 Lucas M. Ascari
Lucas M. Ascari Stephanie C. Rocha1
Stephanie C. Rocha1 Priscila B. Gonçalves
Priscila B. Gonçalves Tuane C. R. G. Vieira
Tuane C. R. G. Vieira Yraima Cordeiro
Yraima Cordeiro- 1Faculty of Pharmacy, Pharmaceutical Biotechnology Department, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 2Institute of Medical Biochemistry Leopoldo de Meis, National Institute of Science and Technology for Structural Biology and Bioimaging, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
Transmissible spongiform encephalopathies (TSEs), also known as prion diseases, arise from the structural conversion of the monomeric, cellular prion protein (PrPC) into its multimeric scrapie form (PrPSc). These pathologies comprise a group of intractable, rapidly evolving neurodegenerative diseases. Currently, a definitive diagnosis of TSE relies on the detection of PrPSc and/or the identification of pathognomonic histological features in brain tissue samples, which are usually obtained postmortem or, in rare cases, by brain biopsy (antemortem). Over the past two decades, several paraclinical tests for antemortem diagnosis have been developed to preclude the need for brain samples. Some of these alternative methods have been validated and can provide a probable diagnosis when combined with clinical evaluation. Paraclinical tests include in vitro cell-free conversion techniques, such as the real-time quaking-induced conversion (RT-QuIC), as well as immunoassays, electroencephalography (EEG), and brain bioimaging methods, such as magnetic resonance imaging (MRI), whose importance has increased over the years. PrPSc is the main biomarker in TSEs, and the RT-QuIC assay stands out for its ability to detect PrPSc in cerebrospinal fluid (CSF), olfactory mucosa, and dermatome skin samples with high sensitivity and specificity. Other biochemical biomarkers are the proteins 14-3-3, tau, neuron-specific enolase (NSE), astroglial protein S100B, α-synuclein, and neurofilament light chain protein (NFL), but they are not specific for TSEs. This paper reviews the techniques employed for definite diagnosis, as well as the clinical and paraclinical methods for possible and probable diagnosis, both those in use currently and those no longer employed. We also discuss current criteria, challenges, and perspectives for TSE diagnosis. An early and accurate diagnosis may allow earlier implementation of strategies to delay or stop disease progression.
Prion Diseases
Prion diseases, also known as transmissible spongiform encephalopathies (TSEs), are a rare group of infectious, fatal neurodegenerative diseases caused by the deposition of misfolded prion protein particles in the brain (Prusiner, 1998; Scheckel and Aguzzi, 2018). The term “prion” was originally coined in 1982 by Stanley Prusiner to define the proteinaceous infectious particles that cause TSEs (Prusiner, 1982). The hallmark event in all TSEs is the conversion of the monomeric cellular prion protein (PrPC) into abnormally folded multimers, collectively termed prion scrapie (PrPSc), which accumulate in the brain and display toxic and aggregation-prone properties (Prusiner, 1998; Requena and Wille, 2017; Baral et al., 2019).
These diseases progress slowly, with a long latency period prior to the manifestation of symptoms, which include ataxia, myoclonus, progressive dementia, depression, and general malaise. In contrast with other progressive neurodegenerative diseases, the period between the onset of symptoms and the TSE patient’s death is usually up to 1 year, making them rapidly progressive dementias (Prusiner and Hsiao, 1994; Chesebro, 2003; Wadsworth et al., 2003). Treatment for human TSEs remains symptomatic and supportive, as no cure is available to date (Geschwind, 2015; Forloni et al., 2019).
Transmissible spongiform encephalopathies are unique in medicine since they can have three origins: spontaneous (sporadic), genetic (familial), and acquired (infectious/transmitted). Human TSEs encompass diseases such as Kuru, Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker syndrome (GSS), and fatal familial insomnia (FFI) (Zanusso et al., 2016; Ironside et al., 2017). Among human TSE cases, 80% to 95% are sporadic CJD (sCJD), 10% to 15% are genetic (often familial), and less than 1% are acquired (Geschwind, 2015). These diseases affect approximately 1–2 people per million worldwide annually (Chen and Dong, 2016). The human TSEs described above are summarized in Table 1.
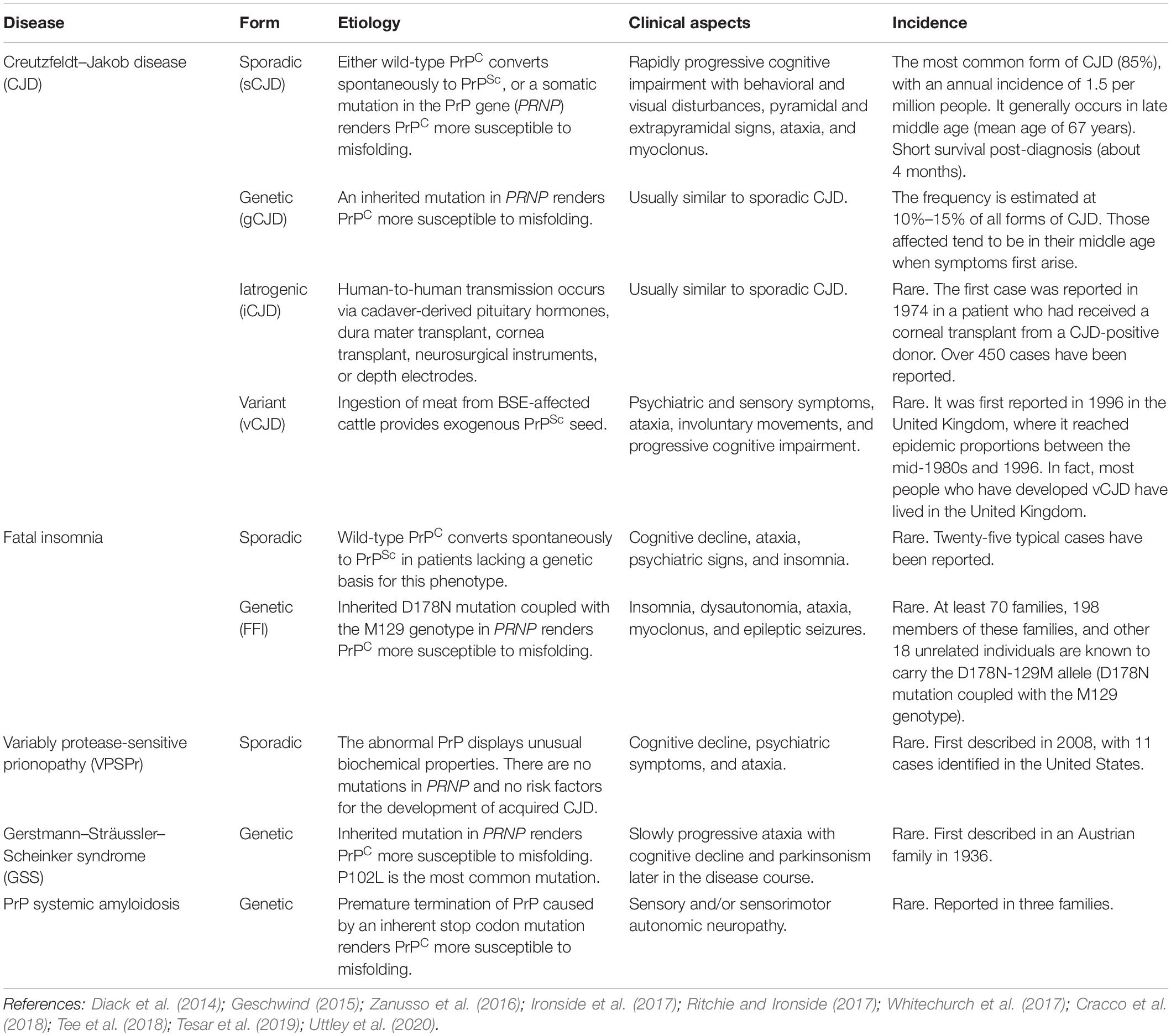
Table 1. Transmissible spongiform encephalopathies (TSEs).
Creutzfeldt–Jakob disease, the most common human prion disease worldwide, occurs mainly in the form of sporadic CJD (sCJD), which was first described in 1920 by German neurologist Hans Gerhard Creutzfeldt in a 23-year-old woman who had experienced fluctuating neuropsychological symptoms since adolescence. Shortly afterward, Alfons Maria Jakob described five other cases between 1921 and 1923, which he found to be similar to Creutzfeldt’s case (Iwasaki, 2017; Tee et al., 2018). There is no known genetic etiology of sCJD, nor are there known links between cases that are suggestive of an acquired disease; the assumption is, therefore, that cases of sCJD arise spontaneously (Ironside et al., 2017).
In 1987, a study described a prion disease in United Kingdom cattle named bovine spongiform encephalopathy (BSE) (Wells et al., 1987). Subsequently, in the 1990s, scientific and public attention was drawn to the appearance in the United Kingdom of a new human prion disease called variant CJD (vCJD), which was caused by the ingestion of meat from BSE-affected cattle (Will et al., 1996). In the late 1990s and early 2000s, some groups pointed to blood as a source of the scrapie agent, which at the time led to restrictions on blood transfusion in the United Kingdom as a way to prevent human-to-human transmission of vCJD (Turner and Ironside, 1998; Hunter et al., 2002; Llewelyn et al., 2004). A more recent study has shown that prion transmission in sheep via blood transfusion is indeed very efficient (Andréoletti et al., 2012).
Prior to the emergence of BSE, there were numerous reports of iatrogenic CJD (iCJD) cases in different countries in the 1970s and 1980s. These transmissions occurred via one of the following routes: treatment with human cadaver-derived growth hormone or gonadotropin, dura mater graft or corneal graft transplantation, and medical operations with neurosurgical instruments or depth electrodes (Will, 2003). Conventional sterilization protocols are ineffective in eradicating the PrPSc content in surgical instruments, and the methods recommended by the World Health Organization (WHO) are damaging to metal instruments and not practical for routine application (Thomas et al., 2013).
Despite the rarity of TSEs, these diseases represent a significant concern, not only because of their puzzling etiological aspects but also because of their considerable threat to public health. The development of non-invasive diagnostic methods for the early stages of TSEs is crucial in order to enable (i) the prevention of PrPSc spread, (ii) the implementation of potential therapies during earlier stages of the disease, and (iii) improvement in patient well-being. The development and validation of diagnostic methods for the detection at a preclinical stage are essential.
The Prion Protein
Mature PrPC is a 208-residue protein constitutively found on the surface of many cells, particularly neurons (Peggion et al., 2017). Before reaching the cell surface, a 253-residue precursor of PrPC undergoes some post-translational modifications: the excision of a 22-residue segment at the N-terminus and a 23-residue segment at the C-terminus, the establishment of a disulfide bond between cysteine residues 179 and 214, and the attachment of a glycosylphosphatidylinositol (GPI) anchor to the C-terminus (Prusiner, 1991). Recent findings have suggested that PrPC plays a central role in a variety of neurodegenerative diseases and could be a common therapeutic target for these disorders (Ayers and Prusiner, 2020; Corbett et al., 2020).
The structure of PrPC consists of an intrinsically unfolded N-terminal domain (residues 23–124) and a globular C-terminal domain (residues 125–230). The latter comprises three α-helices and a small, two-stranded β-sheet (Riek et al., 1996; Zahn et al., 2000; Requena and Wille, 2017) (Figure 1A). There are two glycosylation sites at asparagine residues 181 and 197, such that PrPC exists in diglycosylated, monoglycosylated, and non-glycosylated forms (Prusiner, 1991). Despite its high conservation in mammals, the human PrP gene (PRNP) has over 40 deleterious mutations and a few polymorphisms (Figure 1B). The polymorphism at codon 129 (rs1799990) draws particular attention because it influences disease susceptibility and phenotype (Kim M. O. et al., 2018). Nearly 55% of the world population are homozygous for methionine (MM), while 9% are homozygous for valine (VV) and 36% are heterozygous (MV). The most common allele, M129, has a frequency of 97.5% in East Asians, 76% in South Asians, 67.5% in Europeans, 64.7% in Africans, and 59.4% in Hispanic Americans with Native ancestry (The 1000 Genomes Project Consortium, 2012).
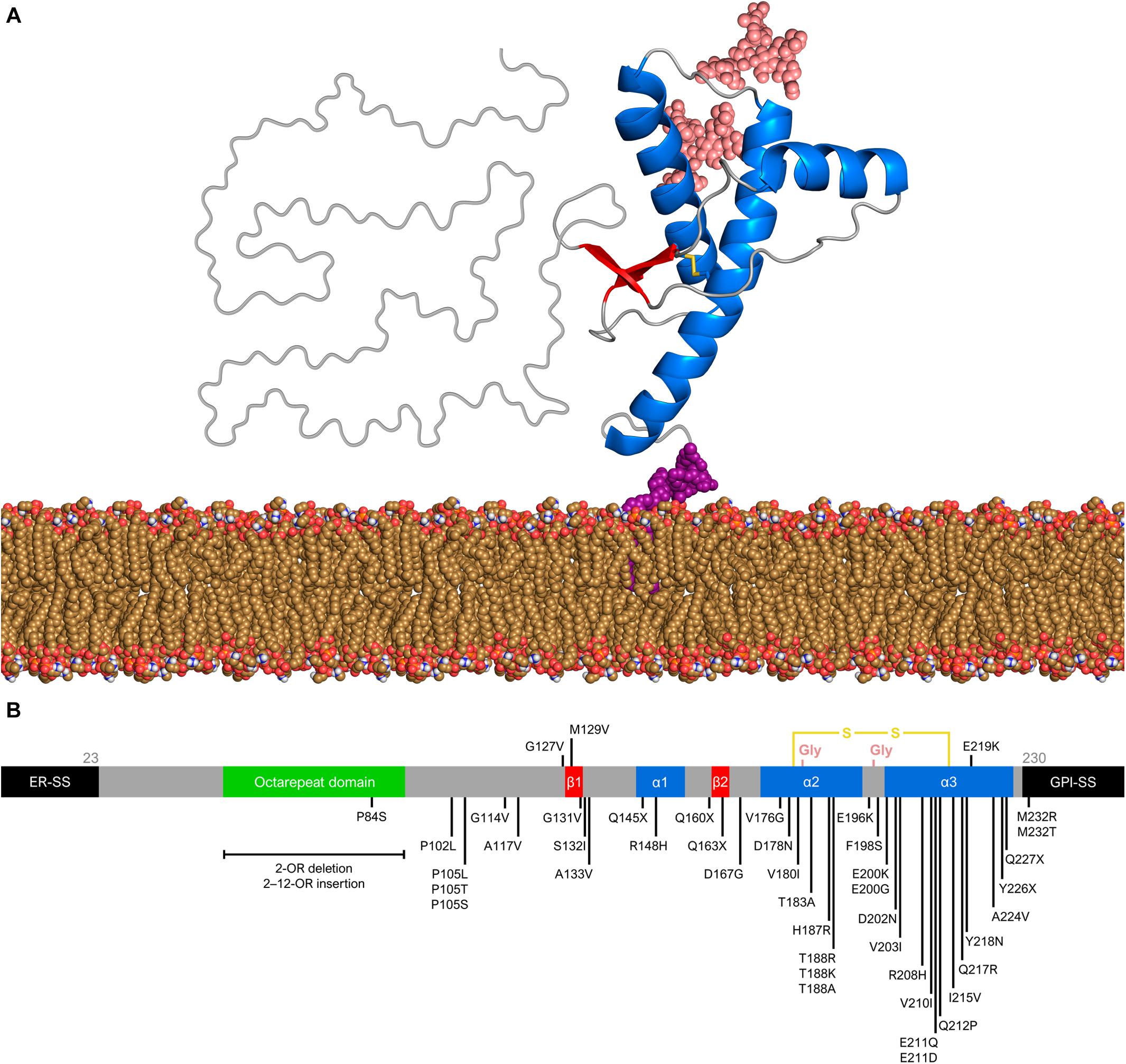
Figure 1. Prion protein structure and genetic variation. (A) Diglycosylated, full-length human PrPC (huPrP23–230) attached to the membrane (yellow brown) by a GPI anchor (purple). The globular C-terminal domain comprises three α-helices (blue) and two small β-strands (red), while the N-terminal is intrinsically flexible. Turns and random coils are represented in gray, and the glycosides are shown in salmon. A disulfide bond (yellow) connects the α-helices 2 and 3. The globular domain structure was determined by nuclear magnetic resonance (PDB code 1QLX). (B) Schematic of human PrP primary structure, with disease-associated mutations (below) and polymorphisms (above). Deleterious mutations include missense and nonsense changes, as well as octarepeat insertions and deletions. The octarepeat domain (green) is a copper-binding segment of 4–5 contiguous repeats of the sequence PHGGGWGQ. The 22-residue endoplasmic reticulum signal sequence (ER-SS) and 23-residue GPI signal sequence (GPI-SS) at the ends are shown in black. The other portions are colored as in panel (A).
While PrPC has a predominantly α-helical structure, PrPSc consists of β-sheet-rich multimeric assemblies (Prusiner, 1998). Experimental evidence combined with computational approaches suggests that PrPSc consists of β-strands and relatively short turns and/or loops that form a β-solenoid architecture with a hydrophobic β-sheet core (Wasmer et al., 2008; Smirnovas et al., 2011; Spagnolli et al., 2019). A remarkable property of the PrPSc particles is that they are self-propagating—i.e., they act like seeds/nuclei and induce the refolding of PrPC units, which are then integrated into PrPSc multimers. Thus, PrPSc particles grow by a nucleation process (Chiti and Dobson, 2006; Caughey et al., 2009; Cobb and Surewicz, 2009) (Figure 2). The pathogenic scrapie agent can spread from cell to cell and can transfer to a new host through the mechanisms outlined earlier (Acquatella-Tran Van Ba et al., 2013; Kraus et al., 2013).
