Abstract
Rheumatic heart valve disease (RHVD) is a post-infectious sequel of acute rheumatic fever resulting from an abnormal immune response to a streptococcal pharyngitis that triggers valvular damage. RHVD is the leading cause of cardiovascular death in children and young adults, mainly in women from low and middle-income countries. It is known that long-term inflammation and high degree of fibrosis leads to valve dysfunction due to anatomic disruption of the valve apparatus. However, since public and private investments in RHVD studies are practically inexistent the number of publications is scarce. This disease shows different natural history and clinical presentations as compared to other degenerative heart valve diseases. Although more than five decades passed after the pioneering studies on the pathogenesis of RHVD, it is still unclear how self-tolerance mechanisms fail in this disease, and how humoral and cellular inflammatory responses are interconnected. Despite that pathological mechanisms have been already proposed for RHVD, none of them are able to explain the preferential involvement of the mitral valve. This review focuses on pathophysiology and underlying mechanisms of RHVD.
Introduction
Rheumatic Heart Valve Disease
Heart valve disease (HVD) is generic term that includes several etiologic entities with different pathophysiologic mechanisms that lead to anatomic disruption of the valve apparatus (1). Functional abnormalities due to alteration in matrix architecture and cellular components impair the proper directionality of blood flow through the heart chambers resulting in heart failure (2).
Overall, HVD are slowly progressive disorders that affect mainly the aging population (>65 years), reaching epidemic proportions worldwide (3). Despite increased life expectancy over the last several decades, calcific aortic valve disease (CAVD), and degenerative mitral valve disease are the two most common types of non-rheumatic valve disease (4). Due to vast health impact in developed world, CAVD has sustained significant research interest and a greater number of studies as compared to the other valvular disorders.
On the other hand, in low and middle-income countries, rheumatic heart valve disease (RHVD) is the leading cause of cardiovascular death in children and young adults (5, 6). Even though the observed progress in research on RHVD pathogenesis with findings that have challenged a variety of historical paradigms, a number of key scientific questions remain. Public and private investments in RHVD studies are low and therefore the number of publications is limited. RHVD could be viewed as a marker of inequality and social injustice for countless populations living in poverty. Over the last years, defense groups are making efforts for identification and removal of barriers to the translation of existing knowledge into policy, programs, and practice to provide high-quality care for patients with RHVD (7).
HVD require a substantial allocation of health resources, and there is no effective drug therapy to prevent or treat these pathologies. In addition, these valvular diseases show different natural histories and clinical presentations. This review focuses on pathophysiology and underlying mechanisms of RHVD.
Pathology of RHVD
Gross Pathomorphological Findings
RHVD is a harmful post-infectious sequel of acute rheumatic fever (ARF) resulting from an abnormal immune response to a streptococcal pharyngitis that triggers valvular damage (8). The first episode of ARF is often associated with only mild manifestations and can occur at any age in genetically predisposed individuals (9). Recurrent Streptococcus pyogenes infection, which boosts immune response leads to RHVD. Thus, although RHVD first occurs in childhood, its incidence peaks in adulthood, usually between the ages of 25–45 years (10). In developing countries, social determinants of the disease such as inadequate housing, lack of access to primary health care, education, and availability of cardiologic diagnostic tools hamper the diagnosis. Most of children are undiagnosed and therefore do not receive antibiotic as secondary prophylaxis to prevent new S. pyogenes increasing their chances to develop RHVD. Women and girls may experience less access to primary and secondary prophylaxis as compared with men and boys in low-income countries, and this could also contribute to differences in RHVD rates between females and males (9). In addition, women have a closer involvement in childcare and therefore higher S. pyogenes exposure.
The mitral valve is affected in almost all RHVD cases, with regurgitation in the early stages, and stenosis in later stages (11). RHVD can also affect aortic valves, however, calcific degeneration is an outcome usually associated with aortic valve. During initial phase of rheumatic disease, echocardiographic exams can detect small verrucous nodules caused by the presence of thrombi along the lines of heart valve closure. These lesions are not able to produce leaflet destruction and therefore valve function is relatively normal. On the other hand, development of long-term inflammation after single or multiple episodes of rheumatic fever can lead to valve dysfunction in untreated genetically predisposed patients. As general pathomorphological findings, mitral valve specimens from patients at end state disease are thick and stiff due to a high degree of fibrosis (Figures 1A,B). As this process stretches over decades, different morphological changes dominate the various phases. While leaflets are usually minimally fibrose and pliable in three quarters of patients younger than 30 years of age, they are scared and ridged in two thirds of patients older than 40 years. Different morphological manifestations also lead to different clinical symptoms. While chordal shortening is dominant in 90% of patients with mitral stenosis, it only occurs in 3% of patients with pure mitral regurgitation (MR). Annular dilatation is found in 90% of patients with pure MR but only in 30% of cases with pure stenosis. In most late cases, the valve commissures are fused and often endothelium surface erosion is observed. Chordae tendineae show fusion and shortening (Figure 1B), which may reduce subvalvular chordal space (12, 13). Calcification occurs in some cases of RHVD (Figures 1C,D), however fibrosis and inflammation are major findings (Figures 2A–C) (14, 15). Accumulating studies had shown that lipids may trigger vascular calcification associated with atherosclerosis. Therefore, it is possible that RHVD patients who develop valve calcification have altered cholesterol profile. It still remains a gap of knowledge in RHVD research.
Figure 1
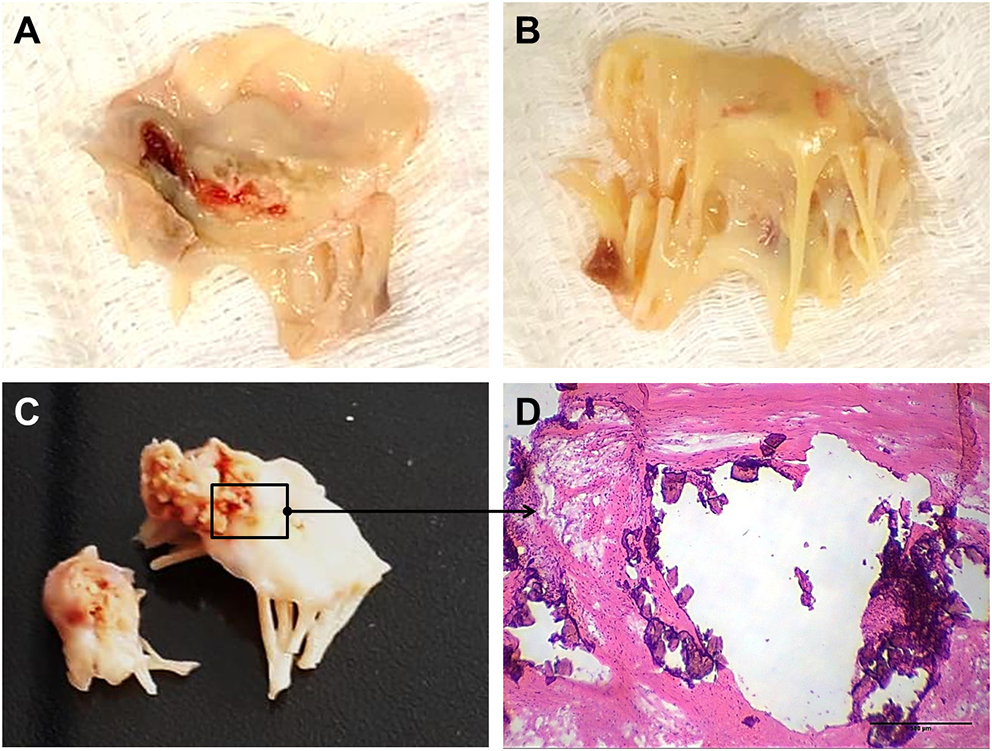
Gross pathology and histological aspects of rheumatic mitral valve at the end stage of RHVD. (A,B) Atrial and ventricular sides of mitral valves excised from female, 49 year-old patient, showing thick leaflets with retraction. (C) Mitral valve excised from male, 61 year-old, showing calcification. (D) Representative Hematoxylin and Eosin staining of anterior mitral valve leaflet showing presence of nodular calcification. Scale bar = 500 μm.
