Department of Physiology, University of Melbourne, Parkville, Australia
Vasoactive intestinal peptide (VIP) immunoreactive secretomotor neurons in the submucous plexus are involved in mediating bacterial toxin-induced hypersecretion leading to diarrhoea. VIP neurons become hyperexcitable after the mucosa is exposed to cholera toxin, which suggests that the manipulation of the excitability of these neurons may be therapeutic. This study used standard intracellular recording methods to systematically characterize slow excitatory postsynaptic potentials (EPSPs) evoked in submucosal VIP neurons by different stimulus regimes (1, 3 and 15 pulse 30 Hz stimulation), together with their associated input resistances and pharmacology. All slow EPSPs were associated with a significant increase in input resistance compared to baseline values. Slow EPSPs evoked by a single stimulus were confirmed to be purinergic, however, slow EPSPs evoked by 15 pulse trains were non-purinergic and those evoked by 3 pulse trains were mixed. NK1 or NK3 receptor antagonists did not affect slow EPSPs. The group I mGluR receptor antagonist, PHCCC reduced the amplitude of purinergic and non-purinergic slow EPSPs. Blocking mGluR1 receptors depressed the overall response to 3 and 15 pulse trains, but this effect was inconsistent, while blockade of mGluR5 receptors had no effect on the non-purinergic slow EPSPs. Thus, although other receptors are almost certainly involved, our data indicate that there are at least two pharmacologically distinct types of slow EPSPs in the VIP secretomotor neurons: one mediated by P2Y receptors and the other in part by mGluR1 receptors.
Vasoactive intestinal peptide (VIP) immunoreactive neurons in the submucous plexus are the key secretomotor neurons in the enteric nervous system (Bornstein and Furness, 1988
). Their importance is shown by their involvement in the hypersecretion produced by cholera toxin and Escherichia coli enterotoxins (Banks et al., 2005
), the fact that they are the only secretomotor neurons to receive input from outside the intestine and their converging input from many different enteric neurons (Bornstein and Furness, 1988
). These neurons exhibit several excitatory and inhibitory potentials identified by their time courses and neurotransmitters (Gwynne and Bornstein, 2007
). These include two types of fast excitatory postsynaptic potentials (EPSPs, duration up to 50 ms), mediated by nicotinic acetylcholine receptors (Monro et al., 2004
; Moore and Vanner, 1998
; Surprenant, 1984
) and P2X purine receptors (Monro et al., 2004
), two inhibitory postsynaptic potentials (IPSPs; Bornstein et al., 1988
; Mihara et al., 1987
), an EPSP with an intermediate time course (250–750 ms) (Bornstein et al., 1986
; Monro et al., 2004
) and slow EPSPs that can last several tens of seconds (Bornstein et al., 1986
; Surprenant, 1984
). The fast EPSPs and IPSPs have been studied extensively, but studies of the slow EPSPs have been contradictory.
Several studies have found that slow EPSPs can be evoked in VIP neurons by a single stimulus pulse applied to an internodal strand (Bornstein et al., 1986
; Monro et al., 2004
; Surprenant, 1984
). This slow EPSP is mediated by P2Y1 receptors (Hu et al., 2003
; Monro et al., 2004
). However, there are radically different reports about the conductance changes underlying these events with early studies indicating that they are due to a decrease in membrane K+ conductance (Bornstein et al., 1986
; Mihara et al., 1985
; Surprenant, 1984
), but a recent report that they are due to increased cation conductance (Hu et al., 2003
). Further, an early study reported that only neurons with the electrophysiological properties of VIP neurons are depolarized by substance P (Surprenant, 1984
), but histochemical studies indicate that VIP neurons do not express tachykinin receptors (Jenkinson et al., 1999
; Moore et al., 1997
). Another complication comes from the report that some submucosal neurons are depolarized by the NK1 receptor agonist septide, but are not immunoreactive for NK1 tachykinin receptors (Moore et al., 1997
).
There is a similar lack of clarity, when one considers another candidate transmitter, glutamate. Group I metabotropic 1α and 5 glutamate receptors (mGluR1α and mGluR5) have been found via immunohistochemistry in submucosal neurons of guinea-pig ileum (Hu et al., 1999
; Liu and Kirchgessner, 2000
). Glutamate, and group I mGluR agonists evoke slow depolarizations sensitive to S-4-carboxyphenylglycine (S-4CPG, group I mGluR antagonist) in submucosal neurons (Hu et al., 1999
; Ren et al., 2000
), but the antagonist did not affect slow EPSPs (Ren et al., 2000
). Others reported that a mGluR5 selective antagonist, 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), and S-4CPG depressed slow EPSPs in submucosal neurons (Liu and Kirchgessner, 2000
), while it has also been reported that glutamate acts via group I mGluRs to suppress slow EPSPs and potentiate slow IPSPs in submucosal neurons (Ren et al., 1999
).
One explanation for these discrepancies is that stimulus regimes differed between studies: trains of stimuli evoke slow EPSPs in submucosal neurons that lack responses to single stimulus pulses. However, the relationship between stimulus regime, input resistance and pharmacology of slow EPSPs is unknown. The present study characterized the input resistance changes and pharmacology of slow EPSPs evoked by different stimulus regimes in VIP submucosal neurons. The roles of P2, NK1 and NK3 tachykinins, and group I mGluR receptors were specifically examined.
Tissue Preparation
Male or female guinea-pigs (200–400 g) were killed by applying a sharp blow to the back of the head and immediately severing the carotid arteries and spinal cord, in accordance with the guidelines of the University of Melbourne Animal Experimentation Ethics Committee (AEEC). A 3–5 cm segment of ileum, 15–30 cm oral to the ileo-caecal junction was removed from the abdominal cavity and placed in physiological saline solution (composition in mM: NaCl 118, NaHCO3 25, D-glucose 11, KCl 4.8, CaCl2 2.5, MgSO4 1.2, NaH2PO4 1.0), bubbled with 95% O2, 5% CO2. The tissue was flushed clean with physiological saline and a 1 × 1.5 cm preparation of submucous plexus was dissected as described elsewhere (Monro et al., 2004
) and pinned flat, serosa side up, in an organ bath. It was then equilibrated for at least 1 h before recording began. The preparation was continually superfused with physiological saline bubbled with 95% O2, 5% CO2 and kept at 33–34°C. Idazoxan hydrochloride was added into the superfusing solution in some experiments; it does not affect slow EPSPs (Foong and Bornstein, 2009
).
Electrophysiology
Intracellular recordings were made from submucosal neurons (Monro et al., 2004
). Submucosal ganglia were viewed at 200× magnification. A monopolar stimulating electrode (50 μm, non-insulated tungsten wire) was placed on an internodal strand leading to the chosen ganglion and synaptic potentials were evoked by focal stimulation. Cells were impaled with glass microelectrodes (100–200 MΩ, 1 M KCl, with or without 2% biocytin). Hyperpolarizing current (0–2 nA) was used to inject biocytin (Sigma Aldrich, Castle Hill, NSW, Australia). Stimuli were delivered in 1 pulse (12 V), 3 pulse (12 V, 30 Hz) and 15 pulse (12 V, 30 Hz) trains to evoke IPSPs and slow EPSPs.
Drugs used
Antagonists were added to the superfusate. These were N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide (PHCCC), BAY 36-7620, MPEP (all Tocris Bioscience, East Brisbane, QLD, Australia), idazoxan hydrochloride, pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid) (PPADS) (both Sigma Aldrich, Castle Hill, NSW, Australia), SR142801 and SR140333 (gifts from Dr Emonds-Alt, Sanofi Recherche, Montpellier, France). All were prepared as stock solutions dissolved in distilled water and/or dimethyl sulfoxide (DMSO), kept in aliquots at −20°C and dissolved to their final working concentrations in physiological saline when used in experiments.
Data analysis and statistics
The mean amplitudes of slow EPSPs and IPSPs were analyzed using Axoscope 9.2.0.12 (Axon Instruments, Inc., Union City, CA, USA) in control, drug and, where possible, after washout (at least 15–30 min). All data are presented as mean ± SEM. A two-tailed or one-tailed Student’s paired t-test, or repeated measures ANOVA were performed. A p < 0.05 was considered to be significant.
Immunohistochemistry
A subset of electrophysiology preparations was fixed in Zamboni’s fixative (2% formaldehyde and 0.2% picric acid in 0.1-M phosphate buffer, pH 7.0) at 4°C overnight. They were then cleared of fixative and permeabilized using 3 × 10 min washes with DMSO followed by 3 × 10 min washes with phosphate buffered saline (PBS) and processed to reveal the presence of VIP and/or biocytin by incubating in rabbit anti-VIP antibody (1:500; Euro-Diagnostica, East Brisbane, QLD, Australia) for 48 h at room temperature, washing with PBS, and incubating with donkey anti-rabbit fluorescein isothiocyanate (FITC) antibody (1:40; Amersham Biosciences Pty Ltd, Castle Hill, NSW, Australia) and Streptavidin-Texas Red (1:200; Amersham Biosciences) for 2.5 h. After removing excess antibody by washing with PBS, preparations were mounted on slides in buffered glycerol (pH 8.7), and viewed under a fluorescence microscope.
A total of 213 neurons were impaled in 67 preparations and 72 neurons were studied in detail as they had stable impalements and displayed slow EPSPs. Pharmacological experiments were conducted on 34 of the 72 neurons characterized.
Slow EPSPs, IPSPs and Immunoreactivity for VIP
IPSPs were evoked by trains (3 or 15 pulse) of focal stimuli in 69 out of the 72 neurons. Idazoxan (2 μM), which depresses IPSPs, was present for two of the other three neurons. IPSPs identify VIP neurons, as they are confined to this neuronal subtype (Bornstein et al., 1986
; Evans et al., 1994
). In our study, 28 of the 69 neurons with IPSPs were filled with biocytin, all were immunoreactive for VIP. Six neurons that lacked IPSPs were filled with biocytin, none were immunoreactive for VIP.
All 34 neurons that were used for pharmacology displayed fast EPSPs, followed by intermediate EPSPs or IPSPs and then slow EPSPs in response to a 1 (22 of 34), 3 (31 of 34) and/or 15 pulse (30 of 34) stimulus (Figures 1
A–C). Slow EPSPs vary in duration, some do not even return to baseline. The 1 pulse evoked slow EPSPs had amplitudes of 7.7 ± 0.6 mV (n = 36) and durations ranging from 4.4 to 26.8 s (n = 29), 3 pulse slow EPSPs had amplitudes of 9.8 ± 0.7 mV (n = 48) and durations from 5.7 to 35.6 s (n = 40) and 15 pulse slow EPSPs had amplitudes of 10.1 ± 0.6 mV (n = 44) and durations from 8.2 to 41.8 s (n = 40).
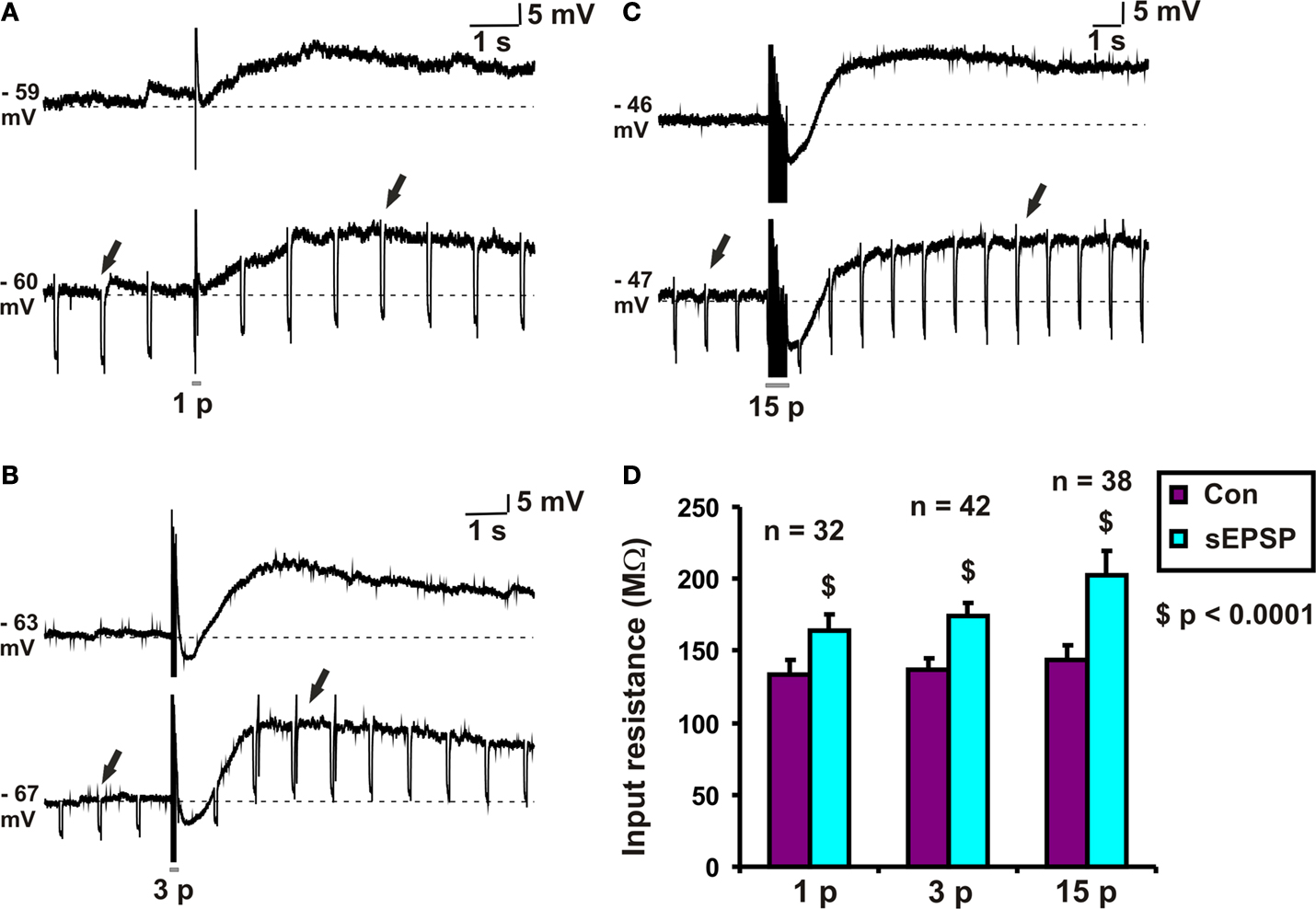
Figure 1. Input resistance associated with slow EPSPs. Examples of (A) 1 pulse, (B) 3 pulse and (C) 15 pulse evoked slow EPSPs in a submucosal neuron. (D) Quantified data comparing input resistance at baseline (arrows) and that associated with 1, 3 and 15 pulse slow EPSPs (arrows).
Slow EPSPs are Associated with an Increase in Input Resistance
Hyperpolarizing pulses (0.1 nA, 60–80 ms, 1 Hz) were used to measure the change in input resistance associated with slow EPSPs. The amplitude of the hyperpolarization evoked by a current pulse at the baseline membrane potential and that of one from the peak of the slow EPSP were measured for each stimulus protocol. Input resistances were calculated from these values and are shown in Figure 1
D. Each stimulus regime produced a significant increase in input resistance: for 1 pulse, the increase was 30 ± 4 MΩ (n = 32, p < 0.0001, Figures 1
A,D); for 3 pulse trains, the increase was 37 ± 4 MΩ (n = 42, p < 0.0001, Figures 1
B,D); for 15 pulse trains, the increase was 58 ± 12 MΩ (n = 38, p < 0.0001, Figures 1
C,D).
PPADS Reversibly Abolished 1 Pulse Slow EPSPs but had no Effect on 15 Pulse Slow EPSPs
PPADS (30 μM) reversibly abolished 1 pulse slow EPSPs (n = 6, p < 0.0001, Figures 2
A,D), but as reported previously (Monro et al., 2004
) had no effect on mean membrane potential in these neurons. The amplitudes of 3 pulse slow EPSPs were significantly smaller in PPADS, but there was a PPADS-insensitive component (n = 4, p < 0.05, Figures 2
B,D) and post hoc analysis showed that the significant difference was between the drug treatment and the washout. The amplitudes of 15 pulse slow EPSPs were unaffected by PPADS (n = 4, p = 0.52, Figures 2
C,D). Thus, we refer to 1 pulse slow EPSPs as purinergic slow EPSPs and 15 pulse slow EPSPs as non-purinergic slow EPSPs.
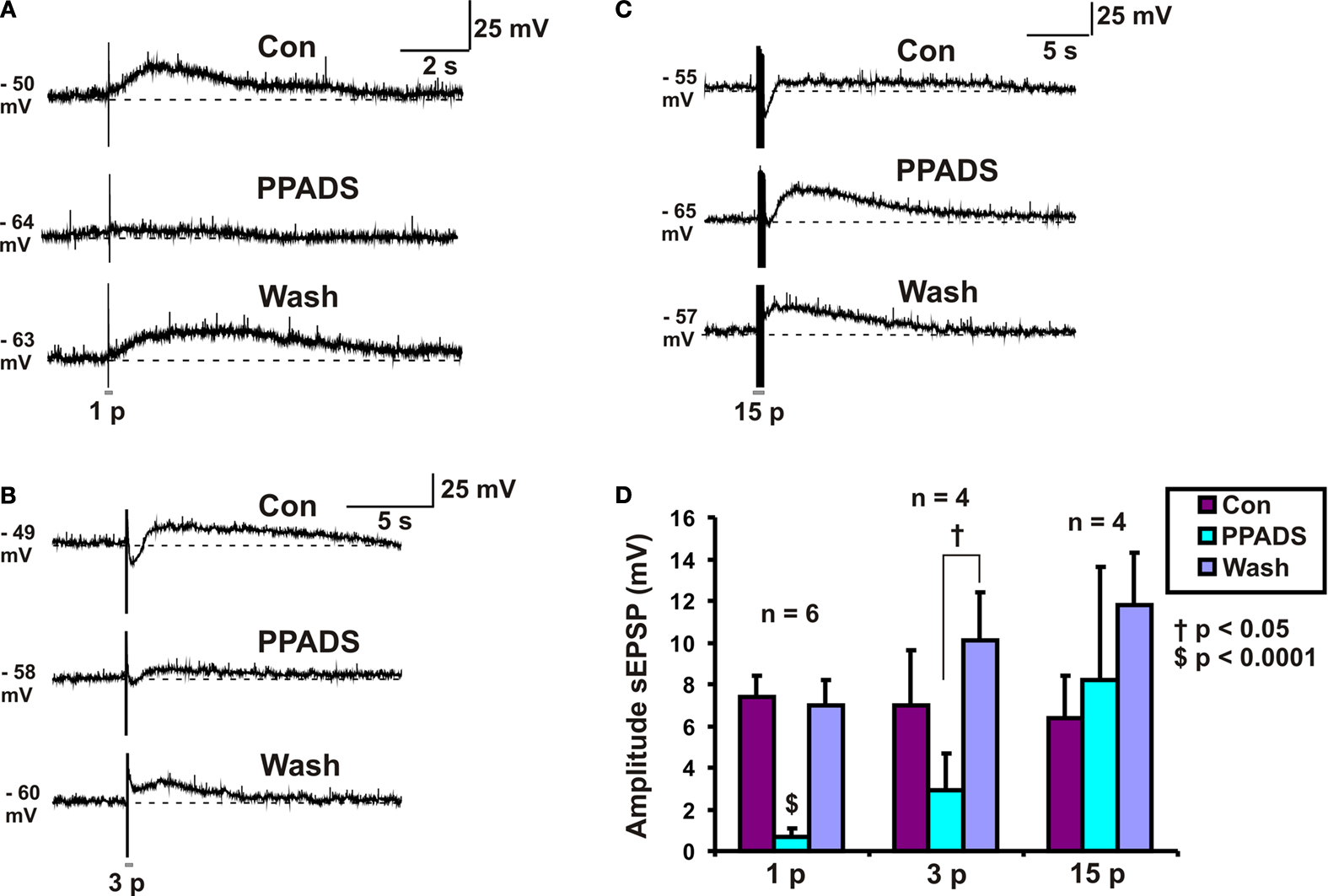
Figure 2. Effects of PPADS (30 μM) on slow EPSPs. (A) 1 pulse (12 V), (B) 3 pulse (30 Hz) and (C) 15 pulse evoked slow EPSPs in control, PPADS and after washout. (D) Quantified data for slow EPSPs evoked by 1, 3 and 15 pulses.
Tachykinin Receptor Antagonists did not Affect Slow EPSP Amplitudes
Possible roles for NK1 tachykinin receptors in slow EPSPs evoked in VIP neurons were tested using the specific NK1 receptor antagonist SR140333 at a concentration (200 nM) that depresses slow EPSPs in myenteric neurons (Johnson and Bornstein, 2004
). SR 140333 had no effect on the purinergic (n = 4, p = 0.12), 3 pulse (n = 4, p = 0.21) or the non-purinergic (n = 4, p = 0.7) slow EPSPs (Figure 3
A).
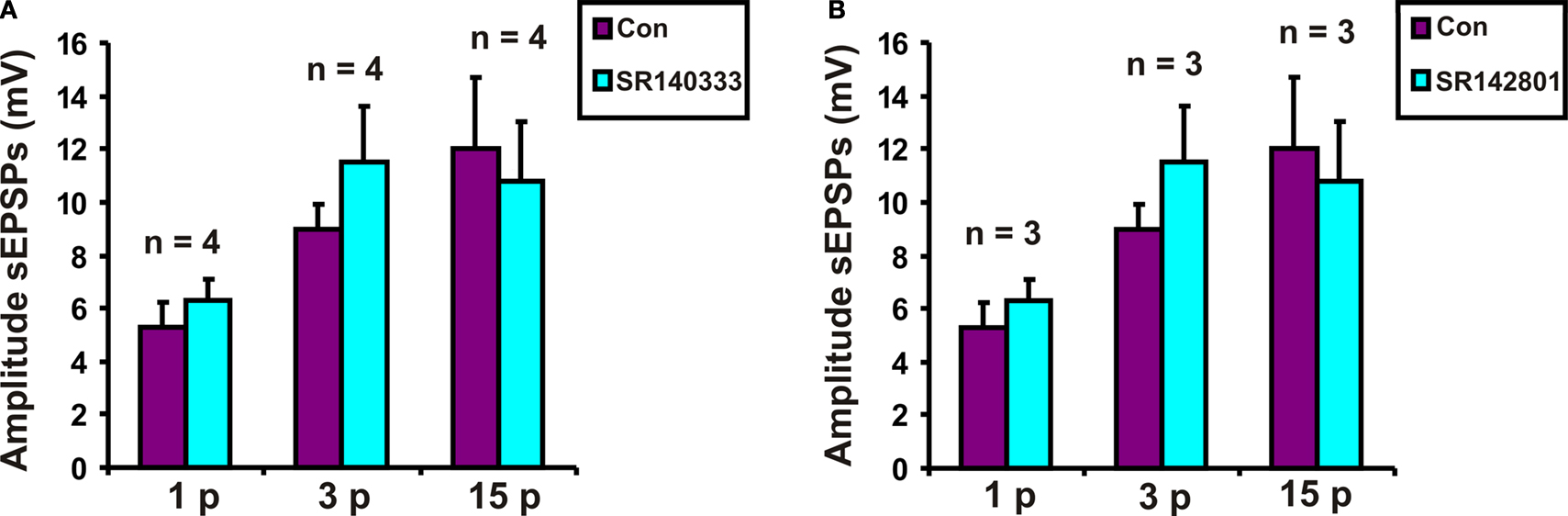
Figure 3. Effects of (A) SR 140333 (200 nM) and (B) SR 142081 (200 nM) on slow EPSPs. Quantified data for purinergic (1 p), 3 pulse (3 p) and non-purinergic (15 p) slow EPSPs.
Possible roles for NK3 tachykinin receptors in the slow EPSPs of VIP neurons were tested using the specific NK3 receptor antagonist SR142801 at a concentration that blocks slow EPSPs in myenteric neurons (Johnson and Bornstein, 2004
). SR 142801 (200 nM) did not affect purinergic (n = 3, p = 0.56), 3 pulse (n = 3, p = 0.07) or non-purinergic (n = 3, p = 0.9) slow EPSPs (Figure 3
B).
PHCCC Reversibly Reduced the Amplitude of Purinergic and Non-Purinergic Slow EPSPs
Roles for group I metabotropic glutamate receptors were initially tested using the group I mGluR antagonist PHCCC, at a concentration that we have found to depress colonic propulsion. PHCCC (30 μM) reversibly reduced the amplitude of purinergic (n = 4, p < 0.05) and non-purinergic (n = 6, p < 0.01) slow EPSPs. However the reduction in the amplitude of 3 pulse slow EPSPs was not significant in the presence of PHCCC (n = 4, p = 0.1) (Figure 4
A).
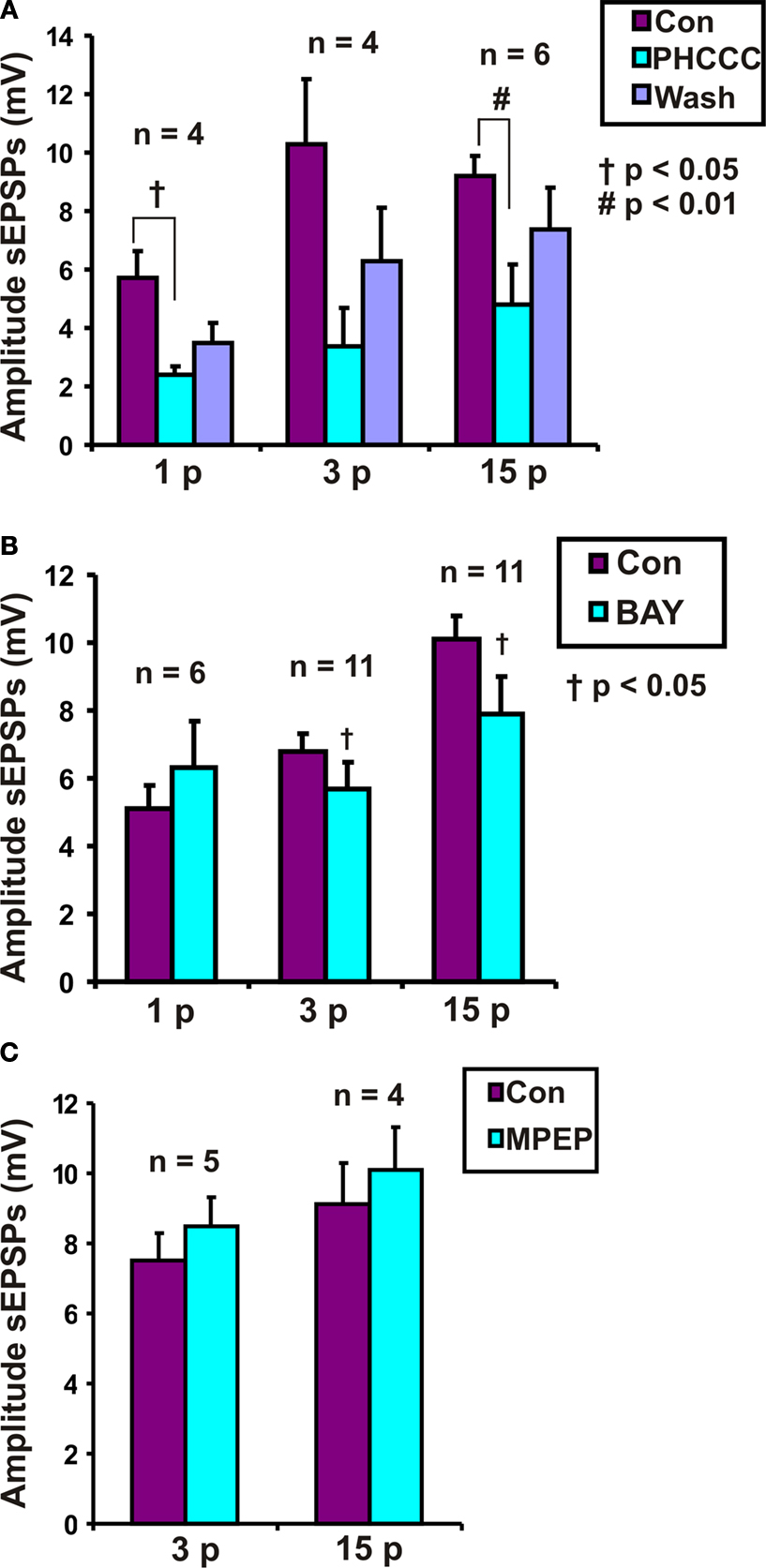
Figure 4. Effects of (A) PHCCC (30 μM), (B) BAY 36-7620 (10 μM) and (C) MPEP (10 μM) on slow EPSPs. Statistical analysis used two-tailed ANOVA for PHCCC and one-tailed paired t-tests for MPEP and BAY 36-7620.
Effects of Specific Antagonists on mGluR1 and mGluR5 Receptors
To determine if mGluR1 receptors play a role in the slow EPSPs of VIP neurons, the specific mGluR1 antagonist BAY 36-7620 (10 μM) was tested against responses to 1, 3 and 15 pulse trains. A one-tailed test was used to minimize the number of neurons analyzed, as the PHCCC results indicated that a decrease in the slow EPSP amplitude was to be expected. BAY 36-7620 did not affect the amplitude of purinergic slow EPSPs (n = 6, p = 0.20), but reduced the amplitudes of 3 pulse slow EPSPs (n = 11, p = 0.048) and non-purinergic slow EPSPs (n = 11, p = 0.037) (Figure 4
B).
When individual neurons were examined, BAY 36-7620 was found to reduce the amplitude of three pulse slow EPSPs in six neurons, but caused a slight increase (two neurons) or had no effect (three neurons) in others. Similarly, its effects on 15 pulse non-purinergic slow EPSPs were variable: six neurons showed a marked reduction in slow EPSP amplitude, but others showing no effect (three neurons) or a slight increase (two neurons). Three neurons whose slow EPSPs were depressed by BAY 36-7620 were held long enough to wash out the drug and in each case the slow EPSPs recovered. Effects on purinergic one pulse slow EPSPs were examined in four of the neurons whose 15 pulse slow EPSPs were depressed and the one pulse responses were increased in two neurons, reduced in one neuron and unchanged in the last.
In contrast, the specific mGluR5 receptor antagonist MPEP (10 μM, previously reported to be effective on submucosal slow EPSPs (Liu and Kirchgessner, 2000
) had no significant effect on either three pulse (n = 5, p = 0.25 one-tailed paired t-test) or non-purinergic (n = 4, p = 0.23) slow EPSPs (Figure 4
C).
This is the first time the input resistance, pharmacology and stimuli regimes have been studied together to characterize slow EPSPs in VIP secretomotor neurons of the submucosal plexus. The results show that sensitivity to blockade of P2 receptors is stimulus regime dependent with single pulses evoking slow EPSPs that appear to be entirely due to P2 receptor activation and 15 pulse trains evoking responses that are insensitive to blockade of these receptors. Both the purinergic slow EPSPs and the non-purinergic slow EPSPs were associated with an increase in input resistance at the peak of the response suggesting they are due to a decrease in K+ conductance. The non-purinergic EPSPs are not mediated by tachykinins acting at either NK1 or NK3 receptors, but a subset are probably mediated by mGluR1 receptors with others mediated by neurotransmitter/receptor combinations that remain to be identified.
The Pharmacology of Slow EPSPs in VIP Neurons Depends on the Stimulus that Evokes them
The finding that slow EPSPs evoked by single pulses applied to an internodal strand are blocked by the P2 receptor antagonist PPADS is consistent with previous reports that such slow EPSPs are mediated by P2Y1 receptors (Hu et al., 2003
; Monro et al., 2004
). We used PPADS rather than a more specific P2Y1 antagonist, because Monro et al. (2004)
had shown that PPADS and the specific P2Y1 antagonist were equally effective against one pulse slow EPSPs in these neurons. Earlier studies showed that only the VIP neurons, i.e. those that exhibit IPSPs, respond to single pulse stimuli with slow EPSPs (Bornstein et al., 1986
; Surprenant, 1984
). Monro et al. (2004)
found that these neurons exhibit fast EPSPs mediated by P2X receptors and a novel class of EPSP with intermediate durations of 250–750 ms that are also mediated by P2Y1 receptors. Thus, the VIP neurons of guinea-pig submucosal plexus receive a prominent purinergic input producing three different classes of EPSP.
Previous studies did not test whether altering the stimulus changes the character of the slow EPSPs. While Bornstein et al. (1986)
showed that increasing the number of pulses in a train increased the amplitude and duration of the slow EPSPs in VIP neurons; they detected no other differences in the characteristics of these slow EPSPs. Similarly, Surprenant (1984)
concluded that responses to trains of stimuli were no different to those evoked by individual stimuli. Hu et al. (2003)
reported that some VIP neurons had purinergic and non-purinergic slow EPSPs, which could be distinguished by underlying conductance changes, but provided no details of stimulus regime. This present study shows that slow EPSPs evoked by long trains of stimuli in VIP neurons differ qualitatively from those evoked by individual stimulus pulses, because they are insensitive to blockade of P2 receptors. As VIP neurons are the major secretomotor neurons in the intestine, this distinction may have important functional consequences.
Both Purinergic and Non-Purinergic Slow EPSPs are Associated with Increased Input Resistance
The present results support earlier findings that all slow EPSPs in VIP neurons involve increased input resistance, due to inactivation of K+ conductance (Bornstein et al., 1986
; Mihara et al., 1985
; Surprenant, 1984
). However, they contrast markedly with the report of Hu et al. (2003)
that purinergic slow EPSPs are mediated by increased non-specific cation conductance, with only non-purinergic slow EPSPs being mediated by decreased K+ conductance. These contrasting findings are very difficult to reconcile, although there are some differences in experimental protocols. For example, Hu et al. (2003)
reported that input resistance was reduced during the depolarizing phase of purinergic slow EPSPs, while we measured input resistance at the peak of the slow EPSPs to avoid contamination from occult IPSPs and intermediate EPSPs. However, this does not account for the distinction between earlier findings that slow EPSPs, including those evoked by one pulse, reverse at about −90 mV (Bornstein et al., 1986
; Surprenant, 1984
) and the report from Hu et al. (2003)
that purinergic slow EPSPs reverse at about −4 mV. One possibility is that P2Y1 receptors produce three distinct conductance changes. They appear to mediate intermediate EPSPs via opening a chloride channel (Monro et al., 2004
) and perhaps they open a non-specific cation channel and close a K+ channel with the contributions of each depending on the background conditions of the experiment. If so, the relevant conditions remain unclear.
Transmitters Mediating the Non-Purinergic Slow EPSPs in VIP Neurons
Substance P (SP) acting on NK1 receptors has often been considered a candidate for a neurotransmitter mediating slow EPSPs in submucosal neurons. Bath application (Surprenant, 1984
) or pressure ejection of SP (Mihara et al., 1985
; Ren et al., 2000
) both depolarize submucosal neurons. Furthermore, although VIP neurons apparently lack immunoreactivity for NK1 receptors (Moore et al., 1997
), NK1 agonists evoke NK1 antagonist sensitive slow depolarizations in submucosal neurons that are not immunoreactive for NK1 receptors (Moore et al., 1997
). Most of these neurons had IPSPs (Moore et al., 1997
) indicating that they were VIP neurons. However, our finding that the non-purinergic slow EPSPs were unaffected by SR 140333 indicates that NK1 receptors do not mediate slow EPSPs in VIP neurons. Similarly, the finding that the NK3 antagonist SR 142801 had no effect on slow EPSPs in VIP neurons indicates that NK3 receptors do not mediate slow EPSPs in these neurons. Although the number of neurons tested with the tachykinin antagonists are small, these data are consistent with an earlier report that slow EPSPs are seen in VIP neurons after all SP-immunoreactive terminals had degenerated after lesions to both extrinsic and intrinsic neural pathways (Bornstein et al., 1988
).
The conclusion that tachykinins do not play a role in synaptic transmission to VIP neurons does not exclude roles for them in transmission to other submucosal neurons. Immunohistochemical studies indicate that both NK1 (Moore et al., 1997
) and NK3 receptors (Jenkinson et al., 1999
) are expressed by cholinergic secretomotor neurons and tachykinin immunoreactive neurons that may be intrinsic sensory neurons. Further, studies of mucosal secretion indicate that tachykinins excite secretomotor neurons (Frieling et al., 1999
, Larsson et al., 2008
).
The other transmitter candidate whose role we investigated was glutamate acting on group I metabotropic glutamate receptors. The data indicate that mGluR1 receptors mediate a component of the non-purinergic slow EPSPs in some VIP neurons. This is consistent with the finding that submucosal neurons express mGluR1 receptors (Hu et al., 1999
). Although submucosal neurons also express mGluR5 receptors (Hu et al., 1999
; Liu and Kirchgessner, 2000
), we found no evidence that these mediate slow EPSPs in VIP neurons. This was surprising because immunoreactivity for mGluR5 has been reported to colocalize with VIP in submucosal neurons (Liu and Kirchgessner, 2000
). However, different groups have reported that activation of mGluRs increase (Liu and Kirchgessner, 2000
; Ren et al., 2000
), decrease (Hu et al., 1999
; Ren et al., 2000
) or did not change the input resistance (Ren et al., 2000
) of submucosal neurons, with no obvious explanation for the discrepancy. Our finding that input resistance is increased during non-purinergic slow EPSPs mediated in part by mGluR1 receptors is consistent with the first observation. However, our observation that MPEP had no effect on slow EPSPs contrasts with the report that this antagonist at a similar concentration depressed submucosal slow EPSPs (Liu and Kirchgessner, 2000
). One possible confounding factor that may explain some, but not all, discrepancies is that focal stimulation of a single internodal strand does not activate every input to the impaled neuron (Bornstein et al., 1986
). This may result in a selective proportion of nerve inputs to be stimulated each time, causing difficulty in interpretation of the data.
Another possible explanation for some of the discrepancies comes from the report that glutamate suppresses slow EPSPs and potentiates slow IPSPs in S-type submucosal neurons via group I mGluRs (Ren et al., 1999
). Glutamate also suppresses slow depolarizations evoked by SP and 5-HT as well as hyperpolarizations evoked by NA (Ren et al., 1999
). This suggests that glutamate may have a modulatory role in neurotransmission.
The conclusion that mGluR1 receptors play a significant role depends on the specificity of BAY 36-7620. This compound has been shown to be highly selective for mGluR1 receptors over other metabotropic glutamate receptors (Carroll et al., 2001
); however, its cross-reactions with other putative transmitters are not known. It appears that PHCCC may cross-react with P2Y1 receptors, because it depressed the purinergic one pulse slow EPSPs. In contrast, BAY 36-7620 had no effect on the purinergic one pulse slow EPSPs, and thus can discriminate between P2Y1 and mGluR1 receptors. Whether it interacts with the variety of other putative transmitter receptors that might be involved (see below) was beyond the scope of this study.
Group I mGluRs are positively coupled to phospholipase C (PLC), while group II (mGluR2 and mGluR3) and group III (mGluR4 and mGluR6–8) are negatively coupled to adenylate cyclase (Coutinho and Knopfel, 2002
; Schoepp et al., 1999
). Group I mGluRs are generally localized postsynaptically, but there is evidence that presynaptic group I mGluRs reduce glutamatergic transmission in the central nervous system (Manzoni and Bockaert, 1995
). Group II and III mGluRs are mostly presynaptic and act as autoreceptors to decrease neurotransmission (Coutinho and Knopfel, 2002
). Group II mGluR-immunoreactivity is present in the submucous plexus of rat jejunum and ileum (Larzabal et al., 1999
). mGluR8 immunoreactivity can be localized to the plasma membrane of VIP immunoreactive neurons in the submucous plexus of the rat ileum (Tong and Kirchgessner, 2003
). Thus, glutamate transmission may be modulated by autoreceptors present in the submucous plexus and hence may have multiple roles.
While the results implicate mGluR1 receptors in some slow EPSPs of VIP neurons, they also clearly indicate that other neurotransmitter receptor combinations are involved. Several potential candidates have been identified in the literature. Pan and Gershon (2000)
suggested that calcitonin gene-related peptide is involved in transmission from intrinsic sensory neurons to submucosal neurons, while Kordasti et al. (2006
) suggested a role for VIP. VIP neurons are depolarized by 5-HT (Surprenant and Crist, 1988
) and are excited by low concentrations of this compound to generate secretory responses in isolated preparations of submucosa/mucosa (Cooke and Carey, 1985
; Johnson et al., 1994
). The slow EPSPs are not mediated by 5-HT7 receptors (Foong and Bornstein, 2009
), but there has been no exhaustive study of the role of 5-HT.
This study shows that the VIP neurons of the submucosal plexus exhibit two qualitatively different slow EPSPs that can be distinguished by their sensitivity to P2 receptor antagonists and the stimulus regimes that evoke them. While there is evidence that P2 mediated slow EPSPs play a role in the regulation of secretion in the colon, whether they play a role in the small intestine is unknown. However, overactivity of VIP neurons plays a critical role in the hypersecretion produced by cholera toxin and some other toxins that produce secretory diarrhoea (Banks et al., 2005
; Gwynne et al., 2009
; Kordasti et al., 2006
). Cholera toxin indirectly causes VIP neurons to become hyperexcitable (Gwynne et al., 2009
), but how the purinergic and non-purinergic slow EPSPs interact with this is unknown. Blockers of P2Y1 receptors or mGluR1 receptors may have interesting effects on cholera-induced diarrhoea and perhaps on other forms of diarrhoea also.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The study was supported by a grant from the National Health and Medical Research Council Australia (NHMRC Grant No. 40053) and an Australian Postgraduate Award (JPPF).
Kordasti, S., Sapnara, M., Thomas, E. A., Lindstrom, E., Forsman, M., Bornstein, J. C., and Sjovall, H. (2006). Effects of cholera toxin on the potential difference and motor responses induced by distension in the rat proximal small intestine in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G948–G958.
Larsson, M. H., Sapnara, M., Thomas, E. A., Bornstein, J. C., Lindstrom, E., Svensson, D. J., and Sjovall, H. (2008). Pharmacological analysis of components of the change in transmural potential difference evoked by distension of rat proximal small intestine in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G165–G173.