 Giuseppe Pepe
Giuseppe Pepe Alba Di Pardo
Alba Di Pardo Vittorio Maglione
Vittorio Maglione- IRCCS Neuromed, Pozzilli (IS), Italy
Genomic instability is a key feature of many neurological disorders, with transposon activation and nucleotide triplet repeat instability playing critical roles. Transposons, which are also referred to as mobile genetic elements, have the potential to destabilize the genome and interfere with gene expression. Conversely, changes in nucleotide triplet sequences, such as expansions or contractions, can lead to the production of abnormal proteins or nonfunctional RNAs. In this perspective, we discussed the intricate relationship between these two forms of genomic instability and their influence on brain disorders. We analyzed the molecular mechanisms that contribute to these phenomena, the shared regulatory systems that govern them, and their role in neurological conditions. Additionally, we provided some insights into the development of potential therapies for brain disorders linked to these genomic alterations.
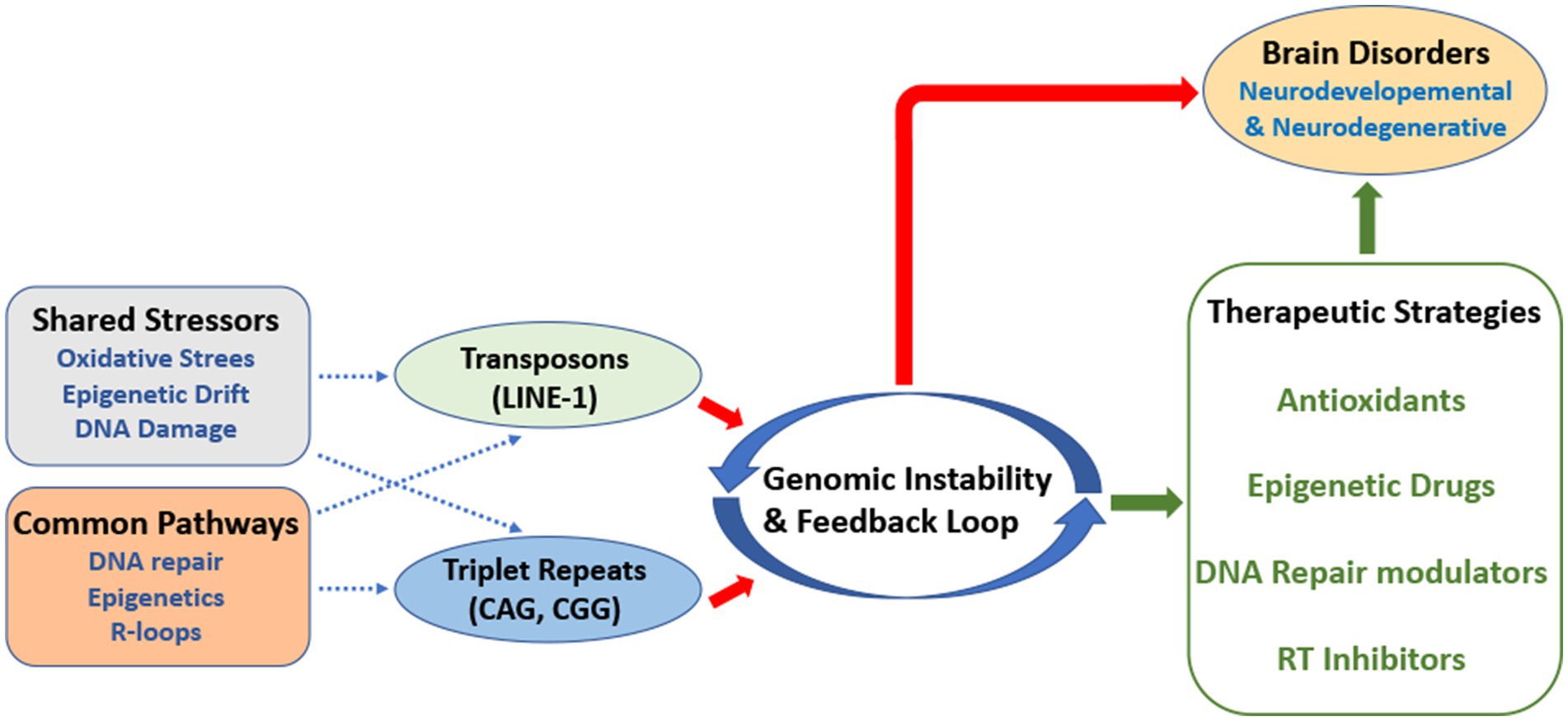
Graphical Abstract.
Introduction
Neurological disorders include a wide range of conditions, often associated with complex genetic and epigenetic changes. The intricate relationship between transposon activity and nucleotide triplet repeat instability represents an intriguing area of research, with the potential to reveal mechanisms that may contribute to disease progression.
Transposons were once considered “junk DNA,” however, they are now recognized as active elements that shape genomic structure and gene expression, playing crucial roles in health and disease (Cordaux and Batzer, 2009). LINE-1 (long interspersed nuclear element-1) elements play an important role during brain development (Suarez et al., 2018). However, while they contribute to neuronal diversity, they also carry risks of genomic instability, highlighting their multifaceted role in brain physiology (Suarez et al., 2018).
In the context of brain disorders, retrotransposons have been implicated in various pathological processes (Zhang et al., 2025). In particular, LINE-1 elements may be reactivated in aging neurons and under stress conditions, leading to DNA damage and transcriptional dysregulation (Gorbunova et al., 2021; Della Valle et al., 2025). Oxidative stress may trigger LINE-1 activation in neuronal models, causing DNA damage and genomic instability (Ravel-Godreuil et al., 2021).
Nucleotide triplet repeat instability, characterized by the abnormal expansion or contraction of specific DNA sequences, is another genomic phenomenon associated with a number of neurodegenerative and psychiatric disorders (La Spada, 1997). Trinucleotide repeat expansions lead to transcriptional dysregulation, toxic RNA foci, and protein aggregation, which represent molecular hallmarks in the disease pathology (Everett and Wood, 2004; Sicot and Gomes-Pereira, 2013; Schwartz et al., 2021).
