 Jeffrey D. Swartz1Medora Lachman1Kelsey Westveer1Thomas O’Neill1Thomas Geary2Rodney W. Kott1
Jeffrey D. Swartz1Medora Lachman1Kelsey Westveer1Thomas O’Neill1Thomas Geary2Rodney W. Kott1 James G. Berardinelli1Patrick G. Hatfield1
James G. Berardinelli1Patrick G. Hatfield1 Jennifer M. Thomson1Andy Roberts2
Jennifer M. Thomson1Andy Roberts2 Carl J. Yeoman1*
Carl J. Yeoman1*- 1Department of Animal and Range Sciences, Montana State University, Bozeman, MT, USA
- 2United States Department of Agriculture-Agricultural Research Service, Miles City, MT, USA
Although a number of common reproductive disorders in livestock involve bacterial infection, very little is known about their normal vaginal microbiota. Therefore, we sought to determine the species composition of sheep and cattle vaginal microbiota. Twenty Rambouillet ewes and twenty crossbred cows varying in age and reproductive status were sampled by ectocervicovaginal lavage. We amplified and sequenced the V3–V4 region of the 16S ribosomal RNA (rRNA) contents yielding a total of 907,667 high-quality reads. Good’s Coverage estimates indicated that we obtained data on 98 ± 0.01% of the total microbial genera present in each sample. Cow and ewe vaginal microbiota displayed few differences. Cow microbiota exhibited greater (P ≤ 0.05) α-diversity compared to the ewe microbiota. Both livestock species differed (P ≤ 0.05) from all previously reported vaginal communities. While bacteria were numerically dominant, Archaea were detected in 95% of cow and ewe samples, mainly of the order Desulfurococcales. Both ewes and cows were predominately colonized by the bacterial phyla Bacteroidetes, Fusobacteria, and Proteobacteria. The most abundant genera were Aggregatibacter spp., and Streptobacillus spp. Lactobacillus spp. were detected in 80% of ewe and 90% of cow samples, but only at very low abundances. Bacteria previously described from culture-based studies as common to the cow and ewe vaginal tract, except for Escherichia, were variably present, and only in low abundance. Ewe and cow pH differed (P ≤ 0.05), with means (±SD) of 6.7 ± 0.38 and 7.3 ± 0.63, respectively. In conclusion, 16S rRNA sequencing of cow and ewe vaginal ectocervicovaginal lavages showed that cow and ewe vaginal microbiota differ from culture-led results, revealing a microbiota distinct from previously described vaginal ecosystems.
Introduction
The human vaginal microbiota is most often dominated by lactobacilli (1). In this system, the lactobacilli are considered important to vaginal homeostasis through their production of lactate, which maintains a low vaginal pH (pH < 4.5) that is inhibitory to many vaginal pathogens (2). A reduction in vaginal lactobacilli, which is typically accompanied by an elevated vaginal pH, is a common feature of bacterial vaginosis (BV), the most common disorder among reproductive-aged women (3). BV is of significant concern, particularly due to its exacerbation of the risks of pre-term birth and spontaneous abortion (4). Given the potential importance of vaginal lactobacilli in diminishing the risks of pregnancy-related complications, it is interesting to note that the vaginal microbiota of some humans (1) and all primates (5) do not display the same Lactobacillus-dominated vaginal ecosystems. Similarly, the few culture-based studies that have been conducted on livestock have reported Lactobacillus spp. at lower abundances than other microbial genera in both the cow and ewe vagina (6–8). Enterococcus spp., Staphylococcus spp., and Streptococcus spp. are more commonly isolated from the cow vagina (6, 7, 9, 10), while Bacillus spp., Corynebacterium spp., Escherichia spp., Staphylococcus spp., and Streptococcus spp. are commonly isolated from the ewe vagina (11–14). To date, no studies have been reported that have utilized culture-independent 16S ribosomal RNA (rRNA) sequencing of the cow or ewe vaginal microbiota. Previous applications of 16S rRNA sequencing techniques have revealed a much greater and historically unrealized diversity of microbiota in various ecosystems (15–17). Other studies have indicated that culture-based approaches may emphasize the rarer members of communities and often miss those microbes that are more abundant (16). The complete reliance on culture has thereby left the true microbial diversity of the cow and ewe vagina hereinto undetermined and the scarcity of lactobacilli uncertain. Therefore, the objective of this study was to elucidate the microbiota present in cow and ewe vaginas using culture-independent 16S rRNA sequencing technology and determine how the composition of the livestock vaginal microbiota compares to other well-described vaginal microbial communities that have been similarly defined in humans and non-human primates (5). This information will provide a basis to evaluate the livestock vaginal microbiota’s potential roles in affecting vaginal health, reproductive outcomes, and perinatal morbidities. Specifically, we seek to develop the necessary background information required to examine the hypotheses that: (i) specific livestock vaginal communities increase or decrease the risks of acquisition of livestock venereal pathogens alike Brucella ovis, Campylobacter fetus, Helicobacter trogontum, or Arcobacter cryaerophilus; (ii) that the vaginal microbiota are the first colonizers of neonatal animals, as has been shown for conventionally born human neonates (18), and, due to the relative importance of this early microbiota (19), directly influence perinatal health and performance; and (iii) that specific vaginal microbial communities of cattle and sheep impact reproductive outcomes, including spontaneous abortion. Well-described infectious agents of the livestock vagina have already been associated with placentitis, abortion, infertility, and the birth of debilitated offspring (20–23). Evidence from human studies indicates that vaginal dysbiosis increases the risks of acquisition and shedding of sexually transmitted infections (24–27), while, a healthy vaginal microbiota is more resistant to venereal infection. Additionally, given the importance of pH to human vaginal health, livestock vaginal pH was also investigated.
Materials and Methods
Ethics Statement
Animal care and use protocols were approved by the Montana State University Agricultural Animal Care and Use (AACUC) committee under protocol number 2012-AA07 dated 09/20/2012.
Sampling
Vaginal lavages were collected from 20 Rambouillet ewes and 20 crossbred beef cows of varied breeding method and pregnancy status (Table 1). Sampled ewes were being maintained at the Bozeman Agricultural Research and Teaching farm, Bozeman, MT, USA, while cows were sampled 286 miles away at Fort Keogh, Miles City, MT, USA. The cows and ewes had never been cohabitated. Samples were collected by injecting 25 ml (ewes) or 50 ml (cows) of sterile saline into the vaginal tract via sterile catheter tubes attached to luer lock 60 ml syringes. Saline was injected in a continuous stream toward the cervix, aspirated 3–5 times, transferred to a sterile 15 ml falcon tube, and stored at −20°C. One milliliter of sample was separated for pH determination. For DNA extraction, up to 4.5 ml of sample was centrifuged for 5 min at 20,000 × g and 4°C.