Figure 2
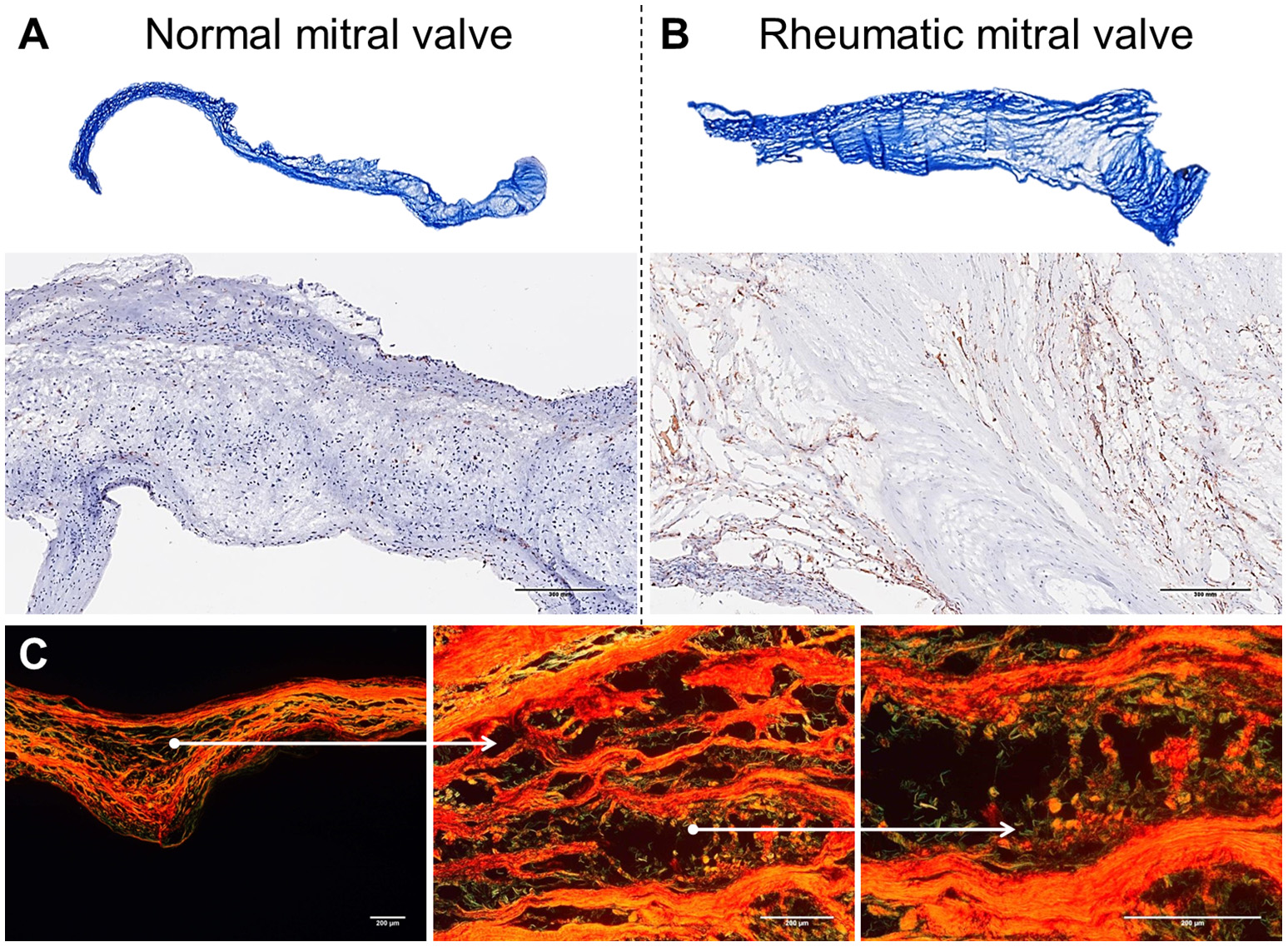
Histological comparison between normal mitral valves and rheumatic mitral valves. (A) Masson trichrome staining showing focal fibrosis and thickening of rheumatic mitral valves. Representative images are shown. (B) Representative immunohistochemistry staining for CD45+ cells evidencing a high frequency of leukocytes. Scale bars = 300 mm. (C) Picrosirius red staining showing uneven collagen deposition. Representative images are shown. Scale bars = 200 μm.
Mechanisms of Mitral Valve Disease
Immune Response in RHVD
Mitral valves are populated by two major types of cells: valvular endothelial cells (VEC), covering the leaflets, on both atrial and ventricular sites; and valvular interstitial cells (VIC), quiescent fibroblast-like cells, which are important in the homeostatic remodeling of matrix constituents (16–19). In disease state, cell composition shifts toward contractile and collagen-producing myofibroblast-like cells leading to fibrotic changes and stiffness of leaflets (Figure 2A) (19). Chronic inflammatory processes are dominant in RHVD resulting in accelerated loss of valve function (Figure 2B). Mitral valve is composed of three layers of specialized connective tissue between the two endothelial layers and its architecture substantially changes during RHVD progression. Anatomical features of mitral valve can be associated with its preferential involvement in this disorder. However, the underlying mechanisms of mitral valve predominant involvement in RHVD are unknown.
Studies related to immunopathogenesis of rheumatic fever as well as of the RHVD have been conducted since 1960 when the presence of autoantibodies in the serum of patients with throat infection by group A β-hemolytic Streptococci has been demonstrated (20). Although more than five decades passed after the pioneering studies on the pathogenesis of RHVD, little progress has been made in respect of the cellular and molecular aspects, which lead to the destruction of the valve tissue. In RHVD patients, the generation of an antibacterial immune response starts in the pharyngeal epithelium by innate immune components such as neutrophils, macrophages, and dendritic cells. These cells recognize and process bacterial antigens and present them to B lymphocytes, culminating in the production of immunoglobulins that are able to recognize epitopes in several host sites and activate T cells.
Still a mystery the mechanisms involved in the loss of self-tolerance in RHVD and how immune system target heart valvular tissue, especially mitral valve. Autoimmune valvular carditis are mainly described in literature as associated with human rheumatic conditions, however, it is possible that autoreactive antibodies can be associated with pathology of other HVD, including CAVD. Ectopic calcification and autoimmunity has been also explored in atherosclerosis (21, 22).
Multiple bacterial antigens are involved in RHVD damage. M, T and R proteins and N-acetylglucosamine (GlcNac), a group A β-hemolytic Streptococci carbohydrate (GAS), are the main epitopes described to be associated with molecular mimicry. These molecules share structural similarity with host cardiac myosin, laminin, vimentin, and tropomyosin. Many studies point to M protein as the most virulent protein (23, 24). Although myosin is present in the myocardium but not in the valvular tissue, anti-myosin antibodies respond against GlcNac epitope due to similar structures of common alpha-helical sequences and glycosylated proteins (25). It is assumed that myosin is an intracellular protein and therefore immunologically inaccessible, and thus does not participate as the initial target of cross-reactive antibodies. However, after initial mild endothelial breakdown, intracellular epitopes may contribute to robust amplification of the immune response due to increased availability of binding sites for anti-myosin antibodies.