The interplay between transposon activity and nucleotide triplet repeat instability may be modulated by shared cellular stressors and regulatory pathways.
Oxidative stress, for instance, is a common trigger that can either activate LINE-1 retrotransposition or exacerbate triplet repeat expansions, through replication stress (Van Meter et al., 2014; Chatterjee et al., 2015; Martins et al., 2021; Ravel-Godreuil et al., 2021). Similarly, deficiencies in DNA repair system have been implicated in both repeat expansion and the mobilization of retrotransposons (Mirkin, 2007; Levin and Moran, 2011; Zhao and Usdin, 2015; Mendez-Dorantes and Burns, 2023). This highlights the interconnection between these types of genomic instabilities.
This perspective aims at unraveling the complex mechanisms underlying these processes, with a focus on their possible crosstalk and the potential pathways that could amplify their effects in brain disorders. Specifically, we argue that these two forms of instability are not merely parallel phenomena; they may engage to each other in a synergistic and deleterious interplay that may ultimately accelerate neuronal dysfunction. By exploring their contributions to brain diseases, we also aimed at highlighting some potential therapeutic strategies.
Understanding this interplay will be crucial for advancing our knowledge of neuronal dysfunction and for improving the development of therapeutic strategies.
Transposon instability in brain health and disease
LINE-1 elements, constituting approximately 17% of the human genome, are retrotransposons capable of replicating and inserting themselves into new genomic locations (Richardson et al., 2014). Their activity is essential during neurodevelopment, where they are a major driver of neuronal genomic mosaicism (Richardson et al., 2014; Upton et al., 2015).
This mosaicism, characterized by diverse genomic configurations among neurons, is hypothesized to underlie individual neuronal functions and adaptability (Richardson et al., 2014; Faulkner and Garcia-Perez, 2017). Such a genomic diversity is crucial for cognitive processes, including learning and memory formation (Bachiller et al., 2017).
While, this activity promotes genomic innovation and adaptation, it may introduce risks, such as insertional mutagenesis and chromosomal instability, that can potentially compromise neuronal integrity (Richardson et al., 2014; Mendez-Dorantes et al., 2024).
The regulation of transposon activity is predominantly regulated by epigenetic mechanisms, including DNA methylation and histone modifications (Freeman et al., 2022; Lanciano et al., 2024). During early development, these mechanisms may act as crucial regulators of transposon activity to maintain genomic stability. Thus, any defect in such mechanisms, with consequent aberrant retrotransposon activity in neuronal precursor cells (NPCs; Jönsson et al., 2019), may be eventually linked to neurodevelopmental disorders such as Schizophrenia and Autism Spectrum Disorders (ASDs; Suarez et al., 2018; Jönsson et al., 2020). For instance, in schizophrenia, LINE-1 mobilization is associated with increased transposition in the neuronal genome (Bundo et al., 2014). Similarly, although the precise underlying mechanisms remain to be fully elucidated, ASDs are characterized by elevated levels of LINE-1 (Shpyleva et al., 2018) which has been suggested to contribute to defective neurodevelopmental processes (Evans and Erwin, 2021).
On the other hand, pathological age-related epigenetic decline (Li and Tollefsbol, 2016) may lead to hypomethylation of LINE-1 sequences, thereby increasing transposition events (Erwin et al., 2014; Senapati et al., 2023; Yang et al., 2023). This decline is particularly evident in neurodegenerative diseases (Brulé et al., 2025), where global hypomethylation, occurring in the adult brain, may potentially coincide with heightened transposon activity.
In this context, dysregulated transposon activity has been identified as a significant contributor to neurodegenerative disorders (Ochoa Thomas et al., 2020). Alzheimer’s disease displays increased LINE-1 activity that could compromise neuronal genome integrity, exacerbating disease progression (Guo et al., 2018; Ochoa Thomas et al., 2020; Roy et al., 2024). In Amyotrophic Lateral Sclerosis (ALS) models, LINE-1 activation has been linked to neuroinflammatory processes (Takahashi et al., 2022), while, in Parkinson’s disease neurons, translocation of LINE ORF1p induces nuclear envelop alterations (Martin, 2006; Znaidi et al., 2025).
Nucleotide triplet repeat instability in brain disorders
Nucleotide triplet repeat instability plays a pivotal role in several brain disorders (Depienne and Mandel, 2021). Among these conditions, Huntington’s disease and Fragile X syndrome exemplify neurodegenerative and neurodevelopmental disorders, respectively (Salcedo-Arellano et al., 2020; Depienne and Mandel, 2021).
Nucleotide triplet repeat instability arises from different mechanisms like DNA replication slippage, defective mismatch repair, and oxidative DNA damage (Mirkin, 2007; Castel et al., 2010; Li and Bonini, 2010; McMurray, 2010).
Either expansions or contractions of specific repeat sequencing (Castel et al., 2010) can occur in both dividing cells and post-mitotic neurons due to the limited DNA repair capacity that may intensify instability over time (Konopka et al., 2022). In Huntington’s disease this leads to a phenomenon known as CAG repeat somatic instability (Cattaneo et al., 2025).
Expanded trinucleotide repeats can disrupt gene transcription, RNA splicing, and translation, leading to significant neuronal dysfunction (Castel et al., 2010; Li and Bonini, 2010; Schwartz et al., 2021). Moreover, repeat expansions can sequester vital RNA-binding proteins, like TDP-43, inducing neurodegeneration (Sun et al., 2023).
Furthermore triplet repeat expansions can affect chromatin dynamics (Frisch et al., 2001; Dion and Wilson, 2009; Nageshwaran and Festenstein, 2015) and change histone acetylation and methylation patterns (Kumari and Usdin, 2009; Wei et al., 2017), potentially modifying the transcriptional landscape of the neurons. In this context, CGG triplet expansions, associated with Fragile X Syndrome, have been linked to histone hypoacetylation and chromatin condensation, which contributes to the inactivation of FMR1 gene (Coffee et al., 2002).