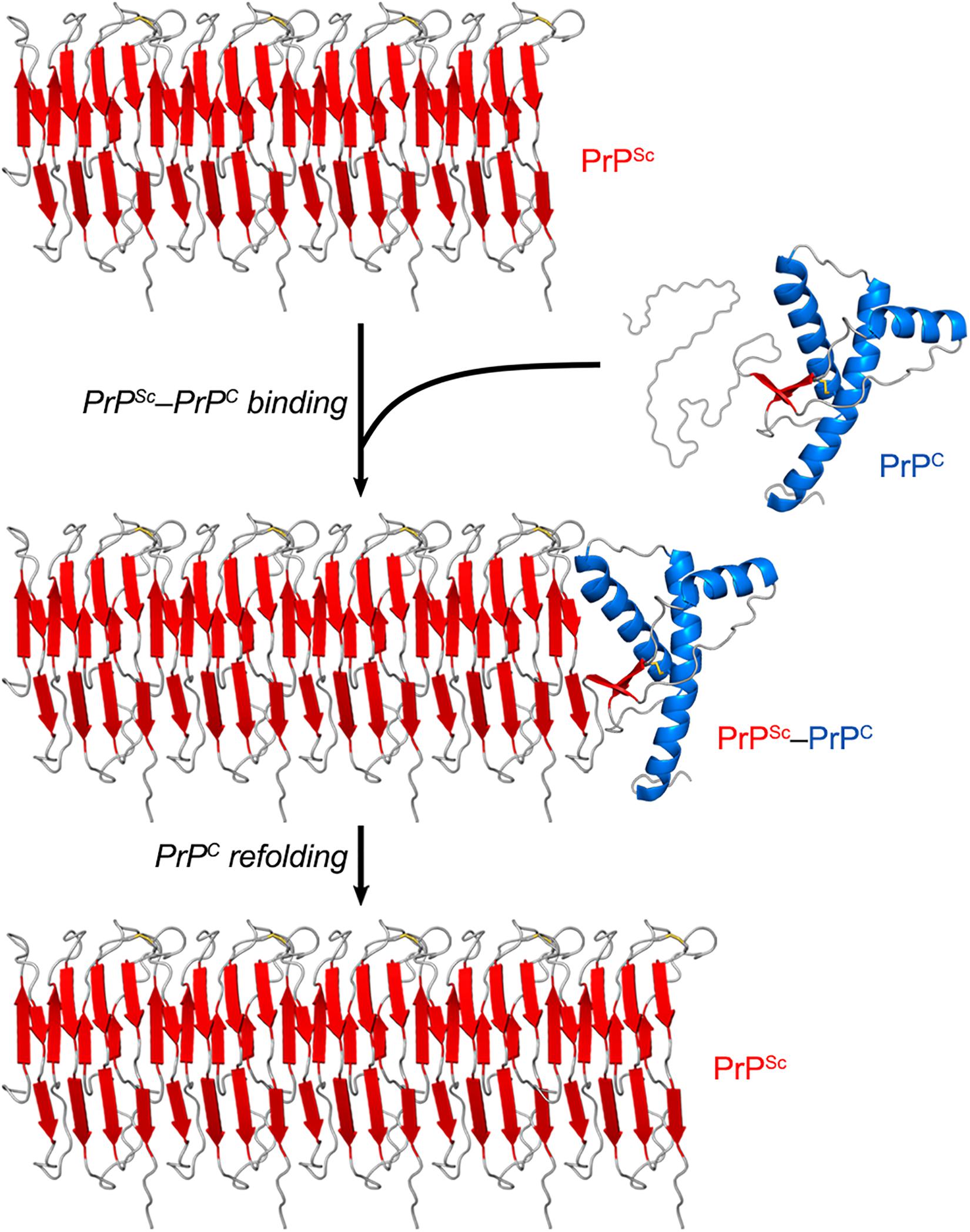
Figure 2. PrPSc amplification from PrPC. PrPSc particles grow by a nucleation process. A unit of PrPC forms a complex with PrPSc, and the latter induces the refolding of the former into a β-sheet-rich structure which then becomes part of the PrPSc multimer. In this self-propagating process, the PrPSc particles act like seeds/nuclei, with PrPC serving as a substrate. The secondary structures are colored as follows: α-helices in blue, β-strands in red, and unstructured portions in gray. The atomistic model of PrPSc was constructed by combining experimental data and molecular dynamics simulations (Spagnolli et al., 2019).
PrPSc particles are not homogenous structures. Indeed, they can adopt different conformations. An intriguing consequence of this fact is that these distinct PrPSc species can be associated with different clinical characteristics of TSEs, including the latency period, brain damage pattern, and symptomatology. The concept of the prion strain has, therefore, been defined to represent this structural, biochemical, and neuropathological diversity (Aguzzi et al., 2007; Rossi et al., 2019). Some groups have even reported the artificial production of prion strains in vitro, and the infectivity and pathogenicity in animal models of these scrapie agents generated de novo (Colby and Prusiner, 2011; Kim C. et al., 2018). Notably, the diversity of PrP primary sequences and PrPSc conformations (strains) greatly affects the susceptibility of an organism to PrPSc from another organism, especially if these organisms belong to different species. It is thus common to talk about a species barrier or strain barrier for prion diseases (Scott et al., 2005; Sharma et al., 2016; Igel-Egalon et al., 2018).
A distinguishing feature between PrPC and PrPSc is that the former has total protease sensitivity, whereas the latter generally has a protease-resistant core comprising the ∼90–230 region; hence, PrPC and PrPSc are also referred to as sensitive PrP (senPrP) and resistant PrP (resPrP), respectively (Prusiner, 1998; Caughey, 2000). Different strain-related PrPSc species can also have particular proteolytic digestion profiles, as they may differ in the length of their protease-resistant core (Bessen et al., 1995; Zou et al., 2003; Kobayashi et al., 2011; Kushnirov et al., 2020). Distinguishing between native PrPC and infectious PrPSc based on their different biochemical properties is considered the “gold standard” approach for TSE diagnosis (Lukan et al., 2013; Haley and Richt, 2017). However, such an approach to discern different prion strains has yet to be exploited for diagnostic purposes.
Diagnostic Approaches for Prion Diseases
At present, the definite diagnosis for TSEs is obtained via at least one of the following options: (i) identification of pathognomonic histological features in brain tissue sections, (ii) detection of resPrP by immunohistochemical staining of brain tissue sections, or (iii) detection of resPrP by immunoblotting of brain homogenate. Clinical evaluation and paraclinical tests only lead to a classification as possible or probable (Brown et al., 2003; CDC’s Diagnostic Criteria for Creutzfeldt–Jakob Disease (CJD), 2018). In the following sections, we will discuss the currently validated tests for definite, possible, and probable diagnosis, as well as other tests.
At this point, it is important to note two key terms in medical diagnosis: sensitivity and specificity. Sensitivity is the ability of a test to correctly identify true positive cases of the disease, whereas specificity is the ability of a test to identify true negative cases of the disease correctly. A high-sensitivity test produces more true positive results than false-negative results, while a high-specificity test produces more true negative results than false-positive results. The ultimate goal for diagnostic testing is to obtain sensitivity and specificity values as close to 100% as possible (Feinstein, 1975).
Brain Tissue Examination
Transmissible spongiform encephalopathy patients’ brain tissue is characterized by vacuolization (spongiosis), neuronal death, dendritic and synaptic loss, astrogliosis, and resPrP deposition. These features are most prominent in the cerebral and cerebellar cortexes. Vacuolization can be observed by histopathological examination. The vacuoles may, in fact, be an artifact generated by tissue processing (fixation, paraffin embedding, and staining). Apoptotic neurons can be identified by staining nuclei with DAPI (4′,6-diamidino-2-phenylindole) and/or immunochemical staining of caspase-3. Dendritic and synaptic loss can be revealed by Golgi silver staining. While astrogliosis is not specific to this group of diseases, it is consistently present and can be detected through immunochemical staining of glial fibrillary acidic protein (GFAP) (Aguzzi et al., 2001; Soto and Satani, 2011). However, these examinations have some drawbacks. Since tissue segments may have been collected from unaffected areas of the brain, these analyses offer limited diagnostic sensitivity and may well lead to false-negative results (Soto, 2004; Safar et al., 2005).
Immunohistochemical staining of brain tissue sections is a validated technique for detecting PrPSc in the brains of TSE-affected patients. To selectively stain for resPrPSc, brain tissue sections are previously subjected to limited proteolysis by proteinase K (PK) to eliminate the senPrP content (Bendheim et al., 1984; Parchi et al., 1999, 2012; Gambetti et al., 2003). This technique allows for higher diagnostic sensitivity than the brain tissue staining techniques mentioned above since it directly probes the etiological agent behind TSEs (Safar et al., 2005). However, the enzymatic pre-treatment does represent a significant drawback in terms of diagnostic sensitivity, since the PrPSc content may have varying degrees of resistance to proteolysis depending on the prion strain. Indeed, there have been reports of strain-associated PrPSc particles so poorly protease-resistant that they have been termed sensitive PrPSc (senPrPSc) (Safar et al., 2005; Pastrana et al., 2006; Sajnani et al., 2012). Limited proteolysis can thus result in digestion of a significant part of the whole PrPSc content, underestimating the total level of PrPSc in the brain (Safar et al., 2005).
One way to circumvent the limitations of the immunohistochemical examination is to skip the PK pre-treatment step and to perform conformation-dependent immunoassays (CDIs) using brain homogenate. CDIs markedly increase diagnostic sensitivity since they detect PrPSc species regardless of their protease resistance level (Safar et al., 1998, 2005; Kim et al., 2011, 2012). CDIs employ antibodies that recognize conformational epitopes rather than linear epitopes and are therefore selective for PrPSc over PrPC (Kascsak et al., 1987; Bellon et al., 2003). Many conformational antibodies with selectivity for PrPSc have already been produced (Saijo et al., 2016). However, CDIs have not been tested in large cohorts and are thus not yet used in routine diagnostics or screening.
Another validated technique that is extensively used for diagnosis is Western blotting (immunoblotting) of brain homogenate, which also employs a pre-treatment with PK to digest the PrPC content. This technique is particularly important because it provides information on several aspects of PrPSc: its degree of proteolytic resistance, the size of the protease-resistant cores, and the ratio between the three different PrP glycoforms (un-, mono-, and diglycosylated). These features are correlated with the properties of individual strains as well as disease genotypes and phenotypes. The electrophoretic migration of the unglycosylated PK-resistant core is used to define two PrPSc types in cases of CJD: type 1, characterized by a ∼21-kDa band, and type 2, characterized by a ∼19-kDa band (Parchi et al., 1999). CJD subtypes are then categorized by codon 129 polymorphism and PrPSc type as MM1, MM2, MV1, MV2, VV1, and VV2. Type-1 PrPSc is mostly correlated with the 129 MM genotype, while type-2 PrPSc is generally correlated with the 129VV and 129MV genotypes; therefore, the most frequent subtypes are MM1, VV2, and MV2 (Parchi et al., 1999; Zerr et al., 2000b). With respect to PrP glycoforms, samples from sCJD patients predominantly contain the monoglycosylated band (Bendheim et al., 1984; Parchi et al., 1999, 2012; Gambetti et al., 2003). Meanwhile, FFI and vCJD are characterized by type-2 PrPSc and abundance of the diglycosylated band (Hill et al., 1997; Rossi et al., 1998).
Brain tissue examination is frequently performed postmortem, but brain biopsies provide an option for antemortem investigation. However, brain biopsies can lead to false-negative results since TSE pathology is variably distributed in the brain, and small samples may not contain pathognomonic histological features and/or detectable levels of PrPSc. Because of their invasiveness, brain biopsies are very uncomfortable for the patients and can cause severe complications, including brain abscesses and hemorrhaging. Furthermore, the medical instruments used in neurosurgery must be appropriately discarded or subjected to WHO-recommended cleaning methods. Therefore, brain tissue examination is generally performed only postmortem (Brown et al., 2003).
Neuropsychiatric Clinical Evaluation
The degeneration of certain areas of the brain, probably caused by PrP conversion and aggregation or by lack of functional PrPC, leads to the development of clinical signs that motivate patients to seek medical attention. Depending on their symptoms, a number of assessments of sCJD patients are possible. These include the cognitive or Heidenhain form of CJD, which accounts for the largest number of sCJD cases and is characterized by cognitive decline, cortical visual disturbances (involvement of the occipital cortex), mild psychiatric symptoms, and myoclonus. Cases can also be classified as ataxic or Brownell-Oppenheimer, a form that is characterized by cerebellar ataxia, cognitive decline, mood changes, and myoclonus at later stages, or as non-CJD phenotypes or Stern, characterized by presenile dementia, ataxia, and sleep disturbance (related to degeneration of the thalamus) (Puoti et al., 2012; Fragoso et al., 2017).
Gerstmann–Sträussler–Scheinker syndrome patients predominantly present cerebellar clinical features, progressive ataxia, incoordination, and late dementia. FFI patients’ main symptoms include sleep-related issues (insomnia, sleep-related dyspnea, sleep-related involuntary movements), and less frequently ataxia and psychiatric symptoms. Behavioral and psychiatric symptoms (excluding sleep disturbance) are also found in vCJD patients, along with persistent painful sensory symptoms, ataxia, myoclonus, and later dementia (Fragoso et al., 2017; Wu et al., 2018).
However, none of these signs alone are enough to diagnose prion diseases definitively. Subacute encephalopathy can be a consequence of infectious, inflammatory, autoimmune, or other causes, leading to diseases that should have their differential diagnosis because they are mostly treatable, unlike TSEs (Annus et al., 2016; Fragoso et al., 2017). For example, Hashimoto’s encephalopathy is a treatable disorder that results in cognitive decline, myoclonus, ataxia, and neuropsychiatric signs (Murray, 2011), similar to TSEs.
To determine a possible or probable TSE diagnosis, clinical symptoms must be consistent with paraclinical tests, which can include magnetic resonance imaging (MRI), electroencephalography (EEG), and CSF analysis (Soto, 2004; Araújo, 2013). However, this combination of clinical evaluation and paraclinical tests is insufficient for a definite TSE diagnosis, since biochemical and imaging alterations are not specific to the various TSE cases, as they are also present in other neurodegenerative conditions.
In vitro Cell-Free PrP Conversion Assays
Based on the self-replicating ability of PrPSc, a cell-free conversion assay called protein misfolding cyclic amplification (PMCA) was published at the beginning of the 2000s (Saborio et al., 2001). This technique consists of mixing PBS-diluted healthy brain homogenate and scrapie-infected brain homogenate (at a much higher dilution) in microtubes and then performing repeated cycles of incubation with orbital shaking at 37°C and sonication. Healthy and scrapie-infected brain homogenates are sources of PrPC/senPrP (the reaction substrate) and PrPSc/resPrP (the infectious seed), respectively. The incubation step allows aggregate growth, while the sonication step fragments these aggregates into smaller particles, which can act as seeds in the following incubation step. The reaction products are subjected to PK treatment and then to Western blotting to reveal their final resPrP content. The result is an exponential increase in the amount of resPrP per cycle (Saborio et al., 2001) (Figure 3).
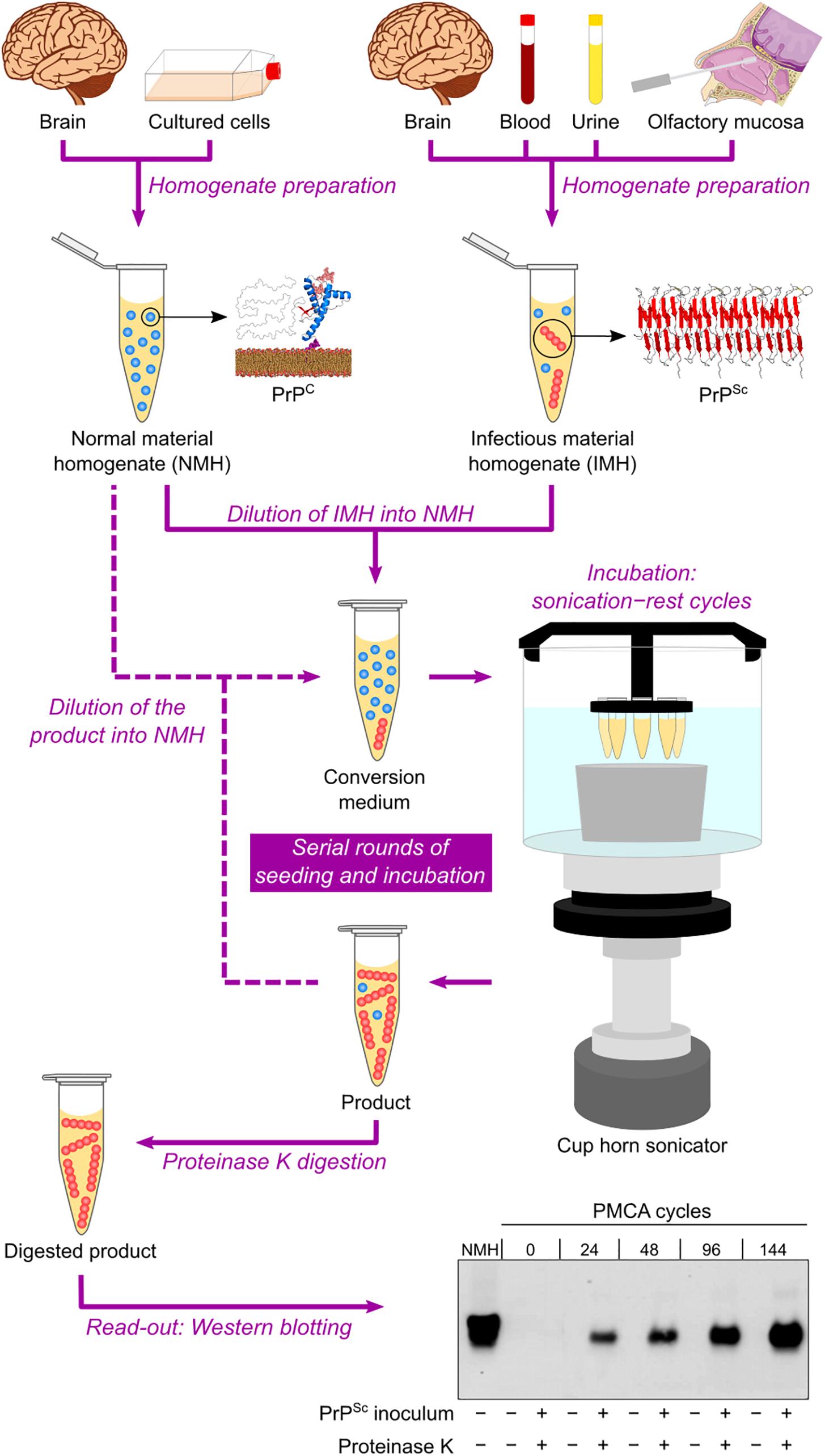
Figure 3. Protein misfolding cyclic amplification (PMCA) in automated fashion. Normal material homogenate (NMH), a source of PrPC (substrate), is prepared from brain sections or cultured cells. Suspected infectious material homogenate (IMH), a possible source of PrPSc (seed), is prepared from brain sections, blood, urine, or olfactory mucosa brushings. Suspected IMH is diluted into NMH, and the resulting mixture is incubated with alternating cycles of sonication and rest. Subsequent rounds can be performed by diluting the product of the previous round into fresh NMH and repeating the incubation step. As a result, the amount of PrPSc is amplified after several sonication–rest cycles and even more after serial rounds of seeding and incubation. The products from different cycles and rounds are treated with proteinase K to eliminate PrPC and then subjected to Western blotting to reveal the growing amount of resPrP. This result indicates a positive TSE diagnosis. The Western blot shown is republished with permission of American Society for Biochemistry and Molecular Biology, from Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification, Saá, Castilla, and Soto, 281, 46, 2006; permission conveyed through Copyright Clearance Center, Inc.
