1
Edmond and Lily Safra International Institute of Neuroscience of Natal (ELS-IINN), Natal, RN, Brazil
2
Federal University of Pará, Belém, PA, Brazil
3
Federal University of Rio Grande do Norte, Natal, RN, Brazil
Nitric oxide (NO) is a versatile messenger molecule first associated with endothelial relaxing effects. In the central nervous system (CNS), NO synthesis is primarily triggered by activation of N-methyl-D-aspartate (NMDA) receptors and has a Janus face, with both beneficial and harmful properties. There are three isoforms of the NO synthesizing enzyme nitric oxide synthase (NOS): neuronal (nNOS), endothelial (eNOS), and inducible nitric oxide synthase (iNOS), each one involved with specific events in the brain. In the CNS, nNOS is involved with modulation of synaptic transmission through long-term potentiation in several regions, including nociceptive circuits in the spinal cord. Here, we review the role played by NO on central pain sensitization.
Nitric oxide (NO) is one of the most intriguing molecules to be found in the brain. This versatile neuronal messenger was first identified as an endothelium-derived relaxing factor (EDRF), responsible for smooth muscle relaxation (Ignarro et al., 1987
; Palmer et al., 1987
). NO is freely diffusible across cell membranes and is synthesized by a group of enzymes, known as nitric oxide synthases (NOS), from L-arginine and different cofactors, depending on the cell type (Hobbs and Ignarro, 1996
; Lipton et al., 1994
; Marletta et al., 1998
). Three members of the NOS family of enzymes have been identified in mammals: neuronal NOS (nNOS), endothelial NOS (eNOS) and inducible NOS (iNOS), which have distinct functional and structural features (see Stuehr et al., 2004
). Both nNOS (also known as Type I) and eNOS (Type III) activities are controlled by Ca2+-calmodulin and are expressed constitutively in the brain (Alderton et al., 2001
); iNOS (Type II), on the other hand, once expressed is always active, being independent of intracellular Ca2+ levels (Stuehr and Griffith, 1992
), and is expressed in response to inflammatory and proinflammatory mediators acting on macrophages, astrocytes and microglia (Marletta, 1994
). Though traditionally only nNOS has been associated with nitrergic signaling in the brain, there have been many evidences showing that eNOS also contributes to the physiological actions of NO in the brain parenchyma (Garthwaite, 2008
). NO is very stable in low, physiological concentrations (Garthwaite, 2008
) and is produced stoichiometrically after the conversion of L-arginine to citrulline in a process requiring both NADPH and O2 as co-substrates, and other cofactors (Marletta, 1993
; Moncada et al., 1989
; Vincent, 1994
) (Figure 1
). It is useful to divide the chemical reactions in which NO takes part as either direct or indirect (Wink et al., 2000
). Direct reactions occur between low concentrations of NO and iron-containing proteins, such as guanylate cyclase (GC), and is responsible for most physiological actions of NO (Wink et al., 2000
). Indirect reactions, conversely, occur when high concentrations of NO are present and generate reactive nitrogen oxide species (RNOS) and reactive oxygen species (ROS), such as the powerful oxidant peroxynitrite (ONOO–), that can cause harmful effects on cells, though nitrosative and oxidative stress (Wink et al., 2000
).
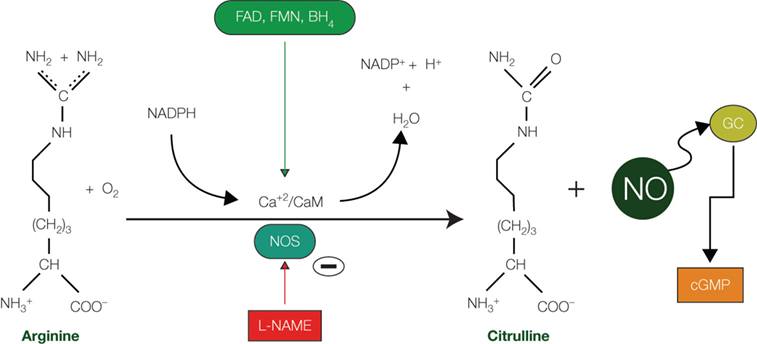
Figure 1. Nitric oxide synthesis. Diagram showing the synthetic pathway responsible for nitric oxide (NO) production from arginine and O2 and catalyzed by the enzyme nitric oxide synthase (NOS). Cofactors include reduced nicotinamide adenine dinucleotide phosphate (NADPH), tetrahydrobiopterin (BH4), flavin mononucleotide (FMN), and flavin adenine dinucleotide (FAD). Synthesis of NO can be pharmacologically abolished by the action of the N-omega-nitro-L-arginine methyl ester (L-NAME), an inhibitor of both nNOS and eNOS. Abbreviations: Ca+2/CaM: Calcium/calmodulin; cGMP: cyclic guanosine monophosphate; GC: guanylate cyclase; PKG: protein kinase G.
This Janus face of NO, representing its potential for both protective and deleterious effects in the brain, depends mostly on its concentration levels and the identity of the acting enzymatic isoform (Calabrese et al., 2007
; Lipton et al., 1993
). The cell signaling activity of NO requires only a low amount generated by both nNOS and eNOS (Esplugues, 2002
). The high levels of NO generated by iNOS are responsible for an increase in the amount of both RNOS and ROS (Wink et al., 2000
). The toxic effects of NO are mostly mediated by its oxidation products, particularly the biological oxidant peroxynitrite (Pacher et al., 2007
). Knockout mice lacking the iNOS gene, for instance, are less affected by ischemic brain injury (Iadecola et al., 1997
). Oxidative stress is a common feature of the pathogenesis of many neurodegenerative disorders, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases (Guix et al., 2005
; Moreira et al., 2005
). Peroxynitrite generation, specifically, has been singled out as an important pathogenic mechanism behind those disorders (Pacher et al., 2007
; Saha and Pahan, 2006
). Interestingly, neurons that express nNOS are notoriously resistant to many types of injury, including intoxication by heavy metals (Freire et al., 2007
) and NO-induced neurotoxicity (Firestein and Bredt, 1999
).
In the Peripheral Nervous System (PNS), NO induces the relaxation of smooth muscle cells in the gastrointestinal tract (Bult et al., 1990
) and the corpus cavernosum (Toda et al., 2005
). In the CNS, NO is a neurotransmitter (Bredt et al., 1991
), albeit an unconventional one due to its free diffusion through aqueous and lipid compartments. NOS is physiologically active at nanomolar concentrations and is remarkably stable within this range (Garthwaite, 2008
). After being synthesized, NO can act on both postsynaptic and presynaptic structures by way of a GC transduction mechanism that eventually increases the intracellular levels of the second messenger cyclic guanosine monophosphate (cGMP) (Vincent, 1994
). There are hints, however, that other transduction mechanisms might be engaged by NO (see Garthwaite, 2008
for review). NO is involved with other important functions in the normal brain, including regulation of blood flow, neurogenesis, and synaptic plasticity (Estrada and DeFelipe, 1998
; Holscher, 1997
; Moreno-Lopez et al., 2000
; Wiesinger, 2001
). It is also associated with the long-term potentiation (LTP) of synaptic connections (Brenman and Bredt, 1996
) and can act as an anti-apoptotic factor when produced in normal concentrations (see Blaise et al., 2005
for a review). Conversely, following injury of the brain, NO acts as a neurodegenerative agent, contributing to neuronal death (Cassina et al., 2002
; Guimarães et al., 2009
; Kawase et al., 1996
). Several neurodegenerative disorders of the CNS are related to the excessive activation of NMDA receptors, including stroke and amyotrophic lateral sclerosis (Lipton and Rosenberg, 1994
). Since NMDA receptor activation is the primary stimulus for NO synthesis, it has been suggested that it might have a role in NMDA-mediated neurodegeneration. This has been confirmed in studies of nNOS knockout mice, which are resistant to excitotoxicity following acute ischemic injury (Huang et al., 1994
).
All three NOS types are dependent on NADPH as a co-factor for their activity (Marletta, 1994
). Since the early 1960’s, it has been known that histochemistry to reveal the activity of the oxidoreductase enzyme nicotinamide adenine dinucleotide phosphate diaphorase (NADPH-d) histochemistry is associated with a sub-population of inhibitory neurons in the mammalian CNS (Thomas and Pearse, 1964
). Any enzyme of the NADPH-d family catalyzes the transfer of electrons from NADPH to the colorless salt nitroblue tetrazolium (NBT), producing a colored, water-insoluble formazan dye which is visible through light microscopy (see Scherer-Singler et al., 1983
for details). Though in unfixed tissue many enzymes show a NADPH-d activity, it was demonstrated that in fixed tissue most of these enzymes are inactivated and only those of the NOS family retain their NADPH-d activity (Dawson et al., 1991
; Hope et al., 1991
).
Accordingly, NADPH-d has been widely used to detect NOS activity in aldehyde-fixed tissue in both the normal (Freire et al., 2005
; Sandell, 1986
; Wiencken and Casagrande, 2000
) and altered CNS (Freire et al., 2007
; Stojkovic et al., 1998
; Wallace et al., 1996
), revealing two distinct types of non-pyramidal cells: a more reactive group, with a Golgi-like appearance (type I cells) and a small and more weakly reactive population (type II cells) (Luth et al., 1994
). Both are co-localized with GABA in cortical neurons (Yan et al., 1996
). Type I neurons have large, darkly stained cell bodies and dendritic trees (Luth et al., 1994
) and are present in the brain of all mammals examined, from monotremes to primates (Hassiotis et al., 2005
; Yan and Garey, 1997
). Type II neurons, conversely, are weakly stained, with small cell bodies and few or no labeled processes (Luth et al., 1994
), and are reported to be especially numerous in the primate brain (Sandell, 1986
; Yan and Garey, 1997
). Both cell groups constitute an important source of NO in the CNS.
The distribution of NADPH-d neurons has already been described in the CNS of several mammalian orders (Barone and Kennedy, 2000
; Luth et al., 1994
; Mizukawa et al., 1989
; Pereira et al., 2000
; Wiencken and Casagrande, 2000
; Xiao et al., 1996
). In the rat brain, for instance, it was shown that NADPH-d neurons amount to approximately 2% of the whole cortical cell population (Valtschanoff et al., 1993
; Vincent and Hope, 1992
). NADPH-d/NOS neurons were also detected in the spinal cord (Anderson, 1992
; Dun et al., 1993
; Marsala et al., 1997
; Valtschanoff et al., 1992b
), mainly in superficial laminae (Valtschanoff et al., 1992a
), with a distribution pattern very similar to PKG (Tao et al., 2000
). The development of hyperalgesia in the rat’s hindpaw is followed by nNOS up-regulation in the spinal cord (Lam et al., 1996
; Yonehara et al., 1997
). In our research paper (Freire et al., 2008
), we characterized the distribution and morphometric aspects of NADPH-d/NO neurons in the spinal cord of the agouti, a medium-sized terrestrial and burrowing rodent with a widespread distribution over Central and South American Neotropical forests (Silvius and Fragoso, 2003
) that rely mostly on a frugivorous diet, usually manipulating seeds with high dexterity (Henry, 1999
).
We showed that in this species, similar to the rat (Valtschanoff et al., 1992a
), NADPH-d/NOS cells are detected mainly in laminae I–IV and X along the entire length of the spinal cord and in the intermediolateral column (IML) of both the thoracic and lumbar segments. In addition, we showed that NOS/NADPH-d neuropil is usually more reactive in the dorsal horn of both the cervical and lumbar enlargements than in the thoracic segments of the agouti. NOS/NADPH-d neurons also concentrate around the central canal (lamina X) (Freire et al., 2008
), which has been shown to receive input from visceral and cutaneous noxious inputs (Wall et al., 2002
; Willis et al., 1999
). Pain information from lamina X ascends to the brain through the dorsal column, blurring the lines separating the “touch” and “pain” pathways (Willis et al., 1999
).
Pain, as a sensation, is notoriously difficult to define in scientific terms, mostly due to its affective-emotional component. The International Association for the Study of Pain (IASP) offers the following definition of pain: “An unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage”. Damage is signaled to the brain by specialized high-threshold receptors spread all over the body, both externally and internally and associated with small, slow-conducting unmyelinated fibers. Pain signals enter the brain through neurons located in ganglia in the spinal cord and the brainstem. In the spinal cord, nociceptors make synapse with projection neurons located in superficial laminae, using neurotransmitters such as glutamate, that can directly activate them, and the neuropeptides substance P (SP) and neurokinin A (NKA), which modulate neuronal output in more subtle ways. Both SP and NKA are expressed through the preprotachykinin A (PPT-A) gene and bind to the neurokinin-1 (NK-1) receptor (Todd et al., 2002
). Most of the spinal cord neurons in lamina I that possess the NK-1 receptor belong to the spinothalamic and spinoparabrachial tracts, that send ascending nociceptive information to the brain.
When subjected to a regime of repetitive stimulation resulting from persistent tissue injury, nerve damage or electrical stimulation, spinal cord circuits are reorganized and pain is exacerbated in a process called nociceptive sensitization (Wall and Woolf, 1984
; Woolf, 1983
). Nociceptive sensitization is perceived as a decreased threshold to both noxious and innocuous stimulation, called hyperalgesia and allodynia, respectively. This effect has the obvious adaptive value of forcing the animal to protect the injured site and avoid further damage. Nociceptive sensitization has both central and peripheral components. Peripheral sensitization results from the release of inflammatory mediators such as prostaglandin E2 (PGE2), bradykinin, nerve growth factor (NGF), and NO in the injured site (Milligan and Watkins, 2009
). These chemicals increase the sensitivity/excitability of sensory terminals leading to localized tissue effects. Central sensitization, on the other hand, can result from the repetitive activation of C-fibers and the consequent temporal summation of dorsal horn neuronal responses, termed “windup” (Mendell and Wall, 1964
). This sensitization operates on mechanisms that are broadly similar to those underlying long-term potentiation (LTP) (see Ji et al., 2003
for a review). As with LTP in the hippocampus, activity-dependent plastic modification of synapses between SP peripheral nociceptor terminals and lamina dorsal horn neurons occurs through phosphorylation of both α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) and N-methyl-D-aspartate (NMDA) ionotropic receptors and the recruitment of AMPA receptors to the postsynaptic membrane (Ji et al., 2003
) (Figure 2
). The complement of second messenger pathways recruited in each case is slightly different (Ji et al., 2003
). Though there is still much debate about specific details of the mechanisms involved, it seems evident that NO plays an important role as a signaling molecule on LTP in the hippocampus and other cortical areas (Haghikia et al., 2007
; Hopper and Garthwaite, 2006
).
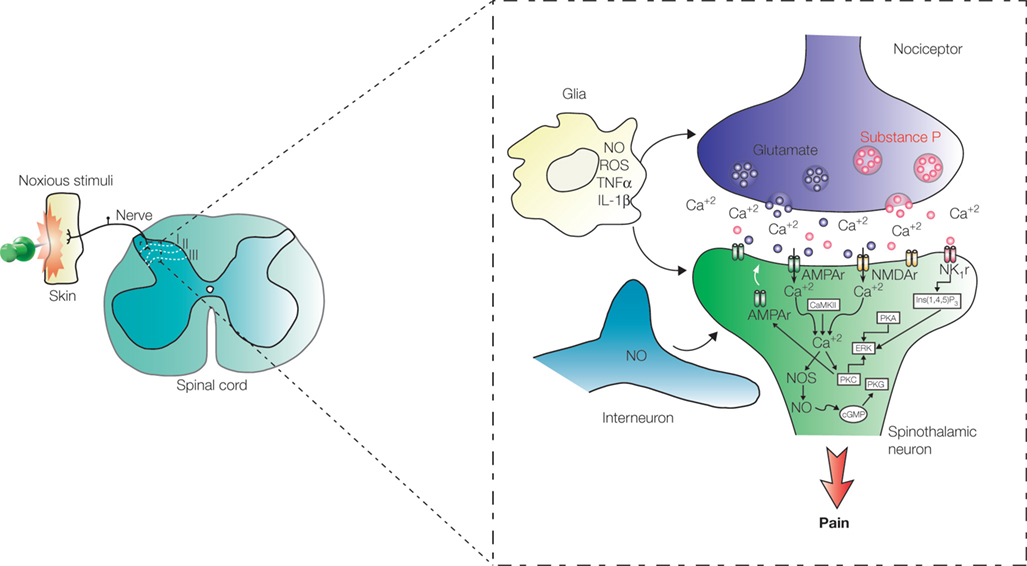
Figure 2. Processing of pain information in the spinal cord. Pain signals are transmitted to the dorsal horn by nociceptive terminals that synapse with neurons located in laminae I, II, and III. Early-onset activity-dependent central sensitization of ascending pathways depends on LTP of the synapse between peripheral terminal containing substance P and spinothalamic projection neurons in lamina I. Pain sensitization depends, in some level, on nitric oxide (NO) released by interneurons located in laminae II and III and also by glial cells. Abbreviations: AMPAr: α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor; NMDAr: N-methyl-D-aspartate receptor; NK1r: neurokinin 1 receptor; Glu: glutamate; Ins(1,4,5)P3: Inositol 1,4,5-triphosphate; CaMKII: calcium/calmodulin-dependent protein kinase II; PKA: protein kinase A; PKC: protein kinase C; PKG: protein kinase G; NOS: nitric oxide synthase; ERK: extracellular signal-regulated kinase; cGMP: cyclic guanosine monophosphate; IL-1β: interleukin 1beta; TNFα: tumor necrosis factor-alfa; ROS: reactive oxygen species.
LTP mediated hyperalgesia occurs with the contribution of projection neurons in lamina I expressing the neurokinin 1 (NK1) receptor (Ikeda et al., 2006
). The presence of NO is essential for induction of LTP in lamina I neurons (Ikeda et al., 2006
). In rats, the main source of NO in lamina I neurons are interneurons located in laminae II and III (Ruscheweyh et al., 2006
). Recently, a study using some specific inhibitors for distinct NOS isoforms (Tanabe et al., 2009
) showed that both nNOS and iNOS are involved with hyperalgesia. However, while nNOS has a definite role in spinal cord circuits, there is some disagreement whether iNOS also participates in central transmission (Tanabe et al., 2009
) or it expressed only peripherally (De Alba et al., 2006
) in both inflammatory and neuropathic pain models. Strong noxious stimuli lead to the activation of downstream serine/threonine protein kinases, including cAMP-dependent protein kinase (PKA), Ca+2/phospholipid-dependent protein kinase (PKC), Ca+2/calmodulin-dependent protein kinase II (CaMKII), and (cGMP)-dependent protein kinase (PKG) (Ji et al., 2003
; Tanabe et al., 2009
; Wu et al., 2005
), some of which with an effective participation of NO. Briefly, the general mechanism by which NO participates in spinal nociceptive processing consists in the activation of soluble GC, which in turn generates cGMP. Intracellular elevation of the cGMP levels may further activate PKG (see Figure 2
). Otherwise, NO may produce peroxynitrite by rapidly reacting with superoxide (Beckman et al., 1990
), whose importance in enhancing phosphorylation of NMDA receptors, and consequently central sensitization, has been recently suggested in animal models of neuropathic and inflammatory pain (Gao et al., 2007
). Besides, the ROS produced by either NO or NMDA-receptor activation, superoxide and peroxynitrite, respectively, also contribute to maintenance of hyperalgesia (Tanabe et al., 2009
), and can stimulate directly the pheripheral nociceptive terminals (Aley et al., 1998
).
The crucial involvement of the NO-GC signaling pathway in inflammatory and neuropathic pain is underscored by the markedly reduced nociceptive behavior presented by a NO-GC knock-out mouse strain (Schmidtko et al., 2008
). Inhibitors of NOS activity have also provided evidence for the role of NO in pain sensitization. Yonehara and coworkers (1997)
, in a chronic inflammatory pain model, reported a decrease in hyperalgesia after the intravenous administration of the arginine analog N-omega-nitro-L-arginine methyl ester (L-NAME), which inhibits the nNOS and eNOS isoforms (Knowles and Moncada, 1994
; Parkinson, 2000
). More recently, however, other groups were also able to show the specific role of iNOS in hyperalgesia by using highly selective inhibitors for this isoform, such as N-(3-[Aminomethyl] benzyl) acetamidine (1400W) (Tang et al., 2007
) and (S)-2-amino-7-acetamidino-5-thioheptanoic acid (GW274150) (De Alba et al., 2006
). Both inhibitors induced a significant reduction in hyperalgesia.
Since its discovery in the 1980’s, NO has been shown to be a key player in many normal and pathological events in the brain. In normal conditions, it is produced in every major region of the CNS by a specific group of NADPH-d positive neurons. NO easily diffuses through cell membranes and gets to the intracellular compartment, where it increases the level of the second messenger cGMP (Qian et al., 1996
) and initiates a cascade of molecular events leading to apoptosis, neurogenesis, LTP, etc. The NO-dependent LTP of nociceptive pathways in the spinal cord is a crucial mechanism behind long-term central pain sensitization. The ensuing hyperalgesia and allodynia warns the animal to the need to protect the affected region on the periphery. NO is released by interneurons in spinal cord’s laminae II and III and affects synapses between peripheral nociceptor terminals and lamina I neurons sending upward projections to the brain. The amount of NOS in this region is upregulated during sensitization and could be responsible for the maintenance of LTP.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supported by grants CNPq 479936/2008-0, 481132/2007-4, and 304422/2006-1 to AP. The authors wish to thank Financiadora de Estudos e Projetos (FINEP), and Associação Alberto Santos Dumont para Apoio à Pesquisa (AASDAP) – Brazil.
LTP: Long-lasting increase in synaptic efficiency following high-frequency stimulation of presynaptic elements. It was discovered in the hippocampus and depends on either N-methyl-D-aspartate (NMDA) receptor-dependent or -independent mechanisms to be evoked and on gene expression and protein synthesis to be maintained.
Hyperalgesia: Heightened sensitivity to pain caused by injury to peripheral tissues or neural elements. It can be elicited by both peripheral and central sensitization mechanisms. Peripheral mechanisms are triggered by inflammatory agents and the release of intracellular molecules by damaged cells. Central sensitization can be of the early and late-onset types. Early-onset sensitization is caused by LTP-like mechanisms, while the late-onset type involves changes in gene expression.
De Alba, J., Clayton, N. M., Collins, S. D., Colthup, P., Chessell, I., and Knowles, R. G. (2006). GW274150, a novel and highly selective inhibitor of the inducible isoform of nitric oxide synthase (iNOS), shows analgesic effects in rat models of inflammatory and neuropathic pain. Pain 120, 170–181.
Freire, M. A. M., Tourinho, S. C., Guimarães, J. S., Oliveira, J. L. F., Picanço-Diniz, C. W., Gomes-Leal, W., and Pereira, A., Jr. (2008). Histochemical characterization, distribution and morphometric analysis of NADPH diaphorase neurons in the spinal cord of the agouti. Front. Neuroanat. 2, 2. doi: 10.3389/neuro.3305.3002.2008.
Guimarães, J. S., Freire, M. A. M., Lima, R. R., Souza-Rodrigues, R. D., Costa, A. M., dos Santos, C. D., Picanço-Diniz, C. W., and Gomes-Leal, W. (2009). Mechanisms of secondary degeneration in the central nervous system during acute neural disorders and white matter damage. Rev. Neurol. 48, 304–310.
Schmidtko, A., Gao, W., Konig, P., Heine, S., Motterlini, R., Ruth, P., Schlossmann, J., Koesling, D., Niederberger, E., Tegeder, I., Friebe, A., and Geisslinger, G. (2008). cGMP produced by NO-sensitive guanylyl cyclase essentially contributes to inflammatory and neuropathic pain by using targets different from cGMP-dependent protein kinase I. J. Neurosci. 28, 8568–8576.
Tanabe, M., Nagatani, Y., Saitoh, K., Takasu, K., and Ono, H. (2009). Pharmacological assessments of nitric oxide synthase isoforms and downstream diversity of NO signaling in the maintenance of thermal and mechanical hypersensitivity after peripheral nerve injury in mice. Neuropharmacology 56, 702–708.