The potential mechanistic interplay between transposons and triplet repeats: a fascinating hypothesis
The pathological impact of these instabilities is age- and cell-type dependent. Transposon activation contributes to neuronal diversity during early neurodevelopment and is largely suppressed in mature neurons (Zhang et al., 2025). Its pathological reactivation occurs under age-related epigenetic erosion, oxidative stress, or neurodegenerative processes in post-mitotic neurons (Ochoa Thomas et al., 2020; Gorbunova et al., 2021).
Conversely, triplet repeat instability can originate from errors during DNA replication in developing cells (germline or somatic) that continues to accumulate in non-dividing neurons throughout life making neurons particularly vulnerable (McMurray, 2010; Konopka et al., 2022; Cattaneo et al., 2025). This may create a scenario where both instabilities can converge in aging and in the diseased neurons, potentially exacerbating genomic stress.
This mechanistic interaction between transposon activity and nucleotide triplet repeat instability, is a fascinating potential event which may reveal that these two forms of genomic instability may influence each other, thereby amplifying their deleterious effects.
Oxidative stress as potential shared mechanism between transposon activation and triplet instability
Oxidative stress may influence both transposon activity and triplet repeat instability (Giorgi et al., 2011; La Rosa et al., 2020). It may induce LINE-1 activation in neuronal models (Giorgi et al., 2011) and reduce global DNA methylation and disrupt histone modifications (Niu et al., 2015; García-Guede et al., 2020), conceivable creating a permissive chromatin environment for LINE-1 activation.
Additionally, oxidative lesions of CAG repeats promote repeat expansions in Huntington’s disease models, suggesting a direct link between triplet instability and oxidative stress (Kovtun et al., 2007).
These findings suggest that oxidative stress could independently and synergistically activate mechanisms that may exacerbate genomic instability.
A bidirectional relationship between oxidative stress and genomic instability may generate a feedback loop involving transposon activity and triplet repeat expansions. For instance, ROS-induced DNA breaks may activate LINE-1 transposition, which in turn could likely introduce additional DNA damage. On the other hand, the generation of oxidative stress, in nucleotide triplet diseases, may create a self-sustaining loop of oxidative damage and DNA instability, which could also affect retrotransposon activity.
Shared pathways in DNA repair system
Transposons and triplet repeats might compete for or interfere with common pathways involved in DNA repair.
Repeat expansions are often linked to DNA repair pathways (McMurray, 2010; Schmidt and Pearson, 2016; Lai et al., 2020). During their active transposition phases, transposons may affect the same repair pathways to support their insertion and stabilization within the genome (Gasior et al., 2006). DNA repair pathway, crucial for correcting replication errors, may be intimately involved in both processes. Proteins like MLH1, MSH2, MSH3 (component of DNA Mismatch Repair-MMR-system) and FAN1 (DNA repair nuclease) are key drivers of somatic CAG repeat expansion (Schmidt and Pearson, 2016; Deshmukh et al., 2021; Bunting et al., 2025). Concurrently, when transposable elements are highly active, their retrotransposition creates DNA double-strand breaks that could potentially hijack the DNA repair machinery (Morrish et al., 2002; Gasior et al., 2006; Mendez-Dorantes and Burns, 2023; Dias et al., 2025).
The condition in which DNA repairing is highly required, may theoretically make triplet repeats more susceptible to instability and expansion.
RNA-mediated mechanisms
An interesting hypothesis is represented by the possibility that RNA may mediate a possible interaction between transposons and triplet repeats.
Triplet repeats can be associated with the formation of RNA–DNA hybrid structures (R-loops), which could potentially interfere with the regulation of gene expression and with DNA repair system and genome stability (Wulfridge and Sarma, 2024). Such a structures have been identified in brain disorders including neurodegenerative diseases (Li et al., 2025).
Interestingly, the existence of LINE1 transposition-derived R-loops has been recently shown (Paul et al., 2025). On the other hand, it has been also proposed that elevated R-loops may reactivate retroelements in some pathological conditions (Lim et al., 2015).
This highlights a potential R-loop-mediated cross talk between trinucleotide repeats and retrotransposons, able to exacerbate the instability of both elements and interfere with genome stability.
Epigenetic interactions between transposons and triplet repeats
Epigenetic modifications, such as DNA methylation and histone modifications are essential in regulating the activities of both transposons and triplet repeats (Dion and Wilson, 2009; Protasova et al., 2021), suggesting a possible indirect interaction, mediated by changes in the epigenetic landscape.
For instance, DNA methylation is a well-documented mechanism that silences transposons, but it may also impact the stability of triplet repeat sequences (Dion and Wilson, 2009; Essebier et al., 2016; Jönsson et al., 2019; Poeta et al., 2020; Lanciano et al., 2024).
On the other hand, activation of transposable elements could themselves modify chromatin organization, DNA methylation state and histone acetylation at their insertion points (Hancks and Kazazian, 2016).
In this scenario, it is plausible that such epigenetic changes can even extend to nearby regions of the genome, possibly altering the dynamics of replication and repair processes for adjacent triplet repeats. In the case of Fragile X syndrome, the expanded CGG repeats, found in the FMR1 gene, correlate with changes in chromatin structure, which include heightened DNA methylation and repressive histone modifications (Yudkin et al., 2015).
These findings underscore the important, although indirect, role that transposons could play in shaping the epigenetic landscape of triplet repeats, potentially affecting gene expression and genome stability.
Therapeutic opportunities: evidence from Huntington’s disease models
Huntington’s disease (HD) is a rare, inherited neurodegenerative disease that affects both the brain and the body, and worsens over time. The expanded CAG nucleotide repeat sequence in HTT gene, leads to the production of an abnormal protein (huntingtin) which gains new toxic functions (Tabrizi et al., 2020).
Instability of this triplet repeat sequence, which expands over generations, is characteristic of HD (Aziz et al., 2008), and is associated with neuronal CAG repeat mosaicism (Cattaneo et al., 2025).
Activation of retroelements has been reported in HD pre-clinical models as well as in human patients (Casale et al., 2021; Floreani et al., 2022).