The PMCA assay was later improved by optimizing brain homogenate preparation, medium composition, and sonication method, as well as by developing an automated option for the incubation–sonication step (Castilla et al., 2004; Saá et al., 2005) (Table 2 and Figure 3). These modifications allowed simultaneous runs of multiple samples with enhanced amplification efficiency (Castilla et al., 2005). The improved PMCA assay was shown to have very high sensitivity, as it significantly amplified as little as 10–18 g of PrPSc (equivalent to a 1012-fold dilution of SHB), as well as high specificity, as it never generated any detectable resPrP in the absence of scrapie-infected brain homogenate inoculum, even after hundreds of PMCA cycles (Saá et al., 2006). When applied to human brain samples (collected either postmortem or by brain biopsy), PrPSc from sCJD and vCJD patients has been successfully amplified by PMCA (Soto et al., 2005; Jones et al., 2007; Oshita et al., 2016).
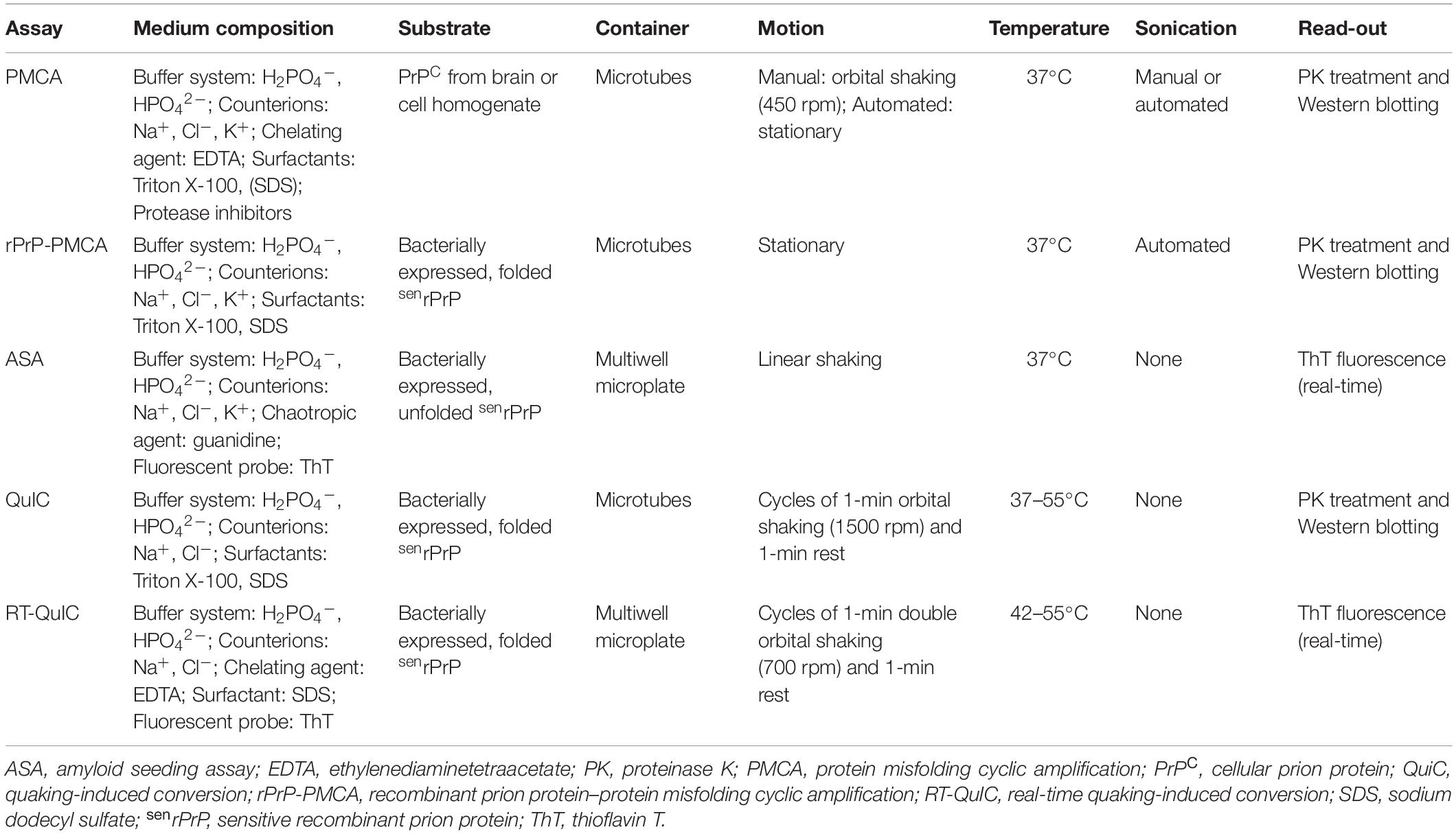
Table 2. In vitro cell-free PrP conversion assays.
A simpler and less expensive version of PMCA, called rPrP-PMCA, was developed by replacing PrPC from brain or cell homogenate with bacterially expressed, folded recombinant senPrP (senrPrP) (Table 2). The substrate concentration could thus be more accurately and efficiently defined in rPrP-PMCA than in classical PMCA (Atarashi et al., 2007). In the same year, another cell-free conversion technique, the amyloid seeding assay (ASA), was published (Colby et al., 2007). This cell-free conversion technique consists of loading a multiwell microplate with (i) guanidine-denatured senrPrP, (ii) PrPSc partially purified from scrapie-infected brain homogenate, (iii) guanidine, and (iv) thioflavin T (ThT). The latter component is a probe that emits fluorescence when it binds to amyloid structures. The mixture is incubated with continuous linear shaking at 37°C in a plate reader, which measures ThT fluorescence emission throughout the reaction, thus revealing the kinetics of aggregate formation (Colby et al., 2007) (Table 2).
Protein misfolding cyclic amplification (and its variation, rPrP-PMCA) and ASA have important differences. While it is challenging to deliver sound energy evenly to samples during sonication, lateral shaking subjects samples to the same motion, making ASA more practical and reproducible than the PMCA assays (Colby et al., 2007). The ASA technique is also faster and much more suitable for high-throughput investigations than PMCA since ASA relies on simple, automated, real-time, and fluorescence-based read-outs rather than time-consuming, immunoblotting-based read-outs (Colby et al., 2007). However, ASA has the disadvantage of frequently producing false-positive results, probably because of the presence of guanidine, a chaotropic agent, in the reaction medium (Colby et al., 2007).
A more refined cell-free conversion assay, the quaking-induced conversion (QuIC), was published more recently than the above techniques (Atarashi et al., 2008). The QuIC assay consists of loading microtubes with folded senrPrP and highly diluted scrapie-infected brain homogenate and then incubating the mixture at a controlled temperature with alternating cycles of orbital shaking and rest. The intermittent shaking results in separate aggregate fragmentation (seed formation) and aggregate growth phases, speeding up the conversion process. The read-out is performed by PK digestion and Western blotting, as in the PMCA assay (Table 2). The QuIC technique had high sensitivity, successfully amplifying as little as 10–16 g of PrPSc from brain homogenate, while also achieving high specificity, as it generated either no PK-resistant band or atypical, low molecular weight PK-resistant bands (Atarashi et al., 2008). The QuIC assay detected PrPSc with high sensitivity in brain homogenates from animals and humans and CSF samples from animals (Atarashi et al., 2008; Orrú et al., 2009).
The QuIC assay was then combined with the ASA read-out method to create the most sophisticated technique currently available for amplifying PrPSc in vitro, namely the real-time quaking-induced conversion (RT-QuIC) (Wilham et al., 2010). RT-QuIC differs from its predecessor in several steps. First, instead of microtubes, the conversion medium is supplemented with ThT and loaded into multiwell microplates. It is then incubated at 42°C with alternating cycles of double orbital shaking and rest and analyzed using real-time ThT fluorescence measurements rather than immunoblotting (Table 2 and Figure 4). The RT-QuIC technique is the fastest, most straightforward, most practical, least expensive, and most suitable cell-free conversion assay for high-throughput surveys. The RT-QuIC assay detected PrPSc at levels between ∼10–15 g and 10–13 g in brain homogenate, nasal fluids, and CSF from different animal species with high specificity (Wilham et al., 2010).
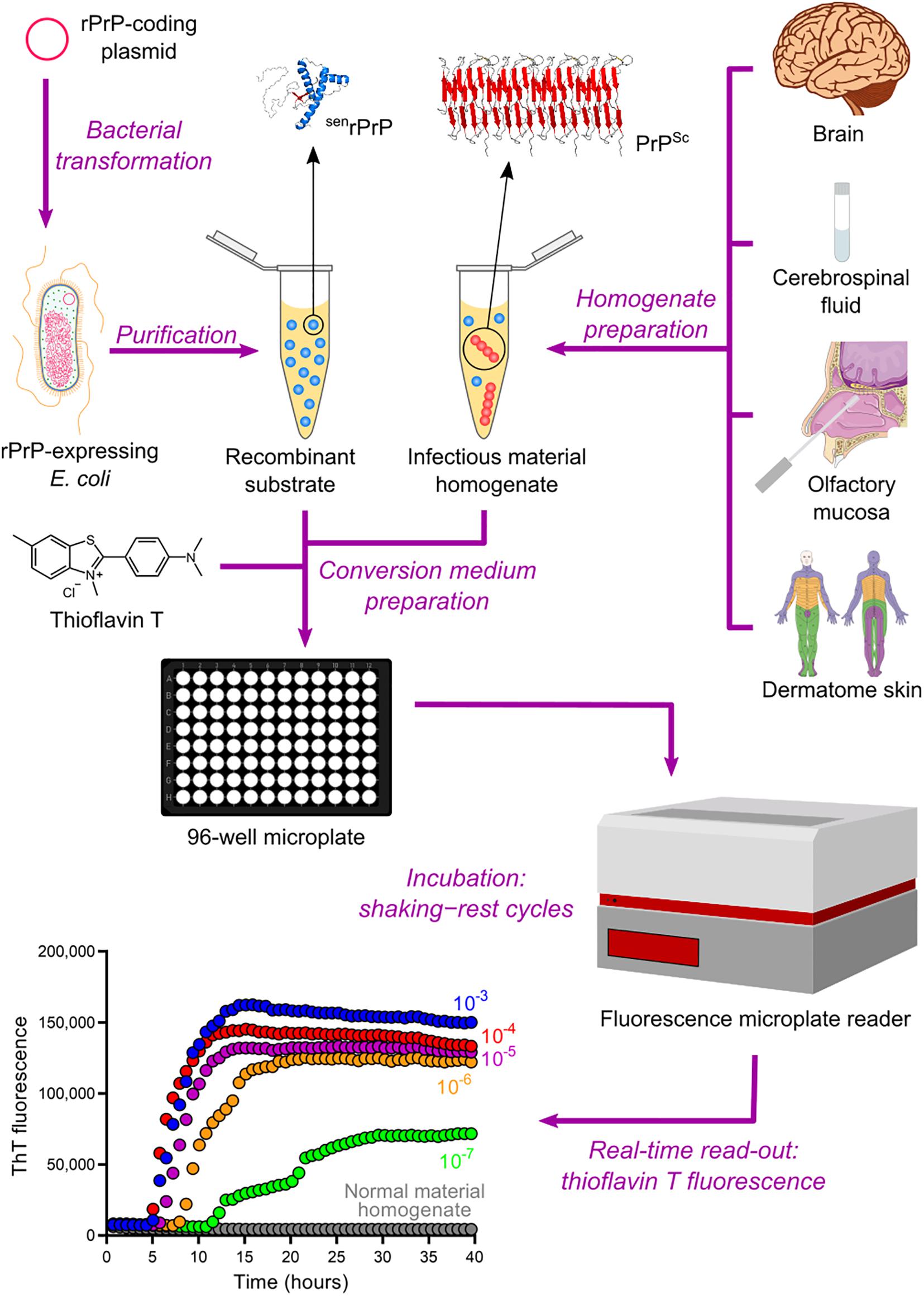
Figure 4. Real-time quaking-induced conversion (RT-QuIC). Recombinant PrP (rPrP) is expressed heterologously in E. coli and purified as folded protease-sensitive rPrP (senrPrP), the substrate for RT-QuIC. Suspected infectious material homogenate, a possible source of PrPSc (seed), is prepared from brain sections, cerebrospinal fluid, olfactory mucosa brushings, and dermatome skin sections. High dilutions of a test sample are mixed with senrPrP, buffer, and thioflavin T (a fluorescent amyloid probe) to produce the conversion medium, which is loaded into a multiwell microplate and then incubated with alternating cycles of shaking and rest in a fluorescence microplate reader. This equipment records thioflavin T fluorescence emission throughout the incubation, thus revealing the kinetics of aggregate formation. A sigmoidal growth in thioflavin T fluorescence indicates a positive TSE diagnosis. The use of normal material homogenate does not lead to any increase in thioflavin T fluorescence, indicating that no conversion is happening. The positive result shown was obtained with brain homogenate from a patient with GSS.
In the last decade, numerous studies have reported the detection of PrPSc in non-brain samples from human TSE patients by PMCA and especially RT-QuIC, as summarized in Table 3. The PMCA assay has been used to amplify PrPSc with high sensitivity and specificity from human samples such as blood (Lacroux et al., 2014; Bougard et al., 2016; Concha-Marambio et al., 2016) and urine (Moda et al., 2014). These studies used homogenates from the following biological materials as substrate sources: healthy human brain (Soto et al., 2005), human platelets (Jones et al., 2007), human PrPC-expressing transgenic mouse brain (Jones et al., 2007; Lacroux et al., 2014; Moda et al., 2014; Bougard et al., 2016; Concha-Marambio et al., 2016), and human PrPC-expressing 293F cells (Oshita et al., 2016). Another study has demonstrated that a CDI can be used as a read-out for PMCA to allow the detection of supposed senPrPSc species (Jones et al., 2007).
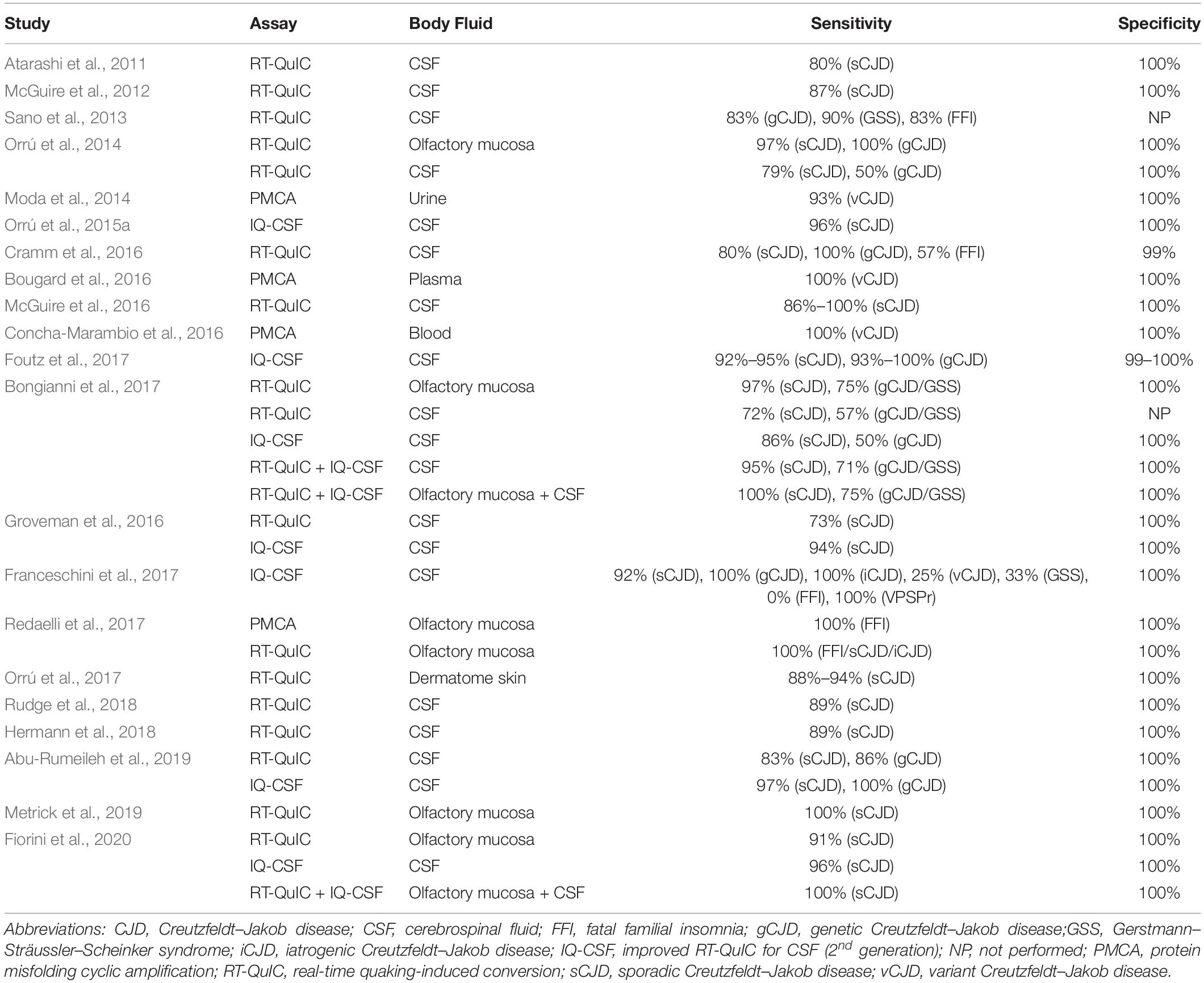
Table 3. Performance of PMCA and RT-QuIC for PrPSc detectionin non-brain samples from human patients with TSEs.