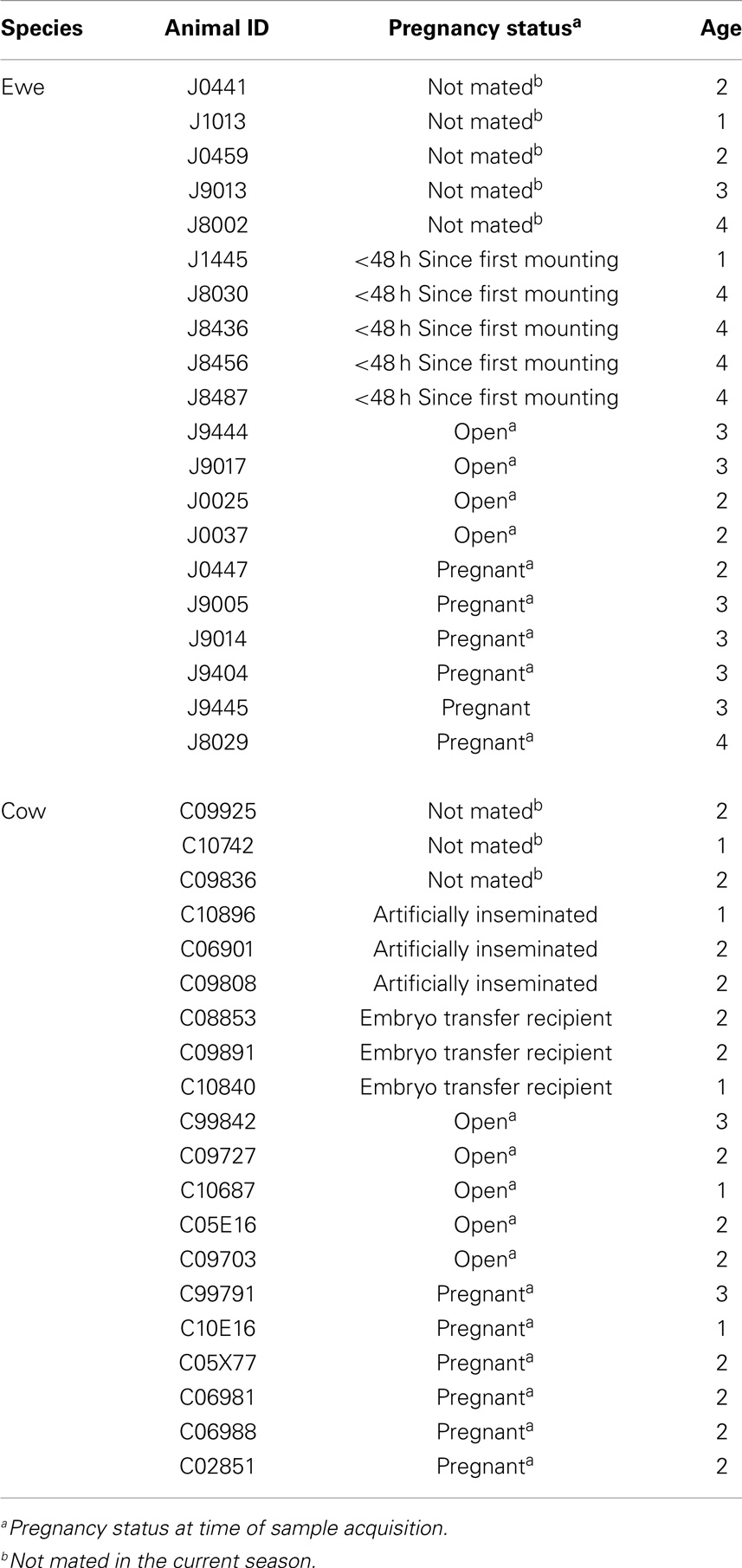
Table 1. Pregnancy status and age of animals used in this trial.
Ph Analyses
A 1 ml subsample from each lavage was assessed for pH using a Ryan 520A pH meter fitted with a ROSS Ultra Electrode (Thermo Scientific, Waltham, MA, USA). All animals were used for analysis of pH except ewes sampled before breeding (Table 1), due to the lavage potentially containing phosphate buffer (Table 1). Mean, SD, boxplots, Shapiro–Wilk test of normality, and two-sided Wilcoxon rank sum test were calculated using R (28).
Sequencing Microbiota
Pellets obtained from up to 4.5 ml of lavage were extracted using MoBio PowerSoil DNA Isolation kits following manufacturer instructions, except a 2 min bead-beating step was used instead of a 10 min vortex. Variable regions three and four of the 16S rRNA genes were amplified using custom primers that included indexes to identify samples after sequencing. The PCR reaction ran for 30 cycles at 94°C for 20 s, 52°C for 30 s, and 72°C for 45 s, using barcoded primers [SeqF(1–8) 5′-AATGATACGGCGACCACCGAGATCTACAC(adaptor)-Index2(1 of 8 different 8 nt codes used to distinguish among samples when pooled for sequencing)-TATGGTAATT(sequencing primer pad)-AT(linker)-CCTACGGGAGGCAGCAG(341f primer)-3′ and SeqR (1-12) 5′-CAAGCAGAAGACGGCATACGAGAT(adaptor)-Index1 (1 of 12 different 8 nt codes used to distinguish among samples when pooled for sequencing)-AGTCAGTCAG(sequencing primer pad)-CC(linker)-GGACTACHVGGGTWTCTAAT(806r primer-3′]. Amplicons were quantified using an Agilent 2200 tape station (Agilent, Santa Clara, CA, USA) and pooled at an equimolar concentration. Pooled amplicons were purified from residual PCR reagents and non-specific amplification products in an agarose gel using a QIAquick gel extraction kit (Qiagen, Valencia, CA, USA) following manufacturers instructions. Purified and pooled amplicons were subsequently quantified using a KAPA Syber quantification kit (KAPABiosystems, Wilmington, MA, USA) as per manufacturer instructions. Purified, quantified, and pooled amplicons were mixed with 5–10% of an equimolar concentration of PhiX and sequenced at 12.5 pM. Sequencing was performed with an Illumina MiSeq using paired-end 2 × 250 nucleotide (nt) dual-index sequencing. Custom primers (R1 5′-CCTACGGGAGGCAGCAG-3′, R2 5′-AGTCAGTCAGCCGGACTAC-3′, and Index 5′-GTAGTCCGGCTGACTGACT-3′) were used for sequencing and indexing. Raw sequence data were deposited within the short read archive under experiment accession number SRX708102, and processed data were available from the researchers upon request.
Data Analyses
The resulting paired-end 16S rRNA gene reads were assembled using the “make.contigs” command in mothur1 (29). Any base call disagreement in the overlapping portions of the paired reads was ascribed to the base with a higher quality score (“deltaq = 1”) or called “N” if both nucleotides were below Q20. Assembled sequences were then pre-processed to remove low quality and chimeric sequence data. Sequences were removed if they were shorter than 400 nt, had a homopolymeric sequence greater than 10 base calls, had more than two ambiguous base calls (1 per 200 nt), or were found to be chimeric sequences by UCHIME (30). Each of the resulting sequence data sets were subsampled to 5,000 reads to enable direct comparison, as per Schloss et al. (31). Reads were preclustered as per Huse et al. (32) to reduce the influence of sequence error. The resulting data were separately clustered to form operational taxonomic units (OTUs) using mothur’s farthest neighbor approach at 95% sequence identity, or taxonomically assessed using mothur’s implementation of the naïve Bayesian classifier, RDP Classifier (33). OTUs representing < 0.1% of the total 16S rRNA reads in a given sample were eliminated as noise, as this has previously been shown to be the approximate level of noise from Illumina sequencing of complex communities (34). Taxonomic assignments were considered supported if bootstrapping values were greater than 70%. Microbial 16S rRNA gene composition and diversity were compared among samples, as well as to sequences from human and primate vaginal systems, using multivariate statistical approaches provided by mothur for measures of γ-, and α-diversity, and using the vegan package of R (35) for β-diversity. Richness measures included genera observed and Chao1 estimates (36) of total genera richness. These measures were derived from genus-level taxonomic classifications due to the low bootstrapping support offered to the majority of sub-genus-level classifications. Diversity measures were determined using Shannon’s diversity index. Normality of data was tested using the Shapiro–Wilk test (37), and significance was determined using a two-sample t-test or Wilcoxon Mann–Whitney test for normally and non-normally distributed data, respectively. Heatmaps were constructed using gplots2 in R with taxonomic data. Within and across microbial communities, inter-individual, and inter-species similarities were determined by pairwise measurements of Bray–Curtis dissimilarity, with significance determined by analysis of similarities (ANOSIM).