Investigations have highlighted an important participation of GlcNac antigens in RHVD pathogenesis, since persistent levels of anti-GAS antibodies were correlated with valvulitis. It is also observed that after valve replacement the serum levels of anti-GAS were reduced (23, 26). In addition, unlike to some of the cross-reactive intracellular proteins, GlcNac are cell-surface antigens that are more exposed/accessible to antibody recognition (27). In non-rheumatic valve disease, the triggering factors leading to cardiovascular calcification, as in CAVD are still under investigation (22).
Although pan-carditis occurs in the early disease stages, it is reversible, and only the valvular damage is permanent, especially in mitral valve. Mitral valve damage is initiated by circulating autoantibodies that bind to the endothelial surface of the valves, leading to increased expression of vascular cell adhesion protein 1 (VCAM-1). The activated endothelium facilitates the infiltration of T lymphocytes into the valvular subendothelium at the endocardium site, leading to edema and elongation of the chordae tendinae (28, 29). Due to tissue injury, components of ECM are exposed and anti-collagen antibodies are produced. These antibodies can deposit in the valve contributing to pro-inflammatory environment. All these changes cause the heart valves to be a vulnerable immune-privileged site for injury. The role of anti-collagen antibodies will be addressed in more details below.
It is important to comment that typically the first streptococcus throat infection does not trigger an episode of rheumatic fever. Recent studies have shown that continuous infections maintain the germinal center reaction and affinity for antibody maturation (30, 31). As such, preexisting immune complexes would capture more immunoglobulin leading to amplification of the immune response, which further favors the recognition of several self-antigens and propagate tissue damage. Thus, repeated infections feed the disease onset (32). To date, no evidence exists that the isolated presence of valve-reactive antibodies in the serum of RHVD patients is sufficient to produce the valve lesion, suggesting the importance of cellular response besides humoral components. In addition, autoantibodies are often found in patients after uncomplicated streptococcal pharyngitis and in healthy individuals.
Humoral and cellular response acts together in autoimmune diseases. It is known that women produce more immunoglobulin than men, and the X chromosome contains over 1,000 genes, while the Y chromosome only has about a 100 (33). Many of the X-linked genes are relate to the immune system, such as CD40L, CXCR, OGT, FOXP3, TLR7, TLR8, IL2RG, BTK, and IL9R. Also, sex hormones can directly or indirectly affect the immune response by modulating gene expression through ERα stimulation (34). Immune cells express estrogen and androgen receptors, and engagement of these receptors affects lymphocyte responses (35). ARF usually occurs during childhood equally in males and females, however, RHVD has higher prevalence in adult women. Thus, is likely that endogenous hormones are key mediators of disease progression. Overexpressed X-linked immune genes and estradiol probably act in a synergistic manner, leading to a greater female-biased predisposition in RHVD.
Emerging evidence suggests that the transfer of T-cell lines from M-protein vaccinated Lewis rats to naïve animals can induce valvulitis in recipient animals. These data support that T cells are sufficient to induce inflammation, not requiring the presence of cross-reactive antibodies to trigger valvulitis (36). Although some studies showed that the presence of antibodies is not crucial in triggering RHVD pathogenesis, it is important to emphasize that the ability of antibodies to become self-reactive will depend on the combination of factors such as genetic background, recurrence of infections, and strain virulence. These variables make it even more challenging to fully understand the mechanisms associated with the development of valve lesions.
The inflammatory infiltrate described in the rheumatic mitral valve of patients in the end state of disease are predominantly composed of mononuclear cells (Figure 3C), mainly helper (Th) - CD4, and cytotoxic—T CD8 lymphocytes, macrophages and B cells (37, 38). The effector function and therefore the contribution of these cells in the pathogenesis of RHVD is largely associated with the profile of cytokines and other soluble mediators produced by them that lead to differentiation of VICs to activated collagen-producing myofibroblast (39).
Figure 3
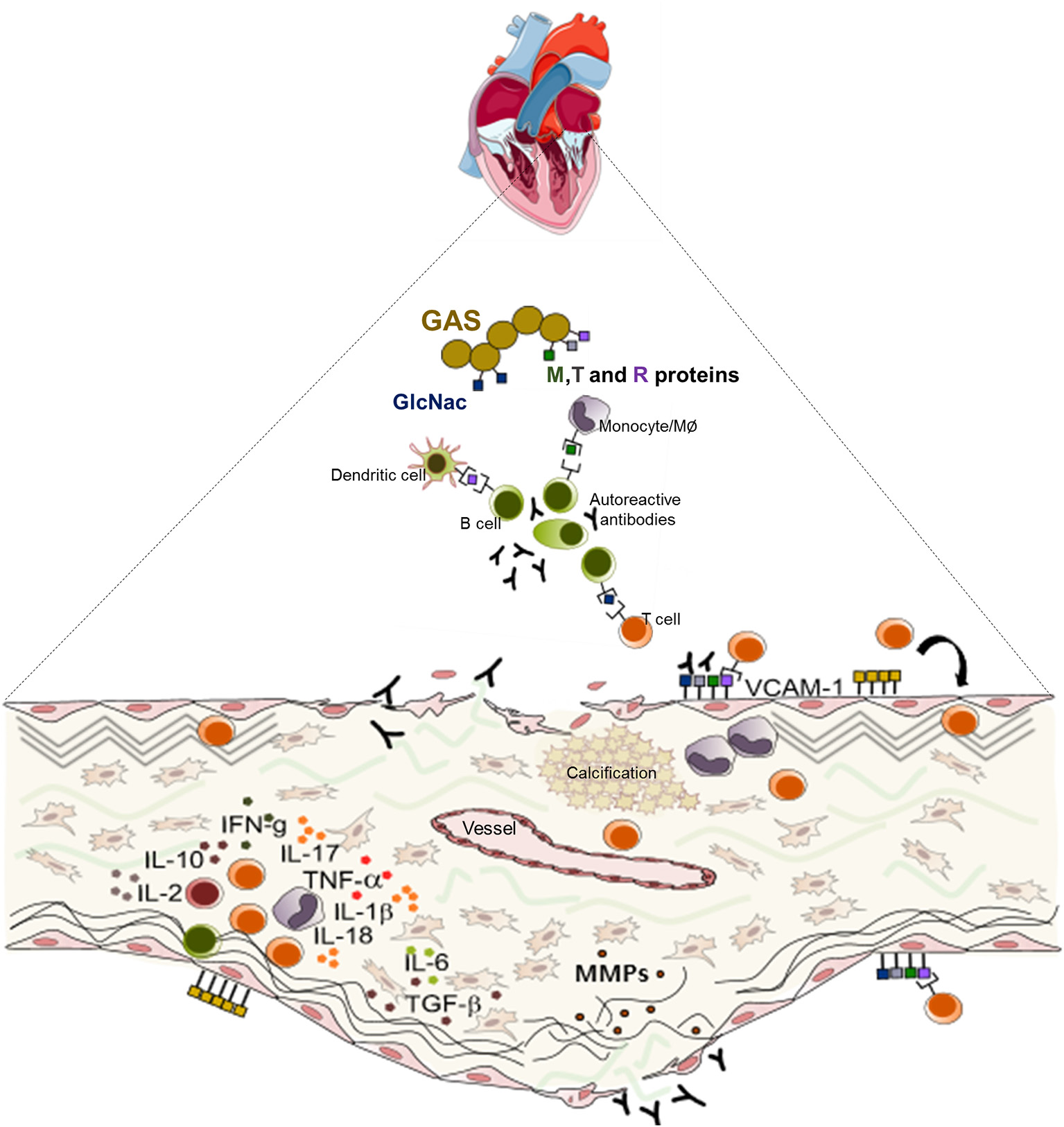
Schematic representation of the mechanisms of the pathogenesis of rheumatic heart valve disease. Following group A Streptococcus (GAS) invasion of the pharyngeal epithelium, GAS recognize and process bacterial antigens and present them to B lymphocytes. Activated B cells produce antibodies that are able to recognize epitopes in several sites in the host and also activate T lymphocytes. In the heart, cross reactive T cell clones and antibodies act against heart valve constituents leading to an intense inflammatory process culminating in valve dysfunction.
