 Thais Vieira1Gisele Lozano Costa1Amanda Yaeko Yamada1Amanda Maria de Jesus Bertani1Marta Ines Cazentini Medeiros1Luisa Zanolli Moreno2
Thais Vieira1Gisele Lozano Costa1Amanda Yaeko Yamada1Amanda Maria de Jesus Bertani1Marta Ines Cazentini Medeiros1Luisa Zanolli Moreno2 Andrea Micke Moreno3Christiane Asturiano Ristori Costa1
Andrea Micke Moreno3Christiane Asturiano Ristori Costa1 Karoline Rodrigues Campos1Marlon Benedito1Claudio Tavares Sacchi1
Karoline Rodrigues Campos1Marlon Benedito1Claudio Tavares Sacchi1 Carlos Henrique Camargo1
Carlos Henrique Camargo1 Monique Ribeiro Tiba1*
Monique Ribeiro Tiba1*- 1Instituto Adolfo Lutz, Bacteriology Center of Bacteriology, São Paulo, Brazil
- 2Department of Pathology, Reproduction, and One Health, School of Agricultural and Veterinarian Sciences, São Paulo State University, Jaboticabal, São Paulo, Brazil
- 3Department of Preventive Veterinary Medicine and Animal Health, School of Veterinary Medicine and Animal Science, University of São Paulo, São Paulo, Brazil
The accurate identification and characterization of Aeromonas species are essential to understand their ecological roles and potential health impacts. This study analyzed 90 Aeromonas isolates from various sources using whole genome sequencing (WGS) and matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS). Species identification by WGS, based on Average Nucleotide Identity (ANI ≥96%), revealed inconsistencies in 12.2% of MALDI-TOF MS results, for species not represented in its database. Phylogenetic analyses using single nucleotide polymorphism (SNP) data were concordant in resolving species-level clusters and revealing intra-species diversity. This study reinforces the value of WGS and complementary genomic approaches as reliable tools for Aeromonas species identification, population structure analysis, and integrated One Health surveillance. The integration of genomic tools into routine diagnostics may enhance the capacity of laboratories, particularly in low and middle income countries to monitor emerging resistance and better understand the evolutionary and epidemiological dynamics of Aeromonas in clinical and environmental settings.
1 Introduction
Species of Aeromonas are increasingly recognized as emerging human pathogens, associated with gastrointestinal, wound, and bloodstream infections in both immunocompromised and immunocompetent individuals (Janda and Abbott, 2010; Fernández-Bravo and Figueras, 2020). Over 95% of clinical cases are linked to four species: A. caviae, A. dhakensis, A. veronii, and A. hydrophila (Janda and Abbott, 2010; Fernández-Bravo and Figueras, 2020), although others such as A. salmonicida have also been implicated in human infections (Janda and Abbott, 2010). Traditionally associated with aquatic habitats and fish diseases, these bacteria are now acknowledged as relevant opportunistic pathogens with global distribution (Janda and Abbott, 2010; Fernández-Bravo and Figueras, 2020).
Aeromonas spp. are widely distributed in freshwater, estuarine environments, and soils, and are frequently isolated from food, wastewater, and animals (Koutsoumanis et al., 2014; Fernández-Bravo and Figueras, 2020). This ecological versatility enables colonization of diverse hosts, including humans, domestic animals, and fish, reinforcing their significance within the One Health framework (Lamy et al., 2022). In tropical and subtropical regions, where exposure to untreated water and aquatic resources is common, Aeromonas infections may be underdiagnosed yet pose a growing burden to public health. Infections are typically acquired via ingestion of contaminated water or food, or contact with contaminated environments, and can range from localized diarrhea and skin lesions to systemic infections, particularly in immunocompromised patients (Parker and Shaw, 2011).
Clinically, accurate identification of Aeromonas at the species level remains challenging. Traditional phenotypic methods often yield ambiguous results due to overlapping biochemical characteristics, leading to frequent misidentification, especially among A. caviae, A. media, A.dhankenis and A. hydrophila (Janda and Abbott, 2010; Fernández-Bravo and Figueras, 2020). Whole genome sequencing (WGS) combined with average nucleotide identity (ANI) analysis has become the gold standard for bacterial species identification and taxonomic resolution. ANI provides an objective and quantitative measure of genomic similarity, with a ≥96% threshold widely accepted to delineate species boundaries in prokaryotes. Unlike phenotypic assays or protein-based approaches, WGS delivers a comprehensive and precise view of the genome, enabling unambiguous discrimination among closely related species. The robustness, reproducibility, and high resolution afforded by WGS-ANI analyses make them indispensable for reliable taxonomic classification. However, the cost, infrastructure requirements, and turnaround time limit its use in routine diagnostics. In contrast, matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) offers a rapid, cost effective alternative with growing accuracy in Aeromonas identification. When paired with curated databases, MALDI-TOF MS enables real time species-level resolution, facilitating timely clinical decisions. Nevertheless, phenotypic overlap and insufficient representation of well-characterized isolates in commercial databases frequently result in misclassification, particularly between A. hydrophila and A. dhakensis (Kitagawa et al., 2022; Bartie and Desbois, 2024; Truong-Ha et al., 2024). Moreover, reference databases are often biased towards human-associated strains and not regularly updated to reflect taxonomic revisions, further reducing discriminatory power. These findings underscore that, while MALDI-TOF is valuable as a first-line tool, its limitations necessitate confirmation with genome-based approaches for reliable species-level resolution within the genus.
This study aims to improve species level identification of Aeromonas through a combined approach using MALDI-TOF MS and WGS. By integrating phenotypic and genotypic methodologies, we seek to overcome diagnostic limitations and provide insights into the taxonomy, resistance markers, and epidemiological relevance of this under recognized group of pathogens, with implications for surveillance and public health.
2 Materials and methods
2.1 Isolates
As part of the routine monitoring and surveillance activities conducted at the Instituto Adolfo Lutz, the public health reference laboratory for the State of São Paulo, Brazil, we included all Aeromonas sp. isolates received over the last ten years (2014-2024). These comprised strains from human clinical specimens, as well from animals, food and waters sources. A total of 90 Aeromonas strains were analyzed (Table 1). All isolates are preserved in the bacterial collection at the Bacteriology Center of the Instituto Adolfo Lutz (IAL).
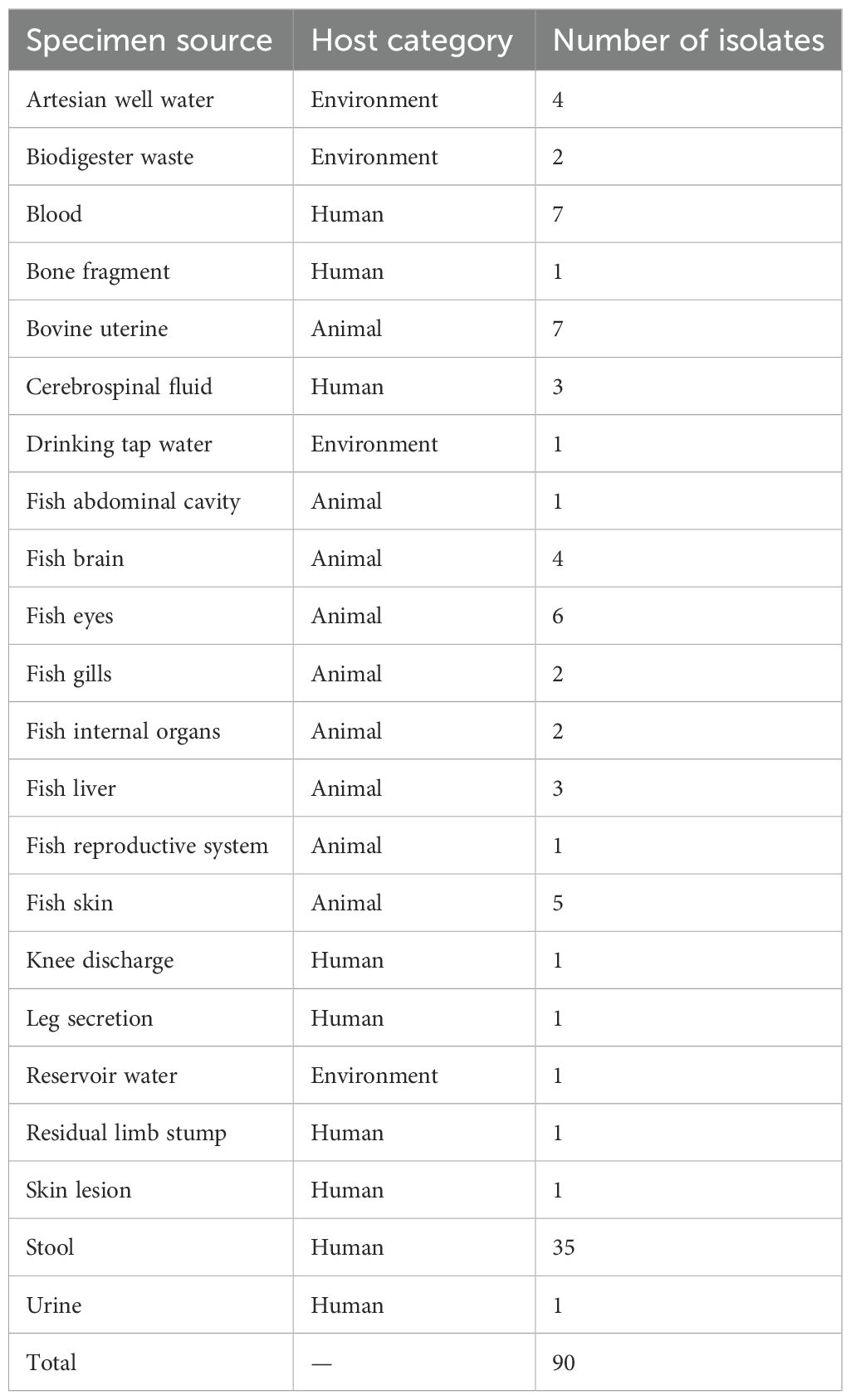
Table 1. Distribution of Aeromonas isolates based on specimen source and host category.
2.2 Identification
The classic phenotypic characteristics of Aeromonas spp. were assessed, including oxidase and catalase positivity, as well as the morphology of Gram-negative rods (Supplementary Table 1). The isolates exhibited facultative anaerobic growth on standard laboratory media such as heart infusion agar, MacConkey’s agar, and blood agar. Additionally, they demonstrated the ability to grow in nutrient broth without NaCl and showed resistance to the vibriostatic agent O/129.
2.3 Identification by MALDI-TOF MS mass spectrometry
Species-level identification of the isolates was initially assessed by MALDI-TOF mass spectrometry (Microflex LT, Bruker Daltonics®, Germany), following the manufacturer’s protocol, which involves direct colony transfer. The isolates were grown for 24 hours on Tryptic Soy Agar (TSA) (Difco, USA), and using a sterile wooden stick, a single colony from each sample was briefly touched to transfer a thin layer of microorganisms to a polished steel plate with 96 wells.
The layer of microorganisms applied to a steel plate was overlaid with 1 µL of formic acid, followed by 1 µL of the matrix solution (10 mg/mL of α-cyano-4-hydroxycinnamic acid in 50% acetonitrile/2.5% trifluoroacetic acid). Each isolate was applied to three wells, and three readings were conducted. The FlexControl software (Bruker Daltonics) was used to capture the protein spectra using the MTB_autoX method within a mass range of 2 to 20 kDa. External calibration of the spectrophotometer was performed using ribosomal proteins from Escherichia coli (BTS - Bruker Daltonics®). For bacterial identification, BioTyper 3.0 software (Bruker Daltonics®) was utilized. The generated mass spectra were analyzed with BioTyper 3.0 software connected to the system and compared to the profiles stored in the system’s library. The criteria for interpreting the patterns used in this study were as follows: scores ≥ 2.0 were considered acceptable for species attribution, while scores ≥ 1.7 and < 2.0 were used exclusively for genus identification, following the manufacturer’s guidelines. In cases where the first and second match species differed, the identification corresponding to the highest score was selected.
Bacterial identification was performed using MALDI-TOF mass spectrometry and analyzed via the MicrobeNet platform (Centers for Disease Control and Prevention, Atlanta, GA, USA; https://www.cdc.gov/microbenet, accessed on 23 July 2025). For each isolate, the XML export generated by MicrobeNet was retrieved and parsed to extract the top-ranked taxonomic identification, confidence score, and database version. Identifications were classified according to predefined score thresholds for species- or genus-level calls.
2.4 Whole genome sequencing
The strains were sequenced using the Ion Torrent S5 platform at the Strategic Laboratory of the Instituto Adolfo Lutz. The genetic material was extracted using the commercial Wizard Genomic DNA Purification Kit (Promega, USA). The integrity, purity, and concentration of the extracted genomic DNA were assessed using agarose gel electrophoresis (1%) at 100 V for 40 minutes, spectrophotometry with a NanoDrop™ One (Thermo Fisher Scientific, USA), and fluorometry with a Qubit™ (Thermo Fisher Scientific, USA), respectively. For the construction of the DNA library, the Ion Xpress™ Plus Fragment Library Kit (Thermo Fisher Scientific, USA) was used according to the manufacturer’s recommendations. The material was applied to the Ion Chip 530 Kit.
Reads were evaluated for quality and GC content using FastQC (version 0.12.1). Contamination screening was performed with Kraken2 (version 1.1.1) via the Galaxy Europe Server (https://usegalaxy.eu/). Genome assembly was carried out using the FASTQ files obtained from short-read sequencing with the CLC Genomics Workbench (version 9.5.3, Qiagen, Venlo, Netherlands). QC metrics (e.g., genome size, N50, N90, contig count, and GC content) were assessed using QUAST (version 5.2.0).
2.4.1 Genome analysis
Average Nucleotide Identity (ANI) was utilized as the gold standard for species identification. Following the methodology established by Richter and Rosselló-Mora (Richter and Rosselló-Móra, 2009), ANI is defined as the percentage of identity observed in the nucleotide sequences of orthologous genes shared between two genomes. The cut-off value for Aeromonas was determined by Colston et al (Colston et al., 2014), who suggested that values of ≥96% indicate that the two strains belong to same species (Maia et al., 2023). ANI analysis was performed in March 2025 using fastANI (Version 1.3) implemented on the Galaxy Europe Server (https://usegalaxy.eu/), with default settings applied. The reference genomes used in this analysis are listed in Supplementary Table 2. The online platform PathogenWatch (https://pathogen.watch/ accessed on 22 April 2025) was also used for species identification.
2.5 Multilocus sequence typing
The assembled genomes were submitted to PubMLST for sequence type (ST) analysis. The MLST (Multilocus Sequence Typing) scheme for Aeromonas was created based on data obtained by Martino et al (Martino et al., 2011). utilizing six genes (gyrB, groL, gltA, metG, ppsA, and recA). Novel alleles and new allele combinations identified in this study were submitted to the PubMLST database for curation and deposition.
2.6 Single nucleotide polymorphisms analysis
Single nucleotide polymorphisms (SNPs) were identified from FASTA sequences using CSI Phylogeny (http://www.genomicepidemiology.org/services/, accessed on 23 July 2025) under default settings. The resulting SNP-based phylogenetic tree was visualized with Microreact (http://microreact.org accessed on 25 July 2025).
The Whole Genome Shotgun project has been deposited in DDBJ/ENA/GenBank under BioProject accession PRJNA1295185. Novel alleles and sequence types (STs) have been deposited in the PubMLST database, and their corresponding accession numbers are listed in the Supplementary Table S3.
2.7 Statistical analysis
Pairwise SNP distances were used to assess genomic diversity and population structure among Aeromonas isolates. Mean pairwise distances were calculated within and between species to quantify intra- and inter-species variation. To evaluate whether genetic distances were significantly associated with species or host/source categories, a permutational multivariate analysis of variance (PERMANOVA) (Herrando-Pérez et al., 2021). PERMANOVA was conducted using 999 permutations (default setting) to test for significant differences among groups.
3 Results
MALDI-TOF MS correctly identified 87.8% of the Aeromonas spp. isolates. Discrepancies between MALDI-TOF MS and genomic identification based on ANI were observed in 12.2% of the cases (Table 2). Differences were also noted between species identification by BDAL (Bruker database), MicrobeNet and the ANI-based approach. For instance, isolates identified as A. rivipollensis by ANI were classified as A. veronii, A. eucrenophila, and A. media by BDAL, and as A. punctata in MicrobeNet corresponding to isolates Aer_91, Aer_92, and Aer_95 respectively. Similarly, isolates identified as A. dhakensis by ANI were assigned to A. hydrophila in both the BDAL database and MicrobeNet (Aer_19, Aer_33, Aer_44, Aer_45, Aer_47, Aer_58, and Aer_64). In addition, one isolate (Aer_89) identified as A. allosaccharophila by ANI was classified as A. veronii in both BDAL and MicrobeNet.
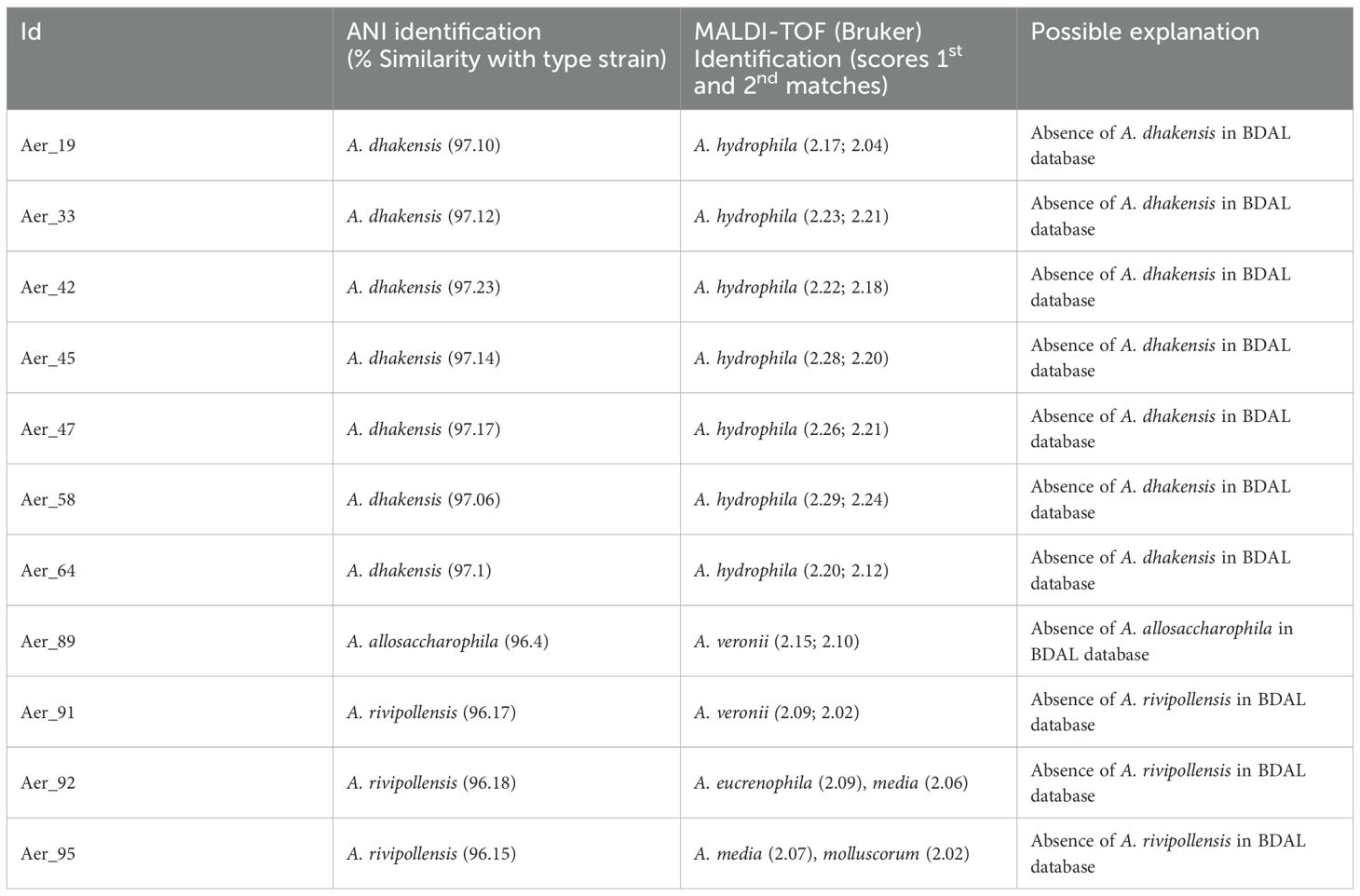
Table 2. Comparison of taxonomic identifications for Aeromonas isolates based on ANI and MALDI-TOF.
The genomic profiles of the Aeromonas spp. isolates analyzed in this study are presented in Supplementary Table S1. The assembled genome sizes ranged from 4.26 to 5.09 Mbp, with an average size of 4.71 Mbp. The average sequencing coverage was approximately 62×, ensuring high quality genome assemblies. The total guanine and cytosine (G + C) content of the Aeromonas spp. genomes ranged from 58% to 62%. Within A. hydrophila, the G + C content varied between 60.9% and 61.7%; in A. caviae, it ranged from 60.8% to 62%; and in A. veronii, from 58% to 58.8% (Supplementary Table S3).
The isolates analyzed originated from diverse sources, including fish, cattle, humans, and environmental samples, revealing distinct species distributions. Fish isolates were predominantly identified as A. caviae, A. veronii, and A. hydrophila, recovered from various tissues such as gills, eyes, brain, and skin. Uterine samples from cattle primarily harbored A. rivipollensis, A. caviae, and A. media, indicating species diversity within this host. Human clinical isolates, obtained from stool, blood, cerebrospinal fluid, and wound samples were largely composed of A. hydrophila and A. caviae, with occurrences of A. veronii, A. dhakensis, and A. jandaei. Environmental isolates, derived from reservoir and well water as well as biodigester waste, were dominated by A. hydrophila, followed by A. dhakensis and A. caviae, highlighting their role as environmental reservoirs.
ANI-based genome comparisons (n=90) showed that A. hydrophila was the most prevalent species (34/90; 37.8%), followed by A. caviae (23/90; 25.5%) and A. veronii (16/90; 17.8%). Less frequent taxa included A. dhakensis (7/90; 7.8%), A. jandaei (4/90; 4.4%), A. rivipollensis (3/90; 3.3%), A. media (2/90; 2.2%), and a single isolate of A. allosaccharophila (1/90; 1.1%). Species identification based on ANI was further supported by results from the PathogenWatch platform, which demonstrated consistent concordance.
Regarding the sources of isolation, A. hydrophila was recovered from animals (8/34; 23.5%), the environment (6/34; 17.6%), and humans (20/34; 58.8%). A. caviae occurred predominantly in humans (17/23; 73.9%), with additional isolates from animals (5/23; 21.7%) and the environment (1/23; 4.3%). A. veronii was mainly animal associated (11/16; 68.8%) with human isolates (5/16; 31.3%). A. dhakensis was mostly human derived (6/7; 85.7%), with a single environmental isolate (1/7; 14.3%). A. jandaei was exclusively recovered from humans (4/4; 100%), while A. rivipollensis (3/3; 100%) and A. media (2/2; 100%) were isolated only from animals. and A. allosaccharophila was represented by a single animal derived isolate (1/1; 100%).
MLST results are presented in the Supplementary Table S3. A large number of isolates (n=61) were found to harbor novel STs not previously reported in the database. The distribution of STs was highly diverse, both across Aeromonas species and among isolates from different sources, including human, animal, and environmental samples. Notably, the ppsA gene, one of the housekeeping loci used in the standard MLST scheme for Aeromonas spp., was not detected in isolate Aer_10. Whole genome sequencing and PCR assays both failed to identify the presence of this gene, confirming its absence.
The SNP distance matrix revealed a wide range of genomic diversity among the Aeromonas isolates, with pairwise SNP differences ranging from 18 to 26,825 across the entire dataset. Intra-species comparisons showed variable patterns of diversity. For A. veronii, SNP distances ranged from 18 to 10,517 SNPs, while A. hydrophila and A. caviae exhibited the greatest intra-species genomic diversity, with SNP distances ranging from 19 to 19,447 and 22 to 7,274 respectively. In contrast, A. dhakensis isolates were more homogeneous, ranging from 11,597 to 12,678 SNPs. These results highlight the coexistence of closely related clonal groups and highly divergent strains, consistent with the phylogenetic analysis. Overall, SNP-based analyses provided detailed, high resolution insights into the evolutionary dynamics within the Aeromonas genus.
SNP-based phylogenetic analysis revealed distinct clustering patterns corresponding to Aeromonas species, with isolates identified as A. caviae, A. hydrophila, and A. veronii forming species-specific clades (Figure 1). Within these clades, A. hydrophila isolates tended to group more closely together, while A.veronii showed a more dispersed distribution across several lineages. A. caviae isolates displayed greater intra-species diversity, compared to the other groups. Integration of metadata indicated that A. hydrophila isolates included both human and environmental isolates, whereas A. veronii isolates of animal origin were mainly grouped in separate lineages.
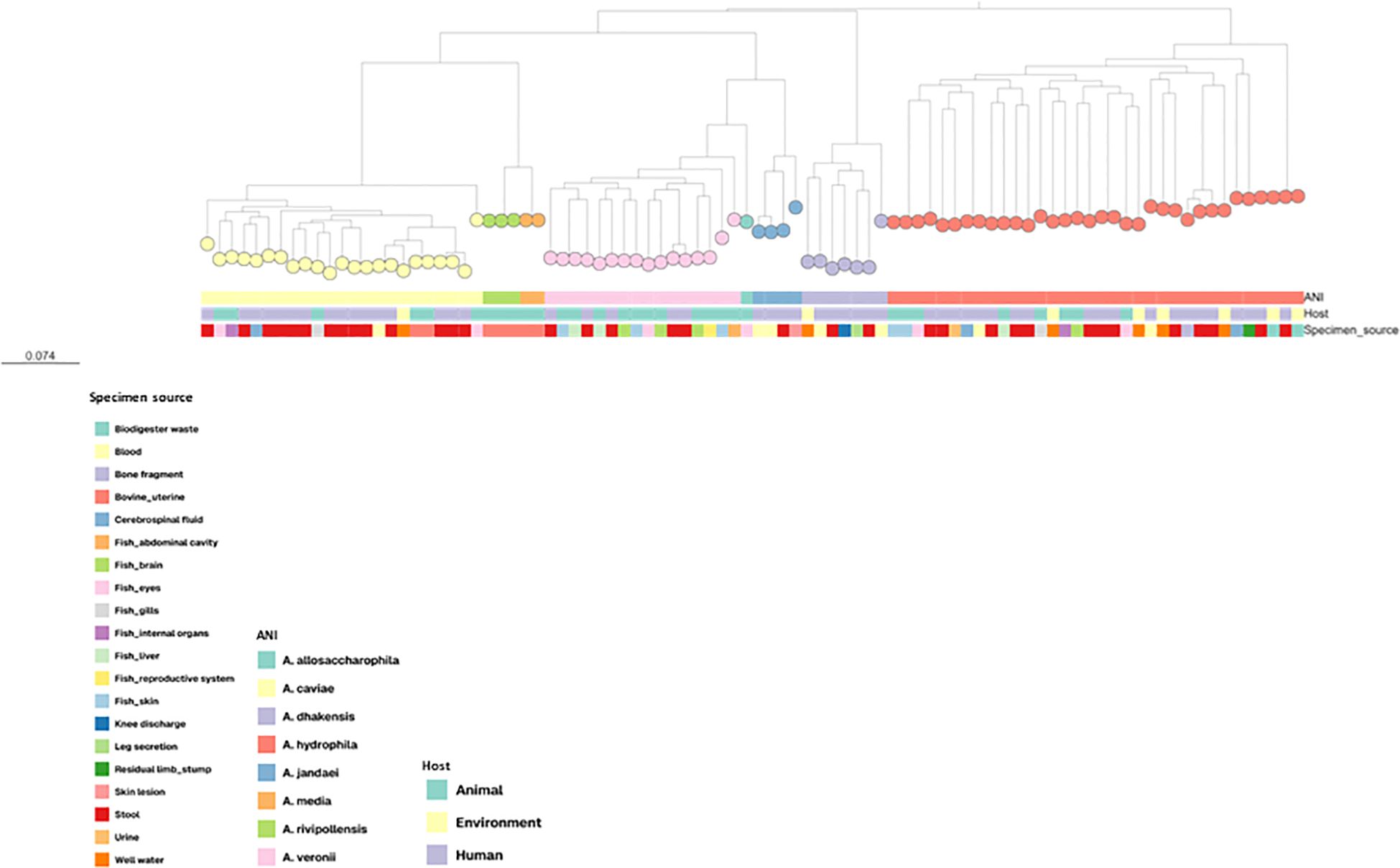
Figure 1. Phylogenetic tree based on SNPs analysis of 90 Aeromonas isolates from human, animal and environmental sources. Colored circles represent individual isolates, grouped according to genetic similarity. The bottom color strip indicates the host (animal, environment and human). ANI (average nucleotide identity) results were used for species identification. The tree reveals species-specific clustering patterns and highlights intra-species diversity, including both closely related subgroups and genetically divergent isolates.
Statistical analysis of the pairwise SNP distance matrix revealed a clear genomic structure among Aeromonas isolates. Most of the genetic variation was explained by species differences (F = 27.54; p = 0.001), confirming strong species-level clustering consistent with the phylogenetic reconstruction. A smaller but statistically significant effect was also detected for host/source categories (F = 2.72; p = 0.003), indicating that, while isolates from human, animal, and environmental origins can share closely related genotypes, their genetic composition varies slightly according to source. In summary, SNP-level diversity in Aeromonas reflects taxonomic differentiation, with minor contributions from ecological or host-related factors.
4 Discussion
While Aeromonas species are increasingly recognized as emerging pathogens with relevance in both clinical and environmental contexts, studies in Brazil integrating isolates from human, animal, and environmental sources remain scarce. Most national investigations have focused on single source isolates and have emphasized the use of MALDI-TOF MS as the primary method for species identification (Cardoso et al., 2021; Teodoro et al., 2022). In contrast, international research has progressively adopted whole genome sequencing (WGS) to improve species-level resolution and explore antimicrobial resistance, virulence, and phylogenetic relationships in Aeromonas (Awan et al., 2018; Klemm et al., 2024; Sagas et al., 2024). However, few global studies have simultaneously compared MALDI-TOF MS and WGS-based approaches across isolates from multiple ecological niches. This study addresses these gaps by employing a combination of MALDI-TOF MS, WGS, average nucleotide identity (ANI), multilocus sequence typing (MLST), and SNP-based phylogenetic analysis to identify and characterize Aeromonas isolates from clinical, animal, and environmental sources. Our findings contribute novel insights into the genetic diversity and ecological relationships of Aeromonas spp., supporting integrated surveillance within a One Health framework.
The WGS-based analysis employing Average Nucleotide Identity (ANI) provided a robust and precise framework for species-level discrimination and in depth characterization of the isolates. ANI, recognized as a gold standard for bacterial taxonomy, allowed classification with high confidence (ANI ≥96%), capturing subtle genetic differences that traditional methods often miss (Martino et al., 2011; Colston et al., 2014; Kim et al., 2014; Awan et al., 2018; Cardoso et al., 2021; Teodoro et al., 2022; Maia et al., 2023; Klemm et al., 2024; Sagas et al., 2024). The identified species exhibited distinct distribution patterns across multiple sources, fish, cattle, humans, and environmental samples, reflecting their diverse ecological niches and potential epidemiological roles (Fernández-Bravo and Figueras, 2020). The concordance of ANI-based identification with the PathogenWatch platform further validates the reliability and reproducibility of this genomic approach, which is easier and faster – only upload the fasta files is enough to retrieve the identification results (PathogenWatch platform. Version 23.4.4).
Overall, MALDI-TOF MS accurately identified the majority of isolates, particularly for species well represented in the instrument’s reference database. However, discrepancies were notably observed for A. dhakensis, A. rivipollensis, and A. allosaccharophila, primarily because these less common species were not included in the MALDI-TOF MS reference library (Fernández-Bravo and Figueras, 2020). Several studies have further demonstrated that A. dhakensis remains particularly prone to misidentification by MALDI-TOF, often being classified as A. hydrophila due to the phenotypic and proteomic overlap between these taxa (Bartie and Desbois, 2024; Truong-Ha et al., 2024). In the study by Truong et al. (2024), MALDI-TOF achieved only 12% concordance with WGS-based ANI assignments, systematically misidentifying A. dhakensis, which emphasizes the critical impact of incomplete databases. This limitation highlights the challenges posed by incomplete spectral databases and reinforces the importance of continually updating and expanding these libraries to improve species coverage and diagnostic accuracy.
While traditional phenotypic methods such as MALDI-TOF MS remain valuable for rapid routine identification, they may lack sufficient resolution to reliably distinguish closely related species. In this context, WGS-based identification using ANI ≥96% provided high resolution classification of 90 Aeromonas isolates and served as a robust reference for evaluating the performance of MALDI-TOF MS (Colston et al., 2014; Kim et al., 2014; Maia et al., 2023). By evaluating the same spectra in another public database (MicrobeNet, developed by the CDC and partners), the results similarly failed to correctly identify the isolates that had been previously misidentified or not identified by the Bruker MALDI-TOF MS library. These findings highlight the limitations of current MALDI-TOF MS platforms in identifying less common Aeromonas species and suggest that integrating complementary methods or relying on high resolution genomic approaches remains essential for accurate species level identification.
In addition to ANI, Multilocus Sequence Typing (MLST) was employed to further characterize the Aeromonas isolates. The MLST analysis revealed considerable genetic heterogeneity with many isolates harboring novel sequence types (STs) not previously reported in the PubMLST database (Navarro and Martínez-Murcia, 2018; Meng et al., 2020; Lu et al., 2025). The identification of these novel STs underscores the ongoing diversification within the genus and provides valuable epidemiological markers for future surveillance and comparative studies. Their inclusion in global databases is essential to improve the resolution of molecular typing frameworks and to facilitate accurate tracking of emerging lineages. This finding underexplored diversity within Aeromonas genus and highlights the necessity to expanded genomic surveillance. The observed variability across isolates from human, animal, environmental sources reflects the ecological versatility of this genus and its potential for circulation between different hosts and environments (Fernández-Bravo and Figueras, 2020). Notably, one isolate (Aer_10) lacked the ppsA housekeeping gene, a result confirmed by both WGS and PCR, suggesting a possible gene loss or genomic rearrangement, an uncommon but significant event that may contribute to our understanding of genome plasticity in Aeromonas.
The overall phylogenetic structure underscores the genomic distinctiveness among Aeromonas species and highlights differences in intra-species diversity, with A. caviae showing broader dispersion compared to other species (Fernández-Bravo and Figueras, 2020; Singh et al., 2025). This wider distribution may reflect its ubiquity across environments, as previously reported, and may indicate higher genomic variability. Moreover, the close phylogenetic proximity of certain A. hydrophila strains from environmental and human samples is consistent with its presence in multiple ecological niches and its occurrence across different hosts. In our dataset, A. caviae was the most frequently isolated species from stool samples, slightly surpassing A. hydrophila. This observation aligns with previous reports identifying A. caviae as a predominant species associated with gastrointestinal infections in humans (Vila et al., 2003; Du et al., 2021; Ruiz de Alegría-Puig et al., 2023; Chong et al., 2024). Together, these findings support the use of SNP-based phylogenomics as a powerful and reliable tool for species-level resolution and for investigating evolutionary and epidemiological patterns in Aeromonas populations.
Our SNP-based phylogeny also revealed distinct and well-supported clades for A. dhakensis and A. jandaei, underscoring the genomic distinctiveness of these species within the genus. The clear separation between A. dhakensis and A. hydrophila corroborates recent findings that have corrected long-standing misclassifications and supports the need for re-evaluation of public health isolates previously identified as A. hydrophila (Bartie and Desbois, 2024; Truong-Ha et al., 2024). Among the species analyzed, A. rivipollensis and A. media exhibited the lowest mean intra-species distances (approximately 31 and 42 SNPs, respectively), followed by A. jandaei (approximately 8,299 SNPs), indicating moderate genomic relatedness among the human isolates analyzed. Given the limited number of A. jandaei isolates analyzed and the lack of environmental or animal representatives, this finding must be viewed within the context of restricted sampling, which limits broader inferences about the population structure, ecological distribution, and potential reservoirs of the species. The statistical analysis of SNP-level variation revealed that the genomic structure of Aeromonas populations is mainly shaped by species-level differentiation. This strong taxonomic signal is consistent with the phylogenetic reconstruction and supports the clear separation of species within the genus, as previously observed in phylogenomic studies of A. media, A. hydrophila, and A. veronii complexes (Bartie and Desbois, 2024; Truong-Ha et al., 2024).
This study highlights the complementary roles of multiple molecular and phenotypic techniques in accurately identifying and characterizing Aeromonas species. WGS and ANI emerged as gold standard methods, offering high resolution taxonomic classification and enabling detailed exploration of phylogenetic relationships and population structure (Fernández-Bravo and Figueras, 2020; Bertran et al., 2021). Their precision was particularly valuable in distinguishing closely related species and in validating other identification tools. However, the study is not without limitations, particularly the uneven distribution of isolates across hosts and environments, as most originated from human stool samples collected through routine public health surveillance, while environmental and animal isolates were only available through specific collaborations. This sampling imbalance may limit the strength of inter-host or ecological comparisons.
This study underscores the complementary roles of molecular and phenotypic techniques in improving the identification and characterization of Aeromonas species. WGS and ANI provided high-resolution taxonomic classification, enabled detailed phylogenetic analysis, and served as a gold standard for validating other tools. Importantly, while WGS-based surveillance may not yet be universally accessible, especially in low and middle-income countries, combining WGS with MALDI-TOF MS and curated online platforms offers a robust framework for accurate identification. Together, these findings underscore the need for integrated diagnostic and surveillance strategies that leverage both advanced genomic approaches and accessible technologies, strengthening One Health surveillance and informing public health interventions to mitigate Aeromonas-associated risks widely distributed worldwide.
Data availability statement
All the sequences generated in this study were deposited in the GenBank (BioProject: PRJNA1295185).
Ethics statement
This study was submitted to and approved by the local ethics committee under CTC-IAL 05-Q/2024.
Author contributions
TV: Conceptualization, Software, Methodology, Investigation, Writing – original draft, Formal Analysis, Validation, Data curation, Visualization. GC: Investigation, Writing – review & editing, Methodology. AY: Writing – review & editing, Methodology, Data curation. AB: Investigation, Software, Formal Analysis, Writing – review & editing, Methodology. MM: Writing – review & editing, Supervision, Conceptualization, Data curation. LM: Methodology, Validation, Writing – review & editing, Formal Analysis, Data curation. AM: Formal Analysis, Validation, Writing – review & editing, Supervision. CARC: Investigation, Data curation, Formal Analysis, Writing – review & editing, Supervision, Validation. KC: Validation, Software, Writing – review & editing, Methodology, Visualization. MB: Data curation, Visualization, Software, Formal Analysis, Writing – review & editing. CS: Methodology, Conceptualization, Software, Writing – review & editing. CHC: Supervision, Visualization, Resources, Formal Analysis, Writing – review & editing, Software, Validation, Writing – original draft. MT: Supervision, Validation, Conceptualization, Funding acquisition, Writing – original draft, Resources, Data curation, Visualization, Project administration, Formal Analysis.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was funded by FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo, grant numbers: 2024/18468-3, 2017/50333-7, 2018/21192-9, 2018/21193-5); CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, grant number: 402563/2021-2. This study was carried out as part of our routine work. MRT is the recipient of a Productivity Research Fellowships from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). FESIMA (Processo n°: 024.00109863/2025-21).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbrio.2025.1708292/full#supplementary-material.
References
Awan F., Dong Y., Liu J., Wang N., Mushtaq M. H., Lu C., et al. (2018). Comparative genome analysis provides deep insights into Aeromonas hydrophila taxonomy and virulence-related factors. BMC Genomics 19, 712. doi: 10.1186/s12864-018-5100-4
Bartie K. L. and Desbois A. P. (2024). Aeromonas dhakensis: a zoonotic bacterium of increasing importance in aquaculture. Pathogens 13, 465. doi: 10.3390/pathogens13060465
Bertran X., Rubio M., Gómez L., Llovet T., Muñoz C., Navarro F., et al. (2021). Taxonomic identification of different species of the genus Aeromonas by whole-genome sequencing and use of their species-specific β-lactamases as phylogenetic markers. Antibiotics 10, 354. doi: 10.3390/antibiotics10040354
Cardoso P. H. M., Moreno L. Z., de Oliveira C. H., Gomes V. T. M., Silva A. P. S., Barbosa M. R. F., et al. (2021). Main bacterial species causing clinical disease in ornamental freshwater fish in Brazil. Folia Microbiol. (Praha). 66, 231–239. doi: 10.1007/s12223-020-00837-x
Chong S. K. T., Liu F., Yuwono C., Tay A. C. Y., Wehrhahn M. C., Riordan S. M., et al. (2024). Analysis of global Aeromonas caviae genomes revealed that strains carrying T6SS are more common in human gastroenteritis than in environmental sources and are often phylogenetically related. Microb. Genom. 10, 1258. doi: 10.1099/mgen.0.001258
Colston S. M., Fullmer M. S., Beka L., Lamy B., Gogarten J. P., and Graf J. (2014). Bioinformatic genome comparisons for taxonomic and phylogenetic assignments using Aeromonas as a test case. mBio 5, e02136–e02114. doi: 10.1128/mBio.02136-14
Du X., Wang M., Zhou H., Li Z., Xu J., Li Z., et al. (2021). Comparison of multiple platforms to identify various Aeromonas species. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.625961
Fernández-Bravo A. and Figueras M. J. (2020). An update on the genus Aeromonas: taxonomy, epidemiology, and pathogenicity. Microorganisms 17, 129. doi: 10.3390/microorganisms8010129
Herrando-Pérez S., Tobler R., and Huber C. D. (2021). smartsnp, an R package for fast multivariate analyses of big genomic data. Methods Ecol. Evol. 12, 2084–2093. doi: 10.1111/2041-210X.13684
Janda J. M. and Abbott S. L. (2010). The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 23, 35–73. doi: 10.1128/CMR.00039-09
Kim M., Oh H. S., Park S. C., and Chun J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Kitagawa D., Suzuki Y., Abe N., Ui K., Suzuki K., Yamashita T., et al. (2022). Comparison of MALDI-TOF mass spectrometry and rpoB gene sequencing for the identification of clinical isolates of Aeromonas spp. Heliyon 13, e11585. doi: 10.1016/j.heliyon.2022.e11585
Klemm E. J., Pickard D., Whiley R., Kingsley R. A., and Dougan G. (2024). Genomic analysis of clinical Aeromonas isolates reveals genetic diversity but little evidence of genetic determinants for diarrhoeal disease. Microb. Genom. 10, 1211. doi: 10.1099/mgen.0.001211
Koutsoumanis K. P., Lianou A., and Sofos J. N. (2014). Food safety: emerging pathogens. Encyclopedia Agric. Food Systems. 21, 250–272. doi: 10.1016/B978-0-444-52512-3.00049-8
Lamy B., Baron S., and Barraud O. (2022). Aeromonas: the multifaceted middleman in the One Health world. Curr. Opin. Microbiol. 65, 24–32. doi: 10.1016/j.mib.2021.09.012
Lu J., Zhang L., Yan C., Lin N., Zhang Y., Sha Y., et al. (2025). Whole-genome sequencing-based species classification, multilocus sequence typing, and antibiotic resistance mechanisms of the clinical Aeromonas complex. Front. Microbiol. 16. doi: 10.3389/fmicb.2025.1473150
Maia J., Silva G., Cunha L., Gouveia G. V., Góes-Neto A., Brenig B., et al. (2023). Genomic characterization of Aeromonas veronii provides insights into taxonomic assignment and reveals widespread virulence and resistance genes throughout the world. Antibiotics 12, 1039. doi: 10.3390/antibiotics12061039
Martino M. E., Fasolato L., Montemurro F., Rosteghin M., Manfrin A., Patarnello T., et al. (2011). Determination of microbial diversity of Aeromonas strains on the basis of multilocus sequence typing, phenotype, and presence of putative virulence genes. Appl. Environ. Microbiol. 77, 4986–5000. doi: 10.1128/AEM.00708-11
Meng S., Wang Y. L., Liu C. G., Yang J., Yuan M., Bai X. N., et al. (2020). Genetic diversity, antimicrobial resistance, and virulence genes of Aeromonas isolates from clinical patients, tap water systems, and food. BioMed. Environ. Sci. 33, 385–395. doi: 10.3967/bes2020.053
Navarro A. and Martínez-Murcia A. (2018). Phylogenetic analyses of the genus Aeromonas based on housekeeping gene sequencing and its influence on systematics. J. Appl. Microbiol. 125, 622–631. doi: 10.1111/jam.14081
Parker J. L. and Shaw J. G. (2011). Aeromonas spp. clinical microbiology and disease. J. Infect. 62, 109–118. doi: 10.1016/j.jinf.2010.12.003
Richter M. and Rosselló-Móra R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U S A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Ruiz de Alegría-Puig C., Fernández-Martínez M., and De Malet Pintos-Fonseca A. (2023). Epidemiology of Aeromonas spp. isolated from stool in a tertiary hospital in Cantabria, Northern Spain, in the last five years. Enferm Infecc Microbiol. Clin. (Engl Ed). 41, 211–214. doi: 10.1016/j.eimce.2021.09.014
Sagas D., Hershko Y., Levitskyi K., Strauss M., Slutzkin M., Chazan B., et al. (2024). Phenotypic and genotypic analysis of antimicrobial resistance and population structure of gastroenteritis-related Aeromonas isolates. Ann. Clin. Microbiol. Antimicrob. 23, 45. doi: 10.1186/s12941-024-00706
Singh A., Liu F., Yuwono C., Wehrhahn M. C., Slavich E., Young A. M., et al. (2025). Age-dependent variations in the distribution of Aeromonas species in human enteric infections. Pathogens 14, 120. doi: 10.3390/pathogens14020120
Teodoro J. R., Carvalho G. G., Queiroz M. M., Levy C. E., and Kabuki D. Y. (2022). Incidence, evaluation of detection and identification methods, and antimicrobial resistance of Aeromonas spp. in ready-to-eat foods. Int. J. Food Microbiol. 379, 109862. doi: 10.1016/j.ijfoodmicro.2022.109862
Truong-Ha M. N., Nguyen Q., Voong P. V., Chau V., Nguyen N. H. T., Nguyen T. H. M., et al. (2024). Genomic characterization of Aeromonas spp. isolates from striped catfish with motile Aeromonas septicemia and human bloodstream infections in Vietnam. Microb. Genom. 10, 1248. doi: 10.1099/mgen.0.001248
Keywords: Aeromonas species, whole genome sequencing (WGS), mass spectrometry, phylogeny, One Health
Citation: Vieira T, Costa GL, Yamada AY, Bertani AMdJ, Medeiros MIC, Moreno LZ, Moreno AM, Costa CAR, Campos KR, Benedito M, Sacchi CT, Camargo CH and Tiba MR (2025) Genomic and proteomic identification of Aeromonas isolates from diverse sources: comparative performance of WGS and MALDI-TOF MS in species-level resolution. Front. Bacteriol. 4:1708292. doi: 10.3389/fbrio.2025.1708292
Received: 18 September 2025; Accepted: 23 October 2025;
Published: 03 November 2025.
Edited by:
Kumaragurubaran Karthik, Tamil Nadu Veterinary and Animal Sciences University, IndiaReviewed by:
Hoang Duc Nguyen, Ho Chi Minh City University of Science, VietnamJoão Gouveia, Universidade Federal do Vale do São Francisco, Brazil
Ying-Wen Chen, National Cheng Kung University Hospital, Taiwan
Aki Sakurai, National Center for Global Health and Medicine, Japan
Copyright © 2025 Vieira, Costa, Yamada, Bertani, Medeiros, Moreno, Moreno, Costa, Campos, Benedito, Sacchi, Camargo and Tiba. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monique Ribeiro Tiba, bW9uaXF1ZS5jYXNhc0BpYWwuc3AuZ292LmJy