Results
Sequencing Overview
Twenty Rambouillet ewes and twenty crossbred cows were sampled to investigate the composition of livestock vaginal microbiota. Samples were used to generate deep V3-V4 16S rRNA gene profiles. A total of 907,667 high-quality reads were obtained following processing, and samples were randomly subsampled to 5,000 reads for direct comparison. Good’s Coverage estimates (38) were not different among cattle and sheep, indicating that this approach obtained data on 98 ± 0.01% of the total microbial genera present in each sample. Consistently, rarefaction curves appear to be trending toward an asymptote (Figure S1 in Supplementary Material).
α-Diversity
Unless otherwise stated, analyses were carried out at genus-level resolution due to low bootstrapping support for species-level taxonomic designations. A greater number of genera (comparative t-test; P ≤ 0.05) were detected in cow vaginal samples compared to ewe samples, with 302 ± 83 and 220 ± 102 genera, respectively. A few outliers existed for each host species as seen in the rarefaction curves (Figure S1 in Supplementary Material), however these were not significantly associated with mating, fertilization method, or reproductive status (all comparative t-tests P > 0.05). Chao1 predicted that there may have been more (comparative t-test; P ≤ 0.05) genera present in cow than ewe vaginal samples, with 394 ± 77 and 310 ± 103 total genera predicted, respectively. However, most genera were observed to be present at very low relative abundances with just 14 ± 4 and 11 ± 4 genera representing greater than 1% of the reads, and 90 ± 41 and 57 ± 45 representing >0.1% of the reads, respectively. Measures of diversity (richness and evenness) were normally distributed (Shapiro–Wilk W = 0.92; P > 0.05) for ewes but not cows (W = 0.9; P ≤ 0.05).
Diversity was measured with Shannon’s diversity index, which indicated low to moderate diversity communities for both ewes and cows at 2.87 ± 1.16 and 3.64 ± 0.96, respectively. Cow vaginal microbiota exhibited greater diversity as measured with Shannon’s diversity index than ewes (Wilcoxon; P ≤ 0.05), humans (Wilcoxon; P ≤ 0.05), and all non-human primates (Wilcoxon; P ≤ 0.05). However, the diversity of ewe vaginal microbiota was not significantly different (P > 0.05) from most non-human primates, although was still more diverse (Wilcoxon; P ≤ 0.05) than the human vagina (Table S1 in Supplementary Material and Figure 1).
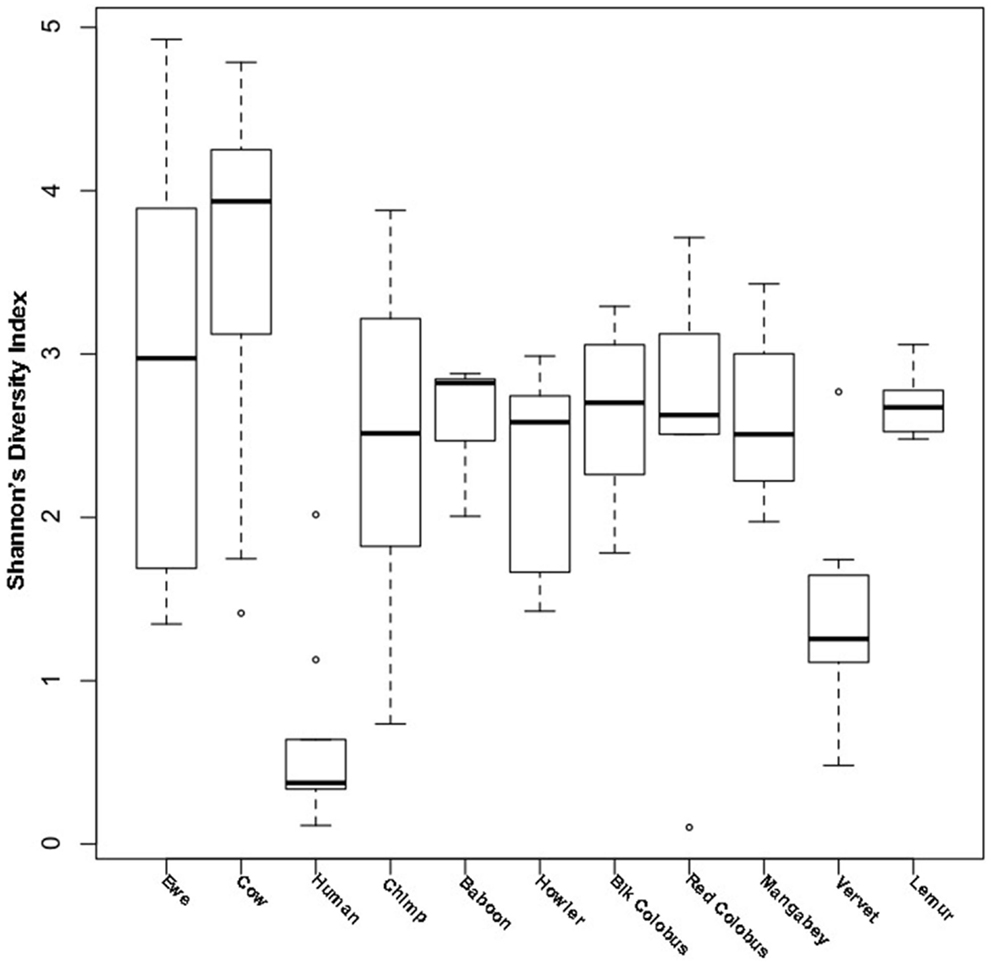
Figure 1. Shannon’s diversity of livestock vaginal microbiota as compared to humans and non-human primates. Boxplots showing the median, quartiles, and extremities of Shannon’s diversity index values calculated for individual ewes, cows, humans, and non-human primates compared in this study.
β-Diversity
Due to low bootstrapping support for species-level taxonomic designations, we employed an OTU approach (described in Section “Materials and Methods”) to compare the compositions of cow and ewe vaginal microbiota. A small but significant difference was determined between the OTU compositions of cow and ewe vaginal microbiota (ANOSIM R = 0.11, P ≤ 0.05). However, no significant difference was seen between the cow and ewe vaginal microbiota when comparing genus-level taxonomic data (ANOSIM R = 0.07, P > 0.05) indicating the small differences observed resulted from sub-genus-level distinctions. No difference was seen in the overall composition with age, even when stratified by host species (ANOSIM R < 0, P > 0.05). Method of fertilization (AI vs. conventional; ANOSIM R = 0.02, P > 0.05) or embryo transplantation in cows (ANOSIM R = 0.04, P > 0.05) did not lead to significant overall differences in vaginal microbiota. At the time of sampling, unmated ewes and cows did not differ from mated (ANOSIM R < 0.2, P > 0.05) or from pregnant animals (ANOSIM R < 0.07, P > 0.05). Unmated ewes were not significantly different from recently (<48 h) mated animals (ANOSIM R = 0.2, P = 0.09). Conventionally bred cows and ewes (both ANOSIM R < 0, P > 0.05) also did not differ among pregnant and mated but open individuals.
The taxonomic composition inferred from 16S rRNA gene sequence data was compared using genus-level resolution to human and non-human primate vaginal microbiota determined by Yildirim et al. (5). Continued use of genus-level resolution was further necessitated by the limited overlap in the regions on the 16S rRNA gene molecule sequenced in this study and by Yildirim et al. (5). RDP Classifier genus-level taxonomy was ordinated by non-metric multi-dimensional scaling (NMDS) using Bray–Curtis dissimilarities (Figure 2). At this level of taxonomic resolution, the host species-specificity of vaginal microbiota among many of the non-human primate species, as previously reported by Yildirim et al. (5), had eroded. However, alike human vaginal microbiota, ewe and cow vaginal microbiota were distinct. Both differed significantly from humans (R = 1, P ≤ 0.05) and non-human primate host species (R > 0.7, P ≤ 0.05).