Understanding how transposon instability relates to triplet repeat expansions could lead to new therapeutic strategies, however, tackling both types of instability at the same time represents unique challenges. Therefore, current strategies often focused on these instabilities separately.
Targeting DNA repair proteins (e.g., MSH3) was effective in reducing somatic expansions in patient-derived iPSC neurons (Bunting et al., 2025). Coherently, FAN1 expression stabilized CAG repeats in HD cell models (Goold et al., 2019).
Furthermore, evidence in a fly model of HD demonstrated elevated transposable element (TE) expression and mobilization, leading to neurodegeneration (Casale et al., 2021). The inhibition of TE mobilization, by reverse transcriptase (RT) inhibitors, rescued disease phenotypes, indicating a potential pathogenic role for TEs, and suggesting a potential therapeutic target for the disease (Casale et al., 2021).
Hypothetically, some approaches could be applied for both transposons and expanded triplets. For example, strategies aimed at targeting oxidative stress, which influences both instabilities (Giorgi et al., 2011; La Rosa et al., 2020), could offer a promising approach to mitigating their possible combined activity. In this context, although not yet investigated, the effect of the antioxidant agent N-acetylcysteine in HD models (Wright et al., 2015, 2016), could potentially address the possible interplay of transposon and triplet repeat instabilities.
On the other hand, strategies based on epigenetic regulation, which has shown a therapeutic potential in HD models (Hyeon et al., 2021; Dai et al., 2024), have been reported to modulate murine retrotransposons and chromatin structure (Brunmeir et al., 2010; Lennartsson et al., 2015).
Discussion
In this perspective, we hypothesized that regulatory pathways, involving chromatin remodeling, DNA repair system and epigenetic marks could regulate both transposon activity and triplet repeat instability. We posit that these phenomena are likely interconnected to each other and create a vicious cycle of genomic damage in vulnerable neurons. Altered histone modifications in neurons, for example, may help activate transposons, while also destabilizing triplet repeats, promoting a greater genomic instability.
On the other hand, environmental triggers, such as oxidative damage may also contribute to generate an ideal “substrate” to maintain this combined instability.
Shared regulatory elements may act as master regulators, timing the activation of transposons and triplet repeats and possibly synchronizing these mechanisms.
Understanding how these factors can favor the possible interaction between transposon activity and triplet repeat instability could pave the way for a better understanding of disease molecular mechanisms and for the development of genome-targeted therapeutic strategies.
In this context, the temporal dynamic may be a critical consideration. While retrotransposon activity can be beneficial during neurodevelopment, its pathological reactivation in post-mitotic neurons, a cell type with limited DNA repair capacity, may be especially destructive. Theoretically, any early activation of transposable elements, occurring in the developing brain, may influence triplet instability and related disease phenotypes in adult neurons.
As shown in the Table 1, all the disease conditions reported, spanning both neurodevelopmental and neurodegenerative disorders, demonstrate the potential relevance of these concepts across the lifespan (developing and adult brain) and across different cell types (NPCs and neurons).
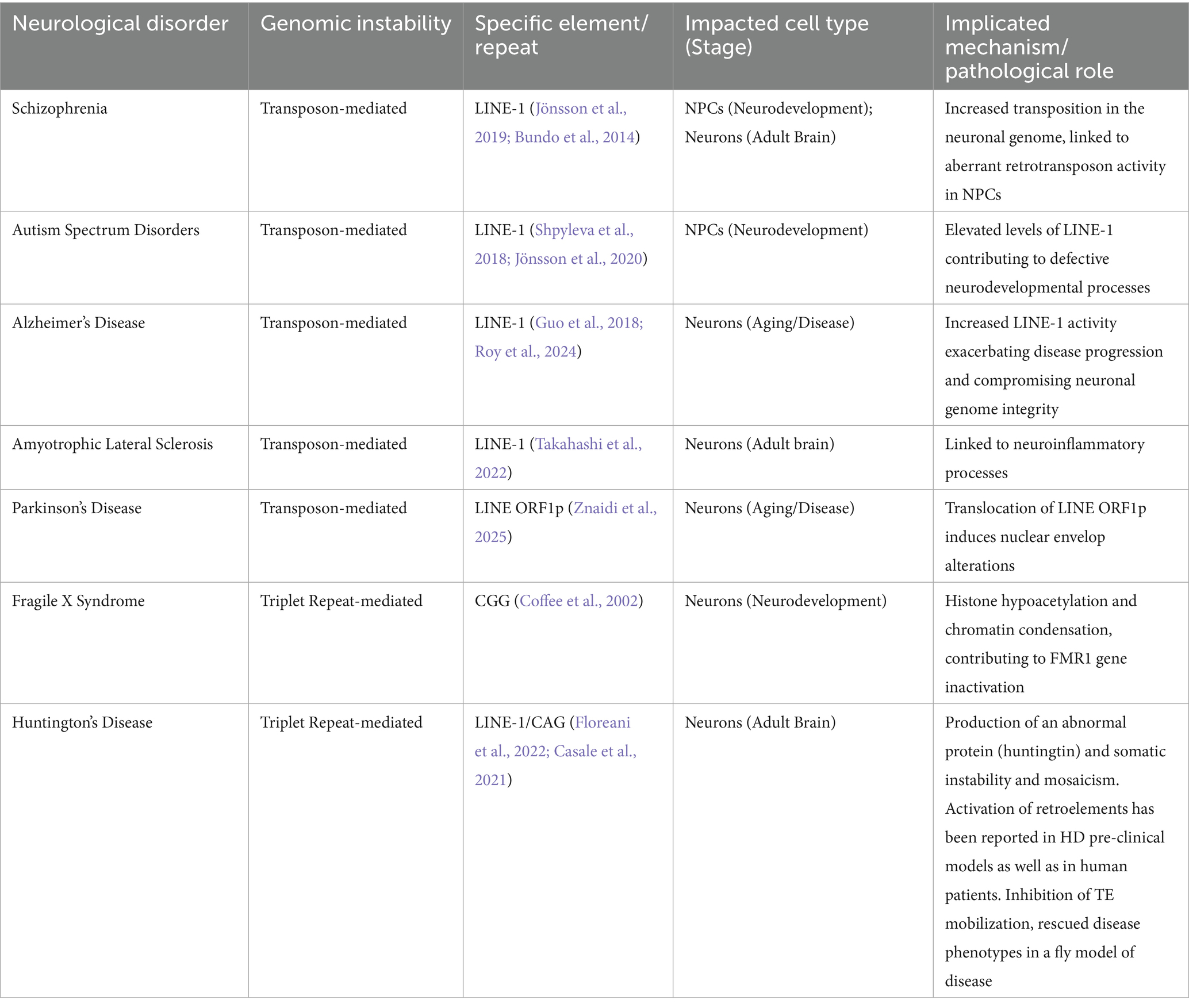
Table 1. Neurological disorders associated with genomic instability.
