 Dongri Piao1
Dongri Piao1 Heng Wang2
Heng Wang2 Dongdong Di3Guozhong Tian1Jiantong Luo2Wenjie Gao2Hongyan Zhao1Weimin Xu2Weixing Fan3
Dongdong Di3Guozhong Tian1Jiantong Luo2Wenjie Gao2Hongyan Zhao1Weimin Xu2Weixing Fan3 Hai Jiang1*
Hai Jiang1*
- 1State Key Laboratory for Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
- 2Hangzhou Center for Disease Control and Prevention (HZCDC), Hangzhou, China
- 3China Animal Health and Epidemiology Center, Qingdao, China
Background: Brucella canis is a pathogenic bacterium that causes brucellosis in dogs, and its zoonotic potential has been increasing in recent years. B. canis is a rare source of human brucellosis in China, where Brucella melitensis has been the major pathogen associated with human brucellosis outbreaks. In late 2011, a case of a B. canis infection was detected in a human patient in Zhejiang Province, China. To compare the genotypes between strains of B. canis isolated from the patient and from dogs, a multiple-locus variable-number tandem-repeat analysis (MLVA-16) was performed. In addition, the lipopolysaccharide-synthesis-related genes were analyzed with the B. canis reference strain RM6/66.
Results: 32 B. canis strains were divided into 26 genotypes using MLVA-16 [Hunter-Gaston Diversity Index (HGDI) = 0.976]. The HGDI indexes for various loci ranged between 0.000 and 0.865. All four Hangzhou isolates were indistinguishable using panel 1 (genotype 3) and panel 2A (genotype 28). However, these strains were distinctly different from other isolates from Beijing, Jiangsu, Liaoning, and Inner Mongolia at Bruce 09. The emergence of a human B. canis infection was limited to an area. Comparative analysis indicated B. canis from canines and humans have no differences in lipopolysaccharide-synthesis locus.
Conclusion: The comprehensive approaches have been used to analyze human and canine B. canis isolates, including molecular epidemiological and LPS genetic characteristics. Further detailed analysis of the whole genomic sequencing will contribute to understanding of the pathogenicity of B. canis in humans.
Introduction
Brucellosis is one of the most common zoonotic diseases worldwide. In north of China, brucellosis is a serious endemic disease mainly caused by Brucella melitensis infection (biovars 1 and 3). Currently, there were 95 human brucellosis surveillance counties nationwide in order to grasp the epidemiological features. In recent years, a few papers about canine brucellosis have published (1–5). The new available data of canine brucellosis showed the outbreak trends in Beijing and other provinces (6–9).
Human brucellosis infection from Brucella canis is not common, probably because B. canis is rough lipopolysaccharide devoid of the O-side. Indeed, in Brucella spp., the smooth lipopolysaccharide, is highly correlated with pathogenesis (10). Some classical Brucella strains, as well as the recently isolated new species, express a smooth phenotype (11, 12). It has been shown that the smooth LPS might play an important role in the invasion and intracellular multiplication of Brucella spp. (13). Several LPS biosynthesis genes have been recognized, most of them clustered in the wbk and wbo genetic regions (14). However, B. ovis is naturally devoid of the wbo region (15). On the other hand, the wbk cluster is present in both the B. ovis and the B. canis (16).
Brucella melitensis was the predominant species associated with humans and animals brucellosis outbreaks in China (9). However, B. canis infection cannot be ignored. The epidemiological data of human canine brucellosis and etiology study have been limited in China. B. canis is known to infect humans, yet only a few cases have ever been reported. On November 10, 2011, a 45-year-old woman who developed fever, back pain, and fatigue was diagnosed with pleurisy at the Jiaxing People’s Hospital, Zhejiang, China. The rough rose bengal plate agglutination test (RRBT) and rough standard tube agglutination test (RSAT) assays were positive. B. canis was also isolated from the blood culture. Because of scarce B. canis infection cases in humans, any potential genetic diversity of the B. canis strain isolated from different hosts should be investigated. Therefore, the aim of this study was to evaluate the epidemiological relatedness among Chinese B. canis strains isolated from dogs and humans. First, variable number of tandem repeats assay was performed to assess the diversity from the different regions, especially in the same geographical area, Zhejiang province. Second, 21 LPS-synthesis related genes were amplified and sequenced, in order to detect genetic mutations between human and canine B. canis strains. This comparative analysis will contribute to knowledge of the Brucella virulence and microevolution.
Materials and Methods
Ethics Approval and Consent to Participate
This study is a retrospective investigation of historical collections strains using modern typing methods and study protocol was approved by the Ethics Committees of National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention. Informed consent was obtained from the patient before diagnosis in the study. We isolated Brucella spp. from patient’ blood sample with his agreement.
Human B. canis Infection Detection
For detection of antibodies against rough Brucella spp., the RRBT and RSAT were performed as previously described (17). Blood culture was performed by biphasic blood bottle (BioMérieux Industry, France) by following the manufacturer’s protocol. 7 days later, some colonies were found in solid phase and inoculated into Brucella Agar Medium.
B. canis Identification and DNA Preparation
Species identification was determined on the basis of phenotype identification procedure (18). 32 B. canis strains isolated from Zhejiang, Beijing, Jiangsu, Liaoning, and Inner Mongolia between 1986 and 2011 were included. The genomic DNA was extracted with the DNeasy Blood & Tissue Kit (Qiagen China Ltd., China) by following the manufacturer’s protocol. Species-level identification was performed using Suis-ladder PCR and real-time PCR assay (19, 20). The procedure of Brucella isolates were carried out in biosafety level 3.
MLVA Typing and Data Analysis
The multiple-locus variable-number tandem-repeat analysis (MLVA-16) was performed as previously described (21–23). Sizes of PCR products were evaluated by capillary electrophoresis on an ABI Prism 3130 automated fluorescent capillary DNA sequencer (Applied Biosystems). Genomic DNA from reference strain B. canis RM6/66 (NC_010103.1 and NC_010104.1) was used as positive control. The cluster analysis was performed using the UPGMA algorithm (Unweighted Pair Group Method Algorithm) with Euclidean distance matrices. Hunter and Gaston diversity indexes were calculated1. Web-based MLVA database2 was utilized to compare different strains.
LPS-Synthesis Characterization of Brucella Strains: PCR Assay and Sequence Analysis
21 LPS-synthesis related genes were analyzed. Primers and amplification were previously described (13). The resultant PCR products were purified and sequenced at Qingke Biosciences Company (Beijing, China). The sequence data were compared by web-based BLAST.3 Reference strain B. melitensis 16M sequences was available in GenBank (AE008917 and AE008918).
LPS Extraction and SDS-Polyacrylamide Gel Electrophoresis (PAGE)
The LPS was extracted using Lipopolysaccharide Extraction Kit (iNtRON Biotechnology Ltd., Korea) by following the manufacturer’s protocol. SDS-PAGE was performed as previously described (24).
Results
Identification of Human Brucellosis by Serological Tests
The serum of the patient was tested by RRBT and RSAT tests for anti-R antibodies, and yielded positive results for antibodies against rough Brucella spp. Blood culture was performed by biphasic blood bottle. A B. canis strain was isolated and showed the same phenotypic characteristics with B. canis RM6/66. The location of B. canis isolated from human and animals between 2006 and 2011 was shown in Figure 1.
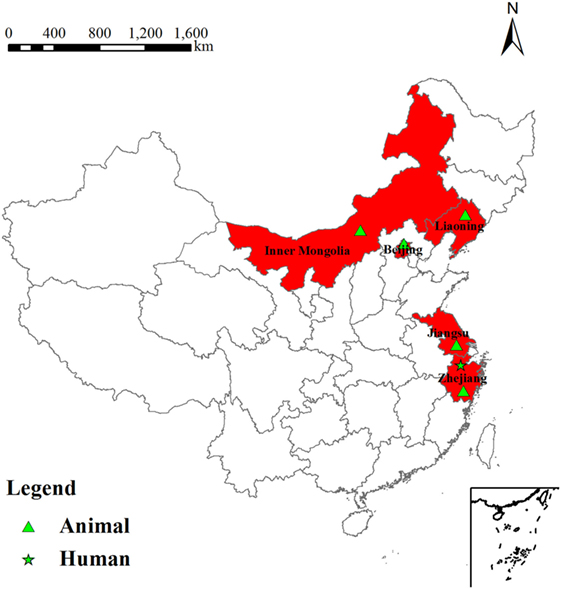
Figure 1. Location of Brucella canis isolated from human and animals between 2006 and 2011.
MLVA Analysis of Human and Canine B. canis Strains
32 B. canis strains were grouped into 26 genotypes using MLVA-16 [Hunter-Gaston Diversity Index (HGDI) = 0.976]. The HGDI indexes ranged between 0.000 and 0.865 (Table 1; Figure 2). 11 loci were of low discriminatory power (HGDI < 0.2). Bruce 09 and Bruce 16 displayed high discriminatory power (HGDI > 0.8), which contained 10 and 7 alleles, respectively. Comparative genetic profiles by MLVA-16 indicated that MLVA-8 and MLVA-11 genotypes of human B. canis strain were identical to B. canis isolates from dogs. Exception of bruce30 loci, four loci (bruce04, bruce07, bruce09, and bruce16) of panel 2B showed diversity (Table 2).
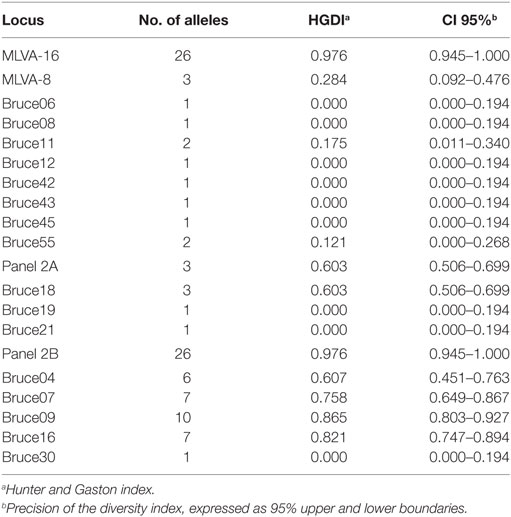
Table 1. Hunter-Gaston Diversity Index (HGDI) for the 32 Chinese Brucella canis isolates.
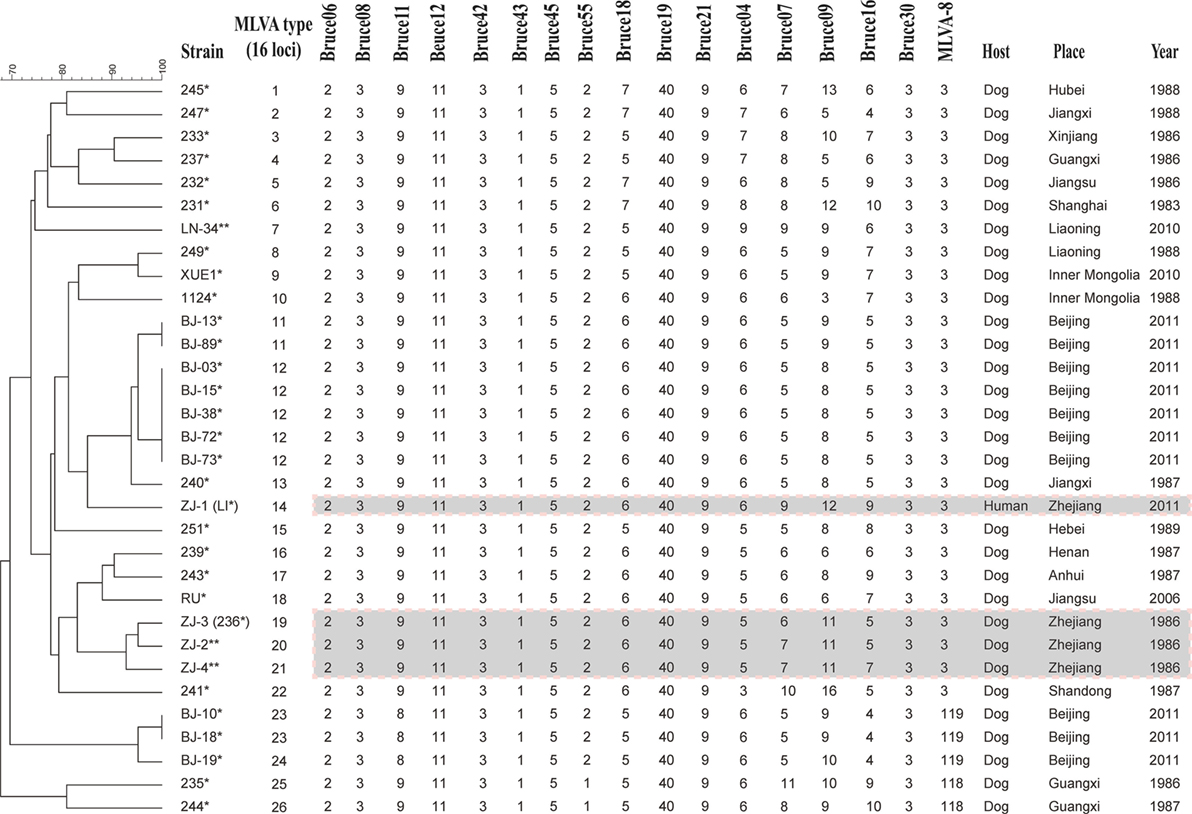
Figure 2. Dendrogram based on the MLVA-16 genotyping assay showing relationships of the four Brucella canis isolates from Hangzhou in this study to other B. canis strains. Strain: strain name in the laboratory in which the DNA extraction was done; year/place: year and country of isolation (when known); * indicates these strains were previously analyzed in Di et al. (23). ** indicates these strains have been analyzed in this study.
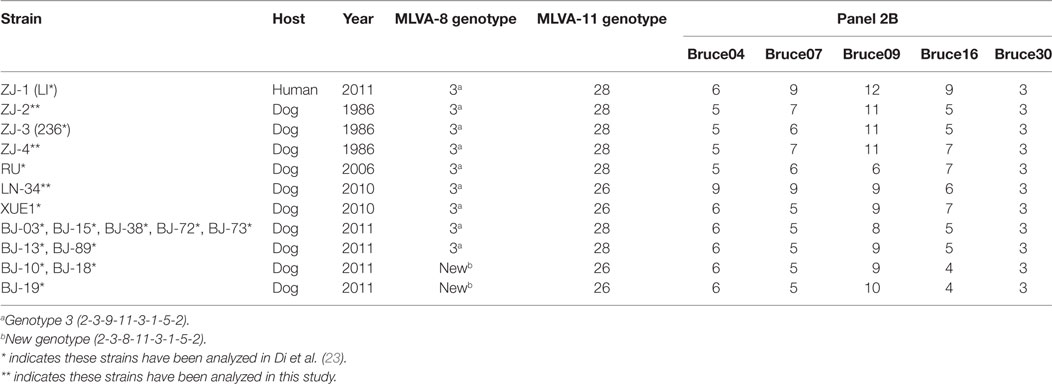
Table 2. MLVA-8 and MLVA-11 genotypes and numbers of tandem-repeat units for panel 2B loci in Brucella canis isolates from Zhejiang province and other regions by MLVA-16.
Genetic Characteristics of Human and Canine B. canis LPS
21 LPS-synthesis genes were analyzed with the reference strain B. canis RM6/66. Comparative analysis indicated B. canis from canines and humans had no differences in LPS-synthesis locus. We next sought to extend the LPS-synthesis genetic characterization between B. canis isolated from human and canines. We also extracted the LPS and performed SDS-PAGE analysis. The results revealed no difference was found between B. canis isolated from human and canines (Figure 3).
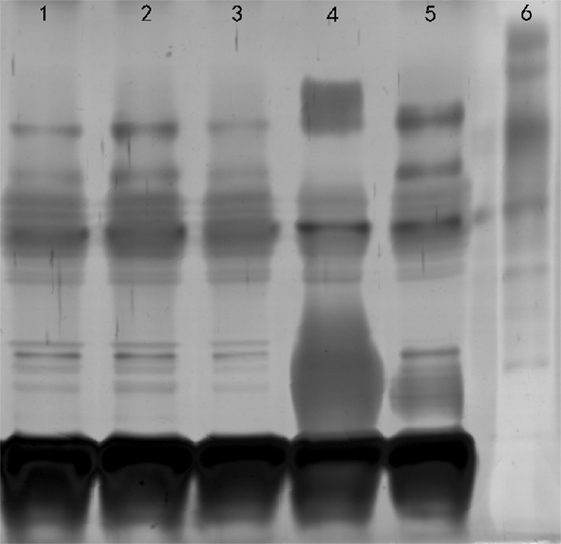
Figure 3. SDS-PAGE analysis of LPS. Lane 1, Brucella canis ZJ-1 (human); lane 2, B. canis ZJ-2 (dog); lane 3, B. canis RM6/66; lane 4, Brucella melitensis 16M; lane 5, B. suis 1330S; lane 6, molecular weight marker.
Discussion
Brucella canis infection was first reported in China in 1984 (6). A nationwide survey on the B. canis infection was conducted in 25 provinces. Epidemic regions were primarily located in south and southeast China during the 1970s and 1980s (6). In 2011, a B. canis outbreak was investigated in a beagle dog breeding farm in Beijing. 48.75% (39/80) was positive by RSAT (7). On November 10, 2011, a 45-year-old woman who developed fever, back pain, and fatigue was diagnosed with pleurisy at the Jiaxing People’s Hospital, Zhejiang, China. Although the epidemiological contact history, brucellosis symptoms were atypical. The RRBT and RSAT tests for anti-R antibodies yielded positive results, considered to be evidence of B. canis infection. On December 12, the patient blood was sent to the Zhejiang Center for Disease Prevention and Control. After several days, an isolate was collected from the blood culture. This isolate displayed rough colony using acridine orange staining. It also showed strong agglutination with anti-R monospecific sera and weak agglutination with anti-Brucella monospecific sera. Surveillance data showed most human brucellosis cases were caused by B. melitensis species (8, 9). There were few reported human brucellosis cases caused by B. canis species. A detailed understanding of genetic diversity of circulating B. canis strains in animals and humans should be achieved to provide appropriate prevention measures for subsequent eradication programs.
Hangzhou city is low prevalence of brucellosis in domestic animals and human populations. We discovered that four B. canis strain were classified into genotype 3 by MLVA-8 analysis. As subset panel 2A loci showed no diversity, the genotypic polymorphism was solely due to the panel 2B loci (Table 2). The lack of genetic diversity in panel 1 and panel 2A suggests that these B. canis strains are highly homogenous. Further analysis showed the three dog MLVA genotypes were only a single-locus variant or double-locus variants in bruce07 (panel 2B) and bruce21 (panel 2A), respectively. However, the human isolate shared only bruce30 (panel 2B) from the dog isolates, which originated from the same breeding farm. These minor mutants probably reflect adaptation or microevolution (21). Although the three dog isolates analyzed in this study were from 25 years earlier, with no reported intermediate infection, it is of interest that the time-distant isolates showed greater homology than geographically distant isolates, consistent with local persistent transmission. It may suggest a recent evolution from a common ancestor in Hangzhou.
Additionally, these four B. canis isolates were compared with some dog isolates from other China provinces: Beijing, Jiangsu, Liaoning, and Inner Mongolia, between 2006 and 2011. Diversities were found with MLVA-16. Previously we observed that B. canis isolates from dogs either separate into a known genotype 3 or a new genotype with MLVA-8 (7). Interestingly, the Hangzhou strains were distinctly different from the above-mentioned strains because of the bruce09. It could be deduced that the Hangzhou strains may have originated from different restricted geographic region compared with the other strains. The differences may be explained by the bacterial adaptation to host and environment that produce genetic changes or polymorphisms. Zhejiang province was an epidemic region where B. melitensis was the dominant epidemic species and B. abortus was an accessory species (unpublished). However, B. canis infection has complicated the current epidemic situation. Nationwide, the true incidence of B. canis in humans remains unknown; therefore, sero-prevalence survey data are needed to determine the risk of B. canis infections. The human canine brucellosis was limited to one area, which is useful in monitoring the source of infection when outbreaks occur in other areas free canine brucellosis.
For LPS-synthesis-related genes, no mutations were detected among these strains, demonstrating the presence of the same LPS genetic loci responsible for rough morphology. We also extracted LPS and performed SDS-PAGE analysis. But, no difference was found between B. canis isolated from human and canine. Importantly, the human B. canis strain is a precious resource for studying Brucella pathogenicity. The whole genome sequencing of the human B. canis strain is ongoing and could be useful for the development of diagnostic tools, in order to reduce health complications of this infection in dogs and humans.
Conclusion
For the first time, comprehensive approaches have been used to analyze human and animal B. canis isolates with molecular epidemiological and genetic characteristics. Further detailed analysis of the whole genomic sequencing will contribute to understanding of the pathogenicity of B. canis in human.
Ethics Statement
After approval by the ethics committee, we included patient data in this study (Ethics Committee, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention).
Author Contributions
HJ and HW designed this study and did most of the typing work. HW, JL, WX, and WG were in charge of the epidemiological investigation and collection of Hangzhou strains. GT, HZ, and DP prepared the DNA samples. HW and DP wrote the report. All authors read and approved the final manuscript. HJ and WF are guarantors of the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was funded by the National Nature Science Foundation (81271900), National Key Program for Infectious Diseases of China (2013ZX10004221, 2013ZX10004805 and 2014ZX10004003), the Science and Technology Basic Work Program (2012FY111000). The funders contributed to the study design and data collection.
Footnotes
References
1. Holst BS, Lofqvist K, Ernholm L, Eld K, Cedersmyg M, Hallgren G. The first case of Brucella canis in Sweden: background, case report and recommendations from a northern European perspective. Acta Vet Scand (2012) 54:18. doi:10.1186/1751-0147-54-18
2. Kang SI, Heo EJ, Cho D, Kim JW, Kim JY, Jung SC, et al. Genetic comparison of Brucella canis isolates by the MLVA assay in South Korea. J Vet Med Sci (2011) 73(6):779–86. doi:10.1292/jvms.10-0334
3. Kulakov I, Tsirel’Son LE, Zheludkov MM. [Molecular-genetic characterization of canine and rangiferine Brucella isolates from different regions of Russia]. Mol Gen Mikrobiol Virusol (2012) 4:28–33. doi:10.3103/s0891416812040052
4. Nomura A, Imaoka K, Imanishi H, Shimizu H, Nagura F, Maeda K, et al. Human Brucella canis infections diagnosed by blood culture. Emerg Infect Dis (2010) 16(7):1183–5. doi:10.3201/eid1607.090209
5. Sayan M, Erdenlig S, Stack J, Kilic S, Guducuoglu H, Aksoy Y, et al. A serological diagnostic survey for Brucella canis infection in Turkish patients with brucellosis-like symptoms. Jpn J Infect Dis (2011) 64(6):516–9.
6. Deqiu S, Donglou X, Jiming Y. Epidemiology and control of brucellosis in China. Vet Microbiol (2002) 90(1–4):165–82. doi:10.1016/S0378-1135(02)00252-3
7. Jiang H, Mao LL, Zhao HY, Li LY, Piao DR, Tian GZ, et al. Reemergence and genetic comparison of Brucella canis in China, using a multiple-locus variable-number tandem-repeat assay. Vet Microbiol (2012) 154(3–4):419–21. doi:10.1016/j.vetmic.2011.07.014
8. Jiang H, Wang H, Xu L, Hu G, Ma J, Xiao P, et al. MLVA genotyping of Brucella melitensis and Brucella abortus isolates from different animal species and humans and identification of Brucella suis vaccine strain S2 from cattle in China. PLoS One (2013) 8(10):e76332. doi:10.1371/journal.pone.0076332
9. Jiang H, Fan M, Chen J, Mi J, Yu R, Zhao H, et al. MLVA genotyping of Chinese human Brucella melitensis biovar 1, 2 and 3 isolates. BMC Microbiol (2011) 11:256. doi:10.1186/1471-2180-11-256
10. Roop RN, Gaines JM, Anderson ES, Caswell CC, Martin DW. Survival of the fittest: how Brucella strains adapt to their intracellular niche in the host. Med Microbiol Immunol (2009) 198(4):221–38. doi:10.1007/s00430-009-0123-8
11. Alton GG, Jones LM, Angus RD, Verger JM. Techniques for the Brucellosis Laboratory. Paris, France: Institut National de la recherche Agronomique (INRA) (1988).
12. Foster G, Osterman BS, Godfroid J, Jacques I, Cloeckaert A. Brucella ceti sp. nov. and Brucella pinnipedialis sp. nov. for Brucella strains with cetaceans and seals as their preferred hosts. Int J Syst Evol Microbiol (2007) 57(Pt 11):2688–93. doi:10.1099/ijs.0.65269-0
13. Adone R, Muscillo M, La Rosa G, Francia M, Tarantino M. Antigenic, immunologic and genetic characterization of rough strains B. abortus RB51, B. melitensis B115 and B. melitensis B18. PLoS One (2011) 6(10):e24073. doi:10.1371/journal.pone.0024073
14. Moriyon I, Grillo MJ, Monreal D, Gonzalez D, Marin C, Lopez-Goni I, et al. Rough vaccines in animal brucellosis: structural and genetic basis and present status. Vet Res (2004) 35(1):1–38. doi:10.1051/vetres:2003037
15. Vizcaino N, Caro-Hernandez P, Cloeckaert A, Fernandez-Lago L. DNA polymorphism in the omp25/omp31 family of Brucella spp.: identification of a 1.7-kb inversion in Brucella cetaceae and of a 15.1-kb genomic island, absent from Brucella ovis, related to the synthesis of smooth lipopolysaccharide. Microbes Infect (2004) 6(9):821–34. doi:10.1016/j.micinf.2004.04.009
16. Cloeckaert A, Grayon M, Verger JM, Letesson JJ, Godfroid F. Conservation of seven genes involved in the biosynthesis of the lipopolysaccharide O-side chain in Brucella spp. Res Microbiol (2000) 151(3):209–16. doi:10.1016/S0923-2508(00)00141-8
17. OIE. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. Paris, France: Office International des Epizooties (2008). p. 624–6.
18. Alton GG, Jones LM, Pietz DE. Laboratory techniques in brucellosis. Monogr Ser World Health Organ (1975) 55:1–163.
19. Gopaul KK, Koylass MS, Smith CJ, Whatmore AM. Rapid identification of Brucella isolates to the species level by real time PCR based single nucleotide polymorphism (SNP) analysis. BMC Microbiol (2008) 8:86. doi:10.1186/1471-2180-8-86
20. Lopez-Goni I, Garcia-Yoldi D, Marin CM, de Miguel MJ, Barquero-Calvo E, Guzman-Verri C, et al. New Bruce-ladder multiplex PCR assay for the biovar typing of Brucella suis and the discrimination of Brucella suis and Brucella canis. Vet Microbiol (2011) 154(1–2):152–5. doi:10.1016/j.vetmic.2011.06.035
21. Le Fleche P, Jacques I, Grayon M, Al DS, Bouchon P, Denoeud F, et al. Evaluation and selection of tandem repeat loci for a Brucella MLVA typing assay. BMC Microbiol (2006) 6:9. doi:10.1186/1471-2180-6-9
22. Al DS, Fleche PL, Nockler K, Jacques I, Grayon M, Scholz HC, et al. Evaluation of Brucella MLVA typing for human brucellosis. J Microbiol Methods (2007) 69(1):137–45. doi:10.1016/j.mimet.2006.12.015
23. Di D, Cui B, Wang H, Zhao H, Piao D, Tian L, et al. Genetic polymorphism characteristics of Brucella canis isolated in China. PLoS One (2014) 9(1):e84862. doi:10.1371/journal.pone.0084862
Keywords: MLVA, LPS biosynthesis, Brucella canis, humans and dogs, epidemiology
Citation: Piao D, Wang H, Di D, Tian G, Luo J, Gao W, Zhao H, Xu W, Fan W and Jiang H (2017) MLVA and LPS Characteristics of Brucella canis Isolated from Humans and Dogs in Zhejiang, China. Front. Vet. Sci. 4:223. doi: 10.3389/fvets.2017.00223
Received: 18 October 2017; Accepted: 05 December 2017;
Published: 18 December 2017
Edited by:
Jiabo Ding, China Institute of Veterinary Drug Control, ChinaReviewed by:
Yuanzhi Wang, Shihezi University Medical School, ChinaDongbo Sun, Heilongjiang Bayi Agricultural University, China
Copyright: © 2017 Piao, Wang, Di, Tian, Luo, Gao, Zhao, Xu, Fan and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hai Jiang, amlhbmdoYWlAaWNkYy5jbg==