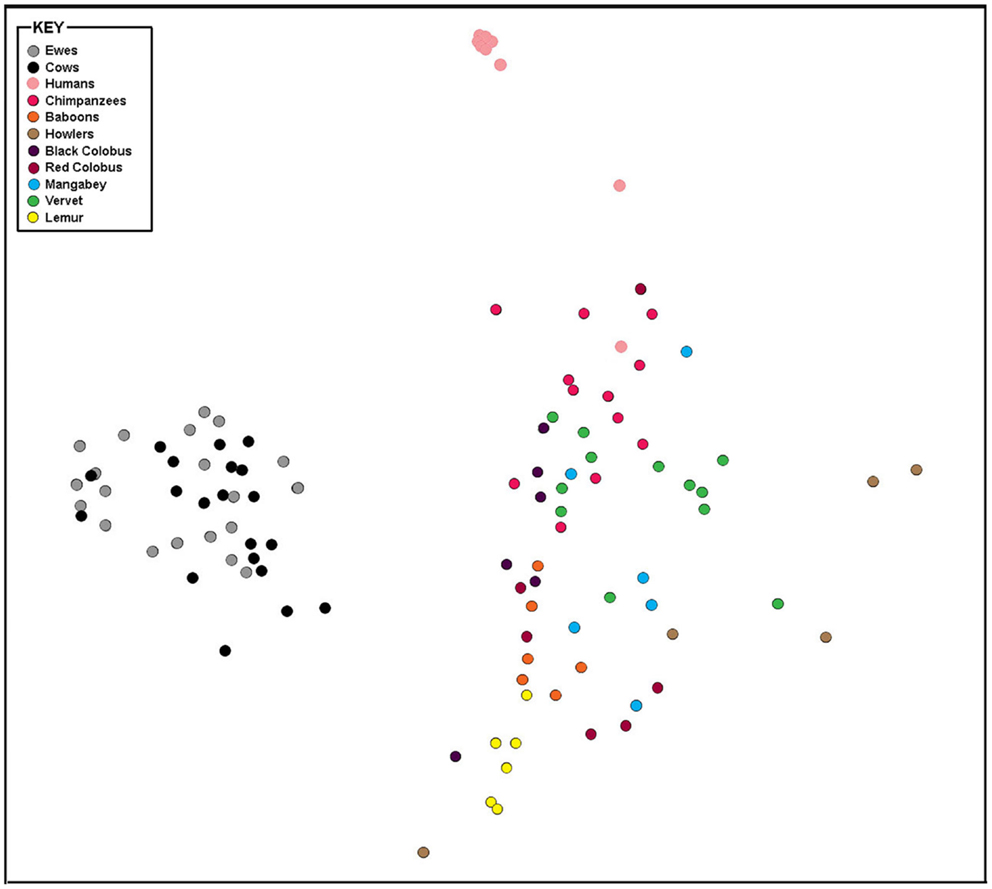
Figure 2. Bray–Curtis relationship among vaginal microbiota of livestock, humans, and non-human primates. Non-metric multi-dimensional scaling plot of Bray–Curtis dissimilarity measures of vaginal microbiota determined from individual ewes, cows, humans, and non-human primates compared in this study.
General Taxonomic Compositional Traits
Our sequencing approach enabled the detection of both bacterial and archaeal microbiota. Bacteria were numerically dominant, representing 98.7 ± 0.02% of 16S rRNA reads in all samples. However, Archaea were detected in 95% (19/20) of vaginal samples collected from both cows and ewes. Archaeal reads were largely assigned to members of the order Desulfurococcales, occurring in 95% of cow and 85% of ewe samples. The bacterial community was most commonly dominated by members of the Proteobacteria (almost exclusively γ-proteobacteria), Fusobacteria, and Bacteroidetes phyla in both ewes and cows (Figure 3). Aggregatibacter spp. and Streptobacillus spp. were typically the most abundant genera in both ewes and cows, while various other genera were observed (Figures 4 and 5). Lactobacilli were common, being detected in 80% (n = 16/20) of ewe, and 90% (n = 18/20) of cow vaginal samples. However, lactobacilli were always found at a low relative abundance (0.36 ± 0.66 and 0.53 ± 0.65%) of the total 16S rRNA gene population determined from both cattle and ewe vaginal samples, respectively. Assignable Lactobacillus species varied among individual animals and were often heterogeneous within samples (Table 2). Species often described in human vaginal microbiota, particularly those defined by Ravel et al. (1) as community state type (CST) IV, and often associated with BV, were observed in ewes and cows. These include Sneathia spp. that were observed in 90% of samples from both ewes and cows at 2.4 ± 4.0 and 1.9 ± 2.3% of the total microbiota, respectively, and Prevotella spp. that were observed in 65% of ewes at 0.5 ± 0.9% of the total microbiota. Paraprevotella spp., a distinct but closely related genera to Prevotella were also observed among both ewe and cow vaginal microbiota.
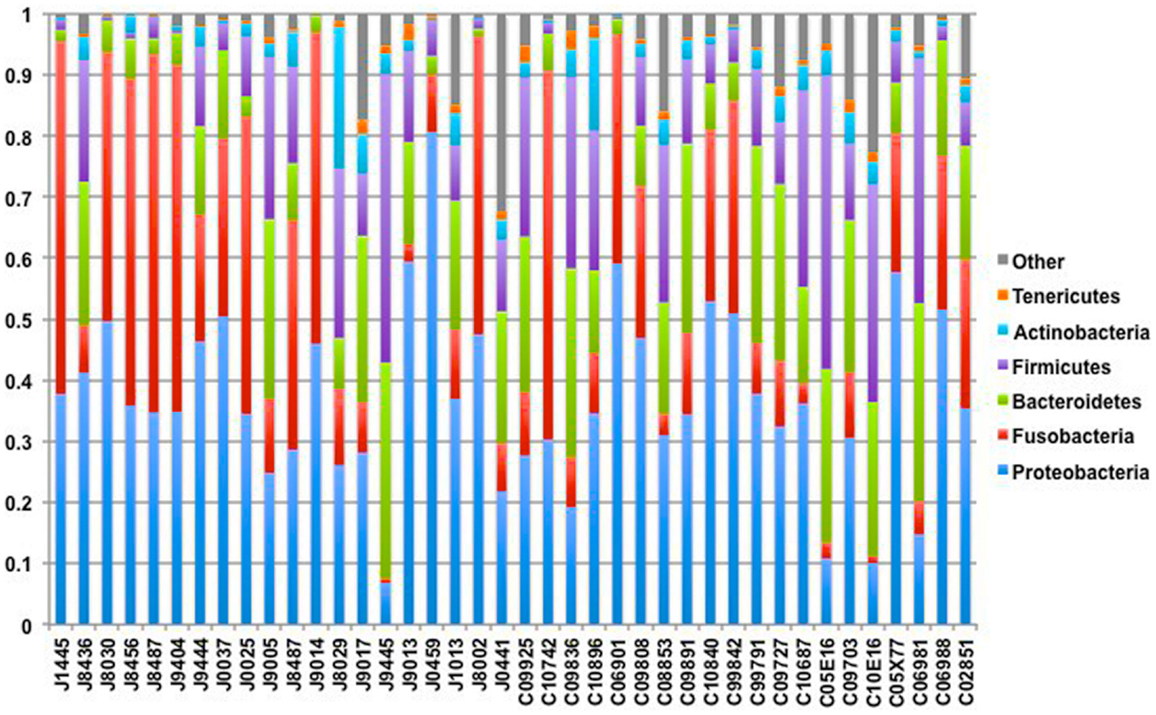
Figure 3. Phylum-level composition among livestock vaginal microbiota. Bar chart showing the proportional distribution of the six most abundant phyla.