Moreover, the possibility that both form of genome instability might share some mechanistic elements suggests a new therapeutic paradigm.
To our perspective, future therapeutic efforts could focus on the master regulatory pathways that govern both genome instabilities, rather than targeting them separately. In this contest, drugs aimed at restoring global epigenetic repression or boosting the efficiency of shared DNA repair pathways could potentially offer a synergistic effective therapeutic approach.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
GP: Writing – review & editing, Writing – original draft. MS: Writing – review & editing. AP: Writing – review & editing. VM: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by Italian Ministry of Health “Ricerca Corrente” funding program to VM and Telethon Grants GGP20101, GJC21157-A and GMR24T2035 to VM. This study was supported by Fondazione Neuromed.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aziz, N. A., van Belzen, M. J., and Ross, R. A. C. (2008). Intergenerational CAG repeat instability is highly heritable in Huntington’s disease. J. Med. Genet. 45:766. doi: 10.1136/JMG.2008.062133
Bachiller, S., del-Pozo-Martín, Y., and Carrión, Á. M. (2017). L1 retrotransposition alters the hippocampal genomic landscape enabling memory formation. Brain Behav. Immun. 64, 65–70. doi: 10.1016/J.BBI.2016.12.018
Brulé, B., Alcalá-Vida, R., Penaud, N., Scuto, J., Mounier, C., Seguin, J., et al. (2025). Accelerated epigenetic aging in Huntington’s disease involves polycomb repressive complex 1. Nat. Commun. 16:1550. doi: 10.1038/S41467-025-56722-Z
Brunmeir, R., Lagger, S., Simboeck, E., Sawicka, A., Egger, G., Hagelkruys, A., et al. (2010). Epigenetic regulation of a murine retrotransposon by a dual histone modification mark. PLoS Genet. 6:e1000927. doi: 10.1371/JOURNAL.PGEN.1000927
Bundo, M., Toyoshima, M., Okada, Y., Akamatsu, W., Ueda, J., Nemoto-Miyauchi, T., et al. (2014). Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron 81, 306–313. doi: 10.1016/J.NEURON.2013.10.053
Bunting, E. L., Donaldson, J., Cumming, S. A., Olive, J., Broom, E., Miclăuș, M., et al. (2025). Antisense oligonucleotide-mediated MSH3 suppression reduces somatic CAG repeat expansion in Huntington’s disease iPSC-derived striatal neurons. Sci. Transl. Med. 17:eadn4600. doi: 10.1126/SCITRANSLMED.ADN4600
Casale, A. M., Liguori, F., Ansaloni, F., Cappucci, U., Finaurini, S., Spirito, G., et al. (2021). Transposable element activation promotes neurodegeneration in a Drosophila model of Huntington’s disease. iScience 25:103702. doi: 10.1016/J.ISCI.2021.103702
Castel, A. L., Cleary, J. D., and Pearson, C. E. (2010). Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol. 11, 165–170. doi: 10.1038/NRM2854
Cattaneo, E., Scalzo, D., Zobel, M., Iennaco, R., Maffezzini, C., Besusso, D., et al. (2025). When repetita no-longer iuvant: somatic instability of the CAG triplet in Huntington’s disease. Nucleic Acids Res. 53:gkae1204. doi: 10.1093/NAR/GKAE1204
Chatterjee, N., Lin, Y., Santillan, B. A., Yotnda, P., and Wilson, J. H. (2015). Environmental stress induces trinucleotide repeat mutagenesis in human cells. Proc. Natl. Acad. Sci. USA 112, 3764–3769. doi: 10.1073/PNAS.1421917112
Coffee, B., Zhang, F., Ceman, S., Warren, S. T., and Reines, D. (2002). Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am. J. Hum. Genet. 71, 923–932. doi: 10.1086/342931
Cordaux, R., and Batzer, M. A. (2009). The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 10, 691–703. doi: 10.1038/NRG2640
Dai, W., Qiao, X., Fang, Y., Guo, R., Bai, P., Liu, S., et al. (2024). Epigenetics-targeted drugs: current paradigms and future challenges. Signal Transduct. Target. Ther. 9:332. doi: 10.1038/S41392-024-02039-0
Della Valle, F., Reddy, P., Aguirre Vazquez, A., and Izpisua Belmonte, J. C. (2025). Reactivation of retrotransposable elements is associated with environmental stress and ageing. Nat. Rev. Genet. 26, 547–558. doi: 10.1038/S41576-025-00829-Y
Depienne, C., and Mandel, J. L. (2021). 30 years of repeat expansion disorders: what have we learned and what are the remaining challenges? Am. J. Hum. Genet. 108, 764–785. doi: 10.1016/J.AJHG.2021.03.011
Deshmukh, A. L., Porro, A., Mohiuddin, M., Lanni, S., Panigrahi, G. B., Caron, M. C., et al. (2021). FAN1, a DNA repair nuclease, as a modifier of repeat expansion disorders. J. Huntingtons. Dis. 10, 95–122. doi: 10.3233/JHD-200448
Dias, M., Moraes, A., Shiroma, T., Pessoa, V., Ermoges, A., Vital, T., et al. (2025). Beyond Trypanosoma cruzi: LINE-1 activation as a driver of chronic inflammation in Chagas disease. Int. J. Mol. Sci. 26:4466. doi: 10.3390/IJMS26104466