Several studies have reported the high-sensitivity and high-specificity amplification of resPrP using RT-QuIC with different human samples, including brain homogenate (Atarashi et al., 2011; Peden et al., 2012), CSF (Atarashi et al., 2011; McGuire et al., 2012, 2016; Sano et al., 2013; Orrú et al., 2014; Cramm et al., 2015, 2016; Hermann et al., 2018; Rudge et al., 2018), olfactory mucosa (Orrú et al., 2014; Bongianni et al., 2017; Fiorini et al., 2020), and dermatome skin (Orrú et al., 2017). Another study coupled immunoprecipitation of PrPSc from human plasma with RT-QuIC, thus avoiding interference from plasma components and increasing sensitivity up to 10–18 g of PrPSc (Orrú et al., 2011).
Since it is common practice to collect CSF samples from patients with suspected CJD in order to rule out other diseases, this bodily fluid is highly valuable for diagnosis (Chitravas et al., 2011). A second-generation RT-QuIC assay (with small modifications in the protocol) has been developed specifically to enhance sensitivity in CSF analyses and shorten reaction times (Orrú et al., 2015a). It was designated as improved RT-QuIC for CSF (IQ-CSF). Subsequent work achieved high-sensitivity diagnosis from human CSF samples by using IQ-CSF (Groveman et al., 2016; Bongianni et al., 2017; Foutz et al., 2017; Franceschini et al., 2017; Abu-Rumeileh et al., 2019; Fiorini et al., 2020).
Some studies have shown that bank voles (BV) have a uniquely weak species/strain barrier for prion transmission, as they are susceptible to multiple PrPSc strains from a wide range of species, including humans, sheep, deer, and hamsters (Nonno et al., 2006; Agrimi et al., 2008; Di Bari et al., 2008, 2013). Another study has shown that BV PrPC-expressing mice are susceptible to many prion strains from eight different species, including humans (Watts et al., 2014). Collectively, these data indicated that BV PrPC was a universal substrate for conversion into PrPSc, thus circumventing the need for suitable senPrP substrates (with different sequences) to identify distinct prion types using RT-QuIC and PMCA.
RT-QuIC reactions using BV senrPrP as substrate have been able to detect multiple human prion strains with high sensitivity in human samples including brain homogenate (Orrú et al., 2015b), olfactory mucosa (Redaelli et al., 2017), and dermatome skin samples (Orrú et al., 2017). These studies observed the generation of BV resPrP species with different PK-resistance profiles, though without any clear correlation between the proteolytic digestion profiles and the different strains used as seed. Similarly, PMCA reactions using BV brain homogenate successfully amplified PrPSc from human olfactory mucosa samples, although the original glycoform ratio and the electrophoretic migration of the unglycosylated PK-resistant core of prion strains were not maintained (Redaelli et al., 2017). Even though the use of BV senPrP cannot provide valuable information on prion strains, this substrate does simplify and lower the cost of routine application of RT-QuIC and PMCA for TSE diagnosis.
An important study has reported the detection of PrPSc in skin samples of prion-infected rodents by PMCA and RT-QuIC at a preclinical stage of disease. Hamsters infected with 263K scrapie strain, which displayed clinical signs from 10 weeks post inoculation (wpi), were positively diagnosed at 2 wpi by PMCA and 3 wpi by RT-QuIC. Human PrPC-expressing mice infected with sCJD agent, which showed clinical signs at 24 wpi, were positively diagnosed at 4 wpi by PMCA and 20 wpi by RT-QuIC (Wang et al., 2019). These findings additionally highlight the enormous potential of in vitro cell-free PrP conversion assays for an early TSE diagnosis.
Protein misfolding cyclic amplification and RT-QuIC have important differences in terms of their end products. When PMCA products were inoculated intraperitoneally in wild-type hamsters, these animals developed the same disease pattern as hamsters inoculated with infectious brain material did, demonstrating that infectivity could also be amplified by PMCA (Saá et al., 2006). Conversely, when RT-QuIC products were inoculated intracerebrally in wild-type hamsters, their brains showed very low seeding activity but no histopathological changes, while Western blotting detected no resPrP. This result indicated that RT-QuIC-generated resPrP was neither infectious nor pathogenic, suggesting that RT-QuIC is much less biohazardous than PMCA for routine use (Groveman et al., 2017). In another study, sCJD-seeded RT-QuIC products inoculated intracerebrally in mice overexpressing human PrPC elicited neither clinical signs of disease nor astrogliosis (Raymond et al., 2020). In fact, PMCA products are more similar to brain-derived PrPSc, while the resPrP species produced by RT-QuIC have shorter PK-resistant cores (McGuire et al., 2012; Groveman et al., 2015, 2017; Kraus et al., 2015), which may explain why PMCA and RT-QuIC end products are distinct in terms of infectivity. Thus, the methodological simplicity and biosafety of RT-QuIC over PMCA is probably the reason why RT-QuIC was the most employed assay for TSE diagnosis in robust studies published in the 2010s (Table 3).
A recent study has shown that the replacement of chloride anions with less hydrated bromide anions in the RT-QuIC medium significantly increased sensitivity and specificity for PrPSc amplification in brain and olfactory mucosa samples from sCJD patients (Metrick et al., 2019). This important finding may enable the RT-QuIC technique to be even more refined and distinguished in the years to come.
The RT-QuIC assay was first offered clinically in 2015 in the United States and became part of the Centers for Disease Control and Prevention (CDC) criteria in 2018. Currently, RT-QuIC, in combination with other techniques, characterizes TSE diagnosis only as probable, not definitive (CDC’s Diagnostic Criteria for Creutzfeldt–Jakob Disease (CJD), 2018). Outstandingly, two studies have reported sCJD diagnosis with 100% sensitivity and 100% specificity by combining RT-QuIC analysis of olfactory mucosa samples and IQ-CSF (Bongianni et al., 2017; Fiorini et al., 2020). Two other studies have shown that the association of IQ-CSF with other techniques—such as MRI, EEG, and 14-3-3 detection (discussed below)—resulted in sCJD diagnosis with at least 97% sensitivity and 99% specificity (Hermann et al., 2018; Rudge et al., 2018). Therefore, it is very plausible that a definitive antemortem diagnosis can be obtained by the combination of RT-QuIC testing of at least two biological samples with analyses by other available techniques.
Surrogate Biomarkers
Although PrPSc is the most specific biochemical marker in TSEs, it is not the sole one. Surrogate biomarkers have been studied for both differential diagnosis and prognosis, especially in CJD cases. The search for biomarkers capable of predicting disease onset is especially important for patients with genetic forms, as well as for the inclusion of patients in clinical trials. Other proteins exploited for TSE diagnosis are 14-3-3 proteins, tau, neuron-specific enolase (NSE), astroglial protein S100B, α-synuclein, and neurofilament light chain protein (NFL) (Table 4). Among these, CSF 14-3-3 is a biomarker already considered in current guidelines (CDC’s Diagnostic Criteria for Creutzfeldt–Jakob Disease (CJD), 2018).
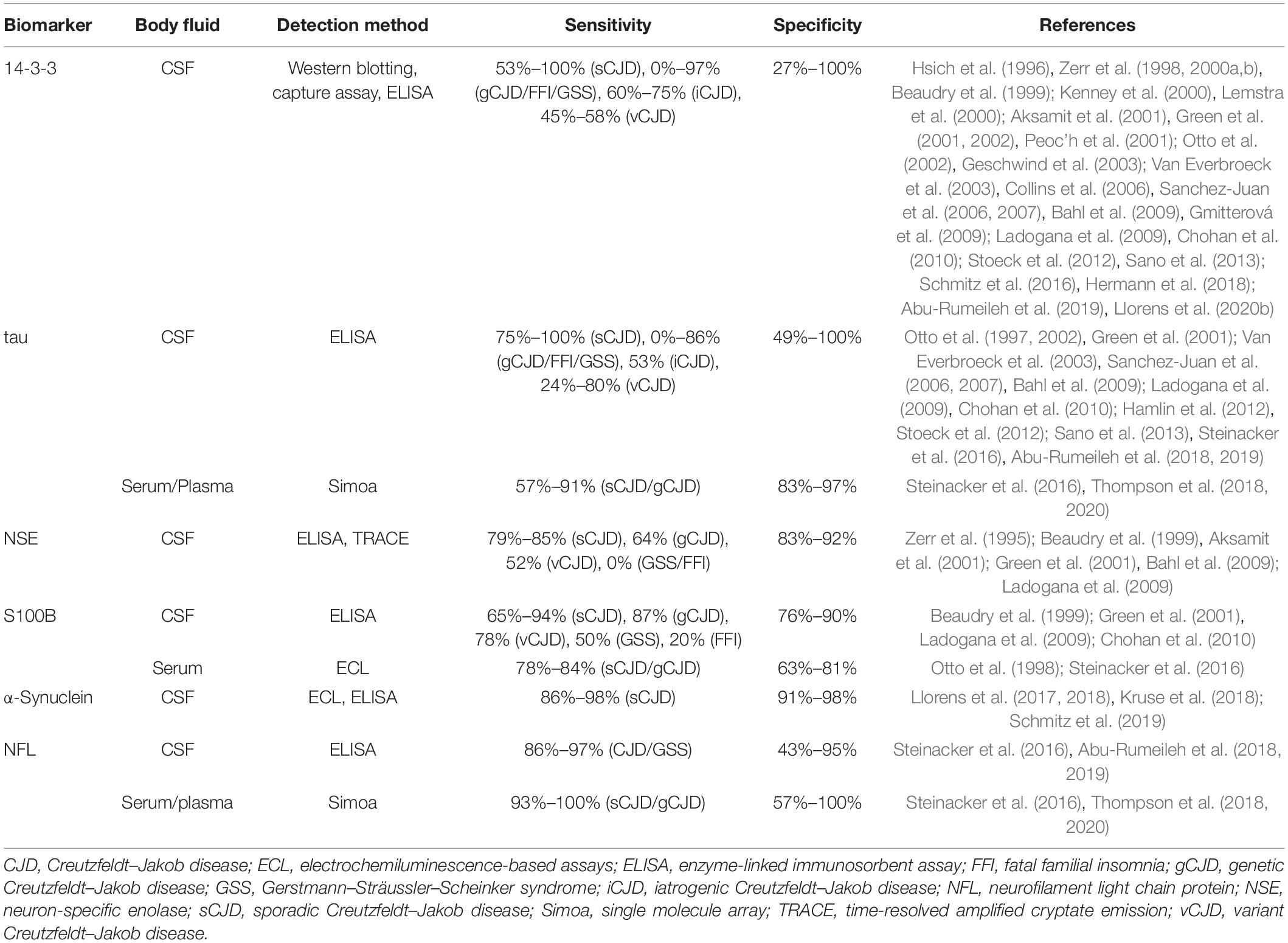
Table 4. Surrogate biomarkers of prion diseases.
14-3-3 proteins are indicators of neuronal cell loss and can be detected in CSF samples by immunoblotting (Hsich et al., 1996), capture assay (Peoc’h et al., 2001), or enzyme-linked immunosorbent assay (ELISA) (Kenney et al., 2000). Measurement of CSF 14-3-3 has sensitivity values of 53%–100% for sCJD, 0%–97% for genetic prion diseases (gCJD, GSS, and FFI), 60%–70% for iCJD, and 45%–58% for vCJD. Numerous features account for these highly variable values: disease form (sCJD, gCJD, vCJD, iCJD, GSS, FFI), disease subtype (MM1, MM2, MV1, MV2, VV1, VV2), patient’s age, disease duration, and method used for quantification (Zerr et al., 2000b; Van Everbroeck et al., 2003; Collins et al., 2006; Gmitterová et al., 2009). Specificity values range from 27% to 100% since 14-3-3 elevation is not specific for TSEs. Acute neurological events (e.g., encephalitis, stroke, and epileptic seizures), brain tumor, and other neurodegenerative diseases also result in neuronal death, leading to an increase in 14-3-3 (Chohan et al., 2010; Stoeck et al., 2012). However, sensitivity and specificity have been shown to increase with disease duration in sCJD (Chohan et al., 2010).
An increase in CSF tau is typically an indicator of neuronal death associated with Alzheimer’s disease and can be measured by ELISA (Jensen et al., 1995). However, CSF tau levels are also increased in CJD and therefore have been extensively used for TSE diagnosis (Otto et al., 1997; Bahl et al., 2009; Stoeck et al., 2012; Abu-Rumeileh et al., 2019). Measurement of CSF tau has sensitivity values of 75%–100% for sCJD, 0%–86% for genetic prion diseases (gCJD, GSS, and FFI), 53% for iCJD, and 24%–80% for vCJD, while specificity values range from 49% to 100%. Similarly to 14-3-3, tau sensitivity is limited by disease form and subtype, patient’s age, and disease progression, while specificity is restricted by overlap with other diseases (Van Everbroeck et al., 2003; Hamlin et al., 2012; Stoeck et al., 2012; Cohen et al., 2016). CSF tau levels—along with information on patient’s age, sex, and codon 129 polymorphism—has been recently used to build the first prognostic model to predict the survival time of sCJD individuals with moderate to good accuracy (Llorens et al., 2020a).
The detection of elevated levels of the proteins NSE and S100B in CSF samples from CJD patients have also been reported (Zerr et al., 1995; Beaudry et al., 1999; Aksamit et al., 2001; Bahl et al., 2009; Ladogana et al., 2009). Measurement of CSF NSE and S100B has respective sensitivity values of 79%–85% and 65%–94% for sCJD, 0%–64% and 20%–87% for genetic prion diseases (gCJD, GSS, and FFI), and 52% and 78% for vCJD, while specificity values are 83–92% for NSE and 76%–90% for S100B. The combination of two or three biomarkers among 14-3-3, tau, NSE, and S100B significantly increased specificity, but at the cost of reduced sensitivity (Aksamit et al., 2001; Bahl et al., 2009; Chohan et al., 2010). However, in relation to 14-3-3 and tau, NSE and S100B alone showed no particular advantage and, therefore, have been much less explored for TSE diagnosis.
More recent studies reported that sCJD patients’ CSF also contains high levels of α-synuclein (Llorens et al., 2017, 2018; Kruse et al., 2018; Schmitz et al., 2019). Measurement of this protein had a sensitivity of 86%–98% and a specificity of 91%–98%, which are consistently high values in comparison to those of 14-3-3, tau, NSE, and S100B. These findings highlight α-synuclein as a very accurate surrogate biomarker in TSEs, but further studies are still necessary to reinforce the diagnostic utility of this protein.
Transmissible spongiform encephalopathy patients present elevated NFL in CSF as a result of neuroaxonal degeneration (Steinacker et al., 2016; Abu-Rumeileh et al., 2018, 2019). Measurement of CSF NFL has a sensitivity of 86%–97% for different TSE forms (sCJD, gCJD, and GSS) and specificity of 43%–95%. Increased tau and NFL levels can also be detected in TSE patients’ serum or plasma (Steinacker et al., 2016; Thompson et al., 2018, 2020). In sCJD and gCJD patients, measurement of blood tau and NFL has a respective sensitivity of 57%–91% and 93%–100% and specificity of 83–97% and 57%–100%. Blood tau and CSF tau display no significant difference in sensitivity and specificity, while blood NFL is slightly more accurate than CSF NFL.
Since serum and plasma samples are more easily obtained than CSF samples, these blood biomarkers also represent very useful tools for TSE diagnosis. Blood tau and NFL levels also increase in other neurological diseases and correlate with worsening of symptoms (Weydt et al., 2016; Mielke et al., 2017; Foiani et al., 2018), but they are generally higher in CJD patients (Thompson et al., 2020). Furthermore, blood tau concentration correlates with disease progression in sCJD patients and, thus, can be used to predict survival time (Thompson et al., 2018; Staffaroni et al., 2019). The analysis of plasma from patients carrying PRNP mutations associated with slowly progressive disease showed that NFL levels increase before the onset of symptoms (2 years before), suggesting that it may constitute a prodromal marker (Thompson et al., 2020). However, this interesting line of research will require additional studies with other populations.
Total PrP (t-PrP) has recently arisen as another potential biomarker. Since the concentration of t-PrP in CSF decreases as disease progresses (Villar-Piqué et al., 2019), it has been suggested as a pharmacodynamic biomarker (Minikel et al., 2019; Vallabh et al., 2019). CSF t-PrP concentration has been shown to be stable during 2 years of follow up, but none of the patients tested developed disease during the time frame of the research (Vallabh et al., 2020). The time point at which t-PrP levels start to decline and PrPSc levels start to increase in CSF is still unknown. Therefore, unveiling these events is of paramount importance for understanding disease progression. Additionally, elevated t-PrP levels have been detected in the plasma of sCJD and genetic TSE patients as a probable consequence of blood-brain barrier impairment (Llorens et al., 2020c). In this study, plasma t-PrP correlated with CSF biomarkers of neuronal death, but not with CSF t-PrP.
The diversity of TSE forms and subtypes poses a great challenge for defining a qualitative and quantitative standard for all the surrogate biomarkers reviewed here. As none of these approaches can undoubtedly distinguish human prion diseases from other neurological disorders, all these biomarkers should be analyzed with caution and always in combination with other methods, such as clinical evaluation, bioimaging (MRI) analysis, and EEG assessment (Hamlin et al., 2012).
