 Elpetra P. M. Timmermans-Sprang
Elpetra P. M. Timmermans-Sprang Ana Gracanin
Ana Gracanin Jan A. Mol
Jan A. Mol- Department of Clinical Sciences of Companion Animals, Utrecht University, Utrecht, Netherlands
Mammary tumors are the most common form of neoplasia in the bitch. Female dogs are protected when they are spayed before the first estrus cycle, but this effect readily disappears and is already absent when dogs are spayed after the second heat. As the ovaries are removed during spaying, ovarian steroids are assumed to play an essential role in tumor development. The sensitivity toward tumor development is already present during early life, which may be caused by early mutations in stem cells during the first estrus cycles. Later on in life, tumors arise that are mostly steroid-receptor positive, although a small subset of tumors overexpressing human epidermal growth factor 2 (HER2) and some lacking estrogen receptor, progesterone receptor (PR), and HER2 (triple negative) are present, as is the situation in humans. Progesterone (P4), acting through PR, is the major steroid involved in outgrowth of mammary tissue. PRs are expressed in two forms, the progesterone receptor A (PRA) and progesterone receptor B (PRB) isoforms derived from splice variants from a single gene. The dog and the whole family of canids have only a functional PRA isoform, whereas the PRB isoform, if expressed at all, is devoid of intrinsic biological activity. In human breast cancer, overexpression of the PRA isoform is related to more aggressive carcinomas making the dog a unique model to study PRA-related mammary cancer. Administration of P4 to adult dogs results in local mammary expression of growth hormone (GH) and wing less-type mouse mammary tumor virus integration site family 4 (Wnt4). Both proteins play a role in activation of mammary stem cells. In this review, we summarize what is known on P4, GH, and Wnt signaling in canine mammary cancer, how the family of HER receptors could interact with this signaling, and what this means for comparative and translational oncological aspects of human breast cancer development.
Introduction
Mammary tumors are the most common neoplasms in intact bitches with an estimated life-time risk of 1:4. About 50% of these tumors are malignant and one-third of these may form life-threatening distant metastases. Breast cancer is also a very common disease in humans with a life-time risk of about 1:8 (1).
Hierarchical cell organization of the mammary gland has recently become more clear in humans and mice and is linked to the heterogeneity of the mammary epithelium, with an outer and inner layer of luminal cells. Both layers have different epithelial cells with differential characteristics. In the outer basal layer of contractile myoepithelial cells, the mammary stem cells (MaSCs) reside, whereas in the layer of ductal and alveolar epithelial luminal cells, progenitor cells are found (2–4).
The main problem in breast cancer treatment is the recurrence of tumor growth and metastases. In both cases, cancer stem cells (CSCs) are thought to play an important role. However, the stem cell for the mammary gland has not yet been identified. The current paradigm is that a common stem cell gives rise to progenitor cells that are intermediates in the lineages of myoepithelial, and epithelial duct or lobular cells (5). In dogs, a subset of mammary carcinomas present as simple carcinomas that may be derived from mutated epithelial progenitor cells and are comparable to the most common form of human breast cancer, the ductal carcinomas. However, more often than in humans, dogs may also present with complex carcinomas that contain various differentiation pathways within a single affected mammary gland. These tumors may be derived from mutations in the most basic and early form of stem cells. The existence of the MaSC and the presence of various forms of progenitors may in part explain the heterogeneity of mammary carcinomas (6).
Hormone-Dependent Mammary Gland Development, Including Normal Roles of Growth Hormone (GH), Wnt, and HER
During embryogenesis, mammary gland development starts with the formation of a mammary placode, and subsequently a mammary bud. The functional development and differentiation of the mammary gland occurs, however, mainly postnatally under hormonal control and is coordinated with further reproductive development. Puberty starts with a trigger from estrogen (E2) and local growth factors to elongate the simple ductal tree by stimulating cell proliferation in the terminal end buds (7–9). Subsequently, fluctuating levels of progesterone (P4) stimulate the process of side branching and development of alveolar buds. During pregnancy, in response to P4 and prolactin (PRL), these alveolar buds can then differentiate into functional milk producing units, alveoli (7–9). P4 is thought to induce these changes in the mammary gland in a paracrine manner by acting on the progesterone receptor (PR)-expressing ductal epithelial cells, to stimulate the expression of growth factors that evoke proliferation of the neighboring PR-negative cells (10). These putative paracrine factors involve GH via signal transducer of activator of transcription (Stat3) (11) and of Janus kinase 2 (Jak2)/Stat5 (12), Wnt, and receptor activator of nuclear factor kappa B ligand (RANKL) (13–16).
Progesterone, together with E2, plays a central role in the outgrowth of the mammary gland by stimulating side branching of the mammary ductal tree during puberty and alveologenesis upon pregnancy. Upon P4 activation, PR-positive epithelial cells secrete GH and Wnt4 that act on MaSCs. In mammary cancer, tumor cells with stem cell properties, such as phenotypical epithelial mesenchymal transition (EMT) and elevated activity of the canonical Wnt pathway, play an important role in regrowth and metastasis (17, 18). In both humans and dogs, most mammary carcinomas are initially hormone dependent [i.e., express receptors for P4 (PR) and E2 (ER)]. The remaining tumors are often categorized as human epidermal growth factor 2 (HER2) positive (overexpressing HER2) or as triple-negative breast cancer (TNBC) (i.e., devoid of PR and ER and no overexpression of HER2) (1, 5, 7–9, 19).
Goal of Review
Both human and canine mammary carcinomas show involvement of common pathways in mammary cell proliferation and migration, such as Wnt and phosphatidyl-3-kinase (PI3K) signaling (1, 19). An important aspect is the hormonal dependence in most human and canine mammary carcinomas and the role of P4. This underscores the relevance of research on canine mammary cancer for both human and veterinary medicine and is a clear example of the one-health/one-medicine principle. Commonly used rodent models develop mammary carcinomas that are either not hormone dependent or do not metastasize as seen in mice and rats. This raises the question of whether the study of canine mammary carcinomas is a valid or even a better model for human breast cancer in comparison to rodents.
This review focuses on what is known of P4, GH, and Wnt signaling in canine mammary carcinomas in relation to what is known in other species, especially human breast cancer. In addition to the many similarities, the differences will also be discussed.
Progesterone
The central role of P4 signaling in breast cancer development gained renewed interest after the large hormone-replacement study of the World Health Initiative in 2003 (20). Since 1986, it has been known that ovarian steroids play an important role in the carcinogenesis of the mammary gland (21). Since then, elaborate research has been done predominantly on the role of E2. The tumorigenic role of P4 has long been underestimated (22) due to the more widespread use of synthetic progestins as inhibitors of tumor growth, even though this effect has been attributed to androgenic side effects since 1977 (23). In relation to reproductive physiology, P4 can have both a protective role or can be a risk factor for breast cancer. The protective role for P4 is seen in women who have an early full-term pregnancy and lactation. Risk factors associated with increased exposure to P4 are a prolonged interval between menarche and age of first childbirth, older age at menopause, early menarche, late menopause, and shorter menstrual cycles (24–27). Some of these factors influence development of a special subtype of breast cancer, for instance, lactation is correlated with TNBC and hormone responsive (HR+) tumors are associated with the length of the period between menarche and first childbirth (25, 26).
An important turning point for the role of P4 came from hormone-replacement studies that showed an increased risk for breast cancer development in women that received (conjugated equine) E2 plus a progestin [medroxyprogesterone acetate (MPA)] (28) compared to the E2 only group, in which there was a protective effect against breast cancer (20, 29). Similarly, in ACI rats that easily develop mammary cancer upon exposure to high dose E2, P4 was shown to be important for hormone-dependent mammary carcinogenesis (30). P4 signaling, therefore, clearly plays a role in breast cancer development, but the challenges are to define its exact role.
PR Signaling
Progesterone signals via the PR. The PR is expressed as two isoforms, progesterone receptor A (PRA) and progesterone receptor B (PRB). Both isoforms are derived from a single gene but regulated by two distinct promoters. In humans, E2 stimulates expression of both PRA and PRB mRNA (31). This is in contrast with ovariectomized mice where PRA expression is stimulated by E2 and inhibited by P4. PRB levels are not affected by E2 alone, but are stimulated by prolonged treatment with P4 or by P4 in combination with E2 (32). Accordingly, in mice, PRA is the main PR isoform expressed during pre-pubertal stages and in adult virgins, while PRB expression increases only during pregnancy (33). Therefore, it has been suggested that in the mouse, the initial proliferative response of the mammary epithelium to P4, leading to side branching is mediated by PRA, while PRB is needed for a proper lobular alveolar development during pregnancy (32). By contrast, in the normal human breast, both PRA and PRB are coexpressed in the same cells implying species-specific regulation of the isoforms (34). The PR isoforms have isoform-specific transcriptional activities on P4-responsive gene promoters, resulting in a distinct target gene profile (35). PRs can activate gene transcription in multiple ways and direct binding to progesterone response elements (PREs) a process usually referred to as classical PR signaling. PR can also tether to other transcription factors, such as Stat5 in the regulation of RANKL expression. Finally, PR can mediate so-called “non-genomic” cytoplasmic signaling through interaction with ERα, thereby activating the Rous sarcoma proto-oncogene/(Ras–Raf–MEK–ERK) cSrc/ERK pathway (36).
In classical signaling, both PR isoforms function as ligand-induced transcription factors and contain distinct activation function (AF) domains essential for their transcriptional activity. Two activation domains are common to both PRA and PRB, AF1 and AF2. PRB, however, has an additional activation function domain-3 (AF3) domain localized within the PRB specific N-terminus (37) making it a stronger transcriptional activator than PRA (Figure 1) (38). Sequence motifs essential for AF3 domain function were shown to be highly conserved across mammalian species (39, 40). We have compared the activities of canine PR (cPR) isoforms to human isoforms (hPR) using luciferase constructs containing classical PREs, mouse mammary tumor virus (MMTV)-luciferase, and PRE2-luciferase. We have shown that canine PRA has an expected hPRA-comparable transcriptional activity, whereas canine PRB (cPRB) has low to absent transactivation potential. No differences were found regardless of background cell type such as Chinese hamster ovary cells, canine mammary cells, or human T47D cells in which the endogenous PR was knocked out. The transactivation potential of cPRB could be restored by replacing the cPRB specific N-terminus with the human sequence in a human B-upstream segment (hBUScPRB) chimera. Next, we made canine mammary cell lines with a doxycycline (dox)-inducible expression of cPRB, hPRB, or the hBUScPRB chimera. Transactivation potential on endogenous target genes was then assessed by gene profiling using canine cDNA microarray. In the absence of dox, no effect of P4 incubation was seen, excluding signaling through P4 membrane receptors. Only the combination of dox plus P4 changed the expression of over 600 genes, both in hPRB- and hBUScPRB-expressing cell lines. Only a minority of these genes were influenced by cPRB (41, 42). These results indicate a very limited transactivation potential of cPRB on endogenous genes, thereby questioning its role in mammary gland development and carcinogenesis.
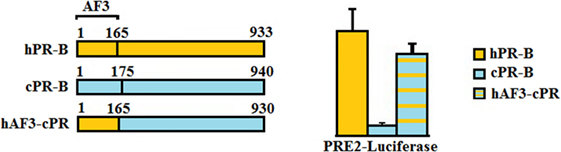
Figure 1. Comparison of the progesterone receptor-B variant of human (hPR-B) and canine (cPRB) species (left panel). The PR-B variant is encoded by the same gene as for the PR-A variant but has, due to an alternative start of translation, an amino-terminal elongation known as activation function domain-3 (AF3). Measurement of transactivation potential, using a PR-sensitive luciferase-reporter construct showed that the activity of the progesterone-stimulated cPRB is almost absent in comparison to the hPR-B (right panel). Replacement of the AF3 domain of humans into the canine sequence almost completely restores the cPRB activity (41).
PR Signaling in Mammary Cancer
Although the PRA and PRB isoforms are usually equally present in epithelial cells of the human mammary gland, in advanced breast cancer, a predominance of PRA is common, indicating that PRB has a protective function. Patients with PRA-rich tumors or tumors with a high PRA:PRB ratio have a much faster recurrence than patients with PRB-rich tumors (43, 44). Predominance of PRA is especially evident in ductal carcinoma in situ (DCIS) and invasive breast lesions. It has been suggested that P4 may also lead to transition of tumors from a luminal toward a basal phenotype (45). Germ line mutations in the genes breast cancer 1 (BRCA1) or breast cancer 2 are associated with a predominance of PRA expression (46). BRCA1 physically interacts with PR and inhibits its activity, in part, by preventing binding of the PR to the PRE and promoting the formation of a corepressor complex (47). Because this activity is lost in the context of BRCA mutant proteins, anti-progestins are recommended for tumor prophylaxis in BRCA mutation carriers (48). Mutation of BRCA1 results, moreover, in stabilization of the PR due to the loss of BRCA1-mediated PR ubiquitination and subsequent degradation (49). Interestingly, loss of BRCA1 also results in increased epidermal growth factor 1 (HER1) expression (50). We hypothesize that the combination of stabilization or enhanced PR expression and active HER1 signaling may stimulate phosphorylation of the PR by mitogen-activated protein kinase (MAPK) and specifically stimulate this mode of P4 signaling.
Within the luminal epithelium of the mammary gland PR-positive cells act as sensors for circulating P4 concentrations (Figure 2). Upon P4 exposure, these cells secrete growth factors (RANKL, Wnt) that may stimulate recruitment and differentiation of stem cells (51). P4 thus induces adult MaSC expansion (52) in mice, and this is also hypothesized to be a major site of carcinogenesis within the human breast (53). As well as an increase in MaSC, P4 was also suggested to act directly on PR-positive cells and convert them to a hormone-receptor negative, more stem-like state (54). Cluster of differentiation (CD) CD44HighCD24Low cells have been reported as breast CSCs since Al-Haij showed that these cells can form tumors in mice (55). In humans, CD44HighCD24Low cells seem to have a higher tumorigenic capacity (56) and even in dogs these cells have a tumor-initiating capacity (57) and stem/progenitor cell properties (58). Cell markers for stem cells and progenitors in the human and murine mammary gland (59, 60) are summarized (Table 1).
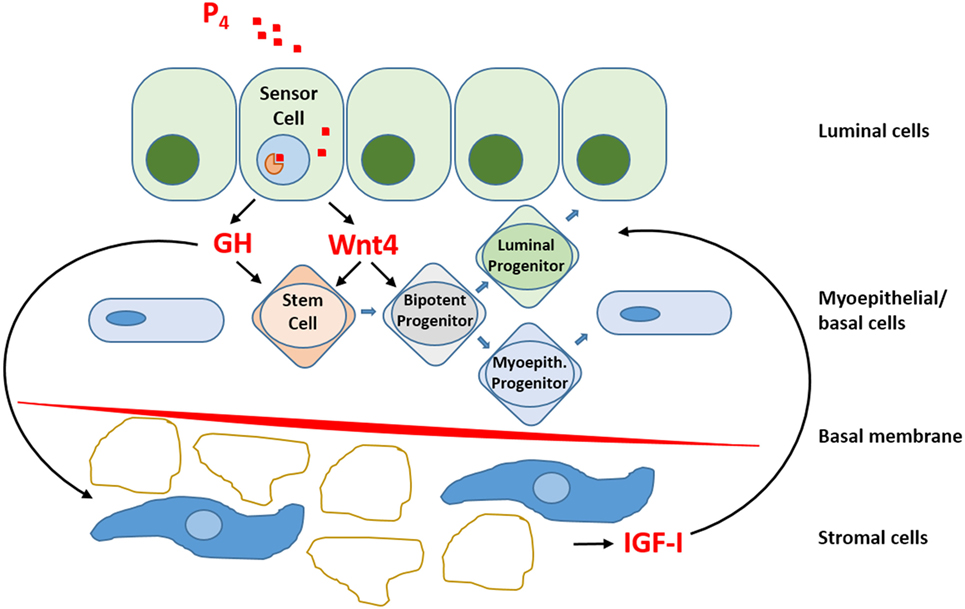
Figure 2. Schematic representation of progesterone (P4) signaling within the mammary gland. So-called “sensor cells” within the luminal epithelium contain progesterone receptors (PRs) that upon exposure to P4 stimulate the production and release of growth hormone (GH) and Wnt4. The local mammary production of GH has both a direct effect on GH receptor (GHR) containing stem cells and an indirect effect by stimulating the production and release of insulin-like growth factor-I (IGF-I) by GHR-containing cells in the stromal compartment. IGF-I stimulates further clonal expansion of activated cells. P4 also stimulates Wnt4 release. The Wnt pathway is essential for stem cell maintenance and activation of stem cells to form progenitor cells. Within the mammary gland, bipotent progenitor cells differentiate into progenitor cells specific for either luminal epithelial or myoepithelial cells.
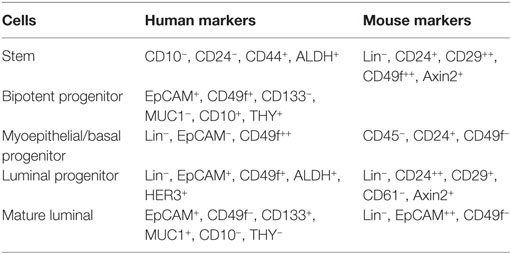
Table 1. Cell markers for stem cells and progenitors in the human and murine mammary gland (59, 60).
These stem cells and progenitor cells are necessary in the mammary gland to ensure proper long-term maintenance of mammary tissue structure and function during puberty, pregnancy, and lactation, which may occur multiple times during the reproductive lifespan of an animal (60). Mouse MaSCs have the ability to resist anoikis and form floating colonies, the so-called mammospheres. Progenitor cells from the inner layer, the luminal cells, have high proliferative potential in colony-forming cell (CFC) assays (61, 62) and also have a high level of PR in humans (63). These high-level PR cells occur in poorly developed lobules of the mammary gland and can be characterized by lineage (Lin−), epithelial cell adhesion molecule (EpCam+), CD49f+, aldehyde dehydrogenase 1 (ALDH+), and human epidermal growth factor 3 (HER3+) (59). ALDH1 is associated with stem/progenitor cell properties in human mammary epithelial cells (HMECs) that lack expression of ER (64, 65). This is in contrast to the mouse where PR+ luminal cells are CD29low, CD49flow, CD24+, and also have ER expression (59). In the canine mammary, tumor cell line (CMT U229) CD49f+, CD24low, CD44+ cells have been identified as tentative stem-like cells (66), with unknown ER/PR status.
Most data on hormone receptor status in the literature come from human mature luminal cells that mostly express ER and PR and mediate the proliferative effects of steroid hormones by paracrine signaling. However, E2 and P4 treatment also increase the MaSC population and in diestrus, when P4 levels are the highest, the number of CD24+/CD29hi mammary repopulating units increases 14 times in mice. Thus, steroids do influence stem cells, in both their proliferative and self-renewal abilities (52, 67). Oakes et al. suggest that a paracrine mediator is responsible for the proliferation and maintenance of human MaSCs, and a likely candidate is P4-regulated RANKL (3).
The PR has two modes of mitogenic action in the mouse mammary gland; a cyclin D1-dependent stimulation of proliferation in PR+ cells and a RANKL-mediated paracrine action on nearby PR− mammary epithelial cells (68). Steroid hormone-receptor positive cells release RANKL to neighboring stem cells in a paracrine manner. In a neighboring PR-negative luminal progenitor cell, RANKL can stimulate the transcription factor E74-like factor 5 and thus promote alveolar development (69). P4 also drives the CD24+/CD29lo/CD61+ luminal progenitor cell population to a CD24+/CD29lo/CD61− mature luminal cell differentiation, resulting in mature alveolar lineage expansion. Another possible mediator of paracrine signaling is neuregulin (Nrg1), which is produced in the basal epithelial cells and is a direct transcriptional target of tumor protein 63 (p63). P63 KO mice have several developmental defects such as no limbs, teeth, and mammary glands. Exogenous Nrg1 rescued the lactation through activation of human epidermal growth factor 4/Stat5 in neighboring mouse luminal epithelial cells (70). MaSCs not only receive signals in a paracrine manner indicating paracrine basal to luminal signaling but they can also signal back to regulate the luminal epithelium. This “teamwork” on breast development and homeostasis is only beginning to be unraveled (71).
Growth Hormone
GH in the Mammary Gland
Similar to steroid hormones P4 and E2, pituitary GH is also necessary for mammary gland development. GH deficiency impairs the mammary development in rats and mice. GH influences alveolar and duct development and limits side branching (72). In humans with Laron syndrome, a mutated GH receptor (GHR) affects mammary gland development. Patients with Laron syndrome are shorter, have no diabetes type II and their risk of getting all kinds of cancer is almost 0 (73). By contrast, patients with hypersecretion of GH (acromegaly) and individuals who are taller in height have a higher cancer incidence (18). A sustained exposure to steroid hormones is the best established risk factor for human breast cancer (74), and this also influences canine mammary gland proliferation and carcinogenesis (75, 76). Sustained exposure leads to nodules of lobular hyperplasia and simple or complex adenomas in the mammary gland (76). The proliferative effect on the mammary gland coincides with increased plasma GH concentrations. Dogs with endogenous high plasma P4 concentrations or treated with exogenous MPA were found to have high plasma GH concentrations. We observed that the canine mammary gland produces GH locally after MPA administration (77). As well as finding immune reactive GH in the mammary gland and a steep decrease in plasma GH concentrations after complete mastectomy, we found expression of GH mRNA in both the canine and feline mammary gland (78). Next, we demonstrated that in the dog, mammary expression of GH was initiated at the same start site as GH from the pituitary (79) but the pituitary-specific POU domain transcription factor was absent from mammary tissue. Analysis of the GH promoter revealed a putative PRE (80) but experiments using a GH-promoter luciferase construct did not show a direct transactivation by P4-activated PR in canine mammary tumor cell lines (81). In tumors, the staining intensity of the PR varied from no staining, normal nuclear staining to remarkable heterogeneous and perinuclear staining and cytoplasmic staining in spindle cells (82). The major pathways that were activated after prolonged MPA exposure in dogs were determined through gene expression studies. Both mammary tissue and cell lines were used to identify carcinoma-related expression profiles (83–85). Autocrine production of GH signals in human MCF7 cells in vitro are associated with a more invasive phenotype and in vivo with more aggressive tumors (86) indicating that GH activates the same pathways in humans as in dogs.
GH Signaling
Growth hormone is a peptide hormone closely related to PRL that exerts its action through GHR present in the cell membrane as constitutively dimerized single transmembrane proteins (87). Upon binding of GH ligands, GHR dimers are transphosphorylated by JAK2 tyrosine kinases that result in activation of multiple downstream signaling pathways, including the STAT pathway, the MAPK pathway, the PI3K pathway, and the protein kinase C (PKC) pathway (88, 89) (Figure 3). Signaling through the JAK/Stat pathway is initiated by phosphorylation of Stat proteins by JAK2. Subsequent dissociation of Stat proteins from GHR is followed by their dimerization and translocation to the nucleus to activate target gene expression. Activation of Stat5A and B isoforms plays an important role in mammary gland development and results in transcriptional activation of multiple target genes including insulin-like growth factor 1 (IGF-1) and serum protease inhibitor 2.1 (spi2.1) (10, 90).
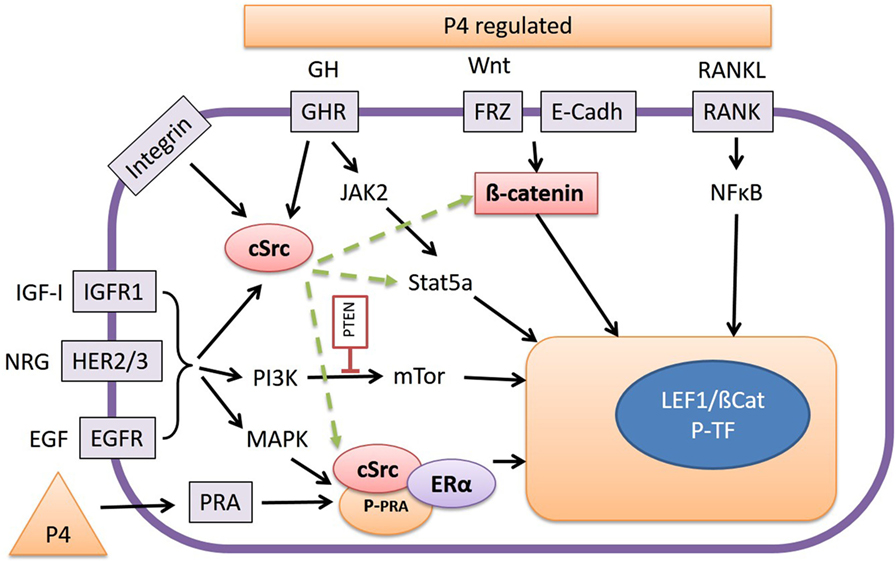
Figure 3. The release of growth hormone (GH), Wnt, or RANKL, as stimulated by progesterone (P4), promotes interaction with various signal transduction pathways. RANKL stimulates the NFκB pathway, and Wnt proteins stabilize cytoplasmic β-catenin, which under basal conditions is bound to E-Cadherin, and both activate Wnt target genes by forming a complex with T-cell transcription factors such as LEF-1. GH signals through the Janus kinase 2/Stat5a pathway but also by activation of cSrc. Inhibition of cSrc with the inhibitor Dasatinib results in inhibition of Wnt reporter activity in our canine cell lines by a yet unknown mechanism. As well as GH, integrins, type-1 IGF receptors (IGFR1), and the HER-family of tyrosine kinase receptors may also be involved in cSrc activation or stimulation of phosphatidyl-3-kinase or mitogen-activated protein kinase pathways. The high Wnt activity in our cell lines is associated with elevated expression of the HER receptors. Finally cSrc may also form a ternary complex with P4 receptor-A (PRA) and E2 receptor-α (ERα) resulting in activation/phosphorylation of nuclear transcription factors.
In the canine CMT-U27 cell line, a GHR-mediated growth effect was found as a consequence of increased cell survival by increased p-ERK 1/2 expression. This resulted in proliferation and an increased number of cells in the S and G2M phase (91). When GH binds to the GHR the tyrosine kinase JAK2 is activated, and this activated JAK2 phosphorylates various signaling mediators with the most important being p-ERK1/2 (MAPK). Activated MAPK signals via myelocytomatosis viral oncogene homolog (MYC) and c-AMP response element-binding protein (CREB) in the nucleus to activate the transcription machinery (91). This transcription machinery is also stimulated by the PI3K/protein kinase B (AKT) pathway where GH indirectly signals via IGF-1 enhancing the PI3K/mammalian target of rapamycin (mTOR) pathway (Figure 3).
GH Signaling in Mammary Cancer
Expression of GH mRNA has also been observed in human breast cancer specimens (18, 92). This GH may interact with GHR-positive cells that are found in some 90% of human DCIS lesions and in 4–19% of normal breast epithelial cells (18). These GHR+ cells can form GH-dependent mammospheres, suggesting that GH may stimulate stem/progenitors to enter the cell cycle. The subpopulation of GHR+ cells also contains more progenitor cells with bipotent and myoepithelial differentiation potential compared to the GHR− cell population (18). Mammosphere-initiating cells from both human and mouse mammary gland can repopulate the cleared mammary fat pad in vivo (93) and both the same mammospheres and T47D human cancer cells can be stimulated by P4 to produce GH. We also found this to be the case in mammary tissue of the dog (18, 82) indicating that there is a link in the mammary gland between P4 stimulation, GH secretion, and GH/GHR activation with GH having a paracrine role (Figure 2). The effect is most likely paracrine because GHR and PR cells differ in EpCam activity in humans. PR-positive cells are Lin−, EpCam+, CD49f+, ALDH+, and HER3+, whereas GHR-positive cells are Lin−, EpCam+/−, CD49fhigh, and ALDH+ (18, 59). EpCam+ cells are luminal progenitor cells and EpCam− cells are stem and early progenitor cells (18, 59). There is also a discrepancy for GH and ALDH+ status. Only a minority of GH-producing cells were ALDH+ in a subset of HMECs, whereas 66% of the sorted ALDH+ cells were GHR+ and CD49high (18). Cell sorting by fluorescence-activated cell sorting, however, is not always representative of the regenerative potential of the selected cells. Cell characteristics and stem cell origin can only be reliably gained from lineage tracing analysis. Using lineage trace experiments, Van Keymeulen demonstrated that the mammary gland in the mouse contains different types of long-lived stem cells that are derived from independent precursors during or prior to the onset of puberty (6). Thus, mammary gland stem cells can have a luminal or basal origin, both having a Lin−, CD24+, CD29+ phenotype (59, 60). For stem cell maintenance, the Wnt/β-catenin signaling can be used (60).
Growth hormone is mainly produced in the differentiated pituitary or placental cells but can also be produced locally within the mammary gland under the influence of P4. GH is responsible for the expansion of mammary stem and progenitor cells when the mammary gland grows, during puberty, pregnancy, and lactation and also in the menstrual cycle when P4 levels are higher. Development of the mammary gland is cumulatively effected by P4 and GH, and this regulatory growth process can eventually lead to changed ratios between progenitor and differentiated cells. Progenitor cells, being more proliferative, have a higher risk of oncogenic hits (18). This undesirable consequence of GH signaling in the mammary gland is not only mediated as a direct effect on GHR-positive cells in the MaSC compartment, but also in the stromal compartment, where indirect GH effects are mediated by the synthesis and release of insulin-like growth factor-I. The PR-positive cells within the mammary epithelium act as sensor cells for P4 signaling and stimulate the local production of not only GH but also RANKL and Wnt4 (17).
Canonical Wnt Signaling
The Wnt signaling pathway is involved in regulation of several processes including cell proliferation, cell polarity, differentiation, and morphogenesis even from very earliest stages of embryogenesis (94). Wnt proteins, in general, are thought to signal through four distinct pathways; the so-called canonical or Wnt/β-catenin pathway and the non-canonical pathways where calcium (Ca2+) acts as a second messenger. These Wnt/Ca2+ pathways involve the PKC, the planar cell polarity (PCP) pathway involving Jun N-terminal kinase (JNK) and a pathway involving protein kinase A (PKA) that functions in muscle myogenesis (95). In the non-canonical Wnt/Ca2+ pathway, binding of Wnt to Frizzled receptor (Fzd) results in an increase of intracellular Ca2+ and the activation of Ca2+/calmodulin-dependent protein kinase II (CAMKII) and PKC, resulting in activated nuclear factors that turn on gene transcription. This Wnt/Ca2+ pathway can interact with the canonical Wnt pathway. CAMKII and PKC are able to phosphorylate β-catenin, thereby serving as a negative regulatory component of the canonical Wnt pathway (96–98). In the PCP pathway, the disheveled protein (DVL1) is recruited to the plasma membrane after binding of Wnt proteins to the Fzd. DVL1 activates small guanosine triphosphatases (GTPases), such as Ras homolog gene family member A (Rho-A) and cell division cycle 42. These GTPases activate Rho-associated kinase and JNK, leading to the transcription of target genes. This pathway functions also in regulation of cell movements and adherence (96). The third non-canonical pathway involves Fzd signaling via heterotrimeric guanosine triphosphate binding proteins (G proteins). G proteins activate phosphatidylinositol signaling via PKA and thus transcription activation (95).
The most important mediator of canonical Wnt signaling is β-catenin (Figure 3). In a cell lacking canonical Wnt activity, the vast majority of β-catenin protein is bound to E-cadherin at the cell membrane where it provides a link between the actin cytoskeleton and cell–cell junctions. The so-called β-catenin destruction complex rapidly degrades the remaining β-catenin in the cytoplasm. In this complex, proteins such as interacting protein (Axin1 and Axin2) and adenomatous polyposis coli (APC) act as scaffolds to bring β-catenin in association with casein kinase I (CK1) and glycogen synthase kinase 3 beta (GSK-3β), which phosphorylate β-catenin at Ser/Thr residues. This phosphorylated β-catenin is then targeted by the ubiquitination complex, resulting in subsequent proteosomal degradation (99). When the canonical Wnt pathway is activated through binding of Wnt ligands to Fzd and low density lipoprotein-related protein 5/6 (LRP5/6) coreceptors, the β-catenin destruction complex dissociates. As a consequence, β-catenin is dephosphorylated by protein phosphatase, resulting in its cytoplasmic stabilization. Stabilized β-catenin is then able to translocate to the nucleus where, in association with T-cell transcription factor/lymphoid enhancer-binding factor 1 transcription factors it regulates expression of target genes (100, 101). Recently, additional mediators of canonical Wnt signal have been identified that modulate stability of Fzd/LRP receptor complex on the cell membrane (i.e., Lgr5 as receptor with R-spondin ligand proteins), thereby enhancing the Wnt ligand signal (102). Experiments with antibodies show that these antibodies bind non-overlapping regions of LRP6 protein, suggesting that LRP6 contains separate binding sites for different classes of Wnt proteins (103). Axin binds preferably to the cytoplasmic tail of LRP6 that is phosphorylated through GSK3 and CK1γ (104). The Wnt-induced LRP6 phosphorylation brings Axin close to the Dvl protein, resulting in degradation of Axin by Dvl. This receptor protein phosphorylation therefore decreases the signal transduction pathway instead of amplifying it as would normally be expected (105). Wnt signaling appears to occur predominantly between cells that are close to each other, for example, in adult stem cell niches thus Wnt signals mediate close range signaling (106). To tightly regulate the canonical pathway activity, cells also express a number of Wnt antagonists, such as Dikkopf (Dkk) and secreted Frizzled-related protein (sFRP) that prevent Wnt proteins from binding to Fzd or LRP5/6. In addition, activation of the canonical Wnt pathway provides a negative feedback through stimulation of Axin2 expression (104, 107, 108).
Wnt Signaling in Mammary Cancer
In the mammary gland, canonical Wnt activity is essential for both embryonic and postnatal development (109). During puberty and pregnancy, Wnt activity has been linked to P4 signaling. P4 has been shown to promote Wnt ligand expression (especially Wnt4) and to activate the downstream signaling in human, mice, and dogs (110, 111). The relevance of Wnt4 is confirmed by the phenotype of conditional knockouts of mammary Wnt4 expression in mice that have impaired ductal side branching, while overexpression of stabilized β-catenin in luminal epithelium results in precocious lobulo-alveolar development, alveologenesis, and neoplasia (112). In addition to its role in normal mammary gland development, deregulation of the canonical Wnt pathway is often associated with tumorigenesis (101). Oncogenic properties of Wnt proteins were first evident in the mammary gland, as Wnt1 and Wnt3 ligand were initially identified as insertion sites for a MMTV (113). Moreover, in human breast cancer, around 60% of examined clinical samples were shown to have elevated levels of nuclear and/or cytoplasmic β-catenin, suggesting an active signaling (114). In addition, in dogs exposed to prolonged MPA, in vivo strong upregulation of Wnt4 mRNA is also found. In dogs with spontaneous mammary carcinomas, we found a 6.8-fold induction of Wnt7a and also 2- to 3-fold changes in Wnt3, 4, 5a, and 5b mRNA (115). In both cases Wnt target genes such as cyclin D1, survivin, axin2, and cMyc were induced. In general, the same common activated pathways were found in canine carcinomas when compared to published human and mouse data (106, 116).
In most human tumors, constitutive activity of the canonical Wnt pathway was shown to be a consequence of mutations in APC or β-catenin (101). By contrast, such mutations are found only rarely in breast cancer (117). Rather, alternative explanations for the canonical pathway activation have been proposed for mammary tumors, including (1) mutations in other components of the pathway, (2) overexpression of Wnt ligands and other activators, (3) loss or downregulation of the antagonists, such as sFRP1, and (4) cross regulation by other deregulated pathways, such as epidermal growth factor (EGF), phosphatase and tensin homolog (PTEN), or tumor protein 53 (p53) signaling (117, 118). Information about canonical Wnt activity in canine mammary tumors is limited. Deregulation of the pathway has been proposed based on elevated β-catenin immunostaining and pathway analysis associated with molecular profiling of normal and tumorous tissue (19, 111, 119–121). None of the studies have, however, quantitatively assessed the activation of canonical Wnt signaling in canine mammary tumors or the underlying mechanism of its activation.
Wnt drives the formation of cells with CSC properties by regulating the expansion and proliferation of MaSCs (60, 122) and Wnt/β-catenin contribute to tumor progression and metastasis and related to all molecular subtypes of invasive breast cancer with a poor clinical outcome (123). The Wnt pathway is therefore a critical component in breast cancer development and a possible therapeutic target across cancer sub types. Wntless (WLS) appears to be required for the release of all Wnt proteins in both the canonical and non-canonical Wnt pathways. WLS is highly overexpressed in 48% in all subtypes of human breast cancer cases, and there is a strong correlation to HER2 overexpression (124). Together with upregulated Wnt signaling, our canine cell lines also have upregulated HER signaling (125). It remains to be shown whether the human and canine high Wnt- and HER-expressing cells point to a comparable stem cell.
HER and the Interaction of Pathways
As previously stated, we found a remarkable association between high intrinsic Wnt activity and high mRNA expression of the four members of the epidermal growth factor receptor (EGFR) family (126) of membrane receptor tyrosine kinases, commonly referred to as ErbB(1–4) or HER(1–4). A main pathway that is activated by the HER family is the PI3K pathway (Figure 4). Activated PI3K leads to phosphorylation of phosphatidylinositol 4,5 bisphosphate to phosphatidylinositol 3,4,4-triphosphate (PIP3). PIP3 is an important transducer of activating downstream components, such as AKT, which is then able to phosphorylate other targets, including the mTOR complex, a key regulator of cell growth, proliferation, survival, and protein synthesis. The mTOR pathway is frequently upregulated in breast cancer specimens. The pathway is stimulated by activation of HER signaling or by the type-1 IGF1-R. The HER2 receptor lacks a ligand-binding domain, whereas HER3 lacks tyrosine kinase activity. Nevertheless, HER2/3 heterodimers are among the strongest activators of the mTOR pathway (127, 128). PI3K/mTOR hyperactivation is linked to resistance to endocrine therapy of breast cancer in humans and dogs, resulting in tumor recurrence (129, 130). Dogs and humans have similarities in breast cancer tumor types and in the distribution of the tumor types. Approximately two-thirds of human breast cancers are steroid hormone receptor (ER/PR) positive and treated with combinations of selective estrogen receptor modulators, gonadotropin-releasing hormone agonists, and/or aromatase inhibitors (131–133), either alone or in combination with third-generation cytotoxic or biological therapies (134, 135). Of the hormone-receptor negative tumors, some 20% are characterized by HER2 amplification and overexpression leading to a dependency on the family of EGF or related growth factors. These breast cancers are treated with various HER2 inhibitors, both therapeutic antibodies and specific tyrosine kinase inhibitors (136–138). HER2 is a truncated receptor that does not bind ligands but has an active tyrosine kinase domain. Homodimers of HER2 cause weak signaling that is greatly enhanced after dimerization with HER1, 3, or 4 (139). In particular, after dimerization with HER3, which binds ligand but has no intrinsic kinase activity, the HER2/HER3 heterodimer is able to stimulate breast tumor cell proliferation (140, 141). The HER2 receptor is stabilized by heat-shock protein 90, but this also limits the capacity of HER2 to recruit HER3 to an active dimer (142). Activated HER3 receptors stimulate the MAPK and PI3K/AKT/mTOR signal transduction pathways (143). The latter is antagonized by PTEN, which dephosphorylates phosphoinositides generated by PI3K and also dephosphorylates focal adhesion kinase (FAK) and thus inhibits cell migration and integrin-mediated cell spreading. Loss of PTEN function by mutation or epigenetic silencing has been found frequently in various cancers including breast cancers (144). Severe PTEN deficiency has been associated with resistance to anti-HER2 therapy but confers susceptibility to inhibitors of the PI3K/AKT/mTOR pathway (145–147). Activation of the PI3K/AKT/mTOR pathway has been associated with resistance to hormone therapy that may be restored using selective mTOR inhibitors as Everolimus (148, 149). Everolimus therapy has recently commenced in human patients with ER+ breast cancer (150). Studies have shown that hormonal therapy combined of Everolimus leads to an increased survival (151, 152). In our in vitro studies with canine cell lines with high basal Wnt activity, a possible negative side effect of Everolimus has been found. Unexpectedly, inhibition of the PI3K/mTOR pathway with Everolimus, or a dual PI3K/mTOR inhibitor, stimulated the Wnt activity measured by reporter constructs. So, although Everolimus inhibited proliferation of the canine mammary cancer cells, it stimulated Wnt activity and thereby potentially their metastatic capacity and recruitment of stem cells. Recently, a new model of EGF receptor signaling in mammary cells was presented. This confirmed the involvement of MAPK, PI3K/AKT/mTOR, and STAT pathways, but due to complex interactions the involvement of cSrc, which is often overexpressed along with the HERs, remained unclear (153).
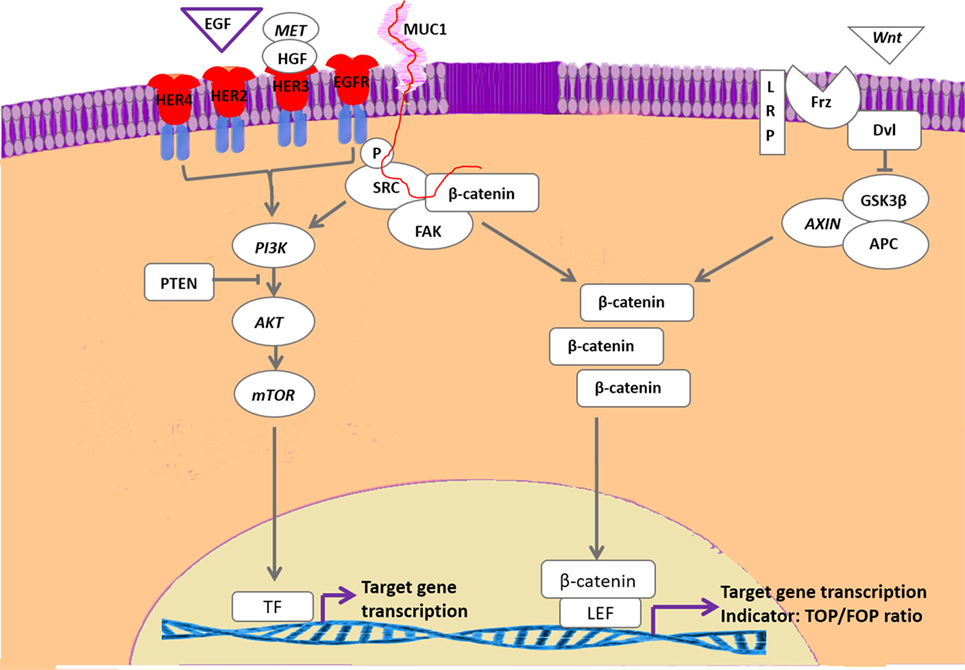
Figure 4. Interaction of HER-activated pathways with canonical Wnt signaling. With the exception of human epidermal growth factor 2, the family of epidermal growth factor receptor-related receptors is activated by epidermal growth factor (EGF) or EGF-related proteins as well as by cMet/HGF. The activated tyrosine kinase activity results in activation of the phosphatidyl-3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway that is downregulated by PTEN activity. Inactivating PTEN mutations are frequently found in mammary carcinomas. The HER pathway may also phosphorylate and activate Src, resulting in stabilization of cytoplasmic β-catenin. Activation of the Wnt pathway also results in stabilization of cytoplasmic β-catenin that stimulates gene transcription by forming a complex with T-cell transcription factors such as lymphoid enhancer-binding factor 1. Both pathways are associated with development of therapy resistance. In our cell lines with high basal Wnt activity, inhibition of the PI3K/mTOR resulted in further increased Wnt activity, whereas SRC inhibition strongly inhibited Wnt activity.
Apart from activation of MAPK and PI3K pathways, it has been proposed that HER dimers may also transduce signals through cSrc/FAK complexes (154). These complexes interact with the extracellular matrix, cell migration signals, integrins, mucin 1 (Muc1), and β-catenin via the Wnt pathway. The Wnt activity decreases when our cells are treated with a FAK inhibitor (125). This Focal-adhesion kinase canonical pathway is also related to resistance to estrogen deprivation and cSrc in ER+ breast cancer. Dasatinib, a pan-Src inhibitor, has shown a mixed success in clinical trials of ER+ patients. Recently, experiments in human MCF-7 cells, modeling resistance to aromatase inhibitors and tamoxifen showed that dasatinib plus endocrine therapy gave a dose-dependent decrease in proliferation and re-sensitized them to the endocrine therapy. Dasatinib also caused an inhibition of the AKT and ERK1/2 downstream pathway and inhibition of cSrc also showed a decrease in cell migration. These data suggest that cSrc produced the endocrine resistant cell in different ways (155). In our canine carcinoma cell lines with a highly upregulated Wnt signaling, we also found a decrease in cell proliferation when we treated these cells with a cSrc inhibitor but more importantly we found that the upregulated Wnt signaling that followed Everolimus treatment was dose-dependently reduced (125) (Figure 4). The fact that breast cancer tumors become therapy resistant is still the most important problem in breast cancer treatment. Major factors associated with resistance are (1) overexpression of EGFR, HER2, insulin-like growth factor 1 receptor (2), loss of ER expression (3), changes in extracellular matrix (4), mutations in PI3K (PTEN) or MAPK pathways, and (5) EMT and CSC processes (Wnt stimulated). However, clinical trials with inhibition of EGF/IGF signaling or PI3K activity are either suboptimal or even disappointing with respect to inhibition of breast cancer progression (156–161). Renoir et al. showed the relation of breast cancer and extra nuclear ERα with PI3K and cSrc. ERα forms complexes with PI3K and cSrc making these pathways potential targets for therapeutic intervention (162).
Conclusion
The heterogeneity of the mammary gland and various breast cancer tumor subtypes make this disease hard to predict and to treat. There is no single-standard treatment, and no way to determine which tumor will respond to therapy even when a predictive biomarker is present. Because the mouse has a different lobular alveolar structure and cannot form spontaneous breast tumors the dog may be an attractive model to study hormone dependence, Wnt and GH signaling. The GH, P4, and Wnt pathways are major players in the development of mammary gland tumors, and this review shows that there are important key players between these pathways and others such as HER overexpression. In vitro studies in canine mammary cancer cell lines show that cSrc influences these pathways and so combination therapies of cSrc and mTOR inhibitors, or direct targeting of P4 or GH signaling are therefore possible new targets for therapeutic interventions.
Author Contributions
ET-S, AG, and JM wrote and approved the final review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Liu D, Xiong H, Ellis AE, Northrup NC, Rodriguez CO Jr, O’Regan RM, et al. Molecular homology and difference between spontaneous canine mammary cancer and human breast cancer. Cancer Res (2014) 74:5045–56. doi:10.1158/0008-5472.CAN-14-0392
2. Visvader JE, Smith GH. Murine mammary epithelial stem cells: discovery, function, and current status. Cold Spring Harb Perspect Biol (2011) 3. doi:10.1101/cshperspect.a004879
3. Oakes SR, Gallego-Ortega D, Ormandy CJ. The mammary cellular hierarchy and breast cancer. Cell Mol Life Sci (2014) 71:4301–24. doi:10.1007/s00018-014-1674-4
4. Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev (2014) 28:1143–58. doi:10.1101/gad.242511.114
5. Rangel MC, Bertolette D, Castro NP, Klauzinska M, Cuttitta F, Salomon DS. Developmental signaling pathways regulating mammary stem cells and contributing to the etiology of triple-negative breast cancer. Breast Cancer Res Treat (2016) 156:211–26. doi:10.1007/s10549-016-3746-7
6. Van Keymeulen A, Rocha AS, Ousset M, Beck B, Bouvencourt G, Rock J, et al. Distinct stem cells contribute to mammary gland development and maintenance. Nature (2011) 479:189–93. doi:10.1038/nature10573
7. Fendrick JL, Raafat AM, Haslam SZ. Mammary gland growth and development from the postnatal period to postmenopause: ovarian steroid receptor ontogeny and regulation in the mouse. J Mammary Gland Biol Neoplasia (1998) 3:7–22. doi:10.1023/A:1018766000275
8. Soyal S, Ismail PM, Li J, Mulac-Jericevic B, Conneely OM, Lydon JP. Progesterone’s role in mammary gland development and tumorigenesis as disclosed by experimental mouse genetics. Breast Cancer Res (2002) 4:191–6. doi:10.1186/bcr451
9. Conneely OM, Mulac-Jericevic B, Lydon JP. Progesterone-dependent regulation of female reproductive activity by two distinct progesterone receptor isoforms. Steroids (2003) 68:771–8. doi:10.1016/S0039-128X(03)00126-0
10. Hennighausen L, Robinson GW, Wagner KU, Liu X. Developing a mammary gland is a stat affair. J Mammary Gland Biol Neoplasia (1997) 2:365–72. doi:10.1023/A:1026347313096
11. Proietti CJ, Rosemblit C, Beguelin W, Rivas MA, Diaz Flaque MC, Charreau EH, et al. Activation of Stat3 by heregulin/ErbB-2 through the co-option of progesterone receptor signaling drives breast cancer growth. Mol Cell Biol (2009) 29:1249–65. doi:10.1128/MCB.00853-08
12. Furth PA, Nakles RE, Millman S, Diaz-Cruz ES, Cabrera MC. Signal transducer and activator of transcription 5 as a key signaling pathway in normal mammary gland developmental biology and breast cancer. Breast Cancer Res (2011) 13:220. doi:10.1186/bcr2921
13. Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci U S A (2003) 100:9744–9. doi:10.1073/pnas.1732707100
14. Mukherjee A, Soyal SM, Li J, Ying Y, He B, DeMayo FJ, et al. Targeting RANKL to a specific subset of murine mammary epithelial cells induces ordered branching morphogenesis and alveologenesis in the absence of progesterone receptor expression. FASEB J (2010) 24:4408–19. doi:10.1096/fj.10-157982
15. Brisken C, Heineman A, Chavarria T, Elenbaas B, Tan J, Dey SK, et al. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev (2000) 14:650–4. doi:10.1101/gad.14.6.650
16. van Garderen E, de Wit M, Voorhout WF, Rutteman GR, Mol JA, Nederbragt H, et al. Expression of growth hormone in canine mammary tissue and mammary tumors. Evidence for a potential autocrine/paracrine stimulatory loop. Am J Pathol (1997) 150:1037–47. doi:10.1101/gad.14.6.650
17. Brisken C, Ataca D. Endocrine hormones and local signals during the development of the mouse mammary gland. Wiley Interdiscip Rev Dev Biol (2015) 4:181–95. doi:10.1002/wdev.172
18. Lombardi S, Honeth G, Ginestier C, Shinomiya I, Marlow R, Buchupalli B, et al. Growth hormone is secreted by normal breast epithelium upon progesterone stimulation and increases proliferation of stem/progenitor cells. Stem Cell Reports (2014) 2:780–93. doi:10.1016/j.stemcr.2014.05.005
19. Uva P, Aurisicchio L, Watters J, Loboda A, Kulkarni A, Castle J, et al. Comparative expression pathway analysis of human and canine mammary tumors. BMC Genomics (2009) 10:135. doi:10.1186/1471-2164-10-135
20. Chlebowski RT, Hendrix SL, Langer RD, Stefanick ML, Gass M, Lane D, et al. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the Women’s Health Initiative randomized trial. JAMA (2003) 289:3243–53. doi:10.1001/jama.289.24.3243
21. Beatson GT. On the treatment of the inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment with illustrative cases. Lancet (1896) 2:104–7. doi:10.1016/S0140-6736(01)72307-0
22. Huggins C, Yang NC. Induction and extinction of mammary cancer. A striking effect of hydrocarbons permits analysis of mechanisms of causes and cure of breast cancer. Science (1962) 137:257–62. doi:10.1126/science.137.3526.257
23. McGuire WL, Horwitz KB. A role for progesterone in breast cancer. Ann N Y Acad Sci (1977) 286:90–100. doi:10.1111/j.1749-6632.1977.tb29408.x
24. MacMahon B, Cole P, Lin TM, Lowe CR, Mirra AP, Ravnihar B, et al. Age at first birth and breast cancer risk. Bull World Health Organ (1970) 43:209–21.
25. Anderson KN, Schwab RB, Martinez ME. Reproductive risk factors and breast cancer subtypes: a review of the literature. Breast Cancer Res Treat (2014) 144:1–10. doi:10.1007/s10549-014-2852-7
26. Islami F, Liu Y, Jemal A, Zhou J, Weiderpass E, Colditz G, et al. Breastfeeding and breast cancer risk by receptor status – a systematic review and meta-analysis. Ann Oncol (2015) 26:2398–407. doi:10.1093/annonc/mdv379
27. Colditz GA, Rosner BA, Chen WY, Holmes MD, Hankinson SE. Risk factors for breast cancer according to estrogen and progesterone receptor status. J Natl Cancer Inst (2004) 96:218–28. doi:10.1093/jnci/djh025
28. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA (2002) 288:321–33. doi:10.1001/jama.288.3.321
29. Stefanick ML. Risk-benefit profiles of raloxifene for women. N Engl J Med (2006) 355:190–2. doi:10.1056/NEJMe068120
30. Blank EW, Wong PY, Lakshmanaswamy R, Guzman R, Nandi S. Both ovarian hormones estrogen and progesterone are necessary for hormonal mammary carcinogenesis in ovariectomized ACI rats. Proc Natl Acad Sci U S A (2008) 105:3527–32. doi:10.1073/pnas.0710535105
31. Huber JC, Ott J. The dialectic role of progesterone. Maturitas (2009) 62:326–9. doi:10.1016/j.maturitas.2008.12.009
32. Aupperlee MD, Haslam SZ. Differential hormonal regulation and function of progesterone receptor isoforms in normal adult mouse mammary gland. Endocrinology (2007) 148:2290–300. doi:10.1210/en.2006-1721
33. Mote PA, Arnett-Mansfield RL, Gava N, deFazio A, Mulac-Jericevic B, Conneely OM, et al. Overlapping and distinct expression of progesterone receptors A and B in mouse uterus and mammary gland during the estrous cycle. Endocrinology (2006) 147:5503–12. doi:10.1210/en.2006-0040
34. Mote PA, Bartow S, Tran N, Clarke CL. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat (2002) 72:163–72. doi:10.1023/A:1014820500738
35. Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem (2002) 277:5209–18. doi:10.1074/jbc.M110090200
36. Ballare C, Uhrig M, Bechtold T, Sancho E, Di Domenico M, Migliaccio A, et al. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Mol Cell Biol (2003) 23:1994–2008. doi:10.1128/MCB.23.6.1994-2008.2003
37. Jacobsen BM, Horwitz KB. Progesterone receptors, their isoforms and progesterone regulated transcription. Mol Cell Endocrinol (2012) 357(1–2):18–29. doi:10.1016/j.mce.2011.09.016
38. Sartorius CA, Melville MY, Hovland AR, Tung L, Takimoto GS, Horwitz KB. A third transactivation function (AF3) of human progesterone receptors located in the unique N-terminal segment of the B-isoform. Mol Endocrinol (1994) 8:1347–60. doi:10.1210/mend.8.10.7854352
39. Takimoto GS, Tung L, Abdel-Hafiz H, Abel MG, Sartorius CA, Richer JK, et al. Functional properties of the N-terminal region of progesterone receptors and their mechanistic relationship to structure. J Steroid Biochem Mol Biol (2003) 85:209–19. doi:10.1016/S0960-0760(03)00197-3
40. Chen C, Opazo JC, Erez O, Uddin M, Santolaya-Forgas J, Goodman M, et al. The human progesterone receptor shows evidence of adaptive evolution associated with its ability to act as a transcription factor. Mol Phylogenet Evol (2008) 47:637–49. doi:10.1016/j.ympev.2007.12.026
41. Gracanin A, van Wolferen ME, Sartorius CA, Brenkman AB, Schoonen WG, Mol JA. Canid progesterone receptors lack activation function 3 domain-dependent activity. Endocrinology (2012) 153:6104–13. doi:10.1210/en.2012-1793
42. Gracanin A, Voorwald FA, van Wolferen M, Timmermans-Sprang E, Mol JA. Marginal activity of progesterone receptor B (PR-B) in dogs but high incidence of mammary cancer. J Steroid Biochem Mol Biol (2014) 144(Pt B):492–9. doi:10.1016/j.jsbmb.2014.08.016
43. Jacobsen BM, Schittone SA, Richer JK, Horwitz KB. Progesterone-independent effects of human progesterone receptors (PRs) in estrogen receptor-positive breast cancer: PR isoform-specific gene regulation and tumor biology. Mol Endocrinol (2005) 19:574–87. doi:10.1210/me.2004-0287
44. Graham JD, Yager ML, Hill HD, Byth K, O’Neill GM, Clarke CL. Altered progesterone receptor isoform expression remodels progestin responsiveness of breast cancer cells. Mol Endocrinol (2005) 19:2713–35. doi:10.1210/me.2005-0126
45. Kariagina A, Aupperlee MD, Haslam SZ. Progesterone receptor isoform functions in normal breast development and breast cancer. Crit Rev Eukaryot Gene Expr (2008) 18:11–33. doi:10.1615/CritRevEukarGeneExpr.v18.i1.20
46. Mote PA, Leary JA, Avery KA, Sandelin K, Chenevix-Trench G, Kirk JA, et al. Germ-line mutations in BRCA1 or BRCA2 in the normal breast are associated with altered expression of estrogen-responsive proteins and the predominance of progesterone receptor A. Genes Chromosomes Cancer (2004) 39:236–48. doi:10.1002/gcc.10321
47. Katiyar P, Ma Y, Riegel A, Fan S, Rosen EM. Mechanism of BRCA1-mediated inhibition of progesterone receptor transcriptional activity. Mol Endocrinol (2009) 23:1135–46. doi:10.1210/me.2008-0347
48. Katiyar P, Ma Y, Fan S, Pestell RG, Furth PA, Rosen EM. Regulation of progesterone receptor signaling by BRCA1 in mammary cancer. Nucl Recept Signal (2006) 4:e006. doi:10.1621/nrs.04006
49. Calvo V, Beato M. BRCA1 counteracts progesterone action by ubiquitination leading to progesterone receptor degradation and epigenetic silencing of target promoters. Cancer Res (2011) 71:3422–31. doi:10.1158/0008-5472.CAN-10-3670
50. Burga LN, Hu H, Juvekar A, Tung NM, Troyan SL, Hofstatter EW, et al. Loss of BRCA1 leads to an increase in epidermal growth factor receptor expression in mammary epithelial cells, and epidermal growth factor receptor inhibition prevents estrogen receptor-negative cancers in BRCA1-mutant mice. Breast Cancer Res (2011) 13:R30. doi:10.1186/bcr2850
51. Brisken C, Duss S. Stem cells and the stem cell niche in the breast: an integrated hormonal and developmental perspective. Stem Cell Rev (2007) 3:147–56. doi:10.1007/s12015-007-0019-1
52. Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature (2010) 465:803–7. doi:10.1038/nature09091
53. Eden JA. Breast cancer, stem cells and sex hormones: part 1. The impact of fetal life and infancy. Maturitas (2010) 67:117–20. doi:10.1016/j.maturitas.2010.05.005
54. Axlund SD, Sartorius CA. Progesterone regulation of stem and progenitor cells in normal and malignant breast. Mol Cell Endocrinol (2012) 357(1–2):71–9. doi:10.1016/j.mce.2011.09.021
55. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A (2003) 100:3983–8. doi:10.1073/pnas.0530291100
56. Liu B, Ordonez-Ercan D, Fan Z, Edgerton SM, Yang X, Thor AD. Downregulation of erbB3 abrogates erbB2-mediated tamoxifen resistance in breast cancer cells. Int J Cancer (2007) 120:1874–82. doi:10.1002/ijc.22423
57. Michishita M, Akiyoshi R, Yoshimura H, Katsumoto T, Ichikawa H, Ohkusu-Tsukada K, et al. Characterization of spheres derived from canine mammary gland adenocarcinoma cell lines. Res Vet Sci (2011) 91:254–60. doi:10.1016/j.rvsc.2010.11.016
58. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi:10.1016/j.cell.2008.03.027
59. Tornillo G, Smalley MJ. ERrrr… where are the progenitors? Hormone receptors and mammary cell heterogeneity. J Mammary Gland Biol Neoplasia (2015) 20:63–73. doi:10.1007/s10911-015-9336-1
60. van Amerongen R, Bowman AN, Nusse R. Developmental stage and time dictate the fate of Wnt/beta-catenin-responsive stem cells in the mammary gland. Cell Stem Cell (2012) 11:387–400. doi:10.1016/j.stem.2012.05.023
61. Stingl J, Raouf A, Eirew P, Eaves CJ. Deciphering the mammary epithelial cell hierarchy. Cell Cycle (2006) 5:1519–22. doi:10.4161/cc.5.14.2983
62. Stingl J. Detection and analysis of mammary gland stem cells. J Pathol (2009) 217:229–41. doi:10.1002/path.2457
63. Arendt LM, Keller PJ, Skibinski A, Goncalves K, Naber SP, Buchsbaum RJ, et al. Anatomical localization of progenitor cells in human breast tissue reveals enrichment of uncommitted cells within immature lobules. Breast Cancer Res (2014) 16:453. doi:10.1186/s13058-014-0453-3
64. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell (2007) 1:555–67. doi:10.1016/j.stem.2007.08.014
65. Liu S, Ginestier C, Charafe-Jauffret E, Foco H, Kleer CG, Merajver SD, et al. BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci U S A (2008) 105:1680–5. doi:10.1073/pnas.0711613105
66. Ferletta M, Grawe J, Hellmen E. Canine mammary tumors contain cancer stem-like cells and form spheroids with an embryonic stem cell signature. Int J Dev Biol (2011) 55:791–9. doi:10.1387/ijdb.113363mf
67. Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, et al. Control of mammary stem cell function by steroid hormone signalling. Nature (2010) 465:798–802. doi:10.1038/nature09027
68. Beleut M, Rajaram RD, Caikovski M, Ayyanan A, Germano D, Choi Y, et al. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc Natl Acad Sci U S A (2010) 107:2989–94. doi:10.1073/pnas.0915148107
69. Lee HJ, Gallego-Ortega D, Ledger A, Schramek D, Joshi P, Szwarc MM, et al. Progesterone drives mammary secretory differentiation via RankL-mediated induction of Elf5 in luminal progenitor cells. Development (2013) 140:1397–401. doi:10.1242/dev.088948
70. Forster N, Saladi SV, van Bragt M, Sfondouris ME, Jones FE, Li Z, et al. Basal cell signaling by p63 controls luminal progenitor function and lactation via NRG1. Dev Cell (2014) 28:147–60. doi:10.1016/j.devcel.2013.11.019
71. Alexander CM, Joshi PA, Khokha R. Fully interlocking: a story of teamwork among breast epithelial cells. Dev Cell (2014) 28:114–5. doi:10.1016/j.devcel.2014.01.011
72. Swanson SM, Unterman TG. The growth hormone-deficient spontaneous Dwarf rat is resistant to chemically induced mammary carcinogenesis. Carcinogenesis (2002) 23:977–82. doi:10.1093/carcin/23.6.977
73. Laron Z, Klinger B. Laron syndrome: clinical features, molecular pathology and treatment. Horm Res (1994) 42:198–202. doi:10.1159/000184193
74. Russo J, Russo IH. The role of estrogen in the initiation of breast cancer. J Steroid Biochem Mol Biol (2006) 102:89–96. doi:10.1016/j.jsbmb.2006.09.004
75. Misdorp W. Canine mammary tumours: protective effect of late ovariectomy and stimulating effect of progestins. Vet Q (1988) 10:26–33. doi:10.1080/01652176.1988.9694142
76. Concannon PW, Spraker TR, Casey HW, Hansel W. Gross and histopathologic effects of medroxyprogesterone acetate and progesterone on the mammary glands of adult beagle bitches. Fertil Steril (1981) 36:373–87. doi:10.1016/S0015-0282(16)45741-9
77. Selman PJ, Mol JA, Rutteman GR, van Garderen E, Rijnberk A. Progestin-induced growth hormone excess in the dog originates in the mammary gland. Endocrinology (1994) 134:287–92. doi:10.1210/en.134.1.287
78. Mol JA, van Garderen E, Selman PJ, Wolfswinkel J, Rijinberk A, Rutteman GR. Growth hormone mRNA in mammary gland tumors of dogs and cats. J Clin Invest (1995) 95:2028–34. doi:10.1172/JCI117888
79. Lantinga-van Leeuwen IS, Oudshoorn M, Mol JA. Canine mammary growth hormone gene transcription initiates at the pituitary-specific start site in the absence of Pit-1. Mol Cell Endocrinol (1999) 150:121–8. doi:10.1016/S0303-7207(99)00010-6
80. Lantinga-van Leeuwen IS, Timmermans-Sprang EA, Mol JA. Cloning and characterization of the 5'-flanking region of the canine growth hormone gene. Mol Cell Endocrinol (2002) 197:133–41. doi:10.1016/S0303-7207(02)00257-5
81. Timmermans-Sprang EP, Rao NA, Mol JA. Transactivation of a growth hormone (GH) promoter-luciferase construct in canine mammary cells. Domest Anim Endocrinol (2008) 34:403–10. doi:10.1016/j.domaniend.2007.11.001
82. Lantinga-van Leeuwen IS, van Garderen E, Rutteman GR, Mol JA. Cloning and cellular localization of the canine progesterone receptor: co-localization with growth hormone in the mammary gland. J Steroid Biochem Mol Biol (2000) 75:219–28. doi:10.1016/S0960-0760(00)00173-4
83. Rao NA, van Wolferen ME, van den Ham R, van Leenen D, Groot Koerkamp MJ, Holstege FC, et al. cDNA microarray profiles of canine mammary tumour cell lines reveal deregulated pathways pertaining to their phenotype. Anim Genet (2008) 39:333–45. doi:10.1111/j.1365-2052.2008.01733.x
84. Krol M, Pawlowski KM, Skierski J, Rao NA, Hellmen E, Mol JA, et al. Transcriptomic profile of two canine mammary cancer cell lines with different proliferative and anti-apoptotic potential. J Physiol Pharmacol (2009) 60(Suppl 1):95–106.
85. Pawlowski KM, Krol M, Majewska A, Badowska-Kozakiewicz A, Mol JA, Malicka E, et al. Comparison of cellular and tissue transcriptional profiles in canine mammary tumor. J Physiol Pharmacol (2009) 60(Suppl 1):85–94.
86. Mukhina S, Mertani HC, Guo K, Lee KO, Gluckman PD, Lobie PE. Phenotypic conversion of human mammary carcinoma cells by autocrine human growth hormone. Proc Natl Acad Sci U S A (2004) 101:15166–71. doi:10.1073/pnas.0405881101
87. Gent J, van Kerkhof P, Roza M, Bu G, Strous GJ. Ligand-independent growth hormone receptor dimerization occurs in the endoplasmic reticulum and is required for ubiquitin system-dependent endocytosis. Proc Natl Acad Sci U S A (2002) 99:9858–63. doi:10.1073/pnas.152294299
88. Chilton BS, Hewetson A. Prolactin and growth hormone signaling. Curr Top Dev Biol (2005) 68:1–23. doi:10.1016/S0070-2153(05)68001-5
89. Waters MJ, Hoang HN, Fairlie DP, Pelekanos RA, Brown RJ. New insights into growth hormone action. J Mol Endocrinol (2006) 36:1–7. doi:10.1677/jme.1.01933
90. Rotwein P. Mapping the growth hormone – Stat5b – IGF-I transcriptional circuit. Trends Endocrinol Metab (2012) 23:186–93. doi:10.1016/j.tem.2012.01.001
91. Pawlowski KM, Popielarz D, Szyszko K, Gajewska M, Motyl T, Krol M. Growth hormone receptor (GHR) RNAi decreases proliferation and enhances apoptosis in CMT-U27 canine mammary carcinoma cell line. Vet Comp Oncol (2012) 10:2–15. doi:10.1111/j.1476-5829.2011.00269.x
92. Mol JA, Henzen-Logmans SC, Hageman P, Misdorp W, Blankenstein MA, Rijnberk A. Expression of the gene encoding growth hormone in the human mammary gland. J Clin Endocrinol Metab (1995) 80:3094–6. doi:10.1210/jcem.80.10.7559904
93. Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell (2009) 138:1083–95. doi:10.1016/j.cell.2009.06.048
94. Smalley MJ, Dale TC. Wnt signaling and mammary tumorigenesis. J Mammary Gland Biol Neoplasia (2001) 6:37–52. doi:10.1023/A:1009564431268
95. Chen AE, Ginty DD, Fan CM. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature (2005) 433:317–22. doi:10.1038/nature03126
96. Turashvili G, Bouchal J, Burkadze G, Kolar Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology (2006) 73:213–23. doi:10.1159/000098207
97. De A. Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Sin (Shanghai) (2011) 43:745–56. doi:10.1093/abbs/gmr079
98. Widelitz R. Wnt signaling through canonical and non-canonical pathways: recent progress. Growth Factors (2005) 23:111–6. doi:10.1080/08977190500125746
99. Kimelman D, Xu W. Beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene (2006) 25:7482–91. doi:10.1038/sj.onc.1210055
100. Hecht A, Kemler R. Curbing the nuclear activities of beta-catenin. Control over Wnt target gene expression. EMBO Rep (2000) 1:24–8. doi:10.1093/embo-reports/kvd012
101. Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta (2003) 1653:1–24. doi:10.1016/S0304-419X(03)00005-2
102. de Lau W, Peng WC, Gros P, Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev (2014) 28:305–16. doi:10.1101/gad.235473.113
103. Gong C, Yao H, Liu Q, Chen J, Shi J, Su F, et al. Markers of tumor-initiating cells predict chemoresistance in breast cancer. PLoS One (2010) 5:e15630. doi:10.1371/journal.pone.0015630
104. He X, Semenov M, Tamai K, Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development (2004) 131:1663–77. doi:10.1242/dev.01117
105. Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell (2010) 143:1136–48. doi:10.1016/j.cell.2010.11.034
106. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell (2012) 149:1192–205. doi:10.1016/j.cell.2012.05.012
107. Finch PW, He X, Kelley MJ, Uren A, Schaudies RP, Popescu NC, et al. Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc Natl Acad Sci U S A (1997) 94:6770–5. doi:10.1073/pnas.94.13.6770
108. Salahshor S, Woodgett JR. The links between axin and carcinogenesis. J Clin Pathol (2005) 58:225–36. doi:10.1136/jcp.2003.009506
109. Incassati A, Chandramouli A, Eelkema R, Cowin P. Key signaling nodes in mammary gland development and cancer: beta-catenin. Breast Cancer Res (2010) 12:213. doi:10.1186/bcr2723
110. Weber-Hall SJ, Phippard DJ, Niemeyer CC, Dale TC. Developmental and hormonal regulation of Wnt gene expression in the mouse mammary gland. Differentiation (1994) 57:205–14. doi:10.1046/j.1432-0436.1994.5730205.x
111. Rao NA, van Wolferen ME, Gracanin A, Bhatti SF, Krol M, Holstege FC, et al. Gene expression profiles of progestin-induced canine mammary hyperplasia and spontaneous mammary tumors. J Physiol Pharmacol (2009) 60(Suppl 1):73–84.
112. Imbert A, Eelkema R, Jordan S, Feiner H, Cowin P. Delta N89 beta-catenin induces precocious development, differentiation, and neoplasia in mammary gland. J Cell Biol (2001) 153:555–68. doi:10.1083/jcb.153.3.555
113. Nusse R, Brown A, Papkoff J, Scambler P, Shackleford G, McMahon A, et al. A new nomenclature for int-1 and related genes: the Wnt gene family. Cell (1991) 64:231. doi:10.1016/0092-8674(91)90633-A
114. Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, et al. Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A (2000) 97:4262–6. doi:10.1073/pnas.060025397
115. Gracanin A, Timmermans-Sprang EP, van Wolferen ME, Rao NA, Grizelj J, Vince S, et al. Ligand-independent canonical Wnt activity in canine mammary tumor cell lines associated with aberrant LEF1 expression. PLoS One (2014) 9:e98698. doi:10.1371/journal.pone.0098698
116. van Amerongen R, Fuerer C, Mizutani M, Nusse R. Wnt5a can both activate and repress Wnt/beta-catenin signaling during mouse embryonic development. Dev Biol (2012) 369:101–14. doi:10.1016/j.ydbio.2012.06.020
117. Howe LR, Brown AM. Wnt signaling and breast cancer. Cancer Biol Ther (2004) 3:36–41. doi:10.4161/cbt.3.1.561
118. Ugolini F, Charafe-Jauffret E, Bardou VJ, Geneix J, Adelaide J, Labat-Moleur F, et al. Wnt pathway and mammary carcinogenesis: loss of expression of candidate tumor suppressor gene SFRP1 in most invasive carcinomas except of the medullary type. Oncogene (2001) 20:5810–7. doi:10.1038/sj.onc.1204706
119. Restucci B, Maiolino P, Martano M, Esposito G, De Filippis D, Borzacchiello G, et al. Expression of beta-catenin, E-cadherin and APC in canine mammary tumors. Anticancer Res (2007) 27:3083–9.
120. De Matos AJ, Lopes CC, Faustino AM, Carvalheira JG, Rutteman GR, Gartner Mde F. E-cadherin, beta-catenin, invasion and lymph node metastases in canine malignant mammary tumours. APMIS (2007) 115:327–34. doi:10.1111/j.1600-0463.2007.apm_544.x
121. Gama A, Paredes J, Gartner F, Alves A, Schmitt F. Expression of E-cadherin, P-cadherin and beta-catenin in canine malignant mammary tumours in relation to clinicopathological parameters, proliferation and survival. Vet J (2008) 177:45–53. doi:10.1016/j.tvjl.2007.05.024
122. Skibinski A, Kuperwasser C. The origin of breast tumor heterogeneity. Oncogene (2015) 34:5309–16. doi:10.1038/onc.2014.475
123. Li S, Li S, Sun Y, Li L. The expression of beta-catenin in different subtypes of breast cancer and its clinical significance. Tumour Biol (2014) 35:7693–8. doi:10.1007/s13277-014-1975-0
124. Stewart J, James J, McCluggage GW, McQuaid S, Arthur K, Boyle D, et al. Analysis of wntless (WLS) expression in gastric, ovarian, and breast cancers reveals a strong association with HER2 overexpression. Mod Pathol (2015) 28:428–36. doi:10.1038/modpathol.2014.114
125. Timmermans-Sprang EP, Gracanin A, Mol JA. High basal Wnt signaling is further induced by PI3K/mTOR inhibition but sensitive to cSRC inhibition in mammary carcinoma cell lines with HER2/3 overexpression. BMC Cancer (2015) 15:545. doi:10.1186/s12885-015-1544-y
126. Wang Z, Longo PA, Tarrant MK, Kim K, Head S, Leahy DJ, et al. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat Struct Mol Biol (2011) 18:1388–93. doi:10.1038/nsmb.2168
127. Zhao M, Ramaswamy B. Mechanisms and therapeutic advances in the management of endocrine-resistant breast cancer. World J Clin Oncol (2014) 5:248–62. doi:10.5306/wjco.v5.i3.248
128. Tokunaga E, Hisamatsu Y, Tanaka K, Yamashita N, Saeki H, Oki E, et al. Molecular mechanisms regulating the hormone sensitivity of breast cancer. Cancer Sci (2014) 105:1377–83. doi:10.1111/cas.12521
129. Wu Y, Ginther C, Kim J, Mosher N, Chung S, Slamon D, et al. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res (2012) 10:1597–606. doi:10.1158/1541-7786.MCR-12-0155-T
130. Qiao L, Liang Y, Mira RR, Lu Y, Gu J, Zheng Q. Mammalian target of rapamycin (mTOR) inhibitors and combined chemotherapy in breast cancer: a meta-analysis of randomized controlled trials. Int J Clin Exp Med (2014) 7:3333–43.
131. Burstein HJ, Prestrud AA, Seidenfeld J, Anderson H, Buchholz TA, Davidson NE, et al. American Society of Clinical Oncology clinical practice guideline: update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Clin Oncol (2010) 28:3784–96. doi:10.1200/JCO.2009.26.3756
132. Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn HJ, et al. Strategies for subtypes – dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol (2011) 22:1736–47. doi:10.1093/annonc/mdr304
133. Polyak K, Metzger Filho O. SnapShot: breast cancer. Cancer Cell (2012) 22:562–562.e1. doi:10.1016/j.ccr.2012.06.021
134. Steelman LS, Navolanic PM, Franklin RA, Bonati A, Libra M, Stivala F, et al. Combining chemo-, hormonal and targeted therapies to treat breast cancer (review). Mol Med Rep (2008) 1:139–60. doi:10.3892/mmr.1.2.139
135. Moreno-Aspitia A. Neoadjuvant therapy in early-stage breast cancer. Crit Rev Oncol Hematol (2012) 82:187–99. doi:10.1016/j.critrevonc.2011.04.013
136. Awada A, Bozovic-Spasojevic I, Chow L. New therapies in HER2-positive breast cancer: a major step towards a cure of the disease? Cancer Treat Rev (2012) 38:494–504. doi:10.1016/j.ctrv.2012.01.001
137. Hurvitz SA, Hu Y, O’Brien N, Finn RS. Current approaches and future directions in the treatment of HER2-positive breast cancer. Cancer Treat Rev (2013) 39:219–29. doi:10.1016/j.ctrv.2012.04.008
138. Nielsen DL, Kumler I, Palshof JA, Andersson M. Efficacy of HER2-targeted therapy in metastatic breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Breast (2013) 22:1–12. doi:10.1016/j.breast.2012.09.008
139. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol (2001) 2:127–37. doi:10.1038/35052073
140. Stern DF. ERBB3/HER3 and ERBB2/HER2 duet in mammary development and breast cancer. J Mammary Gland Biol Neoplasia (2008) 13:215–23. doi:10.1007/s10911-008-9083-7
141. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF III, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A (2003) 100:8933–8. doi:10.1073/pnas.1537685100
142. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer (2012) 12:553–63. doi:10.1038/nrc3309
143. Campbell MR, Amin D, Moasser MM. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res (2010) 16:1373–83. doi:10.1158/1078-0432.CCR-09-1218
144. Ortega-Molina A, Serrano M. PTEN in cancer, metabolism, and aging. Trends Endocrinol Metab (2013) 24:184–9. doi:10.1016/j.tem.2012.11.002
145. DeGraffenried LA, Fulcher L, Friedrichs WE, Grunwald V, Ray RB, Hidalgo M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Ann Oncol (2004) 15:1510–6. doi:10.1093/annonc/mdh388
146. O’Regan R, Hawk NN. mTOR inhibition in breast cancer: unraveling the complex mechanisms of mTOR signal transduction and its clinical implications in therapy. Expert Opin Ther Targets (2011) 15:859–72. doi:10.1517/14728222.2011.575362
147. Zhang S, Yu D. PI(3)king apart PTEN’s role in cancer. Clin Cancer Res (2010) 16:4325–30. doi:10.1158/1078-0432.CCR-09-2990
148. Shtivelband MI. Everolimus in hormone receptor-positive advanced breast cancer: targeting receptor-based mechanisms of resistance. Breast (2013) 22:405–10. doi:10.1016/j.breast.2013.02.003
149. Fedele P, Calvani N, Marino A, Orlando L, Schiavone P, Quaranta A, et al. Targeted agents to reverse resistance to endocrine therapy in metastatic breast cancer: where are we now and where are we going? Crit Rev Oncol Hematol (2012) 84:243–51. doi:10.1016/j.critrevonc.2012.03.004
150. Nagaraj G, Ma C. Revisiting the estrogen receptor pathway and its role in endocrine therapy for postmenopausal women with estrogen receptor-positive metastatic breast cancer. Breast Cancer Res Treat (2015) 150:231–42. doi:10.1007/s10549-015-3316-4
151. Arena F. Clinical implications of recent studies using mTOR inhibitors to treat advanced hormone receptor-positive breast cancer. Cancer Manag Res (2014) 6:389–95. doi:10.2147/CMAR.S56802
152. Chia S, Gandhi S, Joy AA, Edwards S, Gorr M, Hopkins S, et al. Novel agents and associated toxicities of inhibitors of the pi3k/Akt/mtor pathway for the treatment of breast cancer. Curr Oncol (2015) 22:33–48. doi:10.3747/co.22.2393
153. Helikar T, Kochi N, Kowal B, Dimri M, Naramura M, Raja SM, et al. A comprehensive, multi-scale dynamical model of ErbB receptor signal transduction in human mammary epithelial cells. PLoS One (2013) 8:e61757. doi:10.1371/journal.pone.0061757
154. Al Masri A, Gendler SJ. Muc1 affects c-Src signaling in PyV MT-induced mammary tumorigenesis. Oncogene (2005) 24:5799–808. doi:10.1038/sj.onc.1208738
155. Guest SK, Ribas R, Pancholi S, Nikitorowicz-Buniak J, Simigdala N, Dowsett M, et al. Src is a potential therapeutic target in endocrine-resistant breast cancer exhibiting low estrogen receptor-mediated transactivation. PLoS One (2016) 11:e0157397. doi:10.1371/journal.pone.0157397
156. Xiao C, Wu CH, Hu HZ. LncRNA UCA1 promotes epithelial-mesenchymal transition (EMT) of breast cancer cells via enhancing Wnt/beta-catenin signaling pathway. Eur Rev Med Pharmacol Sci (2016) 20:2819–24.
157. Morrison BJ, Schmidt CW, Lakhani SR, Reynolds BA, Lopez JA. Breast cancer stem cells: implications for therapy of breast cancer. Breast Cancer Res (2008) 10:210. doi:10.1186/bcr2111
158. Ellis MJ, Tao Y, Young O, White S, Proia AD, Murray J, et al. Estrogen-independent proliferation is present in estrogen-receptor HER2-positive primary breast cancer after neoadjuvant letrozole. J Clin Oncol (2006) 24:3019–25. doi:10.1200/JCO.2005.04.3034
159. Ellis MJ, Tao Y, Luo J, A’Hern R, Evans DB, Bhatnagar AS, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst (2008) 100:1380–8. doi:10.1093/jnci/djn309
160. Frogne T, Benjaminsen RV, Sonne-Hansen K, Sorensen BS, Nexo E, Laenkholm AV, et al. Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat (2009) 114:263–75. doi:10.1007/s10549-008-0011-8
161. Fox EM, Miller TW, Balko JM, Kuba MG, Sanchez V, Smith RA, et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res (2011) 71:6773–84. doi:10.1158/0008-5472.CAN-11-1295
Keywords: progesterone, growth hormone, Wnt signaling, HER, canine mammary cancer
Citation: Timmermans-Sprang EPM, Gracanin A and Mol JA (2017) Molecular Signaling of Progesterone, Growth Hormone, Wnt, and HER in Mammary Glands of Dogs, Rodents, and Humans: New Treatment Target Identification. Front. Vet. Sci. 4:53. doi: 10.3389/fvets.2017.00053
Received: 01 September 2016; Accepted: 28 March 2017;
Published: 13 April 2017
Edited by:
Tracy Stokol, Cornell University, USAReviewed by:
Joanna Siriol Morris, University of Glasgow, UKFrancisco Ruben Carvallo, University of California Davis, USA
Copyright: © 2017 Timmermans-Sprang, Gracanin and Mol. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan A. Mol, ai5hLm1vbEB1dS5ubA==