Dion, V., and Wilson, J. H. (2009). Instability and chromatin structure of expanded trinucleotide repeats. Trends Genet. 25, 288–297. doi: 10.1016/J.TIG.2009.04.007
Erwin, J. A., Marchetto, M. C., and Gage, F. H. (2014). Mobile DNA elements in the generation of diversity and complexity in the brain. Nat. Rev. Neurosci. 15, 497–506. doi: 10.1038/NRN3730
Essebier, A., Vera Wolf, P., Cao, M. D., Carroll, B. J., Balasubramanian, S., and Bodén, M. (2016). Statistical enrichment of epigenetic states around triplet repeats that can undergo expansions. Front. Neurosci. 10:92. doi: 10.3389/FNINS.2016.00092
Evans, T. A., and Erwin, J. A. (2021). Retroelement-derived RNA and its role in the brain. Semin. Cell Dev. Biol. 114, 68–80. doi: 10.1016/J.SEMCDB.2020.11.001
Everett, C. M., and Wood, N. W. (2004). Trinucleotide repeats and neurodegenerative disease. Brain 127, 2385–2405. doi: 10.1093/BRAIN/AWH278
Faulkner, G. J., and Garcia-Perez, J. L. (2017). L1 mosaicism in mammals: extent, effects, and evolution. Trends Genet. 33, 802–816. doi: 10.1016/J.TIG.2017.07.004
Floreani, L., Ansaloni, F., Mangoni, D., Agostoni, E., Sanges, R., Persichetti, F., et al. (2022). Analysis of LINE1 retrotransposons in Huntington’s disease. Front. Cell. Neurosci. 15:743797. doi: 10.3389/FNCEL.2021.743797
Freeman, B., White, T., Kaul, T., Stow, E. C., Baddoo, M., Ungerleider, N., et al. (2022). Analysis of epigenetic features characteristic of L1 loci expressed in human cells. Nucleic Acids Res. 50, 1888–1907. doi: 10.1093/NAR/GKAC013
Frisch, R., Singleton, K. R., Moses, P. A., Gonzalez, I. L., Carango, P., Marks, H. G., et al. (2001). Effect of triplet repeat expansion on chromatin structure and expression of DMPK and neighboring genes, SIX5 and DMWD, in myotonic dystrophy. Mol. Genet. Metab. 74, 281–291. doi: 10.1006/MGME.2001.3229
García-Guede, Á., Vera, O., and Ibáñez-de-Caceres, I. (2020). When oxidative stress meets epigenetics: implications in cancer development. Antioxidants 9, 1–26. doi: 10.3390/ANTIOX9060468
Gasior, S. L., Wakeman, T. P., Xu, B., and Deininger, P. L. (2006). The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 357, 1383–1393. doi: 10.1016/J.JMB.2006.01.089
Giorgi, G., Marcantonio, P., and Del Re, B. (2011). LINE-1 retrotransposition in human neuroblastoma cells is affected by oxidative stress. Cell Tissue Res. 346, 383–391. doi: 10.1007/S00441-011-1289-0
Goold, R., Flower, M., Moss, D. H., Medway, C., Wood-Kaczmar, A., Andre, R., et al. (2019). FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet. 28, 650–661. doi: 10.1093/HMG/DDY375
Gorbunova, V., Seluanov, A., Mita, P., McKerrow, W., Fenyö, D., Boeke, J. D., et al. (2021). The role of retrotransposable elements in ageing and age-associated diseases. Nature 596, 43–53. doi: 10.1038/S41586-021-03542-Y
Guo, C., Jeong, H. H., Hsieh, Y. C., Klein, H. U., Bennett, D. A., De Jager, P. L., et al. (2018). Tau activates transposable elements in Alzheimer’s disease. Cell Rep. 23, 2874–2880. doi: 10.1016/J.CELREP.2018.05.004
Hancks, D. C., and Kazazian, H. H. (2016). Roles for retrotransposon insertions in human disease. Mob. DNA 7:9. doi: 10.1186/S13100-016-0065-9
Hyeon, J. W., Kim, A. H., and Yano, H. (2021). Epigenetic regulation in Huntington’s disease. Neurochem. Int. 148:105074. doi: 10.1016/J.NEUINT.2021.105074
Jönsson, M. E., Garza, R., Johansson, P. A., and Jakobsson, J. (2020). Transposable elements: a common feature of neurodevelopmental and neurodegenerative disorders. Trends Genet. 36, 610–623. doi: 10.1016/J.TIG.2020.05.004/ASSET/59EC686B-405D-4494-9C13-D44AAC89782A/MAIN.ASSETS/GR3.JPG
Jönsson, M. E., Ludvik Brattås, P., Gustafsson, C., Petri, R., Yudovich, D., Pircs, K., et al. (2019). Activation of neuronal genes via LINE-1 elements upon global DNA demethylation in human neural progenitors. Nat. Commun. 10:3182. doi: 10.1038/S41467-019-11150-8
Konopka, A., Atkin, J. D., and Mitra, J. (2022). Editorial: defective DNA damage response-repair axis in post-mitotic neurons in human health and neurodegenerative diseases. Front. Cell. Neurosci. 16:1009760. doi: 10.3389/FNCEL.2022.1009760
Kovtun, I. V., Liu, Y., Bjoras, M., Klungland, A., Wilson, S. H., and McMurray, C. T. (2007). OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 447, 447–452. doi: 10.1038/NATURE05778
Kumari, D., and Usdin, K. (2009). Chromatin remodeling in the noncoding repeat expansion diseases. J. Biol. Chem. 284, 7413–7417. doi: 10.1074/JBC.R800026200
La Rosa, P., Petrillo, S., Bertini, E. S., and Piemonte, F. (2020). Oxidative stress in DNA repeat expansion disorders: a focus on NRF2 signaling involvement. Biomolecules 10: 702. doi: 10.3390/BIOM10050702
La Spada, A. R. (1997). Trinucleotide repeat instability: genetic features and molecular mechanisms. Brain Pathol. 7, 943–963.