Electroencephalography (EEG)
Transmissible spongiform encephalopathy diagnoses can be supported by the appearance of periodic sharp wave complexes (PSWCs)—typical bilateral periodic discharges with a frequency of 0.5 to 2 per second—in EEG output. However, EEG findings alone are not specific for CJD, since PSWCs have been reported in patients with Alzheimer’s disease, vascular dementia, Lewy body disease, and voltage-gated potassium channel complex antibodies (VGKC-cAbs) encephalitis (Wieser et al., 2006; Savard et al., 2016). PSWCs are found in 60% to 70% of sCJD patients and 75% of fCJD patients, while they are rarely found in GSS and FFI cases and never in vCJD cases. The use of benzodiazepines for epilepsy has been shown to mask EEG findings. PSWCs appear months after the symptoms have started, and patients tend to recover a few weeks after their appearance. Meanwhile, bilateral frontal intermittent rhythmical delta activity (FIRDA) and a slowing of EEG background activity have been recorded in the early stages of CJD (Hansen et al., 1998; Manix et al., 2015). The absence of PSWC in EEG data does not exclude a diagnosis of sCJD, and typical EEG findings may not be observed in the terminal stage of the disease. Patients with CJD can also exhibit diffuse, non-specific slow EEG background patterns, which can also be observed in encephalopathy of various neurodegenerative diseases other than CJD. Hence, the diagnosis of sCJD should depend on a combination of EEG with MRI, CSF analysis, and clinical findings (Kwon and Kwon, 2019).
Bioimaging
Neuroimaging tests are an accessible and non-invasive diagnostic tool for TSEs (Ortega-Cubero et al., 2013). Some neuroimaging techniques to help clinical neurologists provide a definite diagnosis have been extensively evaluated. In particular, CJD diagnosis has employed MRI, computed tomography (CT), radionuclide techniques using positron emission tomography (PET), and single-photon emission tomography (SPECT) with different radiopharmaceuticals (Caobelli et al., 2015). In clinical practice, the probable diagnosis of TSEs relies on clinical features combined with the results of at least one paraclinical test (EEG, CSF analysis, and/or MRI abnormalities).
Diffusion-weighted imaging (DWI), fluid-attenuated inversion recovery (FLAIR), and apparent diffusion coefficient (ADC) are MRI sequences used to image the brain. High-intensity patterns in FLAIR, hyperintensity signals in DWI (the most sensitive sequence), and restricted diffusion in the ADC map gray matter regions give a positive predictive value for TSEs, with sensitivity and specificity values of 80% for detecting CJD (Gibson et al., 2018). CJD diagnostic criteria by bioimaging includes the presence of abnormal signal in the caudate and putamen, and MRI signal hyperintensities in the cortex of at least two lobes, excluding the frontal lobes on DWI and FLAIR for sCJD (Zerr et al., 2009; Bizzi et al., 2020). A recent study included a new criterion (index test) for diffusion MRI for the diagnosis of sCJD, being at least one positive brain region in addition to the current criteria (Bizzi et al., 2020). Analysis of diffusion MRI including the index test from ∼1,400 patients with suspected sCJD was superior to that of current standard criteria and comparable to second generation RT-QuIC (Bizzi et al., 2020).
Between 57% and 80% of sCJD patients show hyperintensity in the basal ganglia or cortex, with some also showing hyperintensity in the thalamus. MRI results are usually normal in patients with GSS and FFI, though some FFI patients show increased quantitative ADC and myoinositol in the thalamus. Quantitative MRI changes were also reported for FFI, characterized by increased mean diffusivity in the thalamus and cerebellum resultant from volumetric and diffusion tensor imaging (DTI) changes in the patient’s brain (Grau-Rivera et al., 2017). In 90% of cases, vCJD shows a pulvinar sign in which the posterior third of the thalamus has a higher signal than other basal ganglia (Manix et al., 2015; Fragoso et al., 2017). MRI signal changes are correlated with disease duration and with the degree of spongiosis (Eisenmenger et al., 2016). The strict application of diagnostic criteria and careful interpretation of MRI output remain the most recommended approach to in vivo diagnosis of sCJD (Fragoso et al., 2017). In the context of clinical suspicion of TSEs, however, CT does remain useful as a first-line test to exclude other diagnoses that may have similar clinical features (Letourneau-Guillon et al., 2012).
Positron emission tomography (PET) is not a paraclinical test included in the guidelines for TSE diagnosis, but it has recently been tested with a small group of patients. CJD patients showed hypometabolism in the frontal and parietal cortex using [18F] fluoro-2-deoxy-D-glucose (FDG-PET), suggesting a correlation between clinical results and PET findings (Renard et al., 2017). In a different group of patients, hypermetabolism was observed in the limbic and mesolimbic systems (Mente et al., 2017). Hypometabolism in brain regions is consistent with neuropathology, but it is not an exclusive characteristic of prion diseases (Higuchi et al., 2000).
Amyloid tracers may also be used with PET to diagnose neurodegenerative diseases, but the selectivity of these tracers remains a topic of discussion and requires improvement. Therefore, this technique is still limited for the differential diagnosis of these diseases, though it has yet to be explored in detail in living patients.
Immunoassays
Immunoassays exploit the structural differences between PrPC and PrPSc and rely on the specific interaction of monoclonal antibodies (mAbs) with PrPSc (Lukan et al., 2013). Although with potential application in diagnosis, immunoassays developed for analyzing body fluids, such as blood, CSF, or urine, have certain limitations. Unlike diagnostic studies using in vitro cell-free PrP conversion assays, reports of immunoassays have been restricted to blood samples not endogenously infected with PrPSc or from symptomatic vCJD patients. Almost all blood-based immunoassays developed to date rely on the detection of PrPSc. The major issue in blood detection systems is an extremely small amount of PrPSc combined with a high background PrPC concentration (Abdel-Haq, 2015). For this reason, a PrPSc enrichment step is usually required in these assays (Lukan et al., 2013). The solid-state binding matrix assay relies on the selective adsorption of PrPSc, thus, no PK pre-treatment of samples is required in this assay (Properzi and Pocchiari, 2013), being also adapted for urine assessment (Luk et al., 2016). This approach has led to the establishment of a prototype blood test for the diagnosis of vCJD with high sensitivity and specificity (Edgeworth et al., 2011), and was shown to be more efficient than immunoprecipitation with antibodies for detecting PrPSc in human whole blood spiked with high dilutions of vCJD-infected brain homogenate (Abdel-Haq, 2015). Although initially promising, this test failed to identify patients with sCJD or other TSEs (Edgeworth et al., 2011) and asymptomatic vCJD individuals (Jackson et al., 2014; Abdel-Haq, 2015). In summary, all these approaches present severe limitations regarding efficient detection of PrPSc in a variety of body fluids and tissues, being not recommended for routine diagnosis of human TSEs.
Guidelines for Diagnosis
The WHO prepared a report in 1998, updated in 2003, defining the diagnostic criteria for CJD. However, much research has been carried out in the intervening years, and new diagnostic tools have been developed and tested with great success. The Centers for Disease Control and Prevention (CDC), an agency of the United States Department of Health and Human Services, updated the clinical diagnostic criteria for sCJD to include MRI brain scans and RT-QuIC analysis for the probable diagnosis of sCJD (Table 5).
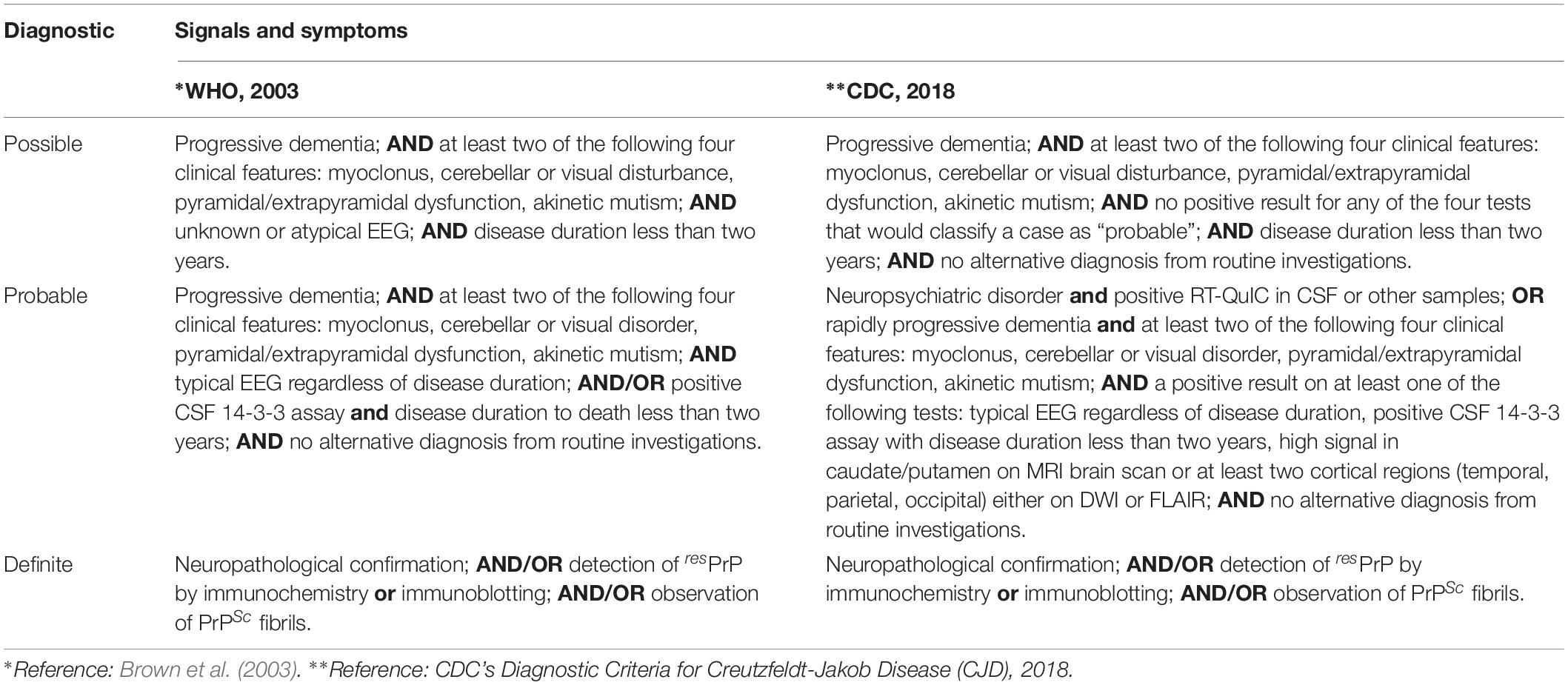
Table 5. Current guidelines for sCJD diagnosis.
The diagnosis of iCJD was defined as a progressive cerebellar syndrome in a patient treated with pituitary hormone receptor derived from human cadavers; or in a patient diagnosed with sporadic CJD with a recognized exposure risk—e.g., previous neurosurgery with dura mater graft. Genetic CJD is classified as definite or probable CJD plus definite or probable CJD in a first-degree relative; and/or neuropsychiatric disorder plus disease-specific PrP gene mutation (Brown et al., 2003; CDC’s Diagnostic Criteria for Creutzfeldt–Jakob Disease (CJD), 2018).
One characteristic that distinguishes vCJD from sCJD is its prominent tropism for lymphoid organs, such as the tonsils. The analysis of PrP extracted from amygdala biopsy tissue appears to provide a sensitive and specific method for the diagnosis of vCJD in the appropriate clinical context (Araújo, 2013). However, due to the invasive nature of this test, it should only be performed in patients who meet the clinical criteria for vCJD, but where MRI data lacks the characteristic pulmonary sign. The detection of 14-3-3 protein in the CSF is not a sensitive marker for vCJD (Araújo, 2013). Definitive cases are diagnosed by visualization of amyloid plaques surrounded by vacuoles in both the cerebellum and cerebrum, called florid plaques, and by PrP deposition shown by immunohistochemistry (CDC’s Diagnostic Criteria for Creutzfeldt–Jakob Disease (CJD), 2018). Suspected cases are identified based on the age of onset of the disease (<50 years), psychiatric symptoms (such as frank pain and/or dysesthesia), neurologic signs (at least two of the following: poor coordination, myoclonus, chorea, hyperreflexia, or visual signs), duration of illness of over 6 months, and no history for iCJD or gCJD.
Because of its genetic character, the diagnosis of GSS is limited to the demonstration of mutations in the PRNP gene. This seems to be a sensitive and highly specific way to diagnose GSS, but neuropathology, although less used in practice, can also be useful in a postmortem scenario (Araújo, 2013).
Finally, the diagnosis of FFI is first suggested by rapidly progressive cognitive impairment (dementia), along with changes in behavior or mood, ataxia, and sleep disorders. Additional diagnosis may include a sleep study. Given its etiology, genetic testing confirms the diagnosis (Wu et al., 2018).
Challenges
Human prion diseases are rare, with an annual incidence of around 1–2 cases per million individuals worldwide, and around five cases per million after age 65 (Holman et al., 2010). These diseases are very intriguing and can have a quite distinct clinical presentation and causes (sporadic, inherited, and acquired). This heterogeneity poses challenges for diagnoses. Interestingly, all prion diseases involve the conversion and aggregation of PrPC into its pathological form, PrPSc. Therefore, it is no wonder that the detection of PrPSc is used as the gold standard for a definite diagnosis.
The detection of PrPSc in patient samples is traditionally performed by neuropathological inspection of brain tissue, which is a criterion for a definite diagnosis (Brown et al., 2003). However, this assessment is done most often postmortem due to the invasiveness of tissue collection. Clinical and paraclinical antemortem evaluations are not considered sufficient to define the final diagnosis. However, with continued technological advances, there has been a marked improvement in several tests in recent years, contributing to the interpretation of cases.
The last decade has seen the development of the RT-QuIC assay, which can now detect the presence of PrPSc. This technique has several advantages, including:
(1) It can be used on more accessible samples, such as CSF;
(2) It exhibits high sensitivity and specificity, making other biochemical tests obsolete;
(3) It can detect PrPSc in samples from different types of TSEs;
(4) It involves a relatively simple method, employing materials and equipment that are more accessible and easier to maintain;
(5) A growing number of centers around the world have independently tested, validated, and used this technique to investigate large cohorts of patients;
(6) It is being adapted for the diagnosis of other neurodegenerative diseases, such as Parkinson’s disease, tauopathies, and others.
Given these advantages, the CDC has included RT-QuIC as a parameter to characterize TSE diagnosis as probable (CDC’s Diagnostic Criteria for Creutzfeldt–Jakob Disease (CJD), 2018). WHO guidelines do not yet include this parameter and should be updated to encourage its implementation in more facilities that can carry out these tests. The combination of (i) clinical evaluation, (ii) RT-QuIC analyses of two biological samples, and (iii) testing with at least one other technique—such as MRI, EEG, or 14-3-3 detection—should be considered internationally for a definite antemortem diagnosis, given that such combinations have provided diagnoses with 100% or nearly 100% sensitivity and specificity (Bongianni et al., 2017; Hermann et al., 2018; Rudge et al., 2018; Fiorini et al., 2020).
Furthermore, the RT-QuIC assay has been adapted in the last few years for detecting α-synuclein aggregates/fibrils and tau filaments in biological samples (Ferreira and Caughey, 2020). Therefore, the growth in RT-QuIC-based TSE diagnosis would certainly pave the way for RT-QuIC utilization in the early diagnosis of other neurodegenerative disorders, such as synucleinopathies (e.g., Parkinson’s disease and dementia with Lewy bodies) and tauopathies (e.g., Alzheimer’s disease and chronic traumatic encephalopathy.
Even with all the advances achieved to date in TSE diagnosis, most patients present the first typical clinical signs of the disease very late, when the degeneration is already quite advanced and irreversible. Regular clinical evaluation is only initiated at this point, resulting in therapeutic options that are, at best, symptomatic. Therefore, a significant challenge is to further increase the sensitivity and accuracy of testing to allow early antemortem diagnosis and to relate the findings to the progression of the disease. Another challenge is to obtain a diagnosis at pre-symptomatic stages of genetic forms of TSEs, which would require the combination of genotyping and the examination of samples using the assays discussed in this review. If an early diagnosis can be achieved, this opens the door to novel therapeutic strategies being tested and used successfully in the future.
Author Contributions
YC, TCRGV, and LMA contributed to the conception and design of the study. SCR wrote the first draft of the manuscript. TCRGV, YC, LMA, and PBG wrote sections of the manuscript. LMA produced all figures. All authors contributed to the manuscript revision and read and approved the submitted version.
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001; by Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ); and by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Natalia C. Ferreira for providing an RT-QuIC result and Pierluigi Gambetti for providing the brain samples used in this assay.
