 Zhao Huang1,2†
Zhao Huang1,2† Zhiying Xu1,2†Haoxuan Cao1,2Fanliang Zeng1,2
Zhiying Xu1,2†Haoxuan Cao1,2Fanliang Zeng1,2 Heng Wang1,2,3
Heng Wang1,2,3 Lang Gong1,2Shengxun Zhang4Sen Cao5
Lang Gong1,2Shengxun Zhang4Sen Cao5 Guihong Zhang1,3,6,7*
Guihong Zhang1,3,6,7* Zezhong Zheng1,3*
Zezhong Zheng1,3*- 1Key Laboratory of Zoonosis Prevention and Control of Guangdong Province, College of Veterinary Medicine, South China Agricultural University, Guangzhou, China
- 2African Swine Fever Regional Laboratory of China, Guangzhou, China
- 3Research Center for African Swine Fever Prevention and Control, South China Agricultural University, Guangzhou, China
- 4Daguang Food Group Co., Ltd., Jiangmen, China
- 5Haifeng Animal Disease Prevention and Control Center, Shanwei, China
- 6Maoming Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Guangdong, China
- 7Key Laboratory of Animal Vaccine Development, Ministry of Agriculture and Rural Affairs, Guangzhou, China
To date, there is no effective vaccine or antiviral therapy available to prevent or treat African swine fever virus (ASFV) infections. ASFV gene deletion strains have been proposed as promising anti-ASFV vaccine candidates. In recent years, most ASFV gene deletion strains worldwide have been recombinant strains expressing EGFP or mCherry as markers. Therefore, in this study, a new triplex real-time PCR (RT-PCR) method was established for the broad and accurate differentiation of ASFV wild-type vs. gene deletion strains. We designed three pairs of primers and probes to target B646L, EGFP, and mCherry, and RT-PCR was used to detect these three genes simultaneously. The detection method prevented non-specific amplification of porcine reproductive and respiratory syndrome virus, porcine epidemic diarrhea virus, circovirus type 2, pseudorabies virus, and classical swine fever virus genes. The minimum copy number of standard plasmid DNA detected using triplex RT-PCR was 9.49, 15.60, and 9.60 copies for B646L, EGFP, and mCherry, respectively. Importantly, of the 1646 samples analyzed in this study, 67 were positive for ASFV, all corresponding to the wild-type virus. Overall, our data show that the triplex RT-PCR method established in this study can specifically identify both ASFV wild-type and gene deletion strains.
Introduction
African swine fever (ASF) is an acute and severe infectious disease caused by the African swine fever virus (ASFV), with a mortality rate of 100%. At present, there is no approved safe and effective vaccine, making ASF prevention and control extremely serious and challenging (1). According to the sequence variation of the C-terminal region of the capsid protein p72 encoded by B646L, ASFV can be classified into 22 genotypes (2). ASF was first reported in China in August 2018, caused by the ASFV genotype II strain (3).
When ASF emerges in a new area or pig population, it is usually accompanied by high mortality and rapid spread among pigs, leading to rapid outbreaks (4). Therefore, viral detection is very important for the rapid implementation of control measures. The viral detection methods available today include virus isolation, PCR detection of the viral genome, enzyme-linked immunosorbent assays (ELISAs), direct immunofluorescence tests (DIF), and the detection of viral antigens (5). Notably, virus isolation is regarded as the gold standard for ASFV detection by the Office International Des Epizooties (OIE) (6); however, this method requires multiple materials, consumes time, and is unsuitable for large-scale detection. On the contrary, PCR detection of ASFV has high efficiency, high sensitivity, and high specificity; therefore, it is widely used, especially in the context of tissues that are not suitable for virus isolation (7, 8).
At present, safe and effective anti-ASFV vaccine is unavailable in the market. However, attenuated vaccines (after ASFV gene deletion) have emerged as the most promising anti-ASFV vaccine candidates (2). For instance, Borca et al. generated the recombinant strain ASFVΔI177L by replacing I177L with mCherry, and showed that its use protected 100% of the pigs infected with the parental strain ASFV-G; of note, these authors further developed a cell line challenged with ASFVΔI177L for high-volume production (9, 10). Additionally, Chen et al. also used mCherry to replace MGF505-1R-MGF505-3R, as well as EGFP, to replace CD2v generating the strain ASFVΔCD2vΔMGF; this new attenuated strain could also protect 100% of pigs infected with the parental strain (11).
The purification of ASFV, particularly genotype II strains, is challenging since plaque formation is difficult in the context of porcine alveolar macrophages. Borca et al. have shown that fluorescent labeling is a suitable tool for the construction of ASFV gene-deficient strains for pathogenesis studies in pigs, studies of virus-macrophage interactions, and large-scale screens that require sensitive high-throughput output (12). Since, EGFP, mCherry, and RFP are commonly used as markers in ASFV gene-deficient strains, we aimed to establish a triplex RT-PCR method for the simultaneous detection of B646L, EGFP, and mCherry, and the consequent differentiation of ASFV wild-type and gene deletion strains. Such a tool might be essential in the future if ASFV gene deletion strains are established as successful anti-ASFV vaccines to prevent the mortality associated with ASF; assays that can broadly differentiate between wild-type ASFV and deletion strains will become very important.
Materials and Methods
Clinical Samples
Samples collected in South China from December 2018 to May 2020 were tested via multiplex PCR at the national ASF reference laboratory; 1646 clinical samples were collected, including 706 blood samples, 336 environmental swabs, 524 oral swabs, 35 tonsil samples, and 45 lymph node samples.
Viral Nucleic Acid Samples
The nucleic acids of ASFV, CD2v- and MGF-deficient ASFV, (ASFVΔCD2vΔMGF), porcine reproductive and respiratory syndrome virus (PRRSV), classical swine fever virus (CSFV), circovirus type 2 (PCV-2), pseudorabies virus (PRV), porcine epidemic diarrhea virus (PEDV) and porcine alveolar macrophages (PAMs) were preserved in the Infectious Diseases Department of the Veterinary College of South China Agricultural University.
Primer and Probe Design
The primers for amplifying B646L, EGFP, and mCherry were designed according to the conserved regions of the respective genes published in GenBank. Notably, the primers for amplifying B646L were designed according to the complete ASFV type I-XXIV sequence. The 5' end modifying group chosen for the B646L probe was FAM and the 3' end quenching group used was MGB. The 5' end modifying group chosen for the EGFP probe was HEX and the 3' end quenching group used was BHQ1. Additionally, the 5' end modifying group chosen for the mCherry probe was Cy5 and the 3' end quenching group used was BHQ3. Table 1 shows the sequences of the designed synthetic primer probes and the expected size of the amplicons.
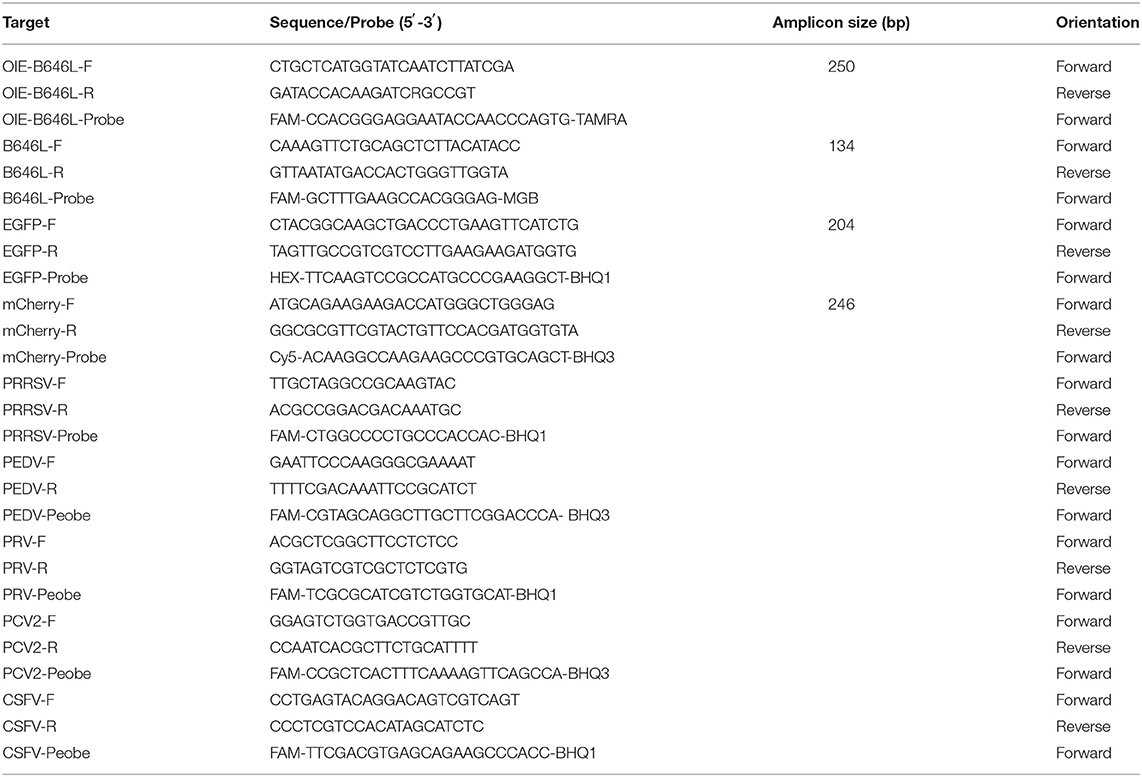
Table 1. List of primers used in this study.
DNA Extraction and Amplification
The tissue samples were ground using a grinding rod and soaked in phosphate buffer solution; the oral and nasal swabs were directly soaked in PBS. The samples were then centrifuged and the supernatants were collected to extract nucleic acids. The nucleic acids were extracted using a nucleic acid kit (Axygen, Hangzhou, China) and then reversely transcribed using a reverse transcription kit (Takara Beijing, China). All nucleic acid samples were stored at −20°C until further use.
Triplex qPCR
Triplex RT-PCR was performed using Bio-Rad CFX Manager 3.1 (Bio-Rad, Shanghai, China). The reaction mixture was composed of 2 μL of viral DNA, 10 μL of 2 × AceQ Universal U+ Probe Master Mix V2 (Vazyme, China), 0.4 μL of forward and reverse primers for B646L, EGFP, and mCherry (10 μM), 0.2 μL of B646L, EGFP, and mCherry TaqMan probe (10 μM), and an appropriate volume of ddH2O to a final volume of 20 μL. The reaction conditions were 40 cycles at 37°C for 2 min, 95°C for 5 min, 95°C for 10 s, and 60°C for 30 s. Importantly, negative controls were used for each test.
Standard Plasmids
The recombinant plasmids pUC57-EGFP, pMD18-B646L, and pUC57-mCherry were obtained from the Department of Infectious Diseases, School of Veterinary Medicine, South China Agricultural University.
Specificity of the Triplex qPCR
The nucleic acids extracted from wild-type ASFV, ASFVΔCD2vΔMGF, PRRSV, PEDV, PCV-2, PRV, CSFV and PAMs (Negative control, NC), were assayed using the triplex RT-PCR.
Sensitivity Verification
The concentration of the pUC57-EGFP, pMD18-B646L, and pUC57-mCherry standard plasmids was determined using the Thermo Nanodrop Lite apparatus (Thermofisher Scientific, Shanghai, China), and then serial dilutions (10 folds) were performed. The diluted plasmids were then subjected to triplex RT-PCR. The copy numbers of pUC57-EGFP, pMD18-B646L, and pUC57-mCherry were determined according to the DNA copy number calculation formula [dsDNA copy number (copies/mL) = 6.02 × 1023(copies/mol) × concentration (g/mL) / DNA length × 660], which were 1.17 × 1011 copies/μL, 1.60 × 1011 copies/μL, and 1.00 × 1011 copies/μL, respectively.
Results
Sequence Alignment of the Primers and Probes
In this study, primers and probes were designed based on the B646L sequence of the 24 ASFV genotypes (Figure 1); notably, the B464L sequence-based genetic evolution was also investigated in the context of the 24 ASFV genotypes (Figure 1A). The black triangles highlight the strains prevalent in China. Importantly, the designed probes were relatively conserved considering the B646L sequence of the 24 genotypes (Figure 1B), while the upstream primers showed mismatches in the the ASFV SPEC/125 genotype (Genotype XIX). Mismatches in the probes were present, particularly in the ASFV MK200 (Genotype V), MOZ/94/1 (Genotype VI), BOT/1/199 (Genotype III), Hinde1/1 (Genotype X), and UGA2003/1 (Genotype IX) genotypes. Furthermore, the downstream primers also showed mismatches in SPEC/260 (Genotype VII), UGA2003/1 (Genotype IX), Hinde/1 (Genotype X), RSA/1/99/W (Genotype IV), SPEC/125 genotype (Genotype XIX), and ETH/1 (Genotype XXIII) genotypes. Remarkably, with respect to the primers/probes designed in this study, only the SPEC/125 genotype (Genotype XIX), Hinde/1 (Genotype X) and UGA/2003/1 (Genotype IX) genotypes showed two mutations; the rest of the strains only showed one base mutation. Importantly, considering the popular Gene II strains in China, the probes/primers designed in this study did not contain any mutation sites.
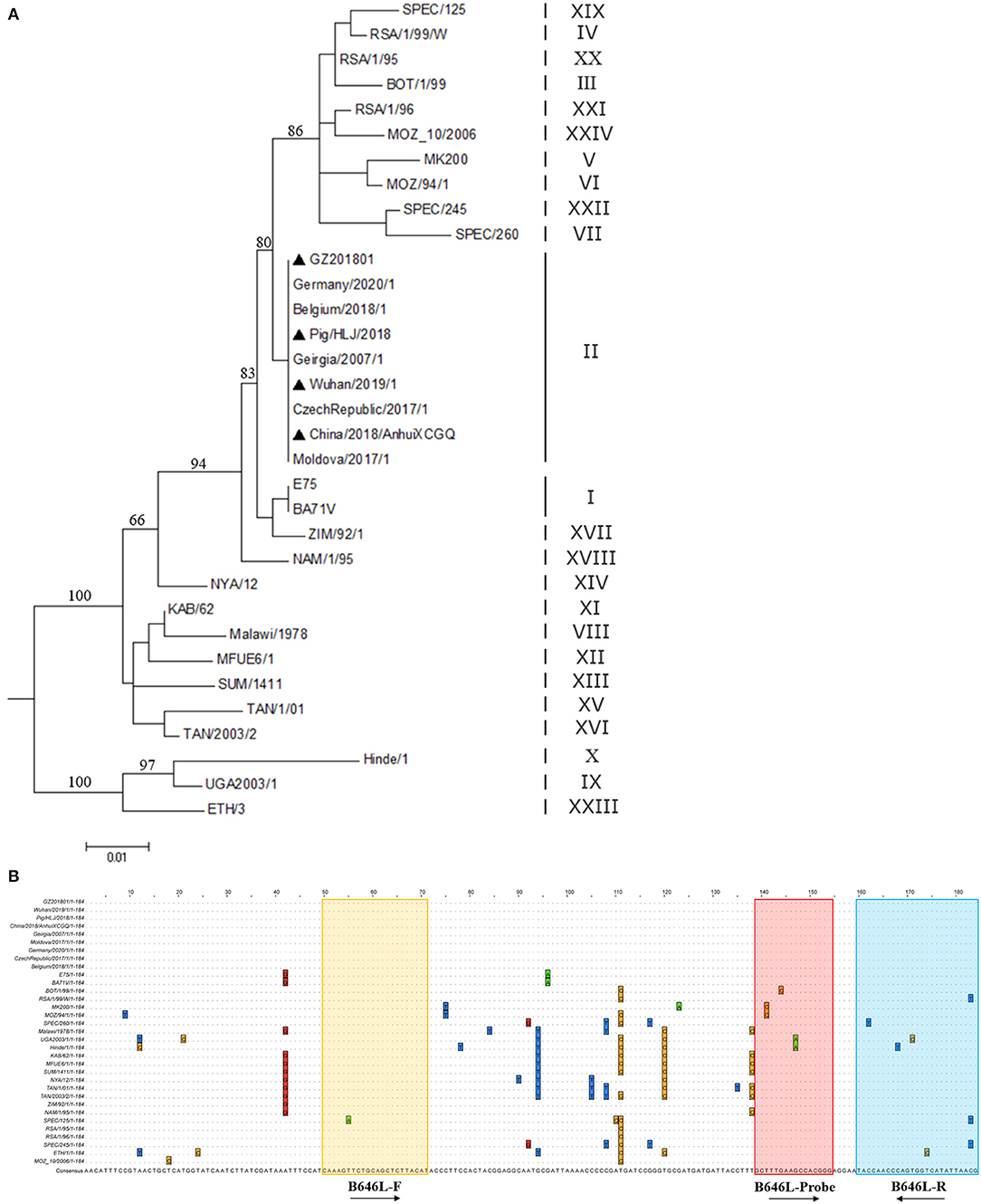
Figure 1. Genetic evolution of different ASFV genotypes and comparison of B646L sequences. (A) According to the B646L sequence of the 24 ASFV genotypes, the genetic evolution tree was constructed using the MEGA7.0 software. The strains marked by ▴ are the popular strains in China. (B) The B646L sequences in 24 ASFV genotypes were analyzed via Clustal W using the Megalig software (DNASTAR, Inc.).
Sensitivity of the Triplex RT-PCR and Establishment of a Standard Curve
Three positive standard samples were serially diluted 12 times for triplex RT-PCR. Amplification was confirmed by diluting pUC57-EGFP, pMD18-B646L, and pUC57-mCherry plasmids (10−4-10−12), thereby obtaining the triplex RT-PCR standard curves. As shown in Figure 2, the slopes of pUC57-EGFP, pMD18-B646L, and pUC57-mCherry standard curves were −3.457, −3.312, −3.615, respectively. The standard curves (R2 > 0.99) showed that each diluted sample had a strong linear relationship. Importantly, the highest dilution (10−11) of the positive plasmid was detected using this method. Additionally, as shown in Figure 3, the minimum copies of pUC57-EGFP, pMD18-B646L, and pUC57-mCherry detected using triplex RT-PCR were 9.49, 15.60 and 9.60, respectively, suggesting that this method had a high detection sensitivity.
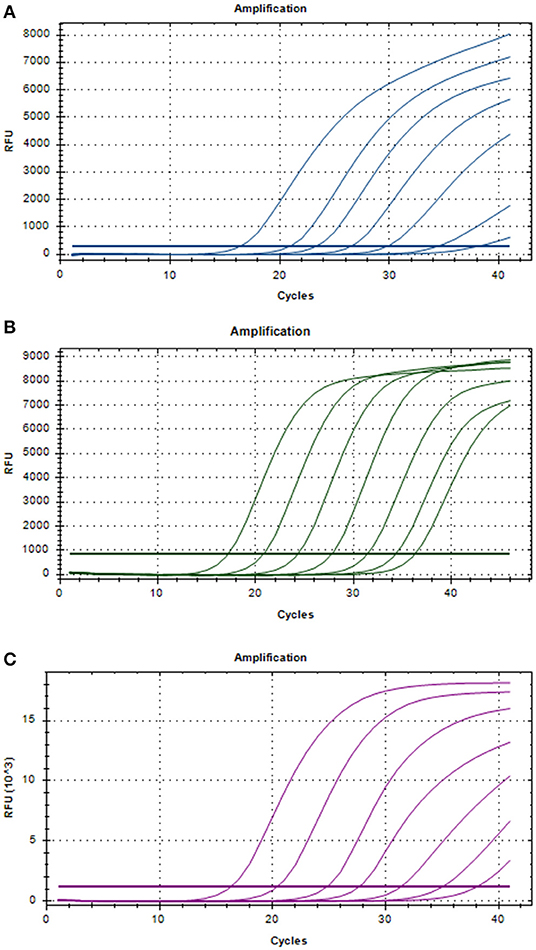
Figure 2. Amplification curves in the sensitivity tests of triplex RT-PCR. (A) Sensitivity test for B646L gene. (B) Sensitivity test for EGFP gene. (C) Sensitivity test for mCherry gene.
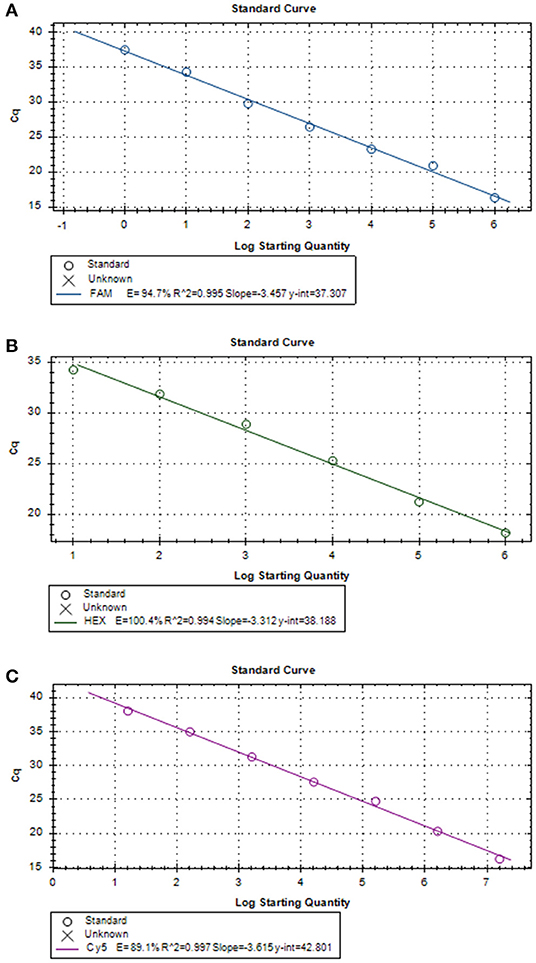
Figure 3. (A) B646L standard curve (y = −3.457x + 37.307; R2 = 0.995). (B) EGFP standard curve (y = −3.312x + 38.188; R2 = 0.994). (C) mCherry standard curve (y = −3.615x + 42.801; R2 = 0.997).
Specificity of the Triplex Real-Time PCR
To test the specificity of the triplex real-time PCR, we used triplex RT-PCR to detect the nucleic acids of wild-type ASFV, ASFVΔCD2vΔMGF, PRRSV, PEDV, PCV-2, PRV, CSFV, and NC. As shown in Figure 4A, detection of WT-ASFV PRRSV, PEDV, PCV-2, PRV, CSFV using triplex RT-PCR, a curve was obtained for ASFV nucleic acid samples (FAM signal), while no amplification curve was obtained for PRRSV, PEDV, PCV-2, PRV, CSFV, and NC nucleic acid. As shown in Figure 4B, detection of ASFVΔCD2vΔMFG PRRSV, PEDV, PCV-2, PRV, CSFV using triplex RT-PCR, in the ASFVΔCD2vΔMFG nucleic acid samples, the FAM, HEX and Cy5 channels all showed amplification curves, corresponding to B646L, EGFP, and mCherry, respectively. Indicated that the EGFP and mCherry were expressed in ASFVΔCD2vΔMGF, which could be identified as a gene deletion virus. As shown in the Figure 4C, specific probe primers were used to detect the nucleic acids of five swine viruses, the nucleic acid samples of the five swine viruses all showed specificity curves, indicating that the nucleic acids of the five swine viruses did exist in the specifically detected samples. As shown in Figure 4D, Using triplex RT-PCR to detect WT-ASFV and ASFVΔCD2vΔMFG simultaneously, WT-ASFV showed amplification curve only in the FAM channel, while ASFVΔCD2vΔMFG showed amplification curve in FAM, HEX and Cy5 channels. The above results suggested that the established triplex real-time PCR has a good specificity without cross-reactivity to other five swine viruses.
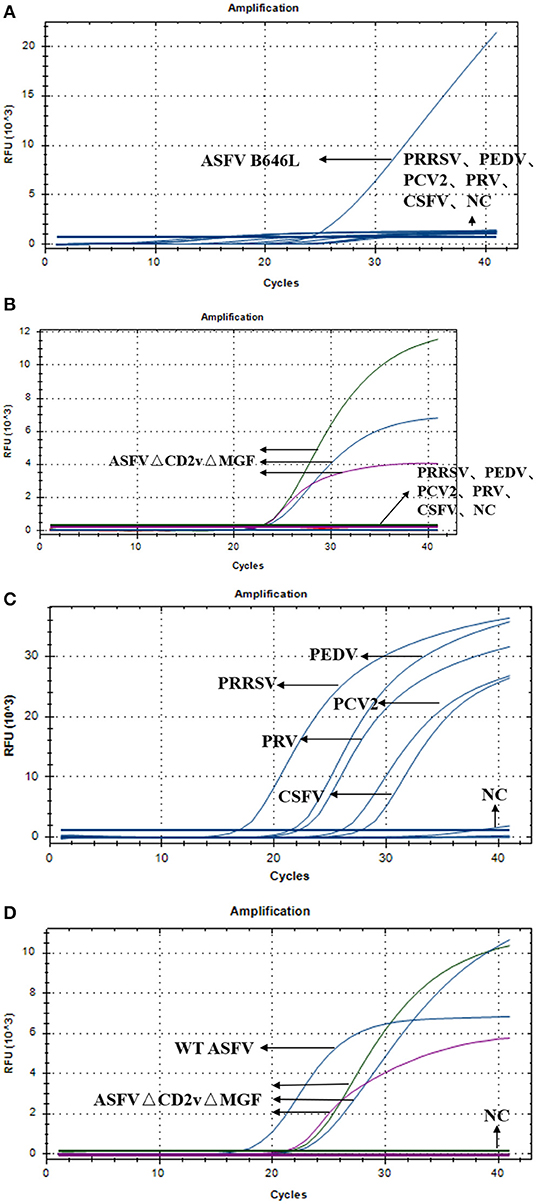
Figure 4. (A) Amplification curves of ASFV, PRRSV, PEDV, PCV-2, PRV, CSFV, and NC. (B) Amplification curves of ASFVΔCD2vΔMGF, PRRSV, PEDV, PCV-2, PRV, CSFV, and NC. (C) Amplification curve of PRRSV, PEDV, PCV-2, PRV, CSFV, and NC. (D) Amplification curves of ASFV, ASFVΔCD2vΔMGF and NC.
Triplex RT-PCR of Clinical Swine Nucleic Acid Samples
A total of 1,646 clinical swine samples were subjected to triplex RT-PCR. The results showed that 67 samples were positive for ASFV, while the remaining 1,579 samples were negative. Notably, the results for all of the 67 positive samples showed that an amplification curve could only be obtained for the FAM channel, indicating that these positive samples were ASFV wild-type strains. This result was expected since the attenuated strains are not yet used as vaccines. Importantly, the same results were obtained using the primers and probes published by OIE (Table 2), validating the triplex RT-PCR designed in this study as a sensitive and specific tool for the identification of ASFV.
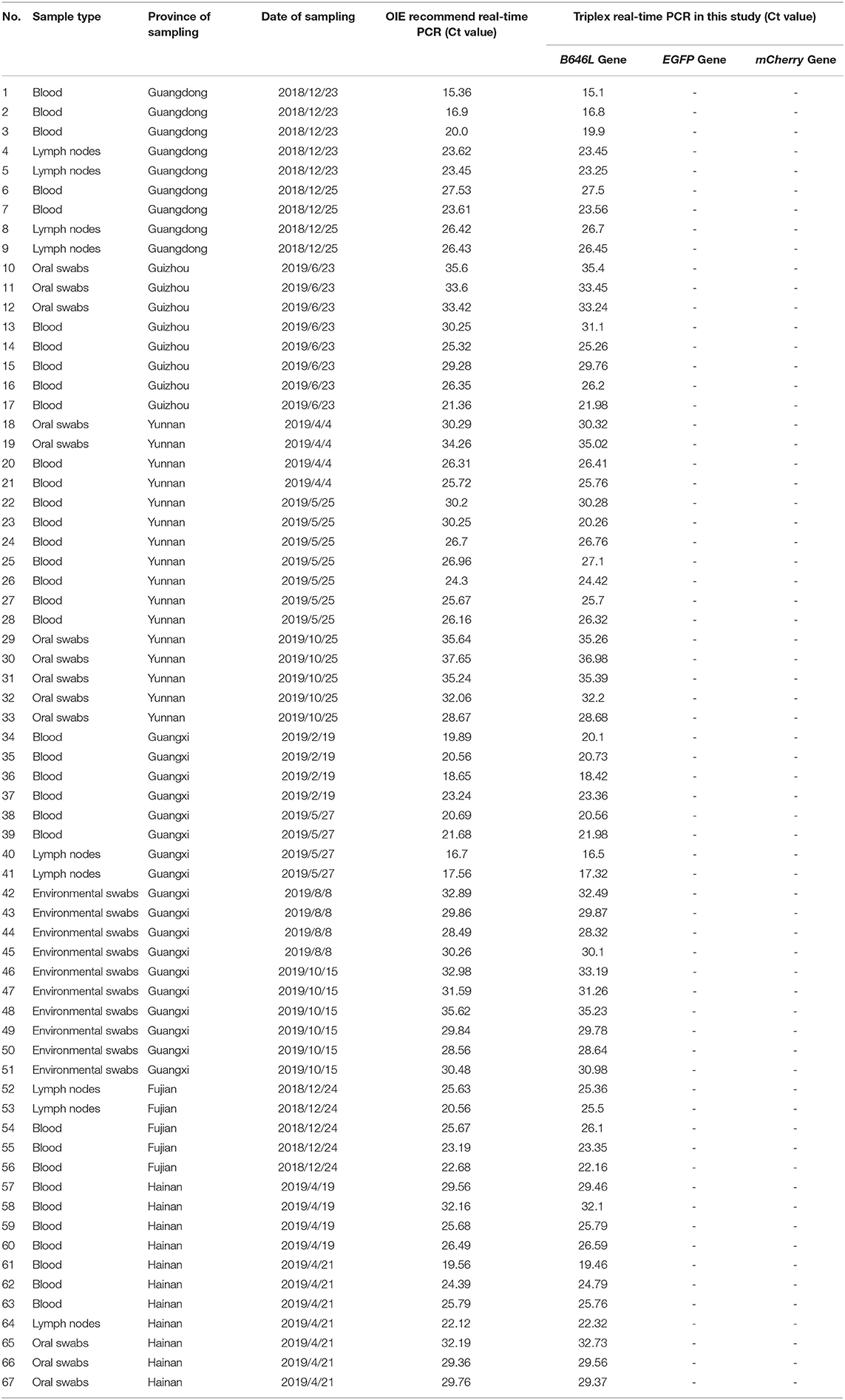
Table 2. Identification of the 67 ASFV clinical positive samples was performed using the ASFV real-time PCR method.
Discussion
Here, a new triplex RT-PCR method was developed to detect B646L, EGFP, and mCherry, and the consequent distinction between wild-type and gene-deleted ASFV strains. If only B646L is detected, the sample must contain only wild-type ASFV. On the contrary, if both B646L and either EGFP or mCherry are detected, the sample must derive from a gene deletion virus strain. It should be noted that B646L must be detected in both wild-type and gene-deleted ASFV strains; otherwise, it would mean that the sample is negative ASFV nucleic acids.
In this study, we designed a pair of primers and probes able to target B646L of all ASFV genotypes for the first time. Notably, a one-base mismatch existed in the genotypes III, V, VI, VII, IV, XIX, and XXIII, and a two-base mismatch was observed considering genotypes IX and X. Ghedira et al. showed that mismatches between primers and templates have different effects on amplification efficiency; mismatches near the 3' end can lead to low amplification efficiency, while mismatches outside the two bases of the 3' end have little effect (13). Importantly, the mismatches observed using the primers/probes designed in this study did not include the two bases at the 3' end. Therefore, we speculated that the amplification efficiency would not be affected and consequently, the triplex RT-PCR designed in this study would amplify B646L of the different ASFV genotypes in circulation.
ASF is a severe infectious disease of swine that causes huge economic losses to the breeding industry. Biosafety and vaccines are the most effective and economically-viable methods for preventing and controlling ASF epidemics. Monitoring whether pigs are infected by pathogens is the most important aspect of biosafety. On the contrary, vaccines are expected to decrease the disease burden. However, to date, there are no vaccines available. Still, gene deletion attenuated ASFV strains have become the most promising anti-ASFV vaccine candidates. In the review part of this paper, we listed some ASFV gene deletion vaccines that can protect pigs from virulent strains, such as ASFVΔMGFΔCD2v and ASFVΔI177L strain. Importantly, if the ASFV gene deletion strains are successfully marketed, it is necessary to distinguish them from the wild-type ASFV accurately.
At present, a variety of fluorescence quantitative PCR methods have been developed to detect ASFV gene deletion strains (14–16). However, most of the above methods are aimed at a single deleted gene, which has great limitations. Therefore, the method developed in this study, which we used to detect a variety of gene deletion strains, can be a valuable new tool with a wider detection range.
Importantly, the specificity results showed that this new method does not lead to non-specific results for common viruses circulating in pig farms. Additionally, the lowest number of copies of the positive standard for the detection of B646L, EGFP, and mCherry were 9.49, 15.60, and 9.60, respectively, and the established standard curves showed a strong linear relationship. Therefore, overall, our experimental data strongly support the accuracy and reliability of the new triplex RT-PCR developed here.
A new triplex fluorescent quantitative PCR method was successfully established for the identification of both wild-type and gene deletion ASFV strains, as well as their distinction. Importantly, the results of this method are consistent with those obtained with the OIE-recommended method. Altogether, our results suggest that the reliable method developed in this study can be used as a technical reserve to identify the ASFV gene deletion vaccines as well as wild-type II ASFV.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
ZH, ZX, HC, and FZ: conceptualization and writing—original draft preparation. ZZ, GZ, HW, and LG: data curation and visualization. ZH, ZX, HC, FZ, and SZ: validation and methodology. ZZ and GZ: funding. All authors have read and agreed to the published version of the final manuscript.
Funding
This research was funded by the Key-Area Research and Development Program of Guangdong Province (2019B020211003), the National Key Research and Development Program of China (2021YFD1800101). Start-up Research Project of Maoming Laboratory (2021TDQD002) and China Agriculture Research System of MOF and MARA (CARS-35).
Conflict of Interest
SZ was employed by Daguang Food Group Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
References
1. Revilla Y, Pérez-Núñez D, Richt JA. African swine fever virus biology and vaccine approaches. Adv Virus Res. (2018) 100:41–74. doi: 10.1016/bs.aivir.2017.10.002
2. Bosch-Camós L, López E, Rodriguez F. African swine fever vaccines: a promising work still in progress. Porcine Health Manag. (2020) 6:17. doi: 10.1186/s40813-020-00154-2
3. Ge S, Li J, Fan X, Liu F, Li L, Wang Q, et al. Molecular characterization of African swine fever virus, China, 2018. Emerg Infect Dis. (2018) 24:2131–3. doi: 10.3201/eid2411.181274
4. Allaway EC, Chinombo DO, Edelsten RM, Hutchings GH, Sumption KJ. Serological study of pigs for antibody against African swine fever virus in two areas of southern Malawi. Rev Sci Tech. (1995) 14:667–76. doi: 10.20506/rst.14.3.864
5. Gallardo C, Fernández-Pinero J, Arias M. African swine fever (ASF) diagnosis, an essential tool in the epidemiological investigation. Virus Res. (2019) 271:197676. doi: 10.1016/j.virusres.2019.197676
6. European Food Safety Authority (EFSA), Boklund A, Cay B, Depner K, Földi Z, Guberti V, et al. Epidemiological analyses of African swine fever in the European Union (November 2017 until November 2018). EFSA J. (2018) 16:e05494. doi: 10.2903/j.efsa.2018.5494
7. King DP, Reid SM, Hutchings GH, Grierson SS, Wilkinson PJ, Dixon LK, et al. Development of a Taqman PCR assay with internal amplification control for the detection of African swine fever virus. J Virol Methods. (2003) 107:53–61. doi: 10.1016/s0166-0934(02)00189-1
8. Agüero M, Fernández J, Romero L, Sánchez Mascaraque C, Arias M, Sánchez-Vizcaíno JM. Highly sensitive PCR assay for routine diagnosis of African swine fever virus in clinical samples. J Clin Microbiol. (2003) 41:4431–4. doi: 10.1128/JCM.41.9.4431-4434.2003
9. Borca MV, Ramirez-Medina E, Silva E, Vuono E, Rai A, Pruitt S, et al. Development of a highly effective African swine fever virus vaccine by deletion of the I177L gene results in sterile immunity against the current epidemic Eurasia strain. J Virol. (2020) 94. doi: 10.1128/JVI.02017-19
10. Borca MV, Rai A, Ramirez-Medina E, Silva E, Velazquez-Salinas L, Vuono E, et al. A cell culture-adapted vaccine virus against the current African swine fever virus pandemic strain. J Virol. (2021) 95:e0012321. doi: 10.1128/JVI.00123-21
11. Borca MV, Ramirez-Medina E, Silva E, Vuono E, Rai A, Pruitt S, et al. ASFV-G-ΔI177l as an effective oral nasal vaccine against the Eurasia strain of Africa swine fever. Viruses. (2021) 13:765. doi: 10.3390/v13050765
12. Chen W, Zhao D, He X, Liu R, Wang Z, Zhang X, et al. A seven-gene-deleted African swine fever virus is safe and effective as a live attenuated vaccine in pigs. Sci China Life Sci. (2020) 63:623–34. doi: 10.1007/s11427-020-1657-9
13. Borca MV, O'Donnell V, Holinka LG, Sanford B, Azzinaro PA, Risatti GR, et al. Development of a fluorescent ASFV strain that retains the ability to cause disease in swine. Sci Rep. (2017) 7:46747. doi: 10.1038/srep46747
14. Ghedira R, Papazova N, Vuylsteke M, Ruttink T, Taverniers I, De Loose M. Assessment of primer/template mismatch effects on real-time PCR amplification of target taxa for GMO quantification. J Agric Food Chem. (2009) 57:9370–7. doi: 10.1021/jf901976a
15. Guo Z, Li K, Qiao S, Chen XX, Deng R, Zhang G. Development and evaluation of duplex TaqMan Real-time PCR assay for detection and differentiation of wide-type and MGF505-2R gene-deleted African swine fever viruses. BMC Vet Res. (2020) 16:428. doi: 10.1186/s12917-020-02639-2
Keywords: African swine fever virus, triplex RT-PCR, ASFV gene deletion strain, differential diagnosis, B646L gene
Citation: Huang Z, Xu Z, Cao H, Zeng F, Wang H, Gong L, Zhang S, Cao S, Zhang G and Zheng Z (2022) A Triplex PCR Method for Distinguishing the Wild-Type African Swine Fever Virus From the Deletion Strains by Detecting the Gene Insertion. Front. Vet. Sci. 9:921907. doi: 10.3389/fvets.2022.921907
Received: 16 April 2022; Accepted: 03 June 2022;
Published: 28 June 2022.
Edited by:
Yogesh Chander, Varigen Biosciences Corporation, United StatesReviewed by:
Shankar Yadav, European Commission for the Control of Foot and Mouth Disease (EuFMD), ItalyNirajkumar Makadiya, Westbrook Veterinary Clinic, Canada
Copyright © 2022 Huang, Xu, Cao, Zeng, Wang, Gong, Zhang, Cao, Zhang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zezhong Zheng, emV6aG9uZ0BzY2F1LmVkdS5jbg==; Guihong Zhang, Z3VpaG9uZ3poQHNjYXUuZWR1LmNu
†These authors have contributed equally to this work