Lai, Y., Beaver, J. M., Laverde, E., and Liu, Y. (2020). Trinucleotide repeat instability via DNA base excision repair. DNA Repair (Amst) 93:102912. doi: 10.1016/J.DNAREP.2020.102912
Lanciano, S., Philippe, C., Sarkar, A., Pratella, D., Domrane, C., Doucet, A. J., et al. (2024). Locus-level L1 DNA methylation profiling reveals the epigenetic and transcriptional interplay between L1s and their integration sites. Cell genomics 4:100498. doi: 10.1016/J.XGEN.2024.100498
Lennartsson, A., Arner, E., Fagiolini, M., Saxena, A., Andersson, R., Takahashi, H., et al. (2015). Remodeling of retrotransposon elements during epigenetic induction of adult visual cortical plasticity by HDAC inhibitors. Epigenetics Chromatin 8:55. doi: 10.1186/S13072-015-0043-3
Levin, H. L., and Moran, J. V. (2011). Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 12, 615–627. doi: 10.1038/NRG3030
Li, L. B., and Bonini, N. M. (2010). Roles of trinucleotide-repeat RNA in neurological disease and degeneration. Trends Neurosci. 33, 292–298. doi: 10.1016/J.TINS.2010.03.004
Li, D., Shao, F., Li, X., Yu, Q., Wu, R., Wang, J., et al. (2025). Advancements and challenges of R-loops in cancers: biological insights and future directions. Cancer Lett. 610:217359. doi: 10.1016/J.CANLET.2024.217359
Li, Y., and Tollefsbol, T. O. (2016). Age-related epigenetic drift and phenotypic plasticity loss: implications in prevention of age-related human diseases. Epigenomics 8, 1637–1651. doi: 10.2217/EPI-2016-0078
Lim, Y. W., Sanz, L. A., Xu, X., Hartono, S. R., and Chédin, F. (2015). Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutières syndrome. eLife 4:e08007. doi: 10.7554/ELIFE.08007
Martin, S. L. (2006). The ORF1 protein encoded by LINE-1: structure and function during L1 retrotransposition. J. Biomed. Biotechnol. 2006:45621. doi: 10.1155/JBB/2006/45621
Martins, S. G., Zilhão, R., Thorsteinsdóttir, S., and Carlos, A. R. (2021). Linking oxidative stress and DNA damage to changes in the expression of extracellular matrix components. Front. Genet. 12:673002. doi: 10.3389/FGENE.2021.673002
McMurray, C. T. (2010). Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 11, 786–799. doi: 10.1038/NRG2828
Mendez-Dorantes, C., and Burns, K. H. (2023). LINE-1 retrotransposition and its deregulation in cancers: implications for therapeutic opportunities. Genes Dev. 37, 948–967. doi: 10.1101/GAD.351051.123
Mendez-Dorantes, C., Zeng, X., Karlow, J. A., Schofield, P., Turner, S., Kalinowski, J., et al. (2024). Chromosomal rearrangements and instability caused by the LINE-1 retrotransposon. bioRxiv Prepr. Serv. Biol. 12.14.628481. doi: 10.1101/2024.12.14.628481
Mirkin, S. M. (2007). Expandable DNA repeats and human disease. Nature 447, 932–940. doi: 10.1038/NATURE05977
Morrish, T. A., Gilbert, N., Myers, J. S., Vincent, B. J., Stamato, T. D., Taccioli, G. E., et al. (2002). DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat. Genet. 31, 159–165. doi: 10.1038/NG898
Nageshwaran, S., and Festenstein, R. (2015). Epigenetics and triplet-repeat neurological diseases. Front. Neurol. 6:262. doi: 10.3389/FNEUR.2015.00262
Niu, Y., Desmarais, T. L., Tong, Z., Yao, Y., and Costa, M. (2015). Oxidative stress alters global histone modification and DNA methylation. Free Radic. Biol. Med. 82, 22–28. doi: 10.1016/J.FREERADBIOMED.2015.01.028
Ochoa Thomas, E., Zuniga, G., Sun, W., and Frost, B. (2020). Awakening the dark side: retrotransposon activation in neurodegenerative disorders. Curr. Opin. Neurobiol. 61, 65–72. doi: 10.1016/J.CONB.2020.01.012
Paul, P., Kumar, A., Parida, A. S., De, A. K., Bhadke, G., Khatua, S., et al. (2025). p53-mediated regulation of LINE1 retrotransposon-derived R-loops. J Biol Chem. 301:108200. doi: 10.1016/j.jbc.2025.108200
Poeta, L., Drongitis, D., Verrillo, L., and Miano, M. G. (2020). DNA hypermethylation and unstable repeat diseases: a paradigm of transcriptional silencing to decipher the basis of pathogenic mechanisms. Genes (Basel) 11:684. doi: 10.3390/GENES11060684
Protasova, M. S., Andreeva, T. V., and Rogaev, E. I. (2021). Factors regulating the activity of LINE1 retrotransposons. Genes (Basel) 12:1562. doi: 10.3390/GENES12101562
Ravel-Godreuil, C., Znaidi, R., Bonnifet, T., Joshi, R. L., and Fuchs, J. (2021). Transposable elements as new players in neurodegenerative diseases. FEBS Lett. 595, 2733–2755. doi: 10.1002/1873-3468.14205
Richardson, S. R., Morell, S., and Faulkner, G. J. (2014). L1 retrotransposons and somatic mosaicism in the brain. Annu. Rev. Genet. 48, 1–27. doi: 10.1146/ANNUREV-GENET-120213-092412
Roy, N., Haq, I., Ngo, J. C., Bennett, D. A., Teich, A. F., De Jager, P. L., et al. (2024). Elevated expression of the retrotransposon LINE-1 drives Alzheimer’s disease-associated microglial dysfunction. Acta Neuropathol. 148:75. doi: 10.1007/S00401-024-02835-6
