 Simone Scherrer
Simone Scherrer Patricia Landolt
Patricia Landolt Natasha Carroli1
Natasha Carroli1 Roger Stephan
Roger Stephan- 1Vetsuisse Faculty, Institute of Veterinary Bacteriology, University of Zurich, Zurich, Switzerland
- 2Vetsuisse Faculty, Institute for Food Safety and Hygiene, University of Zurich, Zurich, Switzerland
Mycobacterium avium subsp. hominissuis (MAH) is an important zoonotic pathogen with raising global health concerns. In humans, MAH is one of the most widespread non-tuberculous mycobacterial species responsible for lung disease. In animals, MAH is frequently isolated from pigs; however, it is also an opportunistic pathogen for other mammals including cattle. To elucidate the genetic diversity of MAH in cattle, a molecular characterization of isolates (n = 26) derived from lymph nodes was performed. Fourteen isolates originated from slaughtered cattle with visible altered lymph nodes at meat inspection, whereas 12 isolates were from lymph nodes without any gross pathological changes of healthy slaughtered cattle. Variable number of tandem repeat (VNTR) analysis was performed at 20 loci to examine genetic differences of isolates and to compare to previously reported VNTR data of human isolates from different countries. Genetic elements IS901, IS1245, IS1311, LSPA17, ITS1 sequevar, and hsp65 code were determined. Interestingly, two bovine MAH isolates harbored ISMav6 and hsp65 code 15, which so far has only been observed in human isolates. We supposed that VNTR data of Swiss samples would show clustering with European samples. Minimum spanning tree and unweighted pair group method using arithmetic averages analyses based on the VNTR data indicated a specific cluster of MAH isolates obtained from lymph nodes without any gross pathological changes of healthy slaughtered cattle. Comparing Swiss isolates with isolates from different other countries, no geographical clustering was observed; however, four Swiss isolates had an identical VNTR profile as human isolates from the Netherlands, the United States, and Japan. These findings indicate a possible public health issue.
Introduction
Non-tuberculous mycobacteria (NTM) are ubiquitous in the environment, mainly in water and soil (1). Recently, more and more new NTM species were discovered, probably because of improved culturing techniques and the development of new molecular methods. Currently, more than 150 Mycobacterium species are known (1). Mycobacterium avium subsp. hominissuis (MAH), a representative of the M. avium complex (MAC), is an environmental bacterium often found in water, soil, dust, or straw, and its main hosts are humans and pigs. It is also an opportunistic pathogen for other mammals including cattle, from which it is one of the most frequently isolated NTM (2, 3). M. avium infection in humans is leading to severe symptoms, whereas pigs are often subclinically infected (4). An experimental infection of goats with MAH performed by Schinköthe et al. demonstrated two different courses of disease with highly heterogenic lesions, systemic spread in goats with severe clinical disease and the development of granulomas of all stages in the surviving goats (5). Recently in Asia, a Japanese black beef steer was discovered to have MAH granulomas in the systemic organs, indicating that MAH can cause systemic mycobacteriosis in cattle (6). Especially in developed countries, MAH is frequently linked to human mycobacteriosis (7–9). Turenne et al. showed that MAH has the highest level of genomic heterogeneity within MAC. They concluded that M. avium subsp. paratuberculosis (MAP), M. avium subsp. avium (MAA), and M. avium subsp. silvaticum (MAS) evolved independently from MAH (10). Zoonotic aspects of MAH have been investigated to explore exposure risk for humans. Recent population wide genetic studies (11–13) have shown that MAH isolates from Japanese patients had a low degree of similarity with the Japanese pig isolates, whereas the isolates from European patients and pigs showed a high genetic relatedness with the Japanese pig isolates. In contrast to the human cases, pig isolates were more homogenous. To compare MAH isolates across geographical regions and in different hosts, variable number of tandem repeat (VNTR) typing analysis based on the eight loci [mycobacterial interspersed repetitive unit (MIRU)–VNTR] was developed (14). Another VNTR panel of 15 loci [M. avium tandem repeats (MATRs)–VNTR] was introduced (15). Both MIRU- and MATR-VNTR panels are useful for global epidemiological studies. VNTR has a high level of reproducibility and has the advantage of allowing a numerical and reproductive digitalization of typing data and therefore enabling an optimal comparison of results between laboratories (16).
To investigate the molecular features of Swiss bovine MAH, a collective of MAC isolates was gathered from two previous studies. Since MAH seemed to be the predominant NTM species in Swiss cattle, the aim of this study is to map molecular characteristics of MAH isolated from lymph nodes of cattle at slaughter. VNTR data were compared to previously reported data from different countries to elucidate genetic relatedness of isolates from Swiss cattle with international human isolates and to assess geographical differences in the genetic diversity of MAH.
Materials and Methods
Ethics Statement
This study was carried out in accordance with the recommendations of Swiss federal regulations (TSV 916.401 and VSFK 817.190). Analysis of animal specimens was carried out within an official context of monitoring bovine tuberculosis and NTM infections, meaning that no animals were killed for the purposes of this research project and ethical approval was not necessary.
Collection of Isolates
Due to some tuberculosis cases in the cattle population in Switzerland, altered lymph nodes from 534 cattle, tested suspicious or positive by either using intradermal tuberculin test alone or in combination with interferon-γ testing, were submitted between March 2013 and November 2014 for mycobacteriological analysis (group A). An average of three lymph nodes was processed from each animal. Lymph nodes were of retropharyngeal, mandibular, bronchial, or mediastinal origin. Mycobacterium tuberculosis complex (MTBC)-negative cultures resulting in 57 NTM isolates were obtained, and of those, 14 (24.6%) isolates were identified to be MAH. Furthermore, 108 lymph nodes without any gross pathological changes were collected from healthy slaughtered cattle in 2015 giving rise to 20 NTM cultures including 12 (60%) MAH isolates (group B). These 26 MAH isolates were included in this study.
Mycobacterial Culture and DNA Extraction
Mycobacterial culture was performed as described previously (17). Genomic DNA was extracted harvesting mycobacteria from 1.5 ml MGIT cultures by centrifugation for 10 min at 13,000 × g. The sediment was suspended in 180 µl ATL buffer (Qiagen, Hilden, Germany), transferred onto a bead beating matrix in a 2-ml microtube (Omni International, Kennesaw, USA), heat inactivated, and subjected to mechanical cell lysis using a TissueLyser II (Qiagen) and enzymatic digestion with Proteinase K (Qiagen). Automated DNA preparation was performed on the QIAcube instrument using the QIAamp cador Pathogen Mini Kit protocol (Qiagen). DNA concentration was measured using a NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific, Reinach, Switzerland) and stored at −20°C until use. Standard biosecurity procedures have been carried out for handling of samples.
Identification and Characterization of MAH
For identification of mycobacteria, 16S rRNA [16S rRNA forward primer: TTGGAGAGTTTGATCMTGGCTC (adapted from CLSI MM-18-A), sequencing primer 259: TTTCACGAACAACGCGACAA (18) and 16S rRNA reverse primer 1492R: TACGGYTACCTTGTTACGACTT (19)], rpoB (20), and hsp65 (21) were sequenced. Sequence analysis of the 3′ fragment of the hsp65 gene was performed by amplifying the near-complete hsp65 gene using primers MAChsp65F and MAChsp65R. To confirm the resulting hsp65 code based on the single-nucleotide polymorphism, Sanger sequencing was performed in duplicate (22). All isolates including four reference strains (MAH ATCC 700898, MAP ATCC 19698, MAA ATCC 25291 and MAS ATCC 49884) were tested by PCR for the presence of the IS901 element as described previously (22). Detection and sequencing of ISMav6 was performed by PCR using specific primer sets designed for MAH (8). Sequencing of the ITS1 region was performed as described previously (23, 24). Results were compared to ITS1 sequences in GenBank including Mav-A (GenBank accession number EF521901.1), Mav-B (GenBank accession number L07856), and Mav-F (GenBank accession number AF315838) sequences (7, 25). Large sequence polymorphisms (LSPs) are molecular markers of genetic diversity in both MTBC (26) and M. avium subspecies (27). LSPA17 was tested on all MAH isolates trying to differentiate presumable MAA being positive for IS901 from MAH, since the absence of LSPA17 is supposed to be characteristic for MAA strains, although not perfectly specific (28). IS1245 and IS1311 were detected according to the study by Johansen et al. (9).
The nucleotide sequences of 16S rRNA, rpoB and hsp65 genes obtained were compared with available sequences by BLAST analysis using the NCBI database.1 Alignment of sequences was performed using CLC Main Workbench 7.6.1 (CLC Bio, Qiagen, Germany).
VNTR Analysis
Variable number of tandem repeat typing was performed on 26 MAH isolates by amplification of 15 MATR (MATR-1, 2, 3, 4, 5, 6, 7, 8, 9, 11, 12, 13, 14, 15, and 16) (15) and 8 MIRU (MIRU-292, X3, 25, 47, 3, 7, 10, and 32) (14) loci as described earlier. Three loci overlapped between MIRU-VNTR and MATR-VNTR: MIRU-292 and MATR-2, MIRU-X3 and MATR-3, and MIRU-10 and MATR-9. Each reaction mixture contained 1× HotStart Taq Master Mix Kit, 1× Q-Solution (Qiagen) (only for loci MATR-5, 8, 9, 11, and 12 and MIRU-47), 0.5 µM of each primer pair and 1 ng purified mycobacterial DNA in a final volume of 10 µl. PCR was performed with an initial 15-min activation step at 95°C followed by 40 cycles of 95°C for 30 s, 60°C for 30 s, 72°C for 30 s, and a final extension step of 72°C for 10 min. 10 µl of each PCR amplification products were analyzed using a capillary electrophoresis device (QIAxcel, Qiagen), applying OH1700 AM10sec method with a QX DNA high-resolution cartridge, QX 15 bp–3 kb alignment marker and QX 100 bp–2.5 kb size marker. Peak size assignment and allele code exportation were performed with the QIAxcel ScreenGel Software version 1.3.0 (Qiagen). As a positive control, four reference strains (MAH ATCC 700898, MAP ATCC 19698, MAA ATCC 25291, and MAS ATCC 49884) were analyzed for each PCR run. For the MATR loci, the number of tandem repeats was determined from the size of amplicons. Repeat numbers (alleles) of MIRU-VNTR loci were assigned according to a previously described allele-calling table and arranged to profiles, called INMVs (INRA Nouzilly MIRU-VNTR), described by Thibault et al. (14). New profiles detected were registered in the MAC-INMV database.2 The allelic diversity index (h) of the different loci was calculated as described by Mazars et al. (29). The Hunter-Gaston Discriminatory Index (HGDI), representing a numerical index for the discriminatory power of a genotyping method, was determined according to the formula described by Hunter and Gaston (30).
Phylogenetic Analysis
Genetic strain lineages were predicted using online tools available from MIRU-VNTRplus website3 (31). A dendrogram based on 20 VNTR loci was generated using the unweighted pair group method using arithmetic averages (UPGMA) algorithm.
Variable number of tandem repeat data from a previous report (11) was retrieved for the geographical comparison of genetic diversity of MAH. A minimum spanning tree (MST) was generated based on 14 loci VTNR genotype data that combined our own data of the 26 Swiss isolates with previously published data using online tools available from MIRU-VNTRplus website. The MST classification into three main subgroups (Cluster A, B, and C) was adopted (11).
Results
Molecular Characterization of MAH
PCR for IS901 yielded positive results for the two reference strains MAA ATCC 25291 and MAS ATCC 49884. The IS901 PCR also produced an amplicon for MAH isolates ZH38 and ZH43 belonging to group B. Sequencing of the obtained PCR product from these isolates revealed a 100% sequence identity with the recently described ISMav6 from Japan (GenBank accession number AB447556.1). PCR for IS901 yielded negative results for all remaining MAH isolates including reference strain MAH ATCC 700898 and MAP ATCC 19698. PCR amplification of the ITS1 region resulted in a single amplicon of 480 bp for all MAC isolates. Sequence analysis revealed three M. avium sequevars, Mav-A, Mav-B, and Mav-F, whereas 21 isolates belonged to Mav-B (80.8%), 3 isolates to Mav-F (11.5%), and 2 isolates to Mav-A (7.7%). The major hsp65 sequevars of MAH isolates from Swiss cattle was code 1 (23.1%) and code 2 (65.4%), less frequently code 15 (7.7%) and a newly identified code N8 (3.8%) (Table 1). LSP analysis showed 12 isolates (46.2%) without LSPA17 and 14 isolates (53.8%) with LSPA17. Insertion element analysis revealed IS1245 positive for all except one isolate and IS1311 positive for all except two isolates (Table 2).

Table 1. Single-nucleotide polymorphisms among 26 Swiss Mycobacterium avium subsp. hominissuis isolates of this study in comparison with Mycobacterium avium 104.
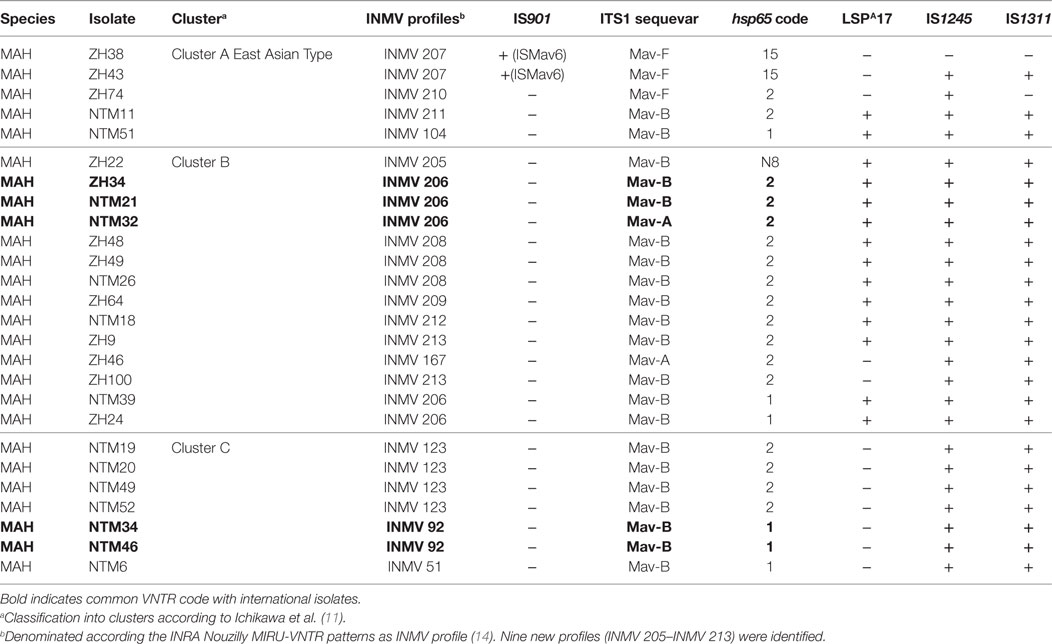
Table 2. Molecular characteristics of 26 Swiss Mycobacterium avium subsp. hominissuis (MAH) isolates evaluated in this study.
By assigning all molecular characteristics (ITS1 sequevar, hsp65 code, LSPA17, IS1245, IS1311) to geographical clusters distinctive features could be observed. Cluster C had most homogenous properties with ITS1 sequevar Mav-B: the absence of LSPA17 and the presence of IS1245 and IS1311 and hsp65 code of 1 or 2. Cluster A was heterogeneous; ITS1 sequevar was Mav-F and Mav-B, hsp65 code was 15, 2, and 1, and LSPA17, IS1245, and IS1311 were present or absent. In Cluster B, the distribution of molecular characteristics was heterogeneous as well; IS1245 and IS1311 were present, ITS1 sequevar was Mav-A or Mav-B, hsp65 code was either N8, 2, or 1, and LSPA17 was mostly present except for two isolates (Table 2).
VNTR Analysis
MIRU-VNTR analysis resulted in 14 profiles, called INMVs. Nine profiles (INMV 205–INMV 213) were newly identified. Diversity of combined genotypes was eight INMV profiles for both groups A and B. Two genotypes were shared (INMV 206 and INMV 208) by both groups. Eleven genotypes were only detected in one or two isolates. The three most frequently detected genotypes in this study were INMV 206, INMV 123, and INMV 208 (Table S1 in Supplementary Material).
Variable number of tandem repeat profiles including both 15 MATR loci and 5 MIRU loci from 26 MAH isolates (Table S1 in Supplementary Material) were used to calculate the allelic diversity (Table 3). In terms of discriminatory hierarchy loci MATR-3, MATR-9, MATR-2, MATR-7, and MATR-5 displayed the highest allelic diversity. In contrast, MATR-13, MIRU-3, MIRU-7, and MIRU-32 were monomorphic (Table 3). Three triplicates of isolates with identical VNTR profiles (ZH34, NTM21, NTM32; ZH48, ZH49, NTM26 and NTM19, NTM20, NTM49) were observed, resulting in 20 distinct allelic VNTR profiles (Figure 1). The HGDI was 0.972 revealing a good discriminatory power of the VNTR analysis.
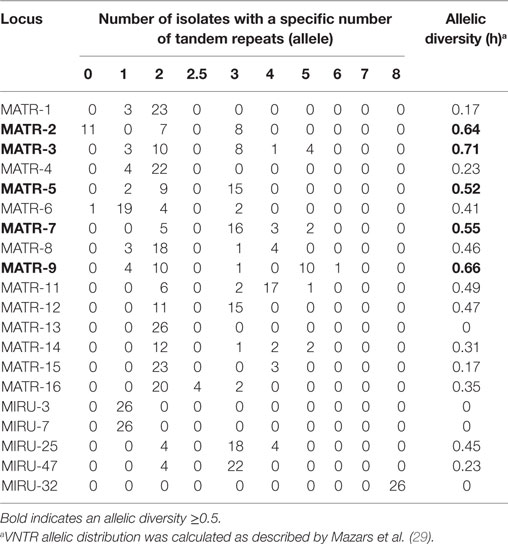
Table 3. MATR/MIRU allelic distribution and diversity among 26 Swiss Mycobacterium avium subsp. hominissuis isolates evaluated in this study.
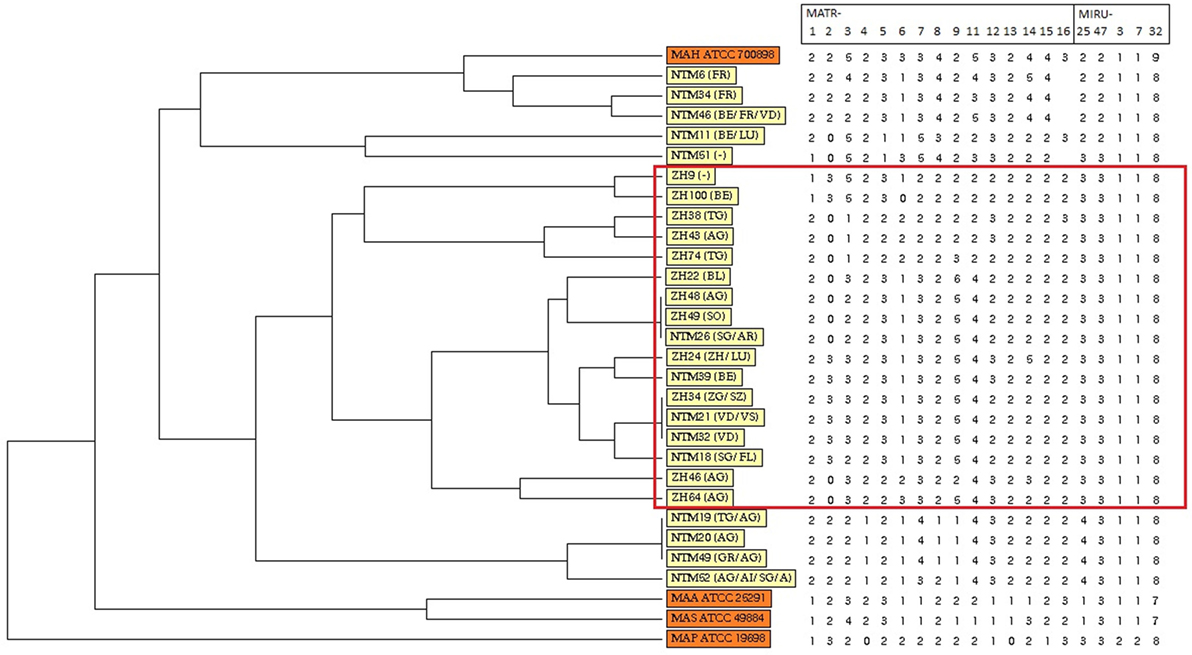
Figure 1. Dendrogram and allele profiles constructed from variable number of tandem repeat typing results of 26 Swiss Mycobacterium avium subsp. hominissuis (MAH) isolates including four reference strains. The dendrogram was generated with the unweighted pair group method using arithmetic average algorithm using tools available from the MIRU-VNTRplus database. Reference strains MAA ATCC 25291, MAS ATCC 49884, MAH ATCC 700898, and MAP ATCC 19698 are colored in orange, whereas all MAH isolates of this study are colored in yellow. The red box highlights a cluster of isolates from lymph nodes without any gross pathological changes of healthy, slaughtered cattle (group B).
Phylogenetic Analysis
Unweighted pair group method using arithmetic average analysis displayed a cluster of isolates deriving from lymph nodes without any gross pathological changes (Figure 1). MST analysis of MAH isolates from six different countries revealed a scattered distribution of Swiss isolates (Figure 2A).
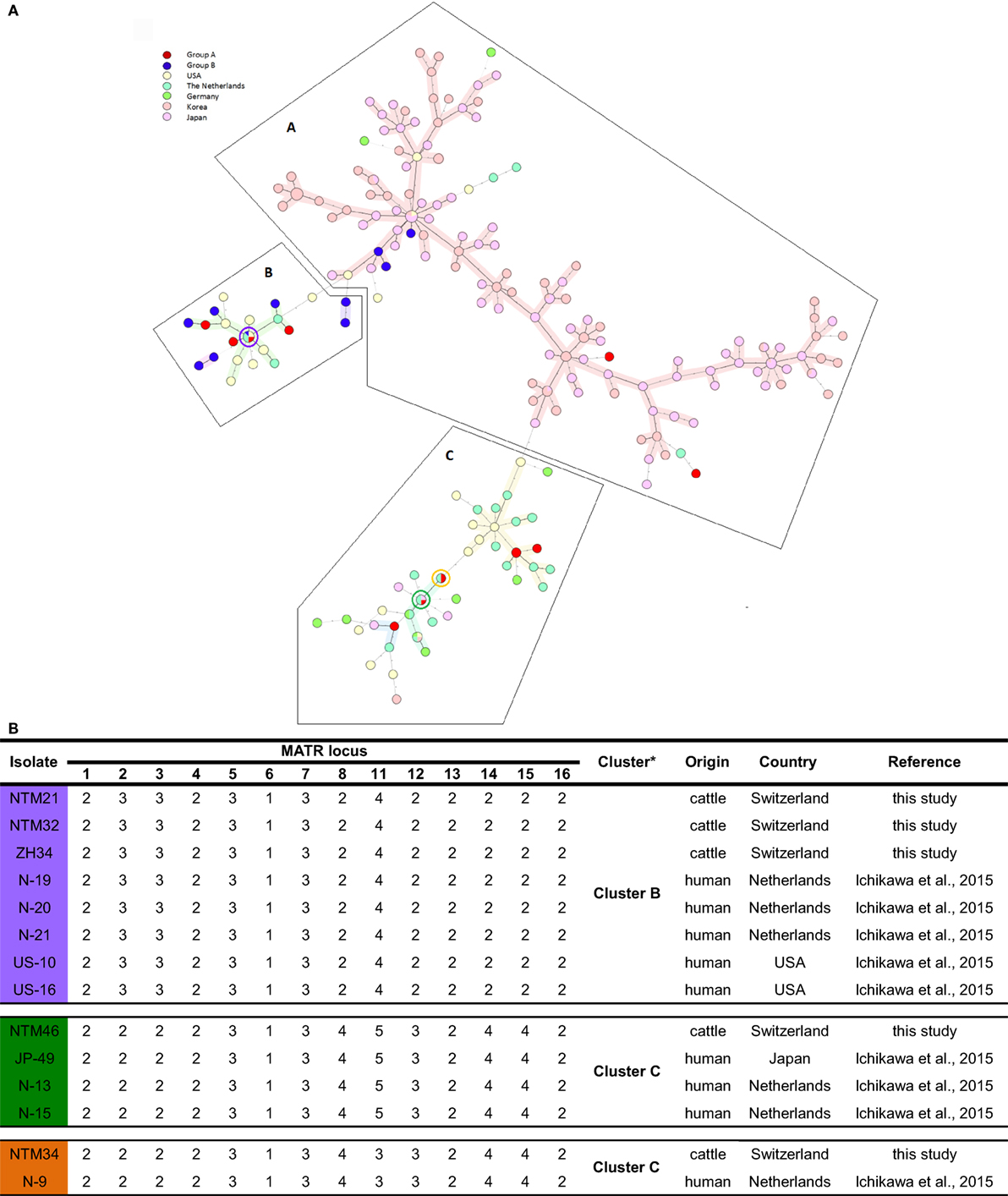
Figure 2. (A) Representation of Swiss Mycobacterium avium subsp. hominissuis (MAH) isolates in an international context using a minimum spanning tree based on the 14-MATR-VNTR genotyping of MAH isolates (26 isolates from Switzerland, 94 isolates from Japan, 98 isolates from Korea, 32 isolates from the United States, 27 isolates from The Netherlands, and 10 isolates from Germany). Red (group A) and blue (group B) represent Swiss isolates. Violet, green, and orange circles represent Swiss isolates with identical profiles to Dutch, American, and Japanese isolates. (B) VNTR profiles of Swiss isolates with common international VNTR profiles. *Classification in clusters based on MATR-VNTR proposed by Ichikawa et al. (11).
Among the population of isolates of group B, a predominant grouping in Cluster A (25%; 3/12) and Cluster B (75%; 9/12) was found. In Cluster C, none of those isolates were found. In contrast, for isolates of group A, a distribution in all three clusters was observed: Cluster A consisted of 14.3% (2/14), Cluster B of 35.7% (5/14), and Cluster C of 50% (7/14) of MAH isolates. In total, 19.2% (5/26) Swiss isolates were grouped into Cluster A, which represents the East Asian Type mainly, and 53.9% (14/26) isolates in Cluster B together with the United States and The Netherlands and 26.9% (7/26) in Cluster C, which is the most heterogeneous cluster harboring isolates from Asia, Europe, and the United States. As anticipated, the two ISMav6-positive isolates (ZH38 and ZH43) were found in Cluster A together with the isolates of Japan and Korea.
Four Swiss isolates showed an identical VNTR profile compared to isolates of different European and East Asian countries. NTM21 and NTM32 were identical to isolates N-19, N-20, and N-21 from the Netherlands and to isolates US-10 and US-16 from the United States; NTM34 was identical to isolate N-9 from the Netherlands, and NTM46 was identical to isolate JP-49 from Japan and to isolates N-13 and N-15 from the Netherlands (Figure 2B).
Discussion
This study showed a good discriminatory power of PCR-based VNTR analysis and the usefulness to compare resulting VNTR genetic profiles using global sample sets. The UPGMA and MST analysis presented a genetically heterogeneous set of Swiss isolates and a widespread geographical distribution compared to other countries.
To investigate the genetic relatedness of MAH isolates in greater detail, UPGMA analysis was preformed based on 20 loci VNTR profile of the isolates. A cluster of MAH isolates deriving from lymph nodes of group B was revealed, indicating a possible relationship between VNTR profile and virulence. Comparing each VNTR locus of isolates of group A and B, it could be observed that allelic distribution of five loci (MATR-4, MATR-8, MATR-15, MIRU-25, and MIRU-47) was monomorphic (Table S1 in Supplementary Material) for isolates from group B in comparison to isolates from group A. However, one limitation of this study is that group A and group B were determined by gross analysis of the lymph nodes. Therefore, no validation of group designations has been performed.
A partial geographical correlation could be observed between the main clusters and the origin of MAH isolates. In one case, animals collected on the same farm (NTM19, NTM20, and NTM49) revealed one distinct VNTR profile. Of cattle showing an identical VNTR profile (NTM21, NTM32, and ZH34), only two cattle (NTM21 and NTM32) were from the same canton, but not from the same farm. Cattle harboring ZH48, ZH49, and NTM26 with an identical VNTR profile were in non-neighboring cantons. On the other hand, there were cattle (NTM6 and NTM34) originating from the same canton but showing different distinct profiles. Furthermore, one case of a geographical cluster with three animals of the same farm (NTM19, NTM20, and NTM49) could be observed. The three isolates showed an identical VNTR profile, indicating a common infection source. The affected cattle were slaughtered at the age of 5 and 6 months and 1 year, respectively, indicating an infection at young age. A fourth cow (NTM52) from the same farm showed a variation of VNTR profile in MATR-7 and MATR-8. This cow had a trading history within Switzerland and Austria. Presumably, the infection source could be within the farm from an animal with a mixed infection of two MAH subtypes or it could arise from a closely related MAH genotype present in the nearby region. Heterogeneity within a sample possibly containing different subtypes was not studied since only one colony of the original culture was subcultured. While studying the trading histories of involved animals, no other clusters reflecting a direct contact between cattle could be discovered. An absence of correlation between allelic profiles and geographical sample source of isolates could be explained in indirect transmission of MAH through common environmental sources, other animals, or humans. Several epidemiological studies of MAH in Europe found no correlation between genetic profiles and geographical origins, as well (32–36).
ISMav6, a homologous insertion sequence to the original bird-type IS901 with 95% sequence identity (8), was found in two MAH isolates comprising hsp65 sequevar code 15. This combination of ISMav6 together with hsp65 sequevar code 15 has been described for Japanese clinical isolates often recovered from sputum (8, 37) and more recently from Korean clinical isolates, where a high prevalence of ISMav6 was observed (38). Adachi et al. compared ISMav6 in human and swine isolates detecting ISMav6 in human samples only (39). In a human patient from Germany, one MAH isolate with hsp65 code 15 harboring ISMav6 was reported (40). In Belgium, three human isolates containing ISMav6 were found, and one of those isolates was IS1245 positive and two were IS1245 negative. However, hsp65 sequevar of those Belgian isolates was code 1 and 2, respectively (41). As IS901 is believed to have an influence on the pathogenicity of MAA (42, 43), ISMav6 may also be related to the pathogenicity of MAH (8). Interestingly, in this study, ISMav6 was detected for the first time from animal origin.
Remarkably, in Cluster B and C, four Swiss isolates showed identical VNTR profiles compared to isolates from the Netherlands, the United States, and Japan. Identical VNTR genetic profiles could be an indication for epidemiological linkage. Or ultimately, homologous genotypes may have evolved in parallel. Among the 5 Swiss MAH isolates belonging to Cluster A, two isolates with similar 14 loci VNTR profiles to East Asian isolates were ISMav6 positive, indicating that MAH isolates could have been introduced to Switzerland form East Asia. Nevertheless, isolates with identical VNTR profiles do not necessarily perfectly reflect phylogenetic relationship because there could be differences in other genetic elements, indicating only distantly related isolates. This could be observed in isolates ZH34, NTM21, and NTM32 sharing the same VNTR copy numbers but different ITS1 sequevar, namely Mav-B for NTM21 and NTM32 and Mav-A for ZH34. Consequently, VNTR could give some insight in the epidemiology of strains evolving worldwide and its connections; however, data should be critically analyzed. To globally analyze VNTR data, it is of advantage to use an extended set of VNTR loci rather than reducing VNTR data to a country specific set of loci with high allelic diversity and to include other molecular characteristics as ITS1 and hsp65 sequevar. We would propose combining MATR and MIRU loci into a single panel of 20 loci VNTR, serving as global standard for international comparison of genotyping data, thereby minimizing false assignment of epidemiological linkage. With such a creation of an international VNTR data base, it could be possible to better understand the global pattern of MAH genetics and therefore achieve better control in MAH infections.
Conclusion
We could show a clustering in the distribution of VNTR profiles of isolates from lymph nodes without any gross pathological changes of healthy slaughtered cattle in comparison to isolates from slaughtered cattle with visible altered lymph nodes indicating a possible link to virulence. Further studies elucidating subtractive differences of the genome of MAH isolates of healthy cattle versus isolates of animals with altered lymph nodes would give rise to some insight into differences in virulence.
The geographical comparison of genetic diversity of MAH indicated no distinct distribution of Swiss isolates related to international clusters. Remarkably, some Swiss isolates showed an identical VNTR code in 14 loci as in human isolates causing pulmonary diseases from the Netherlands, the United States, and Japan. Moreover, the detection of ISMav6, which was observed for the first time in animals and otherwise, is frequently distributed in human isolates in the East Asian part of the world, underlines a great genomic variability of Swiss MAH isolates from cattle.
Author Contributions
SS and RS designed and coordinated the study. PL, NC, and SS performed the experiments. SS and PL interpreted the data. PL sketched the figures. SS drafted the manuscript. RS reviewed and edited the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ella Hübschke and Ute Friedel for the excellent technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fvets.2018.00032/full#supplementary-material.
Footnotes
References
1. Johnson MM, Odell JA. Nontuberculous mycobacterial pulmonary infections. J Thorac Dis (2014) 6:210–20. doi:10.3978/j.issn.2072-1439.2013.12.24
2. Biet F, Boschiroli ML. Non-tuberculous mycobacterial infections of veterinary relevance. Res Vet Sci (2014) 97(Suppl):S69–77. doi:10.1016/j.rvsc.2014.08.007
3. Dvorska L, Matlova L, Bartos M, Parmova I, Bartl J, Svastova P, et al. Study of Mycobacterium avium complex strains isolated from cattle in the Czech Republic between 1996 and 2000. Vet Microbiol (2004) 99:239–50. doi:10.1016/j.vetmic.2004.01.008
4. Agdestein A, Olsen I, Jorgensen A, Djonne B, Johansen TB. Novel insights into transmission routes of Mycobacterium avium in pigs and possible implications for human health. Vet Res (2014) 45:46. doi:10.1186/1297-9716-45-46
5. Schinköthe J, Mobius P, Kohler H, Liebler-Tenorio EM. Experimental infection of goats with Mycobacterium avium subsp. hominissuis: a model for comparative tuberculosis research. J Comp Pathol (2016) 155:218–30. doi:10.1016/j.jcpa.2016.06.008
6. Komatsu T, Inaba N, Kondo K, Nagata R, Kawaji S, Shibahara T. Systemic mycobacteriosis caused by ‘Mycobacterium avium subspecies hominissuis’ in a 14-month-old Japanese black beef steer. J Vet Med Sci (2017) 79:1384–8. doi:10.1292/jvms.17-0204
7. Mijs W, de Haas P, Rossau R, Van der Laan T, Rigouts L, Portaels F, et al. Molecular evidence to support a proposal to reserve the designation Mycobacterium avium subsp. avium for bird-type isolates and ‘M. avium subsp. hominissuis’ for the human/porcine type of M. avium. Int J Syst Evol Microbiol (2002) 52:1505–18. doi:10.1099/00207713-52-5-1505
8. Ichikawa K, Yagi T, Moriyama M, Inagaki T, Nakagawa T, Uchiya K, et al. Characterization of Mycobacterium avium clinical isolates in Japan using subspecies-specific insertion sequences, and identification of a new insertion sequence, ISMav6. J Med Microbiol (2009) 58:945–50. doi:10.1099/jmm.0.008623-0
9. Johansen TB, Djonne B, Jensen MR, Olsen I. Distribution of IS1311 and IS1245 in Mycobacterium avium subspecies revisited. J Clin Microbiol (2005) 43:2500–2. doi:10.1128/jcm.43.5.2500-2502.2005
10. Turenne CY, Collins DM, Alexander DC, Behr MA. Mycobacterium avium subsp. paratuberculosis and M. avium subsp. avium are independently evolved pathogenic clones of a much broader group of M. avium organisms. J Bacteriol (2008) 190:2479–87. doi:10.1128/jb.01691-07
11. Ichikawa K, van Ingen J, Koh WJ, Wagner D, Salfinger M, Inagaki T, et al. Genetic diversity of clinical Mycobacterium avium subsp. hominissuis and Mycobacterium intracellulare isolates causing pulmonary diseases recovered from different geographical regions. Infect Genet Evol (2015) 36:250–5. doi:10.1016/j.meegid.2015.09.029
12. Uchiya KI, Tomida S, Nakagawa T, Asahi S, Nikai T, Ogawa K. Comparative genome analyses of Mycobacterium avium reveal genomic features of its subspecies and strains that cause progression of pulmonary disease. Sci Rep (2017) 7:39750. doi:10.1038/srep39750
13. Yano H, Iwamoto T, Nishiuchi Y, Nakajima C, Starkova DA, Mokrousov I, et al. Population structure and local adaptation of MAC lung disease agent Mycobacterium avium subsp. hominissuis. Genome Biol Evol (2017) 9:2403–17. doi:10.1093/gbe/evx183
14. Thibault VC, Grayon M, Boschiroli ML, Hubbans C, Overduin P, Stevenson K, et al. New variable-number tandem-repeat markers for typing Mycobacterium avium subsp. paratuberculosis and M. avium strains: comparison with IS900 and IS1245 restriction fragment length polymorphism typing. J Clin Microbiol (2007) 45:2404–10. doi:10.1128/jcm.00476-07
15. Inagaki T, Nishimori K, Yagi T, Ichikawa K, Moriyama M, Nakagawa T, et al. Comparison of a variable-number tandem-repeat (VNTR) method for typing Mycobacterium avium with mycobacterial interspersed repetitive-unit-VNTR and IS1245 restriction fragment length polymorphism typing. J Clin Microbiol (2009) 47:2156–64. doi:10.1128/jcm.02373-08
16. Supply P, Lesjean S, Savine E, Kremer K, van Soolingen D, Locht C. Automated high-throughput genotyping for study of global epidemiology of Mycobacterium tuberculosis based on mycobacterial interspersed repetitive units. J Clin Microbiol (2001) 39:3563–71. doi:10.1128/jcm.39.10.3563-3571.2001
17. Ghielmetti G, Scherrer S, Friedel U, Frei D, Suter D, Perler L, et al. Epidemiological tracing of bovine tuberculosis in Switzerland, multilocus variable number of tandem repeat analysis of Mycobacterium bovis and Mycobacterium caprae. PLoS One (2017) 12:e0172474. doi:10.1371/journal.pone.0172474
18. Kirschner P, Bottger EC. Species identification of mycobacteria using rDNA sequencing. Methods Mol Biol (1998) 101:349–61. doi:10.1385/0-89603-471-2:349
19. Lane DJ. 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M, editors. Nucleic Acid Techniques in Bacterial Systematics. New York: John Wiley & Sons (1991). p. 115–75.
20. Adekambi T, Colson P, Drancourt M. rpoB-based identification of nonpigmented and late-pigmenting rapidly growing mycobacteria. J Clin Microbiol (2003) 41:5699–708. doi:10.1128/JCM.41.12.5699-5708.2003
21. Telenti A, Marchesi F, Balz M, Bally F, Bottger EC, Bodmer T. Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J Clin Microbiol (1993) 31:175–8.
22. Turenne CY, Semret M, Cousins DV, Collins DM, Behr MA. Sequencing of hsp65 distinguishes among subsets of the Mycobacterium avium complex. J Clin Microbiol (2006) 44:433–40. doi:10.1128/jcm.44.2.433-440.2006
23. Frothingham R, Wilson KH. Molecular phylogeny of the Mycobacterium avium complex demonstrates clinically meaningful divisions. J Infect Dis (1994) 169:305–12. doi:10.1093/infdis/169.2.305
24. Stout JE, Hopkins GW, McDonald JR, Quinn A, Hamilton CD, Reller LB, et al. Association between 16S-23S internal transcribed spacer sequence groups of Mycobacterium avium complex and pulmonary disease. J Clin Microbiol (2008) 46:2790–3. doi:10.1128/jcm.00719-08
25. Frothingham R, Wilson KH. Sequence-based differentiation of strains in the Mycobacterium avium complex. J Bacteriol (1993) 175:2818–25. doi:10.1128/jb.175.10.2818-2825.1993
26. Mostowy S, Cousins D, Brinkman J, Aranaz A, Behr MA. Genomic deletions suggest a phylogeny for the Mycobacterium tuberculosis complex. J Infect Dis (2002) 186:74–80. doi:10.1086/341068
27. Semret M, Zhai G, Mostowy S, Cleto C, Alexander D, Cangelosi G, et al. Extensive genomic polymorphism within Mycobacterium avium. J Bacteriol (2004) 186:6332–4. doi:10.1128/jb.186.18.6332-6334.2004
28. Semret M, Turenne CY, de Haas P, Collins DM, Behr MA. Differentiating host-associated variants of Mycobacterium avium by PCR for detection of large sequence polymorphisms. J Clin Microbiol (2006) 44:881–7. doi:10.1128/jcm.44.3.881-887.2006
29. Mazars E, Lesjean S, Banuls AL, Gilbert M, Vincent V, Gicquel B, et al. High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc Natl Acad Sci U S A (2001) 98:1901–6. doi:10.1073/pnas.98.4.1901
30. Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J Clin Microbiol (1988) 26:2465–6.
31. Weniger T, Krawczyk J, Supply P, Niemann S, Harmsen D. MIRU-VNTRplus: a web tool for polyphasic genotyping of Mycobacterium tuberculosis complex bacteria. Nucleic Acids Res (2010) 38:W326–31. doi:10.1093/nar/gkq351
32. Turenne CY, Wallace R Jr, Behr MA. Mycobacterium avium in the postgenomic era. Clin Microbiol Rev (2007) 20:205–29. doi:10.1128/cmr.00036-06
33. Radomski N, Thibault VC, Karoui C, de Cruz K, Cochard T, Gutierrez C, et al. Determination of genotypic diversity of Mycobacterium avium subspecies from human and animal origins by mycobacterial interspersed repetitive-unit-variable-number tandem-repeat and IS1311 restriction fragment length polymorphism typing methods. J Clin Microbiol (2010) 48:1026–34. doi:10.1128/jcm.01869-09
34. Tirkkonen T, Pakarinen J, Rintala E, Ali-Vehmas T, Marttila H, Peltoniemi OA, et al. Comparison of variable-number tandem-repeat markers typing and IS1245 restriction fragment length polymorphism fingerprinting of Mycobacterium avium subsp. hominissuis from human and porcine origins. Acta Vet Scand (2010) 52:21. doi:10.1186/1751-0147-52-21
35. Alvarez J, Castellanos E, Romero B, Aranaz A, Bezos J, Rodriguez S, et al. Epidemiological investigation of a Mycobacterium avium subsp. hominissuis outbreak in swine. Epidemiol Infect (2011) 139:143–8. doi:10.1017/s0950268810001779
36. Pate M, Kusar D, Zolnir-Dovc M, Ocepek M. MIRU-VNTR typing of Mycobacterium avium in animals and humans: heterogeneity of Mycobacterium avium subsp. hominissuis versus homogeneity of Mycobacterium avium subsp. avium strains. Res Vet Sci (2011) 91:376–81. doi:10.1016/j.rvsc.2010.10.001
37. Iwamoto T, Nakajima C, Nishiuchi Y, Kato T, Yoshida S, Nakanishi N, et al. Genetic diversity of Mycobacterium avium subsp. hominissuis strains isolated from humans, pigs, and human living environment. Infect Genet Evol (2012) 12:846–52. doi:10.1016/j.meegid.2011.06.018
38. Kim SY, Jeong BH, Park HY, Jeon K, Han SJ, Shin SJ, et al. Association of ISMav6 with the pattern of antibiotic resistance in Korean Mycobacterium avium clinical isolates but no relevance between their genotypes and clinical features. PLoS One (2016) 11:e0148917. doi:10.1371/journal.pone.0148917
39. Adachi T, Ichikawa K, Inagaki T, Moriyama M, Nakagawa T, Ogawa K, et al. Molecular typing and genetic characterization of Mycobacterium avium subsp. hominissuis isolates from humans and swine in Japan. J Med Microbiol (2016) 65:1289–95. doi:10.1099/jmm.0.000351
40. Kolb J, Hillemann D, Mobius P, Reetz J, Lahiri A, Lewin A, et al. Genetic characterization of German Mycobacterium avium strains isolated from different hosts and specimens by multilocus sequence typing. Int J Med Microbiol (2014) 304:941–8. doi:10.1016/j.ijmm.2014.06.001
41. Vluggen C, Soetaert K, Duytschaever L, Denoel J, Fauville-Dufaux M, Smeets F, et al. Genotyping and strain distribution of Mycobacterium avium subspecies hominissuis isolated from humans and pigs in Belgium, 2011-2013. Euro Surveill (2016) 21:30111. doi:10.2807/1560-7917.es.2016.21.3.30111
42. Kunze ZM, Wall S, Appelberg R, Silva MT, Portaels F, McFadden JJ. IS901, a new member of a widespread class of atypical insertion sequences, is associated with pathogenicity in Mycobacterium avium. Mol Microbiol (1991) 5:2265–72. doi:10.1111/j.1365-2958.1991.tb02157.x
Keywords: Mycobacterium avium subsp. hominissuis, variable number of tandem repeat, ISMav6, hsp65 code, ITS1 sequevar, cattle, lymph nodes
Citation: Scherrer S, Landolt P, Carroli N and Stephan R (2018) Molecular Characterization of Mycobacterium avium subsp. hominissuis of Two Groups of Lymph Nodes, Being Intradermal Tuberculin or Interferon-Gamma Test Positive and Negative, Isolated from Swiss Cattle at Slaughter. Front. Vet. Sci. 5:32. doi: 10.3389/fvets.2018.00032
Received: 07 November 2017; Accepted: 15 February 2018;
Published: 05 March 2018
Edited by:
Subhash Verma, Chaudhary Sarwan Kumar Himachal Pradesh Krishi Vishvavidyalaya, IndiaReviewed by:
Sunil Kumar Mor, University of Minnesota Twin Cities, United StatesKaori Sakamoto, University of Georgia, United States
Copyright: © 2018 Scherrer, Landolt, Carroli and Stephan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simone Scherrer, c2ltb25lLnNjaGVycmVyQHZldGJha3QudXpoLmNo