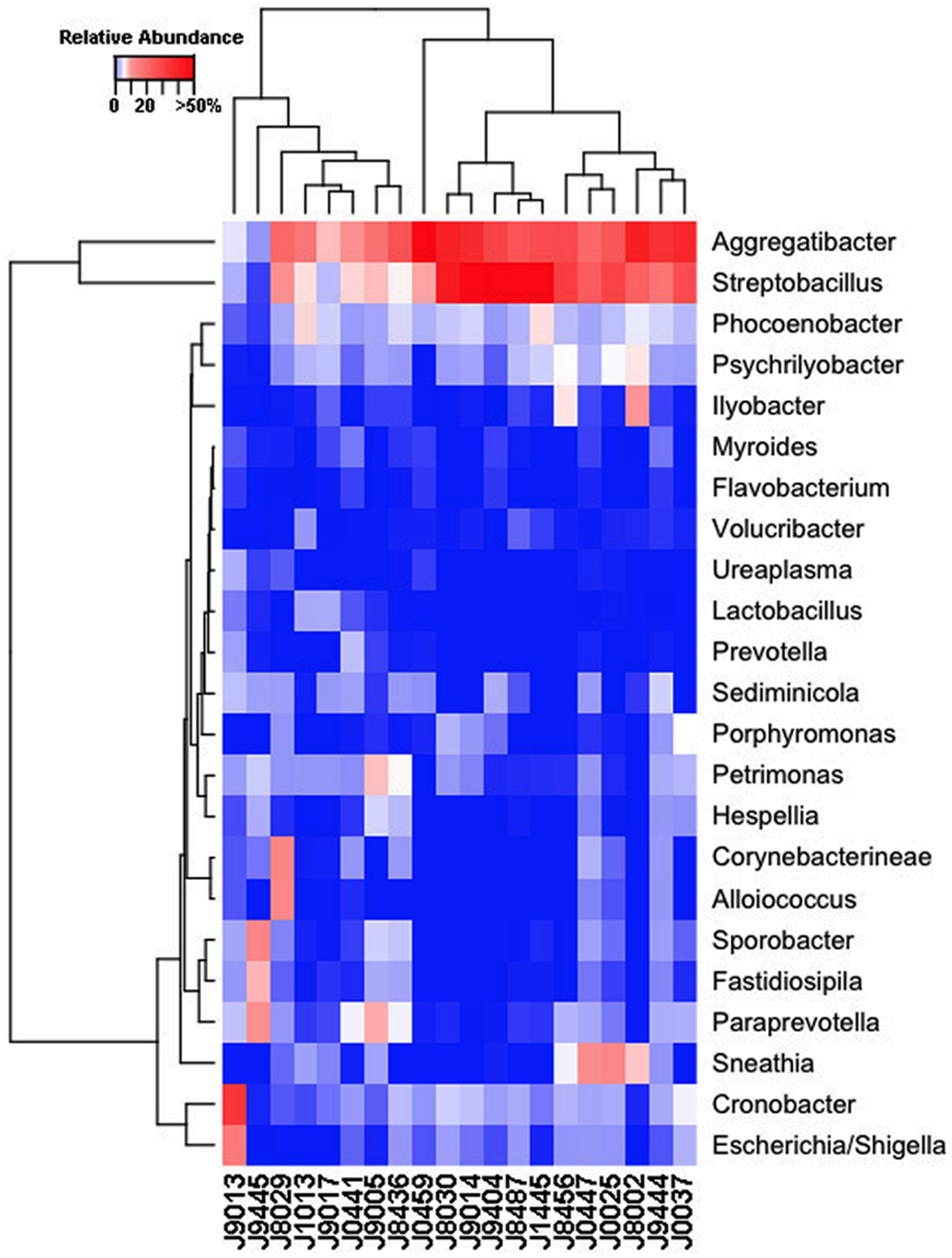
Figure 4. Genus-level composition among ewe vaginal microbiota. Heat map showing the relative abundances of the most abundant genera identified in individual ewe vaginal microbiota. Color breaks in heatmap are adjusted to show genera seen at <1% (blue shades), 1–10% (white shades), and >10% (red shades) relative 16S rRNA gene abundance.
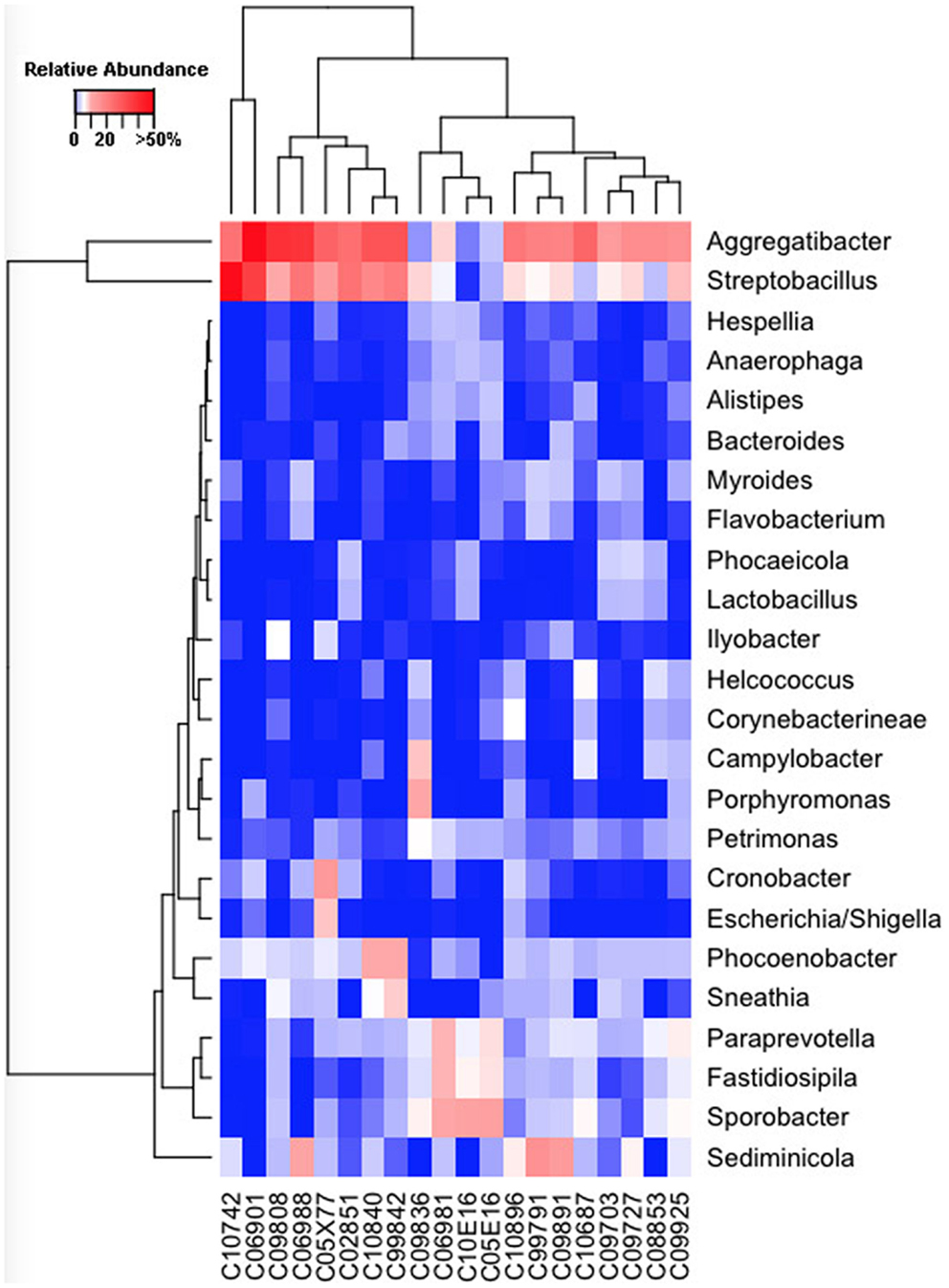
Figure 5. Genus-level composition among cow vaginal microbiota. Heat map showing the relative abundances of the most abundant genera identified in individual cow vaginal microbiota. Color breaks in heatmap are adjusted to show genera seen at <1% (blue shades), 1–10% (white shades), and >10% (red shades) relative 16S rRNA gene abundance.