References
Abdel-Haq, H. (2015). Factors intrinsic and extrinsic to blood hamper the development of a routine blood test for human prion diseases. J. Gen. Virol. 96, 479–493. doi: 10.1099/vir.0.070979-0
Abu-Rumeileh, S., Baiardi, S., Polischi, B., Mammana, A., Franceschini, A., Green, A., et al. (2019). Diagnostic value of surrogate CSF biomarkers for Creutzfeldt–Jakob disease in the era of RT-QuIC. J. Neurol. 266, 3136–3143. doi: 10.1007/s00415-019-09537-0
Abu-Rumeileh, S., Capellari, S., Stanzani-Maserati, M., Polischi, B., Martinelli, P., Caroppo, P., et al. (2018). The CSF neurofilament light signature in rapidly progressive neurodegenerative dementias. Alzheimer’s Res. Ther. 10:3. doi: 10.1186/s13195-017-0331-1
Acquatella-Tran Van Ba, I., Imberdis, T., and Perrier, V. (2013). From prion diseases to prion-like propagation mechanisms of neurodegenerative diseases. Int. J. Cell Biol. 2013:975832. doi: 10.1155/2013/975832
Agrimi, U., Nonno, R., Dell’Omo, G., Di Bari, M. A., Conte, M., Chiappini, B., et al. (2008). Prion protein amino acid determinants of differential susceptibility and molecular feature of prion strains in mice and voles. PLoS Pathog. 4:e1000113. doi: 10.1371/journal.ppat.1000113
Aguzzi, A., Glatzel, M., Montrasio, F., Prinz, M., and Heppner, F. L. (2001). Interventional strategies against prion diseases. Nat. Rev. Neurosci. 2, 745–749. doi: 10.1038/35094590
Aguzzi, A., Heikenwalder, M., and Polymenidou, M. (2007). Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 8, 552–561. doi: 10.1038/nrm2204
Aksamit, A. J., Preissner, C. M., and Homburger, H. A. (2001). Quantitation of 14-3-3 and neuron-specific enolase proteins in CSF in Creutzfeldt-Jakob disease. Neurology 57, 728–730. doi: 10.1212/WNL.57.4.728
Andréoletti, O., Litaise, C., Simmons, H., Corbière, F., Lugan, S., Costes, P., et al. (2012). Highly efficient prion transmission by blood transfusion. PLoS Pathog. 8:e1002782. doi: 10.1371/journal.ppat.1002782
Annus, Á, Csáti, A., and Vécsei, L. (2016). Prion diseases: new considerations. Clin. Neurol. Neurosurg. 150, 125–132. doi: 10.1016/j.clineuro.2016.09.006
Araújo, A. Q. C. (2013). Doenças priônicas. Arq. Neuropsiquiatr. 71, 731–737. doi: 10.1590/0004-282X201301461
Atarashi, R., Moore, R. A., Sim, V. L., Hughson, A. G., Dorward, D. W., Onwubiko, H. A., et al. (2007). Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat. Methods 4, 645–650. doi: 10.1038/nmeth1066
Atarashi, R., Satoh, K., Sano, K., Fuse, T., Yamaguchi, N., Ishibashi, D., et al. (2011). Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat. Med. 17, 175–178. doi: 10.1038/nm.2294
Atarashi, R., Wilham, J. M., Christensen, L., Hughson, A. G., Moore, R. A., Johnson, L. M., et al. (2008). Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat. Methods 5, 211–212. doi: 10.1038/nmeth0308-211
Ayers, J. I., and Prusiner, S. B. (2020). Prion protein — mediator of toxicity in multiple proteinopathies. Nat. Rev. Neurol. 16, 187–188. doi: 10.1038/s41582-020-0332-8
Bahl, J. M. C., Heegaard, N. H. H., Falkenhorst, G., Laursen, H., Høgenhaven, H., Mølbak, K., et al. (2009). The diagnostic efficiency of biomarkers in sporadic Creutzfeldt-Jakob disease compared to Alzheimer’s disease. Neurobiol. Aging 30, 1834–1841. doi: 10.1016/j.neurobiolaging.2008.01.013
Baral, P. K., Yin, J., Aguzzi, A., and James, M. N. G. (2019). Transition of the prion protein from a structured cellular form (PrPC) to the infectious scrapie agent (PrPSc). Protein Sci. 28, 2055–2063. doi: 10.1002/pro.3735
Beaudry, P., Cohen, P., Brandel, J. P., Delasnerie-Lauprêtre, N., Richard, S., Launay, J. M., et al. (1999). 14-3-3 Protein, neuron-specific enolase, and S-100 protein in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Dement. Geriatr. Cogn. Disord. 10, 40–46. doi: 10.1159/000017095
Bellon, A., Seyfert-Brandt, W., Lang, W., Baron, H., Gröner, A., and Vey, M. (2003). Improved conformation-dependent immunoassay: suitability for human prion detection with enhanced sensitivity. J. Gen. Virol. 84, 1921–1925. doi: 10.1099/vir.0.18996-0
Bendheim, P. E., Barry, R. A., Dearmond, S. J., Stites, D. P., and Prusiner, S. B. (1984). Antibodies to a scrapie prion protein. Nature 310, 418–421. doi: 10.1038/310418a0
Bessen, R. A., Kocisko, D. A., Raymond, G. J., Nandan, S., Lansbury, P. T., and Caughey, B. (1995). Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 375, 698–700. doi: 10.1038/375698a0
Bizzi, A., Pascuzzo, R., Blevins, J., Grisoli, M., Lodi, R., Moscatelli, M. E. M., et al. (2020). Evaluation of a new criterion for detecting prion disease with diffusion magnetic resonance Imaging. JAMA Neurol. 77, 1–9. doi: 10.1001/jamaneurol.2020.1319
Bongianni, M., Orrù, C., Groveman, B. R., Sacchetto, L., Fiorini, M., Tonoli, G., et al. (2017). Diagnosis of human prion disease using real-time quaking-induced conversion testing of olfactory mucosa and cerebrospinal fluid samples. JAMA Neurol. 74, 155–162. doi: 10.1001/jamaneurol.2016.4614
Bougard, D., Brandel, J. P., Bélondrade, M., Béringue, V., Segarra, C., Fleury, H., et al. (2016). Detection of prions in the plasma of presymptomatic and symptomatic patients with variant Creutzfeldt-Jakob disease. Sci. Transl. Med. 8:370ra182. doi: 10.1126/scitranslmed.aag1257
Brown, P., Brunk, C., Budka, H., Cervenakova, L., Collie, D., Green, A., et al. (2003). ). WHO Manual for Surveillance of Human Transmissible Spongiform Encephalopathies, Including Variant Creutzfeldt-Jakob Disease. Geneva: World Health Organization.
Caobelli, F., Cobelli, M., Pizzocaro, C., Pavia, M., Magnaldi, S., and Guerra, U. P. (2015). The role of neuroimaging in evaluating patients affected by creutzfeldt-jakob disease: a systematic review of the literature. J. Neuroimaging 25, 2–13. doi: 10.1111/jon.12098
Castilla, J., Saá, P., and Soto, C. (2004). “Cyclic Amplification of Prion Protein Misfolding,” in Techniques in Prion Research, eds S. Lehmann and J. Grassi (Basel: Birkhäuser), 198–213. doi: 10.1007/978-3-0348-7949-1_14
Castilla, J., Saá, P., and Soto, C. (2005). Detection of prions in blood. Nat. Med. 11, 982–985. doi: 10.1038/nm1286
Caughey, B. (2000). Formation of protease-resistant prion protein in cell-free systems. Curr. Issues Mol. Biol. 2, 95–101.
Caughey, B., Baron, G. S., Chesebro, B., and Jeffrey, M. (2009). Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 78, 177–204. doi: 10.1146/annurev.biochem.78.082907.145410
CDC’s Diagnostic Criteria for Creutzfeldt–Jakob Disease (CJD) (2018). Centers Dis. Control Prev. Available online at: https://www.cdc.gov/prions/cjd/diagnostic-criteria.html (accessed September 22, 2020).
Chen, C., and Dong, X. P. (2016). Epidemiological characteristics of human prion diseases. Infect. Dis. Poverty 5:47. doi: 10.1186/s40249-016-0143-8
Chesebro, B. (2003). Introduction to the transmissible spongiform encephalopathies or prion diseases. Br. Med. Bull. 66, 1–20. doi: 10.1093/bmb/dg66.001
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901
Chitravas, N., Jung, R. S., Kofskey, D. M., Blevins, J. E., Gambetti, P., Leigh, R. J., et al. (2011). Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann. Neurol. 70, 437–444. doi: 10.1002/ana.22454
Chohan, G., Pennington, C., Mackenzie, J. M., Andrews, M., Everington, D., Will, R. G., et al. (2010). The role of cerebrospinal fluid 14-3-3 and other proteins in the diagnosis of sporadic Creutzfeldt-Jakob disease in the UK: a 10-year review. J. Neurol. Neurosurg. Psychiatry 81, 1243–1248. doi: 10.1136/jnnp.2009.197962
Cobb, N. J., and Surewicz, W. K. (2009). Prion diseases and their biochemical mechanisms. Biochemistry 48, 2574–2585. doi: 10.1021/bi900108v
Cohen, O. S., Chapman, J., Korczyn, A. D., Warman-Alaluf, N., Nitsan, Z., Appel, S., et al. (2016). CSF tau correlates with CJD disease severity and cognitive decline. Acta Neurol. Scand. 133, 119–123. doi: 10.1111/ane.12441
Colby, D. W., and Prusiner, S. B. (2011). De novo generation of prion strains. Nat. Rev. Microbiol. 9, 771–777. doi: 10.1038/nrmicro2650
Colby, D. W., Zhang, Q., Wang, S., Groth, D., Legname, G., Riesner, D., et al. (2007). Prion detection by an amyloid seeding assay. Proc. Natl. Acad. Sci. U.S.A. 104, 20914–20919. doi: 10.1073/pnas.0710152105
Collins, S. J., Sanchez-Juan, P., Masters, C. L., Klug, G. M., Van Duijn, C., Poleggi, A., et al. (2006). Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 129, 2278–2287. doi: 10.1093/brain/awl159
Concha-Marambio, L., Pritzkow, S., Moda, F., Tagliavini, F., Ironside, J. W., Schulz, P. E., et al. (2016). Detection of prions in blood from patients with variant Creutzfeldt-Jakob disease. Sci. Transl. Med. 8:370ra183. doi: 10.1126/scitranslmed.aaf6188
Corbett, G. T., Wang, Z., Hong, W., Colom-Cadena, M., Rose, J., Liao, M., et al. (2020). PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 139, 503–526. doi: 10.1007/s00401-019-02114-9
Cracco, L., Appleby, B. S., and Gambetti, P. (2018). Fatal familial insomnia and sporadic fatal insomnia. Handb. Clin. Neurol. 153, 271–299. doi: 10.1016/B978-0-444-63945-5.00015-5
Cramm, M., Schmitz, M., Karch, A., Mitrova, E., Kuhn, F., Schroeder, B., et al. (2016). Stability and reproducibility underscore utility of RT-QuIC for diagnosis of Creutzfeldt-Jakob Disease. Mol. Neurobiol. 53, 1896–1904. doi: 10.1007/s12035-015-9133-2
Cramm, M., Schmitz, M., Karch, A., Zafar, S., Varges, D., Mitrova, E., et al. (2015). Characteristic CSF prion seeding efficiency in humans with prion diseases. Mol. Neurobiol. 51, 396–405. doi: 10.1007/s12035-014-8709-6
Di Bari, M. A., Chianini, F., Vaccari, G., Esposito, E., Conte, M., Eaton, S. L., et al. (2008). The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J. Gen. Virol. 89, 2975–2985. doi: 10.1099/vir.0.2008/005520-0
Di Bari, M. A., Nonno, R., Castilla, J., D’Agostino, C., Pirisinu, L., Riccardi, G., et al. (2013). Chronic wasting disease in bank voles: characterisation of the shortest incubation time model for prion diseases. PLoS Pathog. 9:e1003219. doi: 10.1371/journal.ppat.1003219
Diack, A. B., Ritchie, D. L., Peden, A. H., Brown, D., Boyle, A., Morabito, L., et al. (2014). Variably protease-sensitive prionopathy, a unique prion variant with inefficient transmission properties. Emerg. Infect. Dis. 20, 1969–1979. doi: 10.3201/eid2012.140214
Edgeworth, J. A., Farmer, M., Sicilia, A., Tavares, P., Beck, J., Campbell, T., et al. (2011). Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 377, 487–493. doi: 10.1016/S0140-6736(10)62308-2
Eisenmenger, L., Porter, M. C., Carswell, C. J., Thompson, A., Mead, S., Rudge, P., et al. (2016). Evolution of diffusion-weighted magnetic resonance imaging signal abnormality in sporadic Creutzfeldt-jakob disease, with histopathological correlation. JAMA Neurol. 73, 76–84. doi: 10.1001/jamaneurol.2015.3159
Feinstein, A. R. (1975). XXXI. On the sensitivity, specificity, and discrimination of diagnostic tests. Clin. Pharmacol. Ther. 17, 104–116. doi: 10.1002/cpt1975171104
Ferreira, N. D. C., and Caughey, B. (2020). Proteopathic seed amplification assays for neurodegenerative disorders. Clin. Lab. Med. 40, 257–270. doi: 10.1016/j.cll.2020.04.002
Fiorini, M., Iselle, G., Perra, D., Bongianni, M., Capaldi, S., Sacchetto, L., et al. (2020). High diagnostic accuracy of RT-QuIC assay in a prospective study of patients with suspected sCJD. Int. J. Mol. Sci. 21:880. doi: 10.3390/ijms21030880
Foiani, M. S., Woollacott, I. O. C., Heller, C., Bocchetta, M., Heslegrave, A., Dick, K. M., et al. (2018). Plasma tau is increased in frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 89, 804–807. doi: 10.1136/jnnp-2017-317260
Forloni, G., Roiter, I., and Tagliavini, F. (2019). Clinical trials of prion disease therapeutics. Curr. Opin. Pharmacol. 44, 53–60. doi: 10.1016/j.coph.2019.04.019
Foutz, A., Appleby, B. S., Hamlin, C., Liu, X., Yang, S., Cohen, Y., et al. (2017). Diagnostic and prognostic value of human prion detection in cerebrospinal fluid. Ann. Neurol. 81, 79–92. doi: 10.1002/ana.24833
Fragoso, D. C., Gonçalves Filho, A. L., da, M., Pacheco, F. T., Barros, B. R., Littig, I. A., et al. (2017). Imaging of Creutzfeldt-Jakob disease: imaging patterns and their differential diagnosis. Radiographics 37, 234–257. doi: 10.1148/rg.2017160075
Franceschini, A., Baiardi, S., Hughson, A. G., McKenzie, N., Moda, F., Rossi, M., et al. (2017). High diagnostic value of second generation CSF RT-QuIC across the wide spectrum of CJD prions. Sci. Rep. 7:10655. doi: 10.1038/s41598-017-10922-w
Gambetti, P., Kong, Q., Zou, W., Parchi, P., and Chen, S. G. (2003). Sporadic and familial CJD: classification and characterisation. Br. Med. Bull. 66, 213–239. doi: 10.1093/bmb/66.1.213
Geschwind, M. D. (2015). Prion Diseases. Contin. Lifelong Learn. Neurol. 21, 1612–1638. doi: 10.1212/CON.0000000000000251
Geschwind, M. D., Martindale, J., Miller, D., DeArmond, S. J., Uyehara-Lock, J., Gaskin, D., et al. (2003). Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch. Neurol. 60, 813–816. doi: 10.1001/archneur.60.6.813
Gibson, L. M., Chappell, F. M., Summers, D., Collie, D. A., Sellar, R., Best, J., et al. (2018). Post-mortem magnetic resonance imaging in patients with suspected prion disease: pathological confirmation, sensitivity, specificity and observer reliability. A national registry. PLoS One 13:e0201434. doi: 10.1371/journal.pone.0201434
Gmitterová, K., Heinemann, U., Bodemer, M., Krasnianski, A., Meissner, B., Kretzschmar, H. A., et al. (2009). 14-3-3 CSF levels in sporadic Creutzfeldt-Jakob disease differ across molecular subtypes. Neurobiol. Aging 30, 1842–1850. doi: 10.1016/j.neurobiolaging.2008.01.007
Grau-Rivera, O., Calvo, A., Bargalló, N., Monté, G. C., Nos, C., Lladó, A., et al. (2017). Quantitative magnetic resonance abnormalities in creutzfeldt-jakob disease and fatal insomnia. J. Alzheimer’s Dis. 55, 431–443. doi: 10.3233/JAD-160750
Green, A. J. E., Ramljak, S., Müller, W. E. G., Knight, R. S. G., and Schröder, H. C. (2002). 14-3-3 in the cerebrospinal fluid of patients with variant and sporadic Creutzfeldt-Jakob disease measured using capture assay able to detect low levels of 14-3-3 protein. Neurosci. Lett. 324, 57–60. doi: 10.1016/S0304-3940(02)00172-6
Green, A. J. E., Thompson, E. J., Stewart, G. E., Zeidler, M., McKenzie, J. M., MacLeod, M. A., et al. (2001). Use of 14-3-3 and other brain-specific proteins in CSF in the diagnosis of variant Creutzfeldt-Jakob disease. J. Neurol. Neurosurg. Psychiatry 70, 744–748. doi: 10.1136/jnnp.70.6.744
Groveman, B. R., Kraus, A., Raymond, L. D., Dolan, M. A., Anson, K. J., Dorward, D. W., et al. (2015). Charge neutralization of the central lysine cluster in prion protein (PrP) promotes PrPSc-Like folding of recombinant PrP amyloids. J. Biol. Chem. 290, 1119–1128. doi: 10.1074/jbc.M114.619627
Groveman, B. R., Orrú, C. D., Hughson, A. G., Bongianni, M., Fiorini, M., Imperiale, D., et al. (2016). Extended and direct evaluation of RT-QuIC assays for Creutzfeldt-Jakob disease diagnosis. Ann. Clin. Transl. Neurol. 4, 139–144. doi: 10.1002/acn3.378
Groveman, B. R., Raymond, G. J., Campbell, K. J., Race, B., Raymond, L. D., Hughson, A. G., et al. (2017). Role of the central lysine cluster and scrapie templating in the transmissibility of synthetic prion protein aggregates. PLoS Pathog. 13:e1006623. doi: 10.1371/journal.ppat.1006623