So far, T CD4+ lymphocytes have been the most investigated cell population in RHVD since they are present at high frequency in the inflammatory infiltrates and have high cross reactivity against cardiac myosin epitopes. In addition, these cells are able to differentiate into various repertoires of subpopulations producing diverse cytokines leading to the development of different degrees of cross-reactivity (40–43).
Th1 cytokines are pro-inflammatory soluble mediators involved in host defense and also crucial to autoimmunity. In the RHVD context, some studies have evaluated the expression of different inflammatory cytokines in the valvular tissue, however cell sources of these cytokines and contributions of each cellular subpopulation to the cytokine production remain unknown.
Among the Th1 cytokines, TNF-α, IFN-γ, IL-1, IL-2, and IL-6 have already been shown to be associated with disease progression. In vitro studies have demonstrated that TNF-α exhibits a high chemotactic potential promoting cell attraction to the site of inflammation, whereas IFN-γ can induce processing and presentation of autoantigens (38, 44, 45). IL-1 is a key cytokine in autoimmune disease and has also been shown to be associated with inflammatory damage, especially in the acute phase of rheumatic disease (46, 47). In a Brazilian population study, polymorphism in IL-Ra and IL-6 genes was associated with susceptibility to RHVD (48, 49). IL-6 was linked to B cell antibody production and suggested to be involved in RHVD pathogenesis (50). Systemic cytokine levels were associated with severity of RHVD and co-regulated expression of IL-6 and TNF-α was associated with severe valve dysfunction (51).
IL-2 is an essential cytokine initiating generation of regulatory T cells (Tregs), which play a vital role in the maintenance of immune tolerance. Low levels of IL-2 and deficiency of circulating Tregs were associated with rheumatic mitral valve disease (44, 52, 53). Moreover, patients who presented multiple valve impairment showed a greater deficiency in the number of Tregs (54).
Th17 cells are T CD4+ cells producing high amounts of IL-17. This cell subset plays opposite roles as compared to Tregs in autoimmune diseases. In the context of RHVD, it was shown to be associated with progression of the disease toward chronic state in an experimental model (55). Peripheral blood cells from patients with rheumatic mitral valve disease showed an increased number of Th17 cells and high serum levels of IL-17 as compared to healthy individuals (52).
Some cytokines with a typical Th2 response mediating activation and regulation responses against allergen toxins, extracellular parasites and bacteria, have also been studied in RHVD. In these patients, IL-10 is present at high levels and has a direct correlation with T CD8+ lymphocyte response. IL-10 acts as a chemoattractant for these cells and creates a favorable milieu for the growth of its precursors. On the other hand, Th2 cytokines such as IL-4 and IL-5 are present in very low concentrations or not detectable in RHVD (56–58).
The role of cytokines in the site of inflammation is still underexplored since the vast majority of studies were done using peripheral blood, and the expression of cytokines at the lesion site could be underestimated. Analysis of valve tissue is essential for the understanding of inflammatory mechanisms in situ. Studies performed in peripheral blood and valvular tissues from RHVD patients, point to a predominance of T CD4+ cells as compared to the number of T CD8+ cells. However, the proportion of circulating T CD4+ and T CD8+ cells appears to vary between stages of the disease, as evidenced by increased frequency of T CD8+ cells in patients with chronic stage disease (37, 38, 44). The role of T CD8+ cells in the pathogenesis of RHVD, however, remains scant.
B lymphocytes are present in the inflammatory infiltrates of rheumatic mitral valves. While their contribution to the pathogenesis of the disease is often associated with the production of antibodies in the early disease stages, it is likely that these cells also play a role of effector cells participating in chronic lesion development (38, 59).
Macrophages are known to play a high pathogenic role in cardiovascular diseases. A recent study showed that pro-inflammatory macrophages (M1) exhibit effector potential through activation of NLRP3 inflammasome (60) leading to the production of IL-1β and IL-18, an important pathway in the pathogenesis of rheumatic diseases (61, 62). IL-1β in turn, induces release of matrix metalloproteinases (MMPs), recruitment and proliferation of resident fibroblasts, and secretion of TGF-β and IL-6 resulting in the development of fibrosis (63).
Collagen Remodeling and Calcification Process in RHVD
More recent studies have proposed that the immune response in RHVD may not be merely related to molecular mimicry or failure of the immune system, but rather associated with collagen autoimmunity as proposed for Goodpasture and Alport syndromes (64). In these diseases, the production of autoantibodies against basement-membrane collagen (type IV) on host endothelium is the triggering step of pathological processes. In Streptococci infection, M protein binds to CB3 region of collagen IV leading to the formation of a complex that promotes conformational changes in collagen structure initiating an anti-collagen response (65–67). Thus, a ubiquitous protein can become a self-antigen that contributes to imbalance between collagen deposition and collagen degradation, culminating in subsequent fibrosis of the valve apparatus in the RHVD.
Mitral valves of rheumatic patients have a higher deposition of collagens Type I and Type III, evidence of fibrosis, when compared to non-rheumatic mitral valve controls (Figures 2A–C) (68). Among numerous cytokines involved in the inflammatory process in RHVD, the high expression of TGF-β has been shown to be positively associated with valvular fibrosis (69) by contributing to myofibroblast activation and collagen production (39).
MMPs are a major group of proteases that regulate matrix remodeling during fibrogenic process accompanying chronic inflammation. MMP-1 has a high affinity to fibrillar collagens and is able to initiate collagenolysis. High concentrations of MMP-1 in patients' plasma, and gene polymorphism are contributing risk factors for RHVD (68, 70). The majorities of studies addressing the role of MMPs were performed in human plasma and myocardium tissues during acute episodes of rheumatic fever and are scarce in the chronic disease phase, particularly in valves (71–73).
Proinflammatory MMPs also play a role in the modulation of calcification by elastin degradation. Exposure of elastin and matrix-bound cytokine generation after tissue injury create a milieu, which promotes the smooth muscle cell changes into osteoblastic phenotype (74, 75). Calcification is a very common finding in rheumatic mitral valves, however, the cellular mechanisms responsible for the calcification in RHVD are not well-understood (Figure 3B). Previous study reported that mineralization occurs in areas of inflammation and neoangiogenesis, which express vascular endothelial growth factor (VEGF) (15). This molecule is able to regulate bone remodeling by attracting endothelial cells and by stimulating osteoblast differentiation (76). Another potential mechanism involved in mitral valve mineralization in RHVD is through calcification-competent extracellular vesicles derived from smooth muscle cells, VICs or macrophages (77–79). Calcification in RHVD seems to be triggered by inflammatory process as observed in CAVD (2, 22).
Together, the immune response triggered by pharyngeal GAS infection results in a cascade of cellular and humoral events that culminate in the production of antibodies and generate self-reactive clones of lymphocytes capable of interacting with valve components leading to leaflet tissue degeneration. Schematic presentation of the mechanisms of RHVD pathogenesis is shown in Figure 3.