Salcedo-Arellano, M. J., Dufour, B., McLennan, Y., Martinez-Cerdeno, V., and Hagerman, R. (2020). Fragile X syndrome and associated disorders: clinical aspects and pathology. Neurobiol. Dis. 136:104740. doi: 10.1016/j.nbd.2020.104740
Schmidt, M. H. M., and Pearson, C. E. (2016). Disease-associated repeat instability and mismatch repair. DNA Repair (Amst) 38, 117–126. doi: 10.1016/J.DNAREP.2015.11.008
Schwartz, J. L., Jones, K. L., and Yeo, G. W. (2021). Repeat RNA expansion disorders of the nervous system: post-transcriptional mechanisms and therapeutic strategies. Crit. Rev. Biochem. Mol. Biol. 56, 31–53. doi: 10.1080/10409238.2020.1841726
Senapati, P., Miyano, M., Sayaman, R. W., Basam, M., Leung, A., LaBarge, M. A., et al. (2023). Loss of epigenetic suppression of retrotransposons with oncogenic potential in aging mammary luminal epithelial cells. Genome Res. 33, 1229–1241. doi: 10.1101/GR.277511.122
Shpyleva, S., Melnyk, S., Pavliv, O., Pogribny, I., and Jill James, S. (2018). Overexpression of LINE-1 retrotransposons in autism brain. Mol. Neurobiol. 55, 1740–1749. doi: 10.1007/S12035-017-0421-X
Sicot, G., and Gomes-Pereira, M. (2013). RNA toxicity in human disease and animal models: from the uncovering of a new mechanism to the development of promising therapies. Biochim. Biophys. Acta 1832, 1390–1409. doi: 10.1016/J.BBADIS.2013.03.002
Suarez, N. A., Macia, A., and Muotri, A. R. (2018). LINE-1 retrotransposons in healthy and diseased human brain. Dev. Neurobiol. 78, 434–455. doi: 10.1002/DNEU.22567
Sun, Y., Dai, H., Dai, X., Yin, J., Cui, Y., Liu, X., et al. (2023). m1A in CAG repeat RNA binds to TDP-43 and induces neurodegeneration. Nature 623, 580–587. doi: 10.1038/S41586-023-06701-5
Tabrizi, S. J., Flower, M. D., Ross, C. A., and Wild, E. J. (2020). Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 16, 529–546. doi: 10.1038/S41582-020-0389-4
Takahashi, F., Zhang, C., Hohjoh, H., Raveney, B., Yamamura, T., Hayashi, N., et al. (2022). Immune-mediated neurodegenerative trait provoked by multimodal derepression of long-interspersed nuclear element-1. iScience 25:104278. doi: 10.1016/J.ISCI.2022.104278
Upton, K. R., Gerhardt, D. J., Jesuadian, J. S., Richardson, S. R., Sánchez-Luque, F. J., Bodea, G. O., et al. (2015). Ubiquitous L1 mosaicism in hippocampal neurons. Cell 161, 228–239. doi: 10.1016/J.CELL.2015.03.026
Van Meter, M., Kashyap, M., Rezazadeh, S., Geneva, A. J., Morello, T. D., Seluanov, A., et al. (2014). SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 5:5011. doi: 10.1038/NCOMMS6011
Wei, J. W., Huang, K., Yang, C., and Kang, C. S. (2017). Non-coding RNAs as regulators in epigenetics (review). Oncol. Rep. 37, 3–9. doi: 10.3892/OR.2016.5236
Wright, D. J., Gray, L. J., Finkelstein, D. I., Crouch, P. J., Pow, D., Pang, T. Y., et al. (2016). N-acetylcysteine modulates glutamatergic dysfunction and depressive behavior in Huntington’s disease. Hum. Mol. Genet. 25, ddw144–ddw2933. doi: 10.1093/HMG/DDW144
Wright, D. J., Renoir, T., Smith, Z. M., Frazier, A. E., Francis, P. S., Thorburn, D. R., et al. (2015). N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 5:e492. doi: 10.1038/TP.2014.131
Wulfridge, P., and Sarma, K. (2024). Intertwining roles of R-loops and G-quadruplexes in DNA repair, transcription and genome organization. Nat. Cell Biol. 26, 1025–1036. doi: 10.1038/S41556-024-01437-4
Yang, J. H., Hayano, M., Griffin, P. T., Amorim, J. A., Bonkowski, M. S., Apostolides, J. K., et al. (2023). Loss of epigenetic information as a cause of mammalian aging. Cell 186, 305–326. doi: 10.1016/J.CELL.2022.12.027
Yudkin, D. V., Lemskaya, N. A., Grischenko, I. V., and Dolskiy, A. A. (2015). Chromatin changes caused by expansion of CGG repeats in fmr1 gene. Mol. Biol. 49, 179–184. doi: 10.1134/S0026893315010197
Zhang, W., Huang, C., Yao, H., Yang, S., Jiapaer, Z., Song, J., et al. (2025). Retrotransposon: an insight into neurological disorders from perspectives of neurodevelopment and aging. Transl. Neurodegener. 14:14. doi: 10.1186/S40035-025-00471-Y
Zhao, X. N., and Usdin, K. (2015). The repeat expansion diseases: the dark side of DNA repair. DNA Repair (Amst) 32, 96–105. doi: 10.1016/J.DNAREP.2015.04.019
Keywords: LINEs, SINEs, transposon activity, repeat instability, brain diseases
Citation: Pepe G, Storto M, Di Pardo A and Maglione V (2025) Transposon activity and nucleotide triplet instability: new perspectives on their potential interplay in brain disorders. Front. Neurosci. 19:1617315. doi: 10.3389/fnins.2025.1617315
Edited by:
Lucia Carboni, University of Bologna, ItalyReviewed by:
Chetan C. Rawal, University of Southern California, United StatesFabian R. Villagomez, Federico Gómez Children's Hospital, Mexico
Copyright © 2025 Pepe, Storto, Di Pardo and Maglione. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vittorio Maglione, dml0dG9yaW8ubWFnbGlvbmVAbmV1cm9tZWQuaXQ=