytoplasmic DA before neurodegeneration, and (Tolosa et al., 2021) accumulation of autophagosomes in DA synaptic buttons at 3-months, which becomes worse later with the observation of dopaminergic degeneration (Vidyadhara et al., 2023). Interestingly, the auxilin KO mice replicate all three mechanisms united by DAT autophagy, as further explained below.
In summary, several studies show increased DAT levels before dopaminergic degeneration, suggesting that upregulation of DAT might be a part of an early pathological process in PD.
Dopamine toxicity
Dopamine is an essential neurotransmitter that plays a critical role in various physiological processes, including reward, motivation, cognition, and motor function. Dopamine toxicity refers to the excessive accumulation of dopamine within the cytoplasm of dopaminergic neurons in the brain, which can lead to their dysfunction and death. Dysregulation of dopamine signaling is implicated in many neurological and psychiatric disorders, such as PD, schizophrenia, and addiction. The toxic effects of dopamine may involve oxidative stress, mitochondrial dysfunction, and the formation of toxic metabolites. In addition, the accumulation of abnormal protein aggregates in the neurons may contribute to their degeneration (Masato et al., 2019).
Dopamine synthesis, packaging in synaptic vesicles, and reuptake are tightly regulated to prevent its accumulation in the cytoplasm, which could be deleterious. The vesicular monoamine transporter 2 (VMAT2) quickly loads dopamine into synaptic vesicles after its synthesis in the cytoplasm or reuptake in the presynaptic terminals by DAT. In addition to dopamine sequestration, the acidic pH ~5.6 of synaptic vesicles stabilizes dopamine (Mani and Ryan, 2009). Dopamine oxidation results from enzymatic or autooxidation (Umek et al., 2018) and produces reactive oxygen species (ROS), such as hydrogen peroxide, hydroxyl radicals, and superoxide. Dopamine autooxidation produces reactive quinones, which can damage cellular macromolecules and impair neural processes (Stokes et al., 1999). Quinone generation is either spontaneous and could be accelerated by metal ions, such as iron or manganese, or is the product of specific enzyme-catalyzed reactions. Quinone-induced damage to macromolecules and oxidative stress could initiate cell death pathways contributing to neurodegeneration (Mor et al., 2019).
Several dopamine–quinone molecules can undergo oxidative ligation to α-synuclein, selectively inhibiting the protofibril-to-fibril conversion (Conway et al., 2001). α-synuclein protofibrils are likely the toxic form that disrupts neural homeostasis and induces neural cell death through various intracellular targets (Stefanis, 2012). α-synuclein protofibrils can permeabilize synaptic vesicles leading to leakage of stored dopamine into the cytoplasm, further worsening dopamine toxicity (Volles et al., 2001; Lashuel et al., 2002a,b; Volles and Lansbury, 2002).
Monoamine oxidase (MAO) enzymatically metabolizes cytoplasmic dopamine to 3,4-dihydroxyphenylacetaldehyde (DOPAL), a toxic metabolite (Burke et al., 2003). Under physiological conditions, this reaction also produces ROS, such as hydrogen peroxide and hydroxyl radicals. ROS production contributes to oxidative stress and lipid peroxidation inhibiting aldehyde dehydrogenase (ALDH), the main enzyme that eliminates DOPAL, which in turn leads to the accumulation of DOPAL, perpetuating a vicious cycle that contributes to dopaminergic neurodegeneration (Masato et al., 2019). DOPAL levels are elevated in PD postmortem putamen tissue. Further, VMAT2 activity is decreased by 89% and ALDH activity by 70%, likely leading to less synaptic vesicle uptake of cytosolic dopamine and decreased DOPAL detoxification (Goldstein et al., 2013; Pifl et al., 2014). The decrease in VMAT2 activity likely reflects DA neuron terminal loss in PD. On the other hand, VMAT2 deficiency in mice is sufficient to induce cytoplasmic DA toxicity, dopaminergic degeneration in SNpc, and l-DOPA-responsive motor deficits (Caudle et al., 2007). These lines of research inspired the “Catecholaldehyde hypothesis,” depicting the role of DOPAL in SNpc dopaminergic degeneration in PD (Masato et al., 2019).
Autophagy selectively targets DAT for degradation
Recently, our studies have suggested that DAT can be selectively targeted for autophagic degradation (Harraz et al., 2021). Our findings show that cocaine, a potent psychostimulant and addictive substance, induces autophagy in neurons with extraordinary potency. Hence, cocaine induces neural autophagy at sub-nanomolar levels both in vitro and in vivo. The rapid induction of autophagy by cocaine is evident in tyrosine hydroxylase-positive nerve terminals located in the nucleus accumbens (NAc), a brain region implicated in reward, motivation, and addiction. Remarkably, this induction of autophagy can be detected as early as 3 min after systemic cocaine injection. Cocaine administration leads to DAT depletion in dopaminergic nerve terminals in the NAc. The depletion of DAT by cocaine in the nucleus accumbens nerve terminals has been detected using various experimental techniques. Western blot analysis of isolated nerve terminals demonstrates a reduction in DAT protein levels, while immunostaining of brain sections reveals decreased DAT expression in dopaminergic neurons. Furthermore, dopamine uptake activity in isolated nerve terminals, assessed using radiolabeled dopamine (3H-dopamine), shows a reduction in the maximum velocity of dopamine uptake, confirming the depletion of DAT from the nerve terminals’ membranes. Interestingly, pharmacological inhibition of autophagy reverses the depletion of DAT induced by cocaine. This suggests that cocaine-mediated autophagy plays a significant role in modulating DAT levels and dopaminergic neurotransmission (Harraz et al., 2021).
Selective degradation of DAT by autophagy represents a novel mechanism for regulating dopamine signaling and dopaminergic neurotransmission. By modulating DAT levels, autophagy can control dopamine reuptake, its availability in the synaptic cleft, and its potential accumulation in the dopaminergic neurons’ cytoplasm in case of sustained elevated dopamine reuptake. This has important implications for the strength and duration of dopaminergic signaling and dopamine neurotoxicity.
The interplay of dopamine oxidation, DAT density per neuron, and autophagy-lysosome pathway dysfunction
The above-mentioned abnormal mechanisms have been proposed to contribute to PD pathophysiology. These include:
1. Dopamine oxidation-mediated neurotoxicity. The catechol structure of dopamine makes it particularly susceptible to enzymatic and non-enzymatic oxidation, generating dopamine quinones and other ROS. These oxidative species can damage cellular components, disrupt mitochondrial function, and initiate cell death pathways, contributing to the neurodegeneration seen in PD.
2. High expression density of DAT per neuron in SNpc. The extent of dopaminergic neuron loss in PD has been found to correlate strongly with the expression density of DAT per neuron. High DAT density could increase dopamine reuptake and intracellular accumulation, promoting dopamine oxidation and associated neurotoxicity. However, the precise role and regulation of DAT in PD pathogenesis have remained unclear.
3. Dysfunction of the autophagy-lysosome pathway. The ALP has also been implicated in PD. Dysregulation of this pathway can lead to the accumulation of damaged proteins and organelles, including potentially neurotoxic dopamine metabolites. Although the connection between autophagy dysfunction and PD has been established, its implications for dopamine homeostasis and DAT regulation were not fully understood.
However, until recently, these mechanisms have been studied in isolation, with no apparent connection to link them together.
Unifying the concepts: autophagy as a regulator of dopamine homeostasis
Our recent research has provided a unifying framework that integrates these previously separate mechanisms. We have discovered that autophagy is a novel mechanism for controlling dopamine homeostasis, directly linking autophagy-lysosome pathway dysfunction, DAT density, and dopamine oxidation. Autophagy declines with aging (Miki et al., 2018), an observation further complicated by ALP dysfunction in familial and sporadic PD (Kabuta and Wada, 2008; Beilina et al., 2014). Autophagic degradation of DAT is likely reduced due to PD pathology-mediated decline in autophagy. Chronic elevation of DAT density per neuron could lead to a sustained elevation in dopamine reuptake, disrupting the tight regulation of cytoplasmic vs. vesicular dopamine, leading to enhanced dopamine oxidation and neurotoxicity, especially in neurons with already preexisting high DAT expression. This interplay could contribute significantly to dopaminergic neuron loss and the progression of PD (Figures 2A,B).
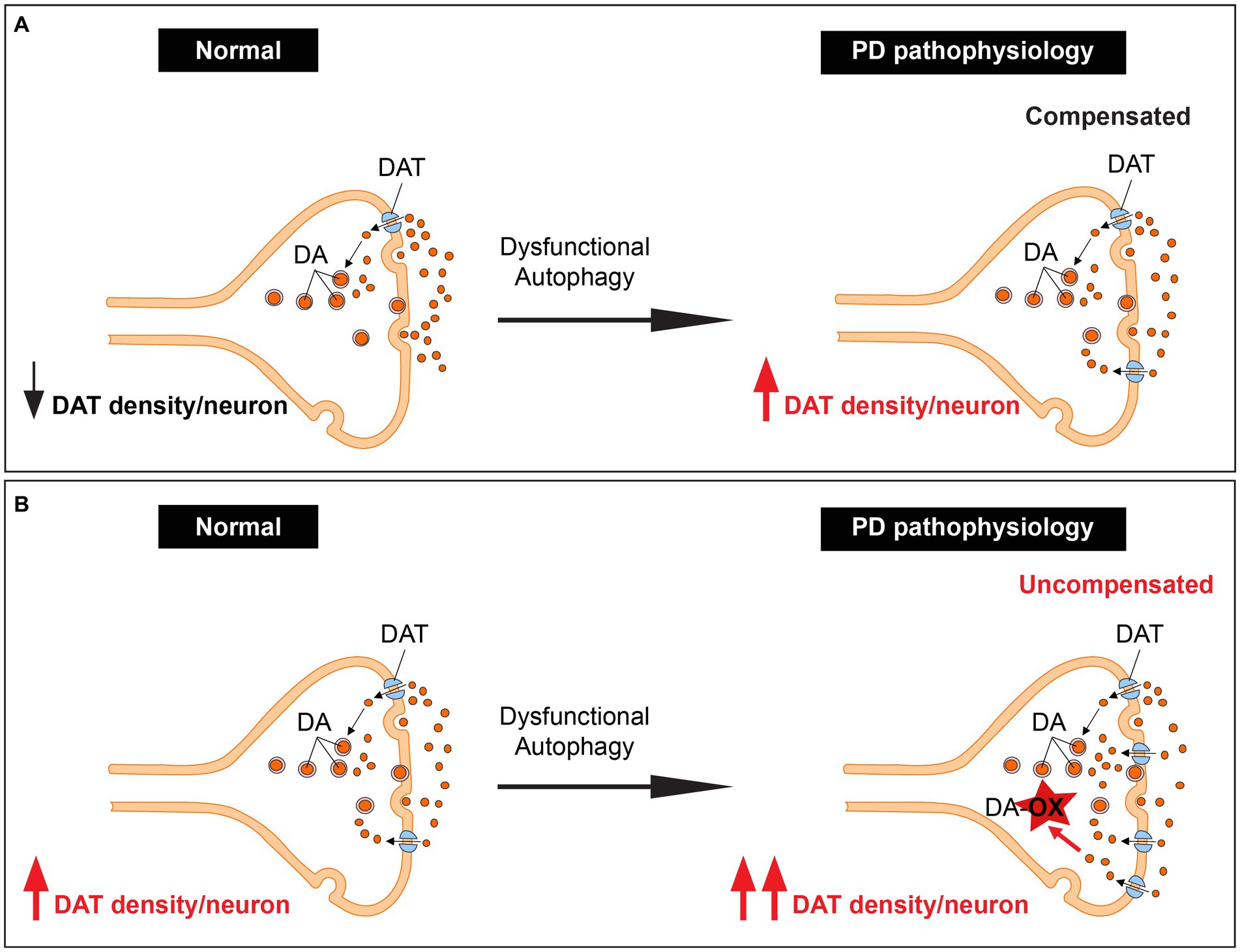
Figure 2. Unifying dopamine oxidation, DAT expression density, and autophagy-lysosome pathway dysfunction by autophagy regulation of dopamine homeostasis. (A) An illustration showing dopaminergic terminals with low basal DAT expression. These terminals are spared in PD since the increase in DAT levels due to dysfunction of the ALP leads to elevated but compensated dopamine reuptake. (B) An illustration showing dopaminergic terminals with high basal DAT expression. These terminals likely degenerate in PD since the increase in DAT levels due to dysfunction of the ALP leads to elevated uncompensated dopamine reuptake.
Conclusion
In conclusion, our research bridges the gap between three previously separate concepts in PD pathophysiology, providing a more comprehensive understanding of aspects of the disease’s pathogenesis. By recognizing autophagy as a novel regulator of dopamine homeostasis, a missing link is recognized between dysfunctional ALP, high expression density of DAT per neuron, dopamine oxidation-mediated neurotoxicity, and correlation with vulnerability of dopaminergic neurons in PD.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Afonso-Oramas, D., Cruz-Muros, I., Alvarez de la Rosa, D., Abreu, P., Giráldez, T., Castro-Hernández, J., et al. (2009). Dopamine transporter glycosylation correlates with the vulnerability of midbrain dopaminergic cells in Parkinson’s disease. Neurobiol. Dis. 36, 494–508. doi: 10.1016/j.nbd.2009.09.002
Andres-Alonso, M., Ammar, M. R., Butnaru, I., Gomes, G. M., Acuña Sanhueza, G., Raman, R., et al. (2019). SIPA1L2 controls trafficking and local signaling of TrkB-containing amphisomes at presynaptic terminals. Nat. Commun. 10:5448. doi: 10.1038/s41467-019-13224-z
Bannon, M. J., and Whitty, C. J. (1997). Age-related and regional differences in dopamine transporter mRNA expression in human midbrain. Neurology 48, 969–977. doi: 10.1212/WNL.48.4.969
Beilina, A., Rudenko, I. N., Kaganovich, A., Civiero, L., Chau, H., Kalia, S. K., et al. (2014). Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. U. S. A. 111, 2626–2631. doi: 10.1073/pnas.1318306111
Bellucci, A., Navarria, L., Falarti, E., Zaltieri, M., Bono, F., Collo, G., et al. (2011). Redistribution of DAT/α-synuclein complexes visualized by “in situ” proximity ligation assay in transgenic mice modelling early Parkinson’s disease. PLoS One 6:e27959. doi: 10.1371/journal.pone.0027959
Bento, C. F., Ashkenazi, A., Jimenez-Sanchez, M., and Rubinsztein, D. C. (2016). The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat. Commun. 7:11803. doi: 10.1038/ncomms11803
Bu, M., Farrer, M. J., and Khoshbouei, H. (2021). Dynamic control of the dopamine transporter in neurotransmission and homeostasis. NPJ Parkinsons Dis. 7, 22–11. doi: 10.1038/s41531-021-00161-2
Burke, W. J., Li, S. W., Williams, E. A., Nonneman, R., and Zahm, D. S. (2003). 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson’s disease pathogenesis. Brain Res. 989, 205–213. doi: 10.1016/S0006-8993(03)03354-7
Butcher, N. J., Marras, C., Pondal, M., Rusjan, P., Boot, E., Christopher, L., et al. (2017). Neuroimaging and clinical features in adults with a 22q11.2 deletion at risk of Parkinson’s disease. Brain 140, 1371–1383. doi: 10.1093/brain/awx053
Cai, S., Wu, Y., Guillén-Samander, A., Hancock-Cerutti, W., Liu, J., and De Camilli, P. (2022). In situ architecture of the lipid transport protein VPS13C at ER-lysosome membrane contacts. Proc. Natl. Acad. Sci. U. S. A. 119:e2203769119. doi: 10.1073/pnas.2203769119
Cao, Y., Li, R., Shen, M., Li, C., Zou, Y., Jiang, Q., et al. (2021). DDRGK1, a crucial player of ufmylation system, is indispensable for autophagic degradation by regulating lysosomal function. Cell Death Dis. 12:416. doi: 10.1038/s41419-021-03694-9
Caudle, W. M., Richardson, J. R., Wang, M. Z., Taylor, T. N., Guillot, T. S., McCormack, A. L., et al. (2007). Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J. Neurosci. 27, 8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007
Chen, L., Cagniard, B., Mathews, T., Jones, S., Koh, H. C., Ding, Y., et al. (2005). Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J. Biol. Chem. 280, 21418–21426. doi: 10.1074/jbc.M413955200
Chen, L., Ding, Y., Cagniard, B., Van Laar, A. D., Mortimer, A., Chi, W., et al. (2008). Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J. Neurosci. 28, 425–433. doi: 10.1523/JNEUROSCI.3602-07.2008
Chu, Y., Muller, S., Tavares, A., Barret, O., Alagille, D., Seibyl, J., et al. (2019). Intrastriatal alpha-synuclein fibrils in monkeys: spreading, imaging and neuropathological changes. Brain 142, 3565–3579. doi: 10.1093/brain/awz296
Conway, K. A., Rochet, J. C., Bieganski, R. M., and Lansbury, P. T. (2001). Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 294, 1346–1349. doi: 10.1126/science.1063522
Cuajungco, M. P., Basilio, L. C., Silva, J., Hart, T., Tringali, J., Chen, C. C., et al. (2014). Cellular zinc levels are modulated by TRPML1-TMEM163 interaction. Traffic 15, 1247–1265. doi: 10.1111/tra.12205
Cuajungco, M. P., and Kiselyov, K. (2017). The mucolipin-1 (TRPML1) ion channel, transmembrane-163 (TMEM163) protein, and lysosomal zinc handling. Front. Biosci. 22, 1330–1343. doi: 10.2741/4546
Cui, Y., Yang, Z., Flores-Rodriguez, N., Follett, J., Ariotti, N., Wall, A. A., et al. (2021). Formation of retromer transport carriers is disrupted by the Parkinson disease-linked Vps35 D620N variant. Traffic 22, 123–136. doi: 10.1111/tra.12779
Daniels, G. M., and Amara, S. G. (1999). Regulated trafficking of the human dopamine transporter. Clathrin-mediated internalization and lysosomal degradation in response to phorbol esters. J. Biol. Chem. 274, 35794–35801. doi: 10.1074/jbc.274.50.35794
Dhanushkodi, N. R., Abul Khair, S. B., Ardah, M. T., and Haque, M. E. (2023). ATP13A2 gene silencing in Drosophila affects Autophagic degradation of A53T mutant α-Synuclein. Int. J. Mol. Sci. 24:1775. doi: 10.3390/ijms24021775
Do, C. B., Tung, J. Y., Dorfman, E., Kiefer, A. K., Drabant, E. M., Francke, U., et al. (2011). Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 7:e1002141. doi: 10.1371/journal.pgen.1002141
Domenicale, C., Mercatelli, D., Albanese, F., Novello, S., Vincenzi, F., Varani, K., et al. (2022). Dopamine transporter, PhosphoSerine129 α-Synuclein and α-Synuclein levels in aged LRRK2 G2019S Knock-in and Knock-out mice. Biomedicine 10:881. doi: 10.3390/biomedicines10040881
Feng, Q., Luo, Y., Zhang, X. N., Yang, X. F., Hong, X. Y., Sun, D. S., et al. (2020). MAPT/tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy 16, 641–658. doi: 10.1080/15548627.2019.1633862
Fleming, A., Bourdenx, M., Fujimaki, M., Karabiyik, C., Krause, G. J., Lopez, A., et al. (2022). The different autophagy degradation pathways and neurodegeneration. Neuron 110, 935–966. doi: 10.1016/j.neuron.2022.01.017
Gallolu Kankanamalage, S., Lee, A. Y., Wichaidit, C., Lorente-Rodriguez, A., Shah, A. M., Stippec, S., et al. (2016). Multistep regulation of autophagy by WNK1. Proc. Natl. Acad. Sci. U. S. A. 113, 14342–14347. doi: 10.1073/pnas.1617649113
Gallolu Kankanamalage, S., Lee, A. Y., Wichaidit, C., Lorente-Rodriguez, A., Shah, A. M., Stippec, S., et al. (2017). WNK1 is an unexpected autophagy inhibitor. Autophagy 13, 969–970. doi: 10.1080/15548627.2017.1286431
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo-San Pedro, J. M., Cecconi, F., et al. (2017). Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. doi: 10.15252/embj.201796697
Gialluisi, A., Reccia, M. G., Modugno, N., Nutile, T., Lombardi, A., Di Giovannantonio, L. G., et al. (2021). Identification of sixteen novel candidate genes for late onset Parkinson’s disease. Mol. Neurodegener. 16:35. doi: 10.1186/s13024-021-00455-2
Giguère, N., Burke Nanni, S., and Trudeau, L. E. (2018). On cell loss and selective vulnerability of neuronal populations in Parkinson’s disease. Front. Neurol. 9:455. doi: 10.3389/fneur.2018.00455
Girault, J. A., and Greengard, P. (2004). The neurobiology of dopamine signaling. Arch. Neurol. 61, 641–644. doi: 10.1001/archneur.61.5.641
Goldberg, M. S., Pisani, A., Haburcak, M., Vortherms, T. A., Kitada, T., Costa, C., et al. (2005). Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial parkinsonism-linked gene DJ-1. Neuron 45, 489–496. doi: 10.1016/j.neuron.2005.01.041
Goldstein, D. S., Sullivan, P., Holmes, C., Miller, G. W., Alter, S., Strong, R., et al. (2013). Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J. Neurochem. 126, 591–603. doi: 10.1111/jnc.12345
Gómez-Suaga, P., and Hilfiker, S. (2012). LRRK2 as a modulator of lysosomal calcium homeostasis with downstream effects on autophagy. Autophagy 8, 692–693. doi: 10.4161/auto.19305
Haber, S. N., Ryoo, H., Cox, C., and Lu, W. (1995). Subsets of midbrain dopaminergic neurons in monkeys are distinguished by different levels of mRNA for the dopamine transporter: comparison with the mRNA for the D2 receptor, tyrosine hydroxylase and calbindin immunoreactivity. J. Comp. Neurol. 362, 400–410. doi: 10.1002/cne.903620308
Haider, A., Elghazawy, N. H., Dawood, A., Gebhard, C., Wichmann, T., Sippl, W., et al. (2023). Translational molecular imaging and drug development in Parkinson’s disease. Mol. Neurodegener. 18:11. doi: 10.1186/s13024-023-00600-z
Hamza, T. H., and Payami, H. (2010). The heritability of risk and age at onset of Parkinson’s disease after accounting for known genetic risk factors. J. Hum. Genet. 55, 241–243. doi: 10.1038/jhg.2010.13
Hancock-Cerutti, W., Wu, Z., Xu, P., Yadavalli, N., Leonzino, M., Tharkeshwar, A. K., et al. (2022). ER-lysosome lipid transfer protein VPS13C/PARK23 prevents aberrant mtDNA-dependent STING signaling. J. Cell Biol. 221:e202106046. doi: 10.1083/jcb.202106046
Harraz, M. M., Guha, P., Kang, I. G., Semenza, E. R., Malla, A. P., Song, Y. J., et al. (2021). Cocaine-induced locomotor stimulation involves autophagic degradation of the dopamine transporter. Mol. Psychiatry 26, 370–382. doi: 10.1038/s41380-020-00978-y
He, N., Tan, H., Deng, X., Shu, L., Qing, B., and Liang, H. (2022). MiR-223-3p-loaded exosomes from bronchoalveolar lavage fluid promote alveolar macrophage autophagy and reduce acute lung injury by inhibiting the expression of STK39. Hum. Cell 35, 1736–1751. doi: 10.1007/s13577-022-00762-w
Herman, A. M., and Moussa, C. E. H. (2011). The ubiquitin ligase parkin modulates the execution of autophagy. Autophagy 7, 919–921. doi: 10.4161/auto.7.8.15814
Hu, M., Li, P., Wang, C., Feng, X., Geng, Q., Chen, W., et al. (2022). Parkinson’s disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes. Cells 185, 2292–2308.e20. doi: 10.1016/j.cell.2022.05.021
Jinn, S., Drolet, R. E., Cramer, P. E., Wong, A. H. K., Toolan, D. M., Gretzula, C. A., et al. (2017). TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc. Natl. Acad. Sci. U. S. A. 114, 2389–2394. doi: 10.1073/pnas.1616332114
Jones, N. (2010). PINK1 targets dysfunctional mitochondria for autophagy in Parkinson disease. Nat. Rev. Neurol. 6:181. doi: 10.1038/nrneurol.2010.19
Joyce, J. N., Smutzer, G., Whitty, C. J., Myers, A., and Bannon, M. J. (1997). Differential modification of dopamine transporter and tyrosine hydroxylase mRNAs in midbrain of subjects with Parkinson’s, Alzheimer’s with parkinsonism, and Alzheimer’s disease. Mov. Disord. 12, 885–897. doi: 10.1002/mds.870120609
Kabuta, T., and Wada, K. (2008). Insights into links between familial and sporadic Parkinson’s disease: physical relationship between UCH-L1 variants and chaperone-mediated autophagy. Autophagy 4, 827–829. doi: 10.4161/auto.6560
Kaushik, S., and Cuervo, A. M. (2012). Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 22, 407–417. doi: 10.1016/j.tcb.2012.05.006
Keller, M. F., Saad, M., Bras, J., Bettella, F., Nicolaou, N., Simón-Sánchez, J., et al. (2012). Using genome-wide complex trait analysis to quantify “missing heritability” in Parkinson’s disease. Hum. Mol. Genet. 21, 4996–5009. doi: 10.1093/hmg/dds335
Koprich, J. B., Johnston, T. H., Huot, P., Reyes, M. G., Espinosa, M., and Brotchie, J. M. (2011). Progressive neurodegeneration or endogenous compensation in an animal model of Parkinson’s disease produced by decreasing doses of alpha-synuclein. PLoS One 6:e17698. doi: 10.1371/journal.pone.0017698
Kuo, S. H., Tasset, I., Cheng, M. M., Diaz, A., Pan, M. K., Lieberman, O. J., et al. (2022). Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci. Adv. 8:eabm6393. doi: 10.1126/sciadv.abm6393
Kuwahara, T., Inoue, K., D’Agati, V. D., Fujimoto, T., Eguchi, T., Saha, S., et al. (2016). LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci. Rep. 6:29945. doi: 10.1038/srep29945
Lashuel, H. A., Hartley, D., Petre, B. M., Walz, T., and Lansbury, P. T. (2002a). Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature 418:291. doi: 10.1038/418291a
Lashuel, H. A., Petre, B. M., Wall, J., Simon, M., Nowak, R. J., Walz, T., et al. (2002b). Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 322, 1089–1102. doi: 10.1016/S0022-2836(02)00735-0
Lohr, K. M., Masoud, S. T., Salahpour, A., and Miller, G. W. (2017). Membrane transporters as mediators of synaptic dopamine dynamics: implications for disease. Eur. J. Neurosci. 45, 20–33. doi: 10.1111/ejn.13357
Longo, F., Mercatelli, D., Novello, S., Arcuri, L., Brugnoli, A., Vincenzi, F., et al. (2017). Age-dependent dopamine transporter dysfunction and Serine129 phospho-α-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol. Commun. 5:22. doi: 10.1186/s40478-017-0426-8
Lonskaya, I., Hebron, M. L., Algarzae, N. K., Desforges, N., and Moussa, C. E. H. (2013). Decreased parkin solubility is associated with impairment of autophagy in the nigrostriatum of sporadic Parkinson’s disease. Neuroscience 232, 90–105. doi: 10.1016/j.neuroscience.2012.12.018
Lu, J., Wu, M., and Yue, Z. (2020). Autophagy and Parkinson’s disease. Adv. Exp. Med. Biol. 1207, 21–51. doi: 10.1007/978-981-15-4272-5_2
Mani, M., and Ryan, T. A. (2009). Live imaging of synaptic vesicle release and retrieval in dopaminergic neurons. Front. Neural Circuits 3:3. doi: 10.3389/neuro.04.003.2009
Manning-Boğ, A. B., Caudle, W. M., Perez, X. A., Reaney, S. H., Paletzki, R., Isla, M. Z., et al. (2007). Increased vulnerability of nigrostriatal terminals in DJ-1-deficient mice is mediated by the dopamine transporter. Neurobiol. Dis. 27, 141–150. doi: 10.1016/j.nbd.2007.03.014
Masato, A., Plotegher, N., Boassa, D., and Bubacco, L. (2019). Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 14:35. doi: 10.1186/s13024-019-0332-6
Masoud, S., Vecchio, L., Bergeron, Y., Hossain, M., Nguyen, L., Bermejo, M., et al. (2015). Increased expression of the dopamine transporter leads to loss of dopamine neurons, oxidative stress and L-DOPA reversible motor deficits. Neurobiol. Dis. 74, 66–75. doi: 10.1016/j.nbd.2014.10.016
Melikian, H. E., and Buckley, K. M. (1999). Membrane trafficking regulates the activity of the human dopamine transporter. J. Neurosci. 19, 7699–7710. doi: 10.1523/JNEUROSCI.19-18-07699.1999
Miki, Y., Holton, J. L., and Wakabayashi, K. (2018). Autophagy in neurodegeneration and aging. Aging (Albany NY) 10, 3632–3633. doi: 10.18632/aging.101652
Miyazaki, M., Hiramoto, M., Takano, N., Kokuba, H., Takemura, J., Tokuhisa, M., et al. (2021). Targeted disruption of GAK stagnates autophagic flux by disturbing lysosomal dynamics. Int. J. Mol. Med. 48:195. doi: 10.3892/ijmm.2021.5028
Moors, T. E., Paciotti, S., Ingrassia, A., Quadri, M., Breedveld, G., Tasegian, A., et al. (2019). Characterization of brain lysosomal activities in GBA-related and sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurobiol. 56, 1344–1355. doi: 10.1007/s12035-018-1090-0
Mor, D. E., Daniels, M. J., and Ischiropoulos, H. (2019). The usual suspects, dopamine and alpha-synuclein, conspire to cause neurodegeneration. Mov. Disord. 34, 167–179. doi: 10.1002/mds.27607
Munson, M. J., Mathai, B. J., Ng, M. Y. W., Trachsel-Moncho, L., de la Ballina, L. R., Schultz, S. W., et al. (2021). GAK and PRKCD are positive regulators of PRKN-independent mitophagy. Nat. Commun. 12:6101. doi: 10.1038/s41467-021-26331-7
Murakami, H., Shiraishi, T., Umehara, T., Omoto, S., and Iguchi, Y. (2023). Recent advances in drug therapy for Parkinson’s disease. Intern. Med. 62, 33–42. doi: 10.2169/internalmedicine.8940-21
Murphy, K. E., and Halliday, G. M. (2014). Glucocerebrosidase deficits in sporadic Parkinson disease. Autophagy 10, 1350–1351. doi: 10.4161/auto.29074
Navarro-Romero, A., Fernandez-Gonzalez, I., Riera, J., Montpeyo, M., Albert-Bayo, M., Lopez-Royo, T., et al. (2022). Lysosomal lipid alterations caused by glucocerebrosidase deficiency promote lysosomal dysfunction, chaperone-mediated-autophagy deficiency, and alpha-synuclein pathology. NPJ Parkinsons Dis. 8:126. doi: 10.1038/s41531-022-00397-6
Ng, J., Cortès-Saladelafont, E., Abela, L., Termsarasab, P., Mankad, K., Sudhakar, S., et al. (2020). DNAJC6 mutations disrupt dopamine homeostasis in juvenile parkinsonism-dystonia. Mov. Disord. 35, 1357–1368. doi: 10.1002/mds.28063
Nguyen, M., Wong, Y. C., Ysselstein, D., Severino, A., and Krainc, D. (2019). Synaptic, mitochondrial, and lysosomal dysfunction in Parkinson’s disease. Trends Neurosci. 42, 140–149. doi: 10.1016/j.tins.2018.11.001
Obergasteiger, J., Castonguay, A. M., Pizzi, S., Magnabosco, S., Frapporti, G., Lobbestael, E., et al. (2023). The small GTPase Rit2 modulates LRRK2 kinase activity, is required for lysosomal function and protects against alpha-synuclein neuropathology. NPJ Parkinsons Dis. 9:44. doi: 10.1038/s41531-023-00484-2
Olgiati, S., Quadri, M., Fang, M., Rood, J. P. M. A., Saute, J. A., Chien, H. F., et al. (2016). DNAJC6 mutations associated with early-onset Parkinson’s disease. Ann. Neurol. 79, 244–256. doi: 10.1002/ana.24553
Orenstein, S. J., Kuo, S. H., Tasset, I., Arias, E., Koga, H., Fernandez-Carasa, I., et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406. doi: 10.1038/nn.3350
Pang, S. Y. Y., Lo, R. C. N., Ho, P. W. L., Liu, H. F., Chang, E. E. S., Leung, C. T., et al. (2022). LRRK2, GBA and their interaction in the regulation of autophagy: implications on therapeutics in Parkinson’s disease. Transl. Neurodegener. 11:5. doi: 10.1186/s40035-022-00281-6
Pifl, C., Rajput, A., Reither, H., Blesa, J., Cavada, C., Obeso, J. A., et al. (2014). Is Parkinson’s disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J. Neurosci. 34, 8210–8218. doi: 10.1523/JNEUROSCI.5456-13.2014
Pradas, E., and Martinez-Vicente, M. (2023). The consequences of GBA deficiency in the autophagy-lysosome system in Parkinson’s disease associated with GBA. Cells 12:191. doi: 10.3390/cells12010191
Pradhan, G., Shrivastva, R., and Mukhopadhyay, S. (2018). Mycobacterial PknG targets the Rab7l1 signaling pathway to inhibit phagosome-lysosome fusion. J. Immunol. 201, 1421–1433. doi: 10.4049/jimmunol.1800530
Robinet, P., Ritchey, B., Lorkowski, S. W., Alzayed, A. M., DeGeorgia, S., Schodowski, E., et al. (2021). Quantitative trait locus mapping identifies the Gpnmb gene as a modifier of mouse macrophage lysosome function. Sci. Rep. 11:10249. doi: 10.1038/s41598-021-89800-5
Salahpour, A., Ramsey, A. J., Medvedev, I. O., Kile, B., Sotnikova, T. D., Holmstrand, E., et al. (2008). Increased amphetamine-induced hyperactivity and reward in mice overexpressing the dopamine transporter. Proc. Natl. Acad. Sci. U. S. A. 105, 4405–4410. doi: 10.1073/pnas.0707646105
Sampaio-Marques, B., Felgueiras, C., Silva, A., Rodrigues, M., Tenreiro, S., Franssens, V., et al. (2012). SNCA (α-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 8, 1494–1509. doi: 10.4161/auto.21275
Sanghera, M. K., Manaye, K. F., Liang, C. L., Lacopino, A. M., Bannon, M. J., and German, D. C. (1994). Low dopamine transporter mRNA levels in midbrain regions containing calbindin. Neuroreport 5, 1641–1644. doi: 10.1097/00001756-199408150-00025
Shrivastava, R., Pradhan, G., Ghosh, S., and Mukhopadhyay, S. (2022). Rabaptin5 acts as a key regulator for Rab7l1-mediated phagosome maturation process. Immunology 165, 328–340. doi: 10.1111/imm.13438
Simon, D. K., Tanner, C. M., and Brundin, P. (2020). Parkinson disease epidemiology, pathology, genetics and pathophysiology. Clin. Geriatr. Med. 36, 1–12. doi: 10.1016/j.cger.2019.08.002
Simuni, T., Uribe, L., Cho, H. R., Caspell-Garcia, C., Coffey, C. S., Siderowf, A., et al. (2020). Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s progression markers initiative (PPMI): a cross-sectional study. Lancet Neurol. 19, 71–80. doi: 10.1016/S1474-4422(19)30319-9
Song, J. X., Lu, J. H., Liu, L. F., Chen, L. L., Durairajan, S. S. K., Yue, Z., et al. (2014). HMGB1 is involved in autophagy inhibition caused by SNCA/α-synuclein overexpression: a process modulated by the natural autophagy inducer corynoxine B. Autophagy 10, 144–154. doi: 10.4161/auto.26751
Sorkina, T., Hoover, B. R., Zahniser, N. R., and Sorkin, A. (2005). Constitutive and protein kinase C-induced internalization of the dopamine transporter is mediated by a clathrin-dependent mechanism. Traffic 6, 157–170. doi: 10.1111/j.1600-0854.2005.00259.x
Stefanis, L. (2012). α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Stokes, A. H., Hastings, T. G., and Vrana, K. E. (1999). Cytotoxic and genotoxic potential of dopamine. J. Neurosci. Res. 55, 659–665. doi: 10.1002/(SICI)1097-4547(19990315)55:6<659::AID-JNR1>3.0.CO;2-C
Storch, A., Ludolph, A. C., and Schwarz, J. (2004). Dopamine transporter: involvement in selective dopaminergic neurotoxicity and degeneration. J. Neural Transm. 111, 1267–1286. doi: 10.1007/s00702-004-0203-2
Tanaka, T., Warner, B. M., Michael, D. G., Nakamura, H., Odani, T., Yin, H., et al. (2022). LAMP3 inhibits autophagy and contributes to cell death by lysosomal membrane permeabilization. Autophagy 18, 1629–1647. doi: 10.1080/15548627.2021.1995150
Threlfell, S., Mohammadi, A. S., Ryan, B. J., Connor-Robson, N., Platt, N. J., Anand, R., et al. (2021). Striatal dopamine transporter function is facilitated by converging biology of α-Synuclein and cholesterol. Front. Cell. Neurosci. 15:658244. doi: 10.3389/fncel.2021.658244
Tolosa, E., Garrido, A., Scholz, S. W., and Poewe, W. (2021). Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 20, 385–397. doi: 10.1016/S1474-4422(21)00030-2
Uhl, G. R., Walther, D., Mash, D., Faucheux, B., and Javoy-Agid, F. (1994). Dopamine transporter messenger RNA in Parkinson’s disease and control substantia nigra neurons. Ann. Neurol. 35, 494–498. doi: 10.1002/ana.410350421
Umek, N., Geršak, B., Vintar, N., Šoštarič, M., and Mavri, J. (2018). Dopamine autoxidation is controlled by acidic pH. Front. Mol. Neurosci. 11:467. doi: 10.3389/fnmol.2018.00467
Velayati, A., DePaolo, J., Gupta, N., Choi, J. H., Moaven, N., Westbroek, W., et al. (2011). A mutation in SCARB2 is a modifier in Gaucher disease. Hum. Mutat. 32, 1232–1238. doi: 10.1002/humu.21566
Vidyadhara, D. J., Somayaji, M., Wade, N., Yücel, B., Zhao, H., Shashaank, N., et al. (2023). Dopamine transporter and synaptic vesicle sorting defects underlie auxilin-associated Parkinson’s disease. Cell Rep. 42:112231. doi: 10.1016/j.celrep.2023.112231
Volles, M. J., and Lansbury, P. T. (2002). Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 41, 4595–4602. doi: 10.1021/bi0121353
Volles, M. J., Lee, S. J., Rochet, J. C., Shtilerman, M. D., Ding, T. T., Kessler, J. C., et al. (2001). Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 40, 7812–7819. doi: 10.1021/bi0102398
Volta, M., Beccano-Kelly, D. A., Paschall, S. A., Cataldi, S., MacIsaac, S. E., Kuhlmann, N., et al. (2017). Initial elevations in glutamate and dopamine neurotransmission decline with age, as does exploratory behavior, in LRRK2 G2019S knock-in mice. elife 6:e28377. doi: 10.7554/eLife.28377
Wang, B., Cai, Z., Tao, K., Zeng, W., Lu, F., Yang, R., et al. (2016). Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy 12, 1215–1228. doi: 10.1080/15548627.2016.1179401
Wang, L., Klionsky, D. J., and Shen, H. M. (2023). The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 24, 186–203. doi: 10.1038/s41580-022-00529-z
Wu, G., Pang, S., Feng, X., Zhang, A., Li, J., Gu, K., et al. (2011). Genetic analysis of lysosomal alpha-galactosidase a gene in sporadic Parkinson’s disease. Neurosci. Lett. 500, 31–35. doi: 10.1016/j.neulet.2011.05.238
Wu, G., Yan, B., Wang, X., Feng, X., Zhang, A., Xu, X., et al. (2008). Decreased activities of lysosomal acid alpha-D-galactosidase a in the leukocytes of sporadic Parkinson’s disease. J. Neurol. Sci. 271, 168–173. doi: 10.1016/j.jns.2008.04.011
Ysselstein, D., Nguyen, M., Young, T. J., Severino, A., Schwake, M., Merchant, K., et al. (2019). LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 10:5570. doi: 10.1038/s41467-019-13413-w
Zavodszky, E., Seaman, M. N. J., Moreau, K., Jimenez-Sanchez, M., Breusegem, S. Y., Harbour, M. E., et al. (2014). Mutation in VPS35 associated with Parkinson’s disease impairs wash complex association and inhibits autophagy. Nat. Commun. 5:3828. doi: 10.1038/ncomms4828
Zeng, Q., Ning, F., Gu, S., Zeng, Q., Chen, R., Peng, L., et al. (2021). The 10-repeat 3’-UTR VNTR polymorphism in the SLC6A3 gene may confer protection against Parkinson’s disease: a meta-analysis. Front. Genet. 12:757601. doi: 10.3389/fgene.2021.757601
Zhao, E., Feng, L., Bai, L., and Cui, H. (2020). NUCKS promotes cell proliferation and suppresses autophagy through the mTOR-Beclin1 pathway in gastric cancer. J. Exp. Clin. Cancer Res. 39:194. doi: 10.1186/s13046-020-01696-7
Zhu, Z., Liu, Y., Li, X., Zhang, L., Liu, H., Cui, Y., et al. (2022). GPNMB mitigates Alzheimer’s disease and enhances autophagy via suppressing the mTOR signal. Neurosci. Lett. 767:136300. doi: 10.1016/j.neulet.2021.136300
Keywords: dopamine transporter, Parkinson’s disease, autophagy lysosome pathway, dopamine toxicity, nigral degeneration
Citation: Harraz MM (2023) Selective dopaminergic vulnerability in Parkinson’s disease: new insights into the role of DAT. Front. Neurosci. 17:1219441. doi: 10.3389/fnins.2023.1219441
Edited by:
Einar M. Sigurdsson, New York University, United StatesReviewed by:
Louis-Eric Trudeau, University of Montreal, CanadaDamiana Leo, University of Mons, Belgium
Copyright © 2023 Harraz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maged M. Harraz, bWhhcnJhekBzb20udW1hcnlsYW5kLmVkdQ==