Neoangiogenesis in RHVD
Neoangiogenesis is a common feature of HVD acting as a facilitator of inflammation by allowing the entry of immune cells and soluble inflammatory factors into the valvular tissue. Besides, vascular networks promote the weakening of valve tissue by changing the normal architecture (80, 81). A variety of growth factors can regulate angiogenesis, including vascular endothelial growth factor (VEGF)-A and MMPs that degrade fibrillar collagen or proteoglycan proteins allowing endothelial cells sprouting vessels by migration (82). Thus, angiogenesis besides contributing to tissue remodeling also can compromise mechanical stability of extracellular matrix. For the first time, we shown that mitral valves from patients with RHVD show an abundant neovascular network characterized by accumulation of large immature vessels, which is lacking in CAVD tissue (Figures 4A,B).
Figure 4
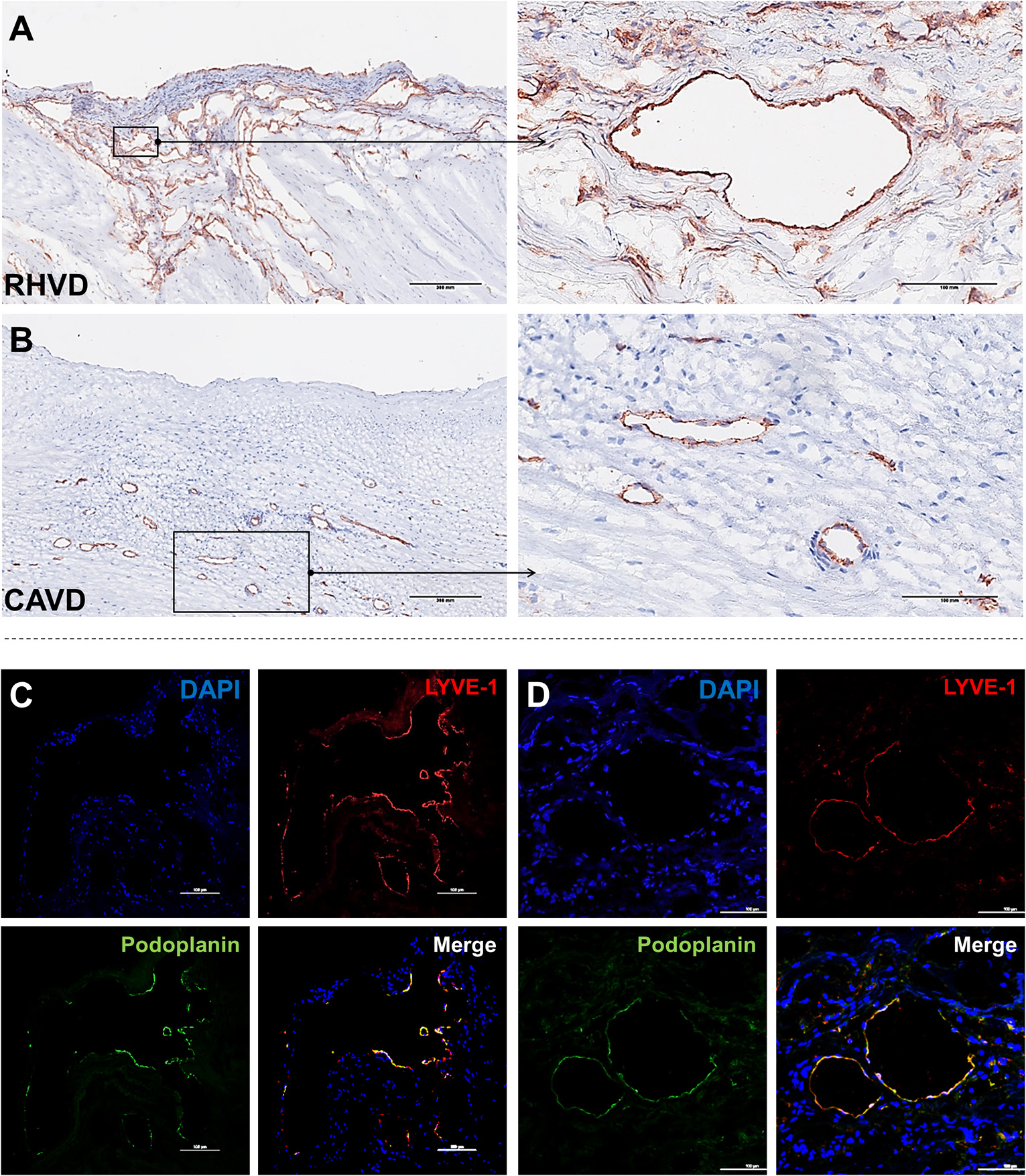
Histological comparison between neoangiogenesis in rheumatic mitral valve and calcific aortic valve. (A,B) Representative immunohistochemistry image for CD31+ staining evidencing presence of immature vessels in RHVD mitral valve and calcific aortic valve interstitium. Scale bars = 300 mm. (C) Representative immunofluorescence image showing cell co-expressing LYVE-1 and podoplanin demonstrating presence of lymphatic vessels in rheumatic mitral valves. Scale bars = 100 μm.
Besides blood vessels, lymphatic vasculature network was also observed in heart valves from patients with end stage RHVD as detected by colocalization of lymphatic endothelial cell receptor (LYVE-1) and podoplanin well-known markers of lymphatic endothelial cells (83) (Figures 4C,D). These vessels play role in lymph transport, tissue interstitial fluid absorption and serve as an entrance point for immune cells, favorable for optimal tissue function. and homeostasis (84). Autopsy results of adult heart valves show that lymphatic vessels in pathologic conditions such as RHVD present in a high (83). It is likely that neolymphogenesis is beneficial in the early phases of disease to maintain tissue homeostasis, but if become uncontrolled during the disease progression, may lead to pathological maladaptation. Persistent local inflammation impairs lymphatic contraction causing altered fluid transport (85). In addition, accumulation of collagen and fibrogenic molecules produced by smooth muscle cells and fibroblasts into the perilymphatic space causes capillary fibrosis and impairs vascular function (86). Thus, although lymphatic vessels are accumulating in lesions in large numbers, they are not able to exert their function properly.
In addition to growth factors and cytokines, hormones also participate in regulating angiogenesis. Estradiol influences hyaluronic acid synthesis, a major ECM ligand for the LYVE-1, suggesting an indirect effect on lymphatic homeostasis (87, 88). Also, while estrogen is described to reduce cardiovascular risk in women, it has been reported as a risk factor for disorders of lymphatic vascular system. Recently it has been shown that estrogen receptor alpha (ERα) directly regulates lymphoangiogenic genes promoting lymphatic endothelial cell migration and sprouting (89). Thus, excessive neolymphoangiogenesis observed in rheumatic mitral valves obtained predominantly from female patients, could be induced by estrogen which in turn can aggravate vessel sprouting in association with proinflammatory milieu.
Final Considerations
No therapies are available to prevent or treat RHVD. Antibiotic prophylaxis is given to prevent repetitive episodes of ARF, and potentially limit the disease progression to severe valve dysfunction. However, there is no robust evidence of efficacy of secondary antibiotic prophylaxis in preventing recurrences of ARF (9). Additionally, there is no treatment to alter the likelihood or the severity of RHVD after an episode of ARF (8). Corticosteroids or intravenous immunoglobulins were tested in clinical trials to reduce the risk of heart valve lesions in patients with ARF, however, little evidence of benefit was found (90). Since RHVD results from an abnormal immune response, therapies targeting immune system could be more effective to avert valvular damage. Therefore, more research is needed to find specific immune components associated with the RHVD pathogenesis that will provide a more precise and effective therapeutical interventions to treat this devastating condition.