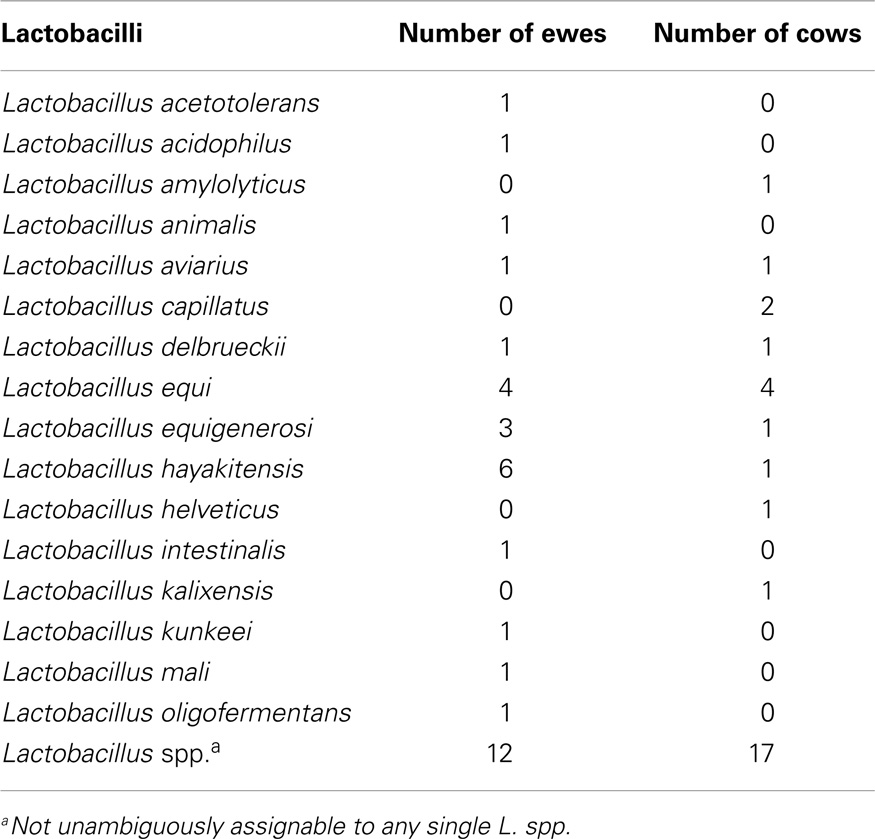
Table 2. Lactobacilli identified in cow and ewe ectocervicovaginal lavages.
Livestock Vagina Maintains a Near-Neutral pH
Consistent with low Lactobacillus spp. abundance in vaginal microbiota, vaginal pH was near-neutral in both cows and ewes. Cow C99842 was noted to have a yellow sample, possibly containing urine. Significance of results was not affected by inclusion or exclusion of this value, except that Shapiro–Wilk test reported that cow pH was not normal (P ≤ 0.05) with C99842 and was normal (P > 0.05) following its removal. Ewe and cow pH differed (P ≤ 0.05) as determined by Welches and Wilcoxon two-sample tests before and after removal of C99842. The pH means (±SD) were 6.7 ± 0.38 and 7.3 ± 0.63, and ewes ranged from 5.6 to 7.1, and cows from 6.5 to 8.7.
Discussion
Despite well-described roles in human reproduction and perinatal health, the vaginal microbiota of very few non-human hosts has been described to date. Thereby their relationships with reproductive outcomes and perinatal morbidity remain to be explored. The human vagina is most commonly dominated by Lactobacillus spp. during their reproductive years (1), however, a recent study of non-human primates (5) has revealed this to be unique among the primate order. Our findings herein indicate the uniqueness of the human vaginal microbiome extends further into Mammalia. Aggregatibacter spp., Streptobacillus spp., Cronobacter spp., Phocoenobacter spp., and Psychrilyobacter spp. were found to be the predominant bacterial genera of the ewe vaginal tract. While, Aggregatibacter spp., Streptobacillus spp., Phocoenobacter spp., Sediminicola spp., and Sporobacter spp. were the major genera in cow samples. Our results contrast with preceding culture-based studies that have more commonly isolated genera such as Bacillus spp., Staphylococcus spp., and Streptococcus spp., from both cow and ewe vaginas as well as Corynebacterium spp., and Escherichia spp., from the ewe vagina and Enterococcus spp. from the cow vagina (6, 7, 9–13, 39). These bacterial genera described in culture-based studies were often detected in our samples but typically with low relative abundances. Escherichia spp. were detected in all but one ewe (19/20; Figure 4), while the rest of the bacteria suggested to be prevalent by culture-based studies were seen at very low abundances in 3–11 of the ewes examined. Similarly, those bacteria commonly cultured from the cow vagina were detected at very low abundance in 0–11 of the 20 cows sampled in this study. A recent 16S rRNA sequencing study of Holstein cow uteri reported the presence of the same bacterial phyla as we observed in the cow and ewe vagina, specifically, Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria, Proteobacteria, and Tenericutes (40). The same study also reported many genera that we detected as prevalent in the ewe and cow vaginal samples, including Escherichia spp., Lactobacillus spp., Porphyromonas spp., Prevotella spp., Sneathia spp., Streptobacillus spp., and Ureaplasma spp. and other genera that were observed specifically in the cow vaginal samples, including Alistipes spp., Bacteroides spp., Campylobacter spp., and Helcococcus spp. (40).
Studies exploring microbial communities with culture-based identification often differ from studies using 16S rRNA gene sequences to assign taxonomy, with the later often revealing a much greater diversity (15). Shade et al. (16) studied the same soil samples using culture and 16S rRNA gene sequencing techniques. These authors reported that cultured organisms were often low, or even absent, from 16S rRNA community profiles. This is similar to the present study, which revealed many more-abundant genera than was evident from previous culture-based studies, although most cultured genera were still detected. While one of the two most dominant genera of our study (Aggregatibacter spp.) was not reported by Machado et al. (40), their study was of uterine bacteria while the present study was of vaginal bacteria. However, given the location of the uterus relative to the vagina, it is likely that microbes enter the uterus via the vagina. In fact, during gestation this “ascending infection” in humans is hypothesized to be an important feature of pre-term birth (41).
The relative dominance of Aggregatibacter spp. and Streptobacillus spp. is interesting and draws parallels to the dominance of lactobacilli often seen in the human vagina (1). Aggregatibacter spp. have previously been observed at very low levels in samples of the human vagina (42), and described members include important human pathogens such as A. aphrophilus and A. actinomycetemcomitans that have been linked to periodontal disease, infective (HACEK) endocarditis, and brain abscess formation (42, 43). Their high relative abundance within the vaginal tracts of livestock may be facilitated by the ability of some members of this genus to adhere to collagen (44), which is a major component of the vaginal wall tissue in cows, and ewes, along with humans. In ewes, collagen accounts for up to 50% of ewe vaginal tissue (45, 46). While it is interesting to note that observed changes in human vaginal microbiota associated with menopause (47) take place at a time when the structure of collagen in the human vagina is also changing (48), the role of collagen in curating the vaginal microbiota is uncertain. The compositional structure and distribution of vaginal collagen in ewes is similar to that of humans and total collagen content is comparable (46). Streptobacillus spp. have also been observed in the human vagina (49). Much of what is known about this genus is based on S. moniliformis, the etiological agent of rat bite fever, which until recently was the only described species (50). Because of the paucity of information presently available on the Streptobacilli it is difficult to speculate on its role or niche within the ewe or cow vagina.
Lactobacillus spp. were prevalent, but at low relative abundances among cow and ewe vaginal microbiota. This is consistent with previously reported culture-driven results of the cow vagina (6–8). Lactobacillus delbrueckii was one Lactobacillus species detected in both cow and ewe vaginas (Table 2), which has previously been isolated from the cow vagina (8). The limited abundance of lactobacilli observed previously using culture-based studies had led Rodriguez et al. (8) to suggest that Lactobacillus spp. have a restricted role in the cow vagina.
The near-neutral pH observed herein is consistent with low Lactobacillus spp. abundance, as this genera is known to be able to produce large quantities of lactate as a metabolic by product (51), and is generally credited with creating low vaginal pH in women. Interestingly, the cow and ewe vaginal microbial communities share several notable genera, namely Sneathia spp., Porphyromonas spp., Prevotella spp., and a low abundance of Lactobacillus spp., with the CST IV described for humans by Ravel et al. (1). This CST corresponded with the highest vaginal pH in women, with a mean of 5.3. Manes et al. (14) reported pH means between 7.0 and 7.6 in ewes prior to synchronization with intravaginal sponges, although they reported a mean as low as 6.8 about 53 h after sponge removal at breeding. Beckwith-Cohen et al. (52) reported a range of 5.52–8.60 for cattle vaginal pH in the literature, and found that the mean vaginal pH of Israeli Holstein multiparous cows was 7.35. The mean cow and ewe pHs of 7.3 and 6.7, respectively, from the present study are comparable to these values.
In conclusion, 16S rRNA sequencing of cow and ewe vaginal ectocervicovaginal lavages revealed that cow and ewe vaginal microbiota are unique from previously described vaginal microbial ecosystems, though similar to one another. Cow microbiota exhibited greater diversity compared to the ewe microbiota, and both differed from humans and non-human primates. Bacteroidetes, Fusobacteria, and Proteobacteria were determined to be the dominant phyla. Archaea and lactobacilli, while prevalent, were not abundant. Culture methods previously employed likely misidentified the most abundant species, with organisms such as Staphylococcus spp. and Streptococcus spp. detected at very low abundance. The two most abundant members of the cow and ewe vaginal microbiota in the present study were Aggregatibacter spp. and Streptobacillus spp. It was confirmed that Lactobacillus spp., in contrast to the human vaginal microbiota, are not an abundant genera. The near-neutral pH observed in both cows and ewes is consistent with the low abundance of Lactobacillus spp. detected. However, it remains to be tested whether the taxa present in livestock vaginal systems are more prevalent or abundant during phases of the estrus cycle, affect reproductive performance, or contribute to perinatal colonization. Considering the great amount of diversity and different taxa identified using 16S rRNA sequencing, it would be valuable to explore these avenues with culture-independent analyses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Robert Brekke and Dr. Glenn Duff for providing access to the BART farm where the study was carried out. This study was supported by the Montana Agricultural Experiment Station project (MONB00113) and the multistate project W-3177 Enhancing the competitiveness of US beef (MONB00195).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fvets.2014.00019/abstract
Footnotes
References
1. Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, et al., Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4680–7. doi: 10.1073/pnas.1002611107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. O’Halan DE, Moench TR, Cone RA. Vaginal pH and microbicidal lactic acid when lactobacilli dominate the microbiota. PLoS One (2013) 8:e80074. doi:10.1371/journal.pone.0080074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Li J, McCormick J, Bocking A, Reid G. Importance of vaginal microbe in reproductive health. Reprod Sci (2012) 19:235–42. doi:10.1177/1933719111418379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Leitich H, Kiss H. Asymptomatic bacterial vaginosis and intermediate flora as risk factors for adverse pregnancy outcome. Best Pract Res Clin Obstet Gynaecol (2007) 21:375–90. doi:10.1016/j.bpobgyn.2006.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Yildirim S, Yeoman CJ, Janga SC, Thomas SM, Ho M, Leigh SR, et al., Primate vaginal microbiomes exhibit species-specificity without universal lactobacillus dominance. ISME J (2014). doi:10.1038/ismeJ.2014.90
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Otero C, Silva de Ruiz C, Ibanez R, Wilde OR, de Ruiz Holgado AAP, Nader-Macias ME. Lactobacilli and enterococci isolated from the bovine vagina during the estrous cycle. Anaerobe (1999) 5:305–7. doi:10.1006/anae.1999.0245
7. Otero C, Saavedra L, Silva de Ruiz C, Wilde O, Holdado AR, Nader-Macias ME. Vaginal bacterial microflora modifications during the growth of healthy cows. Lett Appl Microbiol (2000) 31:251–4. doi:10.1046/j.1365-2672.2000.00809.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Rodriguez C, Cofre JV, Sanchez M, Fernandez P, Goggiano G. Lactobacilli isolated from vaginal vault of dairy and meat cows during progesteronic stage of estrous cycle. Anaerobe (2011) 17:15–8. doi:10.1016/j.anaerobe.2010.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Amin JD, Zaria LT, Malgwi RM. Vaginal aerobic bacterial flora of apparently healthy cattle in various stages of the reproductive cycle in the Sahel region of Nigeria. Bull Anim Health Prod Afr (1996) 44:15–8.
10. Zambrano-Nava S, Boscan-Ocando J, Nava J. Normal bacterial flora from vaginas of Criollo Limonero cows. Trop Anim Health Prod (2011) 43:291–4. doi:10.1007/s11250-010-9701-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Sawyer GJ. Observations on the bacterial population of the os cervix of the ewe before and after embryo death. Aust Vet J (1977) 53:542–4. doi:10.1111/j.1751-0813.1977.tb07942.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Al-Amin JD. Bacteriology of the genital tract of ewes – aetiology of non-specific infections. Bull Anim Health Prod Afr (1988) 36:255–7.
13. Moorthy ARS, Singh SP. Studies on the bacterial flora of female genital tract of sheep. Bull Anim Health Prod Afr (1982) 30:15–8.
14. Manes J, Fiorentino MA, Kaiser G, Hozbor F, Alberio R, Sanchez E, et al., Changes in the aerobic vaginal flora after treatment with different intravaginal devices in ewes. Small Rum Res (2010) 94:201–4. doi:10.1016/j.smallrumres.2010.07.021
15. Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol (1998) 180:4765–74.
16. Shade A, Hogan CS, Klimowicz AK, Linske M, McManus PS, Handelsman J. Culturing captures members of the soil rare biosphere. Environ Microbiol (2012) 14:2247–52. doi:10.1111/j.1462-2920.2012.02817.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Fodor AA, DeSantis TZ, Wylie KM, Badger JH, Ye Y, Hepburn T, et al., The “most wanted” taxa from the human microbiome for whole genome sequencing. PLoS One (2012) 7:e41294. doi:10.1371/journal.pone.0041294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al., Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A (2010) 107:11971–5. doi:10.1073/pnas.1002601107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Yeoman CJ, White BA. Gastrointestinal tract microbiota and probiotics in production animals. Ann Rev Anim Biosci (2014) 2:469–86. doi:10.1146/annurev-animal-022513-114149
20. Prittchard GC, Smith RP, Errington J, Hennon S, Jones RM, Mearns R. Prevalence of Coxiella burnetii in livestock abortion material using PCR. Vet Rec (2011) 169(15):391. doi:10.1136/vr.d4693
21. Bekele M, Demelash B, Fufa A, Regassa A, Godfroid J, Skjerve E. Seroprevalence of brucellosis and its contribution to abortion in cattle, camel and goat kept under pastoral management in Borana, Ethiopia. Trop Anim Health Prod (2010) 43:651–6. doi:10.1007/s11250-010-9748-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Olsen S, Tatum F. Bovine brucellosis. Vet Clin North Am Food Anim Pract (2010) 26(1):15–27. doi:10.1016/j.cvfa.2009.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Dekeyser J. Bovine genital campylobacteriosis. In: Butzler JP editor. Campylobacter Infection in Man and Animals. Boca Ranton, FL: CRC Press (1984). p. 181–91.
24. Martin HL, Richardson BA, Nyange PM, Lavreys L, Hillier SL, Chohan B, et al., Vaginal lactobacilli, microbial flora, and risk of human immunodeficiency virus type 1 and sexually transmitted disease acquisition. J Infect Dis (1999) 180:1863–8. doi:10.1086/315127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Wisenfeld HC, Hilier SL, Krohn SL, Landers DV, Sweet RL. Bacterial vaginosis is a strong predictor of Neisseria gonorrhoeae and Chlamydia trachomatis infection. Clin Infect Dis (2003) 36:663–8. doi:10.1086/367658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Cherpes TL, Melan MA, Kant JA, Cosentino LA, Meyn LA, Hilier SL. Genital tract shedding of herpes simplex virus type 2 in women: effects of hormonal contraception, bacterial vaginosis, and vaginal group B Streptococcus colonization. Clin Infect Dis (2005) 40:1422–8. doi:10.1086/429622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Brotman RM, Klebanoff MA, Nansel TR, Yu KF, Andrews WW, Zhang J, et al., Bacterial vaginosis assessed by Gram stain and diminished colonization resistance to incident gonococcal, chlamydial, and trichomonal genital infection. J Infect Dis (2010) 202:1907–15. doi:10.1086/657320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Core Team R. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing (2013). Available from: http://www.R-project.org/
29. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al., Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol (2009) 75:7537–41. doi:10.1128/AEM.01541-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics (2011) 27:2194–200. doi:10.1093/bioinformatics/btr381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One (2011) 6:e27310. doi:10.1371/journal.pone.0027310
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OUT clustering. Environ Microbiol (2010) 12:1889–98. doi:10.1111/j.1462-2920.2010.02193.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol (2007) 73:5261–7. doi:10.1128/AEM.00062-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Degnan PH, Ochman H. Illumina-based analysis of microbial community diversity. ISME J (2012) 6:183–94. doi:10.1038/ismej.2011.74
35. Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, et al., Community Ecology Package 2.0-5. (2012). Available from: http://vegan.r-forger.r-project.org/
36. Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJM. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol (2001) 67:4399–406. doi:10.1128/AEM.67.10.4399-4406.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Shapiro SS, Wilk MB. An analysis of variance test for normality. Biometrika (1965) 52:591–611. doi:10.1093/biomet/52.3-4.591
38. Good IJ. The population frequencies of species and the estimation of population parameter. Biometrika (1953) 40:237–64. doi:10.1093/biomet/40.3-4.237
39. Quinlivan TD. The bacteriology of cervical mucus of ewes at oestrus following treatment with fluoro-progestagen impregnated intravaginal sponges. Aust Vet J (1970) 46:11–6. doi:10.1111/j.1751-0813.1970.tb14873.x
40. Machado VS, Oikonomou G, Bicalho MLS, Knauer WA, Gilbert R, Bicalho RC. Investigation of postpartum dairy cows’ uterine microbial diversity using metagenomic pyrosequencing of the 16S rRNA gene. Vet Microbiol (2012) 159:460–9. doi:10.1016/j.vetmic.2012.04.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Martius J, Eschenbach DA. The role of bacterial vaginosis as a cause of amniotic fluid infection, chorioamnioitis and prematurity – a review. Arch Gynecol Obstet (1990) 247:1–13. doi:10.1007/BF02390649
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Gonzales-Marin C, Spratt DA, Millar MR, Simmonds M, Kempley ST, Allaker RP. Levels of periodontal pathogens in neonatal gastric aspirates and possible maternal sites of origin. Mol Oral Microbiol (2011) 26:277–90. doi:10.1111/j.2041-1014.2011.00616.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. NØrskov-Lauritsen N. Classification, identification, and clinical significance of Haemophilus and Aggregatibacter species with host specificity for humans. Clin Microbiol Rev (2014) 27:214. doi:10.1128/CMR.00103-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Tang G, Kitten T, Munro CL, Wellman GC, Mintz KP. EmaA, a potential virulence determinant of Aggregatibacter actinomycetemcomitans in infective endocarditis. Infect Immun (2008) 76:2316–24. doi:10.1128/IAI.00021-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Ayen E, Noakes DE. Distribution of collagen in the vaginal wall of ewes. Vet J (1998) 155:213–5. doi:10.1016/S1090-0233(98)80023-3
46. Ulrich D, Edwards SL, Letouzey V, Su K, White JF, Rosamilia A, et al., Regional variation in tissue composition and biomechanical properties of postmenopausal ovine and human vagina. PLoS One (2014) 9:e104972. doi:10.1371/journal.pone.0104972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Brotman RM, Shardell MD, Gajer P, Fadrosh D, Chang K, Silver MI, et al., Association between the vaginal microbiota, menopause status, and signs of vulvovaginal atrophy. Menopause (2014) 21:450–8. doi:10.1097/gme.0b013e3182a4690b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Sridharan I, Ma Y, Kim T, Kobak W, Rotmensch J, Wang R. Structural and mechanical profiles of native collagen fibers in vaginal wall connective tissues. Biomaterials (2012) 33:1520–7. doi:10.1016/j.biomaterials.2011.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Oakley BB, Fiedler TL, Marrazzo JM, Fredricks DN. Diversity of human vaginal bacterial communities and associations with clinically defined bacterial vaginosis. Appl Environ Microbiol (2008) 74:4898. doi:10.1128/AEM.02884-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Woo PCY, Wu AKL, Tsang CC, Leung KW, Ngan AHY, Curreem SOT, et al., Streptobacillus hongkongensis sp. nov., isolated from patients with quinsy and septic arthritis, and emended descriptions of the genus Streptobacillus and Streptobacillus moniliformis. Int J Syst Evol Microbiol (2014) 64:3034–9. doi:10.1099/ijs.0.061242-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Wylie JG, Henderson A. Identity and glycogen-fermenting ability of lactobacilli isolated from the vagina of pregnant women. J Med Microbiol (1969) 2:363–6. doi:10.1099/00222615-2-3-363
Keywords: vaginal microbiota, vaginal pH, Lactobacillus, Aggregatibacter, Streptobacillus
Citation: Swartz JD, Lachman M, Westveer K, O’Neill T, Geary T, Kott RW, Berardinelli JG, Hatfield PG, Thomson JM, Roberts A and Yeoman CJ (2014) Characterization of the vaginal microbiota of ewes and cows reveals a unique microbiota with low levels of lactobacilli and near-neutral pH. Front. Vet. Sci. 1:19. doi: 10.3389/fvets.2014.00019
Received: 24 August 2014; Paper pending published: 15 September 2014;
Accepted: 29 September 2014; Published online: 15 October 2014.
Edited by:
Maureen T. Long, University of Florida College of Veterinary Medicine, USAReviewed by:
Stacey Gilk, Indiana University School of Medicine, USAMd. Tanvir Rahman, Bangladesh Agricultural University, Bangladesh
Copyright: © 2014 Swartz, Lachman, Westveer, O’Neill, Geary, Kott, Berardinelli, Hatfield, Thomson, Roberts and Yeoman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carl J. Yeoman, Montana State University, 321 Animal Biosciences Building, Bozeman, MT 59717, USA e-mail:Y2FybC55ZW9tYW5AbW9udGFuYS5lZHU=