 Kamal R. Acharya
Kamal R. Acharya Navneet K. Dhand
Navneet K. Dhand Richard J. Whittington1,2
Richard J. Whittington1,2 Karren M. Plain
Karren M. Plain- 1Faculty of Science, Sydney School of Veterinary Science, The University of Sydney, Camden, NSW, Australia
- 2School of Life and Environmental Sciences, University of Sydney, Sydney, NSW, Australia
Johne’s disease is a chronic debilitating enteropathy of ruminants caused by Mycobacterium avium subspecies paratuberculosis (MAP). Current abattoir surveillance programs detect disease via examination of gross lesions and confirmation by histopathological and/or tissue culture, which is time-consuming and has relatively low sensitivity. This study aimed to investigate whether a high-throughput quantitative PCR (qPCR) test is a viable alternative for tissue testing. Intestine and mesenteric lymph nodes were sourced from sheep experimentally infected with MAP and the DNA extracted using a protocol developed for tissues, comprised enzymatic digestion of the tissue homogenate, chemical and mechanical lysis, and magnetic bead-based DNA purification. The extracted DNA was tested by adapting a previously validated qPCR for fecal samples, and the results were compared with culture and histopathology results of the corresponding tissues. The MAP tissue qPCR confirmed infection in the majority of sheep with gross lesions on postmortem (37/38). Likewise, almost all tissue culture (61/64) or histopathology (52/58) positives were detected with good to moderate agreement (Cohen’s kappa statistic) and no significant difference to the reference tests (McNemar’s Chi-square test). Higher MAP DNA quantities corresponded to animals with more severe histopathology (odds ratio: 1.82; 95% confidence interval: 1.60, 2.07). Culture-independent strain typing on tissue DNA was successfully performed. This MAP tissue qPCR method had a sensitivity equivalent to the reference tests and is thus a viable replacement for gross- and histopathological examination of tissue samples in abattoirs. In addition, the test could be validated for testing tissue samples intended for human consumption.
Introduction
Ovine Johne’s disease (OJD) is a chronic debilitating gastroenteritis of sheep caused by Mycobacterium avium subspecies paratuberculosis (MAP). OJD-specific enteric lesions cause protein loss and chronic malabsorption of protein (1, 2) leading to a protracted wasting condition in the affected sheep. Consequently, OJD-infected sheep can have poor welfare status and reduced production traits, including quality of the carcass and fleece (3). Control of OJD has been attempted by means of flock vaccination, which is effective to an extent (4–6); however, this strategy has failed to achieve a complete cessation of fecal shedding (5, 6). Fecal shedding of the bacteria by the vaccinated sheep could become a continued source of infection in the flock through faeco-oral transmission to other susceptible animals. Therefore, implementation of a suitable OJD surveillance program is essential for detection and control of MAP spread, both in vaccinated and unvaccinated flocks.
Ovine Johne’s disease surveillance activities include collection and testing of a range of samples from suspected sheep. In Australia, to confirm infection in suspected animals and flocks, fecal culture and/or fecal quantitative PCR (qPCR) (7) may be performed, whereas postmortem confirmation is attained by tissue culture and examination of histopathological lesions within tissue samples such as intestine and mesenteric lymph nodes (MLNs) collected at abattoirs participating in the National Sheep Health Monitoring Program or on farm (8–10).
For confirmation of MAP infection, culture of fecal samples is less reliable than tissue culture of the small intestines and MLN, the major predilection sites of MAP, in both clinically and subclinically affected animals (11–15). This is due to the intermittent fecal shedding of MAP (16). Furthermore, passive fecal shedding of MAP has been noted in animals irrespective of their infection status and animals exposed to MAP showing positive fecal sample test results may never become infected or may clear the infection (17, 18). Like fecal samples, biopsies could also be collected from live animals for the confirmation of MAP infection in sheep (17, 19, 20). Testing of tissue samples for confirmation of MAP infection could also have an application for human health, as MAP has been suggested to be the causative agent of Crohn’s disease in humans (21, 22). MAP has been detected in biopsies from the intestinal tissues of humans (23), including pediatric Crohn’s disease patients (24). Human exposure could be associated with the consumption of animal tissues from infected animals that can contain MAP, due to dissemination of bacteria during systematic infection or through fecal contamination during slaughter procedures (25–28). Hence, a suitable tissue sample collection and testing approach could significantly contribute to the detection of MAP infected sheep.
The culture of tissue homogenate is considered the gold standard test for confirmation of MAP infection in animals. This test has been reported to be more sensitive than examining histological lesions, Ziehl–Neelsen staining and/or immunohistochemistry (29), and fecal culture (14). However, the use of decontaminants reduces the sensitivity of this method (30) and the long incubation time is a further limitation (31). Therefore, development and validation of alternative techniques is essential, especially if the test result is intended for surveillance purposes. Culture-independent methods such as qPCR that require mycobacterial DNA isolated directly from the sample have been used successfully to diagnose nontuberculous mycobacterial infection (32) and to perform whole genome sequencing of Mycobacterium tuberculosis (33). Similar approaches could be used to diagnose as well as rapidly identify the strain and type of MAP, thereby aiding in molecular epidemiology to identify the source of infection. Diagnostic tests based on qPCR are preferred, owing to the speed and high-throughput nature of these assays. In addition, there is some evidence that qPCR may perform better than tissue culture to detect MAP in diaphragm muscle samples of cattle (25).
Several conventional PCR or qPCR methods have been reported for the detection of MAP DNA directly from intestine, MLN, meat, and formalin-fixed paraffin-embedded intestinal tissues (34–41). There are few studies that focus on testing sheep and goat tissues (38, 41, 42). Although some studies have compared qPCR with culture methods using bovine and caprine tissue samples (40, 42), a comparative study using ovine tissue samples is lacking.
Therefore, the objectives of this study were to (a) test the tissue homogenates (intestinal and MLN) from MAP-exposed sheep using a qPCR method with suitable sensitivity, (b) compare qPCR with other relevant diagnostic techniques such as tissue culture and examination of histopathological lesions of the tissue sections, and (c) assess the potential for direct strain typing by means of a restriction endonuclease analysis (REA) IS1311 method.
Materials and Methods
Sample Collection
The tissue samples originated from a trial, in which the animals were infected using validated infection model (43). Approval from the University of Sydney Animal Ethics committee was obtained before the commencement of this study. In brief, approximately 5-month-old lambs received a total dose of 2.74 × 109 viable cells of MAP (S strain, Telford 9.2) orally over a period of 1 month. Necropsy of the animals was performed at 14 months post-inoculation or earlier if there was evidence of significant loss of weight (>10% in a month) or death. Intestinal sections (terminal ileum and middle jejunum) and MLN sections (posterior and middle jejunal MLN) of these sheep were collected at necropsy and stored at −80°C before culture.
For each animal, pooled tissue homogenates of intestinal tissues and MLN tissues were prepared separately. Representative sections of tissues amounting to 2–5 g were finely chopped with a sterile scalpel blade and added to an 80–100 mL stomacher bag (Interscience) containing approximately an equal amount of sterile 0.85% w/v sodium chloride solution. Homogenization was achieved by stomaching at the highest speed in a stomacher (Minimix, Interscience) for 2 min. The homogenate was transferred to 4-mL screw capped tubes avoiding any obvious tissue pieces. Two milliliters of tissue homogenate were used for tissue culture and the rest was stored at −80°C for qPCR. In total, 107 intestinal and 107 MLN homogenates were tested corresponding to 107 MAP-exposed animals. The histopathological test result of intestinal samples of five animals and MLN samples of six animals were not available; these samples were excluded when comparing qPCR with histopathological lesions.
Bacterial Culture
The culture of MAP in tissue homogenates was performed in M7H9C liquid culture media and the growth was confirmed by qPCR, as previously described (44, 45). Briefly, 2 mL of tissue homogenate was transferred to sterile polycarbonate tubes containing 25 mL of 0.75% (w/v) hexadecylpyridinium chloride (HPC), mixed by inverting the tubes and incubated in a dark cupboard at room temperature for 72 h. Without disturbing the HPC in the tubes, 100 µl of the precipitate was transferred to 6 mL of M7H9C liquid media and incubated at 37°C for 12 weeks. MAP growth was confirmed by testing DNA extracted from 200 µL of the culture medium using IS900 qPCR.
Tissue Histopathological Examination
Tissue histopathological examination was performed as reported previously (46). For this study, results from the examination performed previously were accessed. In brief, a 5-µm section of formalin-fixed paraffin-embedded tissue sample was microscopically examined following staining with hematoxylin and eosin, and Ziehl–Neelsen method. The lesions were scored by an experienced pathologist (R. Whittington) following the criteria established by Pérez et al. (47). The intestinal lesions were graded from 0 to 3. The grade 3 lesion was further classified into paucibacillary (3a), multibacillary (3b), or severe paucibacillary (3c) depending on the lesion and number of acid-fast bacteria present within the lesion. The lesions in MLNs were graded as mild focal (1), mild multifocal (2), or severe multifocal to diffuse (3). If an animal showed a range of lesions, the highest grade of the lesion score was considered for the analysis. An animal was considered to be positive if the lesion score was ≥1.
Detection of MAP DNA in Tissue Homogenates
DNA Extraction
The tissue homogenates were thawed and vortexed. A 1-mL aliquot of the homogenate was transferred to 1.5-mL sterile screw cap tubes (Interpath services). To 1,000 µL of the homogenate, 20 µL of proteinase K (40 mg/mL solution, Astral) was added. The enzymatic action was facilitated by incubating in a heat block (Ratek) at 50°C for 1 h. Vortexing for 20 s was performed following the addition of proteinase K at the end of the incubation time.
Following enzymatic digestion, the suspension was centrifuged at 15,000 × g for 15 min and the supernatant was discarded. The pellet was dissolved in lysis buffer (597 µL buffer RLT and 2.8 µL carrier RNA, Qiagen) and transferred to bead tubes containing 0.3 mL of 0.1 mm diameter silica/zirconium beads in 2-mL screw cap tubes. Bead beating was done twice at 30 Hz for 100 s (TissueLyser, Qiagen) with a change in alignment by 180° between two bead beatings, followed by centrifugation at 16,000 × g for 3 min. The supernatant was transferred to 1.5 mL LoBind DNA flip-top tubes (Eppendorf) and stored at −20°C.
DNA Purification
DNA was purified using the Biosprint® 96 One-for-all Vet kit (Qiagen). Briefly, 400 µL of the supernatant obtained above was transferred to a 96-well lysate plate containing 40 µL proteinase K (Qiagen) to which 300 µL of magnetic bead suspension comprising 25 µL MagAttract suspension G (Qiagen) in 300 µL isopropanol (Sigma-Aldrich) was added. Magnetic bead-based purification of DNA was performed using an automated platform (MagMAX Express-96; Life Technologies), which included transfer of the bead-bound DNA through three wash steps using two different wash buffers (Buffer AW1 and RPE, Qiagen) contained in separate 96-well plates (Qiagen), air drying of any wash buffer residue and DNA elution in Buffer AVE (Qiagen). The eluted DNA was stored at −80°C before use. The DNA extract was tested neat and also as a fivefold dilution in Buffer AVE (Qiagen).
IS900 qPCR
Quantification of MAP genomic DNA in the DNA extracts from the tissues was performed as previously described (7). The final volume of 25 µL of reaction mixture comprised 5 µL template DNA, 250 nM final concentration of each forward [MP10-1 (5′-ATGCGCCACGACTTGCAGCCT-3′)] and reverse [MP11-1 (5′-GGCACGGCTCTTGTTGTAGTCG-3′)] primers (48) and 12.5 µL of SensiMix SYBR Low-ROX qPCR mastermix (Bioline). The amplification was performed (initial denaturation at 95°C for 8.5 min; 40 cycles of denaturation at 95°C for 15 s, annealing at 68°C for 30 s, and extension at 72°C for 1 min, and melt curve analysis from 55 to 95°C) using an Mx3000P real-time PCR instrument (Stratagene, Agilent). The quantification of MAP DNA was performed by constructing a standard curve using 10-fold serially diluted MAP genomic DNA, ranging from 10 to 0.001 pg/reaction, included in each DNA amplification plate. The acceptance criteria for a positive test result were (i) MAP-specific amplification (melt temperature in the range of 89.1 ± 1.5°C) and (ii) MAP DNA quantity of ≥0.0005 pg/reaction, which is the analytical limit of detection of the qPCR (7).
Strain Typing Using a Culture-Independent Method
Culture-independent strain typing was performed using DNA extracted directly from the tissues. For this purpose, an IS1311 PCR followed by REA of the PCR product was performed as described (49). Briefly, 50 µL PCR reactions comprised 5 µL of DNA, 250 ng each of forward M56 (GCGTGAGGCTCTGTGGTGAA) and Reverse M119 (ATGACGACCGCTTGGGAGAC) primers, 2 U of Taq polymerase (Roche) and 200 µM of each dNTP in reaction buffer. Amplification conditions were one cycle of denaturation at 94°C for 3 min followed by 37 cycles of 94°C for 30 s, 62°C for 15 s, and 72°C for 1 min. The PCR products were evaluated by electrophoresis at 100 V in 2% agarose gels stained with Redsafe dye (INtRON Biotechnology) with molecular weight marker size VIII (Roche). PCR product was stored at −20°C before REA, performed by digestion with two enzymes (Mse I and Hinf I, New England Biolabs) for 2 h at 37°C according to the manufacturers’ instructions. Visualization of the products (Gel Doc, Bio-Rad) post-enzymatic digestion was performed by electrophoresis at 100 V in 3% agarose gels stained with Redsafe dye.
Case Definitions
An animal was considered to be infected if the tissue culture result of the pooled intestinal homogenate (terminal ileum, middle jejunum) and/or pooled MLN homogenate (posterior and middle jejunal MLN) was positive.
An animal was considered to be diseased if there was a histopathological lesion score of ≥1 in any of the intestinal and/or lymph node sections examined (47). For this purpose, mid to terminal ileum and proximal jejunum and ileocecal, posterior jejunal and mid proximal jejunal lymph node samples were examined.
A positive qPCR result corresponded to a MAP-specific DNA quantity of ≥0.0005 pg/5 µl of the DNA extracted from 1 mL of tissue homogenate.
Statistical Analysis
Comparison of qPCR with Tissue Culture and Histopathology
The numeric result from qPCR was categorized into positive and negative categories according to the cut-point of ≥0.0005 pg, as previously described (7). The categorical results were used to test the agreement between the qPCR test result with reference tests (tissue culture and histopathological lesions). The difference between detection rates of qPCR and reference tests was evaluated by undertaking the McNemar’s chi-square test. If the difference was not significant, the agreement between qPCR with reference tests was measured by means of Cohen’s kappa coefficient. The relative sensitivity of tissue qPCR was computed as a ratio of the number of positive test results from tissue qPCR to the number of positive results by the reference test.
Furthermore, ordinal logistic regression analysis was performed to assess the relationship between log10 DNA quantified by tissue qPCR and histopathological lesion score of the tissues. For this analysis, lesion score 0 and 1 were merged together and treated as lesion score 1 as there were insufficient observations with lesion score 1. In order to perform the log10 transformation, a very small value was added to the samples with zero DNA quantity. Type of tissue (intestine and MLN) was included as a factor in the multivariable model.
The discriminatory ability of numeric qPCR results to differentiate the test positive and negative results of tissue culture and/or histopathology was assessed by means of receiver operating characteristic (ROC) curve analysis and calculation of the area under the curve (AUC). The DNA quantity obtained by tissue qPCR was used to conduct the ROC curve analysis.
Comparisons between Tissue Types
The mean log10 DNA values quantified by qPCR of intestinal and MLN homogenates were assessed using scatter plots and correlation coefficients. The overall difference was also compared using a Bland–Altman plot, which plotted the difference in log10 DNA quantity versus average log10 DNA quantity. The DNA quantity was log10 transformed after adding a very small value (0.0000001 pg) to the quantified DNA.
A two-sided p-value <0.05 was considered statistically significant for all analyses reported in this manuscript. The statistical analyses were conducted using GenStat v 16.2.11713 (VSN International Ltd., Hemel Hempstead, UK) and the graphs were prepared using GraphPad Prism 7 for Windows, Version 7.02 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
The experimentally MAP-exposed sheep included in the study (n = 107) comprised a range of sheep breeds: the majority were Border Leicester (33.7%), followed by White Suffolk cross Merino (33.7%), Poll Dorset (16.9%), and Merino (15.9%) breeds of sheep. The animals were not vaccinated, but shared the same paddock as a group of Merino sheep that were vaccinated against paratuberculosis with Gudair®. Male and female animals were almost equally represented in the study.
General Performance of the qPCR Test
The MAP-specific qPCR method detected 59.9% of intestinal homogenates and 65.5% of MLN homogenates from the exposed sheep as positive for the presence of MAP DNA.
A few of the DNA extracts appeared to contain high amounts of initial MAP DNA, as determined by the raw baseline fluorescence before amplification. Fivefold dilutions of the DNA extracts were also subjected to qPCR with specific amplification seen in both neat and diluted samples. The threshold cycle (CT) values for such samples was determined within the exponential phase of the amplification curve after assessment of the raw amplification plot (data not shown).
Ability of the Tissue qPCR to Confirm MAP Infection in Animals with Clinical Signs and Gross Lesions of Paratuberculosis
The qPCR method was successful in detecting animals that exhibited clinical signs and gross lesions consistent with OJD. All of the 25 of animals that had clinical signs suggestive of the disease (25/107; 23.4%) were confirmed as positive for MAP DNA by qPCR of intestinal homogenates. Histopathological lesion examination and MAP tissue culture of the intestine and MLN at necropsy also confirmed that these were true clinical cases. However, one of the clinical cases was negative by qPCR of the MLN alone. Similarly, qPCR confirmed 37 of the 38 (97.4%) animals that showed gross lesions, suggestive of paratuberculosis disease, on postmortem examination, when considering the results for both the intestinal homogenate and MLN in parallel. The clinical cases were a subset of the sheep with gross lesions.
The ability of qPCR to detect the presence of MAP in animals with gross lesions was comparable or better than the use of a culture result from both the intestinal and MLN homogenate, which detected 35/38 of the animals. When considering the results separately for the intestinal tissues and MLN of animals with gross lesions, qPCR detected 33/34 intestine culture positive animals and 32/35 MLN culture positive animals. In addition, qPCR of intestinal homogenates detected 2/4 intestinal culture negative animals and qPCR of MLN detected 3/3 MLN culture negative animals.
In addition to confirmation of animals showing clinical signs and gross lesions, qPCR of the tissues detected additional animals as positive for MAP DNA: for the intestinal homogenates, an additional 29 exposed animals were detected, whereas qPCR of MLN homogenates detected an additional 35 animals.
Ability of the qPCR to Confirm MAP Presence Compared to Tissue Culture and Histopathological Lesion Examination
The qPCR method detected MAP in a majority of tissues identified as infected by the reference tests. For the same sample type, the qPCR tended to detect more samples as MAP positive than culture and histopathological lesion scoring (Table 1). The proportion of test positive results for any of the reference tests compared to the qPCR test for individual tissues was not significantly different when examined by McNemar’s Chi-square test suggesting equivalence of the tests. The agreement when assessed with Cohen’s kappa statistic showed a good to moderate agreement of qPCR with the reference tests.
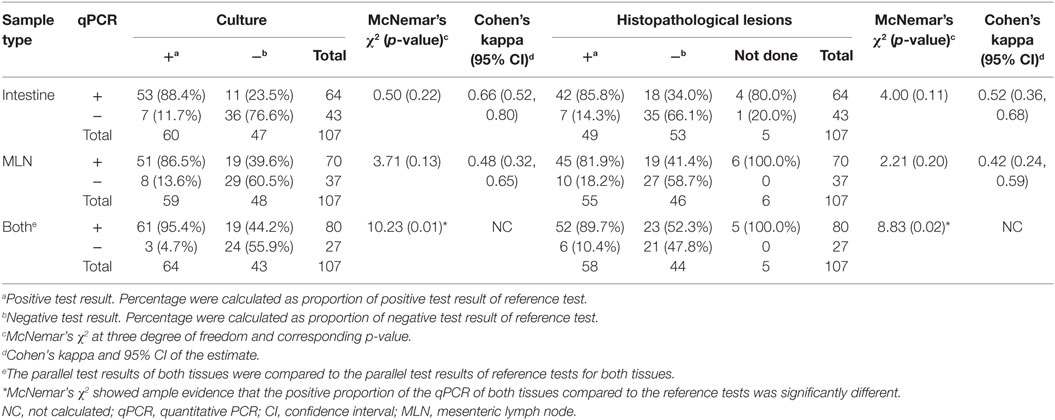
Table 1. Comparison of qPCR test results of tissues (intestine and MLN) with reference test (culture and histopathological lesion score) results.
When the tissue qPCR results for both the intestinal and MLN tissues were considered, tissue qPCR detected most of the animals detected by the parallel interpretation of the reference tests (Table 1). In addition, samples that were not detected by the reference tests were detected by the qPCR. Consequently, there was evidence that the qPCR performed better than the reference tests, when assessed with McNemar’s Chi-square test (Table 1).
Evaluation of Test Performance Parameters of the qPCR Compared to Reference Tests (Culture and Histopathological Lesion Score)
Relative sensitivity of the qPCR assay for intestinal and MLN tissues was determined, assuming tissue culture result or histopathological examination result of the respective tissues as a gold standard (Table 2). The qPCR was more sensitive when assessed against culture than against the histopathological examination result; this was not unexpected as culture and qPCR are both tests for the presence of the MAP organism.
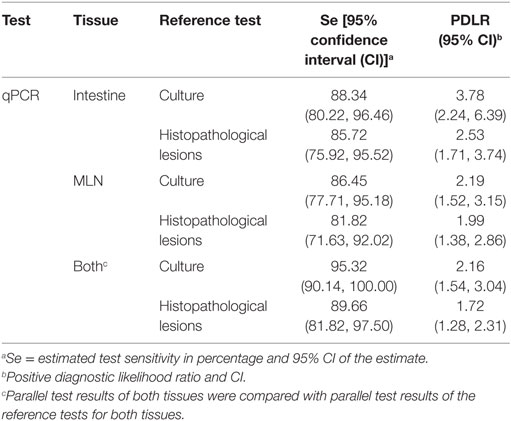
Table 2. Performance characteristics of intestinal tissue and mesenteric lymph node (MLN) quantitative PCR (qPCR) with reference to tissue culture and tissue histopathological lesion results.
A ROC curve analysis showed a good discriminatory power of the qPCR to differentiate between positive and negative results of the reference tests (Figure 1). The positive diagnostic likelihood ratio estimated at the cutoff used for the qPCR (approximately >0.0005 pg) was higher for predicting the culture results compared to the histopathological examination results.
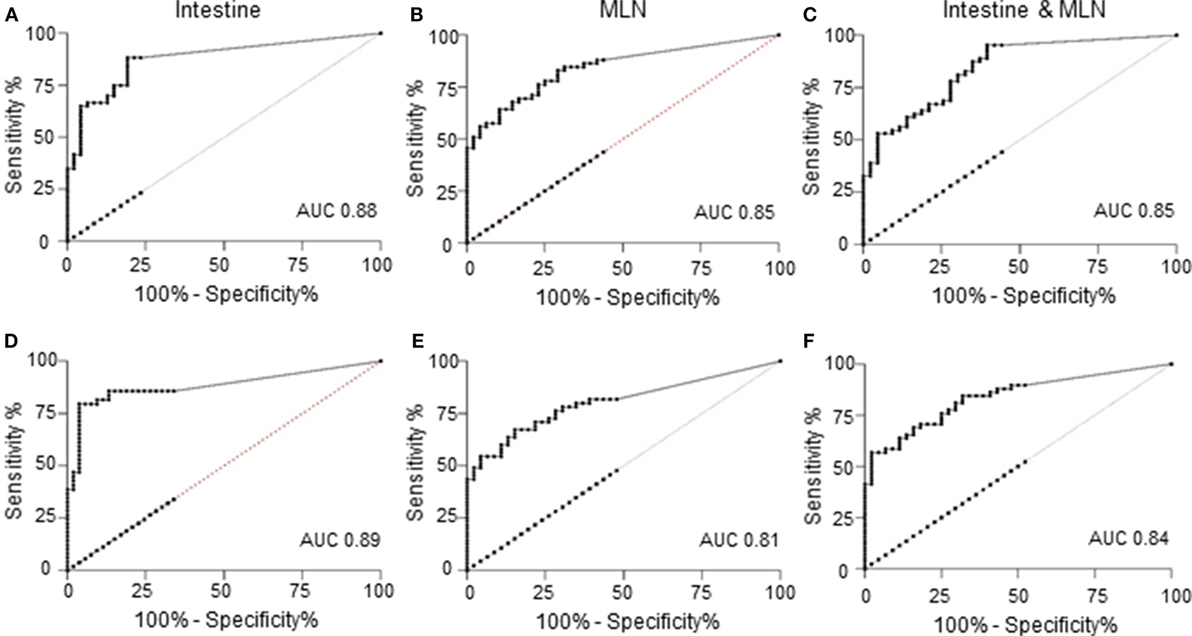
Figure 1. Receiver operating characteristic (ROC) curves of quantitative results of quantitative PCR (qPCR) versus reference tests; tissue culture (A–C) or histopathology (D–F). (A) ROC curve of qPCR of intestine against culture results as reference test. Area under the curve (AUC) was 0.88 [95% confidence interval (CI): 0.82, 0.95], and at a cutoff of >0.00054 pg, the sensitivity was 88.33% (95% CI: 77.43%, 95.18%), and the likelihood ratio was 4.16. (B) ROC curve of qPCR of mesenteric lymph node (MLN) against culture results as reference test. AUC was 0.85 (95% CI: 0.77, 0.92), and at a cutoff of >0.00052 pg, the sensitivity was 86.44% (95% CI: 75.02%, 93.96%), and the likelihood ratio was 2.19. (C) ROC curve of qPCR of intestine and MLN against culture results of both tissues as reference test. AUC was 0.85 (95% CI: 0.78, 0.92), and at a cutoff of >0.00053 pg, the sensitivity was 95.31% (95% CI: 86.91%, 99.02%), and the likelihood ratio was 2.28. (D) ROC curve of qPCR of intestine against histopathological lesion examination as reference test. AUC was 0.89 (95% CI: 0.82, 0.96), and at a cutoff of >0.00054 pg, the sensitivity was 85.71% (95% CI: 72.76%, 94.06%), and the likelihood ratio was 2.68. (E) ROC curve of qPCR of MLN against histopathological lesion examination as reference test. AUC was 0.81 (95% CI: 0.72, 0.89), and at a cutoff of >0.00052 pg, the sensitivity was 81.82% (95% CI: 69.10%, 90.92%), and the likelihood ratio was 1.99. (F) ROC curve of qPCR of intestine and MLN against histopathological lesion examination of both tissues as reference test. AUC was 0.84 (95% CI: 0.77, 0.92), and at a cutoff of >0.00053 pg, the sensitivity was 89.66% (95% CI: 78.83%, 96.11%), and the likelihood ratio was 1.80.
Similarly, when parallel test results of intestinal and MLN qPCR were compared to the parallel test results of culture or histopathological examination, there was a notable increase in sensitivity, with higher sensitivity against culture compared to histopathological lesion score results (Table 2). A ROC curve analysis also showed that parallel use of test results of two tissues was good at discriminating the positive test result of the reference tests when interpreted in parallel (Figures 1C,F).
The comparison of tissue qPCR with histological lesion score results showed that the number of qPCR positives (Table 3) and MAP DNA quantity (Figure 2) increased according to lesion severity. Furthermore, with each log increase in DNA quantity, the odds of histopathological lesion score of the tissues increased by 1.82 times (Table 4). Likewise, MLN tissues had about half the odds of having a more severe lesion than intestinal tissues, suggesting that lesion scores are more severe in intestinal tissue.
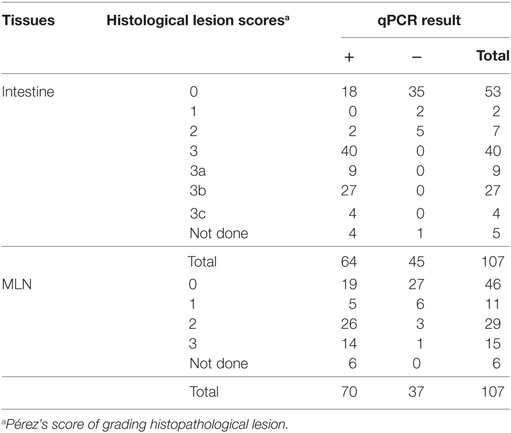
Table 3. Comparison of the test result (cutoff ≥0.0005 pg) of intestine and mesenteric lymph node (MLN) quantitative PCR (qPCR) corresponding to various grades of lesion scores on histopathological examination.
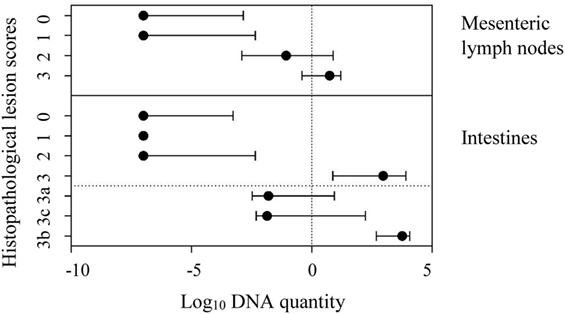
Figure 2. Line plot showing median log10 DNA quantity and interquartile range of Mycobacterium avium subspecies paratuberculosis DNA quantified in mesenteric lymph node and intestinal tissues against the respective histopathological lesion score (according to Pérez et al.) in these tissues. In intestines, the lesion type 3 was further distinguished as subtypes 3a, 3c, and 3b.
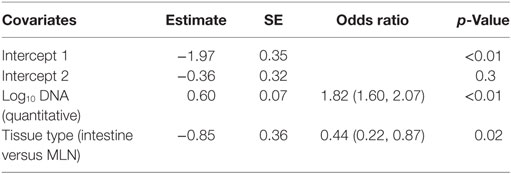
Table 4. Result of ordinal logistic regression with ordinal histopathological lesion score as response and log10 DNA quantified in the quantitative PCR of tissues and types of tissues [intestine and mesenteric lymph node (MLN)] as predictors.
Comparison of Intestinal qPCR with MLN qPCR
A strong positive correlation between DNA quantified by qPCR of intestinal homogenate and MLN homogenates of the same animal was found (Pearson’s correlation coefficient of 0.78, 95% CI: 0.69, 0.84). The DNA quantified by these two tests showed a positive linear relationship (Figure 3A). On an average, MAP DNA levels in the intestine were 2.94 times than those in the associated MLNs. The Bland–Altman graph (Figure 3B) depicts that there was a slight increase in the difference of DNA quantified by intestinal qPCR and MLN qPCR, with an increase in average DNA quantified by these methods.
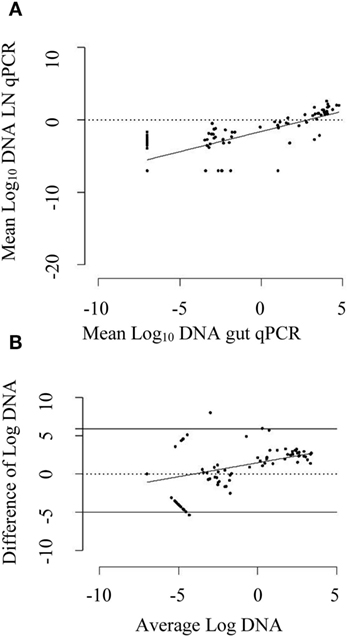
Figure 3. (A) Scatter plot of mean log10 DNA quantified by mesenteric lymph node (MLN) quantitative PCR (qPCR) against log10 DNA quantified by intestinal tissue (gut) qPCR. The solid line is the regression line. A positive correlation coefficient of 0.78 (95% CI: 0.69, 0.84) was observed. (B) Bland–Altman plot depicting difference of log10 DNA quantified by gut qPCR and MLN qPCR against average log10 DNA quantified by them. The dotted line shows the line of perfect agreement whereas two solid lines show the limit of agreement. The DNA quantified by gut qPCR was 2.94 times higher than that quantified by MLN qPCR for the same animal on an average. The solid line is the regression line of average log10 DNA against difference of log10 DNA. An increase in average difference was observed for the increase in DNA quantity in the gut and MLN homogenate.
When the qPCR results from the two tissues (at cutoff point of ≥0.0005 pg) were assessed, the overall agreement between intestine qPCR and MLN qPCR results was good. Although qPCR of MLN homogenates detected more animals compared to qPCR of intestinal homogenates, the proportions of positive test results of the two assays were not significantly different (Table 5). Likewise, the agreement between the two tests was moderate, suggesting the complementary nature of these two tests; animals that went undetected by qPCR test of one sample type were detected by the examination of the other sample type.
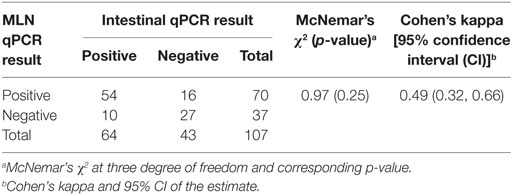
Table 5. Comparison of quantitative PCR (qPCR) results (cutoff of ≥0.0005 pg/5 µl of DNA extract) of intestinal tissue compared to qPCR results of mesenteric lymph node (MLN) samples.
Strain Typing
The good performance of qPCR in identifying infected animals raised the possibility of using the isolated DNA to perform direct strain typing and hence enable rapid molecular epidemiological analysis, essential from a disease control perspective. Strain typing on the directly isolated DNA correctly identified the S-strain of MAP, which was used for the experimental infection of the animals, in the DNA extracted from both intestine and LN tissue of the samples tested (Figure 4).
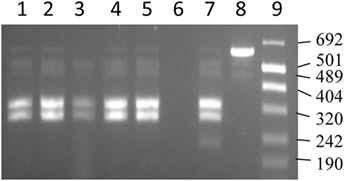
Figure 4. IS1311 PCR-restriction endonuclease analysis profiles for DNA extracted from tissues. Lanes 1, 2, and 4 are IS1311 PCR products from DNA extracted from gut tissues restricted with Mse I and Hinf I enzymes. Lanes 3 and 5 are IS1311 PCR products from DNA extracted from lymph node tissues restricted with Mse I and Hinf I enzymes. Lane 6 is negative control. Lane 7 is IS1311 PCR product from S strain restricted with Mse I and Hinf I enzymes. Lane 8 is the unrestricted IS1311 PCR product from S strain. Lane 9 is the molecular size markers (Number VIII).
Discussion
This study was conducted to investigate whether the relative sensitivity of tissue qPCR compares to tissue culture and histopathological lesion examination to determine the MAP infection status of an animal and predict the potential infectivity of an animal, with a potential application in abattoir surveillance as a replacement test. Furthermore, we investigated whether MAP DNA isolated from intestinal and MLN homogenates could provide rapid information on MAP strain type. The combined information is important in disease investigation and in planning and evaluating OJD management on farms.
It was evident from this study that the qPCR method used gave results that were similar or better than culture results in terms of sensitivity for MAP detection for both of the tissue types assessed. The qPCR test outperformed both culture and histopathological examination tests, when the tests were interpreted in parallel for both tissue types. Moreover, only half of the tissue amount generally used for culture was used for qPCR and the results of qPCR could be obtained within a week compared to 12 weeks required for culture. Both the tests provide similar information on flock health status based on sensitivity and test performance characteristics of the qPCR test. However, the tests could complement each other for the purpose of an individual animal diagnosis, as an animal “missed” by tissue culture could be detected by the tissue qPCR, suggesting that the detection limit and sensitivity of qPCR is better than the culture method, possibly due to the necessary inclusion of a decontamination step in culture methods (30). As has been previously reported, culture tests require the use of a decontamination step that might decrease the diagnostic sensitivity, especially when the contamination level of MAP in the tissue is low (30). Conversely, samples not detected by qPCR, due to low MAP numbers in the tissues, sampling site tested or PCR inhibition, might be detected by culture. PCR inhibition and remediation strategies have been previously reported when assessing fecal sample types (50). Although no obvious evidence of inhibition was found in this study, based on the assessment of the quality of DNA extracts and testing and relief of potential PCR inhibition (50) by performing the qPCR test of fivefold diluted DNA extracts (data not shown), the absence of PCR inhibition cannot be guaranteed as it is a complex phenomenon. Likewise, the inability of qPCR test result to confirm the viability of MAP is a limitation. It was evident in this study that some of the DNA extracts appeared to contain high amounts of initial DNA, possibly target DNA of MAP and other non-target DNA, as determined by the raw baseline fluorescence before amplification. This may be due to the carryover of host DNA, which is expected when bacterial DNA is being extracted from tissues and might result in PCR inhibition (51). However, the qPCR was successful in amplifying the target DNA of MAP, yielding amplification products within the correct melt temperature peak range.
Although the study of histopathological lesions provides different information to that provided by qPCR, the two were on par in their ability to determine MAP infection in the tissues. For this purpose, qPCR detected more animals than histopathological lesion scoring. This is expected as the amount of sample tested in the qPCR is significantly larger compared to that used in preparing the histopathological slides (15). Furthermore, it has also been noted earlier that only in the advanced stage of MAP infection the lesions are diffuse (52, 53), and the sample that goes for DNA extraction and section preparation are mutually exclusive. Likewise, the discrepancy can be contributed to by the disease stage, as was shown when the number of qPCR-positive results were compared by lesion grade; this has been previously noted when comparing tissue culture results with microscopic tissue lesions (15). As lesion severity increased, there was an associated trend toward higher MAP DNA quantities and thus greater likelihood of detection.
The diagnosis of clinical MAP infection is not a problem; most of the available tests can correctly identify animals in the clinical stage of the disease (15). This study also demonstrated that all three types of test (culture, histopathological lesion scoring, and qPCR) were able to diagnose the clinical animal correctly. Although all of the tests were able to detect most of the animals with gross lesions, the qPCR test was similar to or better than the reference tests, including culture and histopathological lesion scoring, for confirming MAP infection, in particular for animals showing the clinical form of the disease or gross lesions on necropsy. However, in addition to the animals showing clinical signs and gross lesions, the qPCR test detected additional non-clinical animals. Presently, slaughterhouse surveillance programs in Australia focus on the examination of histopathological lesions of animals showing gross paratuberculosis lesions in the intestines and MLN (9). This study showed that focusing on gross lesions might miss a lot of animals when assessment of abattoir surveillance was done, as predicted by Abbott and Whittington (8). Similar observations were made in a cattle study (54). This is expected as examination of gross lesions has very low to low diagnostic sensitivity for animal and flock level diagnosis; the flock level sensitivity is particularly low when the flock level prevalence of the disease is very low (8). Furthermore, many animals reaching the slaughterhouse are too young to develop lesions, given the chronic nature of the disease and in particular if the disease prevalence and exposure level is low. It would thus be a good strategy to randomly test intestines and MLNs from slaughtered animals rather than targeting only those animals with gross lesions to obtain a more accurate understanding of MAP infection in a flock or a region. This type of sampling approach would be aided by qPCR techniques that are cost effective and high-throughput and would benefit from a pooling approach subject to validation.
Testing tissue samples from intestines and lymph nodes could complement each other. This study found that, although the agreement between the qPCR results of the two tissue types was very high, it was not perfect. Similar observations of imperfect agreement between the culture results of ileum and MLN have been previously reported (13). This supports the strategy in testing both of these tissues for obtaining broader information on the disease status of the animal. Furthermore, intestinal lesions in naturally and experimentally infected sheep are distributed across the segments of the intestines (52, 53). Likewise, MAP is non-uniformly distributed in the associated MLNs (52). Hence, pooling strategies applied to different sections of intestine and several MLNs, as used in this study, can increase the sensitivity of using intestinal tissue as a sample for diagnosing JD, as observed from our results. As expected, a study in cattle noted an improvement in the detection rate following inclusion of several types of tissues (54). Thus, the pooling strategy and testing of both tissues used in this study detected more animals when compared to testing either of the samples. Furthermore, although the intestinal qPCR result was able to predict the MLN qPCR result, the R2 estimate was only 60.0% in a regression analysis, suggesting that intestinal qPCR does not completely predict MLN qPCR result. Thus, testing both tissues can give complementary information on the MAP infection status of an animal.
Concerns have been raised over the reduced specificity of qPCR methods for MAP detection, particularly those using IS900 as the genetic marker of MAP DNA. This was due to published and unpublished reports of IS900-like insertion elements in some mycobacterial species (7, 55, 56). The specificity of the PCR primers used in this study had been previously validated against a panel of 51 mycobacterial taxa and strains, including 10 isolates that were reported to contain IS900-like elements, with no evidence of false positive test results (7). There have been no other reports of other bacterial species in the gut of sheep that cross react with the IS900 qPCR. Thus, based on the available information, the results obtained by the current qPCR are very specific and the presence of MAP confirmed by qPCR that went undetected by culture and histopathological lesion scoring is likely due to the increased analytical sensitivity of the current qPCR compared to these tests.
Culture-independent methods using DNA isolated directly from samples have been successfully used for whole genome sequencing, diagnosis, as well as strain typing of mycobacterial species (32, 33). However, the success of such procedures depends on the quality and quantity of the target DNA in the extract and on the typing method. The DNA extracted from the tissues using this method was of high quality (data not shown) and hence was successfully used to strain type MAP by performing the REA method targeting the insertion sequence IS1311, previously used to strain type MAP from cultures obtained from tissue sections of sheep (57). The insertion sequence (IS1311) is shared by members of Mycobacterium avium including MAP and polymorphism in this sequence have been successfully utilized by means of restriction enzymes (Mse I and Hinf I) to differentiate MAP from Mycobacterium avium and to differentiate strains of MAP. Although we did not attempt to make a comparison with different strain typing tools available for MAP (58–60), a study reported that several typing methods complement each other with no significant difference in Simpson’s Index of Diversity when the discriminatory power of different typing methods were assessed (61).
As expected, more DNA was quantified in intestinal samples compared to MLN samples, which indicates the possibility of reliably using MAP DNA isolated from intestinal homogenates to provide rapid information on the MAP strain type and hence perform rapid molecular epidemiological analysis. The popularity of culture-independent methods arises from their efficiency in terms of time, especially when dealing with a fastidious organism like MAP. The culture-independent procedures reported in this study would also help to rapidly diagnose and hence address the concerns of cross-species transmission of MAP strains from sheep to cattle (62). The disease management cost would be higher if culture was required to be used instead for the same information. The classification of MAP as cattle and sheep strain, which is provided by IS1311 PCR-REA, is sought by epidemiologist to understand the likely source of MAP infection in Australia as the epidemiology of these two strains are considered distinct (63).
The samples from this study came from an infection trial using an experimental infection model, the outcomes of which have been shown to be comparable to what is observed following natural exposure to MAP via contaminated pasture (12, 46). Tissue samples are frequently collected and tested in an experimental trial to confirm the infection status of an animal, to understand the disease progression, and to study the dissemination of the MAP infection (27, 52). Tissue samples have also been previously shown to facilitate early detection of MAP infection in sentinel sheep grazed in MAP contaminated pasture with varying level of contamination (12). Tests such as culture and histopathological lesion scoring are used for these purposes. This study shows that validated qPCR methods, where the diagnostic capacity and correlation to other test results are understood, could be equally useful or better for these purposes. Such validated qPCR methods as presented here are equally valuable in disease control and trade certification situations, quantitative risk assessment. Although tissue culture, histopathological lesion scoring, and qPCR tests can complement each other to obtain information on an overall status of the flock, the qPCR can provide rapid information which might be more valuable for rapid interventions and decision making in a disease control and trade dispute scenario. Likewise, when parallel results of qPCR tests of intestine and MLN were used, they provided more information than the parallel results of culture or histopathological lesions. Furthermore, there is the possibility of adopting this test for antemortem diagnosis of high value animals by testing tissue samples obtained via biopsy of the animals (17, 20), which is an approach being used in humans (23).
Thus, this study reports a qPCR method for testing intestines and MLNs, validated against the current gold standard method, capable of providing the information currently obtained by testing these sample types using culture and histopathological lesion scoring. As a result, disease status information can be obtained rapidly and used for prompt decision making. Furthermore, the DNA obtained directly from tissues can be used for culture-independent procedures like strain typing. We recommend using both intestine and MLN tissues for testing to improve sensitivity. Further validation of the tissue qPCR method and sampling strategies should be conducted for testing abattoir samples, so that it could replace or complement the currently used surveillance system which is based on gross lesion detection followed by histopathology.
Ethics Statement
This study protocol was approved by the University of Sydney Animal Ethics committee and the study was carried out in accordance with the recommendations of the Australian code of practice for the care and use of animals for scientific purposes.
Author Contributions
KA codesigned the study with other authors, developed the protocol, conducted the experiment, analyzed the data, and drafted the manuscript. KP provided laboratory support. ND provided support in analyzing data. RW provided intellectual input during designing and conducting the study. All authors reviewed and revised the manuscript drafts.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Douglas Begg, Dr. Kumi deSilva, and Dr. Auriol Purdie involved in the experimental trial design and conduct. Likewise, our thanks goes to the laboratory assistance of Ms. Anna Waldron, Ms. Ann-Michele Whittington, Rebecca Maurer, Nicole Carter, and Gina Attard. The assistance from Mr. Craig Kristo, Mr. Nobel Toribio, Mr. Lee white, and Mr. James Dalton obtained during the field work is also acknowledged.
Funding
The first author was funded for the entire study duration by means of scholarship provided by the University of Sydney (reference no. 440074360). This work was supported by Meat and Livestock Australia and by Cattle Council of Australia, Sheepmeat Council of Australia, and WoolProducers Australia through Animal Health Australia. The funding bodies had no role in the design of the study, the collection, analysis, and interpretation of data, or in writing the manuscript.
References
1. Allen WM, Berrett S, Patterson DSP. A biochemical study of experimental Johne’s disease: I. Plasma protein leakage into the intestine of sheep. J Comp Pathol (1974) 84:381–4. doi:10.1016/0021-9975(74)90012-7
2. Allen WM, Berrett S, Patterson DSP. A biochemical study of experimental Johne’s disease: II. An in vitro study of L-histidine uptake by sheep intestinal mucosa. J Comp Pathol (1974) 84:385–9. doi:10.1016/0021-9975(74)90013-9
3. McGregor H, Abbott KA, Whittington RJ. Effects of Mycobacterium avium subsp. paratuberculosis infection on serum biochemistry, body weight and wool growth in Merino sheep: a longitudinal study. Small Ruminant Res (2015) 125:146–53. doi:10.1016/j.smallrumres.2015.02.004
4. Dhand NK, Eppleston J, Whittington RJ, Toribio J-AL. Risk factors for ovine Johne’s disease in infected sheep flocks in Australia. Prev Vet Med (2007) 82:51–71. doi:10.1016/j.prevetmed.2007.05.007
5. Dhand NK, Eppleston J, Whittington RJ, Windsor PA. Changes in prevalence of ovine paratuberculosis following vaccination with Gudair®: results of a longitudinal study conducted over a decade. Vaccine (2016) 34:5107–13. doi:10.1016/j.vaccine.2016.08.064
6. Reddacliff L, Eppleston J, Windsor P, Whittington R, Jones S. Efficacy of a killed vaccine for the control of paratuberculosis in Australian sheep flocks. Vet Microbiol (2006) 115:77–90. doi:10.1016/j.vetmic.2005.12.021
7. Plain KM, Marsh IB, Waldron AM, Galea F, Whittington A-M, Saunders VF, et al. High-throughput direct fecal PCR assay for detection of Mycobacterium avium subsp. paratuberculosis in sheep and cattle. J Clin Microbiol (2014) 52:745–57. doi:10.1128/JCM.03233-13
8. Abbott KA, Whittington RJ. Monte Carlo simulation of flock-level sensitivity of abattoir surveillance for ovine paratuberculosis. Prev Vet Med (2003) 61:309–32. doi:10.1016/j.prevetmed.2003.08.005
9. Bradley TL, Cannon RM. Determining the sensitivity of abattoir surveillance for ovine Johne’s disease. Aust Vet J (2005) 83:633–6. doi:10.1111/j.1751-0813.2005.tb13278.x
10. Department of Agriculture and Water Resources. Approved Tests. Canberra: Commonwealth of Australia (2017). Available from: http://www.agriculture.gov.au/animal/health/laboratories/tests/approved-tests
11. Coussens P, Lamont EA, Kabara E, Sreevatsan S. Host-pathogen interactions and intracellular survival of Mycobacterium avium subsp. paratuberculosis. In: Behr MA, Collins DM, editors. Paratuberculosis: Organism, Disease, Control. Wallingford: CABI (2010). p. 109–25.
12. Reddacliff LA, McGregor H, Abbott K, Whittington RJ. Field evaluation of tracer sheep for the detection of early natural infection with Mycobacterium avium subsp paratuberculosis. Aust Vet J (2004) 82:426–33. doi:10.1111/j.1751-0813.2004.tb11135.x
13. Smith SL, West DM, Wilson PR, de Lisle GW, Collett MG, Heuer C, et al. The prevalence of disseminated Mycobacterium avium subsp. paratuberculosis infection in tissues of healthy ewes from a New Zealand farm with Johne’s disease present. N Z Vet J (2013) 61:41–4. doi:10.1080/00480169.2012.704627
14. Whittington RJ, Marsh I, McAllister S, Turner MJ, Marshall DJ, Fraser CA. Evaluation of modified Bactec 12B radiometric medium and solid media for culture of Mycobacterium avium subsp. paratuberculosis from sheep. J Clin Microbiol (1999) 37:1077–83.
15. Whittington RJ, Sergeant ESG. Progress towards understanding the spread, detection and control of Mycobacterium avium subsp paratuberculosis in animal populations. Aust Vet J (2001) 79:267–78. doi:10.1111/j.1751-0813.2001.tb11980.x
16. Mitchell RM, Schukken Y, Koets A, Weber M, Bakker D, Stabel J, et al. Differences in intermittent and continuous fecal shedding patterns between natural and experimental Mycobacterium avium subspecies paratuberculosis infections in cattle. Vet Res (2015) 46:66. doi:10.1186/s13567-015-0188-x
17. Dennis MM, Reddacliff LA, Whittington RJ. Longitudinal study of clinicopathological features of Johne’s disease in sheep naturally exposed to Mycobacterium avium subspecies paratuberculosis. Vet Pathol (2011) 48:565–75. doi:10.1177/0300985810375049
18. Kralik P, Pribylova-Dziedzinska R, Kralova A, Kovarcik K, Slana I. Evidence of passive faecal shedding of Mycobacterium avium subsp. paratuberculosis in a Limousin cattle herd. Vet J (2014) 201:91–4. doi:10.1016/j.tvjl.2014.02.011
19. Gwozdz JM, Thompson KG, Murray A, West DM, Manktelow BW. Use of the polymerase chain reaction assay for the detection of Mycobacterium avium subspecies paratuberculosis in blood and liver biopsies from experimentally infected sheep. Aust Vet J (2000) 78:622–4. doi:10.1111/j.1751-0813.2000.tb11938.x
20. Smith SL, Wilson PR, Collett MG, Heuer C, West DM, Stevenson M, et al. Liver biopsy histopathology for diagnosis of Johne’s disease in sheep. Vet Pathol (2014) 51:915–18. doi:10.1177/0300985813516644
21. Chiodini RJ, Rossiter CA. Paratuberculosis: a potential zoonosis? Vet Clin North Am Food Anim Pract (1996) 12:457–67.
22. Waddell LA, Rajić A, Stärk K, McEwen SA. Mycobacterium avium ssp. paratuberculosis detection in animals, food, water and other sources or vehicles of human exposure: a scoping review of the existing evidence. Prev Vet Med (2016) 132:32–48. doi:10.1016/j.prevetmed.2016.08.003
23. Scanu AM, Bull TJ, Cannas S, Sanderson JD, Sechi LA, Dettori G, et al. Mycobacterium avium subspecies paratuberculosis infection in cases of irritable bowel syndrome and comparison with Crohn’s disease and Johne’s disease: common neural and immune pathogenicities. J Clin Microbiol (2007) 45:3883–90. doi:10.1128/JCM.01371-07
24. Kirkwood CD, Wagner J, Boniface K, Vaughan J, Michalski WP, Catto-Smith AG, et al. Mycobacterium avium subspecies paratuberculosis in children with early-onset Crohn’s disease. Inflamm Bowel Dis (2009) 15:1643–55. doi:10.1002/ibd.20967
25. Alonso-Hearn M, Molina E, Geijo M, Vazquez P, Sevilla I, Garrido JM, et al. Isolation of Mycobacterium avium subsp. paratuberculosis from muscle tissue of naturally infected cattle. Foodborne Pathog Dis (2009) 6:513–8. doi:10.1089/fpd.2008.0226
26. Antognoli MC, Garry FB, Hirst HL, Lombard JE, Dennis MM, Gould DH, et al. Characterization of Mycobacterium avium subspecies paratuberculosis disseminated infection in dairy cattle and its association with antemortem test results. Vet Microbiol (2008) 127:300–8. doi:10.1016/j.vetmic.2007.08.017
27. Bower KL, Begg DJ, Whittington RJ. Culture of Mycobacterium avium subspecies paratuberculosis (MAP) from blood and extra-intestinal tissues in experimentally infected sheep. Vet Microbiol (2011) 147:127–32. doi:10.1016/j.vetmic.2010.06.016
28. Smith SL, West DM, Wilson PR, de Lisle GW, Collett MG, Heuer C, et al. Detection of Mycobacterium avium subsp. paratuberculosis in skeletal muscle and blood of ewes from a sheep farm in New Zealand. N Z Vet J (2011) 59:240–3. doi:10.1080/00480169.2011.596257
29. Martinson SA, Hanna PE, Ikede BO, Lewis JP, Miller LM, Keefe GP, et al. Comparison of bacterial culture, histopathology, and immunohistochemistry for the diagnosis of Johne’s disease in culled dairy cows. J Vet Diagn Invest (2008) 20:51–7. doi:10.1177/104063870802000109
30. Reddacliff LA, Vadali A, Whittington RJ. The effect of decontamination protocols on the numbers of sheep strain Mycobacterium avium subsp. paratuberculosis isolated from tissues and faeces. Vet Microbiol (2003) 95:271–82. doi:10.1016/S0378-1135(03)00181-0
31. Whittington R. Cultivation of Mycobacterium avium subsp. paratuberculosis. In: Behr MA, Collins DM, editors. Paratuberculosis: Organism, Disease, Control. Wallingford: CABI (2010). p. 244–66.
32. Caverly LJ, Carmody LA, Haig S-J, Kotlarz N, Kalikin LM, Raskin L, et al. Culture-independent identification of nontuberculous mycobacteria in cystic fibrosis respiratory samples. PLoS One (2016) 11:e0153876. doi:10.1371/journal.pone.0153876
33. Brown AC, Bryant JM, Einer-Jensen K, Holdstock J, Houniet DT, Chan JZM, et al. Rapid whole-genome sequencing of Mycobacterium tuberculosis isolates directly from clinical samples. J Clin Microbiol (2015) 53:2230–7. doi:10.1128/JCM.00486-15
34. Englund S, Bölske G, Ballagi-Pordány A, Johansson KE. Detection of Mycobacterium avium subsp. paratuberculosis in tissue samples by single, fluorescent and nested PCR based on the IS900 gene. Vet Microbiol (2001) 81:257–71. doi:10.1016/S0378-1135(01)00348-0
35. Khol JL, Kralik P, Slana I, Beran V, Aurich C, Baumgartner W, et al. Consecutive excretion of Mycobacterium avium subspecies paratuberculosis in semen of a breeding bull compared to the distribution in feces, tissue and blood by IS900 and f57 quantitative real-time PCR and culture examinations. J Vet Med Sci (2010) 72:1283–8. doi:10.1292/jvms.10-0052
36. Lorencova A, Vasickova P, Makovcova J, Slana I. Presence of Mycobacterium avium subspecies and hepatitis E virus in raw meat products. J Food Prot (2014) 77:335–8. doi:10.4315/0362-028X.JFP-13-252
37. Park KT, Allen AJ, Davis WC. Development of a novel DNA extraction method for identification and quantification of Mycobacterium avium subsp. paratuberculosis from tissue samples by real-time PCR. J Microbiol Methods (2014) 99:58–65. doi:10.1016/j.mimet.2014.02.003
38. Preziuso S, Magi GE, Renzoni G. Detection of Mycobacterium avium subsp. paratuberculosis in intestinal and mammary tissues and in lymph nodes of sheep with different techniques and its relationship with enteric lesions. Small Rumininant Res (2012) 105:295–9. doi:10.1016/j.smallrumres.2011.11.015
39. Pribylova R, Slana I, Kralik P, Kralova A, Babak V, Pavlik I. Correlation of Mycobacterium avium subsp. paratuberculosis counts in gastrointestinal tract, muscles of the diaphragm and the masseter of dairy cattle and potential risk for consumers. Int J Food Microbiol (2011) 151:314–8. doi:10.1016/j.ijfoodmicro.2011.09.025
40. Timms VJ, Mitchell HM, Neilan BA. Optimisation of DNA extraction and validation of PCR assays to detect Mycobacterium avium subsp. paratuberculosis. J Microbiol Methods (2015) 112:99–103. doi:10.1016/j.mimet.2015.03.016
41. Whittington RJ, Reddacliff L, Marsh I, Saunders V. Detection of Mycobacterium avium subsp paratuberculosis in formalin-fixed paraffin-embedded intestinal tissue by IS900 polymerase chain reaction. Aust Vet J (1999) 77:392–7. doi:10.1111/j.1751-0813.1999.tb10315.x
42. Singh PK, Singh SV, Singh AV, Sohal JS. Evaluation of four methods of DNA recovery from Mycobacterium avium subspecies paratuberculosis present in intestine tissue of goats and comparative sensitivity of IS900 PCR with respect to culture for diagnosis of Johne’s disease. Indian J Exp Biol (2008) 46:579–82.
43. Begg DJ, Purdie AC, de Silva K, Dhand NK, Plain KM, Whittington RJ. Variation in susceptibility of different breeds of sheep to Mycobacterium avium subspecies paratuberculosis following experimental inoculation. Vet Res (2017) 48:36. doi:10.1186/s13567-017-0440-7
44. Plain KM, Waldron AM, Begg DJ, de Silva K, Purdie AC, Whittington RJ. Efficient, validated method for the detection of mycobacterial growth in liquid culture media using bead-beating, magnetic particle nucleic acid isolation and quantitative PCR. J Clin Microbiol (2015) 53:1121–8. doi:10.1128/JCM.03521-14
45. Whittington RJ, Whittington AM, Waldron A, Begg DJ, de Silva K, Purdie AC, et al. Development and validation of a liquid medium (M7H9C) for routine culture of Mycobacterium avium subsp. paratuberculosis to replace modified Bactec 12B medium. J Clin Microbiol (2013) 51:3993–4000. doi:10.1128/JCM.01373-13
46. Begg DJ, O’Brien R, Mackintosh CG, Griffin JFT. Experimental infection model for Johne’s disease in sheep. Infect Immun (2005) 73:5603–11. doi:10.1128/IAI.73.9.5603-5611.2005
47. Pérez V, Marín JFG, Badiola JJ. Description and classification of different types of lesion associated with natural paratuberculosis infection in sheep. J Comp Pathol (1996) 114:107–22. doi:10.1016/S0021-9975(96)80001-6
48. Kawaji S, Taylor DL, Mori Y, Whittington RJ. Detection of Mycobacterium avium subsp. paratuberculosis in ovine faeces by direct quantitative PCR has similar or greater sensitivity compared to radiometric culture. Vet Microbiol (2007) 125:36–48. doi:10.1016/j.vetmic.2007.05.002
49. Whittington RJ, Marsh I, Choy E, Cousins D. Polymorphisms in IS1311, an insertion sequence common to Mycobacterium avium and M. avium subsp. paratuberculosis, can be used to distinguish between and within these species. Mol Cell Probes (1998) 12:349–58. doi:10.1006/mcpr.1998.0194
50. Acharya KR, Dhand NK, Whittington RJ, Plain KM. PCR inhibition of a quantitative PCR for detection of Mycobacterium avium subspecies paratuberculosis DNA in faeces: diagnostic implications and potential solutions. Front Microbiol (2017) 8:115. doi:10.3389/fmicb.2017.00115
51. Radomski N, Kreitmann L, McIntosh F, Behr MA. The critical role of DNA extraction for detection of mycobacteria in tissues. PLoS One (2013) 8:e78749. doi:10.1371/journal.pone.0078749
52. Begg DJ, de Silva K, Di Fiore L, Taylor DL, Bower KL, Zhong L, et al. Experimental infection model for Johne’s disease using a lyophilised, pure culture, seedstock of Mycobacterium avium subspecies paratuberculosis. Vet Microbiol (2010) 141:301–11. doi:10.1016/j.vetmic.2009.09.007
53. Carrigan MJ, Seaman JT. The pathology of Johne’s disease in sheep. Aust Vet J (1990) 67:47–50. doi:10.1111/j.1751-0813.1990.tb07693.x
54. Elze J, Liebler-Tenorio E, Ziller M, Köhler H. Comparison of prevalence estimation of Mycobacterium avium subsp. paratuberculosis infection by sampling slaughtered cattle with macroscopic lesions vs. systematic sampling. Epidemiol Infect (2013) 141:1536–44. doi:10.1017/S0950268812002452
55. Cousins DV, Whittington RJ, Marsh I, Masters A, Evans RJ, Kluver P. Mycobacteria distinct from Mycobacterium avium subsp. paratuberculosis isolated from the faeces of ruminants possess IS900-like sequences detectable by IS900 polymerase chain reaction: implications for diagnosis. Mol Cell Probes (1999) 13:431–42. doi:10.1006/mcpr.1999.0275
56. Englund S, Bölske G, Johansson KE. An IS900-like sequence found in a Mycobacterium sp. other than Mycobacterium avium subsp. paratuberculosis. FEMS Microbiol Lett (2002) 209:267–71. doi:10.1111/j.1574-6968.2002.tb11142.x
57. Marsh I, Whittington RJ, Cousins D. PCR-restriction endonuclease analysis for identification and strain typing of Mycobacterium avium subsp. paratuberculosis and Mycobacterium avium subsp. avium based on polymorphisms in IS1311. Mol Cell Probes (1999) 13:115–26. doi:10.1006/mcpr.1999.0227
58. Collins DM, Gabric DM, de Lisle GW. Identification of two groups of Mycobacterium paratuberculosis strains by restriction endonuclease analysis and DNA hybridization. J Clin Microbiol (1990) 28:1591–6.
59. Griffiths TA, Rioux K, De Buck J. Sequence polymorphisms in a surface PPE protein distinguish types I, II, and III of Mycobacterium avium subsp. paratuberculosis. J Clin Microbiol (2008) 46:1207–12. doi:10.1128/JCM.02031-07
60. Whittington RJ, Hope AF, Marshall DJ, Taragel CA, Marsh I. Molecular epidemiology of Mycobacterium avium subsp. paratuberculosis: IS900 restriction fragment length polymorphism and IS1311 polymorphism analyses of isolates from animals and a human in Australia. J Clin Microbiol (2000) 38:3240–8.
61. Stevenson K, Alvarez J, Bakker D, Biet F, de Juan L, Denham S, et al. Occurrence of Mycobacterium avium subspecies paratuberculosis across host species and European countries with evidence for transmission between wildlife and domestic ruminants. BMC Microbiol (2009) 9:212. doi:10.1186/1471-2180-9-212
62. Moloney BJ, Whittington RJ. Cross species transmission of ovine Johne’s disease from sheep to cattle: an estimate of prevalence in exposed susceptible cattle. Aust Vet J (2008) 86:117–23. doi:10.1111/j.1751-0813.2008.00272.x
Keywords: Mycobacterium avium subspecies paratuberculosis, abattoir surveillance, quantitative PCR, strain typing, sheep, tissue
Citation: Acharya KR, Dhand NK, Whittington RJ and Plain KM (2017) Culture-Independent Identification of Mycobacterium avium Subspecies paratuberculosis in Ovine Tissues: Comparison with Bacterial Culture and Histopathological Lesions. Front. Vet. Sci. 4:232. doi: 10.3389/fvets.2017.00232
Received: 16 October 2017; Accepted: 13 December 2017;
Published: 22 December 2017
Edited by:
Subhash Verma, Chaudhary Sarwan Kumar Himachal Pradesh Krishi Vishvavidyalaya, IndiaReviewed by:
Aneesh Thakur, University of Copenhagen, DenmarkRalph Goethe, University of Veterinary Medicine Hannover, Germany
Copyright: © 2017 Acharya, Dhand, Whittington and Plain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Navneet K. Dhand, bmF2bmVldC5kaGFuZEBzeWRuZXkuZWR1LmF1