Statements
Author contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
EA was supported by National Institutes of Health (NIH) grants R01HL 136431, R01HL 141917, and R01HL 147095.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Boudoulas KD Ravi Y Garcia D Saini U Sofowora GG Gumina RJ et al . Type of valvular heart disease requiring surgery in the 21st century: mortality and length-of-stay related to surgery. Open Cardiovasc Med J. (2013) 7:104–9. 10.2174/1874192420130902001
2.
Yutzey KE Demer LL Body SC Huggins GS Towler DA Giachelli CM et al . Calcific aortic valve disease: a consensus summary from the alliance of investigators on calcific aortic valve disease. Arterioscler Thromb Vasc Biol. (2014) 34:2387–93. 10.1161/ATVBAHA.114.302523
3.
Iung B Baron G Butchart EG Delahaye F Gohlke-Barwolf C Levang OW et al . A prospective survey of patients with valvular heart disease in Europe: The Euro Heart Survey on Valvular Heart Disease. Eur Heart J. (2003) 24:1231–43. 10.1016/S0195-668X(03)00201-X
4.
Nkomo VT Gardin JM Skelton TN Gottdiener JS Scott CG Enriquez-Sarano M . Burden of valvular heart diseases: a population-based study. Lancet. (2006) 368:1005–11. 10.1016/S0140-6736(06)69208-8
5.
Watkins DA Johnson CO Colquhoun SM Karthikeyan G Beaton A Bukhman G et al . Global, regional, and national burden of rheumatic heart disease, 1990-2015. N Engl J Med. (2017) 377:713–22. 10.1056/NEJMoa1603693
6.
Sliwa K Zilla P . Rheumatic heart disease: the tip of the iceberg. Circulation. (2012) 125:3060–2. 10.1161/CIRCULATIONAHA.112.114199
7.
Zilla P Bolman RM Yacoub MH Beyersdorf F Sliwa K Zuhlke L et al . The cape town declaration on access to cardiac surgery in the developing world. Cardiovasc J Afr. (2018) 29:256–9. 10.5830/CVJA-2018-046
8.
Karthikeyan G Guilherme L . Acute rheumatic fever. Lancet. (2018) 392:161–74. 10.1016/S0140-6736(18)30999-1
9.
Carapetis JR Beaton A Cunningham MW Guilherme L Karthikeyan G Mayosi BM et al . Acute rheumatic fever and rheumatic heart disease. Nat Rev Dis Primers. (2016) 2:15084. 10.1038/nrdp.2015.84
10.
Lawrence JG Carapetis JR Griffiths K Edwards K Condon JR . Acute rheumatic fever and rheumatic heart disease: incidence and progression in the Northern Territory of Australia, 1997 to 2010. Circulation. (2013) 128:492–501. 10.1161/CIRCULATIONAHA.113.001477
11.
Marijon E Mirabel M Celermajer DS Jouven X . Rheumatic heart disease. Lancet. (2012) 379:953–64. 10.1016/S0140-6736(11)61171-9
12.
Veinot JP . Pathology of inflammatory native valvular heart disease. Cardiovasc Pathol. (2006) 15:243–51. 10.1016/j.carpath.2006.04.007
13.
Luo T Han J Meng X . Features of rheumatic mitral valves and a grading system to identify suitable repair cases in China. J Thorac Dis. (2017) 9:3138–47. 10.21037/jtd.2017.08.121
14.
Rajamannan NM Antonini-Canterin F Moura L Zamorano JL Rosenhek RA Best PJ et al . Medical therapy for rheumatic heart disease: is it time to be proactive rather than reactive?Indian Heart J. (2009) 61:14–23.
15.
Rajamannan NM Nealis TB Subramaniam M Pandya S Stock SR Ignatiev CI et al . Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation. (2005) 111:3296–301. 10.1161/CIRCULATIONAHA.104.473165
16.
Shapero K Wylie-Sears J Levine RA Mayer JE Jr Bischoff J . Reciprocal interactions between mitral valve endothelial and interstitial cells reduce endothelial-to-mesenchymal transition and myofibroblastic activation. J Mol Cell Cardiol. (2015) 80:175–85. 10.1016/j.yjmcc.2015.01.006
17.
Levine RA Hagege AA Judge DP Padala M Dal-Bianco JP Aikawa E et al . Leducq Mitral Transatlantic, Mitral valve disease–morphology and mechanisms. Nat Rev Cardiol. (2015) 12:689–710. 10.1038/nrcardio.2015.161
18.
Hinton RB Yutzey KE . Heart valve structure and function in development and disease. Annu Rev Physiol. (2011) 73:29–46. 10.1146/annurev-physiol-012110-142145
19.
Rabkin E Aikawa M Stone JR Fukumoto Y Libby P Schoen FJ . Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. (2001) 104:2525–32. 10.1161/hc4601.099489
20.
Kaplan MH . The concept of autoantibodies in rheumatic fever and in the postcommissurotomy state. Ann N Y Acad Sci. (1960) 86:974–91. 10.1111/j.1749-6632.1960.tb42854.x
21.
Sanjadi M Rezvanie Sichanie Z Totonchi H Karami J Rezaei R Aslani S . Atherosclerosis and autoimmunity: a growing relationship. Int J Rheum Dis. (2018) 21:908–21. 10.1111/1756-185X.13309
22.
Passos LSA Lupieri A Becker-Greene D Aikawa E . Innate and adaptive immunity in cardiovascular calcification. Atherosclerosis. (2020) 306:59–67. 10.1016/j.atherosclerosis.2020.02.016
23.
Ellis NM Kurahara DK Vohra H Mascaro-Blanco A Erdem G Adderson EE et al . Priming the immune system for heart disease: a perspective on group A streptococci. J Infect Dis. (2010) 202:1059–67. 10.1086/656214
24.
Raynes JM Frost HR Williamson DA Young PG Baker EN Steemson JD et al . Serological evidence of immune priming by group A Streptococci in patients with acute rheumatic fever. Front Microbiol. (2016) 7:1119. 10.3389/fmicb.2016.01119
25.
Galvin JE Hemric ME Ward K Cunningham MW . Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. J Clin Invest. (2000) 106:217–24. 10.1172/JCI7132
26.
Ayoub EM Taranta A Bartley TD . Effect of valvular surgery on antibody to the group A streptococcal carbohydrate. Circulation. (1974) 50:144–50. 10.1161/01.CIR.50.1.144
27.
Tashima Y Stanley P . Antibodies that detect O-linked beta-D-N-acetylglucosamine on the extracellular domain of cell surface glycoproteins. J Biol Chem. (2014) 289:11132–42. 10.1074/jbc.M113.492512
28.
Roberts S Kosanke S Terrence Dunn S Jankelow D Duran CM Cunningham MW . Pathogenic mechanisms in rheumatic carditis: focus on valvular endothelium. J Infect Dis. (2001) 183:507–11. 10.1086/318076
29.
Williams RV Minich LL Shaddy RE Veasy LG Tani LY . Evidence for lack of myocardial injury in children with acute rheumatic carditis. Cardiol Young. (2002) 12:519–23. 10.1017/S104795110200094X
30.
Zhang D Patel A Zhu Y Siegel A Zalcman SS . Anti-streptococcus IgM antibodies induce repetitive stereotyped movements: cell activation and co-localization with Fcalpha/mu receptors in the striatum and motor cortex. Brain Behav Immun. (2012) 26:521–33. 10.1016/j.bbi.2012.01.005
31.