Haley, N. J., and Richt, J. A. (2017). Evolution of diagnostic tests for chronic wasting disease, a naturally occurring prion disease of cervids. Pathogens 6:35. doi: 10.3390/pathogens6030035
Hamlin, C., Puoti, G., Berri, S., Sting, E., Harris, C., Cohen, M., et al. (2012). A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease. Neurology 79, 547–552. doi: 10.1212/WNL.0b013e318263565f
Hansen, H. C., Zschocke, S., Stürenburg, H. J., and Kunze, K. (1998). Clinical changes and EEG patterns preceding the onset of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Acta Neurol. Scand. 97, 99–106. doi: 10.1111/j.1600-0404.1998.tb00617.x
Hermann, P., Laux, M., Glatzel, M., Matschke, J., Knipper, T., Goebel, S., et al. (2018). Validation and utilization of amended diagnostic criteria in Creutzfeldt-Jakob disease surveillance. Neurology 91, e331–e338. doi: 10.1212/WNL.0000000000005860
Higuchi, M., Tashiro, M., Arai, H., Okamura, N., Hara, S., Higuchi, S., et al. (2000). Glucose hypometabolism and neuropathological correlates in brains of dementia with Lewy bodies. Exp. Neurol. 162, 247–256. doi: 10.1006/exnr.2000.7342
Hill, A. F., Desbruslais, M., Joiner, S., Sidle, K. C. L., Gowland, I., Collinge, J., et al. (1997). The same prion strain causes vCJD and BSE. Nature 389, 448–450. doi: 10.1038/38925
Holman, R. C., Belay, E. D., Christensen, K. Y., Maddox, R. A., Minino, A. M., Folkema, A. M., et al. (2010). Human prion diseases in the United States. PLoS One 5:e8521. doi: 10.1371/journal.pone.0008521
Hsich, G., Kenney, K., Gibbs, C. J., Lee, K. H., and Harrington, M. G. (1996). The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N. Engl. J. Med. 335, 924–930. doi: 10.1056/NEJM199609263351303
Hunter, N., Foster, J., Chong, A., McCutcheon, S., Parnham, D., Eaton, S., et al. (2002). Transmission of prion diseases by blood transfusion. J. Gen. Virol. 83, 2897–2905. doi: 10.1099/0022-1317-83-11-2897
Igel-Egalon, A., Béringue, V., Rezaei, H., and Sibille, P. (2018). Prion strains and transmission barrier phenomena. Pathogens 7:5. doi: 10.3390/pathogens7010005
Ironside, J. W., Ritchie, D. L., and Head, M. W. (2017). Prion diseases. Handb. Clin. Neurol. 145, 393–403. doi: 10.1016/B978-0-12-802395-2.00028-6
Jackson, G. S., Burk-Rafel, J., Edgeworth, J. A., Sicilia, A., Abdilahi, S., Korteweg, J., et al. (2014). Population screening for variant Creutzfeldt-Jakob disease using a novel blood test: diagnostic accuracy and feasibility study. JAMA Neurol. 71, 421–428. doi: 10.1001/jamaneurol.2013.6001
Jensen, M., Basun, H., and Lannfelt, L. (1995). Increased cerebrospinal fluid tau in patients with Alzheimer’s disease. Neurosci. Lett. 186, 189–191. doi: 10.1016/0304-3940(95)11297-A
Jones, M., Peden, A. H., Prowse, C. V., Gröner, A., Manson, J. C., Turner, M. L., et al. (2007). In vitro amplification and detection of variant Creutzfeldt-Jakob disease PrPSc. J. Pathol. 213, 21–26. doi: 10.1002/path.2204
Kascsak, R. J., Rubenstein, R., Merz, P. A., Tonna-DeMasi, M., Fersko, R., Carp, R. I., et al. (1987). Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J. Virol. 61, 3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987
Kenney, K., Brechtel, C., Takahashi, H., Kurohara, K., Anderson, P., and Gibbs, C. J. (2000). An enzyme-linked immunosorbent assay to quantify 14-3-3 proteins in the cerebrospinal fluid of suspected Creutzfeldt-Jakob disease patients. Ann. Neurol. 48, 395–398. doi: 10.1002/1531-8249(200009)48:3<395::AID-ANA18<3.0.CO;2-A
Kim, C., Haldiman, T., Cohen, Y., Chen, W., Blevins, J., Sy, M. S., et al. (2011). Protease-sensitive conformers in broad spectrum of distinct prp sc structures in sporadic creutzfeldt-jakob disease are indicator of progression rate. PLoS Pathog. 7:e1002242. doi: 10.1371/journal.ppat.1002242
Kim, C., Haldiman, T., Surewicz, K., Cohen, Y., Chen, W., Blevins, J., et al. (2012). Small protease sensitive oligomers of PrPSc in distinct human prions determine conversion rate of PrPC. PLoS Pathog. 8:e1002835. doi: 10.1371/journal.ppat.1002835
Kim, C., Xiao, X., Chen, S., Haldiman, T., Smirnovas, V., Kofskey, D., et al. (2018). Artificial strain of human prions created in vitro. Nat. Commun. 9:2166. doi: 10.1038/s41467-018-04584-z
Kim, M. O., Takada, L. T., Wong, K., Forner, S. A., and Geschwind, M. D. (2018). Genetic PrP prion diseases. Cold Spring Harb. Perspect. Biol. 10:a033134. doi: 10.1101/cshperspect.a033134
Kobayashi, A., Mizukoshi, K., Iwasaki, Y., Miyata, H., Yoshida, Y., and Kitamoto, T. (2011). Co-occurrence of types 1 and 2 PrPres in sporadic Creutzfeldt-Jakob disease MM1. Am. J. Pathol. 178, 1309–1315. doi: 10.1016/j.ajpath.2010.11.069
Kraus, A., Anson, K. J., Raymond, L. D., Martens, C., Groveman, B. R., Dorward, D. W., et al. (2015). Prion protein prolines 102 and 105 and the surrounding lysine cluster impede amyloid formation. J. Biol. Chem. 290, 21510–21522. doi: 10.1074/jbc.M115.665844
Kraus, A., Groveman, B. R., and Caughey, B. (2013). Prions and the potential transmissibility of protein misfolding diseases. Annu. Rev. Microbiol. 67, 543–564. doi: 10.1146/annurev-micro-092412-155735
Kruse, N., Heslegrave, A., Gupta, V., Foiani, M., Villar-Piqué, A., Schmitz, M., et al. (2018). Interlaboratory validation of cerebrospinal fluid α-synuclein quantification in the diagnosis of sporadic Creutzfeldt-Jakob disease. Alzheimer’s Dement. 10, 461–470. doi: 10.1016/j.dadm.2018.06.005
Kushnirov, V. V., Dergalev, A. A., and Alexandrov, A. I. (2020). Proteinase K resistant cores of prions and amyloids. Prion 14, 11–19. doi: 10.1080/19336896.2019.1704612
Kwon, G. T., and Kwon, M. S. (2019). Diagnostic challenge of rapidly progressing sporadic Creutzfeldt-Jakob disease. BMJ Case Rep. 12:e230535. doi: 10.1136/bcr-2019-230535
Lacroux, C., Comoy, E., Moudjou, M., Perret-Liaudet, A., Lugan, S., Litaise, C., et al. (2014). Preclinical detection of variant CJD and BSE prions in blood. PLoS Pathog. 10:e1004202. doi: 10.1371/journal.ppat.1004202
Ladogana, A., Sanchez-Juan, P., Mitrová, E., Green, A., Cuadrado-Corrales, N., Sánchez-Valle, R., et al. (2009). Cerebrospinal fluid biomarkers in human genetic transmissible spongiform encephalopathies. J. Neurol. 256, 1620–1628. doi: 10.1007/s00415-009-5163-x
Lemstra, A. W., Van Meegen, M. T., Vreyling, J. P., Meijerink, P. H. S., Jansen, G. H., Bulk, S., et al. (2000). 14-3-3 testing in diagnosing Creutzfeldt-Jakob disease: a prospective study in 112 patients. Neurology 55, 514–516. doi: 10.1212/WNL.55.4.514
Letourneau-Guillon, L., Wada, R., and Kucharczyk, W. (2012). Imaging of prion diseases. J. Magn. Reson. Imaging 35, 998–1012. doi: 10.1002/jmri.23504
Llewelyn, C. A., Hewitt, P. E., Knight, R. S. G., Amar, K., Cousens, S., MacKenzie, J., et al. (2004). Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 363, 417–421. doi: 10.1016/S0140-6736(04)15486-X
Llorens, F., Kruse, N., Karch, A., Schmitz, M., Zafar, S., Gotzmann, N., et al. (2018). Validation of α-Synuclein as a CSF biomarker for sporadic Creutzfeldt-Jakob disease. Mol. Neurobiol. 55, 2249–2257. doi: 10.1007/s12035-017-0479-5
Llorens, F., Kruse, N., Schmitz, M., Gotzmann, N., Golanska, E., Thüne, K., et al. (2017). Evaluation of α-synuclein as a novel cerebrospinal fluid biomarker in different forms of prion diseases. Alzheimer’s Dement. 13, 710–719. doi: 10.1016/j.jalz.2016.09.013
Llorens, F., Rübsamen, N., Hermann, P., Schmitz, M., Villar-Piqué, A., Goebel, S., et al. (2020a). A prognostic model for overall survival in sporadic Creutzfeldt-Jakob disease. Alzheimer’s Dement. 16, 1438–1447. doi: 10.1002/alz.12133
Llorens, F., Villar-Piqué, A., Hermann, P., Schmitz, M., Goebel, S., Waniek, K., et al. (2020b). Cerebrospinal fluid non-phosphorylated tau in the differential diagnosis of Creutzfeldt–Jakob disease: a comparative prospective study with 14-3-3. J. Neurol. 267, 543–550. doi: 10.1007/s00415-019-09610-8
Llorens, F., Villar-Piqué, A., Schmitz, M., Diaz-Lucena, D., Wohlhage, M., Hermann, P., et al. (2020c). Plasma total prion protein as a potential biomarker for neurodegenerative dementia: diagnostic accuracy in the spectrum of prion diseases. Neuropathol. Appl. Neurobiol. 46, 240–254. doi: 10.1111/nan.12573
Luk, C., Jones, S., Thomas, C., Fox, N. C., and Tze, H. (2016). Prospective diagnosis of sporadic CJD by the detection of abnormal PrP in patient urine. JAMA Neurol. 73, 1454–1460. doi: 10.1001/jamaneurol.2016.3733
Lukan, A., Vranac, T., and Èurin Šerbec, V. (2013). TSE diagnostics: recent advances in immunoassaying prions. Clin. Dev. Immunol. 2013:360604. doi: 10.1155/2013/360604
Manix, M., Kalakoti, P., Henry, M., Thakur, J., Menger, R., Guthikonda, B., et al. (2015). Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg. Focus 39:E2. doi: 10.3171/2015.8.FOCUS15328
McGuire, L. I., Peden, A. H., Orrú, C. D., Wilham, J. M., Appleford, N. E., Mallinson, G., et al. (2012). Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 72, 278–285. doi: 10.1002/ana.23589
McGuire, L. I., Poleggi, A., Poggiolini, I., Suardi, S., Grznarova, K., Shi, S., et al. (2016). Cerebrospinal fluid real-time quaking-induced conversion is a robust and reliable test for sporadic creutzfeldt-jakob disease: an international study. Ann. Neurol. 80, 160–165. doi: 10.1002/ana.24679
Mente, K. P., O’Donnell, J. K., Jones, S. E., Cohen, M. L., Thompson, N. R., Bizzi, A., et al. (2017). Fluorodeoxyglucose positron emission tomography (FDG-PET) correlation of histopathology and MRI in prion disease. Alzheimer Dis. Assoc. Disord. 31, 1–7. doi: 10.1097/WAD.0000000000000188
Metrick, M. A., do Carmo Ferreira, N., Saijo, E., Hughson, A. G., Kraus, A., Orrú, C., et al. (2019). Million-fold sensitivity enhancement in proteopathic seed amplification assays for biospecimens by Hofmeister ion comparisons. Proc. Natl. Acad. Sci. U.S.A. 116, 23029–23039. doi: 10.1073/pnas.1909322116
Mielke, M. M., Hagen, C. E., Wennberg, A. M. V., Airey, D. C., Savica, R., Knopman, D. S., et al. (2017). Association of plasma total tau level with cognitive decline and risk of mild cognitive impairment or dementia in the Mayo Clinic study on aging. JAMA Neurol. 74, 1073–1080. doi: 10.1001/jamaneurol.2017.1359
Minikel, E. V., Kuhn, E., Cocco, A. R., Vallabh, S. M., Hartigan, C. R., Reidenbach, A. G., et al. (2019). Domain-specific quantification of prion protein in cerebrospinal fluid by targeted mass spectrometry. Mol. Cell. Proteomics 18, 2388–2400. doi: 10.1074/mcp.RA119.001702
Moda, F., Gambetti, P., Notari, S., Concha-Marambio, L., Catania, M., Park, K.-W., et al. (2014). Prions in the urine of patients with variant Creutzfeldt–Jakob Disease. N. Engl. J. Med. 371, 530–539. doi: 10.1056/NEJMoa1404401
Murray, K. (2011). Creutzfeldt-jacob disease mimics, or how to sort out the subacute encephalopathy patient. Pract. Neurol. 11, 19–28. doi: 10.1136/pgmj.2010.235721rep
Nonno, R., Di Bari, M. A., Cardone, F., Vaccari, G., Fazzi, P., Dell’Omo, G., et al. (2006). Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2:e12. doi: 10.1371/journal.ppat.0020012
Orrú, C. D., Bongianni, M., Tonoli, G., Ferrari, S., Hughson, A. G., Groveman, B. R., et al. (2014). A test for Creutzfeldt–Jakob Disease using nasal brushings. N. Engl. J. Med. 371, 519–529. doi: 10.1056/nejmoa1315200
Orrú, C. D., Groveman, B. R., Hughson, A. G., Zanusso, G., Coulthart, M. B., and Caughey, B. (2015a). Rapid and sensitive RT-QuIC detection of human creutzfeldt-jakob disease using cerebrospinal fluid. mBio 6:e02451-14. doi: 10.1128/mBio.02451-14
Orrú, C. D., Groveman, B. R., Raymond, L. D., Hughson, A. G., Nonno, R., Zou, W., et al. (2015b). Bank vole prion protein as an apparently universal substrate for RT-QuIC-Based Detection and discrimination of prion strains. PLoS Pathog. 11:e1004983. doi: 10.1371/journal.ppat.1004983
Orrú, C. D., Wilham, J. M., Hughson, A. G., Raymond, L. D., McNally, K. L., Bossers, A., et al. (2009). Human variant Creutzfeldt-Jakob disease and sheep scrapie PrPres detection using seeded conversion of recombinant prion protein. Protein Eng. Des. Sel. 22, 515–521. doi: 10.1093/protein/gzp031
Orrú, C. D., Wilham, J. M., Raymond, L. D., Kuhn, F., Schroeder, B., Raeber, A. J., et al. (2011). Prion disease blood test using immunoprecipitation and improved quaking-induced conversion. mBio 2:e00078-11. doi: 10.1128/mBio.00078-11
Orrú, C. D., Yuan, J., Appleby, B. S., Li, B., Li, Y., Winner, D., et al. (2017). Prion seeding activity and infectivity in skin samples from patients with sporadic Creutzfeldt-Jakob disease. Sci. Transl. Med. 9:eaam7785. doi: 10.1126/scitranslmed.aam7785
Ortega-Cubero, S., Luquín, M. R., Domínguez, I., Arbizu, J., Pagola, I., Carmona-Abellán, M. M., et al. (2013). Structural and functional neuroimaging in human prion diseases. Neurología 28, 299–308. doi: 10.1016/j.nrleng.2011.03.012
Oshita, M., Yokoyama, T., Takei, Y., Takeuchi, A., Ironside, J. W., Kitamoto, T., et al. (2016). Efficient propagation of variant Creutzfeldt-Jakob disease prion protein using the cell-protein misfolding cyclic amplification technique with samples containing plasma and heparin. Transfusion 56, 223–230. doi: 10.1111/trf.13279
Otto, M., Wiltfang, J., Cepek, L., Neumann, M., Mollenhauer, B., Steinacker, P., et al. (2002). Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 58, 192–197. doi: 10.1212/WNL.58.2.192
Otto, M., Wiltfang, J., Schütz, E., Zerr, I., Otto, A., Pfahlberg, A., et al. (1998). Diagnosis of Creutzfeldt-Jakob disease by measurement of S100 protein in serum: prospective case-control study. Br. Med. J. 316, 577–582. doi: 10.1136/bmj.316.7131.577
Otto, M., Wiltfang, J., Tumani, H., Zerr, I., Lantsch, M., Kornhuber, J., et al. (1997). Elevated levels of tau-protein in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Neurosci. Lett. 225, 210–212. doi: 10.1016/S0304-3940(97)00215-2
Parchi, P., De Boni, L., Saverioni, D., Cohen, M. L., Ferrer, I., Gambetti, P., et al. (2012). Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 124, 517–529. doi: 10.1007/s00401-012-1002-8
Parchi, P., Giese, A., Capellari, S., Brown, P., Schulz-Schaeffer, W., Windl, O., et al. (1999). Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 46, 224–233. doi: 10.1002/1531-8249(199908)46:2<224::AID-ANA12<3.0.CO;2-W
Pastrana, M. A., Sajnani, G., Onisko, B., Castilla, J., Morales, R., Soto, C., et al. (2006). Isolation and characterization of a proteinase K-sensitive PrPSc fraction. Biochemistry 45, 15710–15717. doi: 10.1021/bi0615442
Peden, A. H., McGuire, L. I., Appleford, N. E. J., Mallinson, G., Wilham, J. M., Orrú, C. D., et al. (2012). Sensitive and specific detection of sporadic Creutzfeldt-Jakob disease brain prion protein using real-time quaking-induced conversion. J. Gen. Virol. 93, 438–449. doi: 10.1099/vir.0.033365-0
Peggion, C., Bertoli, A., and Sorgato, M. C. (2017). Almost a century of prion protein(s): from pathology to physiology, and back to pathology. Biochem. Biophys. Res. Commun. 483, 1148–1155. doi: 10.1016/j.bbrc.2016.07.118
Peoc’h, K., Schröder, H. C., Laplanche, J. L., Ramljak, S., and Müller, W. E. G. (2001). Determination of 14-3-3 protein levels in cerebrospinal fluid from Creutzfeldt-Jakob patients by a highly sensitive capture assay. Neurosci. Lett. 301, 167–170. doi: 10.1016/S0304-3940(01)01619-6
Properzi, F., and Pocchiari, M. (2013). Identification of misfolded proteins in body fluids for the diagnosis of prion diseases. Int. J. Cell Biol. 2013:839329. doi: 10.1155/2013/839329
Prusiner, S. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144. doi: 10.1126/science.6801762
Prusiner, S. B. (1991). Molecular biology of prion diseases. Science 252, 1515–1522. doi: 10.1126/science.1675487
Prusiner, S. B. (1998). Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383. doi: 10.1073/pnas.95.23.13363
Prusiner, S. B., and Hsiao, K. K. (1994). Human prion diseases. Ann. Neurol. 35, 385–395. doi: 10.1002/ana.410350404
Puoti, G., Bizzi, A., Forloni, G., Safar, J. G., Tagliavini, F., and Gambetti, P. (2012). Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 11, 618–628. doi: 10.1016/S1474-4422(12)70063-7
Raymond, G. J., Race, B., Orrú, C. D., Raymond, L. D., Bongianni, M., Fiorini, M., et al. (2020). Transmission of CJD from nasal brushings but not spinal fluid or RT-QuIC product. Ann. Clin. Transl. Neurol. 7, 932–944. doi: 10.1002/acn3.51057
Redaelli, V., Bistaffa, E., Zanusso, G., Salzano, G., Sacchetto, L., Rossi, M., et al. (2017). Detection of prion seeding activity in the olfactory mucosa of patients with Fatal Familial Insomnia. Sci. Rep. 7, 1–8. doi: 10.1038/srep46269
Renard, D., Castelnovo, G., Collombier, L., Thouvenot, E., and Boudousq, V. (2017). FDG-PET in Creutzfeldt-Jakob disease: analysis of clinical-PET correlation. Prion 11, 440–453. doi: 10.1080/19336896.2017.1387348
Requena, J. R., and Wille, H. (2017). The Structure of the Infectious Prion Protein and Its Propagation. Prog. Mol. Biol. Transl. Sci. 150, 341–359. doi: 10.1016/bs.pmbts.2017.06.009
Riek, R., Hornemann, S., Wider, G., Billeter, M., Glockshuber, R., and Wüthrich, K. (1996). NMR structure of the mouse prion protein domain PrP(121–231). Nature 382, 180–182. doi: 10.1038/382180a0
Ritchie, D. L., and Ironside, J. W. (2017). Neuropathology of Human Prion Diseases. Prog. Mol. Biol. Transl. Sci. 150, 319–339. doi: 10.1016/bs.pmbts.2017.06.011
Rossi, G., Macchi, G., Porro, M., Giaccone, G., Bugiani, M., Scarpini, E., et al. (1998). Fatal familial insomnia: genetic, neuropathologic, and biochemical study of a patient from a new Italian kindred. Neurology 50, 688–692. doi: 10.1212/WNL.50.3.688
Rossi, M., Baiardi, S., and Parchi, P. (2019). Understanding prion strains: evidence from studies of the disease forms affecting humans. Viruses 11:309. doi: 10.3390/v11040309
Rudge, P., Hyare, H., Green, A., Collinge, J., and Mead, S. (2018). Imaging and CSF analyses effectively distinguish CJD from its mimics. J. Neurol. Neurosurg. Psychiatry 89, 461–466. doi: 10.1136/jnnp-2017-316853
Saá, P., Castilla, J., and Soto, C. (2005). Cyclic amplification of protein misfolding and aggregation. Methods Mol. Biol. 2005, 53–65. doi: 10.1385/1-59259-874-9:053
Saá, P., Castilla, J., and Soto, C. (2006). Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 281, 35245–35252. doi: 10.1074/jbc.M603964200
Saborio, G. P., Permanne, B., and Soto, C. (2001). Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411, 810–813. doi: 10.1038/35081095
Safar, J., Geschwind, M., Deering, C., Didorenko, S., Sattavat, M., Sanchez, H., et al. (2005). Diagnosis of human prion disease. Proc. Natl. Acad. Sci. U.S.A. 102, 3501–3506. doi: 10.1073/pnas.0409651102
Safar, J., Wille, H., Itri, V., Groth, D., Serban, H., Torchia, M., et al. (1998). Eight prion strains have PrPSc molecules with different conformations. Nat. Med. 4, 1157–1165. doi: 10.1038/2654
Saijo, E., Hughson, A. G., Raymond, G. J., Suzuki, A., Horiuchi, M., and Caughey, B. (2016). PrP Sc -specific antibody reveals c-terminal conformational differences between prion strains. J. Virol. 90, 4905–4913. doi: 10.1128/jvi.00088-16
Sajnani, G., Silva, C. J., Ramos, A., Pastrana, M. A., Onisko, B. C., Erickson, M. L., et al. (2012). PK-sensitive PrPSc is infectious and shares basic structural features with PK-resistant PrPSc. PLoS Pathog. 8:e1002547. doi: 10.1371/journal.ppat.1002547
Sanchez-Juan, P., Green, A., Ladogana, A., Cuadrado-Corrales, N., Sáanchez-Valle, R., Mitrováa, E., et al. (2006). CSF tests in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 67, 637–643. doi: 10.1212/01.wnl.0000230159.67128.00
Sanchez-Juan, P., Sánchez-Valle, R., Green, A., Ladogana, A., Cuadrado-Corrales, N., Mitrová, E., et al. (2007). Influence of timing on CSF tests value for Creutzfeldt-Jakob disease diagnosis. J. Neurol. 254, 901–906. doi: 10.1007/s00415-006-0472-9
Sano, K., Satoh, K., Atarashi, R., Takashima, H., Iwasaki, Y., Yoshida, M., et al. (2013). Early detection of abnormal prion protein in genetic human prion diseases now possible using real-time QUIC assay. PLoS One 8:e54915. doi: 10.1371/journal.pone.0054915
Savard, M., Irani, S. R., Guillemette, A., Gosselin-Lefebvre, S., Geschwind, M., Jansen, G. H., et al. (2016). Creutzfeldt-Jakob disease-like periodic sharp wave complexes in voltage-gated potassium channel-complex antibodies encephalitis: a case report. J. Clin. Neurophysiol. 33, e1–e4. doi: 10.1097/WNP.0000000000000171
Scheckel, C., and Aguzzi, A. (2018). Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 19, 405–418. doi: 10.1038/s41576-018-0011-4
Schmitz, M., Ebert, E., Stoeck, K., Karch, A., Collins, S., Calero, M., et al. (2016). Validation of 14-3-3 protein as a marker in sporadic Creutzfeldt-Jakob Disease Diagnostic. Mol. Neurobiol. 53, 2189–2199. doi: 10.1007/s12035-015-9167-5
Schmitz, M., Villar-Piqué, A., Llorens, F., Gmitterová, K., Hermann, P., Varges, D., et al. (2019). Cerebrospinal fluid total and phosphorylated α-synuclein in patients with Creutzfeldt–Jakob Disease and synucleinopathy. Mol. Neurobiol. 56, 3476–3483. doi: 10.1007/s12035-018-1313-4
Scott, M. R., Peretz, D., Nguyen, H.-O. B., DeArmond, S. J., and Prusiner, S. B. (2005). Transmission barriers for bovine, ovine, and human prions in transgenic mice. J. Virol. 79, 5259–5271. doi: 10.1128/jvi.79.9.5259-5271.2005
Sharma, A., Bruce, K. L., Chen, B., Gyoneva, S., Behrens, S. H., Bommarius, A. S., et al. (2016). Contributions of the prion protein sequence, strain, and environment to the species barrier. J. Biol. Chem. 291, 1277–1288. doi: 10.1074/jbc.M115.684100
Smirnovas, V., Baron, G. S., Offerdahl, D. K., Raymond, G. J., Caughey, B., and Surewicz, W. K. (2011). Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 18, 504–506. doi: 10.1038/nsmb.2035
Soto, C. (2004). Diagnosing prion diseases: needs, challenges and hopes. Nat. Rev. Microbiol. 2, 809–819. doi: 10.1038/nrmicro1003
Soto, C., Anderes, L., Suardi, S., Cardone, F., Castilla, J., Frossard, M. J., et al. (2005). Pre-symptomatic detection of prions by cyclic amplification of protein misfolding. FEBS Lett. 579, 638–642. doi: 10.1016/j.febslet.2004.12.035
Soto, C., and Satani, N. (2011). The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 17, 14–24. doi: 10.1016/j.molmed.2010.09.001
Spagnolli, G., Rigoli, M., Orioli, S., Sevillano, A. M., Faccioli, P., Wille, H., et al. (2019). Full atomistic model of prion structure and conversion. PLoS Pathog. 15:e1007864. doi: 10.1371/journal.ppat.1007864
Staffaroni, A. M., Kramer, A. O., Casey, M., Kang, H., Rojas, J. C., Orrú, C. D., et al. (2019). Association of blood and cerebrospinal fluid tau level and other biomarkers with survival time in sporadic Creutzfeldt-Jakob Disease. JAMA Neurol. 76, 969–977. doi: 10.1001/jamaneurol.2019.1071
Steinacker, P., Blennow, K., Halbgebauer, S., Shi, S., Ruf, V., Oeckl, P., et al. (2016). Neurofilaments in blood and CSF for diagnosis and prediction of onset in Creutzfeldt-Jakob disease. Sci. Rep. 6:38737. doi: 10.1038/srep38737
Stoeck, K., Sanchez-Juan, P., Gawinecka, J., Green, A., Ladogana, A., Pocchiari, M., et al. (2012). Cerebrospinal fluid biomarker supported diagnosis of Creutzfeldt-Jakob disease and rapid dementias: a longitudinal multicentre study over 10 years. Brain 135, 3051–3061. doi: 10.1093/brain/aws238
Tee, B. L., Longoria Ibarrola, E. M., and Geschwind, M. D. (2018). Prion Diseases. Neurol. Clin. 36, 865–897. doi: 10.1016/j.ncl.2018.07.005
Tesar, A., Matej, R., Kukal, J., Johanidesova, S., Rektorova, I., Vyhnalek, M., et al. (2019). Clinical variability in P102L Gerstmann–Sträussler–Scheinker syndrome. Ann. Neurol. 86, 643–652. doi: 10.1002/ana.25579
The 1000 Genomes Project Consortium (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. doi: 10.1038/nature11632
Thomas, J. G., Chenoweth, C. E., and Sullivan, S. E. (2013). Iatrogenic Creutzfeldt-Jakob disease via surgical instruments. J. Clin. Neurosci. 20, 1207–1212. doi: 10.1016/j.jocn.2013.01.007
Thompson, A., Anastasiadis, P., Druyeh, R., Whitworth, I., Nayak, A., Nihat, A., et al. (2020). Evaluation of plasma tau and neurofilament light chain biomarkers in a 12-year clinical cohort of human prion diseases. medRxiv [Preprint]. doi: 10.1101/2020.07.27.20157594
Thompson, A. G. B., Luk, C., Heslegrave, A. J., Zetterberg, H., Mead, S. H., Collinge, J., et al. (2018). Neurofilament light chain and tau concentrations are markedly increased in the serum of patients with sporadic Creutzfeldt-Jakob disease, and tau correlates with rate of disease progression. J. Neurol. Neurosurg. Psychiatry 89, 955–961. doi: 10.1136/jnnp-2017-317793
Turner, M. L., and Ironside, J. W. (1998). New-variant Creutzfeldt-Jakob disease: the risk of transmission by blood transfusion. Blood Rev. 12, 255–268. doi: 10.1016/S0268-960X(98)90007-8
Uttley, L., Carroll, C., Wong, R., Hilton, D. A., and Stevenson, M. (2020). Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 20, e2–e10. doi: 10.1016/S1473-3099(19)30615-2
Vallabh, S. M., Minikel, E. V., Williams, V. J., Carlyle, B. C., McManus, A. J., Wennick, C. D., et al. (2020). Cerebrospinal fluid and plasma biomarkers in individuals at risk for genetic prion disease. BMC Med. 18:140. doi: 10.1186/s12916-020-01608-8
Vallabh, S. M., Nobuhara, C. K., Llorens, F., Zerr, I., Parchi, P., Capellari, S., et al. (2019). Prion protein quantification in human cerebrospinal fluid as a tool for prion disease drug development. Proc. Natl. Acad. Sci. U.S.A. 116, 7793–7798. doi: 10.1073/pnas.1901947116
Van Everbroeck, B., Quoilin, S., Boons, J., Martin, J. J., and Cras, P. (2003). A prospective study of CSF markers in 250 patients with possible Creutzfeldt-Jakob disease. J. Neurol. Neurosurg. Psychiatry 74, 1210–1214. doi: 10.1136/jnnp.74.9.1210
Villar-Piqué, A., Schmitz, M., Lachmann, I., Karch, A., Calero, O., Stehmann, C., et al. (2019). Cerebrospinal fluid total prion protein in the spectrum of prion diseases. Mol. Neurobiol. 56, 2811–2821. doi: 10.1007/s12035-018-1251-1
Wadsworth, J., Hill, A., Beck, J., and Collinge, J. (2003). Molecular and clinical classification of human prion disease. Br. Med. Bull. 66, 241–254. doi: 10.1093/bmb/dg66.241
Wang, Z., Manca, M., Foutz, A., Camacho, M. V., Raymond, G. J., Race, B., et al. (2019). Early preclinical detection of prions in the skin of prion-infected animals. Nat. Commun. 10:247. doi: 10.1038/s41467-018-08130-9
Wasmer, C., Lange, A., Van Melckebeke, H., Siemer, A. B., Riek, R., and Meier, B. H. (2008). Amyloid fibrils of the HET-s(218-289) prion form a β solenoid with a triangular hydrophobic core. Science 319, 1523–1526. doi: 10.1126/science.1151839
Watts, J. C., Giles, K., Patel, S., Oehler, A., DeArmond, S. J., and Prusiner, S. B. (2014). Evidence That Bank Vole PrP Is a Universal Acceptor for Prions. PLoS Pathog. 10:e1003990. doi: 10.1371/journal.ppat.1003990
Wells, G. A., Scott, A. C., Johnson, C. T., Gunning, R. F., Hancock, R. D., Jeffrey, M., et al. (1987). A novel progressive spongiform encephalopathy in cattle. Vet. Rec. 121, 419–420. doi: 10.1136/vr.121.18.419
Weydt, P., Oeckl, P., Huss, A., Müller, K., Volk, A. E., Kuhle, J., et al. (2016). Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann. Neurol. 79, 152–158. doi: 10.1002/ana.24552
Whitechurch, B. C., Welton, J. M., Collins, S. J., and Lawson, V. A. (2017). Clinical Aspects of Alzheimer’s Disease Chapter 13 Prion Diseases. Adv. Neurobiol. 15, 335–364. doi: 10.1007/978-3-319-57193-5
Wieser, H. G., Schindler, K., and Zumsteg, D. (2006). EEG in Creutzfeldt-Jakob disease. Clin. Neurophysiol. 117, 935–951. doi: 10.1016/j.clinph.2005.12.007
Wilham, J. M., Orrú, C. D., Bessen, R. A., Atarashi, R., Sano, K., Race, B., et al. (2010). Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 6:e1001217. doi: 10.1371/journal.ppat.1001217
Will, R., Ironside, J., Zeidler, M., Estibeiro, K., Cousens, S., Smith, P., et al. (1996). A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347, 921–925. doi: 10.1016/S0140-6736(96)91412-9
Will, R. G. (2003). Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br. Med. Bull. 66, 255–265. doi: 10.1093/bmb/66.1.255
Wu, L. Y., Zhan, S. Q., Huang, Z. Y., Zhang, B., Wang, T., Liu, C. F., et al. (2018). Expert consensus on clinical diagnostic criteria for fatal familial insomnia. Chin. Med. J. Engl. 131, 1613–1617. doi: 10.4103/0366-6999.235115
Zahn, R., Liu, A., Lührs, T., Riek, R., von Schroetter, C., López García, F., et al. (2000). NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. U.S.A. 97, 145–150. doi: 10.1073/pnas.97.1.145
Zanusso, G., Monaco, S., Pocchiari, M., and Caughey, B. (2016). Advanced tests for early and accurate diagnosis of Creutzfeldt-Jakob disease. Nat. Rev. Neurol. 12, 325–333. doi: 10.1038/nrneurol.2016.65
Zerr, I., Bodemer, M., Gefeller, O., Otto, M., Poser, S., Wiltfang, J., et al. (1998). Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jakob disease. Ann. Neurol. 43, 32–40. doi: 10.1002/ana.410430109
Zerr, I., Bodemer, M., Räcker, S., Grosche, S., Poser, S., Weber, T., et al. (1995). Cerebrospinal fluid concentration of neuron-specific enolase in diagnosis of Creutzfeldt-Jakob disease. Lancet 345, 1609–1610. doi: 10.1016/S0140-6736(95)90118-3
Zerr, I., Kallenberg, K., Summers, D. M., Romero, C., Taratuto, A., Heinemann, U., et al. (2009). Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 132, 2659–2668. doi: 10.1093/brain/awp191
Zerr, I., Pocchiari, M., Collins, S., Brandel, J. P., De Pedro Cuesta, J., Knight, R. S. G., et al. (2000a). Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology 55, 811–815. doi: 10.1212/WNL.55.6.811
Zerr, I., Schulz-Schaeffer, W. J., Giese, A., Bodemer, M., Schröter, A., Henkel, K., et al. (2000b). Current clinical diagnosis in Creutzfeldt-Jakob disease: identification of uncommon variants. Ann. Neurol. 48, 323–329. doi: 10.1002/1531-8249(200009)48:3<323::AID-ANA6<3.0.CO;2-5
Keywords: prion diseases, prion protein, diagnostic, cell-free conversion assay, neurodegenerative disease
Citation: Ascari LM, Rocha SC, Gonçalves PB, Vieira TCRG and Cordeiro Y (2020) Challenges and Advances in Antemortem Diagnosis of Human Transmissible Spongiform Encephalopathies. Front. Bioeng. Biotechnol. 8:585896. doi: 10.3389/fbioe.2020.585896
Received: 21 July 2020; Accepted: 28 September 2020;
Published: 20 October 2020.
Edited by:
Jesus R. Requena, University of Santiago de Compostela, SpainReviewed by:
Wenquan Zou, Case Western Reserve University, United StatesFranc Llorens, Center for Biomedical Research on Neurodegenerative Diseases (CIBERNED), Spain
Copyright © 2020 Ascari, Rocha, Gonçalves, Vieira and Cordeiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tuane C. R. G. Vieira, dHVhbmVAYmlvcW1lZC51ZnJqLmJy; Yraima Cordeiro, eXJhaW1hQHBoYXJtYS51ZnJqLmJy; eXJhaW1hY29yZGVpcm9AZ21haWwuY29t