 Ting Wang1
Ting Wang1 Shijia Ouyang1Xueyang Niu2
Shijia Ouyang1Xueyang Niu2 Miaomiao Cheng1
Miaomiao Cheng1 Ying Yang1Yonghua Yang3Quanzhen Tan1Wenwei Liu1
Ying Yang1Yonghua Yang3Quanzhen Tan1Wenwei Liu1 Xiaoling Yang1
Xiaoling Yang1 Yuehua Zhang1*
Yuehua Zhang1*- 1Children’s Medical Center of Peking University First Hospital, Beijing, China
- 2Department of Respiratory, Beijing Children’s Hospital, Capital Medical University, Beijing, China
- 3Department of Pediatrics, the First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, China
Objective: To explore the genotypic spectrum and refine the genotype-phenotype correlation of PPP3CA-related developmental and epileptic encephalopathy (DEE).
Methods: whole-exome sequencing or whole-genome sequencing was performed to all patients. Clinical data of 15 epilepsy patients in current study and 21 epilepsy patients from published studies were collected and analyzed.
Results: In this study, 15 patients were identified with 13 de novo PPP3CA variants. Among these, seven frameshift variants and one gene inversion between intron 11 and intron 13 (including exons 12 and 13) were novel. 80% of patients experiencing seizure onset before the age of one. The seizure types observed included epileptic spasms (93.3%), tonic seizures (46.7%), myoclonic seizures (46.7%), focal seizures (40.0%), atypical absence seizures (13.3%), generalized tonic-clonic seizures (6.7%) and myoclonic atonic seizures (6.7%). All patients exhibited global developmental delay. MRI abnormalities were noticed in 9 patients, including widened subarachnoid space, bilateral ventricular width, poor myelination of white matter, and dysplasia of the corpus callosum. 80% specifically diagnosed with infantile epileptic spasms syndrome (IESS). When combining data from this study and published studies, 66.7% of patients experienced seizure onset before the age of one, and 77.8% were diagnosed with IESS. In patients with variants located in the catalytic domain (CD), 45.4% patients exhibited multiple seizure types, while 45.4% patients presented only with epileptic spasms. In contrast, among patients with variants in regulatory domain (RD), 87% had multiple seizure types and only 8.7% had epileptic spasms alone. Additionally, 45.5% of patients with CD variants had comorbid autism spectrum disorders, compared to 13% patients with RD variants. Recurrent variants included p.His92Arg, p.Asp234Glu, p.Glu282Lys, and p.Ser419Asnfs*31.
Conclusion: This study is the first to report a gene inversion in PPP3CA-related DEE. Patients with only epileptic spasms were more prevalent in those with CD variants, compared to those with RD variants. Conversely, patients with multiple seizure types were more common among those with RD variants. The most frequently diagnosed epileptic syndrome was IESS. Additionally, comorbid ASD were more commonly observed in patients with CD variants than in those with RD variants.
Introduction
Calcineurin, a calcium/calmodulin-regulated serine/threonine protein phosphatase, is a heterodimeric protein composed of two subunits: the catalytic A (CnA) and the regulatory B (CnB) (Aramburu et al., 2004; Creamer, 2020; Karagiota et al., 2019; Rusnak and Mertz, 2000). The PPP3CA gene (MIM:114105), located on chromosome 4q24, encodes the α-isoform of CnA and mediates Ca2+-dependent signal transduction (Cook and Creamer, 2016; Creamer, 2013; Palkowitsch et al., 2011). It is widely distributed in the mammalian brain, particularly enriched in synapses, and is involved in the recycling of synaptic vesicles at nerve terminals (Dhindsa et al., 2015). PPP3CA consists of five domains: the catalytic domain (CD), the calcineurin regulatory subunit-calcineurin binding domain (CnBB), the regulatory domain (RD), the calmodulin binding domain (CaMB) and the auto-inhibitory domain (AID) (Li et al., 2016; Rumi-Masante et al., 2012).
In 2017, Myers et al. (2017) first reported that the variants of PPP3CA are associated with developmental and epileptic encephalopathy (DEE). In 2018, Mizuguchi et al. (2018) identified six heterozygous PPP3CA variants in four patients with DEE and two patients with multiple congenital malformation. Using a yeast model, they identified two functionally distinct types of variants: loss-of-function variants, characterized by decreased calcineurin signaling at the CD, and constitutively activating variants, characterized by increased calcineurin signaling at the AID. Panneerselvam et al. (2021) suggested that PPP3CA truncating variants cluster in the RD domain, leading to more severe early-onset refractory epilepsy. The present study identified novel variants and analyzed the genotype-phenotype correlation in conjunction with published literature.
Materials and methods
Patients
Genetic testing was conducted on all patients diagnosed with epilepsy without acquired factors such as perinatal brain injury, traumatic brain injury, central nervous system infections, etc. From August 2019 to July 2024, a total of 3350 cases of children with epilepsy and single gene variants were enrolled. Among them, 0.45% (15/3350) were found to have PPP3CA heterozygous variants. This study summarized the patients’ seizure onset age, seizure types, developmental milestones, neurological status, family history, and results of ancillary examinations, including electroencephalogram (EEG) and brain magnetic resonance imaging (MRI). The normal value of the extracranial space is based on reference (McArdle et al., 1987; Wang et al., 1996b), and the expansion of the extracranial space is defined as exceeding the average value of the corresponding age group plus 2 times the standard deviation. The determination of myelination process can be found in reference (Wang et al., 1996a). Treatment and prognosis were also followed up in the clinic.
This study was approved by the Ethics Committee of Peking University First Hospital [Approval number 2012 (453)]. The children with epilepsy and PPP3CA variants included in this study were diagnosed according to the 2017 epilepsy classification by the International League Against Epilepsy (ILAE) (Fisher et al., 2017) and 2022 epilepsy syndrome classification of ILAE (Zuberi et al., 2022). All procedures involving human participants in this study adhered to the 1964 Declaration of Helsinki and its later amendments or similar ethical standards. Parental written informed consent was obtained from each patient’s guardian.
Genetic testing
Variant screening of PPP3CA (NM-000944.4) was conducted using next-generation sequencing. Fourteen patients were screened using whole-exome sequencing (WES), while one patient was screened using whole-genome sequencing (WGS). High-throughput sequencing was performed using the Agilent Sure Select Method Exome V6 on the Illumina sequencing platform. The sequencing data were aligned and analyzed using NextGENe® software, and variants were screened and interpreted using the Ingenuity online software system. Gene-specific testing was performed on the parental DNA samples for all PPP3CA variants and on the siblings of the probands for segregation analysis. We have identified the allele frequencies for various genetic variants utilizing data from the Genome Aggregation Database (gnomAD).1 The pathogenicity of variant was assessed according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) (Chen et al., 2018; Richards et al., 2015).
The DNA of the patient 5 and his parents were analyzed using the Qsep100 fully automatic nucleic acid analysis system (BioNer Inc., Korea) (Gan et al., 2014; Zhuang et al., 2015). The Qsep100 system quantifies DNA fragments through fluorescence detection and simulates the band pattern as discrete peaks. The target gene was amplified by the primers listed below and then capillary electrophoresis was performed on the Qsep100 system. The detailed procedure could be obtained in Supplementary Data Sheet 1. The PCR conditions and primer specificity were verified through chip analysis (Primer-blast) and empirical optimization. The sequence of PCR primers was as follows,
PPP3CA-1F GCATTTCTGTTTTGTTGTCTGT,
PPP3CA-1R CATCATATACAAAAGTCAAACATAGCT
PPP3CA-2F CACAGAAAAGAGTTGCATTTGTATT,
PPP3CA-2R TCACATCAACTGCTTATTTTAATGTC.
Protein modeling and analysis
The PPP3CA protein sequences were obtained from NCBI. We utilized AlphaFold2 (V.2.3.2) for the monomer’s prediction (Jumper et al., 2021). The prediction models were evaluated based on each residue’s confidence score and were ranked according to their confidence level, with 0 representing the output file with the highest confidence. File with a residual confidence of 0 was selected as the final prediction for further analysis (pTM = 0.82). AlphaFold2 prediction result was shown in Supplementary Figure S1. The open-source Pymol software (V.2.5, by Schrödinger, New York, United States) was utilized to visualize the three-dimensional protein structure, facilitating the analysis of the variants’ position within the PPP3CA domain.
Literature review
Literature with “PPP3CA” and “Epilepsy” as keywords, published in PubMed and Web of Science from October 2017 to October 2024, was searched. A total of 21 patients with PPP3CA variants were identified across 8 studies. Supplementary Table S1 summarized the age of seizure onset, seizure type, patient development, EEG, brain MRI and other clinical data from the selected reports (Li and Cao, 2022; Mizuguchi et al., 2018; Myers et al., 2017; Panneerselvam et al., 2021; Qian et al., 2018; Rydzanicz et al., 2019; Yang et al., 2020).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8. Data were analyzed using Fisher’s exact tests for significance at the bivariate level. Exact p-values are reported, with significance defined as p < 0.05.
Results
In total, 15 patients with PPP3CA variants were identified, and their genotype and clinical features are shown in Table 1. This study included 11 males and 4 females, with the last follow-up age ranging from 7 months to 6 years.
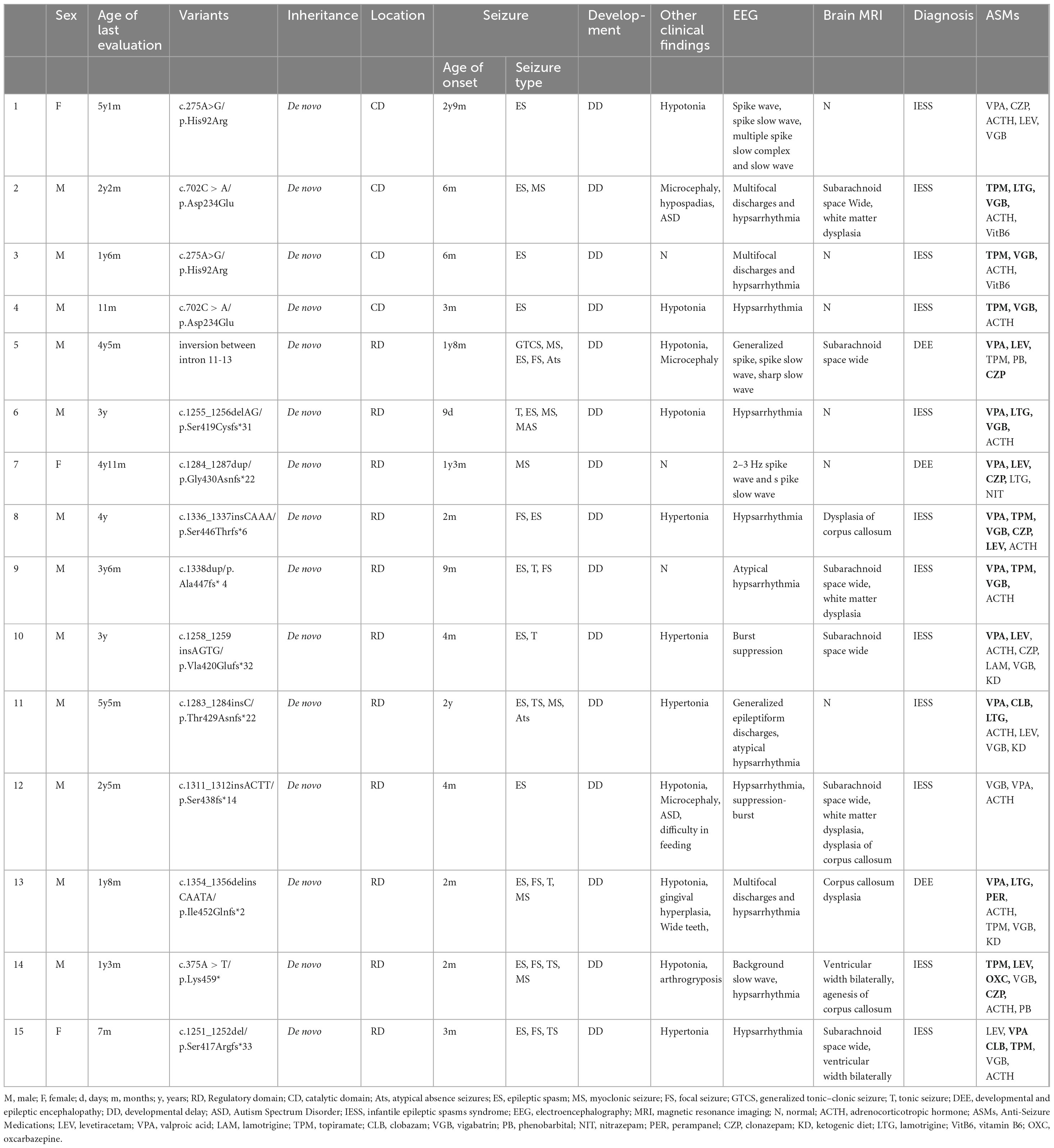
Table 1. The genotype and phenotype of 15 patients with de novo PPP3CA variants in this study.
Genetic analysis
Pathogenic or likely pathogenic of PPP3CA variants were identified in 15 children with epilepsy. The ACMG score results of these variants can be found in Supplementary Table S2. Thirteen unique de novo variants were detected, including 10 frameshift variants, 2 missense variants, and 1 gene inversion located between intron 11 and 13. Two recurrent variants were identified: His92Arg and Asp234Glu, as shown at the top of the Figure 1a and in Table 1. To predict the potential impact of missense mutations on the biological functions of proteins, we use Mutation Taster, polyphen2 and SIFT, etc. to predict pathogenicity. The specific results are shown in Supplementary Table S3 and Supplementary Figure S2. Seven truncating variants (indicated in red in Figure 1a) were reported for the first time, namely p.Gly430Asnfs*22, p.Ser446Thrfs*6, p. Ser438fs*14, p. Ala447fs*4, p.Ile452Glnfs*2, p.Lys459* and p.Ser417Argfs*33. These variants were absent from population frequencydatabases (gnmAD v3.1.2).
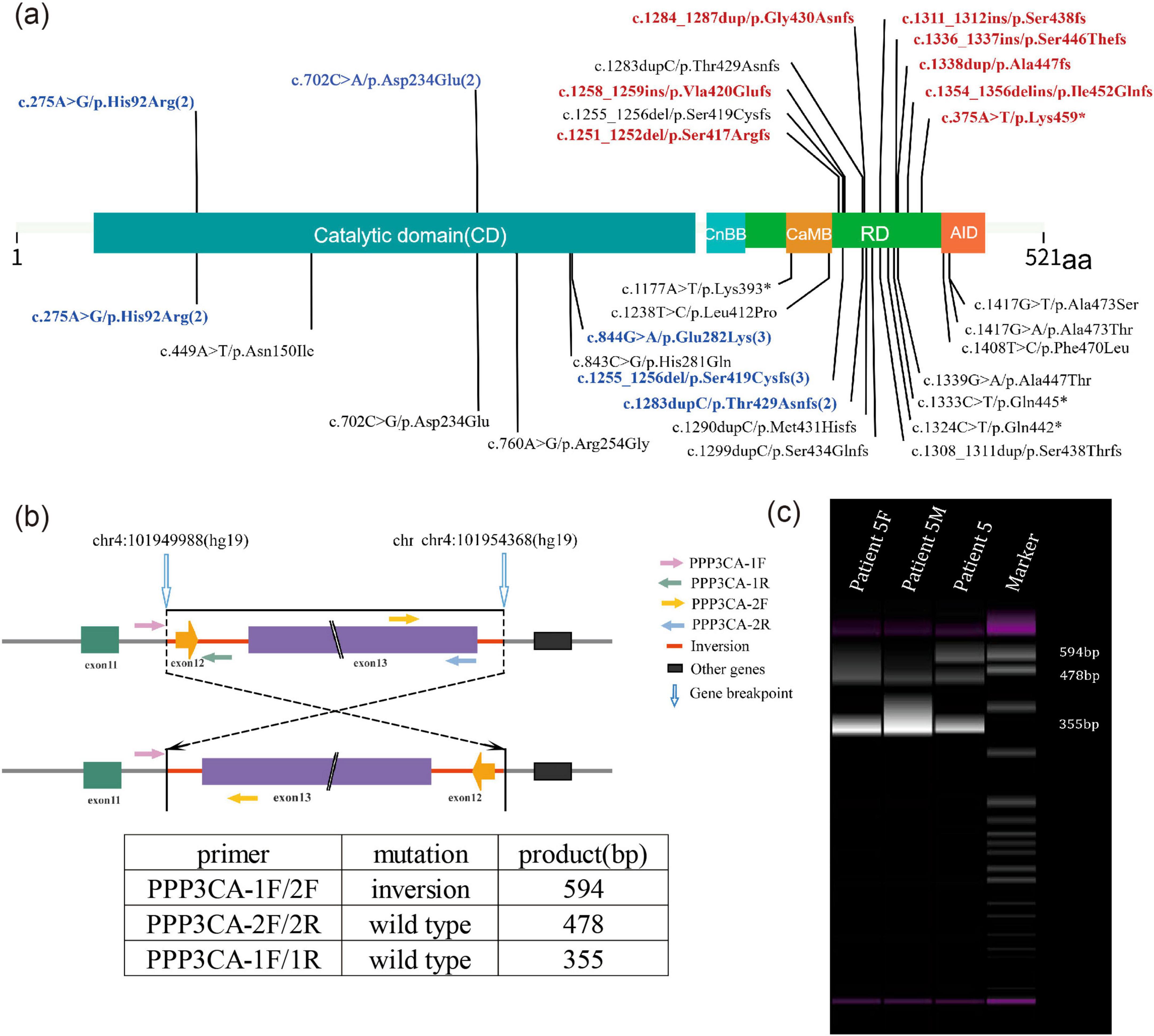
Figure 1. (a) Schematic representation of the location of PPP3CA (NM_000944.4) variants in this study and in existing literature. Variants previously described in the literature are shown at the bottom of the diagram, while those found in this study are shown at the top. Novel variants reported for the first time in this study are indicated in red. Sites with more than 2 variants are indicated in blue. PPP3CA contains five domains: the catalytic domain (CD), the calcineurin regulatory subunit-calcineurin binding domain (CnBB), the regulatory domain (RD), the calmodulin binding domain (CaMB) and the auto-inhibitory domain (AID). (b) Schematic representation of the inversion and primer design for Patient 5. The inversion region is located between introns 11 and 13, including exons 12 and 13. (c) PCR results showing an abnormal product of 594 bp. Patient 5F, DNA sample of patient 5’s father; Patient 5M, DNA sample of patient 5’s mother.
A unique de novo gene inversion was identified in Patient 5, spanning from intron 1l to intron 13 (encompassing exon 12 and exon 13). This inversion was detected through whole-genome sequencing (WGS). Notably, as exon 13 represents the terminal exon of this gene, this structural alteration does not lead to novel protein production from subsequent genetic elements. Furthermore, the mutation does not exert any functional impact on downstream genes through positional effects. The subsequent genes can be queried through NCBI (NC_000004.12). Subsequent PCR validation in the patient revealed an additional abnormal product of 594 bp. In contrast, the normal parental samples exhibited products of 478 and 355 bp (Figures 1b,c).
Variants’ location in protein structure
Figure 2a displays the 3D structural modeling of PPP3CA variants, with 12 identified variants clustering in two distinct regions. The p.Asp234Glu and p.His92Arg variants localize to the catalytic domain’s active site (Figure 2a), both showing reduced hydrogen bonding and altered interactions with surrounding residues (Figures 2b–e). Nine truncating variants occurred in the regulatory domain (Figure 2f).
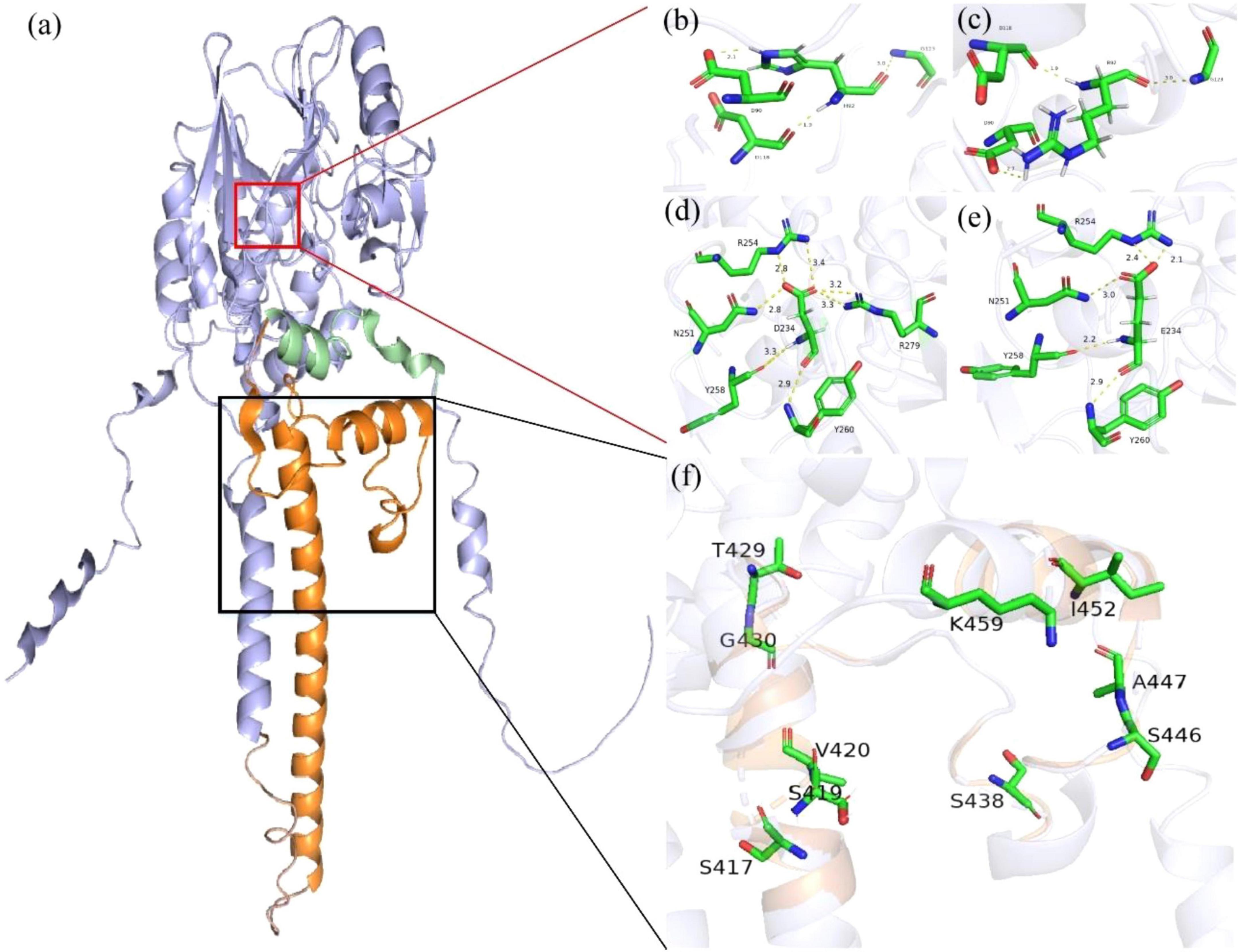
Figure 2. Protein structure of human PPP3CA (Q08209). (a) The regulatory domain (RD) is shown in orange, the auto-inhibitory domain (AID) in green, and the catalytic domain (CD) in purple. (b–e) In the variant model, the hydrogen bonds changed a lot, and the original hydrogen bonds broke, forming new hydrogen bonds. Hydrogen bonds of residues interacting with other amino acids (yellow) and the number represents the direct distance of the hydrogen bond. (f) The detailed structure of the black box highlights the other nine truncating variants that occur in the RD.
Clinical phenotypes
In our study of 15 epileptic patients carrying PPP3CA variants, seizure onset occurred from 9 days to 2 years and 9 months, with 80% (12/15) experiencing seizure onset before the age of one. Epileptic spasms were the predominant seizure type, affecting 93.3% (14/15) of patients. Other seizure types included tonic seizures in 46.7% (7/15), myoclonic seizures in 46.7% (7/15), focal seizures in 40.0% (6/15), atypical absence seizures in 13.3% (2/15), generalized tonic and clonic seizures (GTCS) in 6.7% (1/15), and myoclonic atonic seizures in 6.7% (1/15). Four patients presented only with epileptic spasms, three of whom (patient 1, 3, 4) had variants located in the CD, and one patient (patient 7) presented only with myoclonus.
In this study, all patients exhibited developmental delay and intellectual disability. Twelve patients were unable to walk after the age of 1 year and 6 months. Among those over 2 years old, nine were still unable to walk and speak. Five patients over 3 years old remained unable to walk and speak. Patient 7 was able to walk independently at 13 months and could say limited words by the age of 4 years and 11 months at the last follow-up. Additionally, 10 patients manifested hypotonia, three had microcephaly, two experienced feeding difficulties, one had gingival hyperplasia, and one had hypospadias. Patient 2 was diagnosed with autism spectrum disorder (ASD). The clinical features are summarized in Table 1.
Video EEG and brain imaging
Fifteen patients underwent video EEG for 4–24 h on multiple occasions (Table 1). The VEEG revealed diffuse slow background activity in three patients, hypsarrhythmia in 12 patients, burst suppression in two patients, generalized discharges in three patients, and multifocal discharges in three patients. Epileptic spasms were detected in 13 patients, tonic seizures in five patients, myoclonic seizures in four patients, atypical absence seizures in two patients, tonic-spasms in two patients, and GTCS in one patient. The video EEG of Patient two is shown in Figure 3. His video EEG displayed a slow background. The interictal EEG showed hypsarrhythmia, with numerous generalized and multifocal spikes, as well as multiple spikes and slow waves during both waking and sleeping periods. Epileptic spasms and myoclonic seizures were detected.
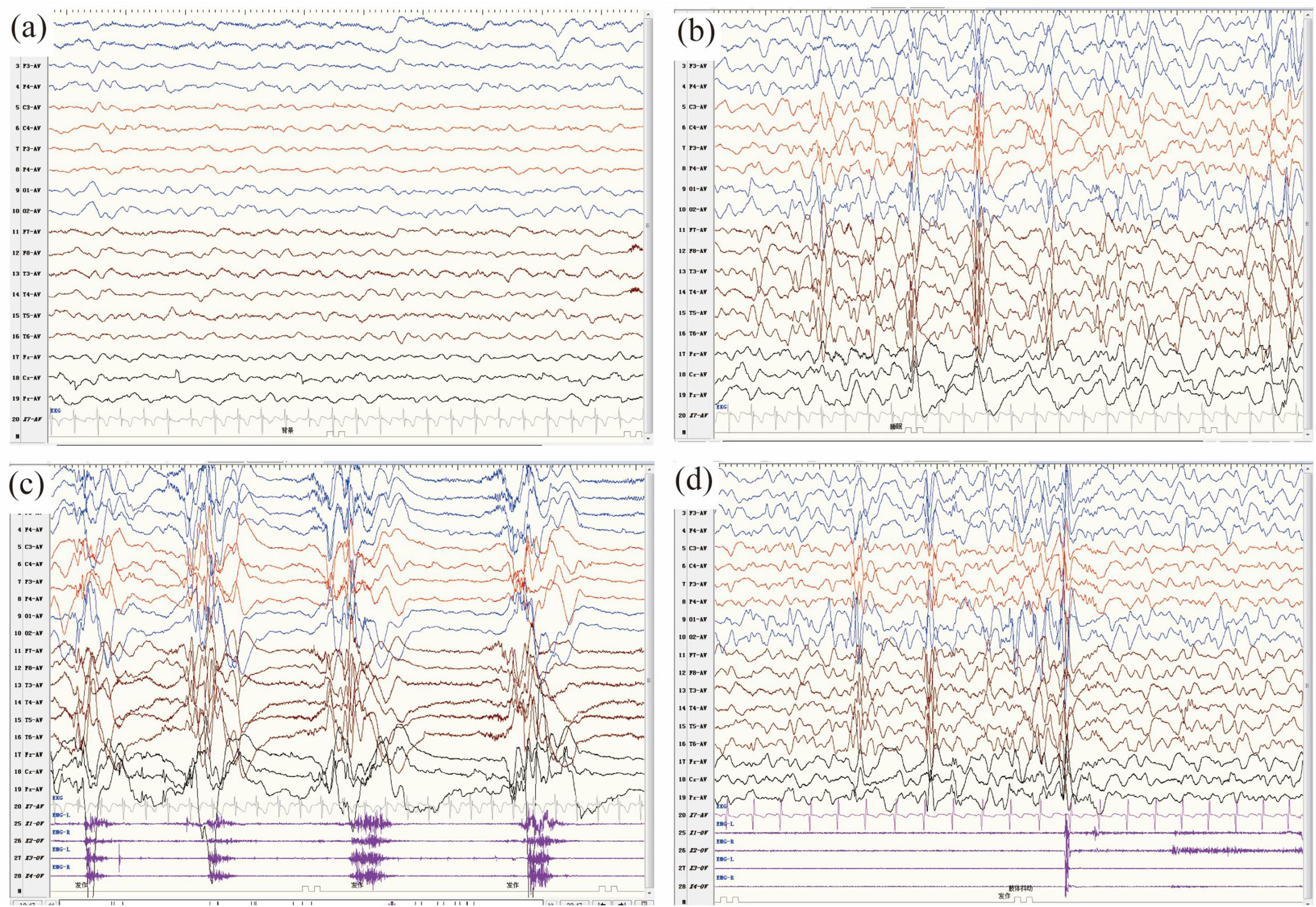
Figure 3. (a–d) Video electroencephalography (VEEG) monitoring of patient 11 in this study at the age of 10 months. (a) The background activity was slow. (b–d) The interictal VEEG demonstrated hypsarrhythmia, characterized by numerous generalized and multifocal spikes, as well as multiple spikes and slow waves, which occurred intermittently or continuously during both waking and sleeping periods. (c,d) The ictal VEEG captured clusters of epileptic spasms (c) and myoclonic seizures (d).
Fifteen patients underwent brain MRI. Abnormalities were detected in the brain of nine patients, including subarachnoid space wide in six patients, corpus callosum dysplasia in three patients, white matter dysplasia in three patients, and enlarged bilateral ventricles in two patients. The brain MRI of patient 4 is shown in Figures 4a–c. His brain MRI showed enlarged bilateral ventricles and agenesis of the corpus callosum. The brain MRI of patient 12 is shown in Figures 4d–f. His brain MRI revealed a widened subarachnoid space, white matter dysplasia, and dysplasia of corpus callosum.
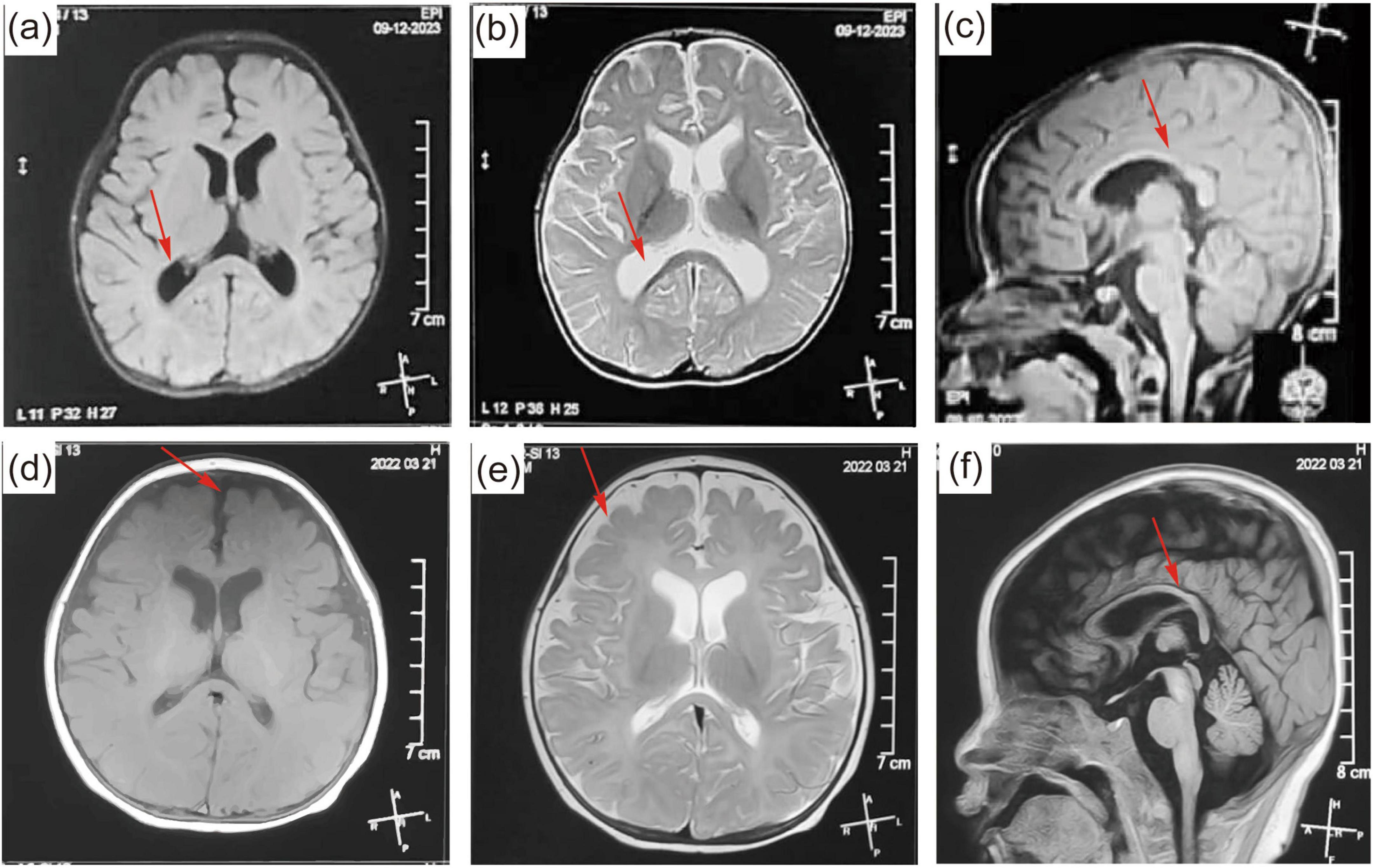
Figure 4. (a–c) Brain magnetic resonance imaging (MRI) of patient 9 at the age of 6 months. The brain MRI showed a widened subarachnoid space, white matter dysplasia, and dysplasia of corpus callosum. (d–f) MRI of patient 13 at the age of 7 months. The brain MRI showed enlarged bilateral ventricles and agenesis of the corpus callosum. The abnormal parts in the brain MRI are pointed out by red arrows.
The diagnosis of epileptic syndrome
In this study, 11 patients were diagnosed with infantile epileptic spasms syndrome (IESS). The remaining three patients were diagnosed with developmental and epileptic encephalopathy (DEE), which could not be further classified into a specific epileptic syndrome.
Literature review and analysis
As of October 2024, 26 patients with PPP3CA variants have been reported in the literature (Jumper et al., 2021; Li and Cao, 2022; Li et al., 2016; Mizuguchi et al., 2018; Myers et al., 2017; Qian et al., 2018; Rydzanicz et al., 2019; Yang et al., 2020). Three patients with variants in the AID exhibited only developmental delay and facial dysmorphic features, and were therefore excluded (Mizuguchi et al., 2018; Panneerselvam et al., 2021). Additionally, two patients (p.Glu282Lys in CD) with ASD but without seizures were excluded (Myers et al., 2017; Panneerselvam et al., 2021). The remaining 21 epileptic patients with variants in the CD, CaMB, and RD were described with detailed clinical manifestations (Table S1). Among these, eight were missense variants (p.His92Arg, p.Asn150Ile, p.Asp234Glu, p.Arg254Gly, p.His281Gln, p.Glu282Lys, p.Leu412Pro, p.Ala447Thr), six were frameshift variants (p.Ser419Cysfs* 31, p.Val420Glufs 32, p.Thr429Asnfs*22, p.Met431Hisfs*20, p.Ser434Glnfs*17, p.Ser438Thrfs*14), and three were nonsense variants (p.Lys393*, p.Gln442*, p.Gln445*). Three variants p.His92Arg (n = 4), p.Glu282Lys (n = 3) and p.Ser419Cysfs* 31 (n = 3) were reported from more than one patient.
In 21 patients with PPP3CA-related epilepsy reported in the literature, the age of seizure onset ranged from 1.5 months to 13 years, with 57.1% (12/21) experiencing their first seizure before the age of one. Multiple seizure types were documented, including epileptic spasms (n = 16), myoclonic seizures (n = 9), tonic seizures (n = 8), generalized tonic-clonic seizures (n = 7), focal seizures (n = 7), atypical absence seizures (n = 5), typical absence seizure (n = 1), and clonic absence (n = 1). Two or more seizure types were identified in 17 patients. All patients exhibited developmental delay, and seven were diagnosed with autism spectrum disorder (ASD). Hypotonia was observed in 73.9% (17/23) of patients.
The VEEG exhibited diffuse slow background activity in 4 patients, hypsarrhythmia in 12 patients, burst suppression in 2 patients, generalized discharges in 2 patients, and multifocal discharges in 9 patients.
All 21 patients underwent brain MRI. Nine patients showed abnormalities, with the main manifestations being generalized prominence of subarachnoid spaces, brain dysplasia, thin corpus callosum, and widened brain interval. Twelve patients had normal brain MRI results, The MRI features of 21 patients were summarized in Supplementary Table S1.
Genotype–phenotype correlation
Genotype-phenotype correlation analysis was performed in 36 patients with PPP3CA variants, including 15 from this study and 21 from published literature. The variants were classified and compared based on their location within different domains (Figure 1a). Eleven patients carried 8 variants in the CD, 2 patients carried 2 variants in the CaMB, and 23 patients carried 17 variants in the RD. Over 60% of the epilepsy-related variants were located in RD, which was more than any other domain. Variants in the CD were exclusively missense variants. In the CaMB, both missense and truncating variants were identified, and 94.1% (16/17) of the variants in the RD were truncating variants, including frameshift variants, nonsense variants and gene inversion.
Figure 5 is the diagram showing the percentage of different seizure types in CD, CaMB and RD. In the CD, 63.6% (7/11) of patients experienced seizures within the first year. 45.4% (5/11) had multiple seizure types, while 45.4% (5/11) presented only with epileptic spasms. 72.7% (8/11) were diagnosed with IESS. MRI results were normal in 8 patients and abnormal in the remaining 3 (27.3%). 54.5% (6/11) of patients had ASD. The variant His92Arg was identified in 4 patients (11.1%), while Asp234Glu and Glu282Lys were found in 3 patients each (8.3%).
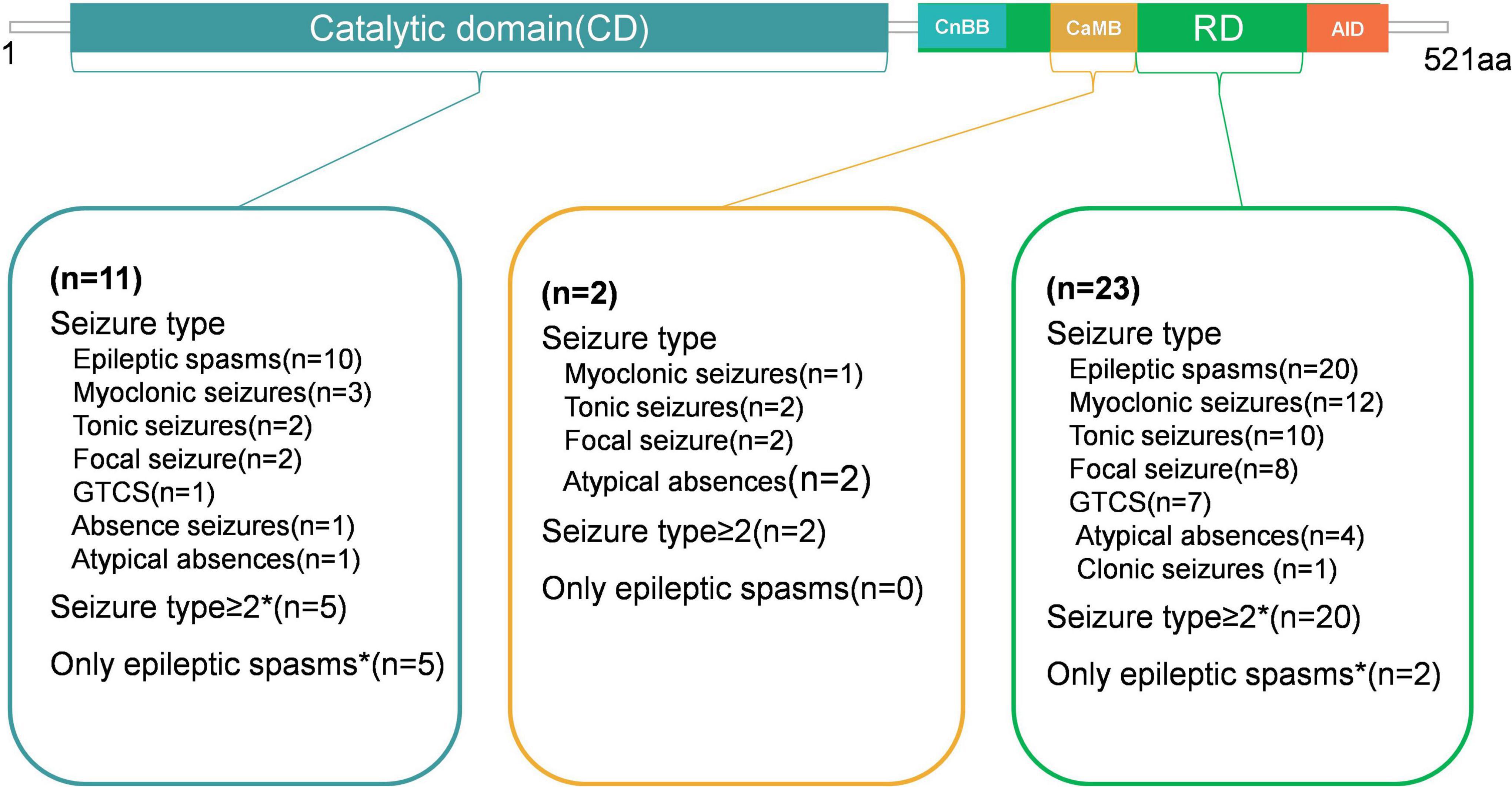
Figure 5. Diagram showing the Percentage of different seizure types in CD, CaMB and RD.
For patients with variants in the CaMB, the age of seizure onset ranged from 3 to 4 years. Both patients exhibited multiple seizure types, and MRI results were normal. In contrast, among patients with variants in the RD, 69.6% (16/23) experienced seizure onset before the age of one. 87% (20/23) had multiple seizure types, while 8.7% (2/23) patients presented only with epileptic spasms. 65.2% (15/23) patients had abnormal MRI results. The variant p.Ser419Cysfs*31 in RD was found in 4 patients (11.1%), and three patients presented epilepsy with ASD.
A comparison of patients with variations in CD and RD revealed. Patients with variants in the RD had a smaller mean age of seizure onset, although this difference was not statistically significant (p > 0.05). Compared to patients with variants in the CD, a significantly higher proportion of patients with variants in the RD had multiple seizure types (p < 0.05), while a significantly lower proportion in the RD presented only with epileptic spasms (p < 0.05). Patients with variants in the CD were more likely to have ASD compared to those with variants in the RD (p < 0.05). The variants. p.His92Arg, p.Asp234Glu Glu282Lys, and p.Ser419Cysfs*31were common variants among patients with PPP3CA-related epilepsy. These features are summarized in Table 2.
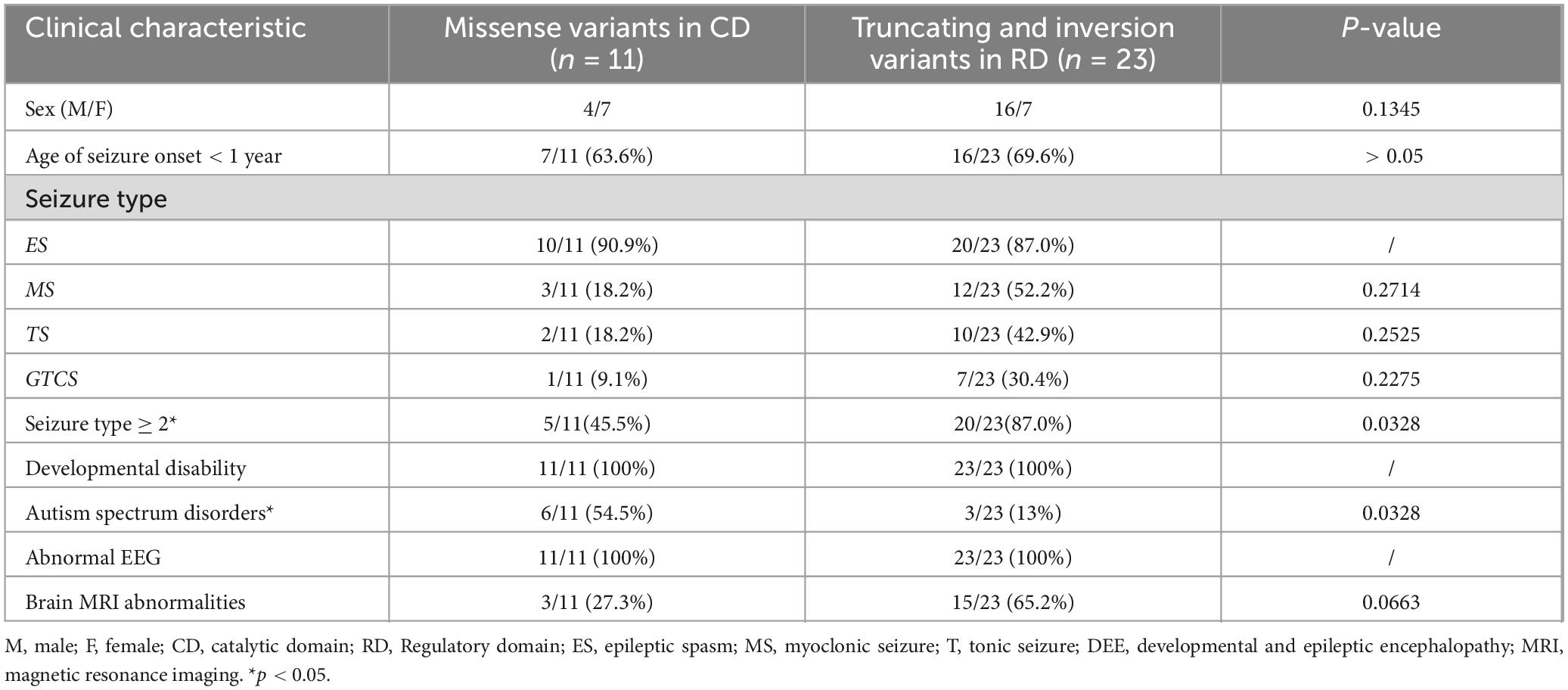
Table 2. Summary of clinical and epilepsy characteristics of 34 patients.
Epilepsy treatment and follow-up
The last follow-up age of our patients ranged from 7 months to 6 years. There was no significant seizure reduction in patients after two or more anti-seizure medications (ASMs) treatment. Thirteen patients with epileptic spasm were treated with adrenocorticotropic hormone (ACTH). For patient 4, epileptic spasms were controlled after ACTH treatment, but focal seizures still occurred intermittently. In patient 7 and 8, epileptic spasms were reduced but not fully controlled. The other 10 patients did not respond to ACTH treatment. Vigabatrin was administered to 13 patients with epileptic spasm, but it showed no reduction in seizures. Three patients were treated with the ketogenic diet for over 3 months, yet their seizures did not decrease, and their cognitive level did not improve significantly. Patient 10 had four seizure types, including epileptic spasm, myoclonic seizure, focal seizure and tonic seizure. His seizures were significantly reduced with valproic acid and lamotrigine, and he became seizure-free after receiving valproic acid, lamotrigine and vagus nerve stimulation (VNS) for 6 months before seizure recurred. Currently, he has 5-6 focal seizures per day. Patient 3, who had only myoclonic seizures was treated with valproic acid, levetiracetam and clonazepam, resulting in a 75% reduction in seizure frequency. At 4 years and 11 months, she could speak simple words and understand simple commands. Nine patients still could not sit unaided and had no language development after the age of 2. Cognitive regression was observed in three patients (patient 6, 8, 12) following seizure onset. The remaining three patients were under 1 years old, all showed unstable head control, lack of visual tracking, and absence of auditory responses.
Discussion
Genetic etiology is increasingly recognized as a significant factor in epilepsy etiologies. Li et al. (2016) reported the first case of a patient with a PPP3CA variant in 2017. As of October 2024, a total of 26 patients with PPP3CA variants have been reported (Favaro et al., 2024; Li and Cao, 2022; Mizuguchi et al., 2018; Myers et al., 2017; Panneerselvam et al., 2021; Qian et al., 2018; Rydzanicz et al., 2019; Yang et al., 2020). Notably, previous studies have shown that pathogenic variants of PPP3CA are concentrated in four domains. These pathogenic variants can be categorized into two types based on clinical phenotype. Variants in the CD, CaMB, and RD result in the developmental and epileptic encephalopathy 91 (DEE91, MIM: 617711). DEE91 is characterized by epileptic spasms accompanied by global developmental delay or regression with intellectual disability, language impairment, and ASD in some patients. However, variants in AID lead to arthrogryposis, cleft palate, craniosynostosis, and impaired intellectual development (ACCIID; MIM: 618265) (Mizuguchi et al., 2018; Panneerselvam et al., 2021).
All PPP3CA variants identified in the patients are de novo, including missense variants, frameshift variants and nonsense variants. Six variants located in the CD were exclusively missense variants. Missense variants in the CD lead to reduced CaN signaling in yeast model, suggesting a loss-of-function mechanism (Mizuguchi et al., 2018). Two variants located in the CaMB were one missense variant and one nonsense variant (Favaro et al., 2024). However, the variants in the RD were predominantly truncating variants, with only one missense variant (Ala447Thr) reported. This missense variant is located at the last nucleotide of exon 12 of PPP3CA. Multiple splicing prediction programs suggested that this change would affect mRNA splicing sites, leading out-of-frame deletion of exon 12 due to exon skipping (Panneerselvam et al., 2021). Some studies have found that nonsense variants mediated mRNA decay, resulting in haploinsufficiency (Mizuguchi et al., 2018). The variant of AID domain disrupts the interaction with the catalytic domain, leading to the failure of self-suppression and thus resulting in gain-of-function (Mizuguchi et al., 2018). Other studies have shown that frameshift variants did not decrease mRNA expression levels but caused protein instability leading to haploinsufficiency (Qian et al., 2018; Rydzanicz et al., 2019).
In this study, all 15 patients had de novo variants, including 2 missense variants, 10 frameshift variants, and 1 gene inversion. The missense variant was located in the CD, consistent with previous literature. AlphaFold2 prediction revealed that the mutated amino acid had reduced interaction with surrounding amino acid residues, indicating a loss-of-function change. However, frameshift variants and gene inversions were located in the RD. A novel variant, gene inversion, was identified for the first time in our study. In addition, seven other novel frameshift variants were identified, further expanding the PPP3CA gene variation spectrum. Currently, truncating variants, including nonsense and frameshift variants, are the most common type of PPP3CA variants in the RD. There are few studies on truncating variants in PPP3CA, so the underlying mechanisms remain unclear.
Combining the patients with epilepsy from this study and those in the literature review, variants were identified in 13 patients in the CD, 2 patients in the CaMB domain and 23 patients in the RD. The variants p.His92Arg, p.Asp234Glu, p.Glu282Lys and p.Ser419Cysfs*33 were recurrent in PPP3CA.
In our study, 80% of patients experienced their first seizure onset before the age of one. In the literature review, among 21 patients with epilepsy, 57.1% had their first seizure before the age of one. Combining the data from our study and the literature review, 66.7% (24/36) of patients had their first seizure within their first year of life. In our study, multiple seizure types were observed, including epileptic spasms, tonic seizures, myoclonic seizures, focal seizures, GTCS, absence seizures, and myoclonic atonic epilepsy. Among the 21 patients reported previously, seizure types included epileptic spasms (16/21), myoclonic seizure (9/21), GTCS (9/21), focal seizure (7/21), tonic (4/21), atypical absence seizure (2/21), and absence seizure (1/21). Combining the results from our study and the literature, the most common seizure type in patients with PPP3CA variants was epileptic spasms (30/36, 83.3%).
In the literature review, epileptic syndromes were diagnosed in approximately 75% (16/21) of patients, which include IESS (n = 14), and Lennox–Gastaut syndrome (LGS, n = 2). In our study, 80% of patients were diagnosed with IESS. This suggests that IESS is the most common phenotype of PPP3CA-related epilepsy.
In the literature review, interictal EEG showed hypsarrhythmia, burst suppression, generalized discharges, and multifocal discharges. Epileptic spasms, focal seizures, myoclonic seizures, generalized tonic–clonic seizures, and absence seizures were observed in some patients (Panneerselvam et al., 2021). In this study, hypsarrhythmia was observed in 80% of patients, multifocal epileptiform discharges in 23.4%, generalized epileptiform discharges in 23.4%, and burst suppression in 13.3%. Epileptic spasms were detected in 13 patients, with patient 2 developing epileptic spasms for the first time nearly 3 years old. These findings suggest that hypsarrhythmia may be a characteristic EEG feature in the majority of patients with PPP3CA-related epilepsy. Among the 21 patients reviewed in the literature, about 50% had abnormal brain MRI, most commonly showing cerebral dysplasia and thin corpus callosum (Li and Cao, 2022; Li et al., 2016; Mizuguchi et al., 2018; Myers et al., 2017; Rydzanicz et al., 2019; Yang et al., 2020). In this study, abnormal brain MRI was observed in 10 patients, including frontotemporal subarachnoid widening, corpus callosum dysplasia, and white matter dysplasia.
Comparison of clinical features among patients with variants located in the CD, RD reveals distinct patterns. Patients with variants in the CD tend to have a relatively late seizure onset age, with nearly half patienting only had epileptic spasm. Additionally, nearly 50% of these patients have comorbid ASD, suggesting that ASD-related examinations should be recommended for patients with variant in the CD. However, patients with variants in the RD experienced an earlier seizure onset age. Approximately 90% of patients with RD variants had multiple seizure types, and about 75% exhibited brain MRI abnormalities, highlighting the importance of enhancing brain MRI for the diagnosis of PPP3CA-related DEE.
In the literature review, 33.3% (7/21) of the patients had epilepsy with ASD, and five of these had variants in the CD between Asn150 and Gln282 site. Additionally, two patients with p.Glu282Lys variants in the CD displayed ASD without seizures. In this study, patient 11, with variants located in CD, exhibited autism-like symptoms. This finding suggests that patients with variants located between Asn150 and Gln282 sites are more likely to have epilepsy with ASD. However, the mechanism of PPP3CA variants causing ASD requires further investigation.
Therapeutic information for PPP3CA related epilepsy is limited. Patients with this condition generally do not respond well to ASMs. In the literature review, one patient with the variant p.Glu282lys experienced seizure onset at 4 years old and became seizure free after the treatment with VPA and LTG (Myers et al., 2017). However, other patients with PPP3CA-related epilepsy showed no significant response to ASMs (Myers et al., 2017). Based on the data from patients in both the literature review and this study, epileptic spasms were controlled by ACTH in only one patient, while the others did not respond to ACTH. Additionally, none of the patients with epileptic spasms responded to VGB. This highlights the urgent need for targeted therapies for PPP3CA-related.
Genetic variations may induce functional alterations in proteins through modifications of distinct structural domains that govern specialized biological roles. These structural and functional distinctions suggest that pathogenic variants at different domain locations may manifest divergent detrimental consequences, ultimately contributing to heterogeneous phenotypic expressions (Liao et al., 2022). The molecular mechanisms underlying epileptogenesis associated with RD domain-truncating mutations remain insufficiently characterized. Notably, clinical observations reveal a distinct phenotypic spectrum among patients with RD domain mutations, characterized by markedly earlier epilepsy onset and polymorphic seizure manifestations compared to individuals harboring mutations in other functional regions. Furthermore, functional domain-specific phenotypic dichotomies demonstrate clear genotype-phenotype correlations: AID domain mutations induce multi-organ developmental anomalies in the absence of epileptic complications, while pathogenic variants consistently present as drug-resistant epilepsy syndromes refractory to conventional anticonvulsant therapies. This domain-dependent phenotypic specificity necessitates the establishment of dual research paradigms: (1) Domain-specific therapeutic targeting of ion channel dysregulation mechanisms and (2) Precision genome editing approaches including CRISPR-based mutation correction or antisense oligonucleotide-mediated transcript modulation. These research directions will fundamentally enhance both mechanistic comprehension and clinical management strategies for this genetically heterogeneous neurological disorder.
There are several strengths in this study. Firstly, we collected the largest single-center study to date. By leveraging this extensive patient samples, we combined the cases from this study with those described in the literature, focusing on detailed phenotypic descriptions. Secondly, this study demonstrated the genotype-phenotype correlation of PPP3CA-related epilepsy. We compared phenotypes across the CD, CaMB and RD and identified clinical phenotypes associated with different PPP3CA domains. Thirdly, this study identified seven novel variants and a gene inversion in PPP3CA-related DEE for the first time, expanding the genotypic spectrum of PPP3CA. Additionally, recurrent variants p.His92Arg, p.Asp234Glu, p.Glu282Lys, and p.Ser419Cysfs*33 were identified.
Admittedly, this study has several limitations. Firstly, the number of patients with PPP3CA variants is still relatively small, and more patients need to be enrolled for a more robust genotype-phenotype correlation analysis. Secondly, this is an observational study, and the observed results may be subject to confounding factors that we could not identify and control in this study. Thirdly, the follow-up period was not long enough to fully assess long-term outcomes.
Conclusion
Over 60% of PPP3CA variants in patients with epilepsy were located in the RD. Variants located in the CD were exclusively missense variants. The majority of variants in the RD were truncating variants. Seizure onset in most cases of PPP3CA-related DEE occurred during infancy. The most common epileptic syndrome was IESS. Nearly 50% of patients with variants in the CD present with epileptic spasms associated with ASD. Most patients with PPP3CA-related epilepsy were drug-resistant. Additionally, our study reported a gene inversion in PPP3CA-related DEE for the first time. The variants p.His92Arg, p.Asp234Glu, p.Glu282Lys and p.Ser419Cysfs* 33 are the most frequently reported PPP3CA variants.
Data availability statement
The data presented in this study are available through Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), with the following accession numbers SCV006082273–SCV006082284. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Peking University First Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
TW: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. SO: Data curation, Formal Analysis, Writing – review & editing. XN: Data curation, Writing – review & editing. MC: Data curation, Writing – review & editing. YiY: Data curation, Writing – review & editing. YoY: Data curation, Writing – review & editing. QT: Data curation, Writing – review & editing. WL: Data curation, Writing – review & editing. XY: Data curation, Writing – review & editing. YZ: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Key R&D Program of China (Nos. 2023YFC2706300 and 2023YFC2706301).
Acknowledgments
We thank the patients and their families who participated in this study. We also thank team staff who assisted in the data collection and statistical analysis. We would like to thank for Xiaodong Wang for assisting in manuscript editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2025.1570997/full#supplementary-material
Footnotes
References
Aramburu, J., Heitman, J., and Crabtree, G. R. (2004). Calcineurin: A central controller of signalling in eukaryotes. EMBO Rep. 5, 343–348. doi: 10.1038/sj.embor.7400133
Chen, X., Gao, Y., Yang, L., Wu, B., Dong, X., Liu, B., et al. (2018). Speech and language delay in a patient with WDR4 mutations. Eur. J. Med. Genet. 61, 468–472. doi: 10.1016/j.ejmg.2018.03.007
Cook, E. C., and Creamer, T. P. (2016). Calcineurin in a crowded world. Biochemistry 55, 3092–3101. doi: 10.1021/acs.biochem.6b00059
Creamer, T. P. (2013). Transient disorder: Calcineurin as an example. Intrinsically Disord Proteins 1:e26412. doi: 10.4161/idp.26412
Dhindsa, R. S., Bradrick, S. S., Yao, X., Heinzen, E. L., Petrovski, S., Krueger, B. J., et al. (2015). Epileptic encephalopathy-causing mutations in DNM1 impair synaptic vesicle endocytosis. Neurol. Genet. 1:e4. doi: 10.1212/01.NXG.0000464295.65736.da
Favaro, J., Iodice, A., Nosadini, M., Asta, F., Toldo, I., Ancona, C., et al. (2024). PPP3CA gene-related developmental and epileptic encephalopathy: Expanding the electro-clinical phenotype. Seizure 121, 253–261. doi: 10.1016/j.seizure.2024.08.017
Fisher, R. S., Cross, J. H., French, J. A., Higurashi, N., Hirsch, E., Jansen, F. E., et al. (2017). Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE commission for classification and terminology. Epilepsia 58, 522–530. doi: 10.1111/epi.13670
Gan, W., Zhuang, B., Zhang, P., Han, J., Li, C. X., and Liu, P. A. (2014). filter paper-based microdevice for low-cost, rapid, and automated DNA extraction and amplification from diverse sample types. Lab. Chip. 14, 3719–3728. doi: 10.1039/c4lc00686k
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Karagiota, A., Mylonis, I., Simos, G., and Chachami, G. (2019). Protein phosphatase PPP3CA (calcineurin A) down-regulates hypoxia-inducible factor transcriptional activity. Arch. Biochem. Biophys. 664, 174–182. doi: 10.1016/j.abb.2019.02.007
Li, J., and Cao, J. (2022). Case report: A novel PPP3CA truncating mutation within the regulatory domain causes severe developmental and epileptic encephalopathy in a Chinese patient. Front. Neurol. 13:889167. doi: 10.3389/fneur.2022.889167
Li, S. J., Wang, J., Ma, L., Lu, C., Wang, J., Wu, J. W., et al. (2016). Cooperative autoinhibition and multi-level activation mechanisms of calcineurin. Cell Res. 26, 336–349. doi: 10.1038/cr.2016.14
Liao, W. P., Chen, Q., Jiang, Y. W., Luo, S., and Liu, X. R. (2022). Editorial: Sub-molecular mechanism of genetic epilepsy. Front. Mol. Neurosci. 15:958747. doi: 10.3389/fnmol.2022.958747
McArdle, C. B., Richardson, C. J., Nicholas, D. A., Mirfakhraee, M., Hayden, C. K., and Amparo, E. G. (1987). Developmental features of the neonatal brain: MR imaging. Part II. Ventricular size and extracerebral space. Radiology 162(Pt 1), 230–234. doi: 10.1148/radiology.162.1.3786768
Mizuguchi, T., Nakashima, M., Kato, M., Okamoto, N., Kurahashi, H., Ekhilevitch, N., et al. (2018). Loss-of-function and gain-of-function mutations in PPP3CA cause two distinct disorders. Hum. Mol. Genet. 27, 1421–1433. doi: 10.1093/hmg/ddy052
Myers, C. T., Stong, N., Mountier, E. I., Helbig, K. L., Freytag, S., Sullivan, J. E., et al. (2017). De novo mutations in PPP3CA cause severe neurodevelopmental disease with seizures. Am. J. Hum. Genet. 101, 516–524. doi: 10.1016/j.ajhg.2017.08.013
Palkowitsch, L., Marienfeld, U., Brunner, C., Eitelhuber, A., Krappmann, D., and Marienfeld, R. B. (2011). The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-kappaB activation. J. Biol. Chem. 286, 7522–7534. doi: 10.1074/jbc.M110.155895
Panneerselvam, S., Wang, J., Zhu, W., Dai, H., Pappas, J. G., Rabin, R., et al. (2021). PPP3CA truncating variants clustered in the regulatory domain cause early-onset refractory epilepsy. Clin. Genet. 100, 227–233. doi: 10.1111/cge.13979
Qian, Y., Wu, B., Lu, Y., Dong, X., Qin, Q., Zhou, W., et al. (2018). Early-onset infant epileptic encephalopathy associated with a de novo PPP3CA gene mutation. Cold Spring Harb. Mol. Case Stud. 4:a002949. doi: 10.1101/mcs.a002949
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rumi-Masante, J., Rusinga, F. I., Lester, T. E., Dunlap, T. B., Williams, T. D., Dunker, A. K., et al. (2012). Structural basis for activation of calcineurin by calmodulin. J. Mol. Biol. 415, 307–317. doi: 10.1016/j.jmb.2011.11.008
Rusnak, F., and Mertz, P. (2000). Calcineurin: Form and function. Physiol. Rev. 80, 1483–1521. doi: 10.1152/physrev.2000.80.4.1483
Rydzanicz, M., Wachowska, M., Cook, E. C., Lisowski, P., Kuźniewska, B., Szymańska, K., et al. (2019). Novel calcineurin A (PPP3CA) variant associated with epilepsy, constitutive enzyme activation and downregulation of protein expression. Eur. J. Hum. Genet. 27, 61–69. doi: 10.1038/s41431-018-0254-8
Wang, X., Chen, L., Yang, H., and Chen, Z. (1996a). MR imaging study on normal neonatal and infant brain maturation (part I): Myelinization. Chin. J. Radiol. 30, 654–658. doi: cnki:sun:zhgs.0.1996-10-001
Wang, X., Chen, L., Yang, H., and Chen, Z. (1996b). MRI study on normal brain development in newborns and infants II. regarding ventricular size and extracranial space. Chin. J. Radiol. 30, 850–854. doi: cnki:sun:zhgs.0.1996-12-016
Yang, S., Shen, X., Kang, Q., Kuang, X., Ning, Z., Liu, S., et al. (2020). Clinical and genetic study on a chinese patient with infantile onset epileptic encephalopathy carrying a PPP3CA null variant: A case report. BMC Pediatr. 20:315. doi: 10.1186/s12887-020-02213-7
Zhuang, B., Gan, W., Wang, S., Han, J., Xiang, G., Li, C. X., et al. (2015). Fully automated sample preparation microsystem for genetic testing of hereditary hearing loss using two-color multiplex allele-specific PCR. Anal. Chem. 87, 1202–1209. doi: 10.1021/ac5039303
Zuberi, S. M., Wirrell, E., Yozawitz, E., Wilmshurst, J. M., Specchio, N., Riney, K., et al. (2022). ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE task force on nosology and definitions. Epilepsia 63, 1349–1397. doi: 10.1111/epi.17239
Keywords: PPP3CA, variants, genotype, phenotype, developmental and epileptic encephalopathy
Citation: Wang T, Ouyang S, Niu X, Cheng M, Yang Y, Yang Y, Tan Q, Liu W, Yang X and Zhang Y (2025) New variants and genotype-phenotype correlation of PPP3CA-related developmental and epileptic encephalopathy. Front. Neurosci. 19:1570997. doi: 10.3389/fnins.2025.1570997
Received: 04 February 2025; Accepted: 12 May 2025;
Published: 06 June 2025.
Edited by:
Rodney C. Samaco, Association of University Centers on Disabilities, United StatesReviewed by:
Christopher McGraw, Northwestern University, United StatesSheng Luo, The Second Affiliated Hospital of Guangzhou Medical University, China
Copyright © 2025 Wang, Ouyang, Niu, Cheng, Yang, Yang, Tan, Liu, Yang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuehua Zhang, emhhbmd5aGRyQDEyNi5jb20=