Gorton D Sikder S Williams NL Chilton L Rush CM Govan BL et al . Repeat exposure to group A Streptococcal M protein exacerbates cardiac damage in a rat model of rheumatic heart disease. Autoimmunity. (2016) 49:563–70. 10.1080/08916934.2016.1217999
32.
Dudding BA Ayoub EM . Persistence of streptococcal group A antibody in patients with rheumatic valvular disease. J Exp Med. (1968) 128:1081–98. 10.1084/jem.128.5.1081
33.
Libert C Dejager L Pinheiro I . The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. (2010) 10:594–604. 10.1038/nri2815
34.
Li J McMurray RW . Effects of estrogen receptor subtype-selective agonists on autoimmune disease in lupus-prone NZB/NZW F1 mouse model. Clin Immunol. (2007) 123:219–26. 10.1016/j.clim.2007.01.008
35.
Rubtsova K Marrack P Rubtsov AV . Sexual dimorphism in autoimmunity. J Clin Invest. (2015) 125:2187–93. 10.1172/JCI78082
36.
Kirvan CA Galvin JE Hilt S Kosanke S Cunningham MW . Identification of streptococcal m-protein cardiopathogenic epitopes in experimental autoimmune valvulitis. J Cardiovasc Transl Res. (2014) 7:172–81. 10.1007/s12265-013-9526-4
37.
Kemeny E Grieve T Marcus R Sareli P Zabriskie JB . Identification of mononuclear cells and T cell subsets in rheumatic valvulitis. Clin Immunol Immunopathol. (1989) 52:225–37. 10.1016/0090-1229(89)90174-8
38.
Guilherme L Cury P Demarchi LM Coelho V Abel L Lopez AP et al . Rheumatic heart disease: proinflammatory cytokines play a role in the progression and maintenance of valvular lesions. Am J Pathol. (2004) 165:1583–91. 10.1016/S0002-9440(10)63415-3
39.
Walker GA Masters KS Shah DN Anseth KS Leinwand LA . Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. (2004) 95:253–60. 10.1161/01.RES.0000136520.07995.aa
40.
Morris K Mohan C Wahi PL Anand IS Ganguly NK . Increase in activated T cells and reduction in suppressor/cytotoxic T cells in acute rheumatic fever and active rheumatic heart disease: a longitudinal study. J Infect Dis. (1993) 167:979–83. 10.1093/infdis/167.4.979
41.
Guilherme L Cunha-Neto E Coelho V Snitcowsky R Pomerantzeff PM Assis RV et al . Human heart-infiltrating T-cell clones from rheumatic heart disease patients recognize both streptococcal and cardiac proteins. Circulation. (1995) 92:415–20. 10.1161/01.CIR.92.3.415
42.
Bhatnagar Grover A Ganguly NK . Superantigen-induced T cell responses in acute rheumatic fever and chronic rheumatic heart disease patients. Clin Exp Immunol. (1999) 116:100–6. 10.1046/j.1365-2249.1999.00853.x
43.
Fae KC da Silva DD Oshiro SE Tanaka AC Pomerantzeff PM Douay C et al . Mimicry in recognition of cardiac myosin peptides by heart-intralesional T cell clones from rheumatic heart disease. J Immunol. (2006) 176:5662–70. 10.4049/jimmunol.176.9.5662
44.
Toor D Vohra H . Immune responsiveness during disease progression from acute rheumatic fever to chronic rheumatic heart disease. Microb Infect. (2012) 14:1111–7. 10.1016/j.micinf.2012.07.003
45.
Ellis NM Li Y Hildebrand W Fischetti VA Cunningham MW . T cell mimicry and epitope specificity of cross-reactive T cell clones from rheumatic heart disease. J Immunol. (2005) 175:5448–56. 10.4049/jimmunol.175.8.5448
46.
Dinarello CA . Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. (2011) 117:3720–32. 10.1182/blood-2010-07-273417
47.
Yegin O Coskun M Ertug H . Cytokines in acute rheumatic fever. Eur J Pediatr. (1997) 156:25–9. 10.1007/s004310050545
48.
Azevedo PM Bauer R Caparbo Vde F Silva CA Bonfa E Pereira RM . Interleukin-1 receptor antagonist gene (IL1RN) polymorphism possibly associated to severity of rheumatic carditis in a Brazilian cohort. Cytokine. (2010) 49:109–13. 10.1016/j.cyto.2009.09.003
49.
Azevedo PM Merriman TR Topless RK Wilson NJ Crengle S Lennon DR . Association study involving polymorphisms in IL-6, IL-1RA, and CTLA4 genes and rheumatic heart disease in New Zealand population of Maori and Pacific ancestry. Cytokine. (2016) 85:201–6. 10.1016/j.cyto.2016.06.029
50.
Dienz O Eaton SM Bond JP Neveu W Moquin D Noubade R et al . The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J Exp Med. (2009) 206:69–78. 10.1084/jem.20081571
51.
Diamantino Soares AC Araujo Passos LS Sable C Beaton A Ribeiro VT Gollob KJ et al . Circulating cytokines predict severity of rheumatic heart disease. Int J Cardiol. (2019) 289:107–9. 10.1016/j.ijcard.2019.04.063
52.
Bas HD Baser K Yavuz E Bolayir HA Yaman B Unlu S et al . A shift in the balance of regulatory T and T helper 17 cells in rheumatic heart disease. J Invest Med. (2014) 62:78–83. 10.2310/JIM.0000000000000023
53.
Moon BI Kim TH Seoh JY . Functional modulation of regulatory T cells by IL-2. PLoS ONE. (2015) 10:e0141864. 10.1371/journal.pone.0141864
54.
Mukhopadhyay S Varma S Mohan Kumar HN Yusaf J Goyal M Mehta V et al . Circulating level of regulatory T cells in rheumatic heart disease: an observational study. Indian Heart J. (2016) 68:342–8. 10.1016/j.ihj.2015.08.009
55.
Wen Y Zeng Z Gui C Li L Li W . Changes in the expression of Th17 cell-associated cytokines in the development of rheumatic heart disease. Cardiovasc Pathol. (2015) 24:382–7. 10.1016/j.carpath.2015.07.006
56.
Rowbottom AW Lepper MA Garland RJ Cox CV Corley EG . Interleukin-10-induced CD8 cell proliferation. Immunology. (1999) 98:80–9. 10.1046/j.1365-2567.1999.00828.x
57.
MacNeil IA Suda T Moore KW Mosmann TR Zlotnik A . IL-10, a novel growth cofactor for mature and immature T cells. J Immunol. (1990) 145:4167–73.
58.
Sharma N Toor D . Interleukin-10: Role in increasing susceptibility and pathogenesis of rheumatic fever/rheumatic heart disease. Cytokine. (2017) 90:169–76. 10.1016/j.cyto.2016.11.010
59.
Bhatia R Narula J Reddy KS Koicha M Malaviya AN Pothineni RB et al . Lymphocyte subsets in acute rheumatic fever and rheumatic heart disease. Clin Cardiol. (1989) 12:34–8. 10.1002/clc.4960120106
60.
He Y Hara H Nunez G . Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. (2016) 41:1012–21. 10.1016/j.tibs.2016.09.002
61.
LaRock CN Nizet V . Inflammasome/IL-1beta responses to Streptococcal Pathogens. Front Immunol. (2015) 6:518. 10.3389/fimmu.2015.00518
62.
Yi YS . Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J Physiol Pharmacol. (2018) 22:1–15. 10.4196/kjpp.2018.22.1.1
63.
Gasse P Riteau N Vacher R Michel ML Fautrel A di Padova F et al . IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS ONE. (2011) 6:e23185. 10.1371/journal.pone.0023185
64.
Hudson BG Tryggvason K Sundaramoorthy M Neilson EG . Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med. (2003) 348:2543–56. 10.1056/NEJMra022296
65.
Dinkla K Talay SR Morgelin M Graham RM Rohde M Nitsche-Schmitz DP et al . Crucial role of the CB3-region of collagen IV in PARF-induced acute rheumatic fever. PLoS ONE. (2009) 4:e4666. 10.1371/journal.pone.0004666
66.
Dinkla K Rohde M Jansen WT Kaplan EL Chhatwal GS Talay SR . Rheumatic fever-associated Streptococcus pyogenes isolates aggregate collagen. J Clin Invest. (2003) 111:1905–12. 10.1172/JCI17247
67.
Dinkla K Sastalla I Godehardt AW Janze N Chhatwal GS Rohde M et al . Upregulation of capsule enables Streptococcus pyogenes to evade immune recognition by antigen-specific antibodies directed to the G-related alpha2-macroglobulin-binding protein GRAB located on the bacterial surface. Microbes Infect. (2007) 9:922–31. 10.1016/j.micinf.2007.03.011
68.
Banerjee T Mukherjee S Ghosh S Biswas M Dutta S Pattari S et al . Clinical significance of markers of collagen metabolism in rheumatic mitral valve disease. PLoS ONE. (2014) 9:e90527. 10.1371/journal.pone.0090527
69.
Kim L Kim DK Yang WI Shin DH Jung IM Park HK et al . Overexpression of transforming growth factor-beta 1 in the valvular fibrosis of chronic rheumatic heart disease. J Korean Med Sci. (2008) 23:41–8. 10.3346/jkms.2008.23.1.41
70.
Hu W Ye Y Yin Y Sang P Li L Wang J et al . Association of matrix metalloprotease 1, 3, and 12 polymorphisms with rheumatic heart disease in a Chinese Han population. BMC Med Genet. (2018) 19:27. 10.1186/s12881-018-0538-4
71.
Wang Z Zhou C Gu H Zheng Z Hu S . Mitral valve repair versus replacement in patients with rheumatic heart disease. J Heart Valve Dis. (2013) 22:333–9.
72.
Wang X Yang L Qi F Et Al . Myocardial matrix metalloproteinase-3 and matrix metalloproteinase inhibitor-1 expression in congestive heart failure. Zhonghua Nei Ke Za Zhi. (2002) 41:453–5.
73.
Zhao Y Zhou X Liao X Yang Z . [Expression and significance of matrix metalloproteinase-1,9, tissue inhibitor of metalloproteinase-4 and extracellular matrix metalloproteinase inducer in the myocardium of congestive heart failure in patients with rheumatic heart diseases]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. (2009) 34:790–5.
74.
Chen NX O'Neill KD Chen X Kiattisunthorn K Gattone VH Moe SM . Activation of arterial matrix metalloproteinases leads to vascular calcification in chronic kidney disease. Am J Nephrol. (2011) 34:211–9. 10.1159/000330175
75.
Qin X Corriere MA Matrisian LM Guzman RJ . Matrix metalloproteinase inhibition attenuates aortic calcification. Arterioscler Thromb Vasc Biol. (2006) 26:1510–6. 10.1161/01.ATV.0000225807.76419.a7
76.
Deckers MM van Bezooijen RL van der Horst G Hoogendam J van Der Bent C Papapoulos SE et al . Bone morphogenetic proteins stimulate angiogenesis through osteoblast-derived vascular endothelial growth factor A. Endocrinology. (2002) 143:1545–53. 10.1210/endo.143.4.8719
77.
Hutcheson JD Goettsch C Bertazzo S Maldonado N Ruiz JL Goh W et al . Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. (2016) 15:335–43. 10.1038/nmat4519
78.
New SE Goettsch C Aikawa M Marchini JF Shibasaki M Yabusaki K et al . Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res. (2013) 113:72–7. 10.1161/CIRCRESAHA.113.301036
79.
Krohn JB Hutcheson JD Martinez-Martinez E Aikawa E . Extracellular vesicles in cardiovascular calcification: expanding current paradigms. J Physiol. (2016) 594:2895–903. 10.1113/JP271338
80.
Pufe T Petersen WJ Mentlein R Tillmann BN . The role of vasculature and angiogenesis for the pathogenesis of degenerative tendons disease. Scand J Med Sci Sports. (2005) 15:211–22. 10.1111/j.1600-0838.2005.00465.x
81.
Yoshioka M Yuasa S Matsumura K Kimura K Shiomi T Kimura N et al . Chondromodulin-I maintains cardiac valvular function by preventing angiogenesis. Nat Med. (2006) 12:1151–9. 10.1038/nm1476
82.
Libby P Schonbeck U . Drilling for oxygen: angiogenesis involves proteolysis of the extracellular matrix. Circ Res. (2001) 89:195–7. 10.1161/res.89.3.195
83.
Kholova Dragneva G Cermakova P Laidinen S Kaskenpaa N Hazes T et al . Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur J Clin Invest. (2011) 41:487–97. 10.1111/j.1365-2362.2010.02431.x
84.
Petrova TV Koh GY . Biological functions of lymphatic vessels. Science. (2020) 369:eaax4063. 10.1126/science.aax4063
85.
O'Melia MJ Lund AW Thomas SN . The biophysics of lymphatic transport: engineering tools and immunological consequences. iScience. (2019) 22:28–43. 10.1016/j.isci.2019.11.005
86.
Wang Y Jin Y Mae MA Zhang Y Ortsater H Betsholtz C et al . Smooth muscle cell recruitment to lymphatic vessels requires PDGFB and impacts vessel size but not identity. Development. (2017) 144:3590–601. 10.1242/dev.147967
87.
Shu YY Maibach HI . Estrogen and skin: therapeutic options. Am J Clin Dermatol. (2011) 12:297–311. 10.2165/11589180-000000000-00000
88.
Jackson DG . Immunological functions of hyaluronan and its receptors in the lymphatics. Immunol Rev. (2009) 230:216–31. 10.1111/j.1600-065X.2009.00803.x
89.
Morfoisse F Tatin F Chaput B Therville N Vaysse C Metivier R et al . Lymphatic vasculature requires estrogen receptor-alpha signaling to protect from lymphedema. Arterioscler Thromb Vasc Biol. (2018) 38:1346–57. 10.1161/ATVBAHA.118.310997
90.
Cilliers Adler AJ Saloojee H . Anti-inflammatory treatment for carditis in acute rheumatic fever. Cochrane Database Syst Rev. (2015) 28:CD003176. 10.1002/14651858.CD003176.pub3
Summary
Keywords
rheumatic heart disease, mitral valve, inflammation, autoimmunity, pathogenesis
Citation
Passos LSA, Nunes MCP and Aikawa E (2021) Rheumatic Heart Valve Disease Pathophysiology and Underlying Mechanisms. Front. Cardiovasc. Med. 7:612716. doi: 10.3389/fcvm.2020.612716
Received
30 September 2020
Accepted
21 December 2020
Published
18 January 2021
Volume
7 - 2020
Edited by
Bruno Ramos Nascimento, Universidade Federal de Minas Gerais, Brazil
Reviewed by
Antonino S. Rubino, University of Campania Luigi Vanvitelli, Italy; Paul “Li-Hao” Huang, Washington University in St. Louis, United States
Updates
Copyright
© 2021 Passos, Nunes and Aikawa.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Aikawa eaikawa@bwh.harvard.edu
This article was submitted to Heart